
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultra-selective 1D clean in-phase correlation spectroscopy†
Daniel A.
Taylor
 a,
Peter
Kiraly
b,
Paul
Bowyer
c,
Mathias
Nilsson
a,
Laura
Castañar
ad,
Gareth A.
Morris
a and
Ralph W.
Adams
*a
a,
Peter
Kiraly
b,
Paul
Bowyer
c,
Mathias
Nilsson
a,
Laura
Castañar
ad,
Gareth A.
Morris
a and
Ralph W.
Adams
*a
aDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: ralph.adams@manchester.ac.uk
bJEOL UK Ltd., Bankside, Long Hanborough, OX29 8LJ, UK
cJEOL UK Ltd., Silver Court, Welwyn Garden City, AL7 1LT, UK
dDepartment of Organic Chemistry, Faculty of Chemical Science, Complutense University of Madrid, Ciudad Universitaria s/n, 28040 Madrid, Spain
First published on 20th April 2023
Abstract
Selective 1D COSY can unambiguously identify coupled spins but is often limited both by lack of selectivity, and by unfavourable multiplet lineshapes. Here, ultra-selective GEMSTONE excitation is employed with CLIP-COSY to provide through-bond correlations for nuclei whose NMR signals overlap. The new method is illustrated using the coccidiostat lasalocid and the immunosuppressant cyclosporin.
NMR spectroscopy has found widespread use in chemistry, for example in chemical analysis and reaction monitoring, wherever an understanding of sample composition is required. It provides detailed structural information on molecules in their solution state conformation(s). For simple molecules, structure elucidation is straightforward, in principle requiring only a simple pulse-acquire 1H spectrum to establish through-bond connectivity from signal multiplicity. Extraction of this information from spectra of complex molecules, or mixtures, is much more difficult due to signal overlap. This is caused by the combination of an abundance of signals dispersed across a narrow chemical shift range, and extensive 1H–1H scalar (J) coupling. Many experiments have been developed for resolving overlapped signals, including multidimensional1 and pure shift NMR methods,2–4 though usually at the expense of increased experiment time and/or loss of structural information.
2D COSY (correlation spectroscopy),1 for instance, correlates signals of spins that share a homonuclear J-coupling. Resolution is increased by dispersing signals into a second frequency dimension, but additional time is required to sample this dimension – several hours where high digital resolution is required to overcome signal overlap. 2D pure shift NMR experiments5–8 can reduce the time needed to sample the second dimension by suppressing signal multiplicity. These methods provide clear and simple correlations, but reduce the chemical information content of the spectrum, and almost always incur a sensitivity penalty. A further limitation of COSY is that the basic spectral lineshape is a mixture of absorption and dispersion mode.9 More desirable absorption mode lineshapes are obtained with a number of COSY variants,10–14 but their anti-phase nature can result in signal cancellation, particularly when J is comparable to, or less than, the linewidth. Double-quantum filtered in-phase cross peak COSY15 and in-phase COSY16 experiments both provide optimal in-phase absorption mode multiplets, but the former is limited by the experiment time required and the latter by the resolution achievable.
The most practical methods for pure absorption mode through-bond correlations are CLIP-COSY (clean in-phase COSY)17 and TOCSY (total correlation spectroscopy).18 TOCSY methods disperse magnetisation throughout the entire spin system during an isotropic mixing period. This relayed correlation of all protons within a given spin system is useful for identifying individual spin systems but complicates the unambiguous assignment of individual resonances. TOCSY is best suited to studies of large molecules, such as proteins, which contain large numbers of small spin systems. In small molecules, which paradoxically tend to have larger spin systems, a series of experiments with different mixing times can be used to distinguish between direct and relayed correlations but is expensive in experiment time. 1D selective COSY methods,19,20 which excite and provide direct correlations to a single signal, are much faster but require the selected signal to be well resolved.
GEMSTONE (gradient-enhanced multiplet-selective targeted-observation NMR experiment)21 spatially encodes an NMR sample to provide chemical shift selective filter22,23 selectivity in a single scan, and enables the selection of a single multiplet even when overlapped by other multiplets with slightly different chemical shifts, provided there is not severe strong coupling. GEMSTONE requires only a chemical shift difference to discriminate between signals. We have previously combined this ultra-selective excitation technique with conventional21 and rotating-frame24 nuclear Overhauser effect spectroscopy and TOCSY25 experiments to identify through-space and through-bond correlations, respectively, to spins whose signals overlap. Here we demonstrate the utility of GEMSTONE in a new experiment, GEMSTONE CLIP-COSY, to identify directly coupled spins when signals are severely overlapped. The conventional selective 1D COSY experiment uses band-selective excitation, which often fails to distinguish between overlapped resonances, and gives broad signals with dispersion-mode character. These limitations have dissuaded many chemists from using selective 1D COSY but both are now overcome by GEMSTONE CLIP-COSY. The new method provides chemical shift selective excitation, and generates spectra with pure in-phase absorption mode lineshapes. The resultant 1D COSY spectra contain clean multiplets that allow easy and accurate measurement of J values, simplifying spectral analysis.
The usefulness of the GEMSTONE CLIP-COSY method is most apparent when signals of spins that form part of a large spin system are severely overlapped, as shown in Fig. 1 for the coccidiostat lasalocid. In this example, the 1H NMR signals (Fig. 1a) of three prochiral methylene protons 17S, 20, and 30 overlap significantly, even at 600 MHz. Conventional selective excitation is not feasible for such overlapping multiplets, even though the pure shift NMR spectrum in Fig. 1b (measured straightforwardly using a simple band-selective method26–29) shows that there are three distinct chemical shifts. The chemical shifts define the excitation frequencies at which to apply GEMSTONE, allowing GEMSTONE CLIP-COSY to reveal the direct couplings to each proton, as shown for 17S in Fig. 1c.
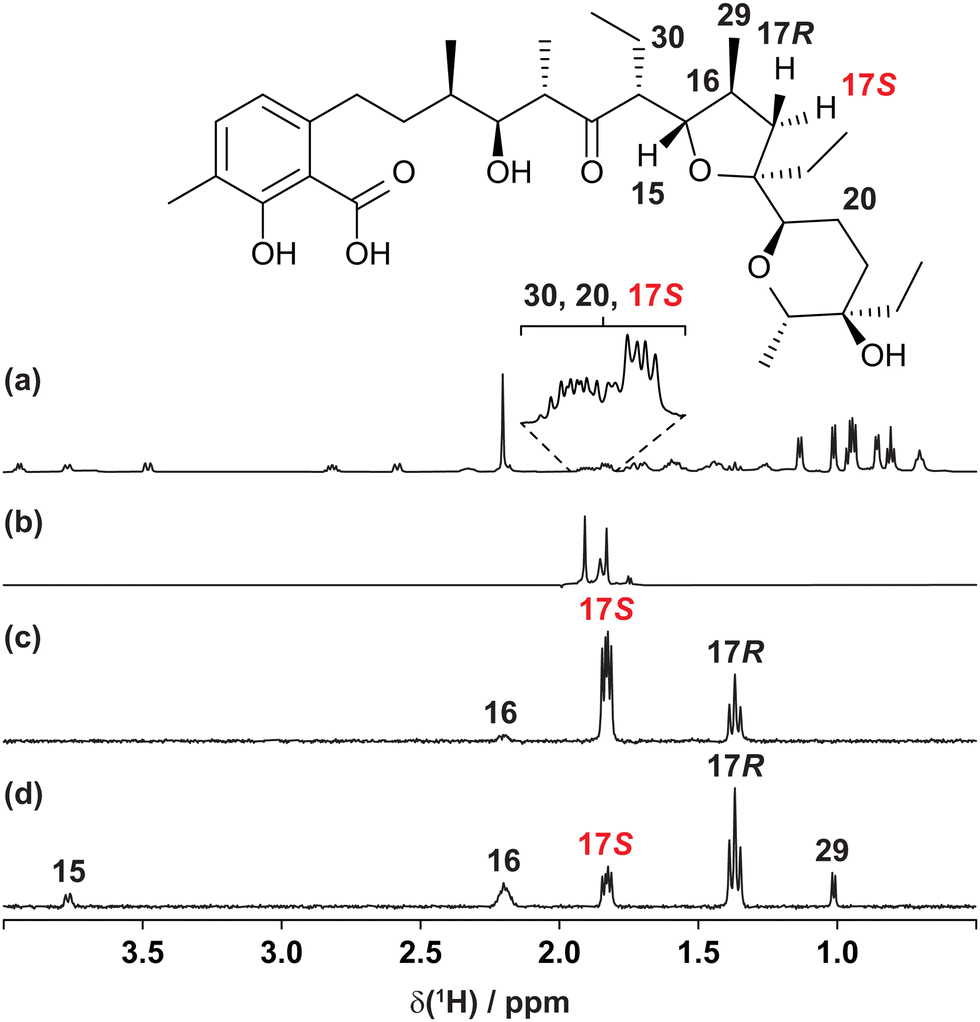 | ||
| Fig. 1 (a) Conventional 600 MHz 1H, (b) interferogram band-selective pure shift, (c) GEMSTONE CLIP-COSY and (d) GEMSTONE TOCSY NMR spectra of lasalocid in CDCl3. In (c) and (d) the transmitter frequency (indicated by the coloured label) was placed on the signal of proton 17S. The CLIP-COSY mixing time (τm) in (c) was 120 ms, whilst the TOCSY mixing time in (d) was 30 ms. Full experimental details are provided in the ESI.† | ||
Short mixing time TOCSY experiments are sometimes used in an attempt to identify direct couplings, but relayed correlations can still arise. This is illustrated in the GEMSTONE TOCSY spectrum of Fig. 1d, where 17S is excited and the brief 30 ms TOCSY mixing period transfers magnetisation not only to the directly coupled spins 16 and 17R but also to the distant spins 15 and 29. Reliable identification of direct couplings using TOCSY is time-consuming, requiring careful analysis of results obtained with multiple mixing times. In contrast, CLIP-COSY spectra show only direct correlations, the choice of mixing time (τm) determining only the amplitudes, not the nature, of the signals observed. In Fig. 1c, for example, a long τm (120 ms) still only transfers magnetisation to directly coupled spins. The advantages of CLIP-COSY over TOCSY are particularly marked when identifying connectivity via small homonuclear couplings, as the long mixing times needed favour relayed magnetisation transfer in TOCSY experiments.
The pulse sequence of the GEMSTONE CLIP-COSY experiment is shown in Fig. 2. The selectivity of GEMSTONE is achieved by application of a pair of frequency-swept RF (radiofrequency) pulses in the presence of a weak magnetic field gradient. The combined effect is to refocus chemical shift evolution for on-resonance signals only; off-resonance signals acquire position-dependent phase shifts so do not contribute to the net signal during acquisition. Selectivity is controlled by the durations (τp) of the adiabatic RF pulses used: longer pulses increase selectivity but reduce sensitivity through relaxational, convectional and diffusional signal loss. Compromise between selectivity and sensitivity is intrinsic to selective excitation, and applies in the same way to traditional selective pulse methods. For the best compromise between selectivity and sensitivity, the gradient amplitude should be matched to the adiabatic sweep width. This sweep width is chosen to be at least twice the frequency difference between the on-resonance signal and the most distant coupled signal, to ensure that both adiabatic RF pulses invert the magnetisation of all coupled spins everywhere in the sample. The conventional band-selective pulse refocuses unwanted J-evolution for the on-resonance signal but does not contribute to the selectivity of the experiment (unlike in conventional selective echo excitation) and need only be selective enough to avoid affecting the resonance of any coupling partner.
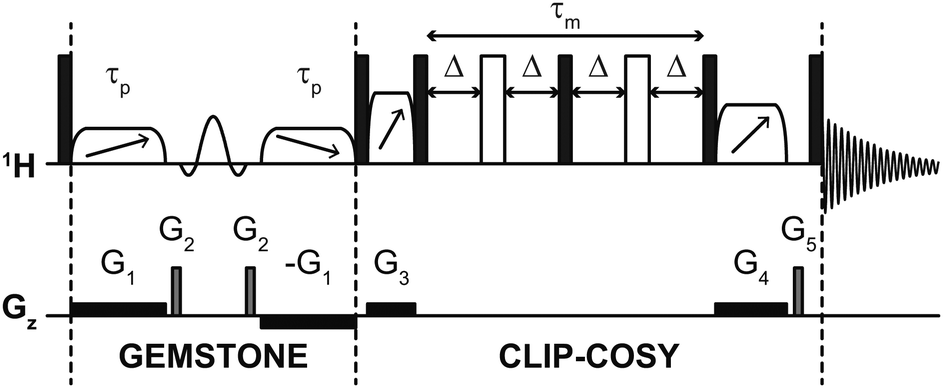 | ||
| Fig. 2 Schematic pulse sequence for GEMSTONE CLIP-COSY. Filled and open rectangles, elements with diagonal arrows, and sinc shapes represent hard 90° and 180°, adiabatic 180° and band-selective 180° pulses, respectively. The first two adiabatic pulses (of duration τp) control the selectivity of GEMSTONE; higher selectivity is achieved with a longer τp. The COSY mixing time is denoted τm and is four times the transfer delay Δ. The pulse program and all data files are available from DOI: https://doi.org/10.48420/21909600. | ||
The CLIP-COSY element17 consists of a perfect echo mixing sequence flanked by zero-quantum suppression elements,30 which ensures in-phase to in-phase coherence transfer between directly coupled spins and removes antiphase and zero-quantum contributions from the detected signal. In a two-spin system, optimal in-phase to in-phase coherence transfer is achieved with transfer delay Δ = 1/4J. For most samples, however, any given spin is J-coupled to multiple other spins. Unless these couplings are identical in magnitude, no single Δ is optimum for complete magnetisation transfer to all coupled spins; a compromise value must be used. A transfer delay of 10 ms, which corresponds to a 40 ms (4Δ) CLIP-COSY mixing time, generally works well for small molecules with typical 3JHH values of up to 10 Hz, and allows the signals of excited and coupled spins to be observed simultaneously. Other COSY variants can also be combined with GEMSTONE (see ESI†).
The GEMSTONE CLIP-COSY method is further illustrated using the cyclic peptide cyclosporin. The conventional 1H NMR spectrum (Fig. 3a) contains signals from 5α, 7α, and 8α between 4.7 and 4.9 ppm. The smallest chemical shift difference is 14 Hz (between 8α and 7α at 300 MHz). The multiplets are over 22 Hz wide, so they overlap. Traditional selective refocusing pulses19,31,32 are therefore unable to discriminate between the three overlapped α-proton signals. To achieve sufficient selectivity with a conventional shaped pulse it would have to be line-, rather than multiplet-, selective. This would excite only a small fraction of the available signal and would also typically lead to lineshape distortions. When multiplet-selective excitation with a shaped pulse is attempted (Fig. 3c), multiple signals are excited simultaneously, leading to uncertainty in signal assignment. In contrast, GEMSTONE excites each individual multiplet independently without perturbing the overlapping resonances. A GEMSTONE CLIP-COSY spectrum is obtained (Fig. 3d–f) for each signal without interference from those nearby, enabling unambiguous identification of through-bond connectivity. The in-phase absorption mode lineshapes provided by the experiment allow the direct measurement of J values, affording insight into dihedral torsion angles through the Karplus equation.33 The method is particularly suited to coupling partners whose signals both lie in overlapped regions of the 1H spectrum. In such cases, GEMSTONE CLIP-COSY provides a particularly significant time saving over its 2D parent. The 1D experiment has the minimum duration of a single increment of the parent 2D experiment, but many increments must be acquired in the latter to resolve the signal overlap.
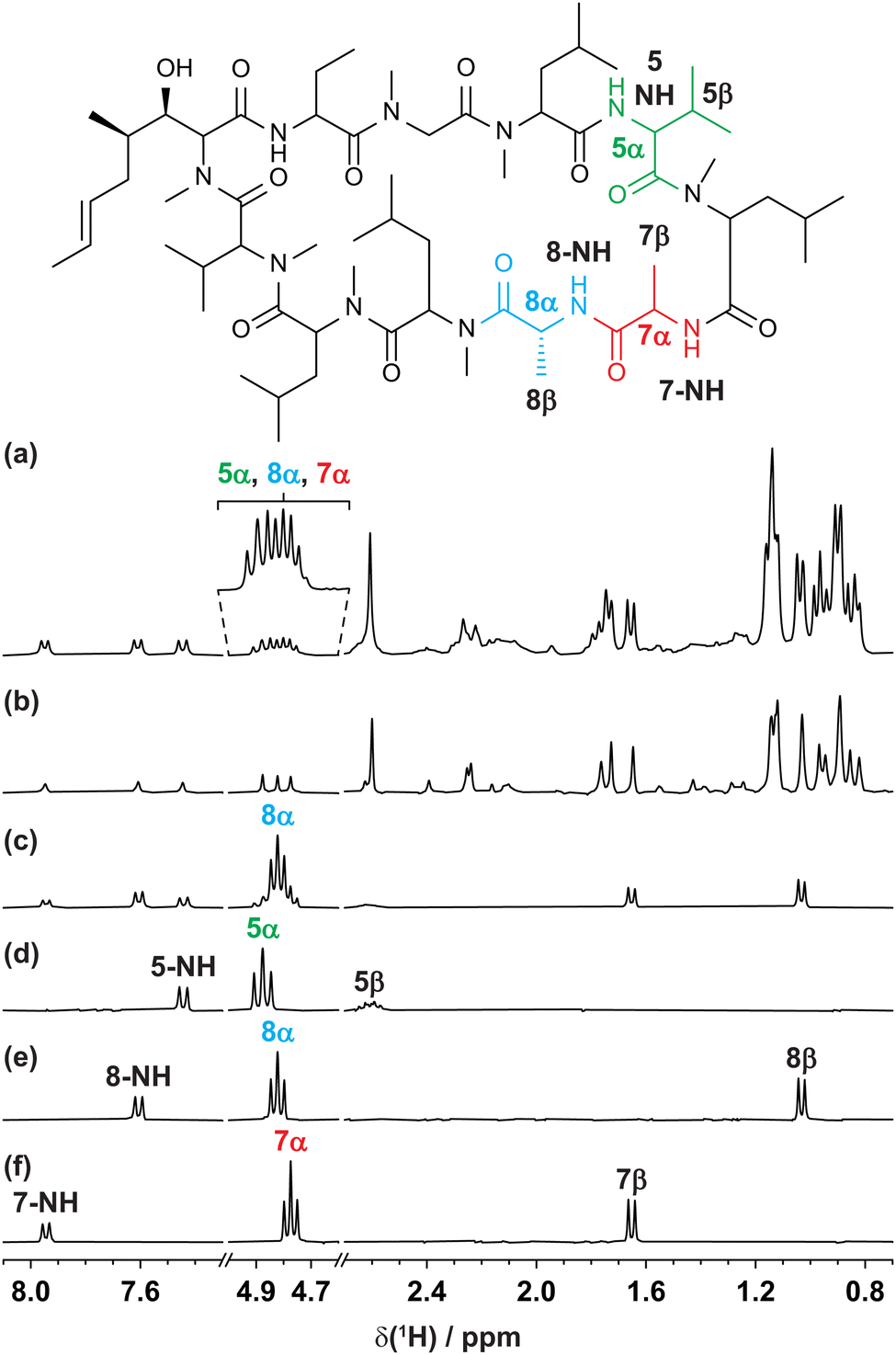 | ||
| Fig. 3 (a) Conventional 300 MHz 1H, (b) interferogram Zangger–Sterk pure shift, (c) conventional selective echo CLIP-COSY, and (d–f) GEMSTONE CLIP-COSY NMR spectra of cyclosporin in C6D6. In each of (c–f) the transmitter frequency (indicated by the coloured label) was placed on a signal of interest identified in the pure shift spectrum (b). The CLIP-COSY mixing time (τm) in each of (c–f) was 40 ms. Conventional selective refocusing in (c) was achieved with a 20 Hz REBURP pulse. Full experimental details are provided in the ESI.† | ||
As demonstrated, combining GEMSTONE and COSY pulse sequence elements allows selective identification of the bonding networks of spins whose 1H NMR signals are overlapped. GEMSTONE CLIP-COSY provides unrivalled selectivity compared with other 1D selective COSY experiments, and requires no additional experiment time. It affords in-phase absorption mode lineshapes for both excited and coupled spin signals, facilitating straightforward measurement of J values. The new method correlates directly coupled spins only, and is complementary to GEMSTONE TOCSY, where both direct and relayed correlations are observed. GEMSTONE CLIP-COSY provides a significant increase in the usefulness of 1D selective COSY methods for structure elucidation, in many cases avoiding the need for long, high-resolution 2D COSY experiments or TOCSY mixing time arrays to deduce through-bond connectivity. GEMSTONE CLIP-COSY is of most benefit when used to provide targeted correlations that are obscured in a low-resolution 2D COSY experiment. Collecting both experiment types and combining their results will allow all correlations to be determined in less time than required for a high-resolution 2D COSY experiment.
DAT: visualisation, writing – original draft; RWA: funding acquisition, project administration; PK, MN, GAM, RWA: supervision; All authors: conceptualisation, data curation, formal analysis, investigation, methodology, resources, writing – review & editing.
This research was supported by the Engineering and Physical Sciences Research Council (EP/V007580/1 and EP/R513131/1, project reference 2297286), Comunidad de Madrid (2022-T1/BMD-24030 to LC) and The University of Manchester (Dame Kathleen Ollerenshaw Fellowship to LC). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising. All experimental data for all the spectra shown, pulse program codes and experimental parameters are freely available at https://doi.org/10.48420/21909600.
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. P. Aue, E. Bartholdi and R. R. Ernst, J. Chem. Phys., 1976, 64, 2229–2246 CrossRef CAS.
- R. W. Adams, eMagRes, 2014, 3, 295–309 CAS.
- K. Zangger, Prog. Nucl. Magn. Reson. Spectrosc., 2015, 86–87, 1–20 CrossRef CAS PubMed.
- L. Castañar, Magn. Reson. Chem., 2017, 55, 47–53 CrossRef PubMed.
- J. A. Aguilar, A. A. Colbourne, J. Cassani, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2012, 51, 6460–6463 CrossRef CAS PubMed.
- V. M. R. Kakita and J. Bharatam, Magn. Reson. Chem., 2018, 56, 963–968 CrossRef CAS PubMed.
- J. A. Aguilar, R. Belda, B. R. Gaunt, A. M. Kenwright and I. Kuprov, Magn. Reson. Chem., 2018, 56, 969–975 CrossRef CAS PubMed.
- S. K. Mishra and N. Suryaprakash, Prog. Nucl. Magn. Reson. Spectrosc., 2023, 136–137, 1–60 CrossRef CAS.
- G. Bodenhausen, R. Freeman, R. Niedermeyer and D. L. Turner, J. Magn. Reson., 1977, 26, 133–164 CAS.
