Double click reaction strategies for polymer conjugation and post-functionalization of polymers
Hakan
Durmaz
a,
Amitav
Sanyal
*b,
Gurkan
Hizal
*a and
Umit
Tunca
*a
aDepartment of Chemistry, Istanbul Technical University, Maslak, 34469, Istanbul, Turkey. E-mail: tuncau@itu.edu.tr; hizal@itu.edu.tr; Web: www.kimya.itu.edu.tr/polimer
bDepartment of Chemistry, Bogazici University, Bebek, 34342, Istanbul, Turkey. E-mail: amitav.sanyal@boun.edu.tr; Web: www.chem.boun.edu.tr/personal/ranasanyal/group/sanyalgroup.html
First published on 2nd December 2011
Abstract
Double click reaction strategies, which are a combination of different type of click reactions, allow the preparation of polymers with various topologies and the post-functionalization of polymers, which cannot be easily achieved by using only one click reaction. The most studied click reaction combinations may be listed as the Cu(I) catalyzed azide-alkyne cycloaddition (CuAAC)–Diels–Alder, and the CuAAC–nitroxide radical coupling reactions for polymer–polymer conjugation and the CuAAC–Diels–Alder, or the CuAAC–thiol-ene reactions for post-modification of polymers.
 Hakan Durmaz | Hakan Durmaz received his BSc in 2002 at the Chemistry Department of the Istanbul Technical University (ITU), Turkey. He received his MSc and PhD degrees in 2004 and 2010 under the supervision of Prof. G. Hizal in the Polymer Science and Technology Program at the ITU, respectively. His research areas mainly include controlled/living polymerizations and “click” coupling methods. He has published 30 papers since 2005. |
 Amitav Sanyal | Amitav Sanyal obtained his PhD in 2001 from Boston University, USA on asymmetric organic synthesis. During post-doctoral research with Prof. Vincent M. Rotello at University of Massachusetts at Amherst, USA, he worked in the area of renewable polymeric coatings and fabrication of polymer-nanoparticle composites. Currently, he is an associate professor at Bogazici University, Istanbul, Turkey. His research focuses on the development of novel reactive polymeric coatings for biomolecular immobilization and thermo-reversible materials using Diels–Alder chemistry. His awards include the young-investigator award (TUBA-GEBIP) (2008) administered by the Turkish Academy of Sciences and the TUBITAK (The Scientific and Technological Research Council of Turkey) Young Researcher Award (2011). |
 Gurkan Hizal | Gurkan Hizal received his MSc (1984) and PhD (1989) degrees in polymer chemistry under the direction of Prof. Y. Yagci at ITU, Turkey. In 1985 and 1986, he spent a year at Tokyo Institute of Technology in Tokyo, Japan with Prof. Y. Doi as a UNESCO fellow. He stayed at Hahn-Meitner Institute in Berlin, Germany as a Humboldt fellow (Prof. W. Schnabel, 1993). He was promoted to Full Professor at ITU in 1998. He was a visiting scientist at Laboratoire de Chimie des Polymères Organiques (LCPO), ENSCPB-CNRS University of Bordeaux working with Prof. Y. Gnanou (1999). His research interests include controlled/living polymerization techniques in polymer synthesis. |
 Umit Tunca | Umit Tunca completed his PhD under supervision of Prof. Y. Yagci in 1990. Since 1998 he has been a professor at the Chemistry Department of ITU, Turkey. He has published over 85 papers and 1 book chapter. His research interests are mainly, controlled/living radical polymerizations and modular ligation reactions. His awards include Monbusho (Japan) (1987), and Alexander von Humboldt (Germany) (1990) research fellowships, and TUBITAK (The Scientific and Technological Research Council of Turkey) Young Researcher (1997). |
Introduction
Contemporary synthetic polymer chemistry aims to enable the design and synthesis of macromolecules with not only precise molecular weight and narrow molecular weight distributions but also well-defined topology and chemical composition. Recent advancements in synthetic strategies allow a fair degree of control over the placement and distribution of functional groups along the macromolecular backbone. Living polymerization techniques such as anionic, cationic and ring opening metathesis (ROMP), and controlled radical polymerizations (CRP) may provide polymers with the above mentioned well-defined properties to a certain extent.1–5 Challenges in employing these polymerization techniques alone manifest themselves when topologically as well as compositionally intricate macromolecular constructs are targeted. The combination of the living and the controlled radical polymerization techniques with highly efficient ‘click’ chemistry based conjugation methodologies expands the toolbox of polymer chemists to realize various complex macromolecular architectures.The ‘click’ chemistry concept introduced by Sharpless and co-workers in 2001 comprises of chemical reactions displaying high stereo- and regio-selectivity with excellent yields under mild reaction conditions. Furthermore, the product of such reactions should be obtainable via simple purification protocols, as a logical expectation from highly efficient reactions. Additionally, these reactions are tolerant to a wide range of functional groups and can be carried out in a wide range of solvents.6 Therefore, in recent years the ‘click’ chemistry concept has found widespread application in the synthesis of various polymeric materials.7 The click chemistry concept has led to a “paradigm shift” toward modular construction approach in the preparative polymer chemistry as noted by Barner-Kowollik and Inglis8 Undoubtedly, the application of click chemistry concept has afforded various macromolecular structures that would otherwise not be achievable easily.8 Recently, Barner-Kowollik et al. evaluated the requirements for click reactions in the light of synthetic polymer chemistry.9 Additional criteria such as the utilization of equimolar amounts of building blocks, a simple large-scale purification process, apart from the expected high yields of products in a reasonable time scale under simple reaction conditions were proposed.9 It must be noted that the introduction of the ‘clickable’ functional groups into the polymeric building blocks should be achievable in a few simple steps otherwise it defeats the overall philosophy of click chemistry.
To date, the most widely utilized ‘click’ reactions in polymer chemistry may be classified as the Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC), the Diels–Alder cycloaddition, the thiol-ene (or thiol-yne), and the nitroxide radical coupling (NRC) reactions. The paragraphs below intend to briefly familiarize the readers with the basic functional groups involved in these reactions. Subsequent sections will highlight the utilization of a combination of these click reactions to yield various macromolecular constructs.
