DOI:
10.1039/C5RA08057F
(Paper)
RSC Adv., 2015,
5, 42653-42662
Adjustable plasmonic optical properties of hollow gold nanospheres monolayers and LSPR-dependent surface-enhanced Raman scattering of hollow gold nanosphere/graphene oxide hybrids†
Received
24th February 2015
, Accepted 5th May 2015
First published on 5th May 2015
Abstract
Colloidal hollow gold nanospheres (HGNs) with adjustable localized surface plasmon resonance (LSPR) properties were synthesized and self-assembled into HGNs monolayers as a proper substrate for investigation of LSPR-dependent surface enhanced Raman scattering (SERS) behavior with Rhodamine B as the probe molecule. The SPR peaks of colloidal HGNs could be tunable over a wavelength region from 540 nm to 630 nm by carefully tailoring HGNs sizes and wall thicknesses. The broadening and red shift of SPR peaks of self-assembled HGNs monolayers were attributed to the electromagnetic coupling of adjacent HGN plasmons and the change of dielectric constant around HGNs when compared to those of colloidal HGNs. The relative SPR peak shift decayed nearly exponentially with the ratio of gap separation to diameter of HGNs, which fitted well with the “plasmon ruler” behavior of solid gold nanospheres and HGN dimeric reported previously. Our results indicated that the SERS performance of HGNs monolayers was highly LSPR-dependent and an obvious enhancement in the Raman signals was collected when the laser excitation line matched with the peak position of LSPR band of HGNs monolayers. Furthermore, the sandwich-structured HGN/graphene oxide/HGN (HGN/GO/HGN) hybrid was constructed through layer-by-layer method with spin-coated GO nanosheets as the interlayer. The cladding structures had better SERS activity than the HGNs monolayer due to the chemical enhancement of GO and coupled electromagnetic enhancement of double HGNs in the horizontal and vertical directions. The designed HGN/GO/HGN hybrid configuration with LSPR-dependent SERS performance exhibited a potential application in chemical sensors, environmental monitoring, disease controlling, and food safety fields.
1 Introduction
Since the surface-enhanced Raman scattering (SERS) effect was first observed on a rough silver electrode in 1974 (ref. 1) and applied on single molecule detection in 1997,2,3 SERS has been regarded as a powerful technique for detection of trace organic and biological molecules in chemistry, biology, food safety and biomedical diagnostics.4–7 Various noble metal nanoparticles, such as nanospheres,8 nanocages,9 nanocubes,10 nanorods,11 nanostars12 and nanoflowers,13 with excellent optical properties in the visible and near-infrared regions have been explored to enhance the Raman signals of determinant since the SERS effect greatly depends on the localized surface plasmon resonance (LSPR) features of nanoparticles and is tunable by controlling the sizes and morphologies of the metal nanostructures.14–18 For example, Zhang et al. successfully regulated the SPR range of hollow gold nanospheres (HGNs) from 550 nm to 820 nm by changing the diameters and wall thicknesses of HGNs when compared to the solid gold nanospheres (SGNs).19–21 Halas et al. also found an obvious SERS enhancement induced by well-designed HGNs, which was comparable to solid nanosphere dimers.22 Chandra et al. adopted HGNs as efficient optical scatterers23 and controlled their SPR features by changing the sizes and morphologies of HGNs.24 Additionally, understanding the optical properties of extended layers of particles is of equal importance. The plasmonic coupling between adjacent particles will result in a red-shifted and broadened LSP band and dramatic enhancement in the Raman signal due to more hot areas generated at the gaps or junctions between aggregated nanoparticles and the coupling of their localized electromagnetic fields.25,26 The fabrication process of aggregated nanoparticle layers, especially nanoparticle monolayer is therefore given great attention for excellent structural control of uniform film to obtain reproducible Raman signals.27 Several methods related to the assembly of highly uniform nanoparticle arrangements, such as the Langmuir–Blodgett technique28–30 and nanoimprint lithography,27 have been adopted. However, these methods require technical apparatus to control the surface pressure or make special stamps. Comparatively speaking, the interface self-assembling technique would be easier and less expensive for large-scale fabrication of SERS active substrates. However, the study on tailorable SPR feature of self-assembled HGNs layers and their application in LSPR-dependent SERS is still limited.
Graphene, a flat monolayer of sp2-hybridized carbon atoms arranged closely in a hexagonal lattice configuration, has been widely studied and confirmed its important role in the enhancement of Raman signal.31,32 It is reported that graphene can provide an SERS enhancement factor of 2–17 when used for aromatic molecules detection, which originates from the quick charge transfer between aromatic molecules and graphene surfaces and called as chemical enhancement effect.33 Moreover, graphene oxide (GO) or graphene or reduced GO (rGO) has high affinity to aromatic molecules and could act as an ideal host material to adsorb many molecular analytes.34 Various graphene–metal composite nanostructures are designed to trap more organic molecules and enhance localized plasmons coupling in nanostructures for promoting the detection sensitivity.35,36 Additionally, Liu et al. fabricated the Au/rGO/Au37 and Ag/rGO/Au38 sandwiched structures through electrochemical deposition and obtained prominent SERS activities via the plasmons transfer in the extra interfaces between metallic plasmons and stable interlayer. Zhu et al.35,39 designed metal/GO/metal cladding structures and investigated the light-matter interactions and SERS behaviors. The metal/GO hybrid structure with adjustable plasmonic absorption and efficient plasmon transfer between extra interfaces might therefore present excellent SERS activity for detection of trace molecules.38
In this work, the HGNs monolayers were self-assembled at the interface between a polar and a nonpolar solvent without the organic addition and were further transferred to the nitrogen doped diamond-like carbon (DLC:N) film substrate. The SERS sensitivity of HGNs monolayers was optimized by tuning the SPR feature of these HGNs. Furthermore, combining the electromagnetic enhancement of HGNs and chemical enhancement of GO, we studied the SERS behavior of sandwiched HGN/GO/HGN structure formed by the self-assembled HGNs layer and spin-coated GO sheets, as shown in Fig. 1. The HGN/GO/HGN hybrid exhibited excellent SERS activities for the detection of organic molecule such as Rhodamine B (RhB) when compared to the HGNs monolayers.
