DOI:
10.1039/D3EY00020F
(Paper)
EES Catal., 2023,
1, 695-703
Hydrogen spillover in Pt5Ru1 nanoalloy decorated Ni3S2 enabling pH-universal electrocatalytic hydrogen evolution†
Received
30th January 2023
, Accepted 18th May 2023
First published on 22nd May 2023
Abstract
As a new type of clean energy, hydrogen receives extensive attention due to its zero-pollution characteristics. Hydrogen produced by electrocatalysis is extensively studied due to its convenient properties. However, most catalysts for catalyzing the hydrogen evolution reaction are noble metals, which hinders their commercialization. Herein, we design a very effective electrocatalyst of Ni3S2 decorated with a small amount of Pt5Ru1 nanoalloy supported on Ni foam (Ni3S2/Pt5Ru1@NF) to enhance its hydrogen evolution reaction kinetics by exploiting the hydrogen spillover effect between Pt5Ru1 and Ni3S2. The designed Ni3S2/Pt5Ru1@NF exhibits excellent HER performance under both acidic and alkaline conditions, achieving a current density of 10 mA cm−2 at only 13 mV and 42 mV, respectively.
Broader context
As a clean energy source with zero pollution, hydrogen has been paid more and more attention. Electrocatalytic hydrogen production is an effective and rapid synthetic method. Most electrocatalytic hydrogen production relies on noble metal catalysts, which increases the cost of hydrogen synthesis. At present, many non-noble metal catalysts have been reported for the hydrogen evolution reaction, but most of them have poor performance. In this work, we designed a Ni3S2 catalyst, which was decorated with a small amount of Pt5Ru1 nanoalloy. This structure can effectively reduce the hydrogen evolution barrier by the hydrogen spillover effect. As a highly efficient hydrogen evolution catalyst, the designed Pt5Ru1 nanoalloy decorated Ni3S2 (Ni3S2/Pt5Ru1@NF) can significantly reduce the hydrogen evolution overpotential. This work could provide theoretical guidance and experimental evidence for designing high catalytic activity and high stability hydrogen evolution electrocatalysts and give fundamental insight into the potential construction of advanced electrocatalysts. We are sure that our work is very valuable to the readers of this journal and will attract a lot of attention.
|
1. Introduction
As the world continues to transition toward carbon emission-free energy technologies, it is necessary for us to reduce the carbon emissions of the chemical production industry.1 Hydrogen, featured by high gravimetric energy density and zero-emission, holds great potential as a sustainable alternative to fossil fuels.2 Electrocatalytic water splitting is an efficient method for producing high-purity hydrogen, but this method is far from cost-competitive with commercial steam reforming for hydrogen production.3 At present, people are devoted to finding electrocatalysts with high catalytic performance and long life in extreme acid–base environments.4,5 In general, catalyst design for electrocatalytic hydrogen evolution reactions is guided by the classical volcano theory, in which the free energy of hydrogen adsorption on the catalyst surface is an important indicator.6 The theory points out that the adsorption energy for protons is neither too strong nor too weak, which should favor the protons gaining electrons at the active sites and easily leaving the surface to form hydrogen gas.7 The elements at the top of the volcano map have thus been a hot research topic. Platinum, a representative of the precious metals, has become the target of choice for the commercial electrocatalytic production of hydrogen.8 However, its high price and weak hydrogen evolution performance under alkaline conditions hinder its further application.9 Therefore, the development of electrocatalysts with no noble metal or low noble metal content is the focus of current research.
Various terrestrially abundant and inexpensive transition metal compounds have been explored, among which sulfides have been extensively studied due to their novel physicochemical properties.10 There are numerous reports on the role of sulfides in electrocatalytic hydrogen evolution, among which the Ni3S2 catalyst stands out due to its unique electronic structure.11–15 However, recent studies have shown that the inherent activity of Ni3S2 is insufficient, and its intrinsic catalytic activity is at least two orders of magnitude worse than that of noble metals,16 which may be related to the unfavorable free energy of hydrogen adsorption on the surface of Ni3S2. Many methods have been utilized to modify the Ni3S2 catalyst, including doping, introduction of vacancy defects, and interfacial engineering, but simply modifying the electronic and geometric structures of the Ni3S2 catalyst cannot effectively compensate for its intrinsically poor activity. Inspired by the phenomenon of hydrogen spillover induced by strong metal–support interactions during thermal hydrogenation, hydrogen spillover has recently emerged as a research frontier in HER electrocatalysts.17,18 This strategy utilizes the difference in the free energy of hydrogen adsorption between metals and supports to kinetically facilitate the adsorption and desorption of protons.19,20 The binary ‘metal–support’ system with the hydrogen spillover effect has great potential to boost the performance of the Ni3S2 catalyst with less metal usage.
Herein, we designed a binary ‘metal–support’ electrocatalyst based on Ni3S2 decorated with Pt and Ru nanoalloys on Ni foam (NF). By introducing a small amount of noble metal elements, the hydrogen evolution performance of Ni3S2@NF in acidic and alkaline solution can be greatly improved. Meanwhile, compared with Pt nanoparticles, the work function difference (Δϕ) between the Pt5Ru1 nanoalloy and Ni3S2 substrate is smaller, which effectively prevents charge accumulation at the interface and enhances the hydrogen overflow process. The unique structure of Ni3S2/Pt5Ru1@NF provides a new idea for designing high catalytic activity and high stability hydrogen evolution electrocatalysts.
