DOI:
10.1039/C6RA01689H
(Paper)
RSC Adv., 2016,
6, 37434-37442
Mesoporous TiO2 encapsulating a visible-light responsive upconversion agent for enhanced sonocatalytic degradation of bisphenol-A†
Received
20th January 2016
, Accepted 21st March 2016
First published on 8th April 2016
Abstract
Herein, we report the integration of a visible-light active upconversion agent (Er:Y2O3) with mesoporous TiO2 via a modified two-step sol–gel coating method. The resultant material possesses a well-defined core–shell structure with a good upconversion property and exhibits uniform worm-like mesopores (∼3.8 nm), a high BET surface area (∼151.5 m2 g−1) and a large pore volume (∼0.23 cm3 g−1). The composite was demonstrated as an advanced sonocatalyst, showing a superior degradation performance for bisphenol-A (BPA). The effect of the erbium dopant content on the upconversion property and sonocatalytic performance was evaluated for the production of the best sonocatalyst. We found that the maximum pseudo first-order reaction rate constant in the presence of the composite with an Er3+ content of 3 wt% in Y2O3 nanocrystals is calculated to be 0.155 min−1, which is 2.9 and 2 times larger than that of US alone (0.054 min−1) and hollow structured mesoporous TiO2 (0.077 min−1), respectively. More importantly, the degradation rate is much higher than that of the sonocatalysts reported previously for treatment of BPA (0.09–0.14 min−1). The superior catalytic activity can be attributed to an intensified cavitation reaction zone and an enhanced amount of photo-generated charges. Moreover, the recycling test shows that a constant catalytic activity is retained even after 4 cycles. This study paves a promising way for the development of a multi-functional catalyst for sonochemical processes.
Introduction
Recently, advanced oxidation processes (AOPs) which involve the generation of highly reactive oxidation species such as hydroxyl radicals, have emerged as a promising technology for the decomposition and mineralization of emerging organic pollutants. Among various AOPs, sonolysis is of great importance because it is environmentally friendly and simple to operate.1–3 When ultrasound (US) is irradiated into an aqueous phase, acoustic cavitation including the nucleation, growth, and violent collapse of microbubbles occurs, leading to the formation of sonoluminescence (SL, λ = 300–600 nm) and “hot spots” with extremely high temperature (5000 K) and pressure (1000 atm).4,5 As a result, water is dissociated to produce ·OH, ·H, and ·OOH, which can mineralize organic pollutants into CO2 and H2O. However, a significant amount of electric energy is typically required to obtain a fast ultrasonic degradation rate using one-fold US owing to the massive loss of input energy in thermal dissipation (>50%), which hinders the wide application of sonolysis for practical water treatment.6 To this end, development of a heterogeneous sonocatalyst is greatly desired. Till now, TiO2-based materials are extensively studied because of their good photocatalytic properties, nontoxicity, and cost-effectiveness. More importantly, it has been demonstrated that TiO2-based materials exhibited high sonocatalytic activities due to the formation of electron–hole pairs excited by SL, which is similar to the photocatalytic mechanism.7,8 However, the efficiency of TiO2 is still limited because it can only be activated by ultraviolet (UV) light. In this perspective, numerous strategies have been reported to extend the absorption spectra of TiO2 to visible-light region such as surface hydrogenation, metal or nonmetal ions doping, narrow band gap semiconductors coupling, etc.9–13 Nonetheless, the overall catalytic performance was sometimes found to decrease due to an increased recombination rate of photo-generated electron–hole pairs. Therefore, it is still a challenge to develop an appropriate way to address this issue. Recently, the incorporation of an upconversion (UC) agent with TiO2 has been extensively investigated as a novel strategy for enhancing the degradation performance of TiO2 under near-infrared light.14–16 However, the application of visible-light active UC agent is still restricted.17,18 In addition, one unique mechanism that governs the sonocatalytic efficiency is the heterogeneous nucleation process, which can be significantly affected by the catalyst surface properties such as roughness, pore size, wettability, etc. To date, substantial research efforts have been made for evaluating the effects of wettability and roughness on heterogeneous cavitation nucleation,19–21 whereas the patterning of a sonocatalyst surface with a nanoporous structure was rarely considered. Thus, to develop a catalyst with a UC capacity for utilizing visible light and a nanoporous surface for facilitating nucleation is highly desirable.
Herein, we report a successive sol–gel coating process to integrate a UC agent with mesoporous TiO2. The resultant material consists of a inner UC core – erbium doped yttrium oxide (Er:Y2O3) that can absorb visible light and then emit UV light, a middle silica layer for preventing dissociation of the core and a mesoporous TiO2 shell with a high surface area (∼151.5 m2 g−1), a large pore volume (0.23 cm3 g−1) and a uniform pore size distribution (∼3.8 nm). The composite is used as an advanced sonocatalyst for degrading bisphenol-A (BPA), exhibiting a superior performance especially when the dopant content of erbium in the Y2O3 nanocrystals is 3 wt%. The high catalytic activity is associated with the intensified cavitation reactive zone resulted from the mesoporous structure and the generation of additional photo-excited charge carriers achieved through UC mechanism. Moreover, the catalyst can be easily recycled and exhibit a constant activity even after 4 cycles. This study paves a promising way for the design and development of multi-functional catalyst for sonochemical process.