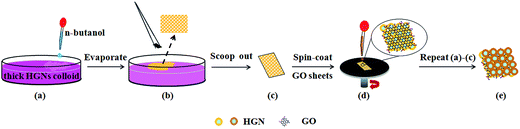 |
| | Fig. 1 Schematic illustration of fabrication process of sandwiched HGN/GO/HGN structure based on the self-assembling technique. | |
2 Materials and experimental methods
2.1 Materials and reagents
Graphite oxide (50 μm) was purchased from Shanghai Carbon Co., Ltd. Chloroauric acid trihydrate (HAuCl4·3H2O, 99%), sodium borohydride (NaBH4) and RhB (purity 98%) were supplied by Sigma, USA. Cobalt chloride hexahydrate (CoCl2·6H2O), trihydrate sodium citrate (TCD, C6H5Na3O7·3H2O, purity 99%) and n-butanol (C4H10O) were supplied by Eagle chemical Reagent Co., Ltd., ZheJiang. All chemicals were of analytical grade and used without further purification. The deionized water used in the synthesis was obtained from Millipore Q purification system (resistivity > 18 MΩ cm).
2.2 Colloidal HGNs synthesis
The colloidal HGNs with tunable SPR properties were synthesized by using a similar technique developed by Zhang,19 in which cobalt nanoparticles were used as the sacrificial templates. Two synthetic methods (fast reaction and slow reaction) were used to make HGNs with different sizes and shell thicknesses in this work. The detailed volumes of reagents used for preparation of different sized HGNs were displayed in Table 1. Notice that we named the HGNs samples with the position of SPR peak at the extinction spectra in the following discussion. In a typical procedure, 75 mL deionized water and a certain amount of TCD (0.1 mol L−1) were first added into a three-necked round-bottom flask at a 35 °C water bath. The mixture solution in the flask was deoxygenated by bubbling with argon gas for 30 min. In the fast reaction process, 100 μL CoCl2 (0.4 mol L−1) was added into the flask with rapid magnetic stirring, and a certain amount of fresh NaBH4 (1 mol L−1) was injected in the solution later. The mixed solution would change from colorless to brown within a few seconds, indicating the reduction of Co2+ to cobalt nanoparticles. The argon flow was continuously introduced into the flask for one hour until the hydrolysis reaction of NaBH4 completed. In the case of slow reaction, 100 μL CoCl2 solution was diluted to 25 mL and added by dropwise into the flask for about 2–3 min, and then the cobalt nanoparticles were produced with the color of mixed solution changing to light brown. After that, 30 mL of the generated cobalt nanoparticles colloid was fast transferred to a stirring solution of HAuCl4 (0.6 mmol L−1) and the HGNs with controllable sizes were obtained immediately. If changing the concentration of HAuCl4 solution from 0.3 mmol L−1 to 1.0 mmol L−1, the color of the mixed solution would turn to cyan, blue, purple and red, respectively, and the HGNs with different shell thicknesses were consequently obtained.
Table 1 Volumes of reagents used for preparation of different-sized HGNs with adjustable SPR properties by the fast or slow reaction process
| HGNs |
TCD |
NaBH4 |
CoCl2 |
Synthetic methods |
HAuCl4 |
SPR peaks |
| HGN 536 |
600 μL |
50 μL |
100 μL |
Fast reaction |
10 mL |
536 nm |
| HGN 548 |
900 μL |
100 μL |
100 μL |
Fast reaction |
10 mL |
548 nm |
| HGN 561 |
700 μL |
100 μL |
100 μL |
Fast reaction |
10 mL |
561 nm |
| HGN 579 |
600 μL |
100 μL |
100 μL |
Fast reaction |
10 mL |
579 nm |
| HGN 593 |
600 μL |
100 μL |
100 μL |
Slow reaction |
10 mL |
593 nm |
| HGN 629 |
400 μL |
100 μL |
100 μL |
Slow reaction |
10 mL |
629 nm |
2.3 Colloidal SGNs synthesis
The SGNs used as a reference were synthesized by the classic Frens' method.40 Briefly, 1.5 mL TCD (the mass fraction of 1%) was added quickly into the boiling solution of 100 mL, 0.25 mmol L−1 HAuCl4 with stirring to react for 15 min. Then the solution was cooled down to the room temperature with stirring and the SGNs colloid was obtained. The way of naming SGNs sample was similar to that of HGNs.
2.4 Preparation of HGN layers and sandwiched HGN/GO/HGN configurations
The 100 nm-thick DLC:N films (4.0 at.% N) were deposited on glass substrates by a filtered cathodic vacuum arc system as described in literature.41 Then the interface self-assembling method described in previous reports42–45 was used to fabricate the first HGNs monolayer and were transferred to the DLC:N surface (Fig. 1). In detail, the n-butanol was slowly dropped in the Petri dish containing thick HGNs colloids protected by TCD, and an agglomeration of HGNs was gradually developed to a monolayer at the oil/water interface within 10 min. (See ESI† movie of the formation of HGNs layers.) After the HGNs layer was scooped out with DLC:N substrate (7 mm × 6 mm), dried at 60 °C and cleaned with ethanol, a 0.5 mg mL−1 exfoliated GO aqueous solution (prepared by the modified Hummers method) was spin-coated on the DLC:N/HGN substrate at 1500 rpm for 30 s. The second HGNs layer was fabricated on the DLC:N/HGN/GO surface as the same way of the first HGNs layer, resulting in a HGN/GO/HGN hybrid.
2.5 Characterization
The morphologies of HGNs, SGNs and their monolayers and hybrids were observed by transmission electron microscopy (TEM, Philips CM 300 FEG) at 200 kV and scanning electron microscopy (SEM, Hitachi S4800). The surface morphology of GO films was characterized by atom force microscopy (AFM, XE-100E) at the tapping mode. The resonant frequency and force constant of cantilever were 300 kHz and 40 N m−1. The X-ray diffraction (XRD) patterns of the gold nanostructures were recorded by a diffractometer (Bruker AXS D8) with an area detector operating under a voltage of 40 kV and a current of 40 mA using Cu Kα radiation (λ = 0.15418 nm). The extinction spectra of gold nanospheres, monolayers and hybrids were measured by the UV-vis spectrophotometer (Hitachi U3900). All samples for SERS measurement were incubated in the RhB aqueous solutions with different concentrations for 24 h and the SERS spectra were acquired using a 633 nm He–Ne laser with 1 mW power and an objective (50×) by using a Thermo Fisher DXR Raman spectrometer. The Raman signal intensity of RhB was normalized by using a silicon peak at ∼1000 cm−1 as the standard.