The CuAAC reaction is a Huisgen-type 1,3-dipolar (3 + 2) cycloaddition reaction that proceeds at room temperature under benign reaction conditions.10 Here, (3 + 2) notation describes the number of atoms in the two components involved in the 1,3-dipolar cycloaddition. Huisgen type reactions are exergonic processes, which combine two (e.g. azide and alkyne) unsaturated reactants and provide heterocycles e.g. a mixture of 1,4- and 1,5-disubstituted-1,2,3-triazole. However the CuAAC click reaction is highly regioselective and gives only 1,4-disubstituted-1,2,3-triazole (Scheme 1).
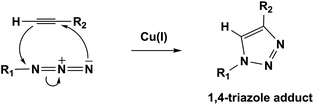 | ||
| Scheme 1 Cu(I) catalyzed azide–alkyne cycloaddition (CuAAC) reaction. | ||
The CuAAC click reaction was the first one to be adapted widely to the field of polymer chemistry,11 because of the tolerance and stability of the functional groups involved with a variety of CRP systems, in addition to the other advantages of click reactions such as high yields under mild conditions.7 The CRP systems, e.g. metal catalyzed controlled radical polymerization often called atom transfer radical polymerization (ATRP), nitroxide-mediated free radical polymerization (NMP) and the reversible addition fragmentation chain transfer (RAFT) polymerization enable the synthesis of various types of polymers with controlled molecular weight, narrow molecular weight distribution and appropriate site-specific branching points and functionalities. Among them, ATRP shares a number of important features with CuAAC including robustness, versatility and excellent tolerance towards many functional groups. Most importantly, polymers obtained via ATRP incorporate halogen end-group at chain terminus, which can be converted efficiently to an azide group using classic organic reactions.7 Here, it should also be mentioned that CuAAC and ATRP use the similar catalytic conditions without detriment to one another and thus can be proceeded to construct various polymeric architectures in a one-pot fashion.12 Furthermore, a wide variety of functional groups including various ‘clickable’ moieties can also be incorporated into the polymer chain ends by utilization of appropriately functionalized initiators.
The Diels–Alder reaction is a [4 + 2] cycloaddition reaction between a conjugated diene (a 4π-electron system) and a dienophile (a 2π-electron system) to yield a 6-membered ring (Scheme 2).13 The most commonly encountered diene/dienophile couples that are extensively employed in synthetic polymer chemistry are the anthracene/maleimide, furan/maleimide and butadiene/electron deficient dithioesters. The temperature range at which the cycloaddition and cycloreversion reaction takes place can be tuned by the selection of an appropriate diene-dienophile combination. The anthracene-maleimide cycloaddition reactions are usually conducted at high temperature, such as 110 °C.14 In the case of the furan/maleimide combination, the cycloaddition can be achieved at lower temperatures but the obtained adduct is not thermally stable over 100 °C, thus limiting its utilization towards the synthesis of polymers with complex architecture. However, the utilization of the furan–maleimide combination enables the design of thermoreversible macromolecular structures.15
![Illustrative diene-dienophile pairs utilized in the Diels–Alder [4 + 2] reactions.](/image/article/2012/PY/c1py00471a/c1py00471a-s2.gif) | ||
| Scheme 2 Illustrative diene-dienophile pairs utilized in the Diels–Alder [4 + 2] reactions. | ||
Another attractive alternative cycloaddition occurs with diene and dithioesters in a hetero Diels–Alder process, which can be carried out at relatively lower temperatures of 50 °C for butadiene or ambient temperature for cyclopentadiene.16 The dithioester end-functionality may be introduced into the polymer backbone via the RAFT process.17 The obtained adducts are not thermally stable over 80 °C for cyclopentadiene/dithioester, thus limiting the applicability of the resulting polymers at high temperatures. However, the same instability conversely enables the design of macromolecular structures with reversible thermoresponsive properties.18 Similar to the CuAAC process, some of the above noted Diels–Alder and hetero Diels–Alder systems fulfil the polymer conjugation click criteria.19
A thiol-ene or thiol-yne reaction involves the free radical addition of thiol to alkene or alkyne via thermal or photochemical process resulting in thioether product with high degree of anti-Markovnikov selectivity (Scheme 3).20,21 In a thiol-yne reaction each alkyne reacts first with a thiol to result in a vinyl sulfide followed by a subsequent reaction of the vinyl sulfide with another thiol to yield the 1,2-disubstituted thioether adduct.20,21
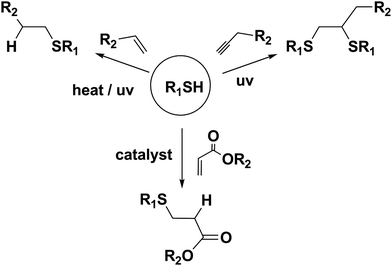
It has recently been noted that radical thiol-ene reactions are more efficient in the ligation of small organic molecules to the polymer than the polymer–polymer conjugation.22 Another commonly used thiol-ene reaction is the Michael addition of thiols to acrylate or maleimide yielding the thioether-ester or -imide functionalized adducts, respectively.
The NRC click reaction proceeds between a halide- and a 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-terminated polymers in the presence of CuBr and ligand at elevated temperature based on the ATRP mechanism (Scheme 4).23
 | ||
| Scheme 4 Nitroxide radical coupling (NRC) reaction. | ||
Huang and coworkers first applied the NRC click reaction together with CuAAC click reaction towards the preparation of linear ABC type triblock terpolymers.23b Later, this strategy was carried out under the single electron transfer controlled radical polymerization (SET-CRP)24 conditions. In this process, an equimolar Cu(0) transferred an electron to a halogen-terminated polymer to provide a macroradical and Cu(I) at ambient temperature by SET-CRP mechanism, which is efficiently trapped by TEMPO-terminated polymer. A wide variety of polymers with different topologies has been prepared using the NRC click reaction strategy.25
The theme of the present review is to highlight the rapidly emerging concept of the utilization of ‘double click reactions’ towards the design and synthesis of novel polymeric materials. The terminology ‘double click reactions’ in this review is strictly limited to strategies based on the utilization of two chemically and mechanistically different click reactions. Polymer conjugations and post-functionalization of the polymers that utilize only one type of the click reactions successively are not considered. The double click reaction strategy for the macromolecular conjugation is classified in this review based on their macromolecular architectures: linear and non-linear structures. Additionally, the post-functionalization of polymers with small organic molecules via the ‘double click reaction’ based strategy is also discussed in the last section of this review.