Experimental section
Materials
ErCl3·6H2O, YCl3·6H2O, urea, tetraethyl orthosilicate (TEOS), ethanol, concentrated ammonia solution (28 wt%), titanium(IV) isopropoxide (TIPO), and bisphenol-A (BPA) were of analytical grade and purchased from Sigma-Aldrich (USA). All chemicals were used as received without further purification. Millipore water was used for all experiments.
Preparation of Er3+ doped Y2O3 NPs
The erbium doped yttrium oxidation (Er:Y2O3) NPs were prepared via a urea assisted co-precipitation method reported previously.22 Briefly, YCl3·6H2O (0.28 g), ErCl3·6H2O (0.5–1.34 mL, 0.05 mmol mL−1), and urea (4.0 g) were dissolved in water (50 mL) with agitation. The mixture was stirred vigorously for 1 h at room temperature and then heated at 90 °C for 3 h. The white products were washed with deionized water and ethanol for 3 times, respectively.
Fabrication of core–shell Er:Y2O3@SiO2 nanospheres
The core–shell Er:Y2O3@SiO2 nanospheres were prepared through a versatile Stöber sol–gel method. For a typical synthesis, an ethanol dispersion of the Er:Y2O3 particles obtained above was added into a three-neck round-bottom flask with a mixture of ethanol (280 mL), deionized water (70 mL) and concentrated ammonia solution (4.0 mL, 28 wt%). The mixed solution was sonicated for 15 min. Then, 2.0 mL of TEOS was added dropwise in 10 min, and the reaction was allowed to proceed for 12 h at room temperature under continuous mechanical stirring (400 rpm). The resultant products were separated and washed with deionized water and ethanol for 3 times, respectively.
Synthesis of core–shell Er:Y2O3@SiO2@mTiO2 nanospheres
The coating of mesoporous TiO2 shell was prepared via a kinetically controlled Stöber method, followed by an ultrasound assisted post-hydrolysis for the formation of mesopores and a calcination process for removing organic species and improving crystallinity.23,24 Typically, core–shell structured Er:Y2O3@SiO2 nanospheres obtained above were dispersed in ethanol (100 mL), and mixed with concentrated ammonia solution (0.40 mL, 28 wt%) under ultrasound for 15 min. Subsequently, 0.75 mL of TIPO was added dropwise in 5 min, and the reaction was allowed to proceed for 24 h at 45 °C under continuous mechanical stirring. The resultant products were separated and collected, followed by an additional ultrasonic post-hydrolysis process in water according to that reported previously. Finally, the resultant samples were collected by centrifuge and calcined at 600 °C for 2 h to remove the organic species and improve crystallinity.
Synthesis of nonporous Er:Y2O3@SiO2@TiO2 nanospheres and hollow structured mesoporous TiO2
As a comparison, nonporous Er:Y2O3@SiO2@TiO2 nanosphere was synthesized without ultrasound assisted post-hydrolysis process. Moreover, the hollow structured mesoporous TiO2 sphere was obtained by etching off the inner Er:Y2O3 core. Typically, the core–shell structured Er:Y2O3@SiO2@mTiO2 nanospheres was dissolved into 10 mL of HCl (0.1 mol L−1) and subjected to ultrasonic treatment for 2 h. The products were washed with H2O and ethanol for several times, and dried at 60 °C for 6 h.
Material characterization
X-ray diffraction (XRD) patterns were recorded on a Bruker D8 X-ray diffractometer with Ni-filtered Cu Kα radiation (40 kV, 40 mA). Nitrogen sorption isotherms were measured at 77 K with a Micromeritics Tristar 3020 analyzer (USA). Prior to measurements, the samples were degassed in a vacuum at 160 °C for 9 h. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas using adsorption data in the relative pressure range P/P0 = 0.05–0.3. Using the Barrett–Joyner–Halenda (BJH) model, the pore size distributions were derived from the adsorption branches of the isotherms, and the total pore volumes were estimated from the adsorbed amount at the relative pressure P/P0 = 0.995. Transmission electron microscopy (TEM) was carried out on a JEOL 2011 microscope (Japan) operated at 200 kV. For TEM measurements, the sample was suspended in ethanol and supported on a holey carbon film on a Cu grid. Time-resolved fluorescence measurements were performed by using a Fluorometer (wavelength range: excitation 200–1100 nm, emission 200–1100 nm; wavelength accuracy: <1.5 nm; wavelength setting repeatability: <0.2 nm; Cary Eclipse, Agilent Technologies). The excitation of samples at 522 nm was achieved by using a Xe pulsed lamp. Powder UV-Vis absorbance spectra were measured using a UV-2600 spectrophotometer (Shimadzu, Japan) equipped with an integrating sphere. The emission spectra were recorded on LS-50B, Perkin Elmer instrument with the excitation wavelength of 350 nm. Scanning electron microscopy (SEM) images were taken on S-4800 II field-emission scanning electron microanalyzer with an accelerating voltage (15 kV).
