
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional poly(phosphoester)s with two orthogonal protective groups†
Laura K.
Müller
ab,
Tobias
Steinbach
a and
Frederik R.
Wurm
*a
aMax-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
bInstitute of Physical Chemistry, Johannes Gutenberg-Universität, Jakob-Welder-Weg 11, 55099 Mainz, Germany
First published on 7th May 2015
Abstract
A novel cyclic phosphate monomer, 2-(2-(benzyloxy)ethoxy)-1,3,2-dioxaphospholane-2-oxide (BnEEP), was developed to generate poly(phosphoester)s containing protected pendant hydroxyl groups by anionic ring-opening polymerization. The hydroxyl-groups were released by a mild catalytic hydrogenation leaving the polymer backbone intact. In addition, the number of pendant hydroxyl groups was varied by copolymerization of BnEEP with ethyl ethylene phosphate (EEP). Furthermore, copolymers of BnEEP with an acetal protected cyclic phosphate, 2-(2,2-dimethyl-1,3-dioxolan-4-yl-methoxy)-2-oxo-1,3,2-dioxaphospholane (GEP), were prepared in order to establish a selective deprotection of the acetal or the benzyl protective groups by acidic hydrolysis or catalytic hydrogenation respectively. No degradation of the polyester backbone was detected under the reported conditions. The novel monomer allows adjustment of the chemical and physical properties of the poly(phosphoester)s and gives access to various side chain functionalities.
Introduction
In materials science, polyvalency is of utmost importance to mimic biological systems and to design elaborate structures, for example for the preparation of polymer therapeutics or polymeric catalyst supports with high loading capacities.1 Several strategies are known to generate polyvalent, i.e. multifunctional, polymers.2 Among those for biomedical applications, especially water-soluble polymers are of high interest in current literature.3–5 Poly(ethylene glycol) (PEG) is the gold standard for drug delivery systems and bioconjugation.6 However, telechelic PEGs, many of them commercially available, exhibit only two functional (end) groups.7 Copolymerization of ethylene oxide with functional epoxide comonomers by anionic copolymerization is an attractive route to generate PEG-like polymers which may be applicable in modern biomedicine.2,8,9 Furthermore, polyglycerols or PEG-methacrylates are currently discussed as potential multifunctional PEG-alternatives.6,10 All of them, however, are not biodegradable, a major disadvantage for many biomedical purposes. Polyesters are the common synthetic answer if degradable materials are demanded. However, well-defined and water-soluble polyesters are rather rare.11 A unique exception is the diverse family of poly(phosphoester)s (PPEs) which are (bio)degradable and are currently attracting interest in several research groups.12–14 PPEs are accessible by different synthetic approaches,15 namely polycondensation, polyaddition, transesterification and ring-opening polymerization (ROP) of cyclic phosphoesters.14,16,17 ROP allows the controlled polymerization with adjustable molecular weights and narrow molecular weight distributions.18 Recent works by Iwasaki, Wang, Wooley, and Lecomte have illustrated that with this approach also multifunctional, biodegradable polymers with high molecular weights are accessible.18–21 An elegant approach by the Wooley lab uses PPEs carrying vinyl-ether side chains which are used for postpolymerization modification into acetals, thioacetals, and by thiol–ene click reaction.20 Side-chain functional PPEs carrying alkyne bonds have also been reported which can be used for post-polymerization functionalization and for the generation of biomimetic nanocarriers. Another example was presented by Ni et al. who prepared PPEs with acrylate side-chains to introduce diverse functionalities via post-functionalization of the polymers via Michael-addition.22 Another advantage of the phosphate unit was presented by Yan and coworkers, who synthesized hyperbranched PPEs by self-condensing ROP.23 We recently prepared polymeric nanoparticles based on hydrophobic PPEs that exhibit a strong interaction to a model bone material24 and used PPEs as polymeric supports in Horner–Wadsworth–Emmons reactions.25 In the last several years, our group has developed various novel monomers and polymerization strategies for new PPEs by olefin metathesis and anionic polymerization.26–30Herein, we present the synthesis of a novel cyclic phosphate monomer carrying a benzyl-protected hydroxyl group in the side chain that can be removed by mild catalytic hydrogenation. This monomer was also used for the synthesis of copolymers to vary the number of hydroxyl groups and to generate orthogonally protected PPEs, with both acetal- and benzyl-protected hydroxyl groups, which are released selectively by the addition of acid or by hydrogenation. Acetal-protected PPEs have been introduced by Song et al.19 To the best of our knowledge this is the first example of PPEs with orthogonal protective groups. The polymers have been investigated with respect to monomer incorporation, sequential deprotection and with respect to their thermal properties. We believe that these polyvalent and water-soluble PPEs will find application in biorelated purposes, such as nanocarriers, which is currently under investigation in our group.
Experimental part
Materials
All chemicals and solvents were merchandised from Sigma Aldrich or Acros Organics and used as received if not otherwise stated. THF and ethanol was dried with sodium and distilled freshly before use; solketal was dried by azeotropic distillation with benzene and stored over molecular sieves. Deuterated solvents were purchased from Deutero GmbH or Sigma Aldrich. For catalytic hydrogenation, an autoclave by Roth (100 mL) was used.Analytical methods and characterization
1H, 13C{H}, and 31P{H} NMR spectra of the monomers were measured on a Bruker AVANCE III 500 spectrometer, 1H NMR spectra of the different polymers were recorded on a Bruker AVANCE 300. All spectra were recorded at 25 °C. As deuterated solvent DMSO-d6 was used and the spectra were calibrated against the solvent signal (2.50 ppm). All SEC measurements were performed in DMF containing 1 g L−1 LiBr. An Agilent Technologies 1260 Infinity instrument with a PSS SecCurity autosampler, a 1260 IsoPump and a GRAM (PSS) 0.8 × 30 cm column (particle size 10 μm and pore sizes 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 1000, 100 Å) was used. FT-IR measurement was done with a Nicolet 5DXC FT-IR. DSC measurements were performed using a Perkin Elmer DSC 823. A heating rate of 10 °C min−1 was employed within a temperature range from −100 to 80 °C in a nitrogen atmosphere.
000, 1000, 100 Å) was used. FT-IR measurement was done with a Nicolet 5DXC FT-IR. DSC measurements were performed using a Perkin Elmer DSC 823. A heating rate of 10 °C min−1 was employed within a temperature range from −100 to 80 °C in a nitrogen atmosphere.
Monomers
2-Chloro-1,3,2-dioxaphospholane and 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) were synthesized according to literature.31 2-Ethoxy-2-oxo-1,3,2-dioxaphospholane (EEP) was synthesized by the esterification of COP with ethanol.31 Briefly, to a solution of COP (72.8 g, 0.55 mol) in 50 mL dry THF was added a mixture of pyridine (43.43 g, 0.55 mol) in 230 mL dry THF and dry ethanol (25.29 g, 0.55 mol) drop wise under an inert atmosphere at −20 °C. The mixture was stirred over night at −4 °C. The precipitated pyridine hydrochloride was removed by filtration under inert conditions and the solvent was evaporated in vacuo. The crude product was purified by distillation. The product was obtained as colorless liquid at 95–98 °C/3 × 10−3 mbar (53.71 g, 0.35 mol, 64%). 1H NMR (300 MHz, DMSO-d6): δ 4.48–4.32 (m, 4H, O–CH2–CH2–O), 4.22–4.01 (m, 2H, O–CH2–CH3), 1.25 (t, 3H, O–CH2–CH3). 13C{H} NMR (125 MHz, DMSO-d6): δ 66.35 (s, 2C, O–CH2–CH2–O), 64.18 (s, 1C, O–CH2–CH3), 15.96 (s, 1C, O–CH2–CH3). 31P{H} NMR (202 MHz, DMSO-d6): δ 16.83.2-(2,2-Dimethyl-1,3-dioxolan-4-yl-methoxy)-2-oxo-1,3,2-dioxaphospholane (GEP)19 was synthesized analogously using dry solketal instead of ethanol. Purification was conducted by filtration to remove most of the precipitated pyridine hydrochloride. The resulting monomer was pure enough for direct use. Yield: quant. Spectra data matched literature values. 1H NMR (300 MHz, DMSO-d6): δ 5.30–4.30 (m, 4H, O–CH2–CH2–O), 4.30–4.20 (m, 2H, O–CH2–CH3), 3.92–4.15 (m, 2H, O–CH2–CH–O2C4H8, 1H, CH–CH2–O–C), 3.59–3.73 (m, 1H, CH–CH2–O–C), 1.34 (s, 3H, C–CH3), 1.27 (s, 3H C–CH3). 13C{H} NMR (125 MHz, DMSO-d6): δ 108.97 (s, 1C, C), 73.80 (s, 1C, CH1), 68.03 (s, 1C, O–CH2–CH–O2C4H8), 66.50 (s, 2C, O–CH2–CH2–O), 26.49 (s, 1C, CH3), 26.25 (s, 1C, CH3). 31P{H} NMR (202 MHz, DMSO-d6): δ 16.94.
(2-(2-(Benzyloxy)ethoxy)-1,3,2-dioxaphospholane-2-oxide) (BnEEP). COP (25.6 g, 180 mmol) was dissolved in 50 mL THF. A solution of 2-(benzyloxy)ethanol (27.3 g, 180 mmol) and pyridine (14.2 g, 180 mmol) in 50 mL THF was added drop wise at −21 °C. After stirring overnight (ca. 12 h) at −4 °C, the hydrochloride salt was removed by Schlenk filtration. The solvent was removed under reduced pressure to obtain the product (40.4 g, 156 mmol, 87%) which can be used directly for polymerization. 1H NMR (300 MHz, DMSO-d6): δ 7.45–7.20 (m, 5H, aromatic), 4.52 (s, 2H, C–CH2–O), 4.49–4.32 (m, 4H, phospholane–CH2–CH2–), 4.22–4.14 (m, 2H, –CH2–O–P), 3.65–3.60 (m, 2H, O–CH2–CH2). 31P{H} NMR (202 MHz, DMSO-d6): δ 17.13.