Linear polymers
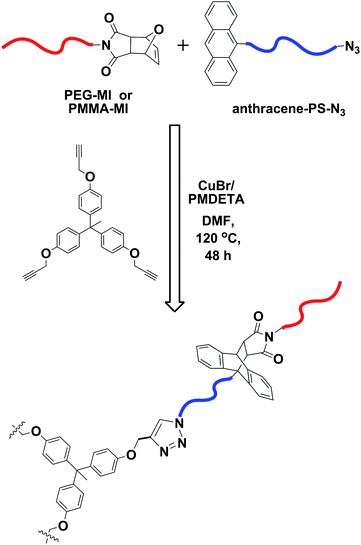
Hizal, Tunca and co-workers utilized the combination of CuAAC and Diels–Alder ‘click’ reaction to synthesize linear ABC triblock copolymers in a one pot reaction.26 The strategy benefits from the orthogonal nature of the azide-alkyne and anthracene-maleimide cycloaddition. A hetero-telechelic polystyrene (PS), containing an anthracene and an azide functional group at the chain termini (anthracene-PS-N3) was utilized as the middle block. Treatment of this polymer with a furan protected-maleimide-terminated poly(methyl methacrylate) (PMMA-MI), and an alkyne-terminated-poly(ethylene glycol) (PEG-alkyne) or -poly(ε-caprolactone) (PCL-alkyne) provided the triblock copolymers, PMMA-b-PS-b-PEG and PMMA-b-PS-b-PCL, respectively, in a modular and efficient manner (Fig. 1). | ||
| Fig. 1 Synthesis of ABC type linear terpolymers via CuAAC–Diels–Alder click reactions combination using a one-pot technique. | ||
In particular, PMMA-MI (Mn = 2600 g mol−1, Mw/Mn = 1.21) and anthracene-PS-N3 (Mn = 6100 g mol−1, Mw/Mn = 1.09) were reacted with a PEG-alkyne (Mn = 3000 g mol−1, Mw/Mn = 1.03) or a PCL-alkyne (Mn = 4000 g mol−1, Mw/Mn = 1.06) in a one-pot fashion to yield the corresponding linear triblock copolymers. The reactions were carried out in the presence of CuBr/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) in N,N-dimethyl formamide (DMF) at 120 °C for 48 h. Gel permeation chromatography (GPC) indicated a clear shift to the higher molecular weight region without any noticeable low molecular weight tail, thus indicating that pure triblock copolymers were obtained. 1H NMR analysis also confirmed the incorporation of all individual polymeric blocks into the corresponding triblock copolymers and the double click reaction efficiency was deduced to be in the range of 89–91%.
Huang and co-workers disclosed an alternative approach towards the one-pot synthesis of linear ABC triblock copolymers by using a combination of CuAAC and atom transfer NRC ‘click’ reactions. They utilized a hetero-telechelic PS containing a α-alkyne- and a ω-bromine end group as the middle block. Reaction of the alkyne-PS-Br polymer with TEMPO-terminated polymers (TEMPO-PEG or TEMPO-PCL) and an azide-terminated poly(tert-butyl acrylate) (PtBA-N3) in the presence of CuBr/PMDETA at elevated temperatures, furnished well-defined linear ABC triblock copolymers (Fig. 2).23b In the NRC reaction, under the ATRP conditions, a bromide-terminated polymer reacts with CuBr to produce the corresponding macroradical, which is efficiently capped with a TEMPO-terminated polymer.23b In particular, alkyne-PS-Br (Mn = 7300–10600 g mol−1, Mw/Mn = 1.10–1.08) and with PtBA-N3 (Mn = 3000–5300 g mol−1, Mw/Mn = 1.13–1.10), both synthesized via ATRP, were clicked with TEMPO-PEG (Mn = 3600–4000 g mol−1, Mw/Mn = 1.20–1.05) or -PCL (Mn,NMR = 4400 g mol−1) to yield linear triblock terpolymers in a one-pot fashion by using a CuBr/PMDETA catalyst system in DMF at 90 °C for 24 h. The corresponding triblock terpolymers, PtBA-b-PS-b-PEG and PtBA-b-PS-b-PCL, were obtained with coupling efficiencies as high as 86%.
 | ||
| Fig. 2 Synthesis of ABC type linear terpolymers via combining CuAAC–NRC click reactions in a one-pot technique. | ||
Barner-Kowollik and co-workers recently applied a diverse radical coupling strategy, namely, the nitrone-mediated radical coupling (NMRC), for the homoconjugation of α,ω-bromo functional poly(isobornyl acrylate)s (Mn,GPC = 7800 g mol−1, Mw/Mn = 1.25) obtained via ATRP.27 Homoconjugation of the polymers in the presence of a trimethylsilyl protected alkyne-functionalized nitrone, followed by the removal of the silyl protecting group was used to obtain a multi-segment polymer containing pendant alkyne moieties. Post-modification of thus obtained polymers was done with 3-mercaptopropionic acid under UV irradiation via the free radical mediated thiol-yne ‘click’ reaction.
Non-linear polymers
Non-linear polymers synthesized using the double click reaction based strategies, discussed in this review are subdivided according to their macromolecular topologies into star, graft, H-shaped, dendrimers and dendronized polymers, and cyclic polymers.Star polymers
Star polymers are branched macromolecules where all the polymer chains as the arm segments are linked to a central unit, referred to as the core. While several methods for their efficient synthesis are available, efficient methods towards their functionalization were quite limited. In recent years click reactions have been exploited towards the post-functionalization of such star polymers. Utilization of double click reaction based strategies has also led to facile multifunctionalization of these constructs.Tunca and co-workers employed the concept of double click reaction to synthesize a three-arm star block copolymer in a one-pot reaction. A trialkyne-functional linking agent, 1,1,1-tris[4-(2-propynyloxy)phenyl]-ethane, was combined with the hetero-bifunctional polymer, anthracene-PS-N3 (Mn,GPC = 5200 g mol−1; Mw/Mn = 1.12) and either PMMA-MI (Mn,GPC = 2500 g mol−1; Mw/Mn = 1.19) or PEG-MI (Mn,GPC = 3200 g mol−1, Mw/Mn = 1.03) in the presence of CuBr/PMDETA at 120 °C in DMF for 48 h to furnish the corresponding star block copolymers (Fig. 3).28 The efficiencies for the star block copolymer formation were calculated to be 82–88% via peak splitting analysis on GPC traces.
 | ||
| Fig. 3 Synthetic strategy for the preparation of a three-arm star block copolymer via CuAAC and Diels–Alder in a one-pot fashion. | ||
The CuAAC–Diels–Alder double click reaction strategy was further extended to produce multiarm core-crosslinked star polymers containing triblock terpolymer arms.29 The (alkyne-PS)m-poly(divinyl benzene) (DVB) multiarm star polymer (number of arms = 23–25) was obtained via ATRP of DVB using α-silyl-alkyne-PS as a linear macroinitiator in the presence of a CuBr/PMDETA catalyst at 110 °C (Fig. 4). Subsequently the silyl protecting groups on the alkynes were removed using tetra-butyl ammonium fluoride (TBAF). Thus obtained (alkyne-PS)m-polyDVB multiarm star polymer was sequentially clicked with a linear hetero bifunctional polymer, anthracene-PMMA-N3, (Mn,GPC = 4100 g mol−1, Mw/Mn = 1.23) and either a PtBA-MI (Mn,GPC = 4100 g mol−1, Mw/Mn = 1.14) or a PEG-MI (Mn,GPC = 550 g mol−1, Mw/Mn = 1.09) polymer to yield the corresponding (PtBA)k-(PMMA)n-(PS)m-polyDVB and (PEG)p-(PMMA)n-(PS)m-polyDVB multiarm star terpolymers. The CuAAC and Diels–Alder click reaction efficiencies were calculated to be over 95% using the NMR and UV data, respectively.
 | ||
| Fig. 4 Preparation of core-crosslinked multiarm star terpolymers using sequential CuAAC–Diels–Alder click reactions. | ||
The sequential CuAAC and Diels–Alder click reaction strategy was also exploited in the synthesis of multi-miktoarm star block copolymers via the ‘arm-first’ approach (Fig. 5).30 The multiarm core-crosslinked star PS with both alkyne and anthracene moieties at the periphery, (alkyne-PS)n–polyDVB–(PS-anthracene)n, was obtained via ATRP of DVB concurrently initiated with linear α-silyl protected alkyne- and α-anthracene-terminated PS macroinitiators (Mn,GPC = 5400 g mol−1, Mw/Mn = 1.10). Sequential CuAAC and Diels–Alder reactions of the multifunctional core-crosslinked star polymer with PtBA-N3 (Mn,NMR = 5100 g mol−1) and PMMA-MI (Mn,GPC = 3900 g mol−1, Mw/Mn = 1.17) resulted in formation of the target multi-miktoarm star block copolymer, (PtBA)m-(PS)n-polyDVB-(PS)n-(PMMA)k, with narrow molecular weight distribution. Molecular weights up to 400000 g mol−1 were measured for these constructs by triple-detection GPC. Analysis of the final star block copolymers by UV spectroscopy revealed that the efficiency of conjugation via the Diels–Alder click reaction was over 95%.
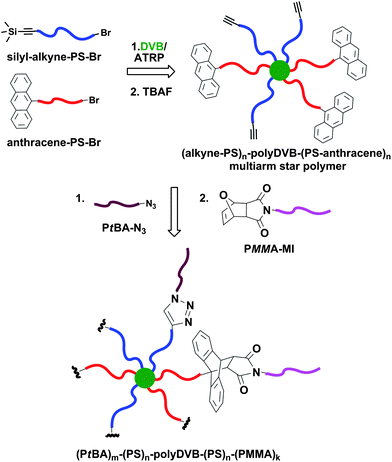 | ||
| Fig. 5 Sequential CuAAC and Diels–Alder click reaction strategy for the synthesis of multi-miktoarm star block copolymers via the ‘arm-first’ approach. | ||
More recently, Yagci and co-workers reported a combination of the thiol-ene and CuAAC reactions for the preparation of a ABC miktoarm star polymer.31 For this purpose, a thiol terminated-PS (Mn,GPC = 3530 g mol−1, Mw/Mn = 1.08) was reacted with the 1-(allyloxy)-3-azidopropan-2-ol core under UV irradiation via thiol-ene click reaction. Thereafter, ROP of ε-CL was initiated from this macroinitiator to yield a diblock copolymer containing an azide functional group at the junction of the two blocks. A PEG-alkyne (Mn = 2000 g mol−1) was clicked via the CuAAC reaction onto the azide functionality to furnish the ABC miktoarm star polymer (Fig. 6).

Graft copolymers
The first example of the synthesis of hetero-graft copolymers via ‘graft-to’ method using the double click reaction was reported by Tunca and co-workers in 2008.32 A hetero-graft terpolymer was obtained using the CuAAC and Diels–Alder reactions in a one-pot technique. A random copolymer of St and p-chloromethylstyrene was prepared via NMP. Subsequent attachment of the anthracene functionality to the preformed copolymer was carried out by the o-etherification procedure followed by conversion of the remaining -CH2Cl into azide functionality.33 Thereafter, a PMMA-MI (Mn,GPC = 3350 g mol−1, Mw/Mn = 1.22) and a PEG-alkyne (Mn = 2000 g mol−1) were efficiently introduced onto the PS polymer bearing the pendant anthryl and azide moieties to yield a PS(-g-PMMA)-g-PEG heterograft terpolymer (Fig. 7).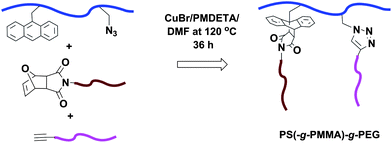 | ||
| Fig. 7 Synthesis of a heterograft terpolymer via a combination of CuAAC–Diels–Alder click reactions using a one-pot technique. | ||
A similar strategy was employed to obtain polynorbornene-based hetero-graft copolymers via the combination of ROMP, and sequential CuAAC and Diels–Alder reaction. Poly(ONB-g-PMMA)-b-poly(ONB-g-PCL) and poly(ONB-g-PtBA)-b-poly(ONB-g-PCL) block brush graft terpolymers were made by sequential CuAAC and Diels–Alder click reactions of PCL-alkyne (Mn,NMR = 2800 g mol−1) and PMMA-MI (Mn,NMR = 3300 g mol−1) or PtBA-MI (Mn,NMR = 2300 g mol−1) respectively with a well-defined ROMP generated polyoxanorbornene (PONB) block (Mn,GPC = 7300 g mol−1, Mw/Mn = 1.06) containing azide and anthryl pendant groups.34 In a related work, the sequential CuAAC and Diels Alder click reactions were successfully employed towards the synthesis of graft terpolymers consisting of a PONB main chain and the PS-b-PMMA (or b-PEG, or b-PtBA) copolymer grafts.35
The double polymer click reactions (CuAAC and Diels–Alder) were also applied to produce regular graft copolymers (Fig. 8).36 In this study, graft copolymers with regular graft points containing PS backbone and PMMA, PtBA, or PEG side chains were simply achieved by a sequential double polymer click reactions.36 The linear PS containing both alkyne and anthracene groups at α-end and azide unit at ω-end (Mn,NMR = 3850 g mol−1) at was produced via ATRP of St. Subsequently, the CuAAC click coupling of this PS to create the linear multiblock PS chain with pendant anthracene sites per PS block (Mn,GPC = 42400 g mol−1, Mw/Mn = 1.40), followed by Diels–Alder click reaction with the PMMA-MI (Mn,NMR = 3300 g mol−1), PtBA-MI (Mn,NMR = 2300 g mol−1), or PEG-MI (Mn,NMR = 750 g mol−1) yielded final PS-g-PMMA, PS-g-PtBA or PS-g-PEG copolymers with regular grafts, respectively. The decrease in the concentration of the anthracene unit due to the Diels–Alder click reaction at 368 nm was found to yield efficiencies over 92% via UV spectroscopy as a function of time.
 | ||
| Fig. 8 Synthesis of regular graft copolymers via sequential CuAAC–Diels–Alder click reactions. | ||
H-shaped polymers
H-shaped polymers possess two polymeric side arms attached to the each end of a polymer backbone. An H-shaped polymer containing pentablocks (H-shaped quintopolymer) with different chemical compositions was obtained by employing the CuAAC and Diels–Alder reaction in a one-pot technique (Fig. 9).37 Herein the H-shaped quintopolymer consists of PEG-PMMA and PCL-PS blocks as side chains and PtBA as a main chain. For the preparation of H-shaped quintopolymer, the diblock copolymers PEG-b-PMMA (Mn,NMR = 8100 g mol−1) and PCL-b-PS (Mn,NMR = 12800 g mol−1) copolymers with maleimide and alkyne functional groups at their block junctions were synthesized. These diblock copolymers were simultaneously subjected to click reactions with a linear anthracene-PtBA-N3 (Mn,NMR = 8700 g mol−1) using CuBr/PMDETA as a catalyst in DMF at 120 °C for 48 h. The double click reaction efficiency was deduced to be 88% by comparing the 1H NMR spectra of the precursor macromolecules and the final H-shaped quintopolymer. | ||
| Fig. 9 Preparation of H-shaped quintopolymer using a combination of CuAAC–Diels–Alder click reactions in a one-pot technique. | ||
Dendrimers and dendronized polymers
Dendrimers are well-defined, monodisperse, highly branched three-dimensional macromolecular structures. Until recently, synthesis of these molecularly precise branched macromolecules has remained a challenging task due to the low reaction yields and tedious purification steps involved. In the past few years, several reports have shown that the utilization of ‘click’ reactions provides an efficient and ‘green’ route towards construction of dendrimers.38a,b Employing the combination of double click reactions not only enables the facile and rapid synthesis of dendrimers but also allows one to introduce diversity at the branching points within these macromolecules.Kakkar and co-workers highlighted the benefits of using the double click reaction based strategy in their design and synthesis of a novel thermally cleavable dendrimer.38c Using the divergent approach for dendrimer construction, the authors utilized the CuAAC and furan-maleimide Diels–Alder reactions sequentially to grow subsequent higher generations (Fig. 10). A bisfuran-azide containing molecule was first reacted with a tris-alkyne core unit using the CuAAC reaction to yield a G1 dendrimer. This was followed by the Diels–Alder reaction with a bis-maleimide-alkyne linker to provide the G2 dendrimer. Iterative sequences of these click reactions provided the higher generations dendrimers. These dendrimers are thermosensitive since the furan-maleimide cycloadduct based junctions are cleaved upon heating via the retro-Diels–Alder cycloreversion reaction.
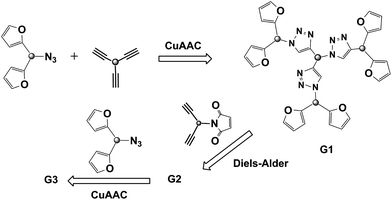 | ||
| Fig. 10 Preparation of a divergent dendrimer structure using the sequential CuAAC–Diels–Alder (furan–maleimide) reactions. | ||
A combination of double click reaction strategy employing both thiol-ene and CuAAC was employed by Hawker and co-workers to accomplish the synthesis of a 6th generation dendrimer in a single day.39 The synthetic strategy facilitates the preparation of higher generation dendrimers via iterative utilization of orthogonally ‘clickable’ monomers containing a thiol-bisazide (SH-(N3)2) and an alkyne-bisalkene (yne-(ene)2) unit (Fig. 11). The efficiency of the double click reaction based protocol enabled the authors to obtain a sixth generation dendrimer in a single day.
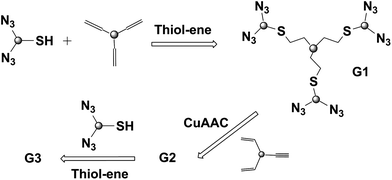 | ||
| Fig. 11 Combination of double click reaction strategy: iterative thiol–ene and CuAAC for the synthesis of a 6th generation dendrimer. | ||
Recently, ‘orthogonally’ functionalizable polyester-based dendrons were reported by Sanyal and co-workers.40 The surface of these dendrons were appended with alkyne groups that can undergo efficient CuAAC reaction while their focal point consisted of a reactive maleimide group that was amenable to facile conjugation with thiol containing molecules via the nucleophilic thiol-ene ‘click’ reaction. These dendrons were reacted with PEG-N3 to obtain water soluble multi-arm polymers containing thiol reactive maleimide unit at their core.
Xiong and Xu reported the synthesis of dendritic polymers with PCL on the periphery using the CuAAC and Diels–Alder click reactions in a one-pot technique.41 A tetra-alkyne functional core, 9-azidomethyl anthracene and maleimide containing mono- and bis-arm PCL polymers in the presence of CuBr/PMDETA in DMF was stirred at room temperature for 12 h, followed by heating at 110 °C for another 12 h. The sequential CuAAC and Diels–Alder reactions yielded the final dendritic polymeric structure.
The first example of a main chain thermoreversible dendronized polymer was recently reported by McElhanon and co-workers.42 The authors applied the combination of CuAAC and furan-maleimide Diels–Alder reactions for the synthesis of linear dendronized polymers (Fig. 12). Three generations of Frechet type poly(aryl ether) dendrons containing an azide moeity at their focal point were reacted with a bisfuran-bisalkyne functional group bearing monomer. Dendronized macromonomers thus obtained were reacted with bismaleimide in CHCl3 at 50 °C to yield the thermoreversible linear dendronized polymers. Recently, the authors reported an elegant extension of this work to synthesize thermally labile dendronized linear AB step-polymers.43 Dendritic AB monomers prepared via the CuAAC reaction were polymerized using a single-component Diels–Alder cycloaddition reaction to afford the main chain thermoreversible polymer.
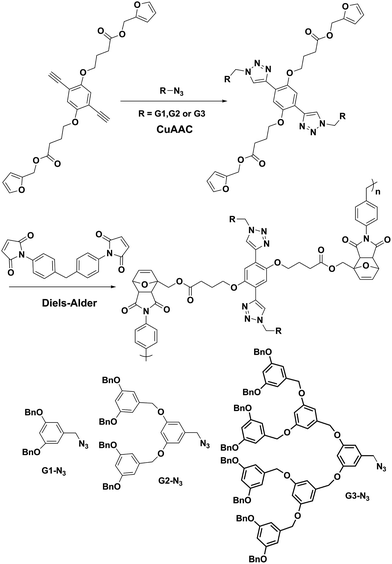 | ||
| Fig. 12 Combination of CuAAC and furan–maleimide Diels–Alder click reactions for the preparation of linear dendronized polymers. | ||
Cyclic polymers
Notably, the click reaction based strategies have clearly advanced the synthesis of cyclic polymers. Cyclic homopolymers and diblock copolymers have been synthesized using the CuAAC reactions.44a Previously, cyclic diblock copolymers were obtained by using an alkyne-azide terminated diblock copolymer precursor that was synthesized via sequential polymerization of monomers using ATRP.44b Recently, Tunca and co-workers reported a modular synthesis of cyclic block copolymers via the sequential use of CuAAC and Diels–Alder reactions.45 The block copolymers, PS-b-PtBA (Mn,GPC = 7400 g mol−1, Mw/Mn = 1.14) and PS-b-PCL (Mn,GPC = 11200 g mol−1, Mw/Mn = 1.05) with α-anthracene and ω-maleimide functional groups were prepared using the highly efficient CuAAC click reaction (Fig. 13). Subsequently, cyclic homo- and block copolymers were obtained from their corresponding linear precursors via the Diels–Alder ‘click’ reaction by refluxing in toluene under dilute conditions. The efficiency of the cyclization reactions was determined from the GPC peak splitting method using the Gaussian function and was found to be in the range of 77–85%. The double click reaction based methodology employed in this work provides an efficient and modular route to obtain cyclic block copolymers with various chemical compositions.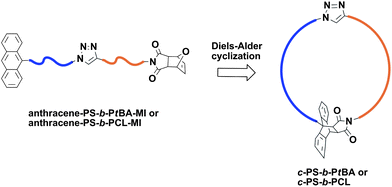 | ||
| Fig. 13 Preparation of cyclic block copolymers via sequential CuAAC and Diels–Alder click reactions. | ||
Post-functionalization of polymers
Chaikof and co-workers first demonstrated the orthogonality of sequential Diels–Alder and CuAAC click reactions for the immobilization of carbohydrates and proteins onto glass surfaces.46 An α-alkyne-ω-cyclopentadiene-PEG linker was immobilized onto a maleimide functionalized glass slide via an aqueous Diels–Alder click reaction. In the subsequent step, thus obtained alkyne terminated surface was then successfully clicked with both azide-containing biotin and lactose via aqueous CuAAC at low temperature. This study demonstrated that use of sequential CuAAC and Diels–Alder reactions provide an effective strategy for the immobilization of a wide range of functionally complex structures onto solid surfaces.Utilization of the combination of the CuAAC and the hydrazone formation click reactions as a tool for modular multifunctionalization of copolymers was reported by Yang and Weck.47 The ROMP generated polynorbornene based random copolymers containing either azide, aldehyde or ketone pendant groups on each repeating units were reacted with molecules containing alkyne and amine groups. Yagci and co-workers used double click reaction strategy combining CuAAC and Diels–Alder cycloaddition reactions in the preparation of macrophotoinitiators containing thioxanthone moieties as side chains.48 In this study a NMP generated PS chain with azide pendant moieties (Mn = 2300 g mol−1) was functionalized via N-propargyl-7-oxanorbornene linker and anthracene-thioxanthone compound using a one-pot CuAAC–Diels–Alder click reactions catalyzed by CuBr/PMDETA in DMF at 120 °C for 36 h. The resulting polymer acts as a photoinitiator due to its thioxanthone chromophoric groups for the polymerization of mono and multifunctional monomers.
A combination of thermal thiol-ene and CuAAC click reactions for the α,ω-functionalization of linear PS was reported by Hawker and coworkers.49 They also tested the orthogonality of thermal thiol-ene with the CuAAC reaction using a telechelic alkene-PS-N3 (Mn,GPC = 6000 g mol−1, Mw/Mn = 1.08), thioglycolic acid and propargyl alcohol. The two possible synthetic pathways: initial thermal thiol-ene followed by CuAAC and vice versa, showed analogous efficiencies and compatibility with quantitative functionalization of both chain ends.
Haddleton and co-workers prepared macromonomers containing two clickable groups (alkyne pendant groups and mono alkene end-functional group), which were subsequently functionalized with thiols and sugar azides using sequential thiol-ene (using dimethylphenylphosphine in acetone at ambient temperature) and CuAAC click reactions (catalyzed by CuBr/bipyridine/triethylamine in DMSO at 60 °C) to yield functionalized glycopolymers in a convenient manner.50
Barner-Kowollik and co-workers reported the functionalization of cross-linked PDVB microspheres containing vinyl groups on their surface using a combination of thiol-ene and CuAAC click reactions.51 The double bonds on the microsphere surface were converted into azide groups using 1-azido-undecane-11-thiol through thermal thiol-ene reaction. In a second step, alkyne terminated poly(hydroxy ethyl methacrylate) (Mn = 21000 g mol−1, Mw/Mn = 1.77) was grafted to the azide modified surface via the CuAAC reaction.
A novel post-functionalization method based on the CuAAC, followed by an addition reaction between electron-rich alkynes (donor) and tetracyanoethylene (TCNE, acceptor) was developed by Michinobu and coworkers.52 Thus, donor–acceptor chromophores are efficiently introduced into the poly(4-azidomethylstyrene) using the CuAAC click reaction, in remarkably high yields. The importance of the reaction order was explained by steric reasons. It was found that the alkyne-TCNE click reaction proceeds even in sterically hindered environments.
Hvilsted and co-workers introduced a second generation of L-lycine and cholestryl groups onto the linear heterotelechelic PCL containing alkyne and alkene terminal groups (Mn = 4200 g mol−1, Mw/Mn = 1.11) using the sequential CuAAC and UV generated thiol-ene click reactions.53
Sumerlin and co-workers applied a combination of the thiol-ene and Diels–Alder click reactions towards the functionalization of a linear polymer.54 The thiocarbonylthio end-groups of a RAFT generated poly(N-isopropylacrylamide) (PNIPAM) (Mn = 3450 g mol−1, Mw/Mn = 1.12) were converted to thiols, and subsequent reaction with an excess of a bismaleimide compound yielded the mono-maleimide terminated PNIPAM under thermal thiol-ene click reaction. Thus obtained maleimide-terminated polymer was further clicked with 9-anthracene methanol through the Diels–Alder reaction.
Conclusions and future perspective
In recent years, double click reaction based strategies have emerged as a powerful methodology that affords polymers with diverse topology and composition, which cannot be prepared by using only one type of click reaction. Needless to say, the strategy also reduces the number of steps and reaction time. More importantly, the double click reaction strategy inspires and invokes the synthesis of novel polymeric material.To date, only a few click reaction combinations, the CuAAC–Diels–Alder and the CuAAC–NRC reactions have been extensively adapted to the double click reaction strategy for polymer–polymer conjugation. Alternative double click reactions, the NMRC–thiol–yne and the CuAAC–tetracyanoethylene combinations have only been used in the post-modification of the polymers via small organic molecules. Due to the easy availability of the clickable groups required for the CuAAC, Diels–Alder and NRC reactions, it is obvious why these three click reactions are so widely adapted to the polymer conjugation. The azide, alkyne, anthracene, and maleimide clickable groups can be easily incorporated into the polymer backbone via functional initiators, monomers, and post-polymerization modification methods using LRP techniques, particularly with ATRP. A TEMPO group can be introduced into the polymer chain via functional initiator using the living ROP technique, or post-functionalization of the polymer.
In the near future, different click reactions combinations other than the CuAAC–Diels–Alder and the CuAAC–NRC may emerge for polymer conjugation, leading to various macromolecular architectures. Moreover, it should be noted that a variety of click reaction combinations containing more than two different click reactions may be of interest for polymer–polymer conjugation and/or the post-modification of the polymers. From this point of view Tunca and co-workers recently applied a triple click reaction concept, a combination of the CuAAC, Diels–Alder and NRC reactions, to the synthesis of linear tetrablock quaterpolymer.55
Finally, one can expect that more challenging polymeric architectures will be obtained in a simple manner by utilizing diverse combinations available from the vast toolbox of click reaction.
However, it should be noted here that each click reaction has its own disadvantages, such as the use of Cu(I) catalyst in the CuAAC, the elevated temperatures in the Diels–Alder, the limitations for polymer–polymer conjugation in the thiol-ene (or thiol-yne), and the limited applicability of polymer precursors in the NRC reactions. In addition to these disadvantages of each click reaction, the selective introduction of the clickable functional groups into the various polymer precursors in multiple complex steps, the deviation from equimolar amounts of polymer building blocks, the low yields of products, the unreasonably longer reaction times, and the complicated purification processes are the most encountered challenges of the double click reaction strategy.
Notes and references
- J. P. Kennedy and S. Jacob, Acc. Chem. Res., 1998, 31, 835 CrossRef CAS.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS.
- A. Hirao, M. Hayashi, S. Loykulnant and K. Sugiyama, Prog. Polym. Sci., 2005, 30, 111 CrossRef CAS.
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS; (b) C. Barner-Kowollik and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5715 CrossRef CAS; (c) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987 CrossRef CAS; (d) C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS; (e) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS; (f) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS; (g) J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901 CrossRef CAS; (h) F. Di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959 CrossRef CAS; (i) V. Percec and B. Barboiu, Macromolecules, 1995, 28, 7970 CrossRef CAS; (j) B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS; (k) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS; (l) L. Barner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539 CrossRef CAS; (m) Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-V C H, Weinheim, Germany, 2008 Search PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- (a) B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS; (b) R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS; (c) K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171 RSC; (d) U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560 RSC; (e) O. Altintas and U. Tunca, Chem.–Asian J., 2011, 6, 2584 CrossRef CAS; (f) P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338 RSC; (g) M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455 CrossRef CAS; (h) C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS; (i) O. Altintas, A. P. Vogt, C. Barner-Kowollik and U. Tunca, Polym. Chem., 2012, 3, 34–45 RSC; (j) D. J. Hall, H. M. Van Den Berghe and A. P. Dove, Polym. Int., 2011, 60, 1149 CrossRef CAS.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- J. A. Opsteen and J. C. M. Van Hest, Chem. Commun., 2005, 57 RSC.
- (a) P. D. Topham, N. Sandon, E. S. Read, J. Madsen, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 9542 CrossRef CAS; (b) J. Yin, Z. Ge, H. Liu and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2608 CrossRef CAS; (c) D. Han, Z. Tong and Y. Zhao, Macromolecules, 2011, 44, 5531 CrossRef CAS; (d) A. J. de Graaf, E. Mastrobattista, C. F. van Nostrum, D. T. S. Rijkers, W. E. Henninka and T. Vermonden, Chem. Commun., 2011, 47, 6972 RSC.
- (a) O. Diels and K. Alder, Ber. Dtsch. Chem. Ges., 1929, 62, 554 CrossRef; (b) H. Kwart and K. King, Chem. Rev., 1968, 68, 415 CrossRef CAS.
- (a) G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4103 CAS; (b) H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 499 CrossRef CAS; (c) H. Durmaz, F. Karatas, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3947 CrossRef CAS; (d) A. Dag, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 302 CrossRef CAS; (e) A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178 CrossRef CAS; (f) O. Altintas, G. Hizal and U. Tunca, Des. Monomers Polym., 2009, 12, 83 CrossRef CAS; (g) O. Gok, H. Durmaz, E. S. Ozdes, G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2546 CrossRef CAS; (h) H. Durmaz, A. Dag, C. Onen, O. Gok, A. Sanyal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4842 CrossRef CAS; (i) A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417 CrossRef CAS.
- M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818 CrossRef CAS.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247 CrossRef CAS.
- A. J. Inglis, T. Paulohrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33 CrossRef CAS.
- (a) A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515 CrossRef CAS; (b) N. Aumsuwan and M. W. Urban, Polymer, 2009, 50, 33 CrossRef CAS; (c) J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Pricec and D. M. Haddleton, Polym. Chem., 2010, 1, 102 RSC.
- (a) H. Durmaz, B. Colakoglu, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1667 CrossRef CAS; (b) A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411 CAS.
- (a) M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS; (b) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- C. E. Hoyle, A. B. Lowe and C. N Bowman, Chem. Soc. Rev., 2010, 39, 1355 RSC.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. DuPrez, C. Barner-Kowollik, W. van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699 CrossRef CAS.
- (a) K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955 CrossRef CAS; (b) W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127 CrossRef CAS; (c) R. Nicolay and K. Matyjaszewski, Macromolecules, 2011, 44, 240 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS.
- (a) Q. Fu, G. Wang, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986 CrossRef CAS; (b) Q. Fu, C. Liu, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6770 CrossRef CAS; (c) G. Wang, Y. Zhang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1633 CrossRef CAS; (d) Y. Li, Y. Zhang, S. Zhai, Y. Deng, H. Xiong, G. Lu and X. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 23 CrossRef CAS; (e) N. Cerit, N. Cakir, A. Dag, O. Sirkecioglu, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2850 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191 CrossRef CAS.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2118 CrossRef CAS.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091 CrossRef CAS.
- H. Durmaz, A. Dag, D. Gursoy, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1557 CrossRef CAS.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621 RSC.
- B. Iskin, G. Yılmaz and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2417 CrossRef CAS.
- A. Dag, H. Durmaz, E. Demir, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6969 CrossRef CAS.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5930 CrossRef.
- A. Dag, H. Sahin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 886 CrossRef CAS.
- H. Durmaz, A. Dag, N. Cerit, O. Sirkecioglu, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5982 CrossRef CAS.
- H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1195 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3409 CrossRef CAS.
- (a) G. Franc and A. Kakkar, Chem. Commun., 2008, 5267 RSC; (b) G. Franc and A. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC; (c) A. Vieyres, T. Lam, R. Gillet, G. Franc, A. Castonguay and A. Kakkar, Chem. Commun., 2010, 46, 1875 RSC.
- P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmstrom, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625 CrossRef CAS.
- M. M. Kose, S. Onbulak, I. I. Yilmaz and A. Sanyal, Macromolecules, 2011, 44, 2707 CrossRef CAS.
- X. Q. Xiong and Y. H. Xu, Polym. Bull., 2010, 65, 455 CrossRef CAS.
- N. W. Polaske, D. V. McGrath and J. R. McElhanon, Macromolecules, 2010, 43, 1270 CrossRef CAS.
- N. W. Polaske, D. V. McGrath and J. R. McElhanon, Macromolecules, 2011, 44, 3202 CrossRef.
- (a) B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202 RSC; (b) D. M. Eugene and S. M. Grayson, Macromolecules, 2008, 41, 5082 CrossRef CAS.
- H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5083 CrossRef CAS.
- X.-L. Sun, C. L. Stabler, C. S. Cazalis and E. L. Chaikof, Bioconjugate Chem., 2006, 17, 52 CrossRef CAS.
- S. K. Yang and M. Weck, Macromolecules, 2008, 41, 346 CrossRef CAS.
- B. Gacal, H. Arat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727 RSC.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Mueller, Macromolecules, 2009, 42, 3707 CrossRef CAS.
- (a) Y. Lia and T. Michinobu, Polym. Chem., 2010, 1, 72 RSC; (b) Y. Li, K. Tsuboi and T. Michinobu, Macromolecules, 2010, 43, 5277 CrossRef CAS.
- I. Javakhishvili, W. H. Binder, S. Tanner and S. Hvilsted, Polym. Chem., 2010, 1, 506 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093 CrossRef CAS.
- H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1962 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |