
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amine-containing block copolymers for efficient catalyst-free hydroamination and preparation of functional metallopolymers†
Till
Rittner
 a,
Kinza
Ghulam
a,
Marcus
Koch
b and
Markus
Gallei
*ac
a,
Kinza
Ghulam
a,
Marcus
Koch
b and
Markus
Gallei
*ac
aPolymer Chemistry, Saarland University, Campus C4 2, 66123 Saarbrücken, Germany. E-mail: markus.gallei@uni-saarland.de
bINM-Leibniz Institute for New Materials, Campus D2 2, 66123, Saarbrücken, Germany
cSaarene, Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
First published on 5th August 2024
Abstract
Cobaltocenium-containing polymers, an emerging class of materials, have historically been challenging to prepare due to their chemical robustness. In this work, we introduce a novel and highly efficient method for their preparation based on methacrylate-containing block copolymers (BCPs), allowing segment-selective introduction of functional moieties. The catalyst-free and quantitative hydroamination reaction we introduce has proven successful for the post-modification of amine-containing polymers with cobaltocenium. To demonstrate the versatility of this method, we successfully synthesized a series of BCPs consisting of polystyrene and a 5 to 20 wt% poly(tert-butyl aminoethyl methacrylate) (PtBAEMA) segment by living anionic polymerization. The selective functionalization with ethynyl-cobaltocenium hexafluorophosphate results in adjustable 5 to 40 wt% cobaltocenium units in the polymer as part of the PtBAEMA block segment. The success was monitored by IR spectroscopy, and the quantitative incorporation of the cobaltocenium moiety was verified by 1H NMR, UV-Vis spectroscopy, and TGA. DSC proved the block-selective cobaltocenium introduction by an additional glass transition temperature at 154 °C, and the strong microphase separation character of the amphiphilic BCPs leads to lamellar structures in the bulk state, as proven by TEM investigations. Finally, the water contact angle on polymer films is compared, showing polarity inversion and tunability upon conversion of hydrophilic amine to hydrophobic cobaltocenium hexafluorophosphate moieties. This successful synthesis and characterization of cobaltocenium-containing BCPs not only paves the way for a new class of metallopolymers but also offers functionalization possibilities for a variety of other responsive moieties, providing access to functional BCPs.
Introduction
Nowadays, metallopolymers are of growing interest due to their unique properties that combine inorganic and organic aspects.1,2 From this combination, numerous applications ranging from electrochromic materials,1 hydrogels,3 and ceramic material4 over ion separation5,6 and PFAS capture7 to materials for controlled wetting8 and many other applications are described.9,10 In this field, ferrocene is the most prominent metallocene.11 It is readily modified, resulting in an abundance of reactive monomers and polymers.12 One major drawback of ferrocene is the unstable ionic state. Here, especially in an aqueous environment, ferrocenium readily oxidizes back to the favored neutral complex, losing the ionic properties essential for further applications.13–15 By replacing the iron with cobalt, this problem can be overcome. The isoelectronic cobaltocenium complex features a permanent and stable cationic state. In contrast, neutral cobaltocene readily oxidizes fast and is exploited for one-electron reduction in organic synthesis and internal standard in cyclic voltammetry.16 This advantage of the stable ionic state over ferrocene, however, also presents a major challenge. Due to the inert ionic nature of cobaltocenium, derivatization is generally difficult. However, there have been significant developments in recent years due to the ever-increasing demand for new metallocene-containing polymers.17,18 In general, there are three routes to obtain cobaltocenium-containing polymers: (i) via main-chain polymerization, (ii) via polymerization of side-chain cobaltocene monomers, and (iii) post-modification methods, each with respective advantages and disadvantages.19 While the first two approaches require the use of specialized techniques, post-modification offers the possibility of using template polymers from almost any preparation method. In general, after polymer synthesis and characterization, cobaltocenium can be introduced by reacting with functionalities in the polymer side-chain. Here, initially, cobaltocene acids and their derivatives were used extensively.20 Later, with the introduction of the acetylene-click reaction, a second approach was introduced, utilizing cobaltocene acetylene.21 Even though both of these methods deliver good results, they still require catalysts for high conversion rates, which can be a hindrance for many applications.19 More recently, a catalyst-free hydroamination reaction of ethynyl cobaltocenium and primary or secondary amines was described by Wang et al. enabling a covalent and efficient cobaltocenium functionalization.22 Using this elegant approach, first macrostructures like dendrimers and homopolymers derived by ring-opening metathesis polymerization (ROMP) could be synthesized in the Astruc group.23,24 These first examples show great potential for many interesting applications. On the other hand, the formation and access to block copolymer (BCP) structures would widen the scope of this metallopolymer class into different fields of application.In this work, we demonstrate a generally applicable method for introducing cobaltocenium moieties into methacrylate-based polymers using tert-butyl aminoethyl methacrylate (tBAEMA) monomer for the formation of block copolymer segments. BCP architectures are prepared by using living anionic polymerization to take advantage of structure formation and morphological evolution. We highlight the convenient control over the amount of introduced cobaltocenium into the BCP segments by different characterization techniques comprising proton nuclear magnetic resonance (1H NMR) spectroscopy, attenuated total reflection infrared spectroscopy (ATR-IR), UV-Vis spectroscopy, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). Finally, to prove the feasibility of metalloblock copolymer design, the microphase separation and influence of the cobaltocenium on the surface polarity control are explored.
Results and discussion
To introduce the amino methacrylate-based functionalization approach and prepare cobaltocenium-containing block copolymers (BCP), a stepwise living anionic polymerization followed by block segment-selective functionalization with ethynyl cobaltocenium hexafluorophosphate was applied. In this case, anionic polymerization was used to access well-defined BCPs with different amounts of amine functionalities. The general procedure used in this work is displayed in Scheme 1. Following the synthesis of the first polystyrene (PS) block by initiation with sec-butyllithium in THF at low temperatures, diphenylethylene (DPE) was utilized for the end-capping reaction of the PS macroinitiator to prepare a sterically demanding reactive macroinitiator.25 This bulky end-group is capable of efficiently initiating the methacrylate double bonds without reacting with the methacrylate ester group, which would otherwise result in the termination of the active polymer chains.26,27 The introduction of DPE is, therefore, essential for the transition from polystyrene to methacrylate units. For the formation of the second block segment, a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of tert-butyl aminoethyl methacrylate (tBAEMA) and methyl methacrylate (MMA) monomers was chosen, whereas tBAEMA is essential for the introduction of the cobaltocenium motive and MMA provides a smooth transition from the first active polystyrene block segment to the polymethacrylate block segment. To facilitate the adaptability of the system, we focused on controlling the total cobaltocenium amount to be 5 to 40 wt% of the polymer. For this purpose, four different BCPs (P1–P4) with tBAEMA contents of 5 to 20 wt% were prepared. An overview of all synthesized and functionalized polymers is given in Table 1.
:1 molar mixture of tert-butyl aminoethyl methacrylate (tBAEMA) and methyl methacrylate (MMA) monomers was chosen, whereas tBAEMA is essential for the introduction of the cobaltocenium motive and MMA provides a smooth transition from the first active polystyrene block segment to the polymethacrylate block segment. To facilitate the adaptability of the system, we focused on controlling the total cobaltocenium amount to be 5 to 40 wt% of the polymer. For this purpose, four different BCPs (P1–P4) with tBAEMA contents of 5 to 20 wt% were prepared. An overview of all synthesized and functionalized polymers is given in Table 1.
 | ||
| Scheme 1 Synthesis of PS-b-(PtBAEMA-co-PMMA) via anionic polymerization initiated by sec-butyllithium (s-BuLi) at low temperatures in tetrahydrofuran (THF) and functionalization to PS-b-(PCoEtBAEMA-co-PMMA) with ethynyl cobaltocenium hexafluorophosphate in a THF/acetonitrile (1:1) mixture at 60 °C for two days. | ||
| Nr. | Polymer |
M
n, SECb |
Đc |
M
n, NMRd |
wt%PCoEtBAEMA(NMR)e |
wt%PCoEtBAEMA(UV-Vis)f |
|---|---|---|---|---|---|---|
| a PS molar masses were determined by SEC in THF (kg mol−1, PS standards) and used to calculate NMR values for the corresponding block copolymers. b Molar masses determined by SEC in DMF (kg mol−1, PMMA standards). c Đ values determined by SEC in DMF. d Molar masses in kg mol−1 determined by 1H NMR data of block copolymer. e Weight content of PtBAEMA or PCoEtBAEMA in respective polymer in wt% calculated by 1H NMR with theoretical values in brackets. f Weight content of PCoEtBAEMA in % calculated by UV-Vis spectroscopy in THF at 489 nm. | ||||||
| PS797a |
77.8 | 1.08 | — | — | — | |
| P1 | PS797-b-(PtBAEMA21-co-PMMA6) | 82.8 | 1.08 | 87.4 | 4.4 (5.0) | — |
| P1* | PS797-b-(PCoEtBAEMA7-co-PMMA8) | 87.8 | 4.5 (11.8) | 6.2 | ||
| PS773a |
76.6 | 1.07 | — | — | ||
| P2 | PS773-b-(PtBAEMA45-co-PMMA13) | 82.6 | 1.08 | 90.1 | 9.2 (10.0) | — |
| P2* | PS773-b-(PCoEtBAEMA31-co-PMMA12) | 98.7 | 17.1 (22.9) | 20.1 | ||
| PS773a |
74.2 | 1.08 | — | — | ||
| P3 | PS773-b-(PtBAEMA71-co-PMMA23) | 83.7 | 1.11 | 95.9 | 13.7 (15.0) | — |
| P3* | PS773-b-(PCoEtBAEMA57-co-PMMA27) | 114.4 | 27.2 (31.7) | 31.1 | ||
| PS776a |
78.4 | 1.09 | — | — | ||
| P4 | PS776-b-(PtBAEMA108-co-PMMA37) | 97.2 | 1.10 | 104.6 | 19.2 (20.0) | — |
| P4* | PS776--b-(PCoEtBAEMA80-co-PMMA30) | 127.0 | 34.0 (41.0) | 40.1 | ||
| P5 | PtBAEMA406-b-PMMA133 | 88.5 | 1.07 | — | 85.0 (85.0) | |
| P5* | PCoEtBAEMA387-b-PMMA133 | 225.5 | 92.6 (94.3) | |||
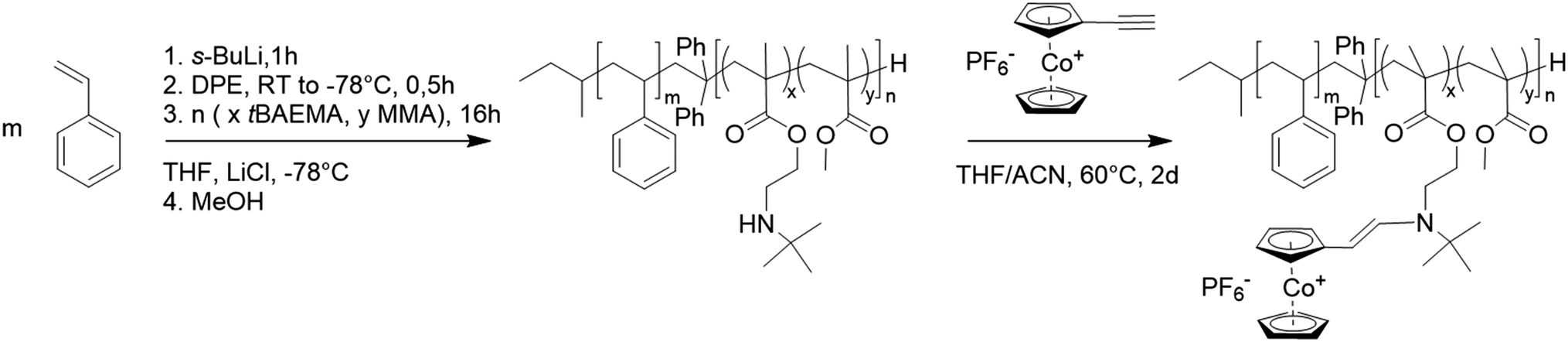
The resulting polymers were first characterized by proton nuclear magnetic resonance spectroscopy (1H NMR) and size exclusion chromatography (SEC). The initial PS block segment was characterized by SEC in THF against PS standards (Fig. 1a). The PS block was kept at a molar mass of 80 kg mol−1 to focus solely on the influence of functionalization of the second block segment. For the PS macroinitiator, SEC analysis showed a similar molecular weight for all initial blocks with polydispersity index values of around 1.03 (Table S1†).
 | ||
| Fig. 1 BCP synthesis data of PS797-b-(PtBAEMA21-co-PMMA6) (P1), PS773-b-(PtBAEMA45-co-PMMA13) (P2), PS773-b-(PtBAEMA71-co-PMMA23) (P3) and PS776-b-(PtBAEMA108-co-PMMA37) (P4); (a) size exclusion chromatography (SEC) data in tetrahydrofuran against polystyrene standards of the first polystyrene blocks; (b) size exclusion chromatography data in dimethylformamide with polymethylmethacrylate standard of P4 and respective first polystyrene block; (c) proton nuclear magnetic resonance (1H NMR) data of P4 in CDCl3 at 500 MHz; (d) attenuated total reflection infrared spectroscopy (ATR-IR) of P1 to P4 with increasing methacrylate second block in comparison. | ||
For better comparison, the PS blocks were additionally measured in dimethylformamide (DMF) against PMMA standards, with very similar results presented in Table 1. Next, the corresponding BCPs were analyzed. Due to the amine functionality in the tBAEMA monomer, SEC in DMF was conducted with LiCl (1 g L−1) against PMMA standards (Fig. 1b). Here, a shift toward higher molecular weights compared to the PS block segment while maintaining a narrow molecular weight distribution was found for all cases, indicating a successful block formation (Table 1). To verify the polymer composition, 1H NMR spectra in CDCl3 were recorded (Fig. 1c). Here, the PS integrals could be used to evaluate the polymethacrylate content and calculate the overall composition of the resulting BCPs. In all cases, the resulting weight ratios of tBAEMA were slightly below the targeted composition (Table 1) but clearly reflected the desired trend. It shall be noted that for all polymers, a water peak at 4.7 ppm could be found, which could be removed via extensive drying. However, the presence of traces of water was not relevant to the ensuing reactions. Next, resulting BCPs were characterized by attenuated total reflection infrared spectroscopy. The resulting spectra (Fig. 1d) showed the PS signals in the C–H (ν = 3100–2700 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (ν = 1400–1520 cm−1) region and increasing CO signals with higher methacrylate content. Additionally, the N–H/O–H region (ν = 3600–3200 cm−1) confirmed the increasing amine moiety content for the respective BCPs. After successful BCP formation, cobaltocenium was introduced via a quantitative and catalyst-free hydroamination reaction. Contrary to the literature, not only acetonitrile but an acetonitrile/tetrahydrofuran (1:1) mixture was used to introduce the ethynyl cobaltocenium to the amine moiety of the poly(tBAEMA) block segment. After workup, the former white polymers featured a bright red color (Fig. 2b) as a first proof of successful cobaltocenium functionalization.23 The metallopolymers were first characterized by ATR-IR spectroscopy. Fig. 2a presents spectra of P4 (PS776-b-(PtBAEMA108-co-PMMA37)) before and after functionalization P4* (PS776--b-(PCoEtBAEMA80-co-PMMA30)) with a cobaltocenium content of 40 wt%. Here, a significant difference between the bands could be found. The former signal intensity of the bands for the N–H region decreased, proving the reaction had taken place at the proposed secondary amine site of the tBAEMA group, and no residual amine was present after the reaction. Additionally, new signals at 839 cm−1 (ν(P–F)) and 559 cm−1 (ν(CP)) confirmed the introduction of the cobaltocenium motive according to the literature.22
C (ν = 1400–1520 cm−1) region and increasing CO signals with higher methacrylate content. Additionally, the N–H/O–H region (ν = 3600–3200 cm−1) confirmed the increasing amine moiety content for the respective BCPs. After successful BCP formation, cobaltocenium was introduced via a quantitative and catalyst-free hydroamination reaction. Contrary to the literature, not only acetonitrile but an acetonitrile/tetrahydrofuran (1:1) mixture was used to introduce the ethynyl cobaltocenium to the amine moiety of the poly(tBAEMA) block segment. After workup, the former white polymers featured a bright red color (Fig. 2b) as a first proof of successful cobaltocenium functionalization.23 The metallopolymers were first characterized by ATR-IR spectroscopy. Fig. 2a presents spectra of P4 (PS776-b-(PtBAEMA108-co-PMMA37)) before and after functionalization P4* (PS776--b-(PCoEtBAEMA80-co-PMMA30)) with a cobaltocenium content of 40 wt%. Here, a significant difference between the bands could be found. The former signal intensity of the bands for the N–H region decreased, proving the reaction had taken place at the proposed secondary amine site of the tBAEMA group, and no residual amine was present after the reaction. Additionally, new signals at 839 cm−1 (ν(P–F)) and 559 cm−1 (ν(CP)) confirmed the introduction of the cobaltocenium motive according to the literature.22
 | ||
| Fig. 2 Compiled data after functionalization of PS776-b-(PtBAEMA108-co-PMMA37 (P4) to PS776--b-(PCoEtBAEMA80-co-PMMA30) (P4*); (a) attenuated total reflection spectra (ATR-IR) of P4 before (top) and after (bottom) functionalization; (b) images of P4 before (left) and after (right) functionalization; (c) proton nuclear magnetic resonance (1H NMR) spectra of P4 before (top) and after (bottom) functionalization; (d) UV-Vis data of 0.05 g L−1 of P4* in tetrahydrofuran with λmax = 486 nm. | ||
It was found that signal intensities increased with increasing cobaltocenium content (Fig. S15†). However, since this method of successful conversion could only be qualitatively proven, 1H NMR spectroscopy was used to evaluate the overall composition further. Here, a strong tendency was observed to form micelles in deuterated chloroform (CDCl3), significantly influencing the signal intensities due to decreased polymer chain mobility. This micellization effect was not found for the non-functionalized polymer, indicating a significant change in solubility behavior attributed to the ionic character of the introduced cobaltocenium moieties. Transmission electron microscopy (TEM) further confirmed micelle formation in chloroform (Fig. S18†). By using a mixture of CDCl3 and deuterated acetonitrile (CD3CN), the micellation could be avoided, and 1H NMR spectra of diluted polymer chains were obtained. To exclude solvent dependencies on the signal intensities, the mixture composition also varied from 25 to 75 vol% CD3CN with CDCl3, and the BCP solutions were investigated using NMR spectroscopy. As a result, only a minimal change of ∼1% change in signal intensity was found in the comparison for these BCP solutions, proving that ACN was suitable for impeding micelle formation. Therefore, a 1:1 mixture of the respective deuterated solvents was used for all the following evaluations on BCP composition. As presented in Fig. 2c, the 1H NMR spectrum of P4 before and after functionalization with cobaltocenium showed a significant difference. The functionalized polymer P4* exhibited new resonances in the range of 5–6 ppm, which could be attributed to the cobaltocenium group.22 Furthermore, an additional shift of the tBAEMA resonance at 2.7 ppm to 3.4 ppm was found, indicating an electronic change in the environment next to the amine moiety, where the functionalization took place. Respective integrals were compared with the aromatic PS signals, and the composition and conversion were calculated (Table 1). In all cases, slightly lower repeating units were found for the functionalized CoEtBAEMA monomer. This was attributed to the difficulties introduced by measuring the 1H NMR in a solvent mixture rather than a change in composition. Nevertheless, the resulting compositions were close to the expected values. Noteworthy, there was no signal found at 2.7 ppm, which would belong to the non-functionalized PtBAEMA, indicating a complete conversion. Only for the functionalized polymer PS797-b-(PCoEtBAEMA7-co-PMMA8) (P1*) with the lowest polymethacrylate segment, a value of 4.5 wt% compared to the predicted 11.8 wt% was found. The reason could not be identified up to this point. In general, it was observed that the functionalization proceeded better with higher tBAEMA content of the used BCP precursor. Different possibilities ranging from suboptimal functionalization environment to micellation effects and the overall workup procedure could be responsible factors for insufficient functionalization. Next, the colored feature of the functionalized BCP was advantageously utilized to evaluate the cobaltocenium content further. The newly formed cobaltocenium functionality exhibited a strong light absorption with a maximum in THF at 486 nm (Fig. 2d). By using a calibration curve (Fig. S11†), the overall concentration of attached cobaltocenium per 1 mg of polymer could be measured, and the resulting weight percentage could be directly calculated. The results for the amount of cobaltocenium functionalization presented in Table 1 show a similar trend compared to the previous results. Except for the above-mentioned PS797-b-(PCoEtBAEMA7-co-PMMA8) (P1*), where the significantly lower degree of functionalization could be confirmed, all samples show almost complete conversion with values close to the expected. Attempts for the SEC measurements of the functionalized polymers were performed, but to date, no solvent/salt/column combination has been found to suppress the strong micellization effect or improve column compatibility. This problem is well-known in the literature for cobaltocenium-containing polymers.23
For a BCP, two separate glass transitions are normally found in differential scanning calorimetry (DSC) measurements when the difference of the respective glass transition temperatures (Tg) is significant. DSC was performed to demonstrate the block-like nature of the synthesized polymers. The DSC measurements from RT to 180 °C in nitrogen are presented in Fig. 3a. Here, no second Tg was found for the non-functionalized polymers, which is presumably an effect of the relatively short block length and an overlap of the Tg for the statistical PMMA copolymer segment with the Tg of PS. In contrast, for the functionalized polymers, a second Tg at 154 °C was found in addition to the expected Tg of PS at 104 °C. A signal intensity increase for the second glass transition with a higher cobaltocenium content was observed. To further prove that the found glass transition temperature stems from the cobaltocenium-containing block segment, a statistical copolymer PCoEtBAEMA387-b-PMMA133 (P5*) with the same composition as the second block segment was synthesized. Here, a Tg of 155 °C was found, further proving the successful functionalization and BCP structure containing cobaltocenium.
 | ||
| Fig. 3 Thermal analysis data of PS797-b-(PtBAEMA21-co-PMMA6) (P1), PS773-b-(PtBAEMA45-co-PMMA13) (P2), PS773-b-(PtBAEMA71-co-PMMA23) (P3), PS776-b-(PtBAEMA108-co-PMMA37) (P4) and PtBAEMA406-b-PMMA133 (P5) with respective functionalized counterparts (*): (a) differential scanning calorimetry (DSC) spectra in the range 30 to 200 °C in nitrogen of cobaltocenium-containing polymers; (b) thermo gravimetric analysis (TGA) data of synthesized polymers before (solid) and after functionalization (dotted) from 30 to 590 °C in nitrogen; (c) Calculated differential thermogravimetric analysis (cDTA) of the calcination process of P4 and P4* in nitrogen atmosphere; (d) linear correlation of residual weight and CoEtBAEMA weight percentage (wt%) in calcinated samples with incline of 0.23%/wt%. | ||
Next, thermogravimetric analysis (TGA) of the synthesized polymers was performed to quantify the cobalt content within the formed BCPs further. Due to the introduction of cobalt to the organic polymer, metal oxides, and other ceramic materials can be formed upon thermal treatment. In a nitrogen atmosphere, mainly metallic cobalt and cobalt–carbon ceramics are formed. In contrast, when synthetic air is used during ceramization, cobalt oxides are formed. In both cases, the residual weight after calcination is increased with increased cobalt content.4,8 In a standard procedure, the polymers were treated from room temperature (RT) up to 590 °C in a nitrogen atmosphere with a heating rate of 10 K min−1. From the resulting Fig. 3b, a strong influence of the introduced cobaltocenium on the overall ceramic yield could be found. Where the non-functionalized polymers resulted in a residual weight of 0.5 to 0.8 wt%, respecting cobaltocenium-containing BCPs with increasing content of 5 to 40 wt% cobaltocenium resulted in 2.2 to 10.1 wt% residual mass. This finding strongly correlated with an increasing degree of cobaltocenium-functionalization, as presented in Fig. 3d. Here, a linear increase of 0.23 wt% residual mass per 1 wt% of cobaltocenium monomer was found for the BCPs. Additionally, a single initial degradation step and a higher decomposition temperature of 380 °C were found for the cobaltocenium-containing polymers. Furthermore, at 440 °C, a second conversion up to 590 °C takes place, further reducing the residual mass. This behavior was not found for the non-functionalized BCPs, where degradation occurred in two steps, starting from 230 °C with the final step at 320 °C. To further illustrate the difference, calculated differential thermogravimetric analysis (cDTA) was performed for PS776-b-(PtBAEMA108-co-PMMA37) (P4) and PS773-b-(PCoEtBAEMA57-co-PMMA27) (P4*) (Fig. 3c), revealing an exothermic peak starting at 450 °C after the initial decomposition step for the cobaltocenium derivative. A second unique feature of block copolymers is their ability to self-assemble into microstructures. Thus, depending on the volume fraction and Flory–Huggins interaction parameter χ, different morphologies can be generally formed, ranging from spheres to cylinders to lamellae and other structures.8,28 This is also known in the literature for neutral cobaltocene polymers but has not been examined for our novel metalloblock copolymer.29 To investigate the morphological evolution of the herein-prepared metalloblock copolymers in the bulk state, polymer films were cast from a THF solution to study the self-assembly capability. As described before, the found glass transition temperature of 104 °C (PS) and 154 °C for varying cobaltocenium content already gave a hint for a microphase separation of the block segments. Moreover, it turned out that the polymers were stable above the glass transition temperature of the metallopolymer segment to a temperature of 210 °C. Therefore, an annealing temperature of 170 °C in a nitrogen atmosphere for two days was chosen. The BCP films were cut into thin slices by ultramicrotomy followed by investigation by transmission electron microscopy (TEM). Due to the high amount of electrons inside the metallopolymer-containing domains, no further contrasting technique was needed, and the metallopolymer morphologies appeared dark during TEM measurements, similar to ferrocene-containing BCP morphologies.30–32 Because of the brittleness after annealing, it was not possible to obtain thin slices from film samples of PS773-b-(PCoEtBAEMA57-co-PMMA27) (P4*). An overview of the other samples is shown in Fig. 4. Generally, the respective polymer volumes are compared. Still, due to the novelty of our system, the PCoEtBAEMA volume has not been measured so far, and therefore, weight ratios are used for comparison. Remarkably, even at a low cobaltocenium content of 5 wt%, a lamellar-like microstructure was found (Fig. 4a and b). Normally, spherical microstructures are expected at lower volume ratios. This normally unexpected behavior is commonly found in metallocene-based polymers. It is proposed that for this system, the normal phase diagram cannot be considered due to the strong interactions, but rather, a “frustrated” layer morphology is observed, not representing the thermodynamical equilibrium.25 Generally, the synthesized BCPs showed a very high tendency to microphase separation and, thus, a high interaction parameter χ. It is assumed that the ionic nature of the cobaltocenium compared to the PS leads to this effect. Similar to before, at a higher cobaltocene content of 17 wt% (Fig. 4c and d), lamellar structures are found, but the uniformity and order are drastically increased. Furthermore, a remote order of several μm can be found, indicating an excellent ordering process. The size of the darker cobaltocene domain increases to 12.9 ± 2.1 nm, which is expected compared to the counterpart with lower cobaltocenium content (∼8 nm). An even larger cobalt domain of 14.7 ± 0.7 nm is found at a cobaltocenium content of 27 wt% (Fig. 4d and f). Here, an even more detailed investigation was possible (Fig. 4f), showing a remarkably narrow cobaltocene domain compared to the polystyrene (69.2 ± 1.3 nm), further facilitating the strong segregation.
 | ||
| Fig. 4 Transmission electron microscopy (TEM) images of thin slices from THF casting solution with respecting CoEtBAEMA content in weight percent (wt%): (a and b) PS797-b-(PCoEtBAEMA7-co-PMMA8), (cand d) PS773-b-(PCoEtBAEMA31-co-PMMA12), (e and f) PS773-b-(PCoEtBAEMA57-co-PMMA27). | ||
Last, the surface polarity of the synthesized polymers was characterized. For this purpose, the water contact angle of the synthesized polymers coated on silica wavers was investigated. After spin coating from a tetrahydrofuran solution, the polymers are compared pre- and post-functionalization with cobaltocenium. In Fig. 5a, the results are shown. By increasing the overall amine amount, the polarity and hydrophilicity increased, resulting in a stepwise decrease in the difference between the plain hydrophilic waver (orange columns). After functionalization, the secondary amine is converted to the rather hydrophobic cobaltocenium hexafluorophosphate derivative. As a result, the water contact angle increased stepwise with the cobalt content. While at low cobaltocenium content, no significant change was visible, a large difference of 40° could be found for higher functionalized species. In future works, we aim to replace the hexafluorophosphate counter-ion with a hydrophilic alternative, interesting for further surface applications. Moreover, it is proposed that this method is not only limited to polymers presented in this study and discussed previously but also to similar amine-containing surfaces. By functionalizing with ethynyl cobaltocenium, not only can the surface polarity be tailored, but an ionic metallocene can also be introduced, opening ways for many other interesting applications.
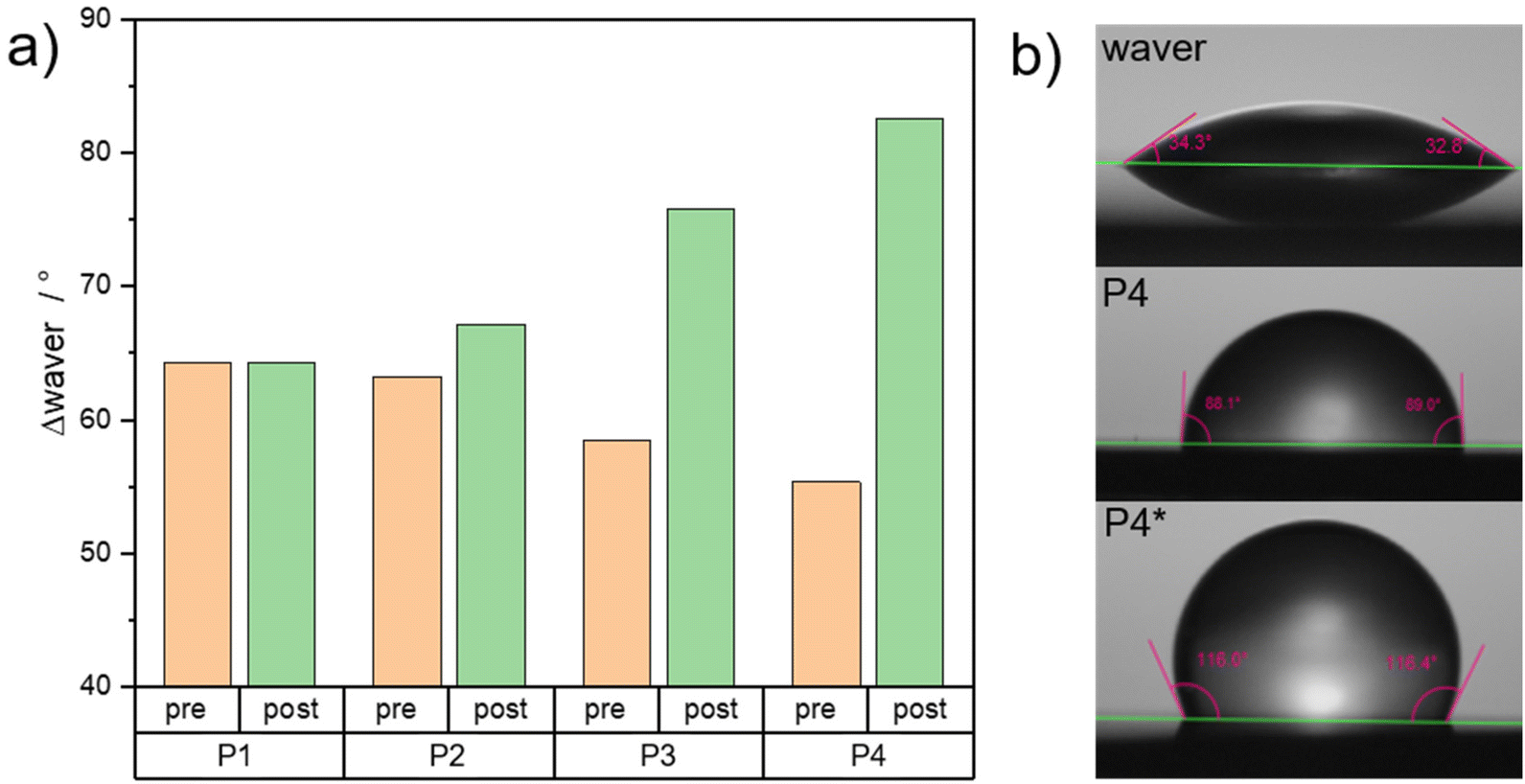 | ||
| Fig. 5 Water contact angle results of spin-coated polymer on silica wavers for synthesized polymers PS797-b-(PtBAEMA21-co-PMMA6) (P1), PS773-b-(PtBAEMA45-co-PMMA13) (P2), PS773-b-(PtBAEMA71-co-PMMA23) (P3) and PS776-b-(PtBAEMA108-co-PMMA37) (P4) with respective functionalized counterparts; (a) Compiled results for water contact angle measurements before and after functionalization of the silica wavers; (b) Exemplary photographs for water contact angle measurements for P4 and P4*. | ||
Conclusions
In this work, we present a new methacrylate-based approach to synthesize cobaltocenium polymers by introducing cobaltocenium into amine-containing polymers via a catalyst-free hydroamination reaction. For this purpose, statistical copolymers, as well as a series of block copolymers (BCP) with 5 to 20 wt% commercially available tert-butyl aminoethyl methacrylate (tBAEMA) and styrene were synthesized by living anionic polymerization. After characterization, the synthesized polymers were functionalized with ethylene-cobaltocenium hexafluorophosphate, resulting in polymers with 5 to 40 wt% cobaltocenium monomer. The overall success was demonstrated by IR spectroscopy, and the quantitative conversion could be verified by 1H NMR and UV-Vis spectroscopy. The thermal properties were investigated by TGA and DSC, where the TGA data demonstrated the linear dependence of the cobaltocenium block length on the residual masses, and a second glass transition temperature at 154 °C became visible, as determined by DSC measurements. Furthermore, the strong phase-separating properties of the synthesized BCPs were utilized to study the self-assembly behavior of the synthesized polymers in thin films for the first time. Remarkably, even at low cobaltocenium content, lamellar structures with good long-range order were found in all cases. Finally, the water contact angle of the synthesized polymers was compared, showing polarity inversion and tunability by conversion of hydrophilic amine moieties to hydrophobic cobaltocenium hexafluorophosphate. Overall, this novel approach of efficiently introducing cobaltocenium to a methacrylate-based polymer structure paves the way for a new class of metallopolymers featuring interesting architectures.Experimental
Material and methods
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance II 500 spectrometer with a 9.4 T Ultrashield Plus Magnet, a BBFO probe, and referenced by using the solvent signals.33 For processing and evaluation of the spectra, MestReNova 14.2.0 was used. Infrared (IR) spectra were collected on a BRUKER ALPHA II FT-IR setup in attenuated total reflection mode (ATR) with spectrum output in transmittance. All spectra were processed with OPUS 8.5 (SP1) software (baseline correction) and Origin2020b (normalized). Thermogravimetric analyses (TGA) were measured on a Netzsch TG 209 F1 Libra with a heating rate of 10 K min−1 and synthetic air as protective and nitrogen as a purge gas with a flow rate of 20 mL min−1 each. Differential scanning calorimetry (DSC) was carried out on a Netzsch DSC 214 Polyma in nitrogen with a heating rate of 10 K min−1. For evaluation, Netzsch Proteus Thermal Analysis 8.0.1 was used. Size exclusion chromatography (SEC) was performed by utilizing a 1260 Infinity II (Agilent Technologies) and two eluents. When using tetrahydrofuran (THF) as the mobile phase (flow rate 1 mL min−1), a PSS SECurity2 RI/UV detector on an SDV column from polymer standard service (PSS) (SDV 1000 Å, 5 μm) was used. Calibration was done using polystyrene (PS) standards from PSS. For dimethylformamide (DMF) as the mobile phase (flow rate 1 mL min−1, containing 1 g L−1 LiBr), a PSS GRAM Analytical column from PSS (103 A) was used at 60 °C. Here, calibration was carried out by using poly(methyl methacrylate) (PMMA) standards from PSS. Transmission electron microscopy (TEM) experiments were performed using a JEOL JEM-2100 electron microscope (200 kV; 0.14 nm resolution) and a Gatan Orius SC1000 camera (Binning 2; 1024 × 1024 pixels) in bright field mode. The camera was computer-aided using the Digital Micro-graph software from Gatan. For thin film preparation, samples were cut by an ultramicrotome from surface to surface at −70 °C using a Reichert ultracut device by Leica Microsystems. Ultra-thin slices with a thickness of 50 nm were prepared using a diamond trim knife cryo-trim 45 and a diamond cutting knife cryo 45° by Diatome. Cryo-temperatures were realized using a cryo chamber equipped with an RMC control unit for cooling with liquid nitrogen. Contact angle measurements were performed using a Hamilton syringe 100 μL in a syringe pump by kdScientific adjusted to 10 μL and a custom XYZ positioning table. Photographs were collected using a Nikon D54000 and digiCamControl 2.1.2. Here, Open drop 3.3.1 was used for evaluation.34 The polymers were coated on double-side polished silicon wafers (N/Phos〈100〉, d = 100 mm, thickness = 600 μm, resistance = 1–10 Ω, Si-Mat, Kaufering, Germany) with an approximate size of 1 × 1 cm. A blank wafer was used as a reference. A SPIN150 spin coater (SPS-Europe, Putten, Netherlands) was used for the formation of polymeric layers on silicon wafers. The following parameters were utilized: 3000 rpm, 500 rpm s−1, and 10 s spinning time. For this, the polymer was dissolved in tetrahydrofuran at a concentration of 10 mg mL−1.The chemicals used were purchased from Sigma Aldrich, Acros Organics, and Alfa Aesar and used as received unless otherwise stated. Ethynyl cobaltocenium hexafluorophosphate was synthesized according to the literature.35
The monomers methyl methacrylate (MMA), tert-butyl aminoethyl methacrylate (tBAEMA), and styrene were passed through basic aluminum oxide and dried over calcium hydride prior to the standard purification procedure for anionic polymerization. Tetrahydrofuran (THF) was dried using diphenyl hexyl lithium (DPHLi) and distilled via glas apparatus directly into the reaction vessel.
Polymer synthesis and functionalization
1H NMR (500 MHz, 300 K, CDCl3, δ in ppm): 0.70–2.30 (backbone + tButyl, m); 2.81 (5, s, 2H); 3.59 (6, s, 3H); 4.06 (4, s, 2H); 6.29–7.23 (1 + 2 + 3, 5H, m).
1H NMR (500 MHz, 300 K, CDCl3, δ in ppm): 0.70–2.12 (backbone, m); 1.12 (3, 9H, s) 2.80 (2, s, 2H); 3.58 (4, s, 3H); 4.04 (4, s, 2H).
P4*: 1H NMR (500 MHz, 300 K, CDCl3/CD3CN (1:1), δ in ppm): 0.40–2.20 (backbone + tButyl, m); 3.12–3.41 (4 + 6, 2H + 3H, m); 3.78 (5, 2H, s); 4.64 (8, 1H, s); 5.08 (9, 5H, s); 5.15–5.60 (10 + 11, 2H + 2H, m); 6.04–7.06 (1 + 2 + 3, 5H, m); 7.13 (7, 1H, s).
P5*: 1H NMR (500 MHz, 300 K, CD3CN, δ in ppm): 0.7–2.25 (backbone + tButyl, m); 1.32 (3, 9H, s); 3.20–3.67 (2 + 4, 2H + 3H, m); 3.99 (1, 2H, s); 4.84 (6, 1H, s); 5.29 (9, 5H, s); 5.15–5.47 (8, 2H, s); 5.55 (7, 5H, m); 7.36 (6, 1H, s).
Data availability
The data that support the findings of this study are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Blandine Boßmann for helping with SEC measurements. M.G. expresses his gratitude for the partial financial support provided by the European Union through the European Regional Development Fund (EFRE) and the State of Saarland, Germany, in the SWIMEMSYS project.References
- C. Deraedt, A. Rapakousiou, Y. Wang, L. Salmon, M. Bousquet and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 8445–8449 CrossRef CAS PubMed.
- N. Kim, W. Oh, K. N. Knust, F. Z. Galetto and X. Su, Langmuir, 2023, 39, 16685–16700 CrossRef CAS PubMed.
- H. Li, P. Yang, J. Hwang, P. Pageni, A. W. Decho and C. Tang, Biomater. Transl., 2022, 3, 162–171 Search PubMed.
- J. von Irmer, S. Vowinkel, D. Scheid, S. Schöttner, C. Rüttiger, M. Appold and M. Gallei, Polymer, 2017, 122, 303–311 CrossRef CAS.
- X. Su, H. J. Kulik, T. F. Jamison and T. A. Hatton, Adv. Funct. Mater., 2016, 26, 3394–3404 CrossRef CAS.
- X. Su, K.-J. Tan, J. Elbert, C. Rüttiger, M. Gallei, T. F. Jamison and T. A. Hatton, Energy Environ. Sci., 2017, 10, 1272–1283 RSC.
- P. Baldaguez Medina, V. Ardila Contreras, F. Hartmann, D. Schmitt, A. Klimek, J. Elbert, M. Gallei and X. Su, ACS Appl. Mater. Interfaces, 2023, 15, 22112–22122 CrossRef CAS PubMed.
- C. Rüttiger, V. Pfeifer, V. Rittscher, D. Stock, D. Scheid, S. Vowinkel, F. Roth, H. Didzoleit, B. Stühn, J. Elbert, E. Ionescu and M. Gallei, Polym. Chem., 2016, 7, 1129–1137 RSC.
- F. Liu, T. Abdiryim and X. Liu, Polymer, 2024, 305, 127170 CrossRef CAS.
- X. Liu, A. Rapakousiou, C. Deraedt, R. Ciganda, Y. Wang, J. Ruiz, H. Gu and D. Astruc, Chem. Commun., 2020, 56, 11374–11385 RSC.
- M. Gallei and J. Elbert, in Functional Metallosupramolecular Materials, ed. J. G. H. a. F. H. Schacher, The Royal Society of Chemistry, 2015, pp. 120–148, 10.1039/9781782622673-00120.
- A. S. Abd-El-Aziz, C. Agatemor and N. Etkin, Macromol. Rapid Commun., 2014, 35, 513–559 CrossRef CAS PubMed.
- J. P. Hurvois and C. Moinet, J. Organomet. Chem., 2005, 690, 1829–1839 CrossRef CAS.
- Y. Li, Z. Xu, Y. Liu, S. Jin, E. M. Fell, B. Wang, R. G. Gordon, M. J. Aziz, Z. Yang and T. Xu, ChemSusChem, 2021, 14, 745–752 CrossRef CAS PubMed.
- D. Schmitt and M. Gallei, Desalination, 2024, 583(117674) CAS.
- N. G. Connelly and W. E. Geiger, Chem. Commun., 1996, 96, 877–910 CAS.
- S. Vanicek, H. Kopacka, K. Wurst, T. Müller, C. Hassenrück, R. F. Winter and B. Bildstein, Organometallics, 2016, 35, 2101–2109 CrossRef CAS.
- S. Vanicek, H. Kopacka, K. Wurst, T. Müller, H. Schottenberger and B. Bildstein, Organometallics, 2014, 33, 1152–1156 CrossRef CAS.
- L. Zhao, X. Liu, L. Zhang, G. Qiu, D. Astruc and H. Gu, Coord. Chem. Rev., 2017, 337, 34–79 CrossRef CAS.
- L. Ren, C. G. Hardy and C. Tang, J. Am. Chem. Soc., 2010, 132, 8874–8875 CrossRef CAS PubMed.
- A. Rapakousiou, Y. Wang, J. Ruiz and D. Astruc, J. Inorg. Organomet. Polym. Mater., 2013, 24, 107–113 CrossRef.
- Y. Wang, A. Rapakousiou, C. Latouche, J. C. Daran, A. Singh, I. Ledoux-Rak, J. Ruiz, J. Y. Saillard and D. Astruc, Chem. Commun., 2013, 49, 5862–5864 RSC.
- Y. Wang, A. Rapakousiou and D. Astruc, Macromolecules, 2014, 47, 3767–3774 CrossRef CAS.
- Y. Wang, A. Rapakousiou, J. Ruiz and D. Astruc, Chem. Europ. J., 2014, 20, 11176–11186 CrossRef CAS PubMed.
- M. Gallei, B. V. Schmidt, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2009, 30, 1463–1469 CrossRef CAS PubMed.
- M. Gallei, S. Tockner, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2010, 31, 889–896 CrossRef CAS PubMed.
- M. Appold, C. Mari, C. Lederle, J. Elbert, C. Schmidt, I. Ott, B. Stühn, G. Gasser and M. Gallei, Polym. Chem., 2017, 8, 890–900 RSC.
- M. Hadadpour, J. Gwyther, I. Manners and P. J. Ragogna, Chem. Mater., 2015, 27, 3430–3440 CrossRef CAS.
- M. Hadadpour, Y. Liu, P. Chadha and P. J. Ragogna, Macromolecules, 2014, 47, 6207–6217 CrossRef CAS.
- H. N. Al-Kharusi, L. P. Wu, G. Whittell, R. Harniman and I. Manners, Polym. Chem., 2018, 9, 2951–2963 RSC.
- C. Rüttiger, H. Hübner, S. Schöttner, T. Winter, G. Cherkashinin, B. Kuttich, B. Stühn and M. Gallei, ACS Appl. Mater. Interfaces, 2018, 10, 4018–4030 CrossRef PubMed.
- C. Rüttiger, M. Appold, H. Didzoleit, A. Eils, C. Dietz, R. W. Stark, B. Stühn and M. Gallei, Macromolecules, 2016, 49, 3415–3426 CrossRef.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- E. Huang, A. Skoufis, T. Denning, J. Qi, R. Dagastine, R. Tabor and J. Berry, J. Open Source Softw., 2021, 6, 2604 CrossRef.
- T. Rittner, J. Kim, A. Haben, R. Kautenburger, O. Janka, J. Kim and M. Gallei, Chem. Eur. J., 2024 DOI:10.1002/chem.202402338.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00780h |
| This journal is © The Royal Society of Chemistry 2024 |