
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unexplored role of the CrOx shell in an elaborated Rh/CrOx core–shell cocatalyst for photocatalytic water splitting: a selective electron transport pathway from semiconductors to core metals, boosting charge separation and H2 evolution†
Tetsu
Kotani‡
a,
Kanta
Ogawa‡
 a,
Hajime
Suzuki
a,
Kosaku
Kato
b,
Osamu
Tomita
a,
Akira
Yamakata
b and
Ryu
Abe
*a
a,
Hajime
Suzuki
a,
Kosaku
Kato
b,
Osamu
Tomita
a,
Akira
Yamakata
b and
Ryu
Abe
*a
aDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto, 615-8510, Japan. E-mail: ryu-abe@scl.kyoto-u.ac.jp
bGraduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushima-naka, Kita-ku, Okayama, 700-8530, Japan
First published on 18th March 2023
Abstract
A core–shell structured Rh/CrOx cocatalyst has endowed various semiconductors with high efficiency in water-splitting photocatalysis, where thin CrOx layers on Rh have been assumed to be physical blockers of O2 to the metal surface to suppress unfavorable reverse reactions (e.g., catalytic H2O formation from H2 and O2). Herein, we propose another unexplored but favorable function of CrOx layers: a selective electron transport pathway from photocatalysts to the Rh core boosting charge separation and H2 production. The subsequent loading of CrOx layers onto Rh increased the rate of visible light H2 evolution of a Bi4NbO8Cl photocatalyst, even in a half reaction with a hole scavenger where O2 does not evolve. Transient absorption spectroscopy revealed that the CrOx layer increases the electron path from Bi4NbO8Cl to Rh. Importantly, the highest H2-evolution activity was obtained by simultaneous photodeposition using CrIII and RhIII precursors, which had not yet been examined. In this sample, Rh nanoparticles were enclosed by an amorphous CrOx shell, where Rh particles were less directly attached to the semiconductor. Therein, CrOx inserted between Bi4NbO8Cl and Rh effectively suppresses undesirable hole transfer from Bi4NbO8Cl to Rh, while such hole transfer partially occurs when they are in direct contact. These results indicated that CrOx functions as a selective electron transport pathway and improves the H2 evolution activity. Although the development strategy of cocatalysts has so far focused on surface redox reactions, this study offers a new approach for the design of highly efficient cocatalysts based on the carrier transfer process, especially at semiconductor–cocatalyst interfaces.
Broader contextWater splitting using a semiconductor photocatalyst enables clean hydrogen production from solar energy. Light irradiation excites electrons and holes in a photocatalyst. After their migration toward the surface of the photocatalyst, they participate in reduction and oxidation at reaction sites. Therefore, not only semiconductor photo-absorbers themselves, but also creating effective reaction sites on their surface is essential. Such a role can be played by “co-catalysts” on photocatalysts, the functions of which are generally (1) capturing either of photoexcited carriers from photocatalysts and (2) catalyzing redox reactions on the surface of photocatalysts. Although vast knowledge in electrocatalysis can be employed for the latter function, a versatile strategy has not yet been established for the former one. Here, we reveal that CrOx in the Rh/CrOx core–shell type cocatalyst helps in electron transfer from photocatalysts to the reaction sites (Rh). Rh/Cr2O3 is a widely used hydrogen evolution reaction cocatalyst in overall water splitting, where CrOx has been considered to be a physical blocker of O2 to the metal surface to suppress reverse reactions. We propose another function of CrOx layers: a selective electron-transport pathway from photocatalysts to Rh, boosting charge separation and H2 production. This study offers a new approach for designing cocatalysts based on the carrier transfer process. |
Introduction
Highly efficient photocatalytic water splitting inevitably requires not only an efficient semiconductor photo-absorber but also effective cocatalysts.1–5 In general, cocatalysts loaded on semiconductor photocatalysts play two crucial roles: (1) capturing photoexcited carriers (either electrons or holes ideally) from the photocatalysts and (2) reducing the activation energy of target redox reactions on the surface of photocatalysts.6 In terms of the latter role, vast knowledge accumulated in electrochemical catalysis for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) has provided us with useful principles for designing effective cocatalyst materials (e.g., Pt for the HER and IrO2 for the OER).7 However, in terms of the former role, a versatile strategy has not yet been established for cocatalysts to capture photoexcited carriers selectively because metal–semiconductor interfacial charge transfer consists of highly complex processes.8,9 Actually, even the most commonly used Pt cocatalyst captures photogenerated holes as well as photoexcited electrons in some cases, which causes undesirable charge recombination on Pt.10–13 Therefore, photocatalytic water splitting systems with high efficiencies have been achieved based on the specially designed photocatalyst particles, where HER and OER cocatalysts are loaded on different crystal facets selectively by leveraging the photocatalytic deposition technique. Specifically, the Al-doped SrTiO3 particle, which is synthesized using a flux method, possesses facet-dependent redox properties, and the photoexcited electrons accumulate on the {100} facet. Thus, it reduces metal cations to produce an HER cocatalyst (i.e., Rh/Cr2O3) on that facet, while the photogenerated holes accumulate on the {110} facet, producing an OER cocatalyst oxidatively (i.e., CoOOH) on it. Therefore, HER and OER cocatalysts selectively capture the photoexcited carriers that are desirable for photocatalytic water splitting. However, this strategy cannot be applied to other systems unless the photocatalyst material possesses a built-in and/or facet-dependent charge separation ability (e.g., decahedral-shaped BiVO414).The abovementioned Rh/Cr2O3 cocatalyst with a core–shell structure is one of the most widely used HER cocatalysts for achieving overall water splitting (i.e., simultaneous evolution of H2 and O2) on various photocatalyst materials owing to its excellent resistance to reverse reactions.4,15,16 Unfavorable reactions involving O2 molecules, such as re-reduction of O2 and/or catalytic formation of H2O from H2 and O2 on conventional metallic HER cocatalysts (e.g., Rh and Pt), are one of the major issues in photocatalytic water splitting. Domen et al. demonstrated that a thin layer of Cr2O3·nH2O (denoted hereafter as CrOx for brevity) coated on Rh (or Pt) effectively suppresses unfavorable reverse reactions on metal species because the layer is permeable to H+ but not to O2, preventing O2 molecules from reaching the metal surface.17
Although the vital role of the CrOx shell as a physical blocker of O2 molecules has gained broad acceptance, we have noticed unaccountable results through our attempt to apply this Rh/CrOx cocatalyst to a layered oxyhalide Bi4NbO8Cl photocatalyst possessing appropriate properties for visible-light water-splitting.18 Specifically, the subsequent loading of CrOx onto Rh increases the rate of H2 evolution on Bi4NbO8Cl in the presence of methanol as a sacrificial electron donor, where O2 does not evolve. In this study, we reveal an unexplored but favorable function of CrOx layers as a selective electron transport pathway from photocatalysts to the Rh core (i.e., reduction site), based on the results of varied Rh/CrOx-type cocatalysts loaded on Bi4NbO8Cl, which were characterized by X-ray absorption near edge structure spectroscopy, X-ray photoelectron spectroscopy, transmission electron microscopy, and time-resolved spectroscopy.