General procedure for the synthesis of (co-)polymers with 1,8-diazabicyclo[5.4.0]-undec-7-ene
The polymerization was conducted in dichloromethane or toluene depending (compare Table S1†). In DCM, the reaction was carried out at room-temperature and in toluene at 40 °C or 50 °C. BnEEP (0.5010 g, 1.94 mmol) and 2-(benzyloxy)ethanol as the initiator (0.0115 g, 0.06 mmol) were introduced into the Schlenk tube via syringe and dissolved in 0.38 mL toluene. A stock solution of DBU in toluene (98 mg in 1 mL toluene) was prepared. After cooling down both solutions to 0 °C, the polymerization was initiated by adding 0.1 mL of the catalysts stock solution to the stirred monomer solution. The ice bath was removed and the reaction mixture heated up to 40 °C. After the reaction time shown in Table S1,† an excess of acetic acid was added. The polymers were purified by precipitation in diethyl ether and centrifuged (10 min, 4500 rpm, 0 °C). The supernatant was decanted and the colorless polymer dried in vacuo. Yields: 85–100%.General procedure for the synthesis of (co-)polymers with Sn(Oct)2
These (co-)polymerizations were carried out in 25 mL flame-dried Schlenk tubes, purged with argon three times prior to use. The reactions were catalyzed by stannous octoate in bulk at 90 °C. The monomers and the initiator were introduced into the Schlenk tube via syringe in desired ratios depending on composition. Then the reaction tube was cooled to 0 °C and the polymerization was initiated by the addition of 0.02 mL stannous octoate (0.06 mmol, 24.3 mg) to the stirred solution. The reaction mixture was heated to 90 °C and allowed to react for 2–20 hours (depending on monomer: 2 hours for PEEP40 and 20 hours for PBnEEP40). Afterwards 2 mL of dichloromethane were added and the crude polymers were purified by precipitation into an excess of diethyl ether. The (co-)polymers were dried in vacuo and obtained as colorless viscous oils. Yields: 85–100%.PEEP32. 1H NMR (DMSO-d6): 4.89 (t, 1H, P–O–CH2–CH2–OH), 4.18–4.15 (m, 62H, O–CH2–CH2–O), 4.07 (m, 30H, O–CH2–CH3), 3.57 (m, 2H, P–O–CH2–CH2–OH), 1.25–1.27 (t, 132H, O–CH2–CH3), 0.82 (m, 3H, CH2–CH2–CH3). 31P NMR (DMSO-d6): −1.247 (s).
PHEEP36. 1H NMR (DMSO-d6): δ 7.17–7.44 (m, 75H, Ar), 4.76–4.80 (t, 1H, P–O–CH2–CH2–OH), 3.91–4.25 (m, 90H, O–CH2–CH2–O), 3.65–3.84 (m, 2H, P–O–CH2–CH2–OH), 3.52–3.64 (m, 30H, Ar-CH2–O), 0.82 (m, 3H, CH2–CH2–CH3). 31P NMR (DMSO-d6): −1.18 (m).
PGEP39. 1H NMR (DMSO-d6): δ 4.83 (t, 1H, OH), 4.13–4.29 (m, 100H, O–CH2–CH2–O), 3.91–4.10 (m, 40H, O–CH2–CH), 3.54–3.64 (m, 2H, Ar-CH2–O–CH2–CH2), 3.35–3.37 (m, 2H, P–O–CH2–CH2–OH), 1.33 (s, 60H, CH3), 1.27 (s, 60H, CH3), 0.82 (m, 3H, CH2–CH2–CH3). 31P NMR (DMSO-d6): −1.23 (m).
Deprotection of PBnEEP via catalytic hydrogenation
In a pressure reactor, 30 mg of the respective polymer was dissolved in 3 mL dry THF. 30 mg of Pd(OH)2 on charcoal was added and the solution was stirred for four hours at 40 bar of hydrogen pressure. After completion, the catalyst was removed by centrifugation (10 min, 4500 rpm, 0 °C) and decantation. THF was evaporated at reduced pressure and the deprotected polymer was dried in vacuo (0.02 g, 96%). 1H NMR (300 MHz, DMSO-d6): δ 4.76–4.80 (t, 16H, P–O–CH2–CH2–OH), 3.91–4.25 (m, 90H, O–CH2–CH2–O), 3.65–3.84 (m, 2H, P–O–CH2–CH2–OH).Deprotection of PGEP via acidic hydrolysis
In a 50 mL round bottom flask, the respective polymer (89.5 mg, 18 nmol) was dissolved in 15 mL 1,4-dioxane. 5 mL aqueous hydrochloric acid (1 M) were added dropwise via a syringe. After stirring the solution for additional 3 hours, the solution was concentrated at reduced pressure and the product was precipitated into cold diethyl ether. Yield: 75 mg, quant. 1H NMR PHEP: (DMSO-d6): δ 7.23–2.32 (m, 5H, Ar), 4.83 (t, 41H, OH), 4.52 (s, 2H, Ar-CH2–O), 4.13–4.29 (m, 100H, O–CH2–CH2–O), 3.91–4.10 (m, 40H, O–CH2–CH), 3.54–3.64 (m, 2H, Ar-CH2–O–CH2–CH2), 3.35–3.37 (m, 2H, P–O–CH2–CH2–OH).Results and discussion
Monomer and polymer synthesis
One striking argument for PPEs is the easy access to water-soluble polyesters. The most prominent dioxaphospholane oxide monomer is probably 2-ethoxy-2-oxo-1,3,2-dioxaphospholane, also called ethyl ethylene phosphate (EEP), carrying ethyl side chains in the resulting PPEs. Some recent works use the pentavalency of phosphorus for the direct introduction of functional side-chains into water-soluble PPEs.20,32,33 The design of functional novel phosphate monomers and the investigation of their polymerization behavior is an attractive field of research for future materials for biomedical applications, because polyvalent PPEs would be accessible. Potentially, also their degradation behavior can be triggered by the nature of the side chain.34 In order to produce PPEs carrying two orthogonally protected side chains, a combination of literature-reported 2-ethoxy-2-oxo-1,3,2-dioxaphospholane (EEP), 2-(2,2-dimethyl-1,3-dioxolan-4-yl-methoxy)-2-oxo-1,3,2-dioxaphospholane (GEP), and a new monomer, namely 2-(2-(benzyloxy)ethoxy)-1,3,2-dioxaphospholane-2-oxide (BnEEP) was investigated. The polymerization and copolymerization was studied as well as the selective deprotection of the pendant groups. GEP carries acid-labile acetal units which can be cleaved under acidic conditions, whereas the benzyl groups in BnEEP are removed by the mild catalytic hydrogenation and remain untouched during acidic treatment. After hydrogenation a single hydroxyl group per repeating unit is released. In contrast, the deprotection of the acetal units of GEP releases two hydroxyl groups per repeating unit. This is an additional trigger to tune the hydrophilicity of these polymers.BnEEP was synthesized via the esterification of 2-chloro-2-oxo-1,3,2-dioxaphospholane (COP) with 2-(benzyloxy) ethanol and obtained as colourless oil in high yield (typically over 85%, Scheme 1).
 | ||
| Scheme 1 Synthetic protocol for the synthesis of 2-(2-(benzyloxy)ethoxy)-1,3,2-dioxaphospholane-2-oxide (BnEEP) (i) THF, triethylamine, −21 °C, 12 h; (ii) O2, benzene, 50 °C, 24 h; (iii) 2-(benzyloxy)ethanol, pyridine, THF, −21 °C to −4 °C, 10 h. | ||
The 1H NMR spectrum of BnEEP shows the characteristic resonances for the benzyl protective group at ca. 7.4 ppm (a in Fig. 1), a sharp singlet for the benzylic methylene group (b), and the remaining signals for the side-chain and ring-methylenes (marked in the spectrum with c–e). The monomer is pure enough after workup of the reaction mixture (13C NMR is shown in the ESI†).
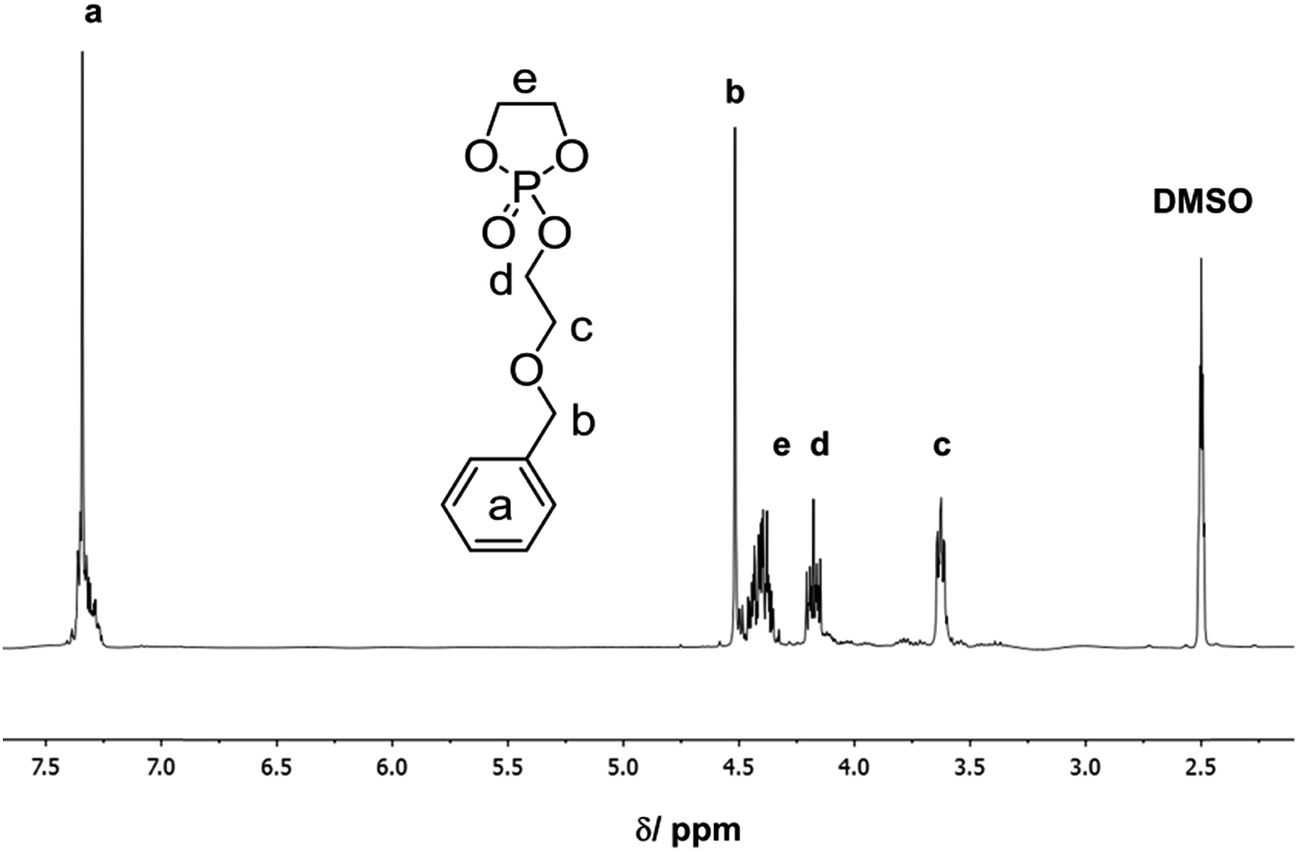 | ||
| Fig. 1 1H NMR spectrum of BnEEP (300 MHz, DMSO-d6, 298 K). | ||
The anionic ROP of BnEEP was investigated with DBU and TBD as organocatalysts and tin(II)-2-ethylhexanoate (Sn(Oct)2) at various conditions (Scheme 2 and Table S1†). The polymerization proceeds with all catalysts, however, high monomer conversion and low molecular weight dispersity were achieved with DBU in toluene at 40 °C over a period of 20 minutes or Sn(Oct)2 in bulk over a period of several hours. The usage of TBD in solution as well as in bulk resulted in low monomer conversion and broad molecular weight distributions (Table S1†). In all cases molecular weights determined by SEC are underestimated compared to values derived from 1H NMR analysis. This underestimation vs. conventional calibration has been reported earlier for other PPEs.34
 | ||
| Scheme 2 Polymerization and deprotection of BnEEP (i) different catalysts: either DBU, 25–40 °C, DCM or toluene, 2 min to 17 h; TBD, 0 °C, DCM, 2 h; Sn(Oct)2, bulk, 90 °C, 2–20 h; polymerization conditions and results are summarized in Table S1.† (ii) H2, Pd(OH)2/C, THF, 40 bar, 4 h. | ||
In contrast to the polymerization of EEP, BnEEP required prolonged reaction times, which can be attributed to the sterical hindrance of the pendant chain next to the electrophilic phosphate center. From the results, stannous octoate is the most promising catalyst for the polymerization of BnEEP. The procedure in bulk led to adjustable molecular weights, with narrow molecular weight distributions within acceptable reaction times (the ESI also shows some representative SEC curves, Fig. S2†).
Fig. 3 shows a 1H NMR spectrum of PBnEEP initiated with hexanol and catalyzed by stannous octoate.
The aromatic protons of the pendant side chain are detectable as a broad resonance at a chemical shift of ca. 7.17–7.44 ppm (signals a in Fig. 2). The signal at 4.31–4.66 is attributed to the benzylic methylene groups b, and the signals at ca. 3.91–4.25 ppm correspond to the alkoxy methylene groups of the polymer backbone (c) as well as the side chain (e). The terminal hydroxyl group f is detected at ca. 4.76–4.80 ppm. The methyl group of the initiator can be detected at ca. 0.8 ppm (g) and allows (together with the signals d or f) the calculation of the number average molecular weight (Mn) from the NMR spectrum.
 | ||
| Fig. 2 Representative 1H NMR of PBnEEP polymerized with Sn(Oct)2 (DMSO-d6, 300 MHz, 298 K). | ||
To release pendant hydroxyl functionalities of PBnEEP, the benzyl protecting groups were removed by hydrogenation catalysed by palladium hydroxide on activated charcoal under different conditions – the effect on stability of the main chain and the deprotection efficiency was monitored.
The polarity of the solvent is of crucial importance as a rather hydrophobic polymer is transformed into a highly hydrophilic material during the process. THF, methanol and dichloromethane–methanol mixtures have been investigated. Methanol-containing mixtures led to partial degradation of the polymer backbone which was indicated by ethylene glycol formation. THF was found to be a good solvent for the hydrogenation procedure with isolated yields that were in most cases quantitative. The successful deprotection of PBnEEP can also be monitored by 1H NMR (Fig. 3) due to the disappearance of the aromatic signals of the benzyl groups. In addition, the signals corresponding to the polymer backbone remain after deprotection.
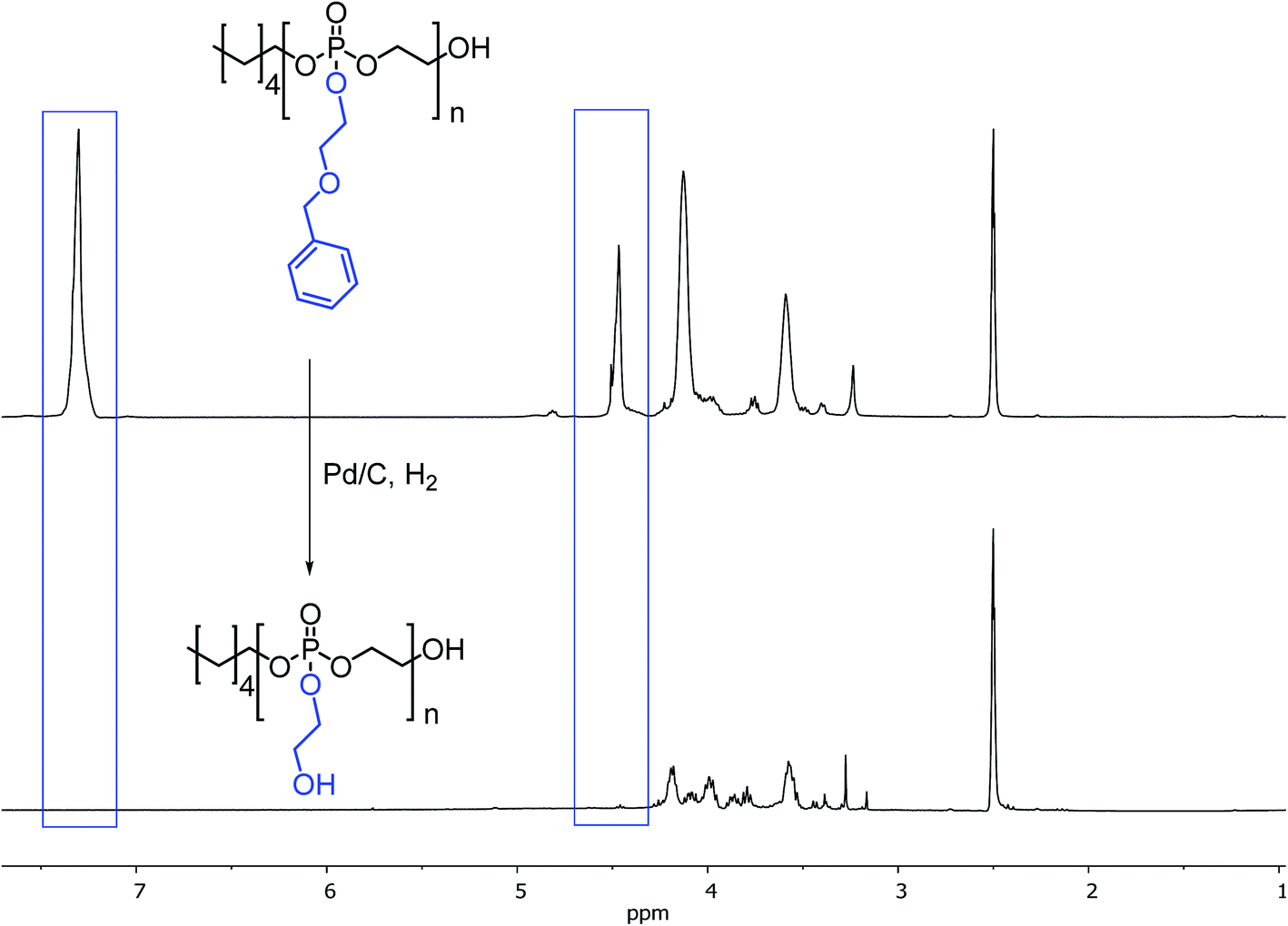 | ||
| Fig. 3 1H NMR spectra of PBnEEP (top) before and after successful hydrogenation (bottom) (DMSO-d6, 300 MHz, 298 K). | ||
For the synthesis of orthogonally protected PPEs with an adjustable number of primary and secondary hydroxyl groups, a series of copolymers of EEP, GEP, and BnEEP were synthesized. Following a similar strategy, Möller and coworkers introduced orthogonally protected linear polyglycerols several years ago.35 All copolymerizations were catalyzed by stannous octoate and conducted at 90 °C in bulk. Hexanol was used as the initiator since the terminal methyl group can be detected in 1H NMR spectra without overlapping with signals of all three monomers. This allows the calculation of the absolute molecular weight from 1H NMR (compare Table S3†) (Scheme 3).
 | ||
| Scheme 3 Protocol for the synthesis of (orthogonally) protected poly(phosphoester)s with subsequent selective deprotection. | ||
The copolymer compositions were calculated from the 1H NMR spectra by comparing the characteristic signals originating from each monomer with the resonances from the initiator. The calculated masses agreed with the theoretical values very well (compare Table S2 and Fig. S3–S6†).
The glass transition temperature (Tg) of all polymers was measured in order to investigate how the monomer composition affects the thermal properties of the copolymers. Table S3† summarizes the results. PEEP has the lowest glass transition temperature (−62 °C) of all three homopolymers, while PBnEEP has a Tg of −39 °C and PGEP has a Tg of −35 °C. With increasing EEP content, the glass-transition temperature of the copolymer decreases as expected. The aromatic side-chains as well as the acetal protected side chain of the other monomers allow electrostatic as well as hydrophilic interactions and have a higher sterical demand than the ethyl side chain and thus limit flexibility of the polymer chains resulting in higher glass-transition states.
In total, a library of 20 homo- and copolymers have been prepared yielding PPEs with an adjustable number of pendant hydroxyl groups: either the copolymerization of EEP and BnEEP or the copolymerization of EEP and GEP produced PPEs with an adjustable number of OH groups in combination with a water-soluble comonomer. The copolymerization of BnEEP and GEP allowed a selective deprotection of the pendant groups. According to Wang and co-workers, the acetal can be cleaved by the addition of aqueous hydrochloric acid to a solution of the protected (co)polymer in 1,4-dioxane. Concentration and precipitation into diethyl ether after three hours led to the product with reasonable yields and no scission of the polymer backbone. Deprotection can be followed by 1H NMR spectroscopy (the 1H NMR spectra before and after acidic hydrolysis of the homopolymers PGEP are shown in Fig. S7†). Fig. 4 shows an overlay of the 1H NMR spectra of PBnEEP22-co-PGEP19 and its selective deprotection. After acidic treatment the acetal groups at ca. 4.5 and 1.2 ppm disappear, while the aromatic benzyl protons remain untouched. Also the polymer backbone is retained under these conditions, however, care has to be taken for longer reaction times or higher temperatures as degradation could occur by transesterification. If hydrogenation is conducted first, the resonances corresponding to the benzyl group at ca. 7.4 ppm disappear, while the acetal signal remain unchanged proving an efficient orthogonal deprotection (Fig. 4, bottom). Currently, these materials are under investigation for subsequent drug loading and the results of biomedical studies will be reported in due course.
 | ||
| Fig. 4 1H NMR spectra (300 MHz, DMSO-d6, 298 K) of the selective deprotection of PGEP-co-PBnEEP by acidic hydrolysis or hydrogenation. | ||
Conclusions
In conclusion, a novel cyclic phosphate monomer for the anionic ring-opening polymerization was presented: 2-(2-(benzyloxy)ethoxy)-1,3,2-dioxaphospholane-2-oxide was prepared from 2-chloro-2-oxo-1,3,2-dioxaphospholane by esterification with 2-(benzyloxy)ethanol. The monomer can be polymerized by controlled polymerization techniques and subsequently deprotected by catalytic hydrogenation to release a single primary hydroxyl group per repeating unit. In addition copolymers with 2-ethoxy-2-oxo-1,3,2-dioxaphospholane (EEP) and the acetal protected 2-(2,2-dimethyl-1,3-dioxolan-4-yl-methoxy)-2-oxo-1,3,2-dioxaphospholane (GEP) were synthesized and the selective removal of the benzyl or the acetal protective groups was proven. These results introduce a novel orthogonal cyclic phosphate monomer to the field of poly(phosphoester)s that will probably find application in future polymer therapeutics.Acknowledgements
F.R.W. and T.S. thank the Max Planck Graduate Center for support. The authors thank Prof. Dr Katharina Landfester (MPIP, Mainz) for continuous support.References
- D. Vance, M. Shah, A. Joshi and R. S. Kane, Biotechnol. Bioeng., 2008, 101, 429–434 CrossRef CAS PubMed.
- B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2012, 64, 302–315 CrossRef PubMed.
- S. Zhang, J. Zou, M. Elsabahy, A. Karwa, A. Li, D. A. Moore, R. B. Dorshow and K. L. Wooley, Chem. Sci., 2013, 4, 2122–2126 RSC.
- F. Wurm and H. Frey, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, vol. 6, pp. 177–198 Search PubMed.
- E. M. Pelegri-O'Day, E.-W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323–14332 CrossRef PubMed.
- M. S. Thompson, T. P. Vadala, M. L. Vadala, Y. Lin and J. S. Riffle, Polymer, 2008, 49, 345–373 CrossRef CAS PubMed.
- C. Mangold, F. Wurm and H. Frey, Polym. Chem., 2012, 3, 1714–1721 RSC.
- R. Klein and F. R. Wurm, Macromol. Rapid Commun., 2015 DOI:10.1002/marc.201500013.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- M. Vert, Biomacromolecules, 2004, 6, 538–546 CrossRef PubMed.
- K. D. Troev, Polyphosphoesters: Chemistry and Application, Elsevier, 2012 Search PubMed.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- S.-W. Huang and R.-X. Zhuo, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 340–348 CrossRef CAS PubMed.
- Z. Zhao, J. Wang, H.-Q. Mao and K. W. Leong, Adv. Drug Delivery Rev., 2003, 55, 483–499 CrossRef CAS.
- S. Penczek, J. Pretula and K. Kaluzynski, Biomacromolecules, 2005, 6, 547–551 CrossRef CAS PubMed.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- B. Clément, B. Grignard, L. Koole, C. Jérôme and P. Lecomte, Macromolecules, 2012, 45, 4476–4486 CrossRef.
- W.-J. Song, J.-Z. Du, N.-J. Liu, S. Dou, J. Cheng and J. Wang, Macromolecules, 2008, 41, 6935–6941 CrossRef CAS.
- Y. H. Lim, G. S. Heo, Y. H. Rezenom, S. Pollack, J. E. Raymond, M. Elsabahy and K. L. Wooley, Macromolecules, 2014, 47, 4634–4644 CrossRef CAS PubMed.
- Y. Iwasaki and E. Yamaguchi, Macromolecules, 2010, 43, 2664–2666 CrossRef CAS.
- H. Shao, M. Zhang, J. He and P. Ni, Polymer, 2012, 53, 2854–2863 CrossRef CAS PubMed.
- J. Liu, W. Huang, Y. Zhou and D. Yan, Macromolecules, 2009, 42, 4394–4399 CrossRef CAS.
- E. Alexandrino, S. Ritz, F. Marsico, G. Baier, V. Mailaender, K. Landfester and F. R. Wurm, J. Mater. Chem. B, 2014, 2, 1298–1306 RSC.
- T. Steinbach, C. Wahlen and F. R. Wurm, Polym. Chem., 2015, 6, 1192–1202 RSC.
- M. Steinmann, J. Markwart and F. R. Wurm, Macromolecules, 2014, 47, 8506–8513 CrossRef CAS.
- T. Steinbach, S. Ritz and F. R. Wurm, ACS Macro Lett., 2014, 3, 244–248 CrossRef CAS.
- T. Steinbach, E. M. Alexandrino, C. Wahlen, K. Landfester and F. R. Wurm, Macromolecules, 2014, 47, 4884–4893 CrossRef CAS.
- F. Marsico, A. Turshatov, R. Pekoz, Y. Avlasevich, M. Wagner, K. Weber, D. Donadio, K. Landfester, S. Baluschev and F. R. Wurm, J. Am. Chem. Soc., 2014, 136, 11057–11064 CrossRef CAS PubMed.
- T. Steinbach, E. M. Alexandrino and F. R. Wurm, Polym. Chem., 2013, 4, 3800–3806 RSC.
- T. Steinbach, R. Schroeder, S. Ritz and F. R. Wurm, Polym. Chem., 2013, 4, 4469–4479 RSC.
- S. Zhang, A. Li, J. Zou, L. Y. Lin and K. L. Wooley, ACS Macro Lett., 2012, 1, 328–333 CrossRef CAS PubMed.
- S. Zhang, H. Wang, Y. Shen, F. Zhang, K. Seetho, J. Zou, J.-S. A. Taylor, A. P. Dove and K. L. Wooley, Macromolecules, 2013, 46, 5141–5149 CrossRef CAS PubMed.
- J. Baran and S. Penczek, Macromolecules, 1995, 28, 5167–5176 CrossRef CAS.
- M. Erberich, H. Keul and M. Möller, Macromolecules, 2007, 40, 3070–3079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07167d |
| This journal is © The Royal Society of Chemistry 2015 |