2. Results and discussion
Microstructure characterization
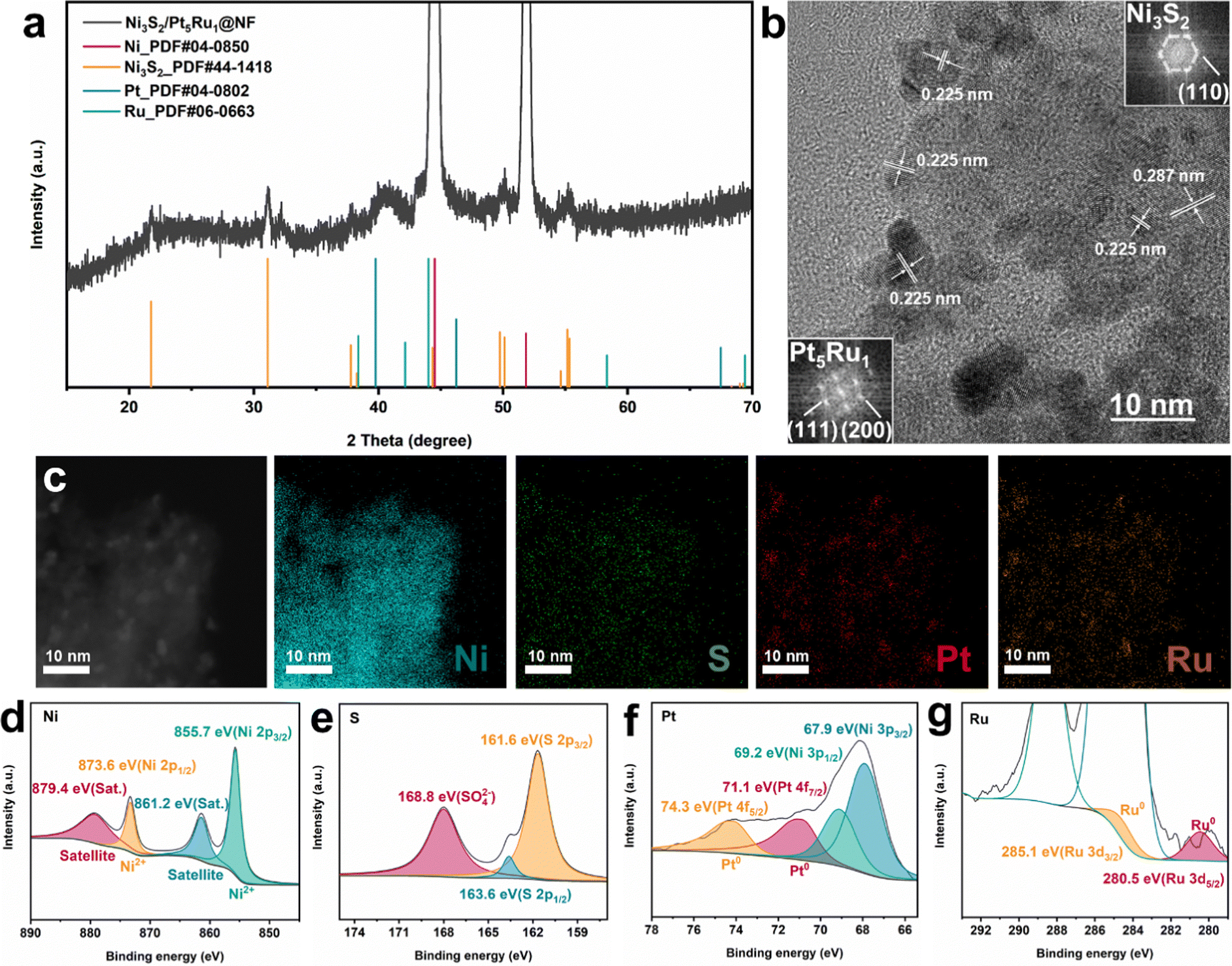
The Ni3S2/Pt5Ru1@NF catalyst was prepared by a one-step hydrothermal method, followed by a chemical reduction reaction. Specifically, Ni3S2 was synthesized in situ on a nickel foam substrate by a hydrothermal method, and then H2PtCl6 and RuCl3 were reduced at room temperature to obtain the Ni3S2/Pt5Ru1@NF heterostructure catalyst. The detailed process is illustrated in the Experimental section. X-ray diffraction (XRD) characterization was employed to identify the crystal structure of the as-prepared Ni3S2/Pt5Ru1@NF sample. As presented in Fig. 1a, two dominant peaks appearing at 2θ of 44.5° and 51.8° are assigned to the pristine Ni foam substrate (PDF#04-0850).21 Additionally, other diffraction peaks are well matched with hexagonal heazlewoodite Ni3S2 (PDF#44-1418).22 Due to the small size and low loading of the catalyst, no XRD peaks for the supported metal are clearly observed. The weight ratio of supported metal catalysts of Pt to Ru was determined to be 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by inductively-coupled plasma mass spectrometry (ICP-MS, Table S1, ESI†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were carried out to investigate the morphology and microstructure of Ni3S2/Pt5Ru1@NF. The SEM images (Fig. S1, ESI†) show that Ni3S2 is densely loaded on the surface of nickel foam, and Pt5Ru1 nanoalloys are grown on Ni3S2. In contrast, the surface of the Ni foam is smoother (shown in Fig. S2a, ESI†) and the surface graininess was increased after loading Ni3S2, as shown in Fig. S2b (ESI†). The TEM image (Fig. 1b) also shows that nanoparticles with an average size of 5 nm are uniformly decorated on the supported submicron particles. The corresponding FFT patterns (insets in Fig. 1b) reveal that the particles really are Pt5Ru1 (Fm
:1 by inductively-coupled plasma mass spectrometry (ICP-MS, Table S1, ESI†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were carried out to investigate the morphology and microstructure of Ni3S2/Pt5Ru1@NF. The SEM images (Fig. S1, ESI†) show that Ni3S2 is densely loaded on the surface of nickel foam, and Pt5Ru1 nanoalloys are grown on Ni3S2. In contrast, the surface of the Ni foam is smoother (shown in Fig. S2a, ESI†) and the surface graininess was increased after loading Ni3S2, as shown in Fig. S2b (ESI†). The TEM image (Fig. 1b) also shows that nanoparticles with an average size of 5 nm are uniformly decorated on the supported submicron particles. The corresponding FFT patterns (insets in Fig. 1b) reveal that the particles really are Pt5Ru1 (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), and Ni3S2 (R32). The interplanar spacings of 0.287 and 0.225 nm correspond to the (110) facet of Ni3S2 and (111) facet of Pt, respectively. Owing to the close interplanar spacing between Ru and Pt, Ru atoms can fill in the Pt lattice to form nanoalloys in a ratio of 5:1, which is proved by line scanning (Fig. S3, ESI†). Additionally, the STEM image (Fig. 1c) and its corresponding elemental mappings prove that Pt5Ru1 nanoalloys are evenly distributed on the surface of the Ni3S2 substrate. The chemical compositions and valence states of Ni3S2/Pt5Ru1@NF were examined by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1d–g, the XPS spectra of Ni3S2/Pt5Ru1@NF testify the presence of elemental Ni, S, Pt and Ru, which is consistent with the EDX results. As for the high-resolution Ni 2p spectrum of Ni3S2/Pt5Ru1@NF in Fig. 1d, two visible peaks located at binding energies of 873.6 and 855.7 eV are assigned to the Ni 2p1/2 and Ni 2p3/2 orbital of Ni2+, respectively.23,24 Compared with Ni3S2@NF (Fig. S4, ESI†), the peaks of Ni in Ni3S2/Pt5Ru1@NF shift to higher energy, indicating that there is interfacial electron transfer between the Pt5Ru1 nanoalloy and Ni3S2, and more electrons flow to the Pt5Ru1 nanoalloy.25 No obvious change has been observed in the peaks of S (Fig. 1e), indicating that the electron transfer mainly occurs between the metal elements. Compared with Pt foil, the Pt in Ni3S2/Pt5Ru1@NF shows a 0.5 eV negative shift in Fig. 1f.15 And a slight negative shift of Ru in Ni3S2/Pt5Ru1@NF is also observed in Fig. 1g compared to Ru foil. The electron transfer process between Pt5Ru1 nanoalloy and Ni3S2 can also be confirmed by the work function calculation below.
m), and Ni3S2 (R32). The interplanar spacings of 0.287 and 0.225 nm correspond to the (110) facet of Ni3S2 and (111) facet of Pt, respectively. Owing to the close interplanar spacing between Ru and Pt, Ru atoms can fill in the Pt lattice to form nanoalloys in a ratio of 5:1, which is proved by line scanning (Fig. S3, ESI†). Additionally, the STEM image (Fig. 1c) and its corresponding elemental mappings prove that Pt5Ru1 nanoalloys are evenly distributed on the surface of the Ni3S2 substrate. The chemical compositions and valence states of Ni3S2/Pt5Ru1@NF were examined by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1d–g, the XPS spectra of Ni3S2/Pt5Ru1@NF testify the presence of elemental Ni, S, Pt and Ru, which is consistent with the EDX results. As for the high-resolution Ni 2p spectrum of Ni3S2/Pt5Ru1@NF in Fig. 1d, two visible peaks located at binding energies of 873.6 and 855.7 eV are assigned to the Ni 2p1/2 and Ni 2p3/2 orbital of Ni2+, respectively.23,24 Compared with Ni3S2@NF (Fig. S4, ESI†), the peaks of Ni in Ni3S2/Pt5Ru1@NF shift to higher energy, indicating that there is interfacial electron transfer between the Pt5Ru1 nanoalloy and Ni3S2, and more electrons flow to the Pt5Ru1 nanoalloy.25 No obvious change has been observed in the peaks of S (Fig. 1e), indicating that the electron transfer mainly occurs between the metal elements. Compared with Pt foil, the Pt in Ni3S2/Pt5Ru1@NF shows a 0.5 eV negative shift in Fig. 1f.15 And a slight negative shift of Ru in Ni3S2/Pt5Ru1@NF is also observed in Fig. 1g compared to Ru foil. The electron transfer process between Pt5Ru1 nanoalloy and Ni3S2 can also be confirmed by the work function calculation below.
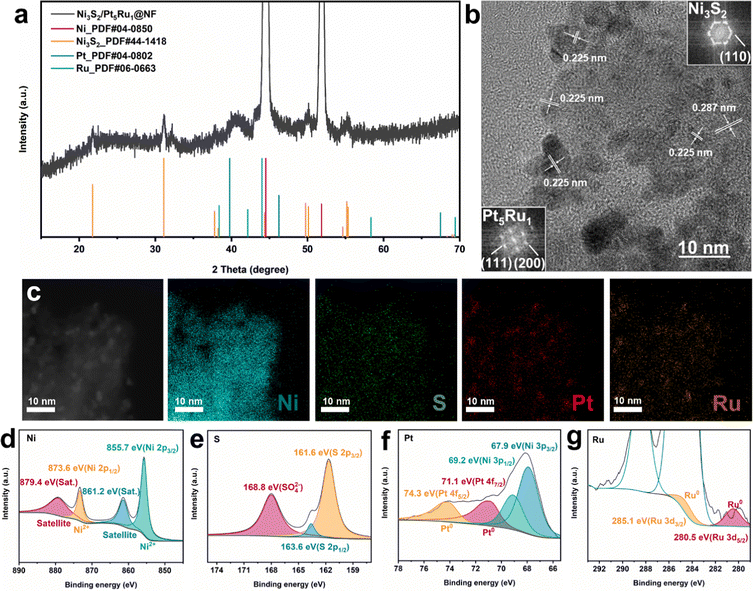 |
| | Fig. 1 Structural and microstructure characterizations of Ni3S2/Pt5Ru1@NF. (a) XRD pattern, (b) HRTEM image and the corresponding FFT patterns, (c) STEM image and the corresponding EDX elemental mapping images, and XPS spectra of (d) Ni 2p, (e) S 2p, (f) Pt 4f, and (g) Ru 3d. | |
Acidic and alkaline HER performance
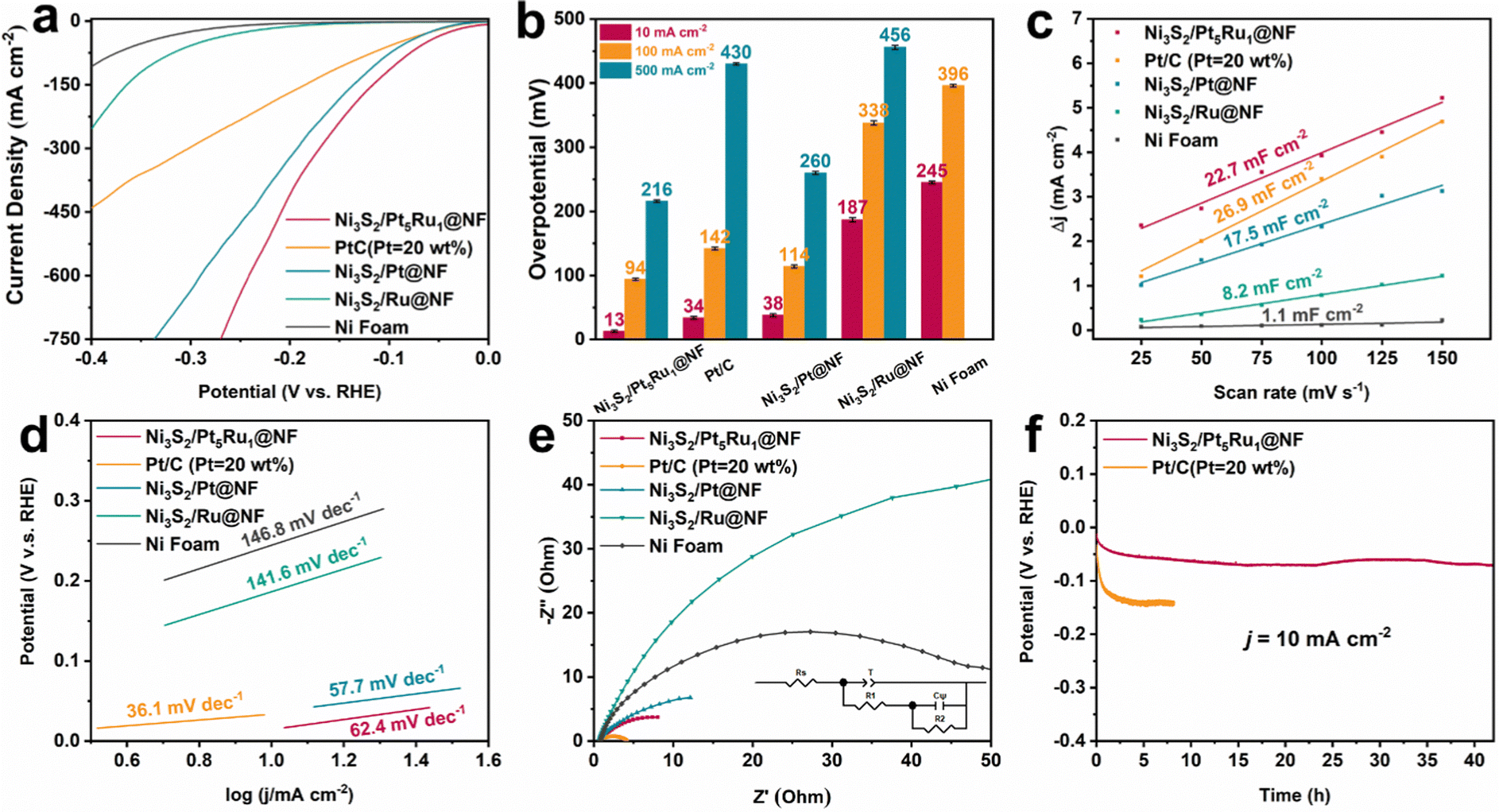
A typical three-electrode system was used to evaluate the HER performance of Ni3S2/Pt5Ru1@NF in 1.0 M KOH and 0.1 M HClO4 solutions. For comparison, we also synthesized control samples of Ni3S2/Pt@NF, Ni3S2/Ru@NF and commercial PtC@NF catalysts with similar noble metal loadings. The performance of the nickel foam substrate was also tested to prove that our catalytic activity mainly comes from the Ni3S2 and nanoalloys. The polarization curves of Ni3S2/Pt5Ru1@NF and the control samples are displayed in Fig. 2a, revealing that Ni3S2/Pt5Ru1@NF can reach a current density of 10 mA cm−2 with only a low overpotential of 13 mV in 1.0 M KOH, exceeding most Ni3S2-based catalysts.15,26,27Fig. 2b compares the overpotential values of Ni3S2/Pt5Ru1@NF and the control group at current densities of 10, 100 and 500 mA cm−2. The results show that Ni3S2/Pt5Ru1@NF has the smallest overpotential values at different current densities, and the performance is still better than that of commercial Pt/C. Electrochemically active surface area (ECSA) is often used to evaluate the actual catalytic performance of electrode materials. Since there is a linear relationship between ECSA and electric double-layer capacitance (Cdl), we utilized cyclic voltammetry (CV) to measure the Cdl of the catalyst at different scan rates, as shown in Fig. 2c. It can be observed that Ni3S2/Pt5Ru1@NF (22.7 mF cm−2) has a Cdl close to that of Pt/C (26.9 mF cm−2), and significantly higher than other control samples. The polarization curves of Ni3S2/Pt5Ru1@NF and control samples based on ECSA-normalization toward the HER in 1.0 M KOH are displayed in Fig. S5b (ESI†). The results show that Ni3S2/Pt5Ru1@NF also has the smallest overpotential values at different current densities. Fig. 2d tests the Tafel slopes of Ni3S2/Pt5Ru1@NF and the control samples. It is found that Ni3S2/Pt5Ru1@NF has a smaller Tafel slope, indicating a faster kinetic process at low potentials. The electrochemical impedance spectroscopy (EIS) of Ni3S2/Pt5Ru1@NF and the control samples was performed and simulated by a double-parallel equivalent circuit model (Fig. 2e and Table S2, ESI†). The first parallel component (T and R1) reflects the charge transfer kinetics, where T is related to the electric double layer capacitance and R1 represents the catalytic charge transfer resistance. Ni3S2/Pt5Ru1@NF has the smallest resistance after fitting, indicating its faster charge transfer process. The second parallel component (Cψ and R2) reflects the hydrogen adsorption behavior on the catalyst surface, where Cψ and R2 represent the hydrogen adsorption pseudo-capacitance and resistance, respectively. It can be observed from the fitting results in Table S2 (ESI†) that Ni3S2/Pt5Ru1@NF has a larger Cψ and a smaller R2, indicating that its unique ‘metal–support’ structure is more conducive to the binding of protons and the desorption of hydrogen on the surface. We also tested the long-term stability by chronopotentiometry measurement (Fig. 2f). At a constant current of 10 mA cm−2, there is basically no obvious attenuation of the overpotential after 40 h for the Ni3S2/Pt5Ru1@NF, which formed a clear contrast with the commercial Pt/C catalyst after 2 h.
