
Intermetallic transfer of unsymmetrical borylene fragments: isolation of the second early-transition-metal terminal borylene complex and other rare species†
Bret B.
Macha
ab,
Debabrata
Dhara
 ab,
Krzysztof
Radacki
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
ab,
Krzysztof
Radacki
ab,
Rian D.
Dewhurst
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 13th November 2020
Abstract
Transition metal borylene complexes of the type [(OC)5M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BN(SiMe3)(tBu)] (M = Cr, Mo, W) have been synthesised by salt elimination of the corresponding dibromoborane and the dianionic metallates Na2[M(CO)5]. The borylene complexes have been characterised by multinuclear solution-state NMR spectroscopy and solid-state molecular structure determination. The group 6 borylene complexes can be used to effectively transfer the borylene ligand to other transition metal complexes by replacing one or two carbonyl ligands upon irradiation of the reaction mixture with UV light. This borylene transfer reaction led to the formation of new terminal and bridging borylene complexes which cannot be formed by the corresponding salt elimination reactions, including a rare example of a bis(terminal borylene) complex and only the second reported terminal borylene complex of an early transition metal (vanadium).
BN(SiMe3)(tBu)] (M = Cr, Mo, W) have been synthesised by salt elimination of the corresponding dibromoborane and the dianionic metallates Na2[M(CO)5]. The borylene complexes have been characterised by multinuclear solution-state NMR spectroscopy and solid-state molecular structure determination. The group 6 borylene complexes can be used to effectively transfer the borylene ligand to other transition metal complexes by replacing one or two carbonyl ligands upon irradiation of the reaction mixture with UV light. This borylene transfer reaction led to the formation of new terminal and bridging borylene complexes which cannot be formed by the corresponding salt elimination reactions, including a rare example of a bis(terminal borylene) complex and only the second reported terminal borylene complex of an early transition metal (vanadium).
Introduction
As ligands, borylenes (:BR) are isolobal with N2 and CO, although the former differ substantially in terms of their binding with transition metals (TMs) and the reactivity of the resulting complexes.1 We reported the first isolation and structural authentication of stable transition metal borylene complexes in 1995, namely the bridging borylene complexes of type I (Fig. 1).2 Since that time, the field of TM-borylene chemistry has expanded significantly,3 while the related advent of base-stabilised (TM-free) borylenes4 has opened up a vast new playground of synthetic possibilities.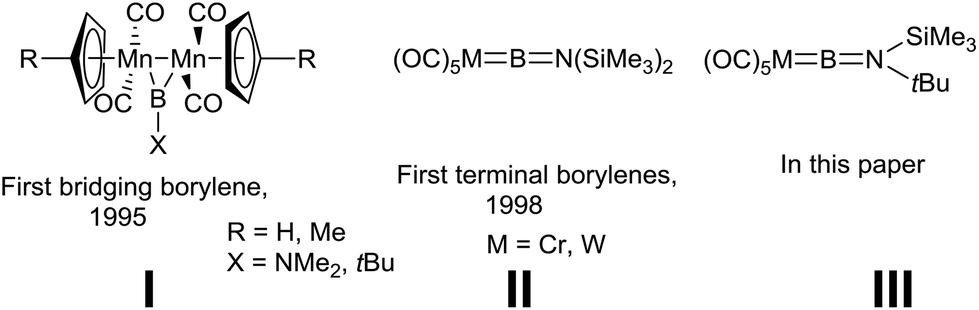 | ||
| Fig. 1 Structures of relevant classes of compounds I–III. | ||
BR ligands, being isoelectronic to CO, can adopt similar coordination modes to TMs (terminal,5 doubly6 and triply bridging,7 and semi-bridging8) and similar bonding patterns to their CO counterparts. Theoretical studies have shown that the highest occupied molecular orbitals (HOMO) of free borylenes are much higher in energy than CO, making them much better σ-donors, whereas they show similar π-accepting properties, thus rationalising the thermodynamically stable metal borylene linkage.9 Several examples of terminal borylene complexes have been reported since their initial discovery in 1998 (II, Fig. 1),10 however, few reliable and generalised methods for their synthesis exist. The most common preparatory route for terminal transition metal borylene complexes from non-borylene starting materials involves halide abstraction3b,11 or salt metathesis reactions.3b,12 The former process allows for the production of a variety of cationic borylenes, most notably of iron, from transition metal haloboryl precursors.11 On the other hand, group 6 terminal borylene complexes are synthesised by salt elimination reactions. Indeed, disodium metallates of the Cr, Mo, and W pentacarbonyl dianions, Na2[M(CO)5], are reported to react with dihaloboranes to furnish the corresponding borylene complexes in reasonable yields.12a To this day, this method is limited to the preparation of [(OC)5MBN(SiMe3)2],10a,12a [(OC)5MB{E(SiMe3)3}] (E = Si, Ge)12b and [(OC)5MBAr],13 where Ar is a very bulky terphenyl moiety. Other more recent and less studied syntheses of borylene complexes include a direct B–H bond activation of a dihydroborane at a ruthenium center,14 as well as the use of silyliron and -ruthenium monoanions for terminal borylene synthesis through subsequent salt and halosilane eliminations.15 Among these “first-generation” borylene complexes (i.e. those not prepared by borylene transfer), the group 6 complexes II (Fig. 1) are unique in their exceptional ligand transfer reactivity, while a few other complexes have also shown limited transfer ability.17f Indeed, the [:BN(SiMe3)2] fragment can be transferred from the group 6 pentacarbonyl complexes to different metal systems through reactions that have parallels in carbene chemistry.16,17 This process has been invaluable in the expansion of TM borylene chemistry as it allows for the preparation of a variety of novel terminal and bridging examples for which no synthetic access exists. It should be noted that the borylene transfer concept is not limited to transition metal acceptors, borylene ligands can also be transferred to organic substrates to form heterocyclic species (e.g. borirenes18) and other hypovalent boron species such as doubly base-stabilised (metal-free) borylenes.10d Like Fischer carbene complexes, terminal borylene complexes can also participate in metathesis chemistry.11d,19
Rather surprisingly considering their rich transfer chemistry,17 aminoborylenes of the group 6 metals remain limited in scope to the [LnMBN(SiMe3)2] family of complexes. One reason for this is the difficulty of obtaining suitable monomeric aminodihaloborane precursors for such borylene complexes. Furthermore, it is well established that steric shielding is an important contributing factor to the stability of terminal borylenes. In this report we present the first synthesis of a family of non-[:BN(SiMe3)2] aminoborylene complexes of the group 6 metals. The rich transfer chemistry of these compounds was investigated, providing a number of new borylene complexes including terminal and bridging mono and bis(borylene) complexes.
Results and discussion
Synthesis of unsymmetrical group 6 borylene complexes
The terminal borylenes [(OC)5MBN(SiMe3)(tBu)] (M = Cr (1a), Mo (1b), W (1c)) were prepared similarly to salt elimination procedures reported for the known [LnMBN(SiMe3)2] complexes (Fig. 2).12 In such a procedure, the freshly synthesised sodium salts of the [M(CO)5]2− (M = Cr, Mo, W) dianions, generated by the reduction of [M(CO)5(NMe3)]20 with sodium naphthalenide, were reacted with Br2BN(SiMe3)(tBu) in toluene at low temperature (−78 °C). Extraction and crystallisation of the reaction products in pentane allowed isolation of all three unsymmetrical borylene compounds as pure products. The yields were found to be moderate in the cases of the chromium, 1a (39%) and tungsten analogues, 1c (35%), but very low for the molybdenum complex 1b (10%), which is an extremely sensitive species. Indeed, while melting points could be measured for 1a and 1c (60 and 73 °C, respectively), compound 1b decomposes above 0 °C. By contrast, 1a and 1c only decompose above ca. 150 °C.
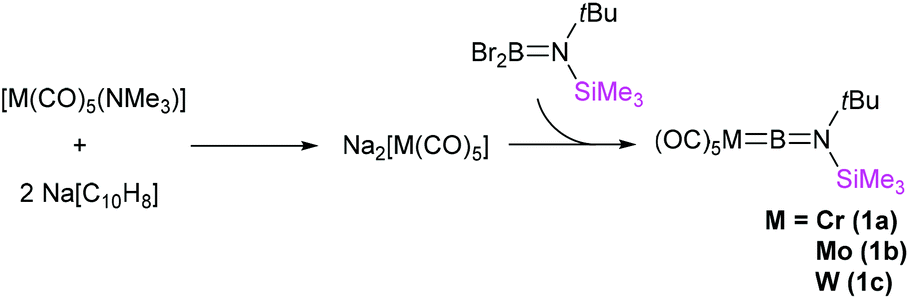 | ||
| Fig. 2 Synthesis of first-generation unsymmetrical group 6 borylene complexes 1a–1c. | ||
All three borylene complexes were characterised by 1H, 11B, 13C, and 29Si NMR spectroscopy. In all cases, the 11B{1H} NMR spectroscopic signal of the unsymmetrical borylene complexes (1a: δ = 97, 1b: δ = 96, 1c: δ = 92) is significantly downfield shifted relative to the signals of the previously reported [(OC)5MBN(SiMe3)2] analogues (δ: I: 92, II: 90 III: 87).12a In the borylene complexes 1a–c, one of the SiMe3 groups is replaced by a tBu group, leading to a more downfield signal for the CH3 protons due to the higher electronegativity of carbon relative to silicon. In their 1H NMR spectra, a sharp singlet is observed in the aliphatic region corresponding to the tBu group (1a: δ = 1.15, 1b: δ = 1.14, 1c: δ = 1.14), along with a singlet for the SiMe3 protons (1a: δ = 0.16, 1JH–Si = 6.7 Hz; 1b: δ = 0.14, 2JH–Si = 6.6 Hz; 1c: δ 0.15, 2JH–Si = 6.8 Hz). The respective signals for the SiMe3 groups can easily be identified by their satellite signals arising from coupling to 29Si nuclei, which have 2JSi–H values of around 6.5 Hz. The 13C{1H} NMR spectra of 1a–c showed the usual low-field shift for four equatorial (cis to borylene) and one axial (trans to borylene) CO ligands, (1a: δ = 218.4 (COax), 217.9 (COeq); 1b: δ = 206.9 (COax), 207.5 (COeq); 1c: δ = 191.2 (COax), 197.7 ppm (COeq). The 29Si{1H} NMR signals of all the three compounds appeared in a similar region to those of their previously reported analogues12a (δ = 3.4 ppm for 1a and 1b, δ = 2.5 ppm for 1c).
The formation of 1a–c was further confirmed by their molecular structures (Fig. 3). Single crystals of suitable quality for X-ray diffraction were grown from saturated hexane solutions. All three complexes crystallised in monoclinic space groups, 1a in P21/n and 1b and 1c in P21/c. As the data quality of 1a is not sufficient to discuss the bond lengths and angles, only those of 1b and 1c will be discussed. All of the metal–boron (1b: 2.1356(19); 1c: 2.143(2) Å) and boron–nitrogen bond distances (1b: 1 1.361(2), 1c: 1.356(3) Å) are in line with those of previously reported group 6 terminal borylene complexes.12a The N–B–M moiety is almost linear with angles of 175.78(15)° (1b) and 176.08(17)° (1c). The Cax–M–B axes are also almost linear (1b: C1–Mo–B 173.59(7)°; 1c: C1–W–B 173.38(8)°). The M–Cax distances of 1b,c (1b: 2.070 (2), 1c: 2.056 (2) Å) are slightly longer than the M–Ceq distances (avg. equatorial M–C distances for 1b and 1c: 2.045 Å) revealing the trans effect of the borylene ligand, as previously observed in transition metal borylene complexes.3,12b,c
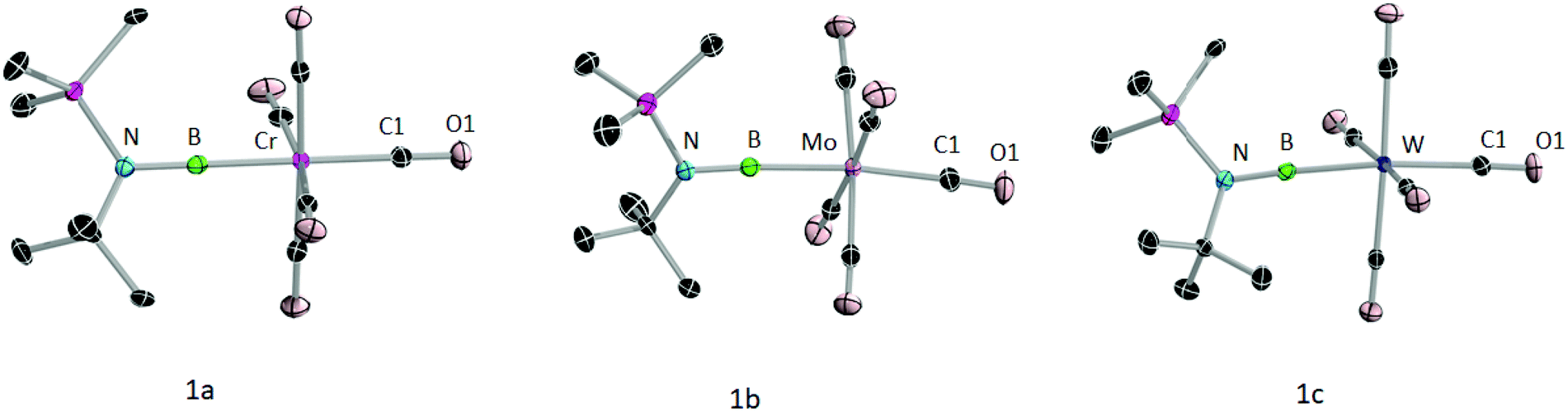 | ||
| Fig. 3 Molecular structures of 1a–c with thermal ellipsoids shown at 50% probability level, all H atoms are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: 1b: B–Mo 2.1356(19); B–N 1.361(2); N–B–Mo 175.78, C1–Mo–B 173.59(7)°. 1c: B–W 1.356(3) (Å), B–N 2.143(2) Å; N–B–W 176.08(17), C1–W–B 173.38(8). Due to poor diffraction quality of the crystals we refrain from discussing the selected bond lengths and angles of 1a. | ||
Synthesis of second-generation borylene complexes by borylene transfer
With these first-generation borylene complexes in hand, we set out to investigate their transfer chemistry, for the synthesis of novel borylene complexes. Our group has previously shown that intermetallic borylene transfer is a viable synthetic route to both terminal as well as bridging borylene complexes.17 In this study, we concentrated on the photolytic transfer of borylene moieties from the chromium and tungsten precursors 1a and 1c, as the molybdenum derivative is highly sensitive and can only be isolated in very low yields. Furthermore, preliminary experiments suggested that 1b decomposes under the applied reaction conditions at a faster rate than any of the transfer processes. It should be noted that the following transfer experiments were first attempted on small scales in NMR tubes, and in each case proceeded regardless of whether 1a or 1c was used. The experiments described below were subsequent reactions performed on larger scales using the precursor that showed the most selective reaction on the NMR scale. Unsymmetrical tungsten borylene 1c reacts readily with vanadium cyclopentadienyl tetracarbonyl complex [(η5-C5H5)V(CO)4] in benzene under UV irradiation. The progress of the reaction can be followed by 11B{1H} NMR, wherein the signal intensity of the starting borylene progressively decreases over 6 h, giving way to a slightly more deshielded resonance (100 ppm) relative to that of its precursor 1c, but in line with that of a previously reported similar half sandwich terminal vanadium borylene complex (δ = 98 ppm).17b After the reaction, removal of the volatiles under vacuum followed by recrystallisation of a silica-filtered hexane solution of the mixture afforded [(η5-C5H5)(OC)3V{BN(SiMe3)(tBu)}] (2) in 84% yield. In a similar manner, two molar equivalents of 1a reacted with [(η5-C5Me5)Ir(CO)2] under UV irradiation, replacing both carbonyl ligands with borylene moieties and providing 5 (Fig. 4) in a 58% yield after fractional crystallisation. The 11B{1H} NMR spectrum of 5 showed a signal (δ = 70.8 ppm) at high field relative to that of its starting material but essentially identical to that of a reported half sandwich bis(borylene) complex of iridium (δ = 69 ppm).17e The solid-state structures of 2 and 5 were ascertained by single-crystal X-ray diffraction (Fig. 5). | ||
| Fig. 4 Synthesis of second-generation unsymmetrical borylene complexes 2–5 by borylene transfer. | ||
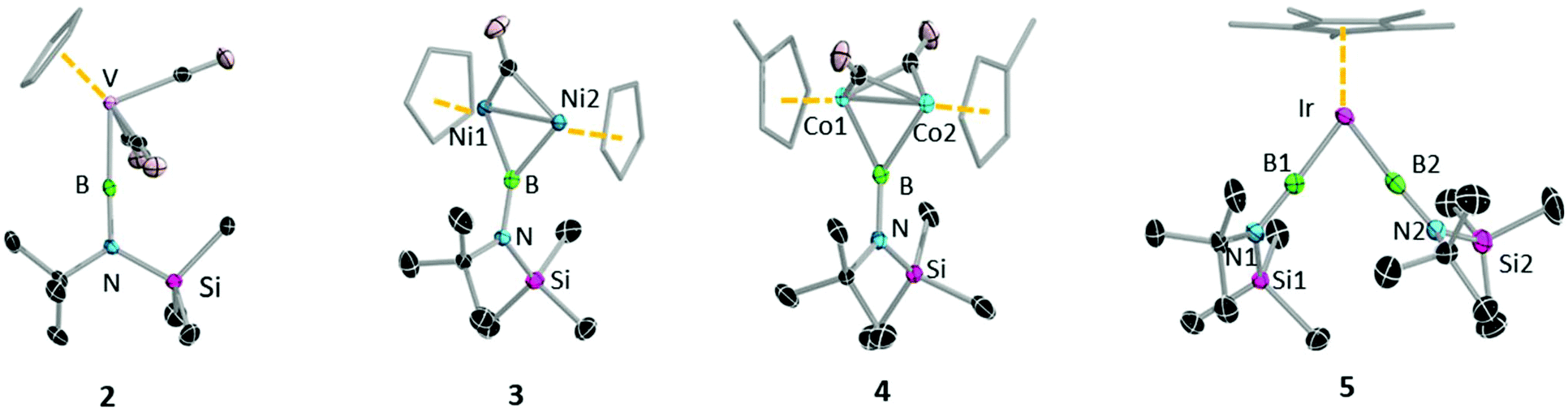 | ||
| Fig. 5 Molecular structure of 2–5 with the thermal ellipsoids shown at 50% probability level, all H atoms are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: 2: B–V 1.964 (2), B–N 1.3714(17); (V–B–N 177.5 (1). 3: B1–Ni1 1.933 (3), B1–Ni2 1.936(3), Ni1–Ni2 2.370 (8); Ni–B–Ni 75.56(11)°. Due to poor diffraction quality of crystal we refrain from discussing the selected bond lengths and angles of 4, 5. | ||
The vanadium half sandwich complex 2 crystallised in the orthorhombic Pbca space group. Its B–V bond distance was found to be 1.964(2) Å, which is similar to that of the previously reported vanadium borylene complex (1.959(6) Å)17b and within the typical range of known terminal M–B distances.3 The V–B–N moiety is almost linear (V–B–N 177.5 (1)°), while the B–N distance 1.3714(17) matches those of related complexes.17b The half-sandwich bis(borylene) complex 5 crystallises in the monoclinic space group P21/n. The primary structure showed the expected “two-legged piano stool” geometry in the solid-state like its published analogues.17e However, due to the poor quality of the crystal data we cannot elaborate further on its structural parameters.
In addition to replacing terminal CO ligands on TM centres through borylene transfer, the borylene moieties of 1a and 1c can also replace bridging CO ligands, providing a complex with a bridging borylene ligand. Thereby, irradiation of a hexane solution of 1c and [(η5-C5H5)2Ni2(CO)2] for 7 h afforded dinickel bridging borylene complex 3 (Fig. 4). After the completion of the reaction, 3 was extracted with hexamethyldisiloxane and crystallised from the same solution as dark red crystals in a 74% yield. In a similar reaction, 1a was irradiated in the presence of 2.5 equivalents of [(η5-C5H4Me)Co(CO)2] in hexane solution at room temperature. Monitoring the reaction mixture by 11B NMR spectroscopy showed the consumption of precursor 1a and appearance of two new peaks at δ = 81 ppm and δ = 104 ppm. These signals likely correspond to the expected terminal and bridging borylene complexes, i.e. [(η5-C5H4Me)(OC)Co{BN(SiMe3)(tBu)}] and [{(η5-C5H4Me)Co}2{μ-(CO)}2{μ-BN(SiMe3)(tBu)}] (4), respectively, although we were unable to isolate the intermediate mononuclear complex due to thermal lability. After complete consumption of 1a (ca. 8 h reaction time) the reaction mixture was evaporated and extracted with hexane and filtered through silica gel, and subsequent crystallisation of the filtrate at −30 °C allowed isolation of 4 in a 65% yield (Fig. 4).
The 11B{1H} NMR spectroscopic signals of bridging borylene complexes 3 and 4 were both significantly broadened, indicating their bridging binding mode, and were found slightly downfield (3: δ = 94.1, 4: δ = 103.6) of those of their starting material. The 1H NMR spectrum of 3 showed a downfield shift of the proton signal of the cyclopentadienyl ring (δ = 5.32 ppm) relative to that of its starting material (δ = 5.15 ppm), suggesting that the σ-donor properties of cyclopentadienyl ring increase in the presence of the borylene ligand.21 The signals for the tBu and TMS protons show a downfield shift with respect to those of the starting materials (δ = 1.18 and 0.24 ppm, respectively). The corresponding spectrum of the dicobalt bridging borylene complex 4 also shows similar phenomena relative to its precursor. The 1H NMR spectrum of 4 showed signals at δ = 4.75, 4.49 and 1.94 ppm for the protons of the methylcyclopentadienyl ligand (η5-C5H4CH3), all significantly deshielded with respect to the corresponding signals of the precursor (δ = 4.53, 4.33, 1.47 ppm).22 The signals for the tBu and SiMe3 protons are also downfield (δ = 1.40 and 0.34 ppm.) of those of 1a.
The solid-state molecular structures of both 3 and 4 (Fig. 5) confirm the presence of a single bridging borylene ligand. However, due to poor quality of the crystal data we refrain from discussing the structural parameters of compound 4. The M–B distances of 3 (B1–Ni1 1.933 (3) Å, B1–Ni2 1.936(3) Å) are comparable to previously reported M–B distances of bridging borylene complexes (avg. Ni–B 1.93(14) Å).23 The Ni–Ni distance (2.370(8) Å) is slightly longer than that of a similar compound (Ni–Ni: 2.341(2) Å)23 and that of the starting material (2.348(2) Å),21 which rules out the possibility of a metal–metal bond. The Ni–B1–Ni angle (75.56(11)°) is furthermore significantly smaller than the Ni–C–Ni angle (80.38(12)°).
Conclusions
Our results herein show that the family of group 6 aminoborylene complexes is amenable to further variation, including complexes with unsymmetrical aminoborylene ligands. These terminal borylene complexes have proven to be excellent precursors for both terminal and bridging mono- and diborylene species through facile borylene transfer reactions with other TM carbonyl complexes, leading to rare examples of nickel borylene complexes, bis(terminal borylene) complexes, and only the second reported terminal borylene complex of an early transition metal (vanadium).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the European Research Council (ERC) under the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 267155).Notes and references
- (a) F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, Wiley, New York, 5th edn, 1988 Search PubMed; (b) J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, 1987 Search PubMed.
- H. Braunschweig and T. Wagner, Angew. Chem., Int. Ed. Engl., 1995, 34, 825–826 CrossRef CAS.
- (a) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS; (b) D. Vidovic, G. A. Pierce and S. Aldridge, Chem. Commun., 2009, 1157–1171 RSC; (c) S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535–559 CrossRef CAS; (d) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC; (e) H. Braunschweig and M. Colling, Coord. Chem. Rev., 2001, 223, 1–51 CrossRef CAS.
- M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef CAS.
- H. Braunschweig and D. Rais, Heteroat. Chem., 2005, 16, 566–571 CrossRef CAS.
- (a) H. Braunschweig and M. Colling, J. Organomet. Chem., 2000, 593, 18–26 CrossRef; (b) H. Braunschweig, K. Radacki, D. Rais, F. Seeler and K. Uttinger, J. Am. Chem. Soc., 2005, 127, 1386–1387 CrossRef CAS.
- H. Braunschweig, C. Burschka, M. Burzler, S. Metz and K. Radacki, Angew. Chem., Int. Ed., 2006, 45, 4352–4355 CrossRef CAS.
- (a) H. Braunschweig, D. Rais and K. Uttinger, Angew. Chem., Int. Ed., 2005, 44, 3763–3766 CrossRef CAS; (b) H. Braunschweig, K. Radacki, D. Rais and K. Uttinger, Organometallics, 2006, 25, 5159–5164 CrossRef CAS.
- (a) A. W. Ehlers, E. J. Baerends, F. M. Bickelhaupt and U. Radius, Chem. – Eur. J., 1998, 4, 210–221 CrossRef CAS; (b) U. Radius, F. M. Bickelhaupt, A. W. Ehlers, N. Goldberg and R. Hoffmann, Inorg. Chem., 1998, 37, 1080–1090 CrossRef CAS; (c) C. Boehme, J. Uddin and G. Frenking, Coord. Chem. Rev., 2000, 197, 249–276 CrossRef CAS.
- (a) H. Braunschweig, C. Kollann and U. Englert, Angew. Chem., Int. Ed., 1998, 37, 3179–3180 CrossRef CAS; (b) A. H. Cowley, V. Lomelí and A. Voigt, J. Am. Chem. Soc., 1998, 120, 6401–6402 CrossRef CAS; (c) H. Braunschweig, P. Brenner, R. D. Dewhurst, I. Krummenacher, B. Pfaffinger and A. Vargas, Nat. Commun., 2012, 3, 1–6 Search PubMed; (d) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS.
- (a) D. L. Coombs, S. Aldridge, C. Jones and D. J. Willock, J. Am. Chem. Soc., 2003, 125, 6356–6357 CrossRef CAS; (b) D. L. Coombs, S. Aldridge, A. Rossin, C. Jones and D. J. Willock, Organometallics, 2004, 23, 2911–2926 CrossRef CAS; (c) D. L. Kays (née Coombs), S. Aldridge, J. K. Day and L.-L. Ooi, Angew. Chem., Int. Ed., 2005, 44, 7457–7460 CrossRef; (d) D. L. Kays (née Coombs), A. Rossin, J. K. Day, L.-L. Ooi and S. Aldridge, Dalton Trans., 2006, 399–410 RSC; (e) S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D. L. Kays (née Coombs), N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118–6122 CrossRef CAS; (f) H. Braunschweig, K. Kraft, T. Kupfer, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2008, 47, 4931–4933 CrossRef CAS; (g) D. Vidovic, M. Findlater, G. Reeske and A. H. Cowley, Chem. Commun., 2006, 3786–3787 RSC; (h) H. Braunschweig, K. Radacki and K. Uttinger, Angew. Chem., Int. Ed., 2007, 46, 3979–3982 CrossRef CAS.
- (a) B. Blank, M. Colling-Hendelkens, C. Kollann, K. Radacki, D. Rais, K. Uttinger, G. R. Whittell and H. Braunschweig, Chem. – Eur. J., 2007, 13, 4770–4781 CrossRef CAS; (b) H. Braunschweig, M. Colling, C. Kollann, K. Merz and K. Radacki, Angew. Chem., Int. Ed., 2001, 40, 4198–4200 CrossRef CAS; (c) H. Braunschweig, K. Radacki, D. Scheschkewitz and G. R. Whittell, Angew. Chem., Int. Ed., 2005, 44, 1658–1660 CrossRef CAS.
- (a) H. Braunschweig, R. D. Dewhurst, C. Hörl, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2013, 52, 10120–10123 CrossRef CAS; (b) M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäfer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem. – Eur. J., 2018, 24, 6843–6847 CrossRef CAS.
- (a) C. Lenczyk, D. K. Roy, J. Nitsch, K. Radacki, F. Rauch, R. D. Dewhurst, F. M. Bickelhaupt, T. B. Marder and H. Braunschweig, Chem. – Eur. J., 2019, 25, 13566–13571 CrossRef CAS; (b) G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878–12879 CrossRef CAS; (c) G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS; (d) H. Braunschweig and R. D. Dewhurst, Angew. Chem., Int. Ed., 2009, 48, 1893–1895 CrossRef CAS.
- (a) H. Braunschweig, Q. Ye and K. Radacki, Chem. Commun., 2012, 48, 2701–2703 RSC; (b) H. Braunschweig, R. D. Dewhurst, K. Radacki, B. Wennemann and Q. Ye, Chem. Commun., 2015, 51, 15465–15468 RSC.
- (a) S.-T. Liu and K. Rajender Reddy, Chem. Soc. Rev., 1999, 28, 315–322 RSC; (b) S.-T. Liu, T.-Y. Hsieh, G.-H. Lee and S.-M. Peng, Organometallics, 1998, 17, 993–995 CrossRef CAS.
- (a) H. Braunschweig, M. Colling, C. Kollann, H. G. Stammler and B. Neumann, Angew. Chem., Int. Ed., 2001, 40, 2298–2300 CrossRef CAS; (b) H. Braunschweig, M. Colling, C. Hu and K. Radacki, Angew. Chem., Int. Ed., 2003, 42, 205–208 CrossRef CAS; (c) H. Braunschweig, M. Forster, K. Radacki, F. Seeler and G. R. Whittell, Angew. Chem., Int. Ed., 2007, 46, 5212–5214 CrossRef CAS; (d) H. Braunschweig, M. Forster and K. Radacki, Angew. Chem., Int. Ed., 2006, 45, 2132–2134 CrossRef CAS; (e) S. Bertsch, H. Braunschweig, B. Christ, M. Forster, K. Schwab and K. Radacki, Angew. Chem., Int. Ed., 2010, 49, 9517–9520 CrossRef CAS; (f) H. Braunschweig, Q. Ye, A. Vargas, R. D. Dewhurst, K. Radacki and A. Damme, Nat. Chem., 2012, 4, 563–567 CrossRef CAS.
- (a) C. E. Anderson, H. Braunschweig and R. D. Dewhurst, Organometallics, 2008, 27, 6381–6389 CrossRef CAS; (b) J. T. Goettel and H. Braunschweig, Coord. Chem. Rev., 2019, 380, 184–200 CrossRef CAS; (c) H. Braunschweig, Q. Ye, K. Radacki and P. Brenner, Inorg. Chem., 2011, 50, 62–71 CrossRef CAS; (d) H. Braunschweig, Q. Ye and K. Radacki, Chem. Commun., 2009, 6979–6981 RSC; (e) H. Braunschweig, T. Herbst, D. Rais and F. Seeler, Angew. Chem., Int. Ed., 2005, 44, 7461–7463 CrossRef CAS.
- (a) H. Braunschweig, M. Burzler, K. Radacki and F. Seeler, Angew. Chem., Int. Ed., 2007, 46, 8071–8073 CrossRef CAS; (b) J. Bauer, H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726–8734 CrossRef CAS; (c) B. Borthakur, H. Braunschweig, A. Deißenberger, T. Dellermann, R. D. Dewhurst, I. Krummenacher, M. Nutz, A. K. Phukan and M. Schäfer, Angew. Chem., Int. Ed., 2017, 56, 7975–7979 CrossRef.
- J. M. Maher, R. P. Beatty and N. J. Cooper, Organometallics, 1985, 4, 1354–1361 CrossRef CAS.
- E. O. Fischer and C. Palm, Chem. Ber., 1958, 91, 1725–1731 CrossRef CAS.
- W. P. Weiner and R. G. Bergman, J. Am. Chem. Soc., 1983, 105, 3922–3929 CrossRef CAS.
- H. Braunschweig, B. Christ, M. Colling-Hendelkens, M. Forster, K. Götz, M. Kaupp, K. Radacki and F. Seeler, Chem. – Eur. J., 2009, 15, 7150–7155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, spectroscopic, crystallographic and computational details. CCDC 2032809–2032813 and 2032815–2032817. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt03557b |
| This journal is © The Royal Society of Chemistry 2020 |