3 Results and discussion
3.1 Microstructures of HGNs and sandwiched HGN/GO/HGN configurations
The TEM images of dispersed HGNs with different sizes including the HGN 536, HGN 579 and HGN 629 are shown in Fig. 2a–c, where a strong contrast difference in all of the HGNs with a bright interior-cavity and a dark shell confirms their hollow architectures. The TEM image of HGN 629 in Fig. 2c indicates a non-uniform gold shell with many surface defects. The outer and inner diameters of HGNs were measured and analyzed statistically from a large amount of HGNs (at least 100 particles). Our results show that the outer diameter and inner one are about 21 ± 1.2 nm and 14 ± 1.1 nm for HGN 536, 34 ± 2.5 nm and 22 ± 2.2 nm for HGN 579 and 63 ± 4.8 nm and 41 ± 4.2 nm for HGN 629, respectively. All HGNs have a similar aspect ratio (inner diameter/outer diameter) near 0.67. The HGNs sizes are determined by the diameters of cobalt templates, thus the fabrication of cobalt templates is a crucial factor in controlling the HGNs sizes. In the present study, by adjusting the volume of reducing agent (NaBH4) and stabilizing agent (TCD) and varying the synthesis method, we can effectively modify the nucleation and growth process of HGNs for further precise controlling of SPR band of HGNs. Comparably, the size of ellipsoidal SGN 524 is 38 ± 5.3 nm in the major axis and 24 ± 4.1 nm in the minor axis, as shown in Fig. 2d. Two kinds of Au facets (111) and (200) are clearly distinguished at the high-resolution TEM image (Fig. 2e) and all five Au facets are obtained at the electron diffraction pattern (Fig. 2f) in which the concentric rings are corresponding to the diffraction from {111}, {200}, {220}, {311} and {222} planes of face-center-cubic (fcc) gold.46 The XRD result of HGNs on glass substrate (Fig. 3) also confirms the fcc-structured gold with fine crystalline.47
 |
| | Fig. 2 TEM images of (a) HGN 536, (b) HGN 579, (c) HGN 629 and (d) SGN 524. (e) High-resolution TEM image and (f) electron diffraction pattern of HGN 579. Images were obtained by drop casting of nanoparticle solutions on the TEM grids. | |
 |
| | Fig. 3 XRD spectrum of HGNs monolayer on the glass substrate. | |
The morphologies of self-assembled HGNs and SGN layers were characterized by TEM and SEM measurements. As shown in Fig. 4, the HGNs with different sizes are close-packed with an interparticle spacing of 2–10 nm statistically analyzed from about 100 particles. Some obvious voids and a few aggregates can be observed due to the disturbance of the oil/water interface after gradual addition of n-butanol and the competition between electrostatic repulsion of negative charged HGNs against the long-range van der Waals interactions.43,45 The SEM images in Fig. 5a, b and S1 (see ESI 1†) also confirm the hollow structured HGNs monolayer with uniform arrangement. Our statistical result based on random ten TEM images indicate that the increscent HAuCl4 enhances the shell thickness (from 9.6 ± 4.3 nm to 30 ± 2.8 nm) and decreases the inner diameter (from 40 ± 4.3 nm to 14.6 ± 2.6 nm) and the aspect ratio (from 0.68 to 0.25) when keeping outer diameter of HGNs (Fig. 4). After the GO nanosheets are spin-coated on the DLC:N/HGN substrate, the HGNs layer underneath the wrinkled and semi-transparent GO sheets is still clearly distinguishable (Fig. 5c) and the GO wrinkles tend to be formed at asperous areas on the surface of HGNs layer. The thickness of GO sheets is about 2.1 nm by the AFM measurement (Fig. S2 in ESI 1†). When the second HGNs layer is fabricated on the DLC:N/HGN/GO with the same method of the first HGNs monolayer, the multilayered structure can be recognized form Fig. 5d. However, the upper HGNs layer is not so flat and symmetrical to some extent, which might be attributed to the wrinkled area of GO sheets maintaining more HGNs.
 |
| | Fig. 4 TEM images of (a) HGN 532, (b) HGN 550, (c) HGN 587, (d) HGN 603, (e) and (f) HGN 629 monolayers self-assembled on the TEM grids. | |
 |
| | Fig. 5 SEM images of (a) and (b) DLC:N/HGN, (c) DLC:N/HGN/GO and (d) DLC:N/HGN/GO/HGN structures fabricated with HGN 579. | |
3.2 SPR properties of HGNs colloids and monolayers
The plasmonic absorption attributed to collective conduction band electron oscillation in response to the electrical field of the electromagnetic radiation of light21,48–50 greatly depends on the structure and morphology of nanoparticles. As shown in the extinction spectra in Fig. 6, there is a single resonance between 520 nm and 550 nm for SGNs with the diameters changing from 20 nm to 80 nm.16 As to HGNs, the SPR band position can be altered over a wavelength region from 540 nm to 630 nm (Fig. 6a), which makes these optically active nanostructures great preponderance in SERS applications compared to the SGNs. The SPR peaks of HGNs are red-shifted with the color changing of colloidal HGNs from red to purple and to blue as the extinction cross-section of gold nanospheres is related to their particle sizes. The SPR properties of HGNs with different shell thicknesses are further investigated and shown in Fig. 6b. The cobalt nanoparticles were prepared referencing to the HGN 579 sample with a gradient concentration of HAuCl4 added in the reaction system. The shell thickness of HGNs increases and the colloid color changes from blue to purple and to red with the increscent HAuCl4 concentration, accompanied by a blue shift of SPR peak of HGNs. This rule is consistent with those reported previously.19,24,50
 |
| | Fig. 6 Extinction spectra of (a) HGNs with different sizes and SGN 524, (b) HGNs with various shell thicknesses, (c) monolayers of HGNs with different sizes and SGN 524 and (d) monolayers of HGNs with different shell thicknesses on glass substrates. The inserted optical images in (a) and (b) show the color change of gold colloids. | |
Furthermore, the extinction spectra of HGNs monolayers self-assembled on glass substrates are measured (Fig. 6c and d) as SERS enhancement greatly depends on the optical properties of HGNs monolayers. Our results indicate that the extinction spectra of HGNs layers are broadened in the visible and near-infrared regions and show a red shift when gold colloids are assembled into films in comparison to those of isolated gold nanoparticles in suspension. Chandra et al. also studied the SPR behavior of HGN dimeric structures with substantial interparticle gaps and observed a large SPR red shift and concomitant band broadening relative to that of monomeric HGN, and attributed the phenomenon to the dielectric screening effects in the gap of adjacent HGN and symmetric surface Plasmon hybridization.24 Therefore, the broadening and red shift of SPR peaks for HGNs monolayers in the present study could be attributed to the large magnitude of electromagnetic coupling of the approached single-particle plasmons and the change of dielectric constant around gold,17 which would be the pivotal factor for the SERS enhancement. Table 2 displays the SPR peak shift (Δλ) of HGNs with different sizes and wall thicknesses. A red shift in the range from 27 nm to 78 nm is observed for HGNs with the same aspect ratio and the red shift also occurs with the increase of aspect ratio or decrease of wall thickness. We also find that all HGNs have larger SPR peak shifts compared to the SGNs. An obvious red shift of HGN 579 compared to SGN 524 with a similar diameter might be ascribed to the roughed surface of HGNs with a stronger electromagnetic field near the surface than SGNs22 and the plasmon resonance would favor the coupling of adjacent gold nanospheres. Additionally, the symmetric coupling between surface and cavity modes of HGNs would yield a large dipole moment and a larger absorption cross section, resulting in the red shift of absorption spectra compared to their solid analogues with similar sizes.24 Moreover, the SPR peak shifts of HGN 532 similar to that of the SGN 524 reveals that the HGN 532 with lower aspect ratio and prepared from a high-concentration HAuCl4 solution would be expected to behave more like a SGN.
Table 2 SPR peak shifts (Δλ) of HGNs with different sizes and wall thicknessesa
| Sample |
λ0 (nm) |
λ (nm) |
Δλ = λ − λ0 |
Δλ/λ0 |
S/D |
| λ0 and λ represent the SPR peak positions of HGNs colloids and monolayers, respectively. S is the gap distance of HGNs in the monolayer and D is the diameter of HGNs. |
| SGN 524 |
524 |
561 |
37 |
0.0706 |
0.2973 |
| HGN 536 |
536 |
563 |
27 |
0.0504 |
0.4762 |
| HGN 561 |
561 |
613 |
52 |
0.0927 |
0.3125 |
| HGN 579 |
579 |
634 |
55 |
0.0950 |
0.2857 |
| HGN 593 |
593 |
671 |
78 |
0.1315 |
0.2600 |
| HGN 629 |
629 |
705 |
76 |
0.1208 |
0.2365 |
| HGN 532 |
532 |
564 |
32 |
0.0602 |
0.3906 |
| HGN 550 |
550 |
599 |
49 |
0.0891 |
0.3232 |
| HGN 587 |
587 |
660 |
73 |
0.1244 |
0.2621 |
| HGN 603 |
603 |
678 |
75 |
0.1244 |
0.2415 |
It is believed that a distance-dependent spectral shift of the dimeric plasmons such as the gold nanodisc pairs51 and HGN dimmers24 exhibits the well-known “plasmon ruler” behavior of solid nanospheres due to the surface plasmon coupling interplay in the dimers with an interparticle gap. The relative SPR peak shift (Δλ/λ0 = (λ − λ0)/λ0), which is related to the SPR peak positions of monomer or isolated colloid λ0 and aggregates λ, decays nearly exponentially with the interparticle gap (S) as  .24 Here the gap distance (S) and diameter (D) of aggregates can be obtained from SEM and TEM measurements. A and B are the simulated parameters. Based on our study, the citrate-stabilized HGNs have a negative surface charge due to the adsorption of citric acid anions. The electrostatic repulsion between the negatively charged HGNs hinders the agglomeration of HGNs in the solution. When n-butanol which is slightly soluble in water is dropped into the colloidal solution, a n-butanol/water interface is formed with the reduction of interfacial energy. The addition of n-butanol also decreases the negative surface charge of HGNs because of competitive adsorption of n-butanol molecules. The reduction of charge density on the HGNs surface is a prerequisite for the formation of HGNs monolayers at the water/oil interface.45 Residual charges at the oil side will lead to Coulombic repulsion between the HGNs. Additionally, a strong lateral capillary attraction between HGNs will also be produced due to the asymmetrical distribution of surface charges on the nanoparticles. The HGNs aggregation will occur when the capillary attraction of interfacial nanoparticles is stronger than the electrostatic repulsion.42–45 Therefore, the interspaces of the HGNs and their conglomeration behavior are directly related to the surface charge and species amount absorbed on the HGNs.45 We design 40 volume% of n-butanol and 10 min aging time, and a close-packed HGNs monolayer with an interparticle spacing about 2–10 nm can be obtained, as measured from 20 TEM images randomly taken. Additionally, according to the extinction spectra in Fig. 6, we obtain the SPR peak shift (Δλ) of HGNs with different sizes and wall thicknesses and listed in Table 2. Our results indicate that the relative SPR peak shift (Δλ/λ0) of HGNs is reliable a function of gap separation/diameter (S/D) and decays nearly exponentially
.24 Here the gap distance (S) and diameter (D) of aggregates can be obtained from SEM and TEM measurements. A and B are the simulated parameters. Based on our study, the citrate-stabilized HGNs have a negative surface charge due to the adsorption of citric acid anions. The electrostatic repulsion between the negatively charged HGNs hinders the agglomeration of HGNs in the solution. When n-butanol which is slightly soluble in water is dropped into the colloidal solution, a n-butanol/water interface is formed with the reduction of interfacial energy. The addition of n-butanol also decreases the negative surface charge of HGNs because of competitive adsorption of n-butanol molecules. The reduction of charge density on the HGNs surface is a prerequisite for the formation of HGNs monolayers at the water/oil interface.45 Residual charges at the oil side will lead to Coulombic repulsion between the HGNs. Additionally, a strong lateral capillary attraction between HGNs will also be produced due to the asymmetrical distribution of surface charges on the nanoparticles. The HGNs aggregation will occur when the capillary attraction of interfacial nanoparticles is stronger than the electrostatic repulsion.42–45 Therefore, the interspaces of the HGNs and their conglomeration behavior are directly related to the surface charge and species amount absorbed on the HGNs.45 We design 40 volume% of n-butanol and 10 min aging time, and a close-packed HGNs monolayer with an interparticle spacing about 2–10 nm can be obtained, as measured from 20 TEM images randomly taken. Additionally, according to the extinction spectra in Fig. 6, we obtain the SPR peak shift (Δλ) of HGNs with different sizes and wall thicknesses and listed in Table 2. Our results indicate that the relative SPR peak shift (Δλ/λ0) of HGNs is reliable a function of gap separation/diameter (S/D) and decays nearly exponentially  with the simulated decay length B about 0.25 (Fig. 7). Notice that a parameter revision for A is carried out when supposing one HGN sphere surrounded by other six HGN spheres in a closely packed HGNs monofilm (six HGN dimeric systems). Based on the hypothesis, our result fits well with that reported by Chandra et al. for a HGN dimeric system, in which fitting function
with the simulated decay length B about 0.25 (Fig. 7). Notice that a parameter revision for A is carried out when supposing one HGN sphere surrounded by other six HGN spheres in a closely packed HGNs monofilm (six HGN dimeric systems). Based on the hypothesis, our result fits well with that reported by Chandra et al. for a HGN dimeric system, in which fitting function  should be revised to
should be revised to  for a close-packed HGNs monofilm.24 Our results confirm that both interparticle gap and shell thickness affect optical properties of HGNs with interparticle spatial separation, for which the optical response might be described by the hybridization of surface plasmon modes in a dielectric gap between screened particles.24
for a close-packed HGNs monofilm.24 Our results confirm that both interparticle gap and shell thickness affect optical properties of HGNs with interparticle spatial separation, for which the optical response might be described by the hybridization of surface plasmon modes in a dielectric gap between screened particles.24
 |
| | Fig. 7 SPR peak shift as a function of ratio of gap distance to particle diameter of HGNs. | |
3.3 SERS responses of RhB molecules on HGNs monolayers with different SPR properties
The broad-tunable SPR property of HGNs is a substantial factor for investigating the SPR-dependent SERS behaviors of HGNs substrate. The SERS-active substrate prepared by self-assembling technique is suitable for SERS detection since the plasmon coupling strongly depends on the hotspots motivated at the gaps between well-patterned particles, which is helpful for achieving reproducible SERS signals. Fig. 8a displays the SERS spectra of 10−7 M RhB absorbed on the monolayers of HGNs with different SPR properties and SGN 524. In order to reduce experimental error in the measurement of Raman spectroscopy, we selected eight points in the middle area of each sample to collect the signals and the obtained results approaching to the average values of Raman intensities of eight measurements for each sample is displayed in Fig. 8. It can be seen that the Raman signals obtained from HGNs possess a stronger response than that from SGNs. Halas22 et al. calculated the integrated quartic electromagnetic fields of isolated SGNs and HGNs by the FDTD method and the results suggested that the electromagnetic field of an isolated HGN was enlarged by a factor of 119 contrast to that of isolated SGN, and for a roughened HGN, the factor could increase to 548 relative to the isolated SGN. What's more, the aggregation of nanospheres in the layers would result in the coupling of electromagnetic fields among the gaps or junctions, leading to the enhancement of electromagnetic field and the formation of more hot areas for SERS.24,51 Aggregation of HGNs could promote the formation of high electromagnetic fields along cavity walls for contact HGNs.24,52 This might contribute to the higher SERS activity of HGNs monolayers when compared to the SGNs one.
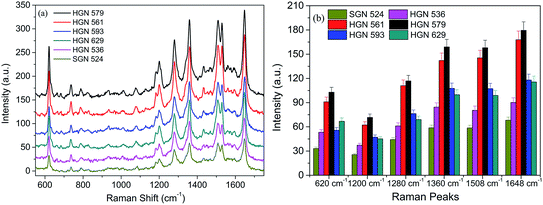 |
| | Fig. 8 SERS spectra of 10−7 M RhB adsorbed on (a) monolayers of SGN 524 and HGNs with different sizes, (b) statistical peak intensity of RhB from eight points of different substrates. The error is the relative standard deviation about 5%. | |
Additionally, the SPR-dependent SERS performance of HGNs monolayers is investigated. The Raman results indicate that the SERS enhancement is greatly affected by the SPR properties of HGNs. It is usually believed that the intensity of Raman signals could be greatly enhanced when the laser excitation wavelength matches or is closed to the peak positions of SPR band.17 The monolayers resulting from HGN 579 and HGN 561 show an outstanding superiority for RhB detection than other HGNs and SGNs monolayers. The statistically average result at each characteristic peak of RhB from eight points of different substrates shows a relative standard deviation about 5% (Fig. 8b). From the extinction spectra in Fig. 5c and the SPR position in Table 2, the wavelength of He–Ne laser at 633 nm matches the SPR peaks of HGN 579 and HGN 561 monolayers very well. The large SERS enhancement is therefore ascribed to the well matching of SPR positions of HGN 579 (634 nm) and HGN 561 (613 nm) monolayer substrates with the excitation wavelength due to the generating of a magnificent enhanced electromagnetic field.17 This also provides deeper insight into the SERS behaviors of HGNs dependent upon the SPR location and field distribution. Thus, we choose proper HGNs with the SPR peak position matching with the excitation wavelength to investigate the SERS behaviors of HGN and GO composite materials in the following discussion.
3.4 SERS responses of RhB molecules on HGNs and GO composite configurations
We chose the HGN 579 to construct the hybridized structures with GO, including DLC:N/HGN/GO, DLC:N/GO/HGN, DLC:N/HGN/HGN and DLC:N/HGN/GO/HGN. After GO is introduced to the experimental system, the Raman signals from DLC:N/HGN/GO and DLC:N/GO/HGN are very close and enlarged than the signals from HGNs monolayer (Fig. 9a). Since the extinction spectra of DLC:N/HGN/GO and DLC:N/HGN have almost the same plasmon peak wavelength (Fig. 10), the larger Raman signal from DLC:N/HGN/GO is therefore attributed to more adsorbed molecules on the GO sheets. The signals from DLC:N/GO/HGN are stronger than those from DLC:N/HGN/GO due to more RhB molecules possibly located at the nanogaps of HGNs on the hybrid surface. For the DLC:N/HGN/HGN sample, the larger intensity of Raman signals of RhB molecules obtained on the double layer could be attributed not only to the coupling of electromagnetic fields at the gaps in the HGNs layer but also to the coupling of electromagnetic fields at the vertical direction between the upper and the bottom HGNs layers. This can be further confirmed from the extinction spectra of HGN/HGN sample self-assembled on glass substrates (Fig. 10). Compared to the HGNs monolayer, the broadened and red-shifted LSP bands of HGN/HGN multilayers could be attributed to light trapping and multiple scattering of incident light in the HGNs multilayer, resulting in the increase of horizontal polarization and vertical polarization. The configuration of DLC:N/HGN/GO/HGN shows the strongest Raman signals of adsorbed RhB molecules due to larger coupled electromagnetic enhancement aroused in the interfaces between HGN–GO and GO–HGN in the DLC:N/HGN/GO/HGN hybrid structure induced by accumulated electron at both sides of GO sheets and stronger interface interactions and charge transfer process. The complex plasmonic behavior of HGNs plasmons on the upper layer with more near contact (Fig. 5d), such as increased contributions from antibonding or higher-order modes and formed charge-transfer plasmon resonance,24 might favor the enhancement of Raman signals. Notice that the peak position change of DLC:N/HGN/HGN and DLC:N/HGN/GO/HGN hybrids may affect the SERS properties when the 633 nm He–Ne laser is used. This part will not be discussed in the present paper.
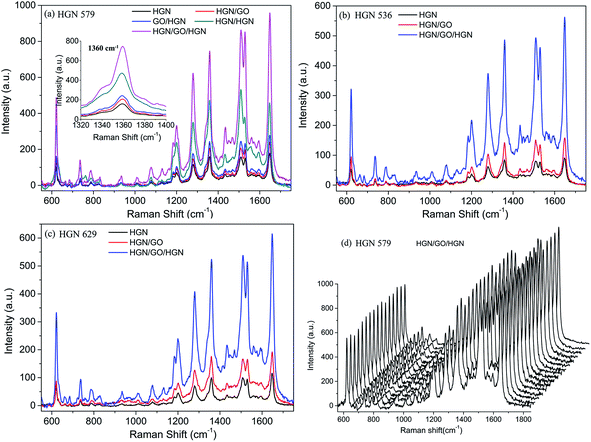 |
| | Fig. 9 SERS spectra of 10−7 M RhB adsorbed on HGNs and GO hybrid structures fabricated with (a) HGN 579, (b) HGN 536 and (c) HGN629. The inset in (a) is the enlarged Raman spectra at 1360 cm−1. (d) 16 SERS spectra collected randomly in the scanned area on the DLC:N/HGN/GO/HGN substrate fabricated with HGN 579. | |
 |
| | Fig. 10 Extinction spectra of HGNs and GO hybrid structures fabricated with (a) HGN 536, (b) HGN 579 and (c) HGN629. | |
Moreover, this synergetic action from GO and HGNs are also confirmed once again from the sandwiched structures assembled using HGN 536 and HGN 629, as shown in Fig. 9b and c, 10a and c. The high homogeneity and reproducibility of SERS signal obtained from our SERS substrates are further demonstrated from the SERS signals of RhB detected randomly from 16 points in the scanned area on the DLC:N/HGN/GO/HGN fabricated from HGN 579 (Fig. 9d) with a relative standard deviation of 8.4%. Such a low relative standard deviation highlights the high reproducibility of SERS response on DLC:N/HGN/GO/HGN substrate for quantitative and trace molecular detection, which is comparably equivalent with other homogeneous SERS substrates.53
3.5 SERS sensitivity of RhB molecules on HGN/GO/HGN hybrid configurations
We further investigate the SERS sensitivity of DLC:N/HGN/GO/HGN hybrid structure fabricated from HGN 579. The Raman signals with a series of gradient RhB concentrations are detected and the Raman intensities of RhB are enlarged with the increasing concentration of RhB (Fig. 11a). When the concentration of RhB is diluted to 10 nM or even to 1 nM, the characteristic peaks of RhB are still clearly distinguished (the inset in Fig. 11a). The Raman response with average intensity of different vibrational modes exhibits a linear dependence on the RhB concentration from 1 nM to 500 nM (R2 = 0.998), as shown in Fig. 11b. The relative standard deviation of SERS intensity is estimated to be 6–9%, indicating the good homogeneity and reproducibility of the DLC:N/HGN/GO/HGN hybrid structure. Considering the ease controlling to the interparticle spacing of HGNs monolayer and reasonable optimization to hybrid configurations, the self-assembled HGN/GO structure might exhibit great potential for preparing highly SERS-active substrates.
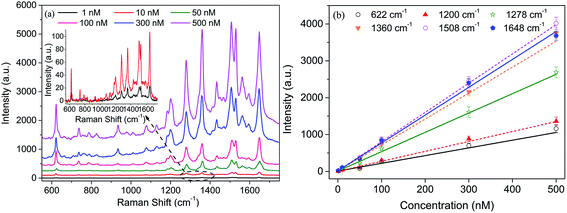 |
| | Fig. 11 (a) SERS spectra of HGN/GO/HGN hybrid from a gradient concentration of RhB and the inset is the enlarged Raman signals of 1 nM and 10 nM RhB. (b) Linear relation of RhB concentration on peak intensity of Raman signals at different vibrational modes. The error is the relative standard deviation from eight point's measurement. | |
4 Conclusions
In this paper, hollow gold nanospheres (HGNs) with adjustable SPR properties were prepared by chemical synthesis and the LSPR-dependent surface-enhanced Raman scattering (SERS) behavior of sandwiched HGN/graphene oxide (HGN/GO) hybrids fabricated by interface self-assembling technique was investigated for detection of RhB molecules. The SPR peaks of colloidal HGNs with tailorable sizes and wall thicknesses showed obvious red shift when forming gold monolayers, which favored the coupling of electromagnetic field and hot spots forming at the gaps or junctions of adjacent HGNs plasmons, resulting in LSPR-dependent Raman enhancement under illumination of incident light with appropriate energy. The relative SPR peak shift decayed nearly exponentially with the value of gap separation/diameter of HGNs and fitted well with the “plasmon ruler” behavior of solid gold nanospheres and HGN dimeric. Furthermore, the sandwiched-structured HGN/GO/HGN hybrid could arouse surface plasmons coupling and charge accumulation at both sides of GO plane, motivating higher local electromagnetic field for further SERS enhancement. The designed HGN/GO/HGN hybrid configuration showed good homogeneity and reproducibility of Raman signals and high detection sensitivity for RhB at the concentration low to 1 nM. The novel, simple, fast and low-cost fabrication process for SERS-active substrate favors a potential application of plasmonic metallic nanostructure with adjustable LSPR performances for nano-optical devices and molecule-level detectors and sensors.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos 51272237 and 11372280), the 521 Talent Project of Zhejiang Sci-Tech University, the Scientific Research Foundation for the Returned Overseas Chinese Scholars (State Education Ministry) and the Technology Foundation for Selected Overseas Chinese Scholar of China.
Notes and references
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667 CrossRef CAS.
- P. S. Waggoner and H. G. Craighead, Lab Chip, 2007, 7, 1238–1255 RSC.
- L. He, N. Kim, H. Li, Z. Hu and M. Lin, J. Agric. Food Chem., 2008, 56, 9843–9847 CrossRef CAS PubMed.
- G. Braun, S. J. Lee, M. Dante, T. Nguyen, M. Moskovits and N. Reich, J. Am. Chem. Soc., 2007, 129, 6378–6379 CrossRef CAS PubMed.
- X. Zhang, M. A. Young, O. Lyandres and R. P. Van Duyne, J. Am. Chem. Soc., 2005, 127, 4484–4489 CrossRef CAS PubMed.
- H. Wang, C. S. Levin and N. J. Halas, J. Am. Chem. Soc., 2005, 127, 14992–14993 CrossRef CAS PubMed.
- M. Rycenga, K. K. Hou, C. M. Cobley, A. G. Schwartz, P. H. C. Camargo and Y. Xia, Phys. Chem. Chem. Phys., 2009, 11, 5903–5908 RSC.
- J. M. R. Tan, J. J. Ruan, H. K. Lee, I. Y. Phang and X. Y. Ling, Phys. Chem. Chem. Phys., 2014, 16, 26983–26990 RSC.
- C. J. Orendorff, L. Gearheart, N. R. Jana and C. J. Murphy, Phys. Chem. Chem. Phys., 2006, 8, 165–170 RSC.
- C. G. Khoury and T. Vo-Dinh, J. Phys. Chem. C, 2008, 112, 18849–18859 CAS.
- H. Liang, Z. Li, W. Wang, Y. Wu and H. Xu, Adv. Mater., 2009, 21, 4614–4618 CrossRef CAS PubMed.
- T. Y. Olson, A. M. Schwartzberg, C. A. Orme, C. E. Talley, B. O'Connell and J. Z. Zhang, J. Phys. Chem. C, 2008, 112, 6319–6329 CAS.
- A. L. Gonz A Lez and C. Noguez, J. Comput. Theor. Nanosci., 2007, 4, 231–238 CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- T. Tan, C. Tian, Z. Ren, J. Yang, Y. Chen, L. Sun, Z. Li, A. Wu, J. Yin and H. Fu, Phys. Chem. Chem. Phys., 2013, 15, 21034–21042 RSC.
- Y. Tanoue, K. Sugawa, T. Yamamuro and T. Akiyama, Phys. Chem. Chem. Phys., 2013, 15, 15802–15805 RSC.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 19935–19944 CrossRef CAS PubMed.
- S. Adams, D. Thai, X. Mascona, A. M. Schwartzberg and J. Z. Zhang, Chem. Mater., 2014, 26, 6805–6810 CrossRef CAS.
- J. Z. Zhang and C. Noguez, Plasmonics, 2008, 3, 127–150 CrossRef CAS.
- C. E. Talley, J. B. Jackson, C. Oubre, N. K. Grady, C. W. Hollars, S. M. Lane, T. R. Huser, P. Nordlander and N. J. Halas, Nano Lett., 2005, 5, 1569–1574 CrossRef CAS PubMed.
- M. Chandra, A. Dowgiallo and K. L. Knappenberger Jr, J. Phys. Chem. C, 2010, 114, 19971–19978 CAS.
- M. Chandra, A. Dowgiallo and K. L. Knappenberger Jr, J. Am. Chem. Soc., 2010, 132, 15782–15789 CrossRef CAS PubMed.
- D. Lim, K. Jeon, H. M. Kim, J. Nam and Y. D. Suh, Nat. Mater., 2009, 9, 60–67 CrossRef PubMed.
- H. Xu, J. Aizpurua, M. K. A. Ll and P. Apell, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4318–4324 CrossRef CAS.
- N. Pazos-Perez, W. Ni, A. Schweikart, R. A. Alvarez-Puebla, A. Fery and L. M. Liz-Marzan, Chem. Sci., 2010, 1, 174–178 RSC.
- Y. Park, S. Yoo and S. Park, Langmuir, 2007, 23, 10505–10510 CrossRef CAS PubMed.
- B. Kim, S. L. Tripp and A. Wei, J. Am. Chem. Soc., 2001, 123, 7955–7956 CrossRef CAS.
- P. Pienpinijtham, X. X. Han, S. Ekgasit and Y. Ozaki, Phys. Chem. Chem. Phys., 2012, 14, 10132–10139 RSC.
- X. Ling, L. G. Moura, M. A. Pimenta and J. Zhang, J. Phys. Chem. C, 2012, 116, 25112–25118 CAS.
- W. Park and Z. H. Kim, Nano Lett., 2010, 10, 4040–4048 CrossRef CAS PubMed.
- B. N. J. Persson, K. Zhao and Z. Zhang, Phys. Rev. Lett., 2006, 96, 207401–207404 CrossRef CAS.
- X. Liu, L. Cao, W. Song, K. Ai and L. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 2944–2952 CAS.
- Y. Zhao, W. Zeng, Z. Tao, P. Xiong, Y. Qu and Y. Zhu, Chem. Commun., 2015, 51, 866–869 RSC.
- X. Zhou, X. Huang, X. Qi, S. Wu, C. Xue, F. Y. Boey, Q. Yan, P. Chen and H. Zhang, J. Phys. Chem. C, 2009, 113, 10842–10846 CAS.
- A. Liu, T. Xu, Q. Ren, M. Yuan, W. Dong and W. Tang, Electrochem. Commun., 2012, 25, 74–78 CrossRef CAS PubMed.
- A. Liu, T. Xu, J. Tang, H. Wu, T. Zhao and W. Tang, Electrochim. Acta, 2014, 119, 43–48 CrossRef CAS PubMed.
- Y. Zhao, X. Li, Y. Du, G. Chen, Y. Qu, J. Jiang and Y. Zhu, Nanoscale, 2014, 6, 11112–11120 RSC.
- S. T. Lin, M. T. Franklin and K. J. Klabunde, Langmuir, 1986, 2, 259–260 CrossRef CAS.
- A. Liu, E. Liu, G. Yang, N. W. Khun and W. Ma, Pure Appl. Chem., 2010, 82, 2217–2229 CrossRef CAS.
- K. M. Schulz, S. Abb, R. Fernandes, M. Abb, A. G. Kanaras and O. L. Muskens, Langmuir, 2012, 28, 8874–8880 CrossRef CAS PubMed.
- Y. J. Li, W. J. Huang and S. G. Sun, Angew. Chem., Int. Ed., 2006, 45, 2537–2539 CrossRef CAS PubMed.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS PubMed.
- M. H. Wang, J. W. Hu, Y. J. Li and E. S. Yeung, Nanotechnology, 2010, 21, 145608 CrossRef PubMed.
- P. R. Selvakannan and M. Sastry, Chem. Commun., 2005, 1684–1686 RSC.
- L. Tan, D. Chen, H. Liu and F. Tang, Adv. Mater., 2010, 22, 4885–4889 CrossRef CAS PubMed.
- V. Myroshnychenko, J. Rodriguez-Fernandez, I. Pastoriza-Santos, A. M. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzan and F. J. G. de Abajo, Chem. Soc. Rev., 2008, 37, 1792–1805 RSC.
- X. Huang and M. A. El-Sayed, J. Adv. Res., 2010, 1, 13–28 CrossRef PubMed.
- H. Liang, L. Wan, C. Bai and L. Jiang, J. Phys. Chem. B, 2005, 109, 7795–7800 CrossRef CAS PubMed.
- Z. Zhang, P. Yang, H. Xu and H. Zheng, J. Appl. Phys., 2013, 113, 33102 CrossRef PubMed.
- S. M. Stagg Jr, K. L. Knappenberger, A. Dowgiallo and M. Chandra, J. Phys. Chem. Lett., 2011, 2, 2946–2950 CrossRef.
- Q. Zhang, Y. H. Lee, I. Y. Phang, C. K. Lee and X. Y. Ling, Small, 2014, 10, 2703–2711 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08057f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






 .24 Here the gap distance (S) and diameter (D) of aggregates can be obtained from SEM and TEM measurements. A and B are the simulated parameters. Based on our study, the citrate-stabilized HGNs have a negative surface charge due to the adsorption of citric acid anions. The electrostatic repulsion between the negatively charged HGNs hinders the agglomeration of HGNs in the solution. When n-butanol which is slightly soluble in water is dropped into the colloidal solution, a n-butanol/water interface is formed with the reduction of interfacial energy. The addition of n-butanol also decreases the negative surface charge of HGNs because of competitive adsorption of n-butanol molecules. The reduction of charge density on the HGNs surface is a prerequisite for the formation of HGNs monolayers at the water/oil interface.45 Residual charges at the oil side will lead to Coulombic repulsion between the HGNs. Additionally, a strong lateral capillary attraction between HGNs will also be produced due to the asymmetrical distribution of surface charges on the nanoparticles. The HGNs aggregation will occur when the capillary attraction of interfacial nanoparticles is stronger than the electrostatic repulsion.42–45 Therefore, the interspaces of the HGNs and their conglomeration behavior are directly related to the surface charge and species amount absorbed on the HGNs.45 We design 40 volume% of n-butanol and 10 min aging time, and a close-packed HGNs monolayer with an interparticle spacing about 2–10 nm can be obtained, as measured from 20 TEM images randomly taken. Additionally, according to the extinction spectra in Fig. 6, we obtain the SPR peak shift (Δλ) of HGNs with different sizes and wall thicknesses and listed in Table 2. Our results indicate that the relative SPR peak shift (Δλ/λ0) of HGNs is reliable a function of gap separation/diameter (S/D) and decays nearly exponentially
.24 Here the gap distance (S) and diameter (D) of aggregates can be obtained from SEM and TEM measurements. A and B are the simulated parameters. Based on our study, the citrate-stabilized HGNs have a negative surface charge due to the adsorption of citric acid anions. The electrostatic repulsion between the negatively charged HGNs hinders the agglomeration of HGNs in the solution. When n-butanol which is slightly soluble in water is dropped into the colloidal solution, a n-butanol/water interface is formed with the reduction of interfacial energy. The addition of n-butanol also decreases the negative surface charge of HGNs because of competitive adsorption of n-butanol molecules. The reduction of charge density on the HGNs surface is a prerequisite for the formation of HGNs monolayers at the water/oil interface.45 Residual charges at the oil side will lead to Coulombic repulsion between the HGNs. Additionally, a strong lateral capillary attraction between HGNs will also be produced due to the asymmetrical distribution of surface charges on the nanoparticles. The HGNs aggregation will occur when the capillary attraction of interfacial nanoparticles is stronger than the electrostatic repulsion.42–45 Therefore, the interspaces of the HGNs and their conglomeration behavior are directly related to the surface charge and species amount absorbed on the HGNs.45 We design 40 volume% of n-butanol and 10 min aging time, and a close-packed HGNs monolayer with an interparticle spacing about 2–10 nm can be obtained, as measured from 20 TEM images randomly taken. Additionally, according to the extinction spectra in Fig. 6, we obtain the SPR peak shift (Δλ) of HGNs with different sizes and wall thicknesses and listed in Table 2. Our results indicate that the relative SPR peak shift (Δλ/λ0) of HGNs is reliable a function of gap separation/diameter (S/D) and decays nearly exponentially  with the simulated decay length B about 0.25 (Fig. 7). Notice that a parameter revision for A is carried out when supposing one HGN sphere surrounded by other six HGN spheres in a closely packed HGNs monofilm (six HGN dimeric systems). Based on the hypothesis, our result fits well with that reported by Chandra et al. for a HGN dimeric system, in which fitting function
with the simulated decay length B about 0.25 (Fig. 7). Notice that a parameter revision for A is carried out when supposing one HGN sphere surrounded by other six HGN spheres in a closely packed HGNs monofilm (six HGN dimeric systems). Based on the hypothesis, our result fits well with that reported by Chandra et al. for a HGN dimeric system, in which fitting function  should be revised to
should be revised to  for a close-packed HGNs monofilm.24 Our results confirm that both interparticle gap and shell thickness affect optical properties of HGNs with interparticle spatial separation, for which the optical response might be described by the hybridization of surface plasmon modes in a dielectric gap between screened particles.24
for a close-packed HGNs monofilm.24 Our results confirm that both interparticle gap and shell thickness affect optical properties of HGNs with interparticle spatial separation, for which the optical response might be described by the hybridization of surface plasmon modes in a dielectric gap between screened particles.24