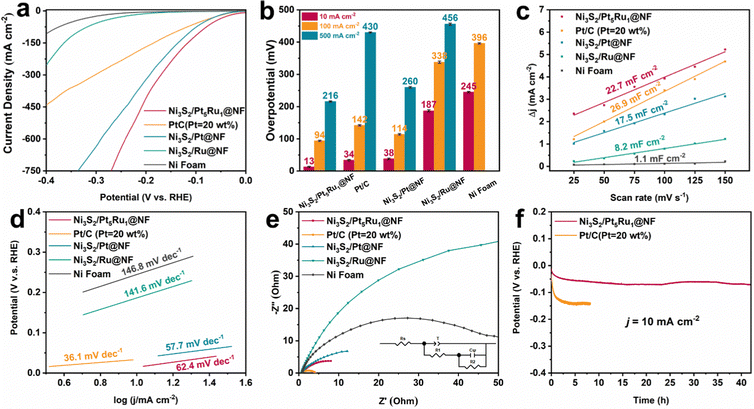 |
| | Fig. 2 Electrocatalytic HER performance in 1.0 M KOH solution. (a) Polarization curves of the electrocatalysts for the HER (with 95% iR compensation), (b) the resultant overpotentials at current densities of 10, 100 and 500 mA cm−2 for the as-prepared electrocatalysts, (c) calculated electrochemical double-layer capacitances, (d) the corresponding Tafel plots, (e) Nyquist plots under open-circuit voltage, and (f) long-term stability test at current densities of 10 mA cm−2. | |
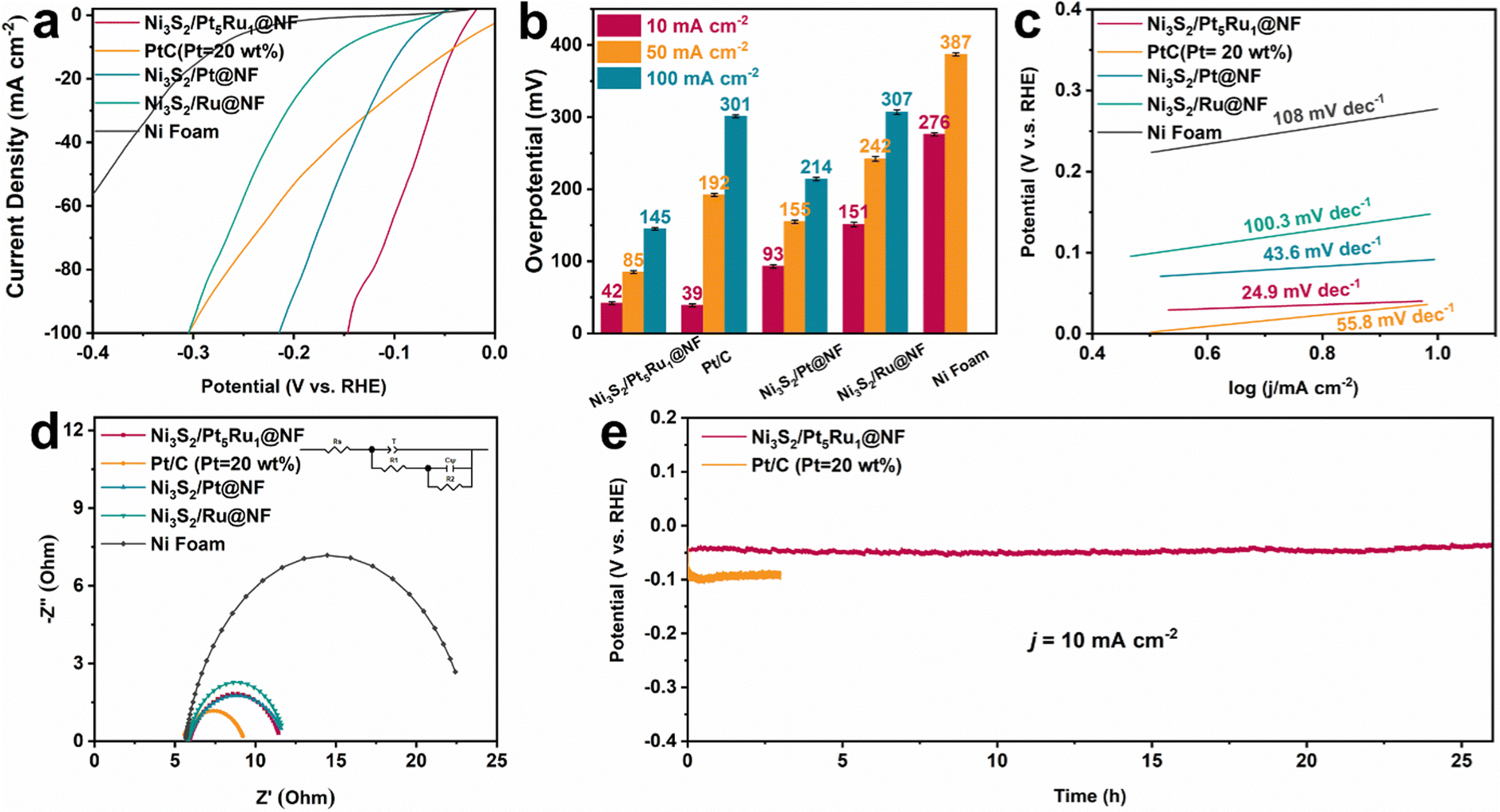
Likewise, the HER performance of Ni3S2/Pt5Ru1@NF in 0.1 M HClO4 was also investigated. From the polarization curves in Fig. 3a and the comparison of the overpotential at different current densities in Fig. 3b, Ni3S2/Pt5Ru1@NF has similar overpotential to commercial Pt/C at a current density of 10 mA cm−2. However, with the increase of the current density, the HER performance of Ni3S2/Pt5Ru1@NF gradually exceeds that of commercial Pt/C, and the overpotential of Ni3S2/Pt5Ru1@NF is even only half that of commercial Pt/C at a large current density of 100 mA cm−2. This may be related to the extremely fast kinetic process of Ni3S2/Pt5Ru1@NF in acidic solution, which can also be demonstrated by the Tafel slope in Fig. 3c. Likewise, the polarization curves of Ni3S2/Pt5Ru1@NF and the control samples based on ECSA-normalization toward the HER in 0.1 M HClO4 show a similar regularity (Fig. S5a, ESI†). The EIS of Ni3S2/Pt5Ru1@NF and control samples under acidic conditions were also fitted, as shown in Fig. 3d and Table S3 (ESI†). Like the results in 1.0 M KOH, the unique structural design of Ni3S2/Pt5Ru1@NF provides a good conductive network for the rapid transfer of protons and desorption of hydrogen. More excitingly, Ni3S2/Pt5Ru1@NF exhibits excellent durability in 0.1 M HClO4 with almost no decay of the overpotential at a current density of 10 mA cm−2 (Fig. 3e), indicating that Ni3S2/Pt5Ru1@NF can exist stably for a long time in acidic solution. Compared with other nickel-based chalcogenide catalysts, the overpotential of Ni3S2/Pt5Ru1@NF is in a leading position, as shown in Table S4 (ESI†).
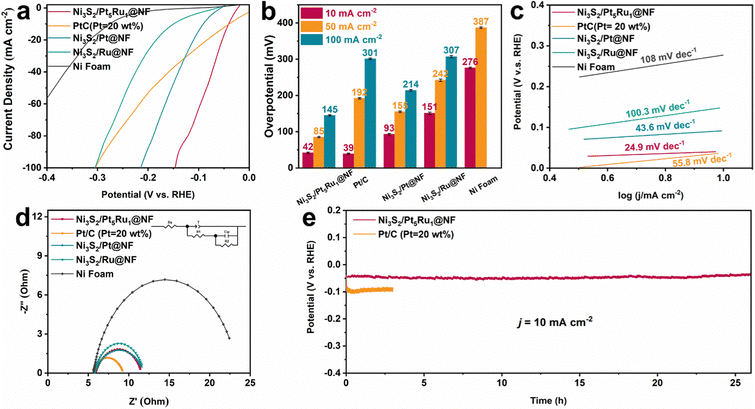 |
| | Fig. 3 Electrocatalytic HER performance in 0.1 M HClO4 solution. (a) Polarization curves of the electrocatalysts for the HER (with 95% iR compensation), (b) the resultant overpotentials at current densities of 10, 50 and 100 mA cm−2 for the as-prepared electrocatalysts, (c) the corresponding Tafel plots, (d) Nyquist plots under open-circuit voltage, and (e) long-term stability test at current densities of 10 mA cm−2. | |
Experimental evidence for hydrogen spillover
The hydrogen spillover process is widespread on catalyst surfaces with at least two active sites, usually from sites with more negative ΔGH to those with more positive ΔGH. Generally, the HER involves a two-electron process, and the reaction pathway is as follows:
Acidic solution:
| | | H+ + e− + H* → * + H2↑ | (2) |
Alkaline solution:
| | | H2O + e− + * → H* + OH− | (3) |
| | | H2O + e− + H* → * + OH− + H2 | (4) |
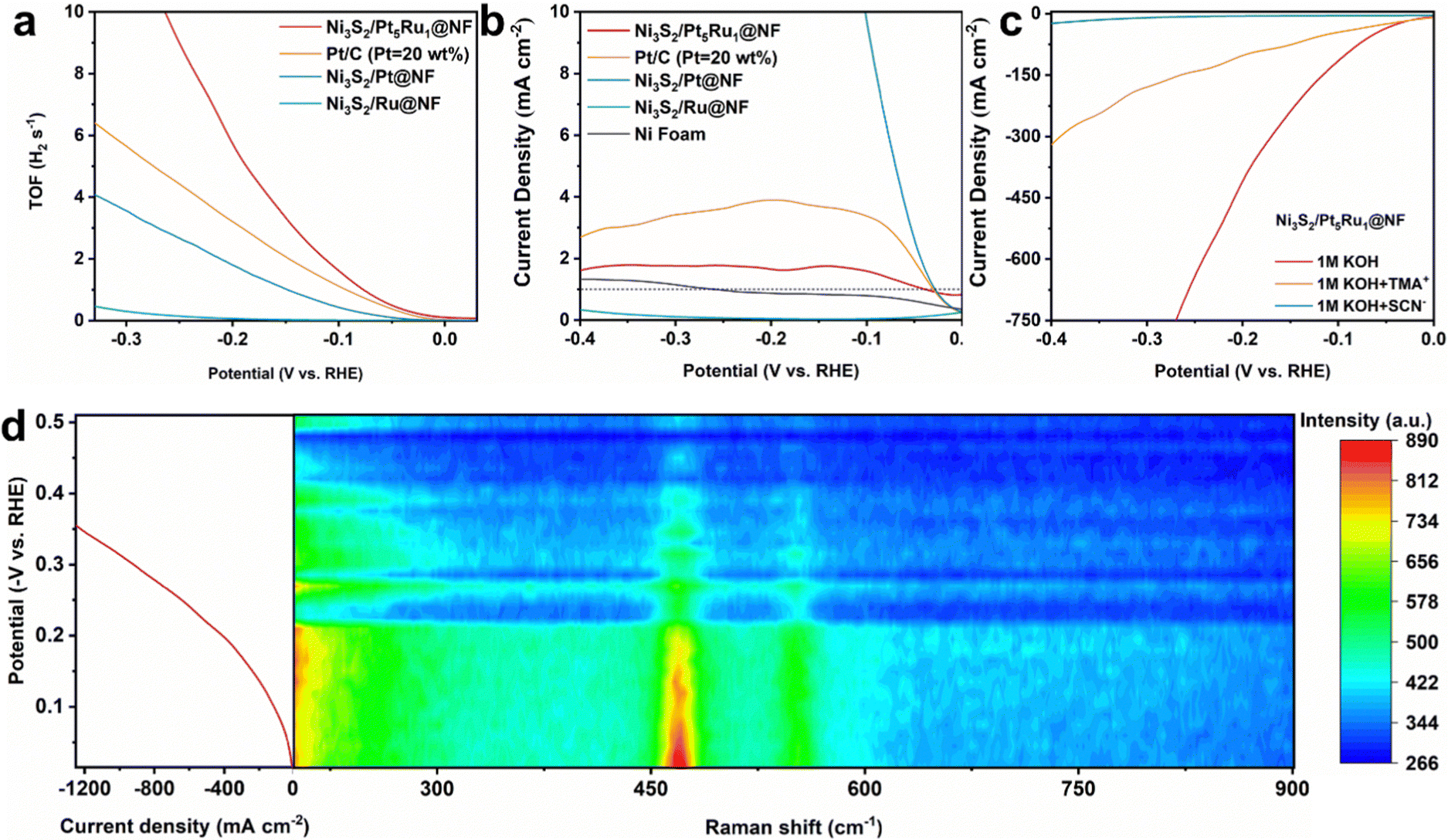
In acidic solution, H+ is preferentially adsorbed on the surface of the active site, taking electrons from the bulk to form protons. Proton and proton combine to form H2 leaving the active site. For alkaline solutions, the first step is often the dissociation of H2O in the active site. Herein, if it is necessary to prove the occurrence of the hydrogen spillover process, two points must be understood: (i) the intrinsic activity of the active site, and (ii) the change of the surface adsorption state. Therefore, we first tested the turnover frequency (TOF) of Ni3S2/Pt5Ru1@NF.28 It can be seen from Fig. 4a that Ni3S2/Pt5Ru1@NF has the highest TOF value compared to the control samples, indicating that the active sites of Ni3S2/Pt5Ru1@NF have high intrinsic activity. The kinetic isotope effect (KIE) is normally used to estimate the involvement of proton transfer in the rate-determining step of an electrocatalytic reaction.29 As shown in Fig. 4b, the KIE value of Ni3S2/Pt5Ru1@NF is over 1, implying that protons are involved in the HER reaction.30 Compared with control samples, Ni3S2/Pt5Ru1@NF has the smallest KIE value, which indicates that Ni3S2/Pt5Ru1@NF has an extremely fast proton transfer kinetic process.31 In order to further confirm the active sites of Ni3S2/Pt5Ru1@NF for the HER, poisoning tests were performed by adding thiocyanate (SCN−) and tetramethylammonium cation (TMA+) ions into acidic and alkaline solutions.32 It has been reported that TMA+ has adsorption properties for O-group elements.33 In Ni3S2/Pt5Ru1@NF, it can be adsorbed on the S site to block the path of hydrogen overflow. As a commonly used ion in poisoning experiments, SCN− can be adsorbed on the surface of the Pt5Ru1 alloy to inactivate it. As shown in Fig. 4c, the addition of SCN− and TMA+ affects the HER response to varying degrees. TMA+ blocks the path for hydrogen spillover, making the transfer of protons between active sites difficult, and SCN− adsorption on the surface of Pt5Ru1 alloy poisons the catalyst, dropping the activity of Ni3S2/Pt5Ru1@NF almost to zero. The above results indicate that both Ni3S2 and Pt5Ru1 are involved in the HER as active sites. By the extent of the decrease in activity in Fig. 4c, we predict that Pt5Ru1 preferentially undergoes the Volmer step to generate protons due to its higher activity.
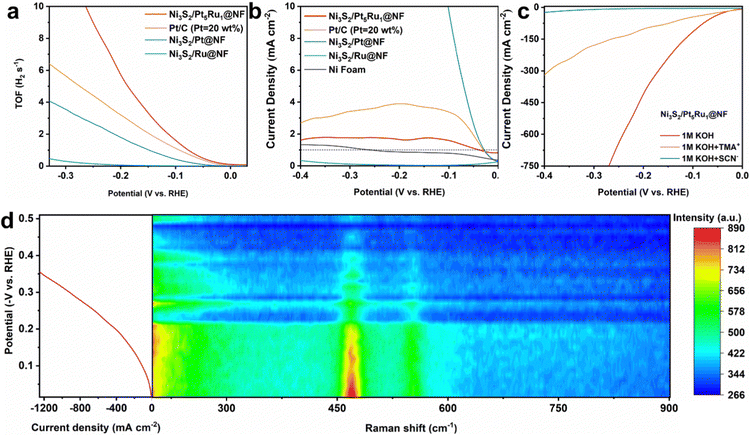 |
| | Fig. 4 Mechanism study of Ni3S2/Pt5Ru1@NF. (a) The potential-dependent TOF curves for Ni3S2/Pt5Ru1@NF, Pt/C, Ni3S2/Pt@NF and Ni3S2/Ru@NF, (b) calculated KIE values (JH2O/JD2O) from Ni3S2/Pt5Ru1@NF, Pt/C, Ni3S2/Pt@NF, Ni3S2/Ru@NF and Ni foam, (c) polarization curves for Ni3S2/Pt5Ru1@NF before and after the addition of SCN− and TMA+ ions in N2-saturated 1.0 M KOH, and (d) operando Raman spectra of Ni3S2/Pt5Ru1@NF in 1.0 M KOH under HER potential. | |
Although the above electrochemical methods can reveal the existence of active sites and the strength of activity in Ni3S2/Pt5Ru1@NF, the real hydrogen spillover process is hard to identify. The hydrogen spillover process itself is dynamic, and transient imaging technology with ultra-high spatial resolution and nanometer size should be used to monitor the hydrogen spillover process in real time.34 Unfortunately, there is no available technology to visually observe the hydrogen spillover process. Considering that the catalytic reaction generally occurs at the surface interface, a surface-sensitive technique can be used to infer the occurrence of the hydrogen spillover process. Herein, we attempt to instantaneously record the HER of Ni3S2/Pt5Ru1@NF by operando Raman spectroscopy, and speculate the hydrogen spillover process via bonding changes on its surface.35,36 The Raman spectra of Ni3S2/Pt5Ru1@NF and control samples were thus studied to obtain the information of surface bonding. As shown in Fig. S6a (ESI†), Raman signals appeared at 192, 224, 305, 326 and 353 cm−1 for Ni3S2/Pt5Ru1@NF and the control samples correspond to the E4, E3, E2, A1 and E1 vibrational states of Ni3S2, respectively, indicating that the Ni3S2 is abundant in the samples.27 The signals of Pt and Ru are not detected, which may be related to the insensitivity of Raman spectroscopy to metal elements. Fig. S6b (ESI†) compares the signals of 1.0 M KOH and Ni3S2/Pt5Ru1@NF in 1.0 M KOH, indicating that the new peaks appearing in Ni3S2/Pt5Ru1@NF are not the signals of KOH. Fig. S6c (ESI†) compares the spectral information of Ni3S2/Pt5Ru1@NF before and after adding 1.0 M KOH. After adding 1.0 M KOH, three new peaks appeared at 468, 549 and 1059 cm−1 correspond to Ni–O and two Ni–S bonds, respectively. A broad peak appears at 1635 cm−1, associated with the H–OH bending vibration of adsorbed water. The above results indicate that after the addition of 1.0 M KOH, OH− and H2O in the solution will be adsorbed on the surface of Ni sites. Therefore, the hydrogen spillover process can be indirectly observed by change of the Ni–O bond on the surface of the Ni site at 468 cm−1. As shown in Fig. 4d, the peak intensity of the Ni–O bond gradually weakens as the potential increases. This is because with the increase of the potential, the hydrogen overflow process of Ni3S2/Pt5Ru1@NF is continuously enhanced, and more and more protons are generated on the surface of the Pt5Ru1 alloy and overflow to the Ni sites in the Ni3S2 to form H2. The adsorption state on the Ni surface gradually changed from Ni–O bonds to the adsorption of protons at Ni sites. This result is quite consistent with the hydrogen spillover process from the Pt5Ru1 alloy to Ni3S2 as we conceived.
DFT calculations for hydrogen spillover
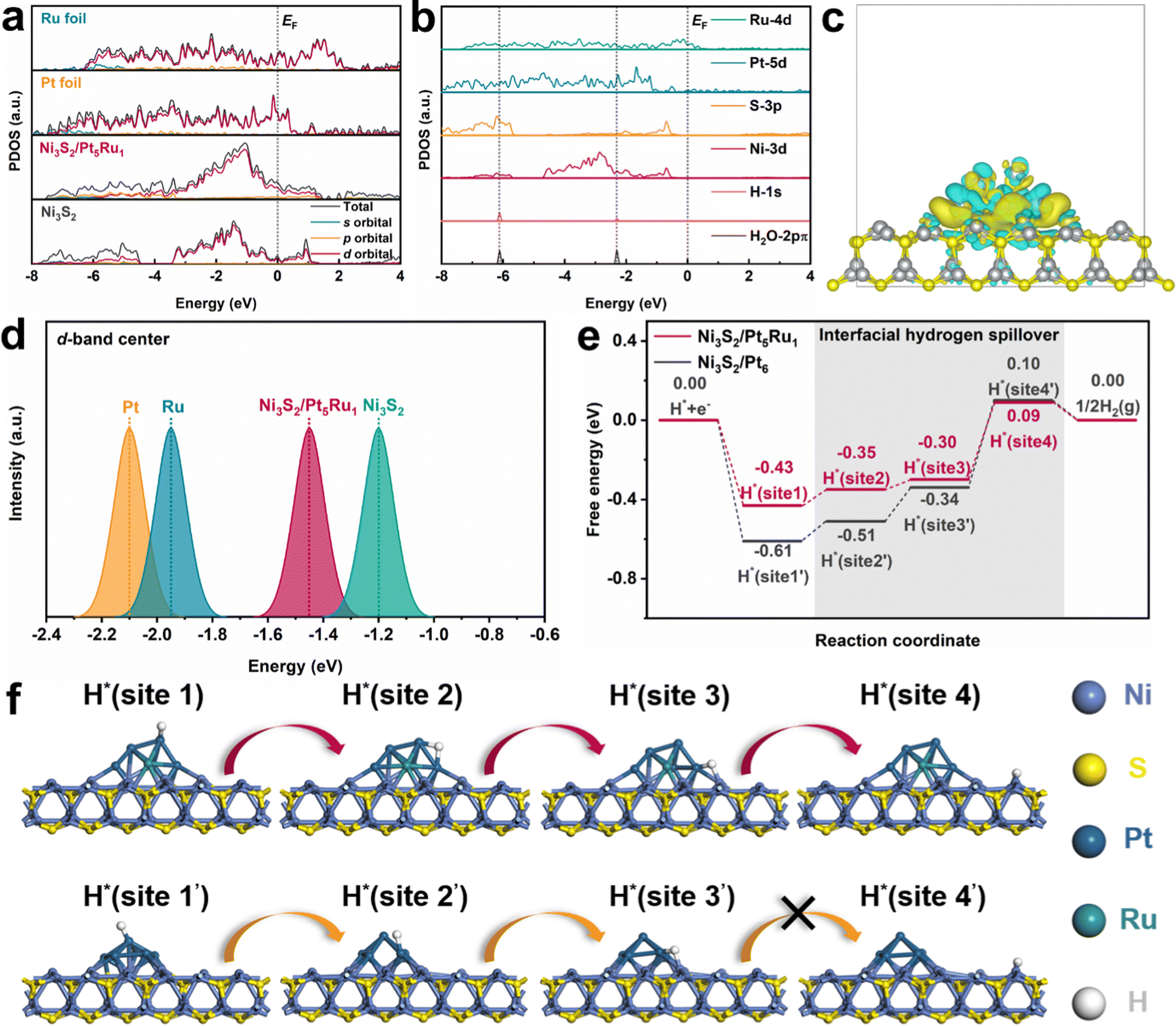
Density functional theory (DFT) calculations were further performed to gain insights into the hydrogen spillover process. Combining XRD and HRTEM results, we choose the (110) facet of Ni3S2 as the substrate, and Pt5Ru1 clusters on it as the nanoalloy.37 Details of the simulation models and DFT calculations are illustrated in the ESI.†Fig. 5a calculates the DOS and PDOS of Ni3S2/Pt5Ru1, Ni3S2, Ru foil and Pt foil. The results show that Pt5Ru1 increases the occupied state of Ni3S2 near the Fermi level, which means that Ni3S2/Pt5Ru1@NF can more easily obtain electrons from the bulk for the HER.38Fig. 5b calculates the PDOS of Ni3S2/Pt5Ru1 from the angle of Pt, Ru, S and Ni elements. The 1s orbital of H and the 2pπ orbital of H2O overlap well with Ni3S2/Pt5Ru1. This means that H2O or protons can make good contact with various elements at the interface of Ni3S2/Pt5Ru1@NF and transfer electrons during the reaction, which also explains the lower charge transfer resistance of Ni3S2/Pt5Ru1@NF in Fig. 2e and 3d. To intuitively describe the interaction between Ni3S2 substrate and Pt5Ru1 nanoalloy, we calculated the charge density difference of Ni3S2/Pt5Ru1. As shown in Fig. 5c, the Pt5Ru1 nanoalloy and Ni3S2 surface have good electron transfer, which means that Pt5Ru1 can be stably supported on Ni3S2 without being detached from the surface.26 The d-band centers of Ni3S2/Pt5Ru1, Ni3S2, Pt foil and Ru foil were calculated by DOS, as shown in Fig. 5d. The lower the d-band center, the lower the position of the antibonding orbital and the lower the adsorption energy. According to previous literature reports, Pt and Ru have the most excellent adsorption energy, allowing protons to react and forming H2 off the surface.39,40 Compared with Ni3S2, Ni3S2/Pt5Ru1 has a d-band center closer to Pt and Ru, so protons are more likely to be adsorbed and desorbed on the surface of Ni3S2/Pt5Ru1. The above calculations indicate that Ni3S2/Pt5Ru1@NF itself has good electrical conductivity and moderate hydrogen adsorption energy, and is an excellent catalyst for the HER.
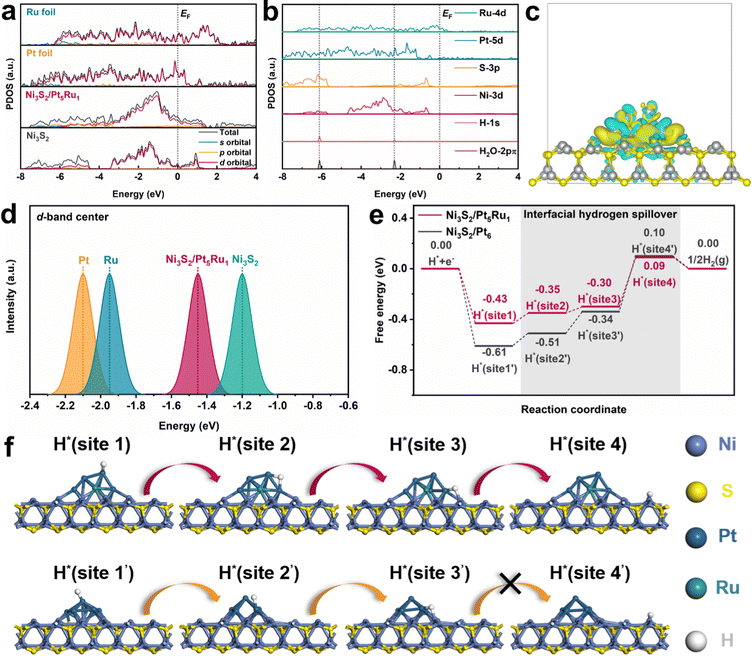 |
| | Fig. 5 DFT calculation, (a) calculated PDOS of Ni3S2/Pt5Ru1, Ni3S2, Ru foil and Pt foil with aligned Fermi level EF, (b) the d–p orbital alignment of the surface Ru, Pt, S and Ni sites for Ni3S2/Pt5Ru1, (c) the local electron density distribution of the Pt5Ru1 cluster in Ni3S2/Pt5Ru1, (d) calculated d-band center (Ed) of Ni3S2/Pt5Ru1, Ni3S2, Ru foil and Pt foil, (e) calculated free energy diagram for the HER on Ni3S2/Pt5Ru1 paradigm and Ni3S2/Pt6 benchmark, and (f) the optimized H* adsorption structures at various sites. | |
The above evidence, however, is not enough to prove that Ni3S2/Pt5Ru1@NF can generate hydrogen spillover. In theory, two conditions are required to produce hydrogen spillover: (i) There is a difference in the free energy of hydrogen evolution (GH) between the active sites. The ions preferentially undergo the Volmer step to form protons at the more negative ΔGH sites, and the protons then transfer to the more positive ΔGH sites to undergo the Tafel step to form the H2. (ii) The transfer between active sites should have a smaller migration energy barrier to reduce obstruction of hydrogen spillover. For the convenience of comparison, we chose Ni3S2/Pt6 as the control sample, and took the HER reaction mechanism under acidic conditions as the overall design framework. The ΔGH of Ni3S2/Pt5Ru1 and Ni3S2/Pt6 was calculated in an attempt to construct a hydrogen spillover path. As shown in Fig. 5e and f, the optimized hydrogen spillover path reveals that the uniquely ‘metal–support’ structured Ni3S2/Pt5Ru1 has a suitable ΔGH compared with Ni3S2/Pt6. H+ preferentially adsorbs to site 1 of the Pt5Ru1 alloy with a ΔGH of −0.43 eV, and gains electrons from the Pt5Ru1 alloy site to form protons. It is then transferred to the interface of the alloy and substrate through site 2 and site 3 of Pt5Ru1, and finally H2 leaves the surface with a ΔGH of 0.09 eV at the Ni site of Ni3S2. On the other hand, it is well known that due to the difference in work function between the metal and the support, a Schottky barrier is inevitably formed, which traps protons at the interface and hinders hydrogen spillover. Therefore, reducing the work function difference (Δϕ) as much as possible is an effective method to enhance the hydrogen spillover process. Here, we calculated Δϕ between Ni3S2 and Pt (Pt5Ru1). The calculated results are shown in Fig. S7 and S8 (ESI†). Δϕ between Pt5Ru1 and Ni3S2 is 0.5678 eV, which is significantly smaller than 0.8442 eV between Pt and Ni3S2. That is to say, the Schottky barrier at the Ni3S2/Pt5Ru1 interface is smaller, and protons are not easily captured at the interface, which is favorable for the occurrence of the hydrogen spillover process.
3. Conclusions
In summary, through a simple two-step synthesis, we designed a ‘metal–support’ structured Ni3S2/Pt5Ru1@NF catalyst with excellent catalytic performance under both acidic and basic conditions. Through the combination of experimental verification and theoretical calculation, the mechanism of the hydrogen spillover process is explained in detail. But as a new theory, the hydrogen spillover process itself needs more characterization methods to clarify its physical and chemical processes. As a successful case, Ni3S2/Pt5Ru1@NF provides strong proof for the development of this theory.
Author contributions
Y. Y. and X. R. conceived and supervised the research. Y. Y. and Z. Y. designed the experiments. Z. Y. performed the experiments and data analysis. Z. Y. conducted DFT calculations. All the authors discussed the results and commented on the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (Grant 2022YFA1504100), the National Natural Science Foundation of China (No. 51925207, U1910210 and 52161145101), the “Transformational Technologies for Clean Energy and Demonstration” Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDA21000000), the National Synchrotron Radiation Laboratory (KY2060000173), the Fundamental Research Funds for the Central Universities (Wk2060140026), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (Grant No. YLU-DNL Fund 2021002).
Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS PubMed.
- S. Hammes-Schiffer and G. Galli, Nat. Energy, 2021, 6, 700–705 CrossRef.
- S. L. Hu and W. X. Li, Science, 2021, 374, 1360 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- M. F. Li, K. N. Duanmu, C. Z. Wan, T. Cheng, L. Zhang, S. Dai, W. X. Chen, Z. P. Zhao, P. Li, H. L. Fei, Y. M. Zhu, R. Yu, J. Luo, K. T. Zang, Z. Y. Lin, M. N. Ding, J. Huang, H. T. Sun, J. H. Guo, X. Q. Pan, W. A. Goddard, P. Sautet, Y. Huang and X. F. Duan, Nat. Catal., 2019, 2, 495–503 CrossRef CAS.
- X. S. Wang, Y. Zheng, W. C. Sheng, Z. C. J. Xu, M. Jaroniec and S. Z. Qiao, Mater. Today, 2020, 36, 125–138 CrossRef CAS.
- B. Zhao, D. Y. Shen, Z. C. Zhang, P. Lu, M. Hossain, J. Li, B. Li and X. D. Duan, Adv. Funct. Mater., 2021, 31, 2105132 CrossRef CAS.
- Z. P. Lin, B. B. Xiao, Z. P. Wang, W. Y. Tao, S. J. Shen, L. A. Huang, J. T. Zhang, F. Q. Meng, Q. H. Zhang, L. Gu and W. W. Zhong, Adv. Funct. Mater., 2021, 31, 2102321 CrossRef CAS.
- S. Jeong, H. D. Mai, T. K. Nguyen, J. S. Youn, K. H. Nam, C. M. Park and K. J. Jeon, Appl. Catal., B, 2021, 293, 120227 CrossRef CAS.
- D. Reynard, B. Nagar and H. Girault, ACS Catal., 2021, 11, 5865–5872 CrossRef CAS.
- B. Geng, F. Yan, X. Zhang, Y. He, C. Zhu, S. L. Chou, X. Zhang and Y. Chen, Adv. Mater., 2021, 33, e2106781 CrossRef.
- K. L. Zhou, C. B. Han, Z. Wang, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Adv. Sci., 2021, 8, 2100347 CrossRef CAS PubMed.
- Z. Wang, M. T. Tang, A. Cao, K. Chan and J. K. Nørskov, J. Phys. Chem. C, 2022, 126, 5151–5158 CrossRef CAS.
- R. Prins, Chem. Rev., 2012, 112, 2714–2738 CrossRef CAS PubMed.
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS PubMed.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, Nat. Commun., 2021, 12, 3502 CrossRef CAS PubMed.
- Z. W. Wei, H. J. Wang, C. Zhang, K. Xu, X. L. Lu and T. B. Lu, Angew. Chem., Int. Ed., 2021, 60, 16622–16627 CrossRef CAS.
- G. Ren, Q. Hao, J. Mao, L. Liang, H. Liu, C. Liu and J. Zhang, Nanoscale, 2018, 10, 17347–17353 RSC.
- H. Du, R. Kong, F. Qu and L. Lu, Chem. Commun., 2018, 54, 10100–10103 RSC.
- N. Chen, Y. X. Du, G. Zhang, W. T. Lu and F. F. Cao, Nano Energy, 2021, 81, 105605 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, eScience, 2021, 1, 69–74 CrossRef.
- X. W. Lv, P. Kannan, S. Ji, X. Y. Wang and H. Wang, CrystEngComm, 2020, 22, 6517–6528 RSC.
- B. Fei, Z. L. Chen, J. X. Liu, H. B. Xu, X. X. Yan, H. L. Qing, M. Chen and R. B. Wu, Adv. Energy Mater., 2020, 10, 2001963 CrossRef CAS.
- S. C. Huang, Y. Y. Meng, Y. F. Cao, F. Yao, Z. J. He, X. X. Wang, H. Pan and M. M. Wu, Appl. Catal., B, 2020, 274, 119120 CrossRef CAS.
- X. Li, Y. Fang, J. Wang, H. Fang, S. Xi, X. Zhao, D. Xu, H. Xu, W. Yu, X. Hai, C. Chen, C. Yao, H. B. Tao, A. G. R. Howe, S. J. Pennycook, B. Liu, J. Lu and C. Su, Nat. Commun., 2021, 12, 2351 CrossRef CAS PubMed.
- C. Pasquini, I. Zaharieva, D. Gonzalez-Flores, P. Chernev, M. R. Mohammadi, L. Guidoni, R. D. L. Smith and H. Dau, J. Am. Chem. Soc., 2019, 141, 2938–2948 CrossRef CAS PubMed.
- Y. Kong, Y. Li, X. Sang, B. Yang, Z. Li, S. Zheng, Q. Zhang, S. Yao, X. Yang, L. Lei, S. Zhou, G. Wu and Y. Hou, Adv. Mater., 2022, 34, e2103548 CrossRef PubMed.
- X. Wang, X. Sang, C. L. Dong, S. Yao, L. Shuai, J. Lu, B. Yang, Z. Li, L. Lei, M. Qiu, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2021, 60, 11959–11965 CrossRef CAS PubMed.
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2019, 31, e1808173 CrossRef PubMed.
- Z. F. Huang, J. J. Song, Y. H. Du, S. B. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. C. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- T. Kosmala, A. Baby, M. Lunardon, D. Perilli, H. Liu, C. Durante, C. Di Valentin, S. Agnoli and G. Granozzi, Nat. Catal., 2021, 4, 850–859 CrossRef CAS.
- J. K. Li and J. L. Gong, Energy Environ. Sci., 2020, 13, 3748–3779 RSC.
- X. Liu, J. Meng, J. Zhu, M. Huang, B. Wen, R. Guo and L. Mai, Adv. Mater., 2021, 33, e2007344 CrossRef PubMed.
- X. Guo, X. Wan, Q. Liu, Y. Li, W. Li and J. Shui, eScience, 2022, 2, 304–310 CrossRef.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS.
- Y. Zhao, P. V. Kumar, X. Tan, X. Lu, X. Zhu, J. Jiang, J. Pan, S. Xi, H. Y. Yang, Z. Ma, T. Wan, D. Chu, W. Jiang, S. C. Smith, R. Amal, Z. Han and X. Lu, Nat. Commun., 2022, 13, 2430 CrossRef CAS PubMed.
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H. T. Wang, C. W. Pao, A. R. Head, C. H. Wang, X. Tong, C. J. Sun, W. F. Pong, J. Luo, J. C. Zheng and H. L. Xin, Nat. Mater., 2022, 21, 681–688 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Yan
Yu
b and
Yan
Yu