Sonocatalytic degradation experiment
The sonoreactor consisted of a double-layered cylindrical container with a capacity of 1.25 L (Φ 10.0 × 16.0 cm), and was equipped with a cup-horn-type ultrasonic transducer (Mirae Ultrasonic MEGA-100). The frequency of 300 kHz and electric power of 100 W were applied for carrying on the sonocatalytic reaction. A 800 mL reactor (Φ 7.0 × 20.0 cm) was emerged into the container loaded with 400 mL of water for the reaction. The solution temperature was measured using a thermometer (Tecpel DTM-318) and maintained with a water jacket. A retort stand was used to fix the emerged reactor. The distance between the bottom of the reactor and container was about 6.0 cm. Aqueous suspensions (100 mL) of BPA (5 mg L−1) and catalyst (0.5 g L−1) were used for the sonocatalytic degradation. At given time intervals, 0.5 mL of the suspension was removed using a 2 mL syringe and filtered by a membrane with a pore size of ∼0.45 μm. The BPA concentration in the resultant filtrate was analyzed on a High-Performance Liquid Chromatography (HPLC, Agilent 1260) with a Eclipse XDB C18 column (4.6 × 250 mm, 5 μm) and a diode array UV detector (G4212B 1260 DAD, λ = 210 nm). Mobile phase consisted of 60 vol% aqueous acetonitrile (ACN) solution (A) and 100 vol% ACN solution (B). In the recycling test, the used catalyst was separated by centrifuging (4000 rpm) and washed with water for three times. The obtained catalyst was dried at 60 °C for 12 h and collected for further recycling test.
Sonochemiluminescence observation
Sonochemiluminescence (SCL) was used to visualize the sonochemical reaction zone in the presence of various catalysts. The luminol solution was prepared by mixing 0.1 g L−1 luminol (3-aminophthalhydrazide) with 1 g L−1 sodium hydroxide. Luminol reacts with hydroxyl radicals generated during the collapse of cavitation bubbles to produce aminophthalate anions that exhibit blue fluorescence. SCL images were recorded for an exposure time of 30 second using a digital camera (Canon EOD 400D equipped with a Tamron AF 17–50 mm lens) in a dark room.
Results and discussion
The synthesis strategy for the core–shell structured Er:Y2O3@SiO2@mesoporous TiO2 (mTiO2) nanospheres is depicted in Fig. 1. First, the uniform Er3+ doped Y2O3 nanoparticles (NPs) with various weight ratio (1, 3, and 5 wt%) were synthesized via a urea assisted co-precipitation method and coated with a silica layer through a sol–gel approach in the presence of tetraethyl orthosilicate (TEOS). Then, a kinetically-controlled sol–gel coating process was utilized to deposit a titania shell onto the silica layer, leading to a core–shell–shell structured amorphous Er:Y2O3@SiO2@TiO2 nanospheres. Finally, an ultrasound assisted post-hydrolysis process was used to assist the formation of mesopores on the TiO2 shell, after which the resultant material was calcined at 550 °C to remove the organic species and improve the crystallinity. The resultant composites with different Er3+ loadings in Y2O3 nanocrystals were denoted as Er:Y2O3-1@SiO2@mTiO2, Er:Y2O3-2@SiO2@mTiO2, and Er:Y2O3-3@SiO2@mTiO2, respectively.
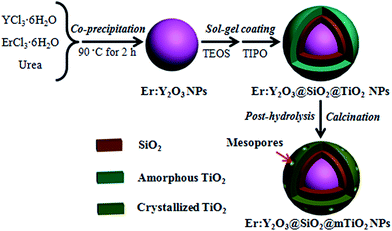 |
| | Fig. 1 Schematic illustration for combining the UC agent with mesoporous TiO2. | |
The transmission electron microscopy (TEM) images reveal that the obtained Er:Y2O3 NPs possess a uniform spherical shape with an average diameter of ∼260 nm (Fig. 2a and b). After the first sol–gel coating process, the TEM images clearly show that a smooth silica layer with a thickness of ∼30 nm is uniformly coated onto the UC core, resulting in a well-defined core–shell structure (Fig. 2c). A subsequent sol–gel coating process leads to the deposition of a titania shell onto the surface of Er:Y2O3@SiO2 nanospheres. Followed by an ultrasound assisted post-hydrolysis and a calcination process, typical sandwich-like core–shell–shell structured Er:Y2O3@SiO2@mTiO2 nanospheres with an average diameter of 380 nm are obtained (Fig. 2d and e), indicating the presence of ∼30 nm thick TiO2 layer. In addition, both the TEM and SEM image (Fig. 2f and g) show that the TiO2 shells exhibit highly mesoporous structures, which are resulted from the voids between the aggregated TiO2 oligomers. The high resolution TEM (HRTEM) image (Fig. 2h) clearly displays that the TiO2 shells are well crystallized with a size of ∼5.5 nm and a d-spacing of 0.35 nm, well-matching to the d101 of anatase TiO2.25 Energy-dispersive X-ray (EDX) analysis of the sample Er:Y2O3-2@SiO2@mTiO2 (Fig. 2i) taken within the red spot exhibits the characteristic peaks of Ti, Y, Er, Si and O, suggesting the possible coexistence of Er:Y2O3, silica and titania. In addition, the quantitative result indicates that the weight percentage of Er and Y in the resultant composites is 0.34 and 9.7 wt%, which is similar with the adding ratio. Moreover, hollow structured mesoporous TiO2 can be obtained by etching off the inner UC core with HCl solution (Fig. 3, insert). EDX analysis only displays the characteristic peaks of O, Si, and Ti, revealing that the inner UC core was well removed (Fig. 3).
 |
| | Fig. 2 TEM images of the uniform erbium doped Y2O3 nanospheres with dopant content of 3 wt% prepared via a urea assisted co-precipitation method (a and b), core–shell structured Er:Y2O3@SiO2 nanospheres obtained by a Stöber sol–gel coating method (c), mesoporous Er:Y2O3@SiO2@mTiO2 nanospheres through a kinetically controlled sol–gel method (d, e, and g), SEM image of Er:Y2O3@SiO2@mTiO2 nanospheres (f), HRTEM image of mesoporous Er:Y2O3-2@SiO2@mTiO2 nanospheres (h), and EDX analysis taken within the red circle (i). | |
 |
| | Fig. 3 EDX analysis for hollow structured mesoporous TiO2. Insert is the TEM image. | |
N2 sorption isotherm of the Er:Y2O3-2@SiO2@mTiO2 composites (Fig. 4A) shows a characteristic IV curve with a hysteresis loops close to H1-type, further suggesting that the outer TiO2 shells contain uniform mesopores. The BET surface area and pore volume of the composites are calculated to be 151.5 m2 g−1 and 0.23 cm3 g−1, respectively. Correspondingly, the pore size distribution (Fig. 4B) derived from the adsorption branch using the Barrett–Joyner–Halenda (BJH) method reveals a uniform pore size centered at ∼3.8 nm. The X-ray diffraction patterns (XRD) of the Er:Y2O3@SiO2@mTiO2 nanospheres (Fig. 5) exhibit a sharp and strong diffraction peak at 2θ = 25.3°, which can be well indexed as the 101 reflection of anatase TiO2 crystalline phase (JCPDS card no. 01-073-1764). Moreover, the size of TiO2 nanocrystals (Table 1) in the Er:Y2O3@SiO2@mTiO2 composites was calculated to be ∼5.5 nm using Scherrer formula, which is similar to the HRTEM result. Additional diffraction peaks were observed in the patterns of all materials, which are typical for Y2O3 (JCPDS card no. 00-043-1036). No diffraction peak of erbium oxide was detected, indicating that the Er3+ ion was well doped into the nanocrystals of Y2O3.
 |
| | Fig. 4 N2 sorption isotherms (A) and pore size distribution (B) of Er:Y2O3-2@SiO2@mTiO2 nanosphere. | |
 |
| | Fig. 5 XRD patterns of (a) Er:Y2O3-2, (b) Er:Y2O3-2@SiO2, (c) Er:Y2O3-1@SiO2@mTiO2, (d) Er:Y2O3-2@SiO2@mTiO2, and (e) Er:Y2O3-3@SiO2@mTiO2. | |
Table 1 Physicochemical properties of the resultant core–shell nanospheresa
| Sample name |
DA (nm) |
D (nm) |
SBET (m2 g−1) |
V (cm3 g−1) |
| DA, the particle size of anatase nanocrystals calculated by Scherrer formula; SBET, BET surface area; D, pore-size diameter; V, total pore volume. |
| Er:Y2O3-1@SiO2@mTiO2 |
5.4 |
3.8 |
150.8 |
0.23 |
| Er:Y2O3-2@SiO2@mTiO2 |
5.5 |
3.8 |
151.5 |
0.23 |
| Er:Y2O3-3@SiO2@mTiO2 |
5.7 |
3.7 |
153.2 |
0.24 |
UV-Vis absorption spectra (Fig. 6A) of the Er:Y2O3@SiO2@mTiO2 nanospheres with varied Er3+ contents reveal that all the samples possess an absorption edge of ∼420 nm, corresponding to the bandgap energy of anatase TiO2 (3.2 eV). Moreover, a peak at 522 nm was clearly observed and the intensity increases with rising Er3+ concentration, which is due to the energy transition from 4I15/2 to 2I11/2 in Er3+ dopant.16 To prove the UC properties of the resultant composites, the fluorescence analysis was carried out (Fig. 6B). When the samples were excited with a 522 nm laser, the UC signals were clearly shown at the wavelengths of 339, 363, 394, 403, and 423 nm, which can be assigned to 2P3/2 → 4I15/2, 2K15/2 → 4I15/2, 4G11/2 → 4I15/2, 2H9/2 → 4I15/2, and 4H3/2 → 4I15/2 transitions of Er3+ ions, respectively.26,27 The UC could be triggered by the excited state absorption (ESA) mechanism which refers to that Er3+ in the excitation state can still absorb photon(s) to transit to higher energy levels. Afterwards, the UC luminescence would be generated when the high-energy leveled Er3+ ion relaxes to the ground state (Fig. S1†).28 In addition, it was found that the maximum intensity of luminescence was obtained with an Er3+ content of 3 wt% in the Y2O3 nanocrystals. This is due to the fact that erbium ions in a lower concentration (1 wt%) are randomly distributed in the host lattice and the average distance between two Er3+ ions are too far apart to interact efficiently. However, at a higher concentration (5 wt%) the average Er3+–Er3+ distance are shortening, thus leading to the formation of Er3+ cluster and concentration quenching of Er3+ ions. As a result, the non-radiative decay will become prominent in the whole UC process, causing the loss of energy and the degradation of emission intensities.29,30 To further evaluate the UC property of the resultant composites, a metal halide lamp (150 W) with a 420 cutoff filter (λ > 420 nm) was employed as the light source to examine the photocatalytic degradation activity for BPA over commercially available TiO2 NPs (P25) and Er:Y2O3-2@SiO2@mTiO2 composites (Fig. 7). Before initiation of the reaction, the mixture was mechanically stirred in dark for 60 min to reach the adsorption/desorption equilibrium between the catalyst and pollutants. It was found that the P25 catalyst did not show any visible photocatalytic activity and the concentration change of BPA in the system fluctuated possibly owing to the adsorption/desorption equilibrium. Among the UC catalysts tested, Er:Y2O3-2@SiO2@mTiO2 exhibits the best degradation performance (16% decomposition of BPA in 2 h), which is higher than that of previous UC based photocatalyst (10% degradation of methyl blue in 2 h).31
 |
| | Fig. 6 UV-Vis absorption spectra (A) and emission spectra (B) of Er:Y2O3@SiO2@mTiO2 nanospheres with various erbium concentrations excited using a Xe flash source. Black: Er:Y2O3-1@SiO2@mTiO2, red: Er:Y2O3-2@SiO2@mTiO2, and green: Er:Y2O3-3@SiO2@mTiO2. | |
 |
| | Fig. 7 (A) Visible-light photocatalytic degradation profile of BPA in the presence of (a) commercial TiO2 nanoparticles (P25), (b) Er:Y2O3-1@SiO2@mTiO2, (c) Er:Y2O3-2@SiO2@mTiO2 and (d) Er:Y2O3-3@SiO2@mTiO2 and (B) the intensity distribution of the light source utilized. Before initiation of the reaction, the mixture was mechanically stirred in dark for 60 min to reach the adsorption/desorption equilibrium between the catalyst and pollutants. | |
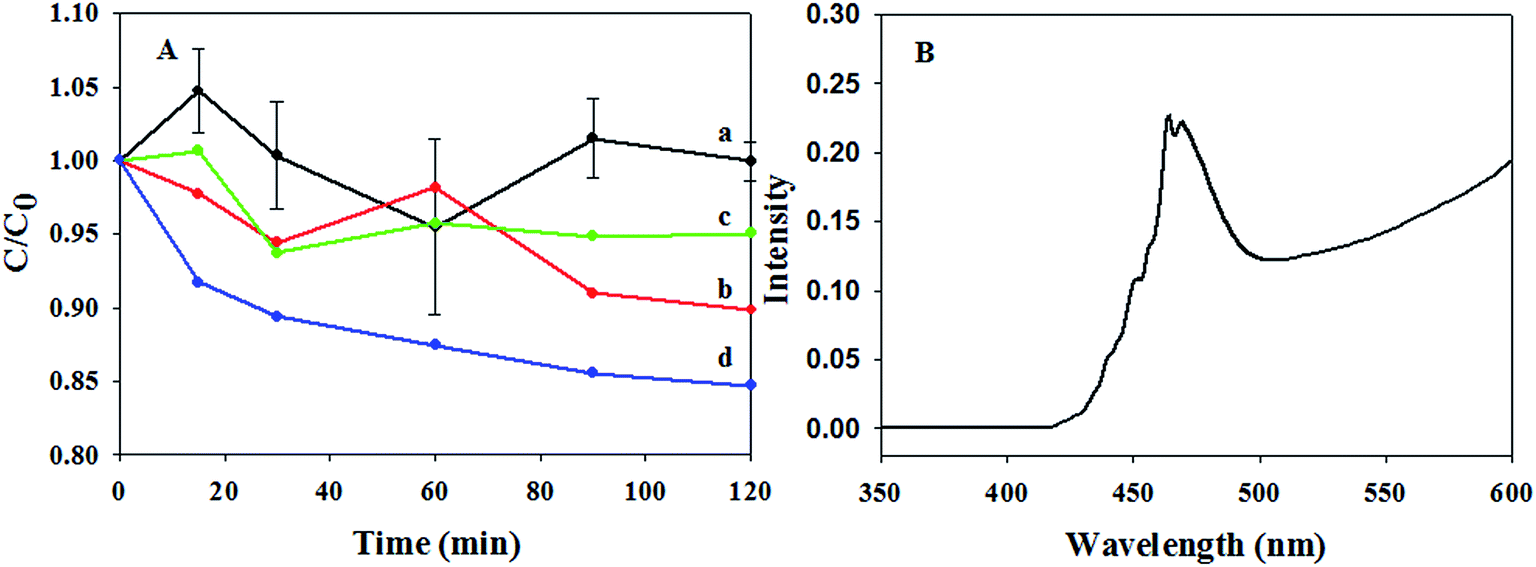
The performance of the resultant core–shell materials on the sonocatalytic degradation of BPA was examined using an US with a frequency of 300 kHz and an applied power of 100 W (Fig. 8 and S2†). As a control, the degradation performances of US alone, Er:Y2O3-2 and Er:Y2O3-2@SiO2 were also tested, exhibiting the degradation rate constants of 0.054, 0.086 and 0.066 min−1, respectively, when a pseudo first-order reaction was assumed. The higher degradation rate of Er:Y2O3-2 nanoparticles than Er:Y2O3-2@SiO2 core–shell nanospheres could be due to the fact that the former one possesses a smaller particle size and a larger surface area, thus providing more nucleation sites for the cavitation bubble formation. Notably, the degradation performance was greatly improved when Er:Y2O3@SiO2@mTiO2 composites were utilized as the sonocatalysts. The sonocatalytic degradation rate of BPA (Fig. 8A and B) first increased with the rise of Er3+ content in the Y2O3 nanocrystals. A maximum sonocatalytic degradation rate was observed in the presence of the sample Er:Y2O3-2@SiO2@mTiO2 (0.155 min−1), which was 2.9 and 2 times higher than that of US alone and hollow structured mesoporous TiO2 (0.077 min−1), respectively. More importantly, the degradation rate is much faster than of the sonocatalyst reported previously for treatment of BPA (0.09–0.14 min−1).32,33 In addition, BPA can be completely removed in 30 min. Further increasing Er3+ content to 5% did not lead to any further increase in the sonocatalytic reaction rate, and even caused a decrease, which is due to the reduced UC emission intensity (Fig. 6B). The trend of sonocatalytic activity with respect to different doping content is same as that of the intensity of luminescence, revealing the close relationship between the sonocatalytic performance and luminescence property of the catalyst. Note that the morphologies and surface areas for all the mesoporous Er:Y2O3@SiO2@mTiO2 samples (Fig. S3,† Table 1) are similar, indicating that the effects of morphology and surface area on the behavior of photo-generated charges for these samples are negligible. Interestingly, the nonporous Er:Y2O3-2@SiO2@TiO2 composites synthesized without a post-hydrolysis process exhibited an apparently slower degradation rate (0.096 min−1) than mesoporous one (0.155 min−1), suggesting the priority of mesoporous structure in the resultant composites. The remarkable sonocatalytic performance of the resultant catalyst can be attributed to two possible reasons, as illustrated in Fig. 9. On one hand, the presence of mesopores accelerates the nucleation rate for the formation of cavitation bubbles, producing superior cavitation effects to the nonporous material because pore corners in porous material can provide energetically preferred binding sites at which the new phase can be easier hold.34–36 The SCL images (Fig. S4†) recorded with an exposure time of 30 s using a digital camera were taken to visualize the change of cavitation reactive zone in the presence of nonporous and porous catalyst.33,37 It clearly shows that the sonochemical reaction zone becomes wider and scattered after adding nonporous Er:Y2O3-2@SiO2@TiO2 catalyst because the presence of solid impurities could increase the nucleation rate to some extent by altering the nucleation from the aqueous phase to the solid–liquid boundary.38 However, when the mesoporous Er:Y2O3-2@SiO2@mTiO2 catalyst was used, the blue color in SCL image becomes more intense and bright, further illustrating the priority of mesoporous structure. On the other hand, the UC core could absorb the visible light photons from SL (Fig. S5†) and then emit UV light photons through an energy transfer, which can excite TiO2 nanocrystals to form additional electron–hole pairs on the surface, increasing the concentration of reactive oxidation species in the reaction system.39,40 Note that although the TiO2 fabricated in this study possessed mesoporous structures and high surface areas, the adsorption effect is not significant possibly owing to the low affinity of TiO2 towards BPA (Fig. S5†). Thus, the physical adsorption effect can be neglected compared with the high degradation performance. Therefore, the high catalytic activity of Er:Y2O3@SiO2@mTiO2 catalyst is related to the intensified cavitation reactive zone resulted from the mesoporous structure and the generation of additional photo-excited charge carriers achieved through UC mechanism. The recycle test of the core–shell structured Er:Y2O3-2@SiO2@mTiO2 was also examined (Fig. 10). After four recycles, a constant degradation performance was retained, indicating the excellent reusability of this material. In addition, TEM images (Fig. 11) clearly showed that the defined core–shell structure is well retained, suggesting the excellent mechanical stability of the resultant composites. However, SEM images revealed that the surface of the recycled sample became a little bit rougher relative to fresh one and a few nanospheres were broken, which was possibly resulted from the continuous asymmetrical pitting by the shock waves (Fig. S6†). The reusability tests further confirm that Er:Y2O3@SiO2@mTiO2 composites are promising candidates as sonocatalysts.
 |
| | Fig. 8 The sonocatalytic degradation performance (A) and pseudo first-order kinetic constant (B) of BPA with varied sonocatalysts. (a) US alone, (b) Er:Y2O3-1@SiO2@mTiO2, (c) Er:Y2O3-2@SiO2@mTiO2, (d) Er:Y2O3-3@SiO2@mTiO2, (e) hollow structured mesoporous TiO2, and (f) nonporous Er:Y2O3-2@SiO2@TiO2 composites. In (B), the value above the vertical bar is the coefficient of determination (R2), which is defined as the ratio of the explained variation to the total variation. | |
 |
| | Fig. 9 Schematic illustration for the sonocatalytic mechanism in the presence of Er:Y2O3@SiO2@mTiO2 catalyst. On one hand, the presence of mesopores serves as nucleation sites for accelerating the formation of cavitation bubbles. On the other hand, the UC agent absorbs the visible-light photons from SL and then emits the UV light photons through ESA mechanism, which can further excite anatase TiO2 to form electron–holes pairs. G: ground state, E1 and E2: excitation state 1 and 2, CB: conduction band, VB: valence band, and SL: sonoluminescence. | |
 |
| | Fig. 10 Recycle test for the Er:Y2O3-2@SiO2@mTiO2 sonocatalyst. | |
 |
| | Fig. 11 TEM images of recycled Er:Y2O3-2@SiO2@mTiO2 catalyst (a and b). | |
Conclusion
In summary, we report the integration of a visible-light active UC agent with mesoporous TiO2 via a versatile two-step sol–gel coating strategy. The resultant composites possess a high BET surface area (∼151.5 m2 g−1), a large pore volume (∼0.23 cm3 g−1) and uniform mesopores (∼3.8 nm) as well as an excellent UC property. When evaluated as an advanced sonocatalyst for degradation of BPA, the catalyst with an erbium dopant content of 3 wt% in the Y2O3 nanocrystals is found to exhibit the best degradation performance. The pseudo first-order degradation rate constant is calculated to be 0.155 min−1, which is 2.9 and 2 times larger than that of US alone (0.054 min−1) and hollow structured mesoporous TiO2 NPs (0.077 min−1), respectively. The superior catalytic activity is ascribed to an intensified cavitation reaction zone and an enhanced amount of photo-generated charges. In addition, the catalyst can be easily recycled and exhibit an excellent reusability. This study gives an important insight into the design and synthesis of multi-functional catalyst for sonochemical process.
Acknowledgements
We thank the Korea Institute of Energy Technology Evaluation and Planning (KETEP, 20152510101820) and Korea Mine Reclamation Corporation (MIRECO, Q1512631) for financial support.
References
- C. D. Vecitis, Y. Wang, J. Cheng, H. Park, B. T. Mader and M. R. Hoffmann, Environ. Sci. Technol., 2010, 44, 432–438 CrossRef CAS PubMed.
- R. Xiao, Z. Wei, D. Chen and L. K. Weavers, Environ. Sci. Technol., 2014, 48, 9675–9683 CrossRef CAS PubMed.
- P. Chowdhury and T. Viraraghavan, Sci. Total Environ., 2009, 407, 2474–2492 CrossRef CAS PubMed.
- K. S. Suslick, S. J. Doktycz and E. B. Flint, Ultrasonics, 1990, 28, 280–290 CrossRef CAS PubMed.
- S. Hilgenfeldt, S. Grossmann and D. Lohse, Nature, 1999, 398, 402–405 CrossRef CAS.
- Z. Eren, J. Environ. Manage., 2012, 104, 127–141 CrossRef CAS PubMed.
- Y. L. Pang, A. Z. Abdullah and S. Bhatia, Appl. Catal., B, 2010, 100, 393–402 CrossRef CAS.
- M. Farshbaf Dadjour, C. Ogino, S. Matsumura, S. Nakamura and N. Shimizu, Water Res., 2006, 40, 1137–1142 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- Y. Wang, D. Zhao, W. Ma, C. Chen and J. Zhao, Environ. Sci. Technol., 2008, 42, 6173–6178 CrossRef CAS PubMed.
- N. Ghows and M. H. Entezari, J. Hazard. Mater., 2011, 195, 132–138 CrossRef CAS PubMed.
- Y. Li, B. P. Bastakoti, M. Imura, S. M. Hwang, Z. Sun, J. H. Kim, S. X. Dou and Y. Yamauchi, Chem.–Eur. J., 2014, 20, 6027–6032 CrossRef CAS PubMed.
- M. Zhao, B. P. Bastakoti, Y. Li, H. Xu, J. Ye, Z. Liu and Y. Yamauchi, Chem. Commun., 2015, 51, 14582–14585 RSC.
- W. Qin, D. Zhang, D. Zhao, L. Wang and K. Zheng, Chem. Commun., 2010, 46, 2304–2306 RSC.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584–2599 CrossRef CAS PubMed.
- J. Xu, T. J. K. Brenner, Z. Chen, D. Neher, M. Antonietti and M. Shalom, ACS Appl. Mater. Interfaces, 2014, 6, 16481–16486 CAS.
- E. L. Cates, M. Cho and J. H. Kim, Environ. Sci. Technol., 2011, 45, 3680–3686 CrossRef CAS PubMed.
- Z. Zhang, W. Wang, W. Yin, M. Shang, L. Wang and S. Sun, Appl. Catal., B, 2010, 10, 68–73 CrossRef.
- V. Belova, D. A. Gorin, D. G. Shchukin and H. Möhwald, ACS Appl. Mater. Interfaces, 2011, 3, 417–425 CAS.
- V. Belova, D. A. Gorin, D. G. Shchukin and H. Möhwald, Angew. Chem., Int. Ed., 2010, 49, 7129–7133 CrossRef CAS PubMed.
- R. E. A. Arndt and A. T. Ippen, J. Basic Eng., 1968, 90, 249–261 CrossRef.
- J. G. Li, X. Li, X. Sun and T. Ishigaki, J. Phys. Chem. C, 2008, 112, 11707–11716 CAS.
- W. Li and D. Zhao, Adv. Mater., 2013, 25, 142–149 CrossRef CAS PubMed.
- P. Qiu, W. Li, B. Thokchom, B. Park, M. Cui, D. Zhao and J. Khim, J. Mater. Chem. A, 2015, 3, 6492–6500 CAS.
- W. Li, J. Yang, Z. Wu, J. Wang, B. Li, S. Feng, Y. Deng, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2012, 134, 11864–11867 CrossRef CAS PubMed.
- I. Kamma, M. Mbila, K. E. Steege Gall and B. R. Reddy, Opt. Mater. Express, 2013, 3, 884–892 CrossRef CAS.
- S. Dong, X. Zhang, F. He, S. Dong, D. Zhou and B. Wang, J. Chem. Technol. Biotechnol., 2015, 90, 880–887 CrossRef CAS.
- V. D. Rodríguez, V. K. Tikhomirov, J. J. Velázquez, M. V. Shestakov and V. V. Moshchalkov, Adv. Opt. Mater., 2013, 1, 747–752 CrossRef.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, Chem. Mater., 2003, 15, 3650–3655 CrossRef CAS.
- S. Obregóna and G. Colón, Chem. Commun., 2012, 48, 7865–7867 RSC.
- Y. Tang, W. Di, X. Zhai, R. Yang and W. Qin, ACS Catal., 2013, 3, 405–412 CrossRef CAS.
- J.-S. Park, N. Her, J. Oh and Y. Yoon, Sep. Sci. Technol., 2011, 78, 228–236 CAS.
- N. Her, J.-S. Park, J. Yoon, J. Sohn, S. Lee and Y. Yoon, Ind. Eng. Chem. Res., 2011, 50, 6638–6645 CrossRef CAS.
- L. O. Hedges and S. Whitelam, Soft Matter, 2012, 8, 8624–8635 RSC.
- S. I. Madanshetty and R. E. Apfel, J. Am. Chem. Soc., 1991, 90, 1508–1514 CAS.
- P. Qiu, W. Li, K. Kang, B. Park, W. Luo, D. Zhao and J. Khim, J. Mater. Chem. A, 2014, 2, 16452–16458 CAS.
- E. Kim, M. Cui, M. Jang, B. Park, Y. Son and J. Khim, Ultrason. Sonochem., 2014, 21, 1504–1511 CrossRef CAS PubMed.
- H. B. Marschall, K. A. Mørch, A. P. Keller and M. Kjeldsen, Phys. Fluids, 2003, 15, 545–553 CrossRef CAS.
- J. Q. Gao, R. Z. Jiang, J. Wang, B. X. Wang, K. Li, P. L. Kang, Y. Li and X. D. Zhang, Chem. Eng. J., 2011, 168, 1041–1048 CrossRef CAS.
- J. Wang, S. Y. Zhou, J. Wang, S. G. Li, J. Q. Gao, B. X. Wang and P. Fan, Ultrason. Sonochem., 2014, 21, 84–92 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01689h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.