- U. Piantini, O. W. Sørensen and R. R. Ernst, J. Am. Chem. Soc., 1982, 104, 6800–6801 CrossRef CAS.
- C. Griesinger, O. W. Sørensen and R. R. Ernst, J. Am. Chem. Soc., 1985, 107, 6394–6396 CrossRef CAS.
- H. Oschkinat, A. Pastore, P. Pfändler and G. Bodenhausen, J. Magn. Reson., 1986, 69, 559–566 CAS.
- A. Kumar, R. V. Hosur and K. Chandrasekhar, J. Magn. Reson., 1984, 60, 143–148 CAS.
- D. Marion and A. Bax, J. Magn. Reson., 1988, 80, 528–533 CAS.
- S. Talluri and H. A. Scheraga, J. Magn. Reson., 1990, 86, 1–10 CAS.
- Y. Xia, G. Legge, K. Y. Jun, Y. Qi, H. Lee and X. Gao, Magn. Reson. Chem., 2005, 43, 372–379 CrossRef CAS PubMed.
- M. R. M. Koos, G. Kummerlöwe, L. Kaltschnee, C. M. Thiele and B. Luy, Angew. Chem., Int. Ed., 2016, 55, 7655–7659 CrossRef CAS PubMed.
- L. Braunschweiler and R. R. Ernst, J. Magn. Reson., 1983, 53, 521–528 CAS.
- C. Bauer, R. Freeman, T. Frenkiel, J. Keeler and A. J. Shaka, J. Magn. Reson., 1984, 58, 442–457 CAS.
- H. Kessler, H. Oschkinat, C. Griesinger and W. Bermel, J. Magn. Reson., 1986, 70, 106–133 CAS.
- P. Kiraly, N. Kern, M. P. Plesniak, M. Nilsson, D. J. Procter, G. A. Morris and R. W. Adams, Angew. Chem., Int. Ed., 2021, 60, 666–669 CrossRef CAS PubMed.
- L. D. Hall and T. J. Norwood, J. Magn. Reson., 1988, 76, 548–554 CAS.
- L. D. Hall and T. J. Norwood, J. Magn. Reson., 1988, 78, 582–587 CAS.
- E. L. Gates, M. J. Smith, J. P. Bradley, M. Johnson, G. W. Widmalm, M. Nilsson, G. A. Morris, R. W. Adams and L. Castañar, Chem. Commun., 2023 10.1039/D3CC00550J.
- P. Kiraly, M. Nilsson, G. A. Morris and R. W. Adams, Chem. Commun., 2021, 57, 2368–2371 RSC.
- L. Byrne, J. Solà, T. Boddaert, T. Marcelli, R. W. Adams, G. A. Morris and J. Clayden, Angew. Chem., Int. Ed., 2014, 53, 151–155 CrossRef CAS PubMed.
- L. Castañar, P. Nolis, A. Virgili and T. Parella, Chem. – Eur. J., 2013, 19, 17283–17286 CrossRef PubMed.
- J. Ying, J. Roche and A. Bax, J. Magn. Reson., 2014, 241, 97–102 CrossRef CAS PubMed.
- R. W. Adams, L. Byrne, P. Király, M. Foroozandeh, L. Paudel, M. Nilsson, J. Clayden and G. A. Morris, Chem. Commun., 2014, 50, 2512–2514 RSC.
- M. J. Thrippleton and J. Keeler, Angew. Chem., Int. Ed., 2003, 42, 3938–3941 CrossRef CAS PubMed.
- H. Geen and R. Freeman, J. Magn. Reson., 1991, 93, 93–141 Search PubMed.
- Ē. Kupče, J. Boyd and I. D. Campbell, J. Magn. Reson., Ser. B, 1995, 106, 300–303 CrossRef PubMed.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, pulse program codes, simulation results. See DOI: https://doi.org/10.1039/d3cc01333b |
| This journal is © The Royal Society of Chemistry 2023 |