Results and discussion
Photodeposition of Rh and CrOx on Bi4NbO8Cl
Bi4NbO8Cl was synthesized using a flux method (Fig. S1, ESI†).19 Rh and Cr species were loaded on Bi4NbO8Cl via photodeposition using Na3RhCl6, Cr(NO3)3·9H2O (CrIII), and K2CrO4 (CrVI) as precursors. Notably, the conventional core–shell Rh/CrOx cocatalysts were prepared mostly via stepwise photoreduction by photoexcited electrons, where RhIII species (i.e., Na3RhCl6) were first reduced to form core Rh metal particles on the photocatalyst surface, and then CrVI species (K2CrO4) were reduced to CrIII species by the electrons that accumulate on the Rh to form a shell on the Rh surface. It has been suggested that the shell layers formed on Rh mainly consist of a mixture of Cr2O3·nH2O, while we herein describe them as CrOx for simplicity, as in previous reports.20 In addition to the high-valent CrVI precursor, we employed another precursor, Cr(NO3)3, containing low-valent (CrIII) species. This is based on the expectation that the hydroxide ions (OH−) produced via the photocatalytic reduction of NO3− on Rh (NO3− + H2O + 2e− → NO2− + 2OH−) can react with the CrIII species to produce Cr(OH)3, finally forming CrOx-like species on the surface of the Rh core, as demonstrated in the coating of photocatalyst surface with CrOx-like layers.21 The samples prepared via the stepwise method using CrIII and CrVI will be hereafter denoted as Rh/CrIII and Rh/CrVI, respectively. Herein, we also employed simultaneous deposition in which the photocatalyst particles were irradiated in a solution containing both Rh and Cr precursors (denoted as Rh + CrIII or Rh + CrVI). To the best of our knowledge, simultaneous deposition using CrIII as a precursor has not been investigated so far, while that with CrVI has been reported.22Fig. 1 shows the Rh–K and Cr–K edge XANES spectra of the prepared samples. All the samples, except for Rh + CrVI, were confirmed to consist of the Rh metal species based on a comparison with the spectrum of the Rh metal foil. The spectrum of the Rh + CrVI sample resembles that of the reference Rh2O3, indicating the insufficient reduction of the Rh precursor by photoexcited electrons. This is probably due to the preferential reduction of CrVI (CrO42− + 5H+ +3e− ⇄ Cr(OH)3 + H2O; E° = 1.386 V). The Cr–K spectra of all prepared samples were in good agreement with that of the reference Cr2O3·nH2O (Fig. 1b) sample, as well as with previous data on CrOx loaded GaN:ZnO.17,23 The EXAFS spectra further supported this conclusion (Fig. S2, ESI†). A trivalent Cr species is also observed by the X-ray photoelectron spectroscopy (XPS) analysis (Fig. S3, ESI†).
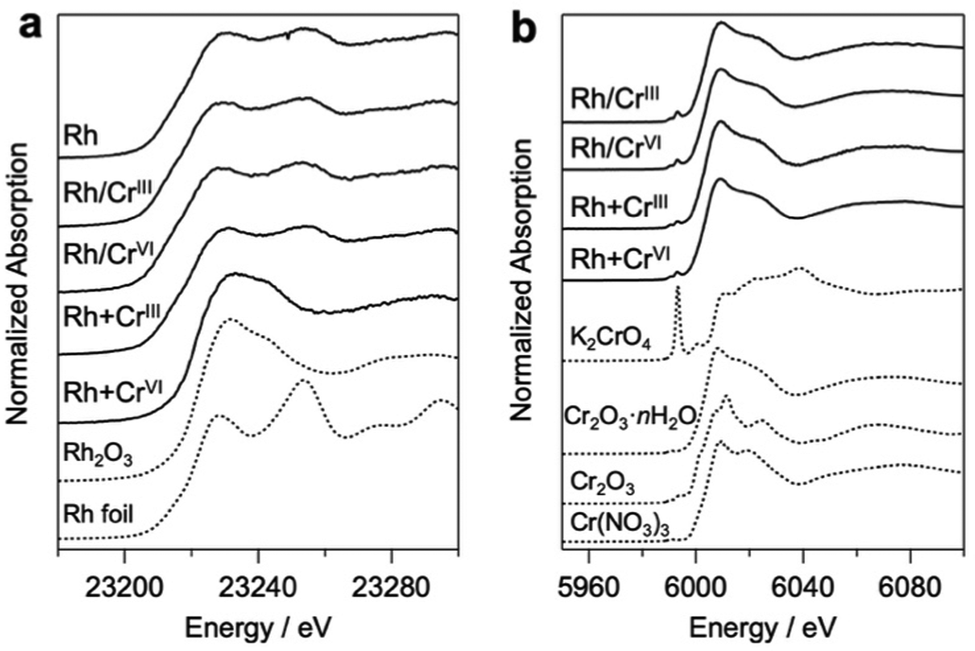 | ||
| Fig. 1 (a) Rh–K and (b) Cr–K edge XANES spectra of Rh and Cr-loaded Bi4NbO8Cl prepared via stepwise (Rh/CrIII, Rh/CrVI) and simultaneous deposition (Rh + CrIII, Rh + CrVI). The reference samples are shown as dotted lines. Cr2O3·nH2O was obtained via a precipitation reaction where NaOH was added to a Cr(NO3)3 solution. | ||
As shown in Fig. 2a, Rh particles were deposited as agglomerates with a size of approximately 20 nm when Rh was solely loaded on Bi4NbO8Cl; the lattice fringes ensured the zero-valent state of Rh (Fig. S4, ESI†). Stepwise loading of the Cr precursor (Rh/CrIII and Rh/CrVI) formed an amorphous CrOx layer (Fig. 2b and c) while maintaining the morphology and size of the Rh core. HAADF-STEM and EDX elemental mapping images confirmed the Rh/Cr core/shell structure (Fig. S5 and S6, ESI†). In stark contrast, the simultaneous deposition of the CrIII precursor (Rh + CrIII) provided differently shaped particles, where Rh of a few nanometer size was enclosed by an amorphous CrOx shell (Fig. 2d–f and Fig. S7, ESI†). In each case, an easier reduction reaction on Rh than on the bare surface of Bi4NbO8Cl may contribute to CrOx growth around Rh nanoparticles. Although some large particles were also observed (Fig. S8, ESI†), their number was small (less than 5.2% of the observed 200 particles). As for Rh + CrVI, only amorphous particles were deposited (Fig. S4, ESI†), which may be RhIII–CrIII mixed-oxides by considering the XANES spectra (Fig. 1a).22,24
 | ||
| Fig. 2 HR-TEM images of cocatalysts loaded on (a) Rh, (b) Rh/CrIII, (c) Rh/CrVI, and (d) Rh + CrIII samples. (e) HAADF-STEM and (f) EDX elemental mapping images of the Rh + CrIII sample. | ||
Impact of the CrOx shell on the H2 evolution activity of the Rh-loaded Bi4NbO8Cl photocatalyst
H2 evolution activities of the prepared samples were evaluated in an aqueous methanol solution (MeOH aq.) where the photoexcited electrons and holes are expected to react with water (or H+) and methanol, respectively. Fig. 3 shows the time courses of H2 evolution on various samples under visible light irradiation (λ > 400 nm). The first noteworthy fact is that the coating of Rh with the CrOx shell enhanced H2 evolution; the Rh/CrIII and Rh/CrVI samples showed higher rates than Rh. The sample with CrOx randomly deposited via the impregnation method instead of the photodeposition method showed a lower H2 evolution activity, which supports the function of the CrOx layer on Rh (Fig. S9, ESI†). The second and most important finding of this study is that the Rh + CrIII sample prepared via the unexplored simultaneous photodeposition of RhIII and CrIII precursors showed a significantly higher activity than the Rh-solely loaded and other CrOx-coated samples. The negligible activity of Rh + CrVI prepared via simultaneous photodeposition of RhIII and CrVI can be due to the insufficient reduction of the RhIII precursor, as indicated by the Rh K-edge XANES spectra (see Fig. 1).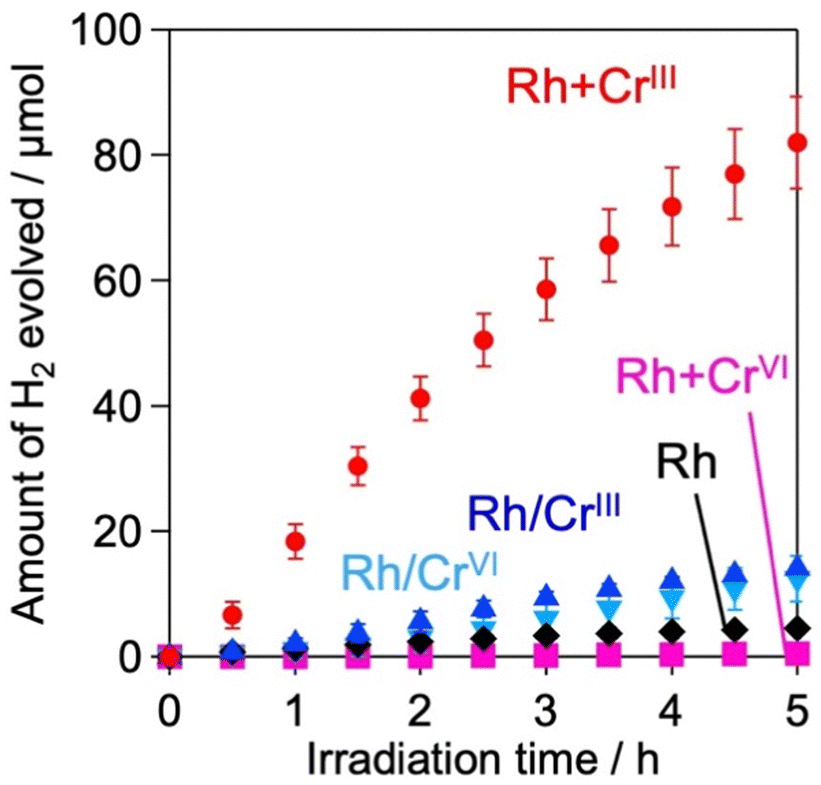 | ||
| Fig. 3 Time courses of H2 evolution of Rh (black), Rh/CrIII (blue), Rh/CrVI (light blue), Rh + CrIII, (red), and Rh + CrVI (pink) samples. The reactions were conducted in an aqueous methanol solution (20 vol%, 250 mL) under visible light irradiation (λ > 400 nm). Error bars signify the standard deviation. | ||
The enhancement in the H2-evolution reaction by CrOx coating on Rh cannot be explained by the widely accepted role of the CrOx shell as a physical blocker of O2 molecules because O2 evolution should not take place in the presence of an efficient hole scavenger, methanol. One possible factor is the deactivation of the Rh metal surface by some intermediates generated from methanol, such as formaldehyde, as suggested in the case of the Pt cocatalyst loaded on TiO2.25 However, H2 evolution on the Rh-loaded sample remained unchanged even in the presence of formaldehyde (Fig. S10, ESI†) in MeOH (aq.), which eliminates this possibility. The Bi4NbO8Cl sample loaded solely with CrOx species (via photodeposition of the CrVI precursor) did not show detectable H2 evolution from MeOH (aq.) under visible light irradiation (Fig. S11, ESI†), indicating negligible activity of CrOx as an HER cocatalyst.20,26 In addition, no H2 was generated over Rh-loaded Cr2O3·nH2O (i.e., in the absence of the Bi4NbO8Cl photocatalyst, see Fig. S12, ESI†), confirming the negligible activity of CrOx itself as a semiconductor photocatalyst under the present reaction conditions. The promotion of H2 evolution from MeOH (aq.) by CrOx-coating on Rh (prepared via simultaneous photodeposition of RhIII and CrIII precursors) was also observed on a conventional water-splitting photocatalyst SrTiO3 under UV light irradiation (Fig. 6), which demonstrates that this unexplored effect of the CrOx shell is not specific to the Bi4NbO8Cl photocatalyst. Previous studies have reported enhanced H2 evolution from MeOH (aq.) by CrOx shells on Rh for GaN:ZnO and SrTiO3, while the detailed mechanism for enhancement has not been discussed.17,27 These facts strongly suggest the presence of a previously unknown function of the CrOx shell in enhancing H2 evolution (i.e., reduction of H+ and/or water), in addition to its function as a physical blocker.
Spectroscopic elucidation of the unexplored function of the CrOx shell
Based on these results and previous reports that CrOx does not to promote the catalytic HER,20,26 we focused on the effect of the loaded CrOx on the charge carrier (either electrons or holes) transfer process rather than the catalytic process in photocatalysis.20,26 We employed transient absorption (TA) spectroscopy28–30 to evaluate the effect of CrOx on the carrier dynamics between a photocatalyst (Bi4NbO8Cl) and its catalytic site (Rh). Fig. 4a shows the TA spectra of bare Bi4NbO8Cl, where bandgap excitation induces two absorption increases in the infrared (IR) (2000 cm−1) and visible (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 cm−1) regions. Referring to previous results (e.g., TiO2,31–33 α-Fe2O334 and LaTiO2N28), the IR features have been attributed to photogenerated free electrons in the conduction band (CB) and/or shallowly trapped electrons,35,36 while the visible features have been associated with photogenerated holes.29,30
800 cm−1) regions. Referring to previous results (e.g., TiO2,31–33 α-Fe2O334 and LaTiO2N28), the IR features have been attributed to photogenerated free electrons in the conduction band (CB) and/or shallowly trapped electrons,35,36 while the visible features have been associated with photogenerated holes.29,30
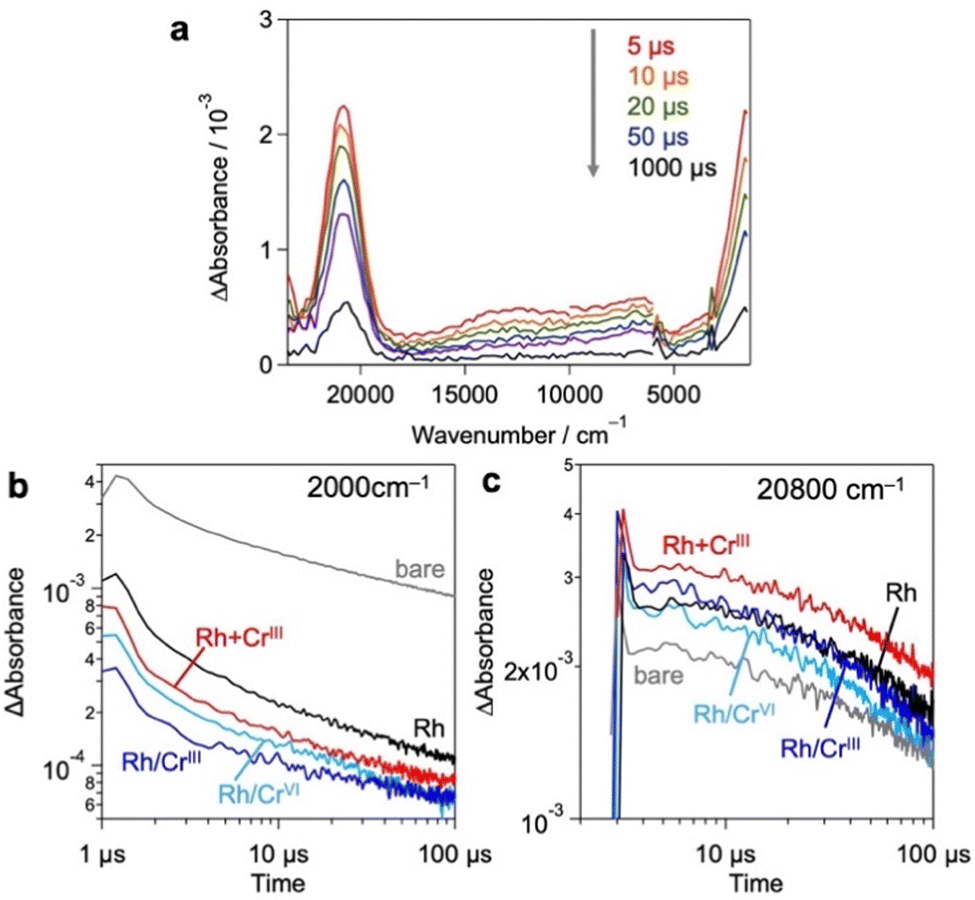 | ||
| Fig. 4 (a) TA spectra of bare Bi4NbO8Cl after excitation. (b and c) Decay kinetics of the transient absorption in the μ-second regions measured at (b) 2000 cm−1 and (c) 20800 cm−1 in N2 (20 torr). The samples were excited by UV laser pulses (355 nm). | ||
Fig. 4b and c show the decay kinetics of the IR (2000 cm−1 for electrons) and visible (20800 cm−1 for holes) regions, respectively, for the various samples. Compared to the bare Bi4NbO8Cl, the sole loading of Rh metal species decreased the signal intensity of electrons but increased that of holes, indicating the preferential capture of photoexcited electrons by Rh, as reported previously.13 The deposition of CrOx shells on the Rh-loaded Bi4NbO8Cl (Rh/CrIII and Rh/CrVI) further decreased the electron signal but had little impact on the hole signal. Given the fact that the CrOx shells on the pre-loaded Rh increased the rate of H2 evolution despite the inertness of CrOx for the HER,20,26 it appears that CrOx captures the photoexcited electrons from Bi4NbO8Cl, transfers them to the Rh core, and consequently increases the number of electrons for the HER. More importantly, in the Rh + CrIII sample, which shows the highest H2 evolution activity, the signal of holes was further increased compared to that of the Rh-loaded sample, whereas the electron signal was decreased as with the case of Rh/CrIII and Rh/CrVI. This phenomenon suggests that the charge separation between the holes on Bi4NbO8Cl and the electrons on Rh was further improved by the CrOx species loaded via the unexplored simultaneous photodeposition of the RhIII and CrIII precursors, which is consistent with the highest H2 evolution activity of this sample. In other words, the function of CrOx, selective electron transfer from Bi4NbO8Cl to Rh, was more apparent in this sample. A similar tendency was also observed in an earlier process on a pico-second scale (Fig. S13, ESI†).
The electron-selective capturing ability of the CrOx shell was further supported by the TA values of the bare and CrOx-loaded Bi4NbO8Cl samples. As shown in Fig. S15 (ESI†), CrOx loading on the bare Bi4NbO8Cl surface significantly decreased the IR signal but increased the visible signal. These results on TA measurements strongly suggest that the CrOx species can selectively capture photoexcited electrons from Bi4NbO8Cl, prolonging the lifetime of the holes by promoting the spatial separation of electrons and holes.
Mechanism for significant enhancement in H2 production on the Rh/CrOx cocatalyst
Based on the above results (physicochemical properties, photocatalytic activity, and carrier dynamics), we now propose a previously unknown function of the CrOx shell as an electron transport layer between the semiconductor and the Rh core, as illustrated in Fig. 5.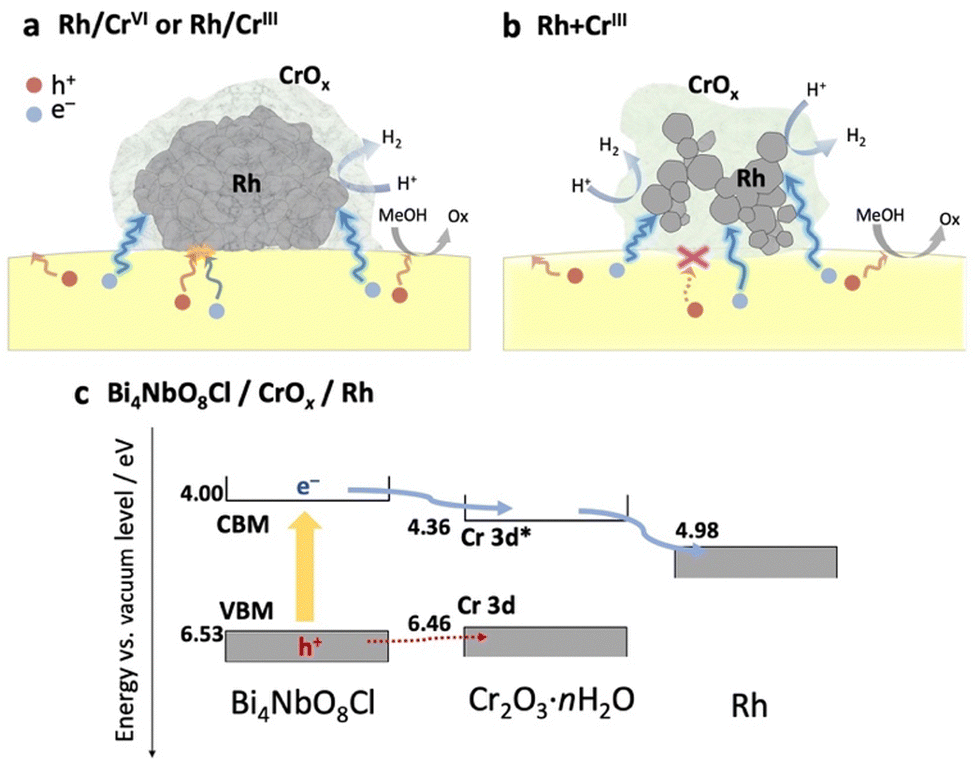 | ||
| Fig. 5 Schematic of the role of CrOx in the carrier transfer process from the semiconductor photocatalyst to Rh in (a) Rh/CrIII (or Rh/CrVI) and (b) Rh + CrIII. (c) Energy level diagram for Bi4NbO8Cl, Cr2O3·nH2O, and Rh. | ||
In the Rh/CrIII and Rh/CrVI samples, where CrOx was subsequently photodeposited on the pre-loaded Rh, the CrOx layers, as well as the Rh directly attached to Bi4NbO8Cl, capture the photoexcited electrons from Bi4NbO8Cl, serving as an electron path between Bi4NbO8Cl and Rh (Fig. 5a). Therefore, the CrOx layer on Rh facilitated electron transfer to Rh, increasing the number of electrons participating in H+ reduction on Rh, enhancing H2 evolution. Notably, a much higher H2 evolution rate was observed for the Rh + CrIII sample, where most of the Rh nanoparticles were enclosed by an amorphous CrOx shell, and the contact area between Rh and Bi4NbO8Cl was much smaller than that of the other samples (Fig. 2 and Fig. S8, ESI†). Given the TA results indicating the electron-selective capturing ability of CrOx, the CrOx intermediate between Bi4NbO8Cl and Rh in Rh + CrIII selectively passed electrons, thus boosting H2 evolution (Fig. 5b). Although undesirable hole transfer from Bi4NbO8Cl to Rh partially occurs when they are in direct contact with each other, as in the case of Pt,10–13 the insertion of the electron-selective pathway of CrOx effectively suppresses this hole transfer and therefore the problematic charge recombination at the semiconductor–metal interfaces. The function of CrOx as an electron-selective pathway was further supported by the enhanced H2 evolution on the sample where CrOx was first deposited on Bi4NbO8Cl followed by the Rh species (Fig. S16, ESI†). The sample showed a much higher activity than the Rh-loaded one despite the incomplete reduction to metal and agglomerated morphology of the Rh species, indicating the effectiveness of CrOx for improving the carrier transfer process.
The function of the CrOx shell as a “selective” electron transport layer is similar to those employed in solar cells to suppress charge recombination.37 Indeed, some chromium oxide species have been reported to serve as an electron transport layer between a semiconductor photo-absorber (e.g., lead-halide perovskite solar cells) and an electrode (e.g., fluorine-doped tin oxide (FTO) and Ag substrates).38,39 Generally, the function of an electron transport layer can be explained based on the band alignment among three components (i.e., photo-absorber, electron transport layer, and conductive electrode). The band levels of the CrOx species were estimated using a combination of diffuse reflectance spectroscopy and photoelectron yield spectroscopy (Fig. S17, ESI†). Cr2O3·nH2O particles were prepared via a precipitation method in which OH− was added to the solution containing Cr3+ ions to imitate the photodeposition process of CrOx from Cr3+ on Bi4NbO8Cl. The Cr–K edge XANES of the sample confirmed the formation of almost the same species as that loaded on Bi4NbO8Cl. Fig. 5c shows the estimated band alignments for Bi4NbO8Cl, Cr2O3·nH2O, and Rh. The CBM (Cr-3d*)40 position of Cr2O3·nH2O was favorable for receiving electrons generated in the CBM of Bi4NbO8Cl and injecting them into Rh. Given the band alignment, the holes, not only electrons, can transfer to Cr2O3·nH2O. However, the driving force (energy difference between the VBM and Cr-3d) is much smaller than that for the electrons (from the CBM to Cr-3d*), which is probably one of the origins of the preferred electron transfer in addition to other factors such as donor or acceptor density of the present CrOx species. In other words, the driving force of hole transfer (work function difference) from Bi4NbO8Cl to Rh (about 1.55 eV) can be reduced to 0.07 eV by introducing the CrOx layer, which can suppress the undesirable hole transfer from the photocatalyst to Rh.
From these results, we conclude that CrOx functions as a selective electron transport layer, promoting charge separation between the photocatalyst and noble metal reaction sites, thus boosting H2 evolution. The significant improvement in H2 evolution indicates that the CrOx layers formed via the present procedure (i.e., simultaneous photodeposition) possess substantial permeability to H+ (or other related species such as H3O+), as proposed in previous reports on conventional Rh/CrOx cocatalysts.4,15,16 Another important question arises: whether the CrOx layers formed via the present procedure can function as a physical blocker against O2 molecules, which will be discussed later.
Applicability of the CrOx layers formed via simultaneous deposition to other systems
The significant impact of CrOx layers on H2 evolution was also confirmed for other metal cores and semiconductors, as it was not specific to the combination of the Rh core and Bi4NbO8Cl. As for metal cores, we have previously revealed that conventional Pt cocatalysts cannot boost H2 production on Bi4NbO8Cl because Pt captures both electrons and holes, acting as a recombination center13 as shown in Fig. S18 (ESI†). When Pt and Cr species were simultaneously loaded on Bi4NbO8Cl (Pt + CrIII), most of the Pt particles were encapsulated by amorphous CrOx (Fig. S19 and S20, ESI†), as in the case of Rh + CrIII. The Pt + CrIII sample showed much improved H2 evolution compared to the Pt sample, which strongly suggests that the CrOx layer also helps selective electron transfer from Bi4NbO8Cl to Pt (Fig. S21, ESI†).The simultaneous photodeposition of CrOx and Rh on SrTiO3 (Rh + CrIII) also provides a higher H2 evolution rate from MeOH (aq.) compared to Rh-loaded SrTiO3 (Fig. S22, ESI†), whereas the enhancement was moderate compared to that of Bi4NbO8Cl (Fig. 6 and Table S3, ESI†). In contrast, the co-deposition of CrOx significantly lowered the H2 evolution rate of Rh-loaded rutile TiO2 (Fig. S24, ESI†) from MeOH (aq.). This stark contrast may support the abovementioned electron-transfer mechanism. Since the CBM level of SrTiO3 (ca. 4.2 eV against the vacuum level) is almost the same as that of Bi4NbO8Cl and higher than that of CrOx, the electron transfer from SrTiO3 to Rh through CrOx layers can be rationalized (see Fig. S25, ESI†). On the other hand, the CBM of rutile TiO2 is lower (ca. 4.37 eV against the vacuum level) than that of Bi4NbO8Cl and quite similar to that of CrOx, making the driving force of the electron transfer through the conduction band of CrOx almost negligible.
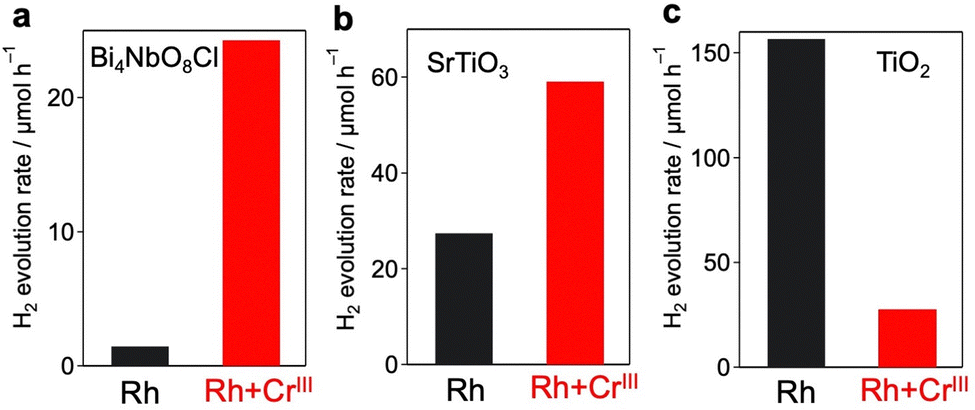 | ||
| Fig. 6 Effect of co-deposition of CrOx with Rh on the photocatalytic activity of (a) Bi4NbO8Cl (λ > 400 nm), (b) SrTiO3 (λ > 300 nm), and (c) rutile TiO2 (λ > 300 nm). | ||
As noted in the Introduction, the main role of the CrOx layer has been regarded as a physical blocker of O2 molecules to suppress reverse reactions such as water formation (from H2 and O2) and O2 reduction on noble metal cocatalysts. To investigate the role of the CrOx layers prepared via simultaneous photodeposition (i.e., Rh + CrIII), overall water splitting was performed using the SrTiO3 photocatalyst. As shown in Fig. S26 (ESI†), while the Rh-loaded SrTiO3 without CrOx does not show stoichiometric water splitting due to the undesirable reaction with O2 molecules on the exposed Rh,6 simultaneous CrOx deposition provided stoichiometric water splitting, which confirms the role of the CrOx shell as a physical blocker. Notably, the sample prepared using the present method (Rh + CrIII) showed slightly higher photocatalytic activity than the sample prepared using the conventional method (Rh/CrVI). Unfortunately, we have not yet succeeded in splitting pure water into H2 and O2 using the Bi4NbO8Cl samples loaded with these Rh/CrOx cocatalysts, probably because of the insufficient optimization of other factors such as the loading of an effective O2-evolution cocatalyst, which will be our future work.
Conclusions
In summary, we demonstrated that CrOx facilitates the transfer of photoexcited electrons from a photocatalyst to a noble metal reduction site. The CrOx layer on the pre-loaded Rh served as an electron path, facilitating electron transfer from the Bi4NbO8Cl photocatalyst to Rh, which resulted in improved H2 evolution activity under visible light. Notably, when Rh and CrOx are loaded simultaneously via photodeposition using a Cr3+ precursor, CrOx lies between Rh and Bi4NbO8Cl. Therein, the CrOx layer passes only electrons to Rh, suppressing undesirable hole transfer to Rh, which further improves the activity. Thus far, the facilitation of surface reactions (e.g., the HER and OER) has mainly been investigated as a function of the cocatalysts. However, we revealed the crucial role of CrOx in the carrier transfer process from the photocatalyst to the catalyst (reaction site). We believe that the precise control of the physicochemical properties of Cr species by doping,41 making solid solutions with other transition metal compounds,40 and employing other ligands (anions)42,43 will reveal more detailed dynamics in the carrier transport process from photocatalysts to noble metals to suppress the undesirable charge recombination more effectively. Employing other semiconductors having appropriate band edge positions as electron transport layers will also be an effective choice. This study offers a new approach for designing cocatalysts based on their carrier-capturing abilities.Experimental
Materials
Bi2O3 (99.99%), BiOCl (95.0%), Nb2O5 (99.9%), NaCl (99.5%), CsCl, methanol (99.8%), Rh2O3 (98.0%), K2CrO4 (99.0%), CrO3(99.5%), H2PtCl6·6H2O (99.9%), PtO2 (98.0%), and BN were purchased from FUJIFILM Wako Pure Chemicals. Na3RhCl6·nH2O (80.0%), Cr(NO3)3·9H2O (98.0%), and Cr2O3 (98.5%) were purchased from Kanto Chemical. SrTiO3 (99%) was purchased from Sigma-Aldrich. TiO2 (rutile, 99.99%) was purchased from High Purity Chemicals. Formaldehyde solution (37%) was purchased from Nacalai Tesque. The compound described as Cr2O3·nH2O was obtained from the precipitate formed by the addition of NaOH to the Cr(NO3)3 solution.Synthesis of Bi4NbO8Cl using the flux method
Bi4NbO8Cl was synthesized using the flux method, as reported in our previous work.19 Briefly, a stoichiometric mixture of Bi2O3, BiOCl, and Nb2O5 was mixed with flux (NaCl and CsCl in the molar ratio of 35:65) at 1 mol%. The mixture (25 g) was placed in a 30 mL alumina crucible, heated at a rate of 50 °C h−1 to 650 °C, and held at the final temperature for 10 h. The product was naturally cooled to room temperature, thoroughly washed with distilled water, filtered, and air-dried. The obtained powder was confirmed to be Bi4NbO8Cl by X-ray diffraction (XRD).
Loading of the co-catalyst
The prepared Bi4NbO8Cl (0.2 g) was dispersed in an aqueous methanol solution (20 vol%, 250 mL). Depending on the preparation conditions, the Rh precursor (Na3RhCl6·nH2O), Pt precursor (H2PtCl6·6H2O), and/or Cr precursor (Cr(NO3)3·9H2O or K2CrO4) were added to the solution. The amount of precursor added was 1.0 wt% for Rh (or Pt) and 1.5 wt% for Cr as a metal cation (Fig. S20, ESI†). After degassing, the solution was irradiated with visible light (λ > 400 nm) for 5 h at room temperature under continuous stirring. Irradiation was conducted using a 300 W Xe lamp equipped with a CM1 cold mirror and an L-42 cutoff filter. The product was filtered, washed thoroughly with distilled water, and air-dried. Absorption spectroscopy ensured that precursors for the Rh were fully consumed during the PD process, while the loading amounts of Cr were approximately 0.6, 1.3, and 1.3 wt% for Rh/CrVI, Rh/CrIII, and Rh + CrIII, respectively (Fig. S28, ESI†).CrOx was also loaded using the impregnation method. A certain amount of Rh/Bi4NbO8Cl powder was immersed in an aqueous solution containing Cr(NO3)3 (1.5 wt%). The suspension was evaporated under constant stirring until achieving complete dryness, followed by heating under an Ar flow at 150 °C.
Characterization
The prepared samples were studied by X-ray absorption fine structure (XAFS) spectroscopy, X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), and scanning electron microscopy (SEM). XAFS measurements were carried out at the AR-NW10A, BL9C, and BL12C beamlines of the Photon Factory (High Energy Accelerator Research Organization, Tsukuba, Japan). The X-ray energy was varied using a Si(111) double-crystal monochromator. The reference samples were diluted in boron nitride, compressed to form pellets (except for Rh foil), and measured in the transmission mode. Cocatalyst-loaded samples were measured in fluorescence mode using a multichannel solid-state detector. XPS measurements were carried out with 5500MT (ULVAC-PHI) using Mg Kα as the X-ray source. The spectra were calibrated with the 4f7/2 peak (84.0 eV) of Au deposited on the sample surface using a magnetron sputtering device (MSP-1S). TEM and SEM were conducted using a JEM-2100F (JEOL) and an Nvision 40 (Zeiss), respectively.Photocatalytic reaction
The photocatalytic reactions were performed in a closed circulation system. The sample (0.1 g) was dispersed in an aqueous methanol solution (20 vol%, 250 mL) in a Pyrex reaction vessel. After degassing, the solution was irradiated using a 300 W Xe lamp at room temperature with continuous stirring. In the case of Bi4NbO8Cl, the sample was irradiated (visible light (λ > 400 nm)) using a CM1 cold mirror and an L-42 cutoff filter. The evolved gases were analyzed using a gas chromatograph (GC-8A, SHIMADZU) with a thermal conductivity detector (TCD) using Ar as the carrier gas.Transient absorption (TA) measurements
TA measurements were performed using a custom-built spectrometer as previously described. Briefly, for microsecond measurements, samples fixed on a CaF2 plate were excited by a 355 nm laser pulse (0.5 mJ pulse energy, 0.5–1 Hz repetition rate). As a probe light, visible to near-IR light (25000–6000 cm−1) was irradiated from a halogen lamp and mid-IR light (6000–1000 cm−1) was irradiated from the MoSi2 coil. The transmitted or diffuse reflected probe light monochromated using a spectrometer was detected using Si, InGaAs, and MCT detectors in the visible, near-IR, and mid-IR regions, respectively. Picosecond measurements were performed using a Ti:sapphire laser system (Spectra-Physics, Solstice & TOPAS Prime; 90 fs duration; 500 Hz repetition rate). To excite the samples, 355 nm pulses (6 μJ pulse energy) were used. The probes 20800 cm−1 and 2000 cm−1 were detected using a photomultiplier and an MCT detector, respectively. All the TA measurements were performed under N2 (20 torr). The bare Bi4NbO8Cl sample used here was irradiated in a methanol solution in advance to obtain a light irradiation history similar to that of other photodeposited samples.
Author contributions
Tetsu Kotani: data curation, investigation, methodology visualization, and writing – original draft. Kanta Ogawa: conceptualization, investigation, methodology, and writing – original draft. Hajime Suzuki: writing – review & editing. Kosaku Kato: investigation. Osamu Tomita: investigation. Akira Yamakata: resources and data curation. Ryu Abe: resources, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the JST-CREST project, the JSPS KAKENHI Grant-in-Aid for Scientific Research (A) (JP20H00398), the JSPS KAKENHI Grant Number 17H06439 for Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion (I4LEC),” the JSPS Research Fellowship (Grant Number 19J23357), and the JSPS Core-to-Core Program (JPJSCCA20200004). This work was also supported by the Iketani Science and Technology Foundation and the TEPCO Memorial Foundation. A part of this work was supported by the Advanced Research Infrastructure for Materials and Nanotechnology in Japan (JPMXP1222KU0026). We acknowledge Prof. Shunsuke Nozawa of KEK for his helpful support in XAFS measurement. We are also grateful to Takaaki Toriyama of Kyushu University for his helpful support in the STEM analysis.Notes and references
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000–1030 CrossRef CAS.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- R. Bala Chandran, S. Breen, Y. Shao, S. Ardo and A. Z. Weber, Energy Environ. Sci., 2018, 11, 115–135 RSC.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. Shang, T. Wang, Y. Xiao, Z. Dong, X. Li and B. Li, J. Alloys Compd., 2021, 875, 159998 CrossRef CAS.
- K. Maeda and K. Domen, Bull. Chem. Soc. Jpn., 2016, 89, 627–648 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- K. Wu, H. Zhu, Z. Liu, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218–13219 CrossRef CAS PubMed.
- S. Schäfer, S. A. Wyrzgol, R. Caterino, A. Jentys, S. J. Schoell, M. Hävecker, A. Knop-Gericke, J. A. Lercher, I. D. Sharp and M. Stutzmann, J. Am. Chem. Soc., 2012, 134, 12528–12535 CrossRef PubMed.
- F. Yan, Y. Wang, J. Zhang, Z. Lin, J. Zheng and F. Huang, ChemSusChem, 2014, 7, 101–104 CrossRef CAS PubMed.
- K. Ogawa, R. Sakamoto, C. Zhong, H. Suzuki, K. Kato, O. Tomita, K. Nakashima, A. Yamakata, T. Tachikawa, A. Saeki, H. Kageyama and R. Abe, Chem. Sci., 2022, 13, 3118–3128 RSC.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- Z. Wang, Y. Inoue, T. Hisatomi, R. Ishikawa, Q. Wang, T. Takata, S. Chen, N. Shibata, Y. Ikuhara and K. Domen, Nat. Catal., 2018, 1, 756–763 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 CrossRef CAS PubMed.
- K. Ogawa, A. Nakada, H. Suzuki, O. Tomita, M. Higashi, A. Saeki, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 5642–5650 CrossRef CAS PubMed.
- M. Qureshi, T. Shinagawa, N. Tsiapis and K. Takanabe, ACS Sustainable Chem. Eng., 2017, 5, 8079–8088 CrossRef CAS.
- S. Chu, R. T. Rashid, X. Liu and Z. Mi, Chem. Commun., 2019, 55, 6305–6308 RSC.
- K. Maeda, D. Lu, K. Teramura and K. Domen, Energy Environ. Sci., 2010, 3, 471–478 RSC.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem. – Eur. J., 2010, 16, 7750–7759 CrossRef CAS PubMed.
- K. Maeda, D. Lu, K. Teramura and K. Domen, J. Mater. Chem., 2008, 18, 3539 RSC.
- H. Huang, J. Feng, S. Zhang, H. Zhang, X. Wang, T. Yu, C. Chen, Z. Yi, J. Ye, Z. Li and Z. Zou, Appl. Catal., B, 2020, 272, 118980 CrossRef CAS.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- Y.-J. Cho, G. Moon, T. Kanazawa, K. Maeda and W. Choi, Chem. Commun., 2016, 52, 9636–9639 RSC.
- A. Yamakata, M. Kawaguchi, N. Nishimura, T. Minegishi, J. Kubota and K. Domen, J. Phys. Chem. C, 2014, 118, 23897–23906 CrossRef CAS.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- K. Murofushi, K. Ogawa, H. Suzuki, R. Sakamoto, O. Tomita, K. Kato, A. Yamakata, A. Saeki and R. Abe, J. Mater. Chem. A, 2021, 9, 11718–11725 RSC.
- D. W. Bahnemann, M. Hilgendorff and R. Memming, J. Phys. Chem. B, 1997, 101, 4265–4275 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- Y. Tamaki, A. Furube, M. Murai, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2006, 128, 416–417 CrossRef CAS PubMed.
- M. Barroso, C. A. Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi, K. Sivula, M. Grätzel, D. R. Klug and J. R. Durrant, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15640–15645 CrossRef CAS PubMed.
- J. I. Pankove, Optical Processes in Semiconductors, Dover Publication, New York, 1975 Search PubMed.
- P. K. Basu, Theory of Optical Processes in Semiconductors, Oxford University Press, New York, 1997 Search PubMed.
- K. Wang, S. Olthof, W. S. Subhani, X. Jiang, Y. Cao, L. Duan, H. Wang, M. Du and S. (Frank) Liu, Nano Energy, 2020, 68, 104289 CrossRef CAS.
- S. Zheng, W. Li, T. Su, F. Xie, J. Chen, Z. Yang, Y. Zhang, S. Liu, M. P. Aldred, K. Y. Wong, J. Xu and Z. Chi, Sol. RRL, 2018, 2, 1700245 CrossRef.
- J. Dong, J. Wu, J. Jia, X. He, Z. Lan, L. Fan, J. Lin and M. Huang, ChemSusChem, 2018, 11, 619–628 CrossRef CAS PubMed.
- Y. Wang, K. Lopata, S. A. Chambers, N. Govind and P. V. Sushko, J. Phys. Chem. C, 2013, 117, 25504–25512 CrossRef CAS.
- E. Arca, A. B. Kehoe, T. D. Veal, A. Shmeliov, D. O. Scanlon, C. Downing, D. Daly, D. Mullarkey, I. V. Shvets, V. Nicolosi and G. W. Watson, J. Mater. Chem. C, 2017, 5, 12610–12618 RSC.
- S. Ohkoshi, K. Nakagawa, K. Tomono, K. Imoto, Y. Tsunobuchi and H. Tokoro, J. Am. Chem. Soc., 2010, 132, 6620–6621 CrossRef CAS PubMed.
- L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin, Z. Yu and X. Wang, Nat. Catal., 2020, 3, 649–655 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM and TEM images, photocatalytic activity, and TA spectroscopy data. See DOI: https://doi.org/10.1039/d2ey00109h |
| ‡ T. K. and K. O. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |