
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomically precise rhodium–indium carbonyl nanoclusters: synthesis, characterization, crystal structure and electron-sponge features†
Guido
Bussoli
a,
Alberto
Boccalini
a,
Marco
Bortoluzzi
 b,
Cristiana
Cesari
a,
Maria Carmela
Iapalucci
a,
Tiziana
Funaioli
c,
Giorgia
Scorzoni
a,
Stefano
Zacchini
a,
Silvia
Ruggieri
*ad and
Cristina
Femoni
*a
b,
Cristiana
Cesari
a,
Maria Carmela
Iapalucci
a,
Tiziana
Funaioli
c,
Giorgia
Scorzoni
a,
Stefano
Zacchini
a,
Silvia
Ruggieri
*ad and
Cristina
Femoni
*a
aDepartment of Industrial Chemistry “Toso Montanari”, University of Bologna, Via Gobetti 85, 40129 Bologna, Italy. E-mail: cristina.femoni@unibo.it; silvia.ruggieri@univr.it
bDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30170 Mestre, VE, Italy
cDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via Moruzzi 13, 56124 Pisa, Italy
dLaboratory of Luminescent Materials, Department of Biotechnology, University of Verona and INSTM, UdR Verona, Strada Le Grazie 15, 37134 Verona, Italy
First published on 28th August 2024
Abstract
In this paper we present the investigation of the reactivity of [Rh7(CO)16]3− with InCl3, with the aim of expanding the more general study that allowed us to obtain, among other species, the icosahedral [Rh12E(CO)27]n− (n = 4 when E = Ge or Sn; n = 3 when E = Sb or Bi) family of clusters. Indeed, the study resulted in the isolation and characterization of the analogous In-centred icosahedral [Rh12In(CO)28]3− nanocluster (1), which is isoelectronic and isostructural with the [Rh12E(CO)27]n− congeners. During the course of the reaction two more new species, namely the octahedral [Rh6(CO)15InCl3]2− (2) and the dimeric [{Rh6(CO)15InCl2}2]2− (3) have also been identified. The reaction between [Rh7(CO)16]3− and InCl3 proved to be poorly selective; nevertheless, by fine tuning some reaction parameters it was possible to drive the reaction more towards one product or the other. Alternatively, [Rh6(CO)15InCl3]2− can be more selectively prepared by reacting either [Rh5(CO)15]− or, less efficiently, [Rh6(CO)15]2− with InCl3. As for the dimeric [{Rh6(CO)15InCl2}2]2− species, this was only isolated by carrying out the reaction with [Rh7(CO)16]3− under inert atmosphere, as opposed to under CO. All clusters were characterized by IR spectroscopy and ESI-MS, and their molecular structures were fully established by single-crystal X-ray diffraction studies. The [Rh12In(CO)28]3− species was also analysed by EDS via SEM, and further investigated through in situ infrared spectroelectrochemistry and CV experiments to check its multivalence nature. Indeed, [Rh12In(CO)28]3− can reversibly undergo two monoelectronic oxidation and one bi-electronic reduction processes, behaving like an electron sponge and, thus, giving rise to the further [Rh12In(CO)28]n− derivatives (n = 1, 2 and 5). These results parallel the findings for the [Rh12E(CO)27]n− series. The geometry variations of the metal framework associated with the changes in the cluster negative charge were investigated by means of DFT calculations.
Introduction
In the last two decades, atomically-precise metal nanoclusters (APMNCs) have been increasingly emerging as nanomaterials characterized by a well-defined structure and a molecular-like behaviour,1 with interesting properties that made them suitable in various applications such as in biomedicine,2 optoelectronics,3 and catalysis.4 Furthermore, they offer the possibility of combining the specific properties of different metals within the same NCs, thus expanding their application range. For instance, in the literature there are several examples of Rh nanoparticles also containing Cu,5 Ag,6 Au,7 Ni,8 Pd,9 and Pt10 with enhanced catalytic activity.Transition-metal carbonyl clusters, whose existence dates back to the Sixties,11 rightly gained their position in the category of APMNCs, as well as in that of intermetalloid compounds,12 having a size which has increased more and more over the years yet keeping their molecular nature.13
In the case of rhodium carbonyl clusters, whose interest also lies in their application as catalyst precursors,14 the strength of the Rh–Rh and Rh–CO bonds15 particularly favours the formation of nanosized compounds.16 The combination of Rh with other transition or post-transition metals may also enhance this feature.17
In the last few years, we have investigated the synthesis and properties of several transition homo- and heterometallic nanoclusters, also with p-block elements,18 as well as Rh nanoclusters with post-transition metals. More specifically, we have fruitfully utilized the redox condensation method19 to prepare a wide number of CO-stabilized atomically precise Rh nanoclusters containing Sn,20 Ge,21 Sb,22 and Bi.23 This method consists of reacting a cluster precursor in a negative oxidation state, in this case [Rh7(CO)16]3− (ref. 24) (Rh oxidation state = −3/7), with another cluster or compound in a higher one, that is either less negative, or positive. In those specific examples, EXn halides of the desired elements were employed, where E = Sn, Ge, Sb and Bi, X = Cl, Br and n = 2–4. The resulting products of such redox reactions usually consist of higher-nuclearity heterometallic clusters (thus the name “condensation”) containing both elements but in a different oxidation state with respect to the original ones. This method relies on the reactant's reduction potentials to drive the reaction; therefore, it can be carried out at room temperature. Furthermore, by employing a carbonyl cluster precursor the CO ligands are provided without need of high pressure of carbon monoxide.25 It is important to underline that the detailed reaction mechanism that leads to the formation of large APMNCs is still hard to be determined, as they can be found in a variety of architectures whose stability is a combination of several parameters including metal–metal bond strength, ligand type, atmosphere, and electronic requirements.
Among the library of obtained Rh–E heterometallic products, which vary depending on the employed heteroatom,26 there is a particular one shared by all studied systems, that is the E-centred icosahedral [Rh12E(CO)27]n− compound. These clusters show an exceptional stability,27 most likely due to their electron count and geometry. In fact, they all possess the same metal structure, consisting of an icosahedron of Rh atoms inside which the E atom is encapsulated, and have the same number of Cluster Valence Electrons (CVEs), that is 170, perfectly in compliance with the Wade–Mingos theory for a metal cluster with such geometry.28
We decided to fill the void and explore the reaction between the [Rh7(CO)16]3− cluster precursor and InCl3. Nanoclusters composed of rhodium and indium have yet to be explored, even if the combined use of those metals in catalysis has been deeply exploited.29 When referring to carbonyl cluster compounds, to our knowledge, in the literature only [Rh6(CO)15InCp*] is reported,30 although indium clusters stabilized by various ligands have been prepared alone31 and in combination with other metals, such as Au,32 Fe,33 Ni34 and Pd.35
The investigation we carried out revealed a quite complex and poorly selective bimetallic system, nevertheless we are herein reporting the isolation of the targeted icosahedral [Rh12In(CO)28]3− nanocluster, which is isostructural and isoelectronic with the [Rh12E(CO)27]n− congeners, as well as the synthesis of the new octahedral [Rh6(CO)15InCl3]2− and the dimeric [{Rh6(CO)15InCl2}2]2− species. All clusters have been characterized by infrared (IR) spectroscopy, Electrospray Ionization Mass Spectrometry (ESI-MS), and single-crystal X-ray diffraction (SC-XRD) analyses. To unambiguously ascertain its metal ratio, [Rh12In(CO)28]3− was also analysed through energy dispersive X-ray spectrometry (EDS) via Scanning Electron Microscopy (SEM), and the results confirmed the crystallographic outcomes. Finally, the electron-sponge like properties of [Rh12In(CO)28]3− have been unravelled by electrochemical studies through in situ infrared spectroelectrochemistry (SEC) and cyclic voltammetry (CV) experiments. The geometries of the [Rh12In(CO)28]n− clusters with different negative charges (n = 1, 2, 3, 5) were investigated by means of Density Functional Theory (DFT) calculations.
Results and discussion
Synthesis
The reaction between the homometallic [Rh7(CO)16]3− cluster precursor and InCl3 was studied under different experimental conditions, more specifically by changing the stoichiometric ratio between the reactants, reaction time, solvent, counterion and atmosphere (N2, CO). However, the key reaction factors were mostly represented by the employed atmosphere and, to a lower extent, by the reactant ratio, as the other parameters did not significantly affect the reaction course.The synthetic strategy was based on the redox condensation method, as it proved to be very successful in preparing many Rh–E heterometallic nanoclusters (E = Ge, Sn, Sb, Bi). In the present case, the targeted icosahedral [Rh12In(CO)28]3− (1) was obtained by reacting [Rh7(CO)16]3− with 1 equivalent of InCl3 under CO atmosphere, whereas the same reaction under N2 only led to traces of it. When the former reaction was repeated but adding 1.5 equivalents of InCl3, the main product was [Rh6(CO)15InCl3]2− (2), even if 1 was found in the crude mixture. Further additions beyond two equivalents of InCl3 mostly resulted in the degradation of the cluster precursor, similarly to what observed for the Rh–E systems. When the reaction was carried out under N2 atmosphere we isolated the dimeric [{Rh6(CO)15InCl2}2]2− (3) but also obtained the [Rh(CO)2Cl2]− complex, both undetected under CO. The presence of the latter is in agreement with the more reducing atmosphere brought by the carbon monoxide, which seems to prevent the formation of Rh(I) complexes. In all reactions, in addition to the mentioned products, the final mixture contained homometallic Rh clusters, both under CO and N2 atmosphere, albeit of different type (vide infra). This occurrence often happens in condensation reactions because, even though the oxidant agent is, here, In3+, the oxidation of [Rh7(CO)16]3− can also be brought about by the intrinsic acidity of the metal cation, thus generating larger homometallic Rh species.36
For the sake of the readers, we collected the isolated species with their identifying numbers and molecular structures in Scheme 1.
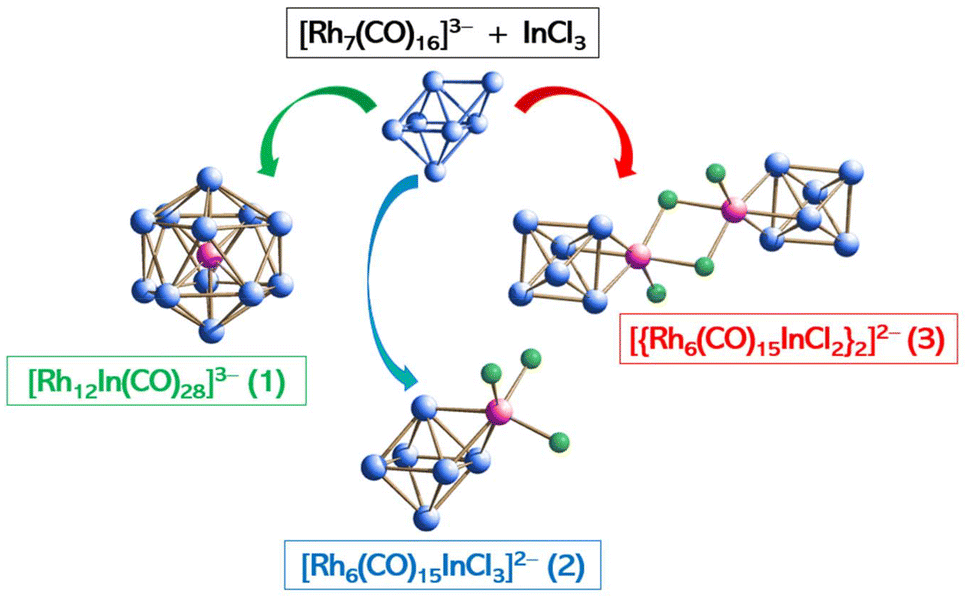 | ||
| Scheme 1 Summary of the characterized clusters with their identifying numbering sequence and molecular structure. Carbonyl ligands have been omitted for clarity. | ||
Going into more details, we initially investigated the reaction between [Rh7(CO)16]3− in its [NEt4]+ salt with 1 equivalent of InCl3, under CO atmosphere and in acetonitrile, as these conditions were those that previously allowed the isolation of the [Rh12E(CO)27]n− derivatives, monitoring its path via IR spectroscopy. We let the reaction run overnight and the day after we dried the solution under vacuum, washed the solid residue with water to eliminate the ammonium salts, then extracted the different species present in the crude mixture by exploiting different and more and more solubilizing solvents: ethanol (EtOH), tetrahydrofuran (THF), acetone. The first extraction in EtOH isolated a product whose IR was very similar to the one related to the already known [Rh6(CO)15InCp*], suggesting the obtainment of a similar species,30 but its fairly low amount made any further characterization ineffective. The subsequent extraction in THF evidenced the presence of the homometallic [Rh5(CO)15]− species, which has been known since 1980.37 The final extraction in acetone isolated a compound whose IR signals were not ascribable to any known Rh carbonyl cluster. By layering hexane onto the acetone solution, we were able to obtain crystals suitable for X-ray diffraction and to characterize the new species as the targeted [Rh12In(CO)28][NEt4]3 (1[NEt4]3). We repeated the same procedure but increasing the amount of InCl3 up to 1.5 equivalents. The EtOH extraction isolated the yet uncharacterized compound, but this time in larger amount. Therefore, we dried it and dissolved it in acetonitrile and after crystallization, achieved by layering di-isopropyl ether on top of the solution, X-ray diffraction analysis allowed us to characterize the new cluster as [Rh6(CO)15InCl3][NEt4]2·CH3CN (2[NEt4]2·CH3CN), which possesses the same metal structure as the analogue with Cp* (see the Crystal structures section), thus confirming our initial hypothesis. In acetone we extracted cluster 1; this time, no homometallic species were isolated. When the reaction was repeated with 1.8 equivalents of InCl3, the same species were obtained but in mixture with other ones, which we were not able to completely characterize, but whose nuclearity, from preliminary structural data, seems to be higher than that of 1.
While the obtaining of 1 was expected and sought after, the isolation of 2 was a surprise, as no such species had been observed before when [Rh7(CO)16]3− was reacted with Ge, Sn, Sb and Bi halides. A similar one, [Rh7Sn4Cl10(CO)14]5−, was obtained in very low yields when adding 1.3 equivalents of SnCl2 to the cluster precursor, similarly to the present case, but its metal structure is not related to the one of 2.20 Interestingly, when the reaction of [Rh7(CO)16]3− with one equivalent of InCl3 was carried out in DMF (dimethylformamide) under CO, and left under stirring for a week, little to no traces of 2 were obtained, unlike what happened in acetonitrile, and the subsequent extractions isolated [Rh5(CO)15]− and cluster 1. At present, this finding is still puzzling as both solvents possess similar characteristics. In fact, they can completely dissolve the reactants and final products, as well as possessing coordinating properties. The only difference stands in the fact that at the end of the reaction in acetonitrile, the mother solution is dried under vacuum to recover the solid mixture, while in DMF the recovery of the crude solid is carried out by precipitation with an aqueous solution of [NEt4]Cl. One possible consequence could be that, upon increase of the concentration during the drying process of the acetonitrile solution, [Rh5(CO)15]− reacts with some residual InCl3 to yield cluster 2.
We paralleled the first two above-described reactions in acetonitrile but under N2 atmosphere. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction followed a different path as it gave rise to more oxidated homometallic products, as mentioned before. In fact, the extraction of the crude reaction mixture in ethanol isolated both [Rh(CO)2Cl2]− and the already known homometallic [Rh12(CO)30]2−,38 as they present similar solubilities. Conversely, in THF and acetone we extracted the same species previously obtained under CO, that is 2 and 1, respectively. By repeating the reaction but adding 1.5 equivalents of InCl3 to the starting [Rh7(CO)16]3−, we were able to isolate the new dimeric species [{Rh6(CO)15InCl2}2][NEt4]2·2THF·2H2O (3[NEt4]2·2THF·2H2O), which was extracted in THF and crystallized by layering hexane onto the solution. The rest of the mixture contained both clusters 1 and 2 as well as traces of unknown species that we were not able to characterize.
:1 reaction followed a different path as it gave rise to more oxidated homometallic products, as mentioned before. In fact, the extraction of the crude reaction mixture in ethanol isolated both [Rh(CO)2Cl2]− and the already known homometallic [Rh12(CO)30]2−,38 as they present similar solubilities. Conversely, in THF and acetone we extracted the same species previously obtained under CO, that is 2 and 1, respectively. By repeating the reaction but adding 1.5 equivalents of InCl3 to the starting [Rh7(CO)16]3−, we were able to isolate the new dimeric species [{Rh6(CO)15InCl2}2][NEt4]2·2THF·2H2O (3[NEt4]2·2THF·2H2O), which was extracted in THF and crystallized by layering hexane onto the solution. The rest of the mixture contained both clusters 1 and 2 as well as traces of unknown species that we were not able to characterize.
In view of the obtained results, which unravelled a very complicated redox system, we tried to overcome the evident poor selectivity of the reactions by finding possible alternative routes to prepare the new clusters.
Since [Rh6(CO)15InCl3]2− could be seen as a direct addition product between the Lewis acid InCl3 and the Lewis base [Rh6(CO)15]2−, which is already known in the literature,39 we directly made the two react in a 1:1 molar ratio under CO. This, indeed, led to a more selective formation of 2. However, as the preparation of [Rh6(CO)15]2− is rather laborious when compared to that of [Rh7(CO)16]3−, this synthetic route for 2 is overall not advantageous. Moreover, despite this apparent straightforward synthesis, some unknown by-products were also detected, suggesting that the reaction of InCl3 with Rh carbonyl clusters involves numerous parallel/subsequent redox reactions, irrespective of the starting cluster. Finally, we tried to prepare 2 by reacting [Rh5(CO)15]− directly with InCl3, exploiting its recovery from the reaction carried out under CO. Indeed, by dissolving the homometallic cluster in THF, still under CO, and making it react with InCl3 in a 1.5:1 molar ratio, respectively, the [Rh6(CO)15InCl3]2− cluster was obtained alongside with some metallic residue. Finally, in terms of the larger [Rh12In(CO)28]3−, we tried to prepare it by thermal treatment of 2, exploiting the knowledge that an alternative way to obtain larger carbonyl clusters is to deplete them from their ligands, so to form unsaturated fragments that would condense in higher nuclearity species.40 As a matter of fact, by dissolving 2 in ethanol and let it under reflux for around two hours, it was possible to obtain cluster 1 in a more selective way, albeit some yet uncharacterized species were formed.
All above experimental findings let us to draw some conclusions in terms of the reaction path. First of all, the reaction between [Rh7(CO)16]3− and InCl3 is significantly different from those with Sn, Sb and Bi halides. In such cases, the most stable and straightforwardly obtainable compound was the icosahedral [Rh12E(CO)27]n−, to the point that it could serve as starting material for subsequent reactions.26 Here, conversely, the low-nuclearity [Rh6(CO)15InCl3]2− seems to be the most recurrent one. One possible explanation could be that the Lewis acidity of InCl3 drives the reaction more towards the formation of an adduct other than a proper redox condensation, that would originate larger heterometallic Rh–In clusters. Second of all, we can assert that over the course of the reaction with InCl3, [Rh7(CO)16]3− not only yields a mixture of Rh–In products, but part of it is oxidized to the homometallic [Rh12(CO)30]2−,38 whose presence was detected in the crude mixture under N2. However, [Rh12(CO)30]2− is known to quickly degrade to [Rh5(CO)15]− under CO pressure,41 hence its absence when the reaction is carried out in such atmosphere. Moreover, as we proved that [Rh5(CO)15]− reacts with InCl3 to give 2, this explains why some of it was left unreacted when working in a 1:1 molar ratio, and why we did not isolate it when working in excess of InCl3 with respect to [Rh7(CO)16]3−.
Characterization
All cluster compounds above described were characterized by IR spectroscopy and ESI-MS (see ESI†), and their molecular structure (discussed in the following section) was determined by single-crystal X-Ray diffraction analysis.Cluster 1 was also analysed with energy dispersive X-ray spectrometry (EDS) through Scanning Electron Microscopy (SEM), to confirm the Rh/In atomic ratio.
In this respect, a few crystals of 1 were mapped in various areas and the resulting mean atomic ratio between Rh and In derived from the EDS analysis was 11.9886:1.0114, being the experimental atomic percentage for Rh and In equal to 92.22 and 7.78%, respectively, corresponding to weight percentage values of 91.40 and 8.60% (see Fig. S1b and Table S1a in the ESI document†). The theoretical values in 1 for Rh and In, translated in atomic percentage, are 92.31 and 7.69%, respectively, while those for the weight percentage are 91.49 and 8.51%; therefore, the EDS results are in complete agreement with the formulated stoichiometry based on the SC-XRD analysis.
In the case of the cluster characterization with infrared spectroscopy, to the contrary of what one may expect based on the experience on carbonyl complexes, the IR spectra of high-nuclearity carbonyl clusters are rather simple. In fact, they usually consist of one intense peak related to the terminal CO ligands, approximately in the range of 2100–1950 cm−1, and a second broader and less intense peak signalling the edge- or face-bridging carbonyls (around 1850–1750 cm−1).42 All clusters presented herein show IR spectra in line with what stated above, and they are presented in the ESI document.†
As for the ESI-MS analysis, carbonyl clusters are quite fragile and need mild experimental conditions. Nevertheless, they do always present losses of CO ligands, therefore it is quite rare to observe their intact molecular ion. Actually, this occurrence is desired as essential to correctly assign the ion charge of the various peaks, because the negative charge resulting from the analysis does not always coincide with the original one. Fragmentation products may also be observed. In this specific case, clusters 1 and 2 showed enough robustness to resist under the experimental conditions, so the observed peaks are always related to the starting species. Conversely, cluster 3 broke in half, giving rise to the [Rh6(CO)15InCl2]− ion. This occurrence is not unknown in dimeric cluster species.23
Crystal structures of 1, 2 and 3
The molecular structure of [Rh12In(CO)28]3− consists of a slightly distorted icosahedron defined by the twelve Rh atoms centred by the unique In atom; the metal framework is then stabilized by 28 carbonyl ligands, of which 12 terminally bonded, 15 edge-bridging, and 1 face-bridging CO equally disordered over two positions. Fig. 1 illustrates the molecular structure of 1, pointing out the central position occupied by the In atom inside the icosahedral cavity, which perfectly mimes the geometrical array observed in the [Rh12E(CO)27]n− family of clusters (E = Sn, Ge when n = 4; E = Sb, Bi when n = 3), thus making them all isostructural as far as the metal skeleton is concerned.
 | ||
| Fig. 1 Metal skeleton (left), molecular structure (centre) and space-filling model (right) of [Rh12In(CO)28]3− (1). Rh is depicted in blue, In in magenta, C in grey, and O in red. | ||
This compound is also isoelectronic with the icosahedral [Rh12E(CO)27]n− series of clusters. The only difference is that 1 possesses one extra carbonyl ligand, but the two electrons brought by the CO are perfectly in comply with the electronic stability predicted by the Wade–Mingos theory, stating that icosahedral clusters should possess 170 CVEs. As a matter of fact, in the present case the electron counting is as follows: 12 × 9 (from the Rh atoms) + 28 × 2 (from the CO ligands) + 3 (from the In atom) + 3 (from the negative charge) = 170 CVEs. Notably, the presence of an extra CO ligand may affect the metal–metal bond lengths when comparing them with those of the other E-centred Rh-icosahedral derivatives. For instance, CO ligands in bridging position may tighten the metal–metal distances. In 1, the number of bridging carbonyls is higher than that found in the other congeners (16, vs. 14 in the RhSn, and 15 in the RhGe, RhSb and RhBi clusters). In line with this, the average Rh–Rh bond length in [Rh12In(CO)28]3− is 2.936 Å, very close to that of [Rh12Ge(CO)27]4− (2.935 Å) but shorter than that of [Rh12Sn(CO)27]4− (2.978 Å), [Rh12Sb(CO)27]3− (2.982 Å) and [Rh12Bi(CO)27]3− (3.024 Å), hence in misalignment with the increasing radius of Ge, Sn, Sb, In and Bi atoms, respectively. Conversely, the Rh–In average distance of 2.826 Å seems more in line with the increasing size of E (Table 1).
| Bond lengths (Å) | [Rh12Ge(CO)27]4− | [Rh12Sn(CO)27]4− | [Rh12Sb(CO)27]3− | [Rh12In(CO)28]3− | [Rh12Bi(CO)27]3− |
|---|---|---|---|---|---|
| Rh–Rh average | 2.935 | 2.978 | 2.982 | 2.936 | 3.024 |
| Rh–Rh longest – shortest | 3.336–2.808 | 3.349–2.810 | 3.368–2.821 | 3.012–2.773 | 3.470–2.812 |
| Rh–E average | 2.788 | 2.830 | 2.823 | 2.826 | 2.859 |
| Rh–E longest – shortest | 2.929–2.645 | 2.933–2.741 | 2.941–2.707 | 2.885–2.767 | 2.978–2.736 |
Cluster 1 possesses nanometric dimensions, being the average distance between the outermost oxygen atoms equal to around 1.0 nm.
The molecular structure of [Rh6(CO)15InCl3]2−, depicted in Fig. 2, consists of an octahedron defined by the six Rh atoms stabilized by 15 CO ligands and capped by a InCl3 moiety. Therefore, it can be seen as the Lewis base-acid addiction product of the hexa-nuclear [Rh6(CO)15]2− with the In(III) halide. Out of the 15 ligands, 12 are terminally coordinated, two per each Rh atom, and 3 are face-bridging; the [InCl3] fragment caps a fourth triangular face, so to create and alternated occupancy of the four triangular faces of the octahedron out of the eight available ones. This ligand arrangement is exactly like the one observed in the [Rh6(CO)15InCp*] parent compound or in the analogue [Rh6(CO)15GaCp*] species.43
 | ||
| Fig. 2 Metal skeleton (left), molecular structure (centre) and face-bridging ligand arrangement (right) of [Rh6(CO)15InCl3]2− (2). Rh is depicted in blue, In in magenta, Cl in green, C in grey, and O in red. | ||
The Rh–Rh distances are in line with those observed for similar clusters. Conversely, the average Rh–In bond length in 2 is 2.8447 Å, longer than that in the InCp* congener where the average value is 2.711 Å, and both values are significantly longer than that found in 1.
As for the electron count, the Wade–Mingos theory predicts 86 CVEs for an octahedral cluster. Indeed, [Rh6(CO)15InCl3]2− has 6 × 9 (from the Rh atoms) + 15 × 2 (from the CO ligands) + 2 (from the negative charge) = 86 CVEs. Note that, being InCl3 a Lewis acid, it brings no electrons to the cluster, thus behaving like a [AuPPh3]+ ligand.44
In terms of size, [Rh6(CO)15InCl3]2− shows a maximum length of around 0.9 nm, therefore it could be defined as a quasi-nanocluster.
The molecular structure of [{Rh6(CO)15InCl2}2]2− is reported in Fig. 3. It consists of two [Rh6(CO)15]2− octahedral clusters, each face-capped by a [InCl2]+ unit, joint together through two intermolecular In–Cl bonds, so to create two {In–Cl–In} bridging links between the cluster units. Hence, two chloride atoms remain terminally coordinated to just one In atom, in trans position with respect to each other. Alternatively, the molecular structure of 3 can be seen as a dimer of [Rh6(CO)15InCl2]− that in turn is the chloride-deficient analogue of 2, or, in essence, as two [Rh6(CO)15]2− clusters held together by a [In2Cl4]2+ bridging fragment. This way, it is easier to see that this cluster too is in comply with the Wade–Mingos rules for the electron count. The two octahedral Rh6 moieties are eclipsed with each other, but not the corresponding ligands. This can be seen by the misalignment of the two In atoms and their coordinated chlorides along the axis of the eclipsed view (Fig. 3(d)).
 | ||
| Fig. 3 Metal skeleton (a), space-filling model (b), molecular structure (c) and eclipsed view of the two octahedral Rh6 moieties (d) of [{Rh6(CO)15InCl2}2]2− (3). Rh is depicted in blue, In in magenta, Cl in green, C in grey, and O in red. | ||
As expected, the average intermolecular In–Cl bond distance (2.637 Å) is slightly longer than the intramolecular one (2.507(3) Å). As for the Rh–Rh bond lengths, their average value is 2.7705 Å, in line with those found in cluster 2 (2.7721 Å). Conversely, the Rh–In distances are slightly shorter (average 2.7896 Å) than those in 2 (average 2.8447 Å), possibly due to a stronger interaction between the anionic [Rh6(CO)15]2− and the cationic [InCl2]+, with respect to the interactions involving the neutral In(III) trihalide. Finally, the maximum length of cluster 3 reaches around 1.8 nm, while the size measured along the other two dimensions remains below 1 nm.
The crystallographic data derived from the single-crystal X-ray diffraction studies of clusters 1–3, as well as their detailed bond lengths, can be found in the ESI document.†
Electrochemical and spectroelectrochemical studies of [Rh12In(CO)28]3− (1)
The icosahedral [Rh12In(CO)28]3− was characterized via CV experiments and in situ infrared spectroelectrochemical experiments (IR SEC), to assess whether it possessed the same multivalence features as its Rh-E congeners (E = Ge, Sn, Sb, Bi).27The cyclic voltammetries were recorded at a glassy carbon (GC) electrode, with a scan rate = 0.1 V s−1; potentials are reported vs. Ag/AgCl, KCl sat. The in situ IR SEC experiments were conducted in an OTTLE cell,45 under CO atmosphere in CH3CN/[NnBu4][PF6] solution (0.1 mol dm−3), resolution of 4 cm−1, with a scan rate of 1 mV s−1; potentials are measured and reported vs. Ag pseudoreference electrode.
As it is shown by the cyclic and hydrodynamic voltammetries (Fig. 4), two electrochemically and chemically reversible and diffusion-controlled oxidations were detected in the anodic region, while the reduction at −1.78 V appeared in the CV as a chemically irreversible two-electrons process. The corresponding formal electrode potentials for the observed redox changes are reported in Table 2.
 | ||
| Fig. 4 Cyclic (bottom) and hydrodynamic (top) voltammetric responses recorded at a GC electrode in a CH3CN solution of [Rh12In(CO)28]3− under CO atmosphere. [NnBu4][PF6] (0.1 mol dm−3) as the supporting electrolyte. Scan rate = 0.1 V s−1. | ||
| Redox steps | |||
|---|---|---|---|
| 1−/2−a | 2−/3−a | 3−/5−b | |
| a One-electron process, as inferred from IR SEC. b Two-electron process, as inferred from hydrodynamic voltammetry at a rotating disk electrode and IR SEC. c Peak potential value for electrochemically quasi-reversible processes as inferred from IR SEC. | |||
| [Rh12In(CO)28]3− | −0.10 (75) | −0.30 (75) | −1.78c |
The in situ IR SEC experiments confirmed that [Rh12In(CO)28]3− undergoes two consecutive oxidations. This is showed by the progressive shift of the IR stretching frequencies of terminal and bridging CO ligands towards higher values, upon increasing of the potential from −0.40 to +0.50 V (Fig. 5a). Based on the relative IR absorbance maxima and the i/E profile curve, the complete IR spectra sequence was separated in two groups, each belonging to one different oxidation process (Fig. 5b and c). The observed shifts of 20 and 24 cm−1 of the stretching frequencies of terminal COs for the first and the second oxidation, respectively, were taken as an indication of their monoelectronic nature, as in line with what previously observed in the [Rh12E(CO)27]n− family of clusters and in several other large carbonyl compounds.27,46
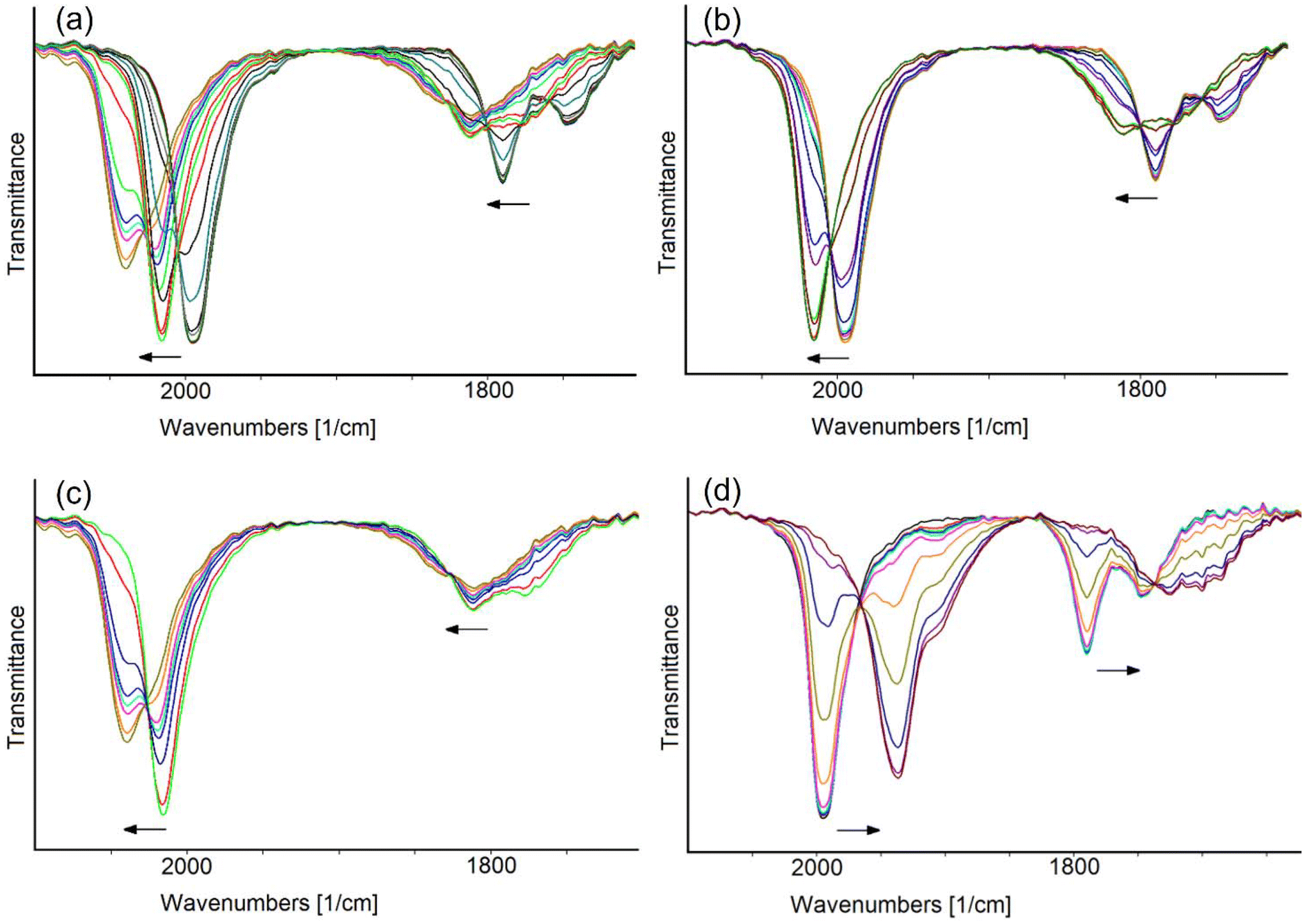 | ||
| Fig. 5 IR spectral changes of a CH3CN solution of [Rh12In(CO)28]3− recorded every 60 s in an OTTLE cell during the progressive (scan rate 1 mV s−1) increase of the potential from −0.40 to +0.50 V (a); progressive increase of the potential from −0.40 to +0.10 V (b); progressive increase of the potential from +0.10 to +0.50 V (c); progressive decrease of the potential from −0.40 to −1.85 V (d). The solvent and supporting electrolyte absorptions have been subtracted. | ||
Well-defined and withstanding isosbestic points are present in the IR spectral sequence of the first oxidation, and the complete stability of the 169-CVE [Rh12In(CO)28]2− cluster, in the experiment time-scale, was confirmed by the quantitative restoration of the IR bands of the tri-anion by reversing the direction of the potential scan after the first oxidation. The second oxidation process, however, is complicated by a slow decomposition of the 168-CVE [Rh12In(CO)28]− species, that did not allow the complete recovery of the starting cluster in the backward reduction scan (Fig. S1d†).
A shift of the CO stretching frequencies of [Rh12In(CO)28]3− towards lower values was observed when the potential was decreased from −0.40 to −1.85 V (Fig. 5d).
This was attributed to a two-electron reduction, since the frequency downshifts of the terminal CO band (58 cm−1) was around twice those observed for the oxidation steps (considering the resolution of 4 cm−1), leading to a −5 cluster charge. Well-defined isosbestic points are present in the IR spectra sequence and, remarkably, when the potential scan was reversed, the chemical reversibility of the electron transfer was observed: the IR bands of the starting cluster were, in fact, almost quantitatively restored in the backward oxidation step (Fig. S1e†).
Based on the results of the IR SEC analysis, the reduction peak at −1.78 V in the CV of Fig. 4 can be described as an electrochemically quasi-reversible step, the reduced species being oxidated back at −1.22 V.
In conclusion, the in situ IR SEC study of [Rh12In(CO)28]3− allowed us to identify four negative charge states in which cluster can exist, namely −1, −2, −3 (the initial one) and −5, with different stability, the −2 and −3 being the more stable ones. Their IR spectra are shown in Fig. 6 and the related CO stretching frequencies are reported in Table 3. Unfortunately, due to their limited stability in the time-scale of the chemical experiments, we were unable to isolate and crystallize any of the differently charged derivatives other than the tri-anion.
 | ||
| Fig. 6 Selected infrared spectra of [Rh12In(CO)28]n− as a function of the cluster charge n in CH3CN containing 0.1 mol dm−3 [NnBu4][PF6]. The absorptions of the solvent and supporting electrolyte have been subtracted. The red arrow indicates the initial spectrum. | ||
| Cluster charge n | ν tCO (cm−1) | ν bCO (cm−1) |
|---|---|---|
| n = 1 | 2040 | 1831, 1811 |
| n = 2 | 2016 | 1812, 1778(br) |
| n = 3 | 1996 | 1790, 1747 |
| n = 5 | 1938 | 1725, 1700 (br) |
Computational simulations
The changes in the IR spectra observable in Fig. 6 associated to the redox processes were simulated by means of GBSA/GFN1-xTB calculations, considering acetonitrile as continuous medium. The computed spectra are shown in Fig. S1f† with Lorenzian interpolation. Despite the limits of the tight-binding method used, the trend of the νCO stretching was confirmed. In particular, the computed νtCO wavenumbers for [Rh12In(CO)28]n− are 2101 cm−1 (n = 1), 2081 cm−1 (n = 2), 2043 cm−1 (n = 3) and 2007 cm−1 (n = 5), that can be superimposed with the experimental data using a scaling factor of around 0.97.Further calculations on the [Rh12In(CO)28]n− clusters were carried out with the hybrid-GGA PBEh-3c method. The optimized geometry of 1 is in good agreement with the experimental data, the root mean square deviation (RMSD) being 0.234 Å. No meaningful variation in the geometry of the metal cage or in the coordination mode of the carbonyl ligands was detected. Despite the fact that Continuous Shape Measures47 confirmed the icosahedral disposition of the Rh atoms surrounding the In centre (CShM = 0.32), the Atoms-In-Molecules (AIM) analysis of the Rh–In bonds indicated a lower degree of symmetry from an electronic point of view. The twelve Rh–In (3,−1) bond critical points (BCPs) are all characterized by negative energy density and positive Laplacian of the electron density, in line with Bianchi's definition of metal–metal bond.48 On considering the electron density (ρ) and the potential energy density (V) values at the BCPs, the Rh–In interactions can be divided in two groups, each composed by six bonds. The first one is characterized by an average ρ at Rh–In BCP around 0.044 a.u. (corresponding average V equal to −0.038 a.u.), and the Rh atoms involved formally describe a trigonal antiprism. The second group has average ρ and V values at BCP equal to 0.036 and −0.027 a.u., respectively. These Rh–In interactions are thus weaker with respect to the first group, and the Rh atoms involved formally cap the previously described trigonal antiprism. Fig. 7a shows the optimized geometry of the {Rh12In} core of 1 with the two different types of Rh–In bonds highlighted.
 | ||
| Fig. 7 DFT-optimized {Rh12In} core of 1. The Rh atoms are connected to the central In (magenta) to highlight the outcomes of the AIM analysis. The Rh atoms involved in the strongest interactions are blue coloured, while the others are light blue coloured (a). DFT-optimized structure of [Rh12In(CO)28]2− with spin density surface (isovalue = 0.002 a.u., green tones) (b). {Rh12In} cores of the DFT-optimized structures of [Rh12In(CO)28]n− (n = 5, 3, 2, 1) (c). Colour map: Rh, blue; In, magenta; C, grey; O, red. | ||
Geometry optimizations at PBEh-3c level were extended to [Rh12In(CO)28]n− clusters having different global charge (n = 1, 2, 5). Given the even number of electrons, [Rh12In(CO)28]− and [Rh12In(CO)28]5− were treated as diamagnetic species, while in the case of [Rh12In(CO)28]2− the odd number of electrons prompted to better investigate the possible multiplicity of the complex. After the geometry optimization of the compound with doublet multiplicity, subsequent single point calculations revealed that the quartet and sextet states are meaningfully less stable, respectively by about 42.3 and 93.2 kcal mol−1. The correct electronic configuration appears therefore related to the presence of only one unpaired electron. The spin density plot, shown in Fig. 7b, indicates that the unpaired electron density is asymmetrically delocalized on the Rh cage, with strong participation of the carbonyl ligands. On the other hand, the participation of In-centred orbitals to the spin density surface is poor.
According to the DFT outcomes, the monoelectronic oxidation of 1 should cause scarce variations in the geometry of the cluster. The RMSD deviation between [Rh12In(CO)28]2− and 1 is in fact very small, 0.079 Å. The carbonyl ligands maintain their coordination mode and the computed Rh–In average bond length is 2.841 Å, very close to the value obtained for 1 (2.826 Å). The negligible changes are in line with the experimentally observed electrochemical reversibility. Similar considerations are valid also focusing the attention on the oxidation process affording [Rh12In(CO)28]−. The DFT-optimized structure of this cluster is similar to that of 1 and [Rh12In(CO)28]2− (RMSD equal to 0.312 Å in both cases), without variations in the coordination modes of the ancillary ligands. The computed average Rh–In distance is the same obtained for [Rh12In(CO)28]2−, 2.841 Å. It must be therefore concluded that the geometry of 1 is poorly altered by the previously described reversible oxidation processes. On considering the reduction leading to the formation of [Rh12In(CO)28]5−, the DFT-optimized geometry remains comparable to that of 1 (RMSD = 0.368 Å), with the same distribution of the carbonyl ligands around the {Rh12} icosahedron. The excess of electrons (172 CVEs) causes an elongation of the average Rh–In distance, equal to 2.927 Å. Continuous Shape Measures indicated that the spatial disposition of the Rh atoms of [Rh12In(CO)28]2− and [Rh12In(CO)28]− is close to the icosahedral geometry (CShM values equal to 0.31 and 0.55, respectively), while the degree of distortion is higher in [Rh12In(CO)28]5− (CShM = 1.76). The lower reversibility of the bi-electronic reduction of 1 is thus probably related to the more relevant geometry changes occurring during the process. The {Rh12In} cores of the DFT-optimized geometries are compared in Fig. 7c.
Experimental section
General procedures
All reactions were performed using standard Schlenk technique under nitrogen (or carbon monoxide, when stated). Solvents were dried and degassed before use; THF was dehydrated with Na-benzophenone and distilled under nitrogen. Ammonium salts and InCl3 were commercial products. The [Rh7(CO)16]3− cluster precursor was prepared according to the literature.24 IR spectra were recorded on a PerkinElmer Spectrum One interferometer in CaF2 cells. Positive/negative-ion mass spectra were recorded in CH3CN solutions on a Waters Micromass ZQ 4000 using electrospray ionization. Experimental conditions: 2.56 kV ES-probe voltage, 10 V cone potential, 250 L h−1 flow of N2 spray-gas, incoming-solution flow 20 μL min−1.EDS experiments were performed on a SEM Zeiss EVO 50 equipped with EDS Detector Oxford Model INCA 350 working at 20 kV of acceleration energy.
Materials and apparatus for electrochemistry and IR SEC have been described elsewhere.49 Voltammetric and IR SEC measurements were performed on CO-saturated CH3CN/[NnBu4][PF6] 0.1 M solutions of [Rh12In(CO)28]3− (1.1–1.6 mM). Glassy-carbon electrodes, used as working electrodes for the voltammetric experiments, were polished, prior to measurements, according to the following procedure: manual rubbing with 0.3 μm Al2O3 slurry in water (eDAQ) for 2 min, then sonication in ultrapure water for 10 min, manual rubbing with 0.05 μm Al2O3 slurry in water (eDAQ) for 2 min, then sonication in ultrapure water for 10 min. After polishing, the electrodes were rinsed with acetone and air-dried. Hydrodynamic voltammetry with the renewal of the diffusion layer made use of a rotating disk electrode Metrohm 628–10, consisting of a glassy carbon surrounded with an insulating Teflon.
Computational simulations
Geometry optimizations and IR simulations were carried out with the extended tight-binding GFN1-xTB method50 in combination with the GBSA solvation model, considering acetonitrile as continuous medium.51 Further geometry optimizations were performed using the PBEh-3c method, which is a reparametrized version of PBE (with 42% HF exchange)52 that uses a split-valence double-zeta basis set (def2-mSVP) with relativistic effective core potentials on Rh and In atoms.53 The method adds three corrections considering dispersion, basis set superposition, and other basis set incompleteness effects.54 In the case of an odd number of electrons, the lack of spin contamination was verified by comparing the computed 〈S2〉 values with the theoretical ones. The software used was ORCA version 5.0.3.55 The output was elaborated using MultiWFN, version 3.8.56 Cartesian coordinates of the DFT-optimized structures are collected in a supplementary DFT coordinates for Rh12In.xyz file.Molecular structure determination
Single-crystal X-ray diffraction experiments were performed at 100 K on a Bruker Apex II diffractometer, equipped with a CMOS detector, by using Kα-Mo radiation. Data were corrected for Lorentz polarization and absorption effects (empirical absorption correction SADABS).57 Structures were solved by direct methods and refined by full-matrix least-squares based on all data using F2.58 Hydrogen atoms were fixed at calculated positions and refined by a riding model. All non-hydrogen atoms were refined with anisotropic displacement parameters, including disordered atoms. In the case of cluster 1, crystal was twinned with a twin matrix of −1 0 0 0 −1 0 0 0 1, and a BASF value of 0.028(4). The face-bridging CO ligand composed of the atoms C(6) and O(6) is disordered over two equally-populated and symmetry-related positions. Anisotropic displacement parameter restraints were applied to other CO ligands, to achieve a better structural model. Structure drawings were made with SCHAKAL99,59 crystal packings with Diamond.Synthesis of [Rh12In(CO)28]3− (1)
An acetonitrile solution of InCl3 (0.159 g, 0.718 mmol) was slowly added to a solution of [Rh7(CO)16][NEt4]3 (1.120 g, 0.718 mmol) in the same solvent, under CO atmosphere, and in a 1:1 molar ratio. After 12 hours, the resulting brown solution was dried under vacuum and the solid was washed with water (60 mL), ethanol (100 mL) and THF (30 mL). The residue was extracted with 20 mL of acetone and hexane was layered on top, to afford black crystals of 1[NEt4]3 (yield around 30% based on Rh). [Rh12In(CO)28][NEt4]3 is soluble in acetone, acetonitrile and DMF and stable, but not soluble, in water. An alternative method to obtain cluster 1 is the thermal treatment of [Rh6(CO)15InCl3][NEt4]2. In particular, an ethanol solution of 2[NEt4]2 was heated at reflux temperature for 2 hours, (until the IR signals of the starting material completely disappeared). The dark brown solid in the reaction mixture was filtered and extracted with 30 mL of acetone (yield around 40% based on Rh). The IR spectrum of 1[NEt4]3 recorded in CH3CN shows νCO absorptions at 1998(s), 1811(w), 1792(m) and 1745(wbr) cm−1. ESI-MS analysis on [Rh12In(CO)28][NEt4]3 in CH3CN shows three main groups of signals. The first one starts at 1113 m/z and it can be assigned to the {[Rh12In(CO)22][NEt4]2}2− ion, accompanied by signals due to CO loss (1099, 1085 and 1072 m/z for the {[Rh12In(CO)21-20-19][NEt4]2}2− ions, respectively). The second group starts at 1048 m/z, attributable to the {[Rh12In(CO)22][NEt4]}2− ion; again, peaks due to subsequent CO losses could be identified down to 1006 m/z ({[Rh12In(CO)19][NEt4]}2−). The last group of signals starts at 983 m/z, attributable to the [Rh12In(CO)22]2− ion, and ends at 899 m/z, characteristic of the [Rh12In(CO)16]2− ion.
Synthesis of [Rh6(CO)15InCl3]2− (2)
An acetonitrile solution of InCl3 (0.130 g, 0.588 mmol) was slowly added to a solution of [Rh7(CO)16][NEt4]3 (0.611 g, 0.390 mmol) in the same solvent, under CO atmosphere, in a 1.5:1 molar ratio, respectively. After 12 hours, the resulting brown solution was dried under vacuum and the solid was washed with water (60 mL). The residue was extracted with 30 mL of ethanol, redissolved in acetonitrile and di-isopropyl ether was layered on top of the solution, to afford black crystals of 2[NEt4]2·CH3CN (yield around 60% based on Rh). Alternatively, 2 can be prepared by adding 0.0280 g of InCl3 powder (0.127 mmol) to a THF solution containing 0.200 g of [Rh5(CO)15][NEt4] (0.188 mmol) under carbon monoxide atmosphere. After under vigorous stirring overnight, by layering the solution with hexane, crystals of 2 can be obtained. [Rh6(CO)15InCl3][NEt4]2 is soluble in ethanol, THF, acetone, acetonitrile and DMF and stable, but not soluble, in water. Its IR spectrum recorded in EtOH shows νCO absorptions at 2092(w), 2055(vs), 2017(w) and 1779(ms) cm−1. ESI-MS analysis on [Rh6(CO)15InCl3][NEt4]2 in CH3CN shows a group of signals starting at 1223 m/z, that can be assigned to the [Rh6(CO)15-14-13-12-11-10InCl2]− ions.
Synthesis of [{Rh6(CO)15InCl2}2]2− (3)
An acetonitrile solution of InCl3 (0.110 g, 0.498 mmol) was slowly added to a solution of [Rh7(CO)16][NEt4]3 (0.517 g, 0.332 mmol) in the same solvent, under N2 atmosphere, in a 1.5:1 molar ratio, respectively. After 12 hours, the resulting brown solution was dried under vacuum and the solid was washed with water (60 mL) and ethanol (80 mL). The residue was extracted with 20 mL of THF:hexane was layered on top to afford black crystals of 3[NEt4]2·2THF·2H2O (yield around 30% based on Rh). [{Rh6(CO)15InCl2}2][NEt4]2 is soluble in THF, acetone, acetonitrile and DMF and stable, but not soluble, in water. The IR spectrum of the dimeric cluster in CH3CN shows νCO absorptions at 2056(w), 2020(vs), 1870(sh) and 1835(m) cm−1. ESI-MS analysis on crystals of 3[NEt4]2·2THF·2H2O displays two groups of peaks attributable to species deriving from its fragmentation. The first one, less intense, starts at 1486 m/z and is related to the [Rh9(CO)20]− ion (and its subsequent CO losses), a decomposition product formed under the experimental conditions. The main group of peaks, starting at 1222 m/z, is attributable to the [Rh6(CO)15-14-13-12-11InCl2]− ions, of which [Rh6(CO)15InCl2]− represents the molecular cluster's halved moiety.
Conclusions
In this paper we present the synthesis and characterization of three new Rh–In carbonyl clusters, namely [Rh12In(CO)28]3− (1), [Rh6(CO)15InCl3]2− (2), and [{Rh6(CO)15InCl2}2]2− (3), that originated by the same redox-condensation reaction between the homometallic [Rh7(CO)16]3− and InCl3, in different stoichiometric ratios and under either CO or N2 atmosphere. The size of 1, being 1.0 nm, makes it eligible to be inserted in the atomically-precise nanocluster category, while 3 could be labelled as a quasi-nanocluster, as its maximum size is just under 1 nm of length. All compounds were fully characterized by IR spectroscopy, ESI-MS, SC-XRD; EDS analysis was performed on 1 to confirm the Rh:In ratio. The multivalence properties of [Rh12In(CO)28]3− were investigated via CV and IR SEC experiments; indeed, they allowed us to identify four different negative charges the nanocluster can hold, namely −1, −2, −3 and −5, reciprocally obtaining under electrochemical reduction and oxidation conditions, yet fully retaining its molecular structure. Computational calculation results confirmed that the geometry of [Rh12In(CO)28]3− is poorly altered under the oxidation process, which generates [Rh12In(CO)28]2− and [Rh12In(CO)28]−, while the excess of electrons in [Rh12In(CO)28]5− causes a slight elongation of the average Rh–In distance.
This interesting multivalent feature is shared with all the parent isostructural [Rh12E(CO)27]n−, confirming once more the extra stability imparted by the central heteroatom under oxidizing and reducing conditions, at least under the electrochemical experimental conditions. It is worth underlining that such electron-sponge behaviour is more generally presented by atomically-precise nanoclusters possessing interstitial atoms, even non-metallic, being this one of the key factors for possessing multivalence properties.60 Among the now five members of the heterometallic icosahedral [Rh12E(CO)27/28]n− family (E = In, Ge, Sn, Sb, Bi), under the same spectroelectrochemical conditions, the one that showed the smoothest multivalence behaviour is the antimony congener, [Rh12Sb(CO)27]3−, as the redox steps were all monoelectronic and fully reversible, giving rise to five well-defined spectra corresponding to cluster charges going from −2 to −6. The less redox active species appears to be [Rh12Sn(CO)27]4−, for which only one stable oxidation step was observable, making up for just two differently charged clusters; upon reduction, in fact, it was not possible to properly isolate the different congeners. As for the bismuth and germanium derivatives, [Rh12Ge(CO)27]4− and [Rh12Bi(CO)27]3−, the former showed quite a remarkable multivalence feature upon oxidation, releasing up to two electrons over two monoelectronic processes, whereas the latter only possessed one stable oxidation step. However, under reduction, in both cases only the even-electron derivatives could be isolated ([Rh12Ge(CO)27]6− and [Rh12Bi(CO)27]5−/7−, respectively), and the intermediate odd-electron cluster spectra were obtained through spectral deconvolution. This behaviour is the same observed in [Rh12In(CO)28]3− where, upon reduction, the SEC experiments were able to isolate the pentaanion [Rh12In(CO)28]5− but not the intermediate odd-electron [Rh12In(CO)28]4− derivative. Fig. 8 summarises the redox behaviour for the [Rh12E(CO)27/28]n− family of clusters (E = In, Ge, Sn, Sb, Bi) illustrating the corresponding outcomes of their SEC experiments.
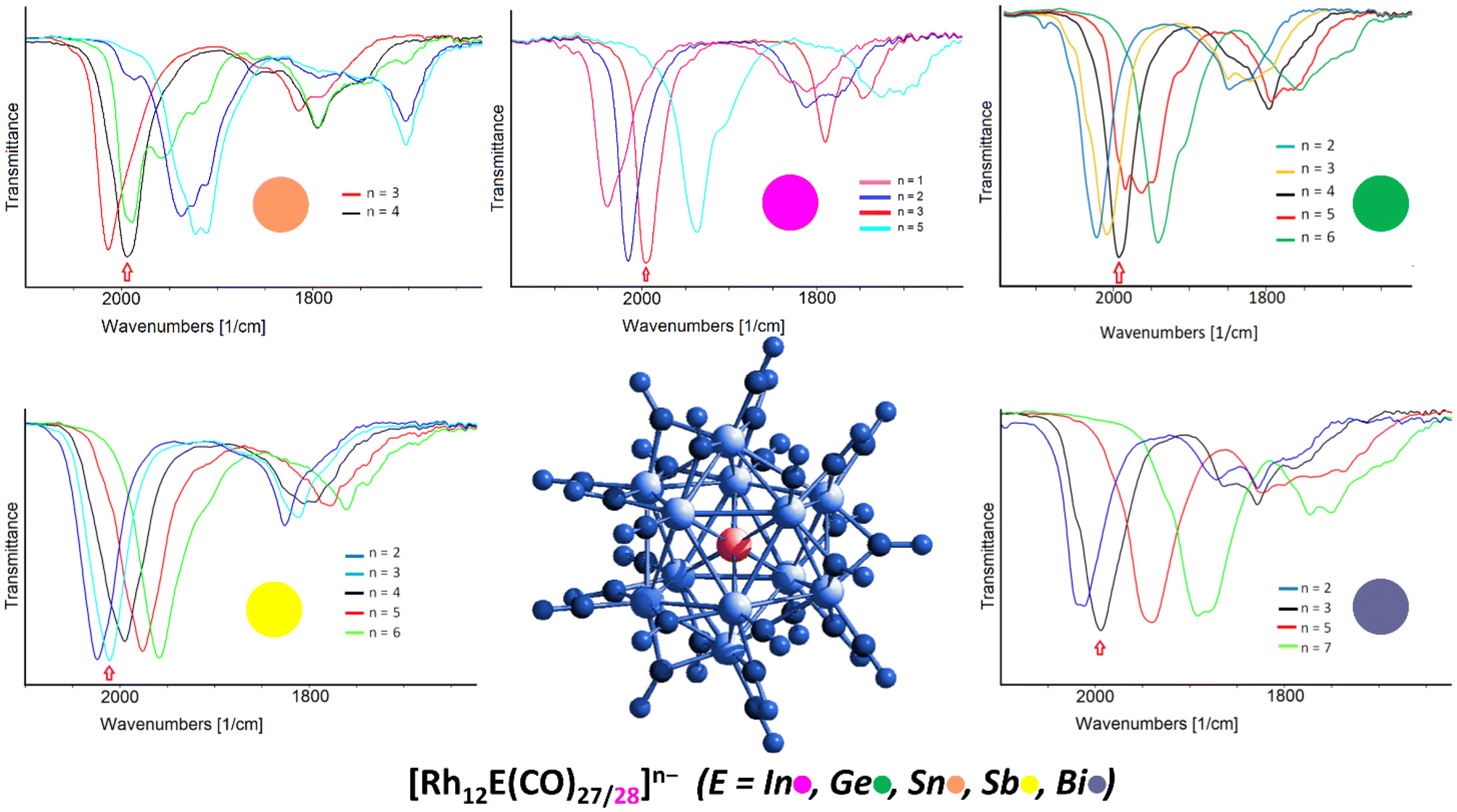 | ||
| Fig. 8 Multivalence behaviour of the [Rh12E(CO)27/28]n− family of clusters (n = 3 when E = In, Sb, Bi; n = 4 when E = Ge, Sn) as obtained by spectroelectrochemical experiments.27 | ||
The investigation of the Rh–In chemical system showed significant differences with the Rh–E ones (E = Ge, Sn, Sb, Bi), but also some similarities. More specifically, in the latter cases (with the exception of Ge27) the most straightforwardly obtainable product is [Rh12E(CO)27]n− when the reaction is conducting with a ca. 1:1 ratio with [Rh7(CO)16]3−, under CO. Conversely, [Rh12In(CO)28]3− can be isolated but in low yields, and in mixture with other products (cluster 2 and [Rh5(CO)15]−), albeit with the same stoichiometric ratio. The addition of more than 1 equivalents of EXn led to larger heterometallic Rh-E nanoclusters, for instance [Rh23Ge3(CO)41]5−, [Rh18Sn3Cl2(CO)33]4−, [Rh28−xSbx(CO)44]6− and [Rh17Bi(CO)33Bi2]4−. Likewise, in the present case we have preliminary evidence of the formation of one higher-nuclearity species, however this is still currently under investigation. Another remarkable difference is represented by the obtaining of cluster 2. We previously isolated the [Rh7Sn4Cl10(CO)14]5− species from the reaction of [Rh7(CO)16]3− with SnCl2, whose structure consists of two Rh4 tetrahedral units sharing one vertex and held together by CO ligands as well as by {SnCl3} and {SnCl2} units.20 However, that was an elusive reaction intermediate; besides, no other similar clusters had been obtained within the investigation of the Rh–Ge, Rh–Sb and Rh–Bi systems. This could be due to the Lewis acidity and to the coordination properties of InCl3, partly shared with Sn(II) chloride, that may act as a ligand as such, while the corresponding Ge(II), Ge(IV), Sb(III) and Bi(III) halides released the anions and got inserted within the Rh metal framework. Finally, and most importantly, the reaction of [Rh7(CO)16]3− with InCl3 has proven to be overall poorly selective. Surely, further investigations will be needed to shed more light on the Rh–In system. Fig. 9 shows the growth trend of heterometallic carbonyl clusters for the various Rh–E systems as a function of the added equivalents of EXn (X = Cl, Br; n = 2–4). Only some key examples are reported, together with their optimized stoichiometric ratios.
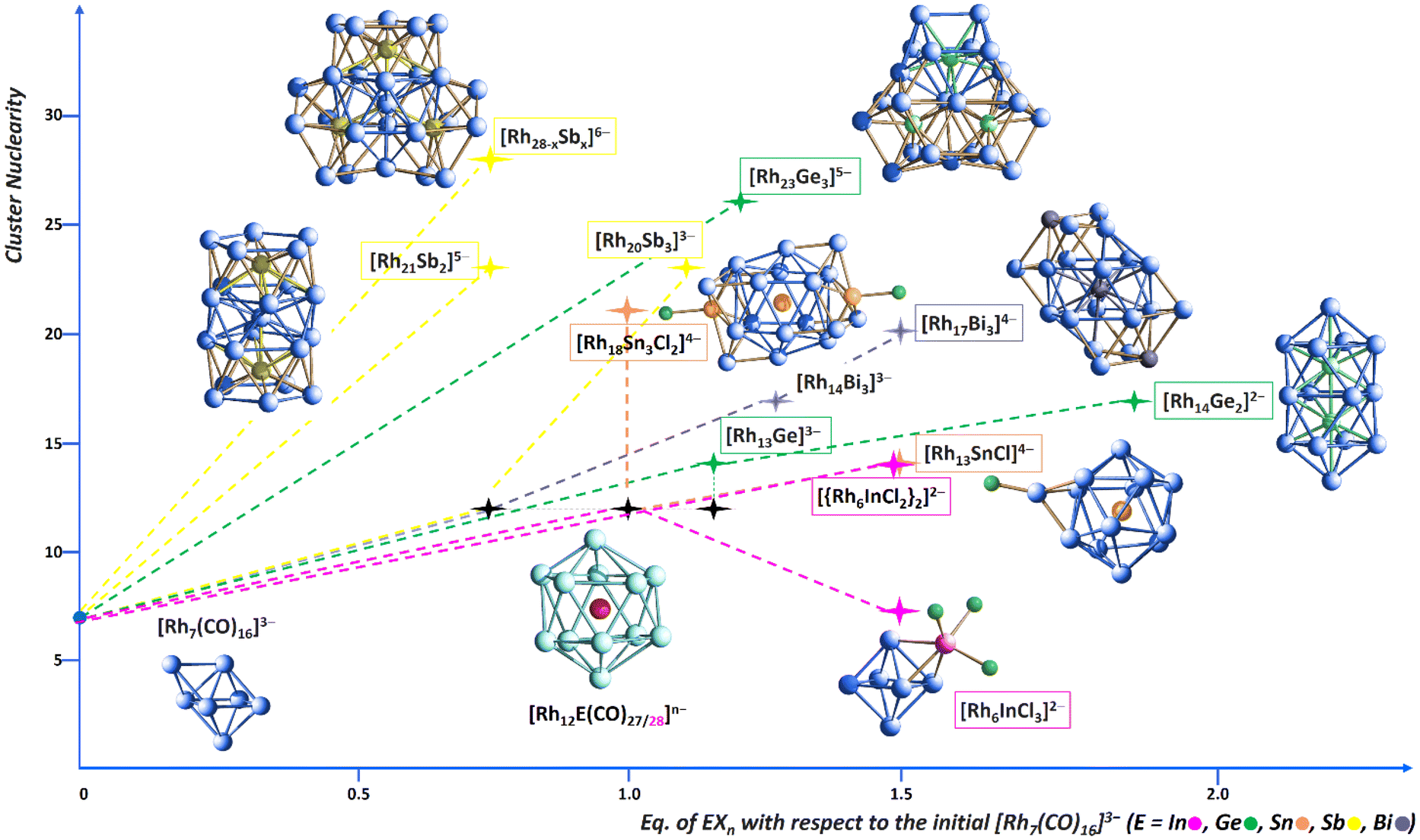 | ||
| Fig. 9 Growth trend of heterometallic carbonyl clusters for the various Rh–E systems as a function of the added equivalents of EXn (X = Cl, Br; n = 2–4). Carbonyl ligands have been omitted in the stated formulas, for sake of clarity. Rh atoms are depicted in blue, in magenta, Ge in bright green, Sn in orange, Sb in yellow, Bi in violet. | ||
Data availability
IR spectra of 1–3; ESI-MS spectra of 1–3; EDS results for 1; IR spectra related to CV experiments on 1; carbonyl regions of the simulated IR spectra for 1; crystallographic data and bond lengths of 1–3; solid state packing views of 1, 2 and 3.CCDC 2067535, 2067536, 2067538, 2067539 contain the supplementary crystallographic data for this paper.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by PRIN 2022: “COntrolled Shuttling inside artificial MOlecular tubes” (COSMO) – Code 20227JMTLE, CUP J53D23008520006 by Ministero dell'Università e della Ricerca (MUR); by National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3 - Call for tender No. 1561 of 11.10.2022 of Ministero dell’Università e della Ricerca (MUR); funded by the European Union – NextGenerationEU. Project code PE0000021, Concession Decree No. 1561 of 11.10.2022 adopted by Ministero dell’Università e della Ricerca (MUR), CUP J33C22002890007, Project title “Network 4 Energy Sustainable Transition – NEST”; and by the Università di Bologna. Authors gratefully thank Mr Luca Zuppiroli for the ESI-MS and Dr Fabrizio Tarterini for the EDS and SEM analyses.References
- S. Biswas, P. Sun, X. Xin, S. Mandal and D. Sun, Atom-Precise Cluster-Assembled Materials: Requirement and Progresses, in Atomically Precise Nanochemistry, ed. R. Jin and D. Jiang, © John Wiley & Sons Ltd, 2023, ch. 15, pp. 453–478 Search PubMed.
- A. S. Schwartz-Duval, C. J. Konopka, P. Moitra, E. A. Daza, I. Srivastava, E. V. Johnson, T. L. Kampert, S. Fayn, A. Haran, L. W. Dobrucki and D. Pan, Intratumoral generation of photothermal gold nanoparticles through a vectorized biomineralization of ionic gold, Nat. Commun., 2020, 11, 4530 CrossRef CAS PubMed; Y. Su, T. Xue, Y. Liu, J. Qi, R. Jin and Z. Lin, Luminescent metal nanoclusters for biomedical applications, Nano Res., 2019, 12, 1251–1265 CrossRef.
- C. Zhang, W.-D. Si, Z. Wang, A. Dinesh, Z.-Y. Gao, C.-H. Tung and D. Sun, Solvent-Mediated Hetero/Homo-Phase Crystallization of Copper Nanoclusters and Superatomic Kernel-Related NIR Phosphorescence, J. Am. Chem. Soc., 2024, 146, 10767–10775 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Toward Active-Site Tailoring in Heterogeneous Catalysis by Atomically Precise Metal Nanoclusters with Crystallographic Structures, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- T. Komatsu, H. Kobayashi, K. Kusada, Y. Kubota, M. Takata, T. Yamamoto, S. Matsumura, K. Sato, K. Nagaoka and H. Kitagawa, First-Principles Calculation, Synthesis, and Catalytic Properties of Rh-Cu Alloy Nanoparticles, Chem. – Eur. J., 2017, 23, 57–60 CrossRef CAS PubMed.
- Y. Jang, H. Nam, J. Song, S. Lee, J.-P. Ahn and T. Yu, Synthesis RhAg bimetallic composite nanoparticles for improved catalysts on direct synthesis of hydrogen peroxide generation, Korean J. Chem. Eng., 2019, 36, 1417–1420 CrossRef CAS; H. Min, H. Miyamura, T. Yasukawa and S. Kobayashi, Heterogeneous Rh and Rh/Ag bimetallic nanoparticle catalysts immobilized on chiral polymers, Chem. Sci., 2019, 10, 7619–7626 RSC.
- Z. Duan, J. Timoshenko, P. Kunal, S. D. House, H. Wan, K. Jarvis, C. Bonifacio, J. C. Yang, R. M. Crooks, A. I. Frenkel, S. M. Humphrey and G. Henkelman, Structural characterization of heterogeneous RhAu nanoparticles from a microwave-assisted synthesis, Nanoscale, 2018, 10, 22520–22532 RSC.
- L. Wang, L. Huang, C. Jiao, Z. Huang, F. Liang, S. Liu, Y. Wang and H. Zhang, Preparation of Rh/Ni bimetallic nanoparticles and their catalytic activities for hydrogen generation from hydrolysis of KBH4, Catalysts, 2017, 7, 125 CrossRef CAS; Z. Zhang, S. Zhang, Q. Yao, G. Feng, M. Zhu and Z.-H. Lu, Metal-organic framework immobilized RhNi alloy nanoparticles for complete H2 evolution from hydrazine borane and hydrous hydrazine, Inorg. Chem. Front., 2018, 5, 370–377 RSC.
- M. O. Sergeev, A. A. Revina, O. A. Boeva, K. N. Zhavoronkova and V. I. Zolotarevskii, Synthesis of Pd−Rh Bimetallic Nanoparticles with Different Morphologies in Reverse Micelles and Characterization of Their Catalytic Properties, Prot. Met. Phys. Chem. Surf., 2020, 56, 63–74 CrossRef CAS; V. V. Yanilkin, N. V. Nastapova, G. R. Nasretdinova, Y. N. Osin, V. G. Evtjugin, A. Y. Ziganshina and A. T. Gubaidullin, Structure and catalytic activity of ultrasmall Rh, Pd and (Rh + Pd) nanoparticles obtained by mediated electrosynthesis, New J. Chem., 2019, 43, 3931–3945 RSC.
- B. Kilbas and B. Y. Kara, Eco-friendly hydrogenation of aryl azides to primary amines on graphene oxide-decorated bimetallic Rh-Pt nanoparticles (RhPt@GO NPs), Catal. Commun., 2019, 122, 33–37 CrossRef CAS; H. Miyamura, A. Suzuki, T. Yasukawa and S. Kobayashi, Polysilane-Immobilized Rh-Pt Bimetallic Nanoparticles as Powerful Arene Hydrogenation Catalysts: Synthesis, Reactions under Batch and Flow Conditions and Reaction Mechanism, J. Am. Chem. Soc., 2018, 140, 11325–11334 CrossRef.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, Platinum Carbonyl Clusters Chemistry: Four Decades of Challenging Nanoscience, J. Cluster Sci., 2014, 25, 115–146 CrossRef CAS.
- K. Mayer, J. Weßing, T. F. Fässler and R. A. Fischer, Intermetalloid Clusters: Molecules and Solids in a Dialogue, Angew. Chem., Int. Ed., 2018, 57, 14372–14393 CrossRef CAS PubMed; T. F. Fässler and S. D. Hoffmann, Endohedral Zintl Ions: Intermetalloid Clusters, Angew. Chem., Int. Ed., 2004, 43, 6242–6247 CrossRef.
- E. G. Mednikov, M. C. Jewell and L. F. Dahl, Nanosized (μ12-Pt)Pd164−xPtx(CO)72(PPh3)20 (x ≈ 7) Containing Pt-Centered Four-Shell 165-Atom Pd-Pt Core with Unprecedented Intershell Bridging Carbonyl Ligands: Comparative Analysis of Icosahedral Shell-Growth Patterns with Geometrically Related Pd145(CO)x(PEt3)30 (x ≈ 60) Containing Capped Three-Shell Pd145 Core, J. Am. Chem. Soc., 2007, 129, 11619–11630 CrossRef CAS PubMed; C. Femoni, C. Cesari, M. C. Iapalucci, S. Ruggieri and S. Zacchini, 8.04 - Group 9 and 10 Carbonyl Clusters, in Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O'Hare, Elsevier, 2022, pp. 205–270. DOI:10.1016/B978-0-12-820206-7.00151-7 (chapter). ISBN 9780323913508 (book).
- A. Fasolini, S. Ruggieri, C. Femoni and F. Basile, Highly Active Catalysts Based on the Rh4(CO)12 Cluster Supported on Ce0.5Zr0.5 and Zr Oxides for Low-Temperature Methane Steam Reforming, Catalysts, 2019, 9, 800 CrossRef CAS.
- K. Wade, Metal-metal and metal-carbon bond energy terms for the rhodium carbonyl clusters Rh4(CO)12 and Rh6(CO)16, Inorg. Nucl. Chem. Lett., 1978, 14, 71–74 CrossRef CAS; A. K. Hughes and K. Wade, Metal–metal and metal–ligand bond strengths in metal carbonyl clusters, Coord. Chem. Rev., 2000, 197, 191–229 CrossRef.
- J. L. Vidal, R. C. Schoening and J. M. Troup, Cs9(18-crown-6)14]9+[Rh22(CO)35Hx]5−[Rh22(CO)35Hx+1]. Synthesis, Structure, and Reactivity of a Rhodium Carbonyl Cluster with Body-Centered and Cubic Arrangement of Metal Atoms, Inorg. Chem., 1981, 20, 227–238 CrossRef CAS; D. Collini, F. Fabrizi De Biani, D. S. Dolznikov, C. Femoni, M. C. Iapalucci, G. Longoni, C. Tiozzo, S. Zacchini and P. Zanello, Synthesis, Structure, and Spectroscopic Characterization of [H8−nRh22(CO)35]n− (n = 4, 5) and [H2Rh13(CO)24{Cu(MeCN)}2]− Clusters: Assessment of CV and DPV Techniques to Circumstantiate the Presence of Elusive Hydride Atoms, Inorg. Chem., 2011, 50, 2790–2798 CrossRef PubMed; D. S. Dolzhnikov, M. C. Iapalucci, G. Longoni, C. Tiozzo, S. Zacchini and C. Femoni, New High-Nuclearity Carbonyl and Carbonyl-Substituted Rhodium Clusters and Their Relationships with Polyicosahedral Carbonyl-Substituted Palladium and Gold-Thiolates, Inorg. Chem., 2012, 51, 11214–11216 CrossRef.
- A. Fumagalli, S. Martinengo, G. Ciani, N. Masciocchi and A. Sironi, Mixed Platinum-Rhodium Carbonyl Clusters. The Isolation of [Pt4Rh18(CO)35]4−, an Example of a Cherry-like Cluster with a Semiexposed Tetraplatinum Core, Inorg. Chem., 1992, 31, 336–340 CrossRef CAS.
- C. Capacci, C. Cesari, C. Femoni, M. C. Iapalucci, F. Mancini, S. Ruggieri and S. Zacchini, Structural diversity in molecular nickel phosphide carbonyl nanoclusters, Inorg. Chem., 2020, 59, 16016–16026 CrossRef CAS PubMed.
- W. O. Hieber and E. H. Schubert, Absorptionsmessungen an Carbonylferrat-Lösungen im Sichtbaren und UV-Gebiet, Z. Anorg. Allg. Chem., 1965, 338, 32–36 CrossRef CAS; P. Chini, Large Metal Carbonyl Clusters (LMCC), J. Organomet. Chem., 1980, 200, 37–61 CrossRef.
- G. Bussoli, C. Cesari, C. Femoni, M. C. Iapalucci, S. Ruggieri, C. Tiozzo and S. Zacchini, Atomically precise Rhodium Nanoclusters: Synthesis and Characterization of the Heterometallic [Rh18Sn3Cl2(CO)33]4− and [Rh7Sn4Cl10(CO)14]5− Carbonyl Compounds, Results Chem., 2022, 4, 100435 CrossRef CAS.
- A. Boccalini, P. J. Dyson, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Insertion of germanium atoms in high-nuclearity rhodium carbonyl compounds: synthesis, characterization and preliminary biological activity of the heterometallic [Rh13Ge(CO)25]3−, [Rh14Ge2(CO)30]2− and [Rh12Ge(CO)27]4− clusters, Dalton Trans., 2018, 47, 15737–15744 RSC.
- C. Femoni, I. Ciabatti, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Alternative synthetic route for the heterometallic CO-releasing [Sb@Rh12(CO)27]3− icosahedral carbonyl cluster and synthesis of its new unsaturated [Sb@Rh12(CO)24]4− and dimeric [{Sb@Rh12Sb(CO)25}2Rh(CO)2PPh3]7− derivatives, Prog. Nat. Sci.: Mater. Int., 2016, 26, 461–466 CrossRef CAS; C. Femoni, T. Funaioli, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Rh-Sb Nanoclusters: Synthesis, Structure, and Electrochemical Studies of the Atomically Precise [Rh20Sb3(CO)36]3− and [Rh21Sb2(CO)38]5− Carbonyl Compounds, Inorg. Chem., 2020, 59, 4300–4310 CrossRef PubMed.
- C. Femoni, G. Bussoli, I. Ciabatti, M. Ermini, M. Hayatifar, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Interstitial Bismuth Atoms in Icosahedral Rhodium Cages: Syntheses, Characterizations, and Molecular Structures of the [Bi@Rh12(CO)27]3−, [(Bi@Rh12(CO)26)2Bi]5−, [Bi@Rh14(CO)27Bi2]3−, and [Bi@Rh17(CO)33Bi2]4− Carbonyl Clusters, Inorg. Chem., 2017, 56, 6343–6351 CrossRef CAS PubMed.
- S. Martinengo and P. Chini, Synthesis and characterization of the [Rh6(CO)15]2− and [Rh7(CO)16]3− anions, Gazz. Chim. Ital., 1972, 102, 344–354 CAS.
- J. L. Vidal and R. C. Schoening, Rhodium Carbonyl Clusters Chemistry under High Pressures of Carbon Monoxide and Hydrogen. 5. Synthesis and Reactivity of [Rh15(CO)27]3−, Inorg. Chem., 1982, 21, 438–441 CrossRef CAS; J. L. Vidal and J. M. Troup, [Rh12Sb(CO)27]3−. An example of encapsulation of antimony by a transition metal carbonyl cluster, J. Organomet. Chem., 1981, 213, 351–363 CrossRef.
- C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, From Mononuclear Complexes to Molecular Nanoparticles: The Buildup of Atomically Precise Heterometallic Rhodium Carbonyl Nanoclusters, Acc. Chem. Res., 2018, 51, 2748–2755 CrossRef CAS PubMed; G. Bussoli, C. Cesari, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Atomically Precise Heterometallic Rhodium Nanoclusters Stabilized by Carbonyl Ligands, in Atomically Precise Nanochemistry, ed. R. Jin and D. Jiang, © John Wiley & Sons Ltd, 2023, ch. 10, pp. 309–330. DOI:10.1002/9781119788676.ch10. Print ISBN:9781119788645 Online ISBN:9781119788676.
- C. Cesari, C. Femoni, T. Funaioli, M. C. Iapalucci, I. Rivalta, S. Ruggieri and S. Zacchini, Heterometallic rhodium clusters as electron reservoirs: Chemical, electrochemical, and theoretical studies of the centered-icosahedral [Rh12E(CO)27]n− atomically precise carbonyl compounds, J. Chem. Phys., 2021, 155, 104301 CrossRef CAS PubMed.
- D. M. P. Mingos, Polyhedral skeletal electron pair approach, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- H. Guo, Z. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. Yu, Rational Design of Rhodium−Iridium Alloy Nanoparticles as Highly Active Catalysts for Acidic Oxygen Evolution, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS; T. Takanashi, M. Tamura, Y. Nakagawa and K. Tomishige, Structure of catalytically active Rh–In bimetallic phase for amination of alcohols, RSC Adv., 2014, 4, 28664–28672 RSC; J. T. Moore and C. C. Lu, Catalytic Hydrogenolysis of Aryl C−F Bonds Using a Bimetallic Rhodium−Indium Complex, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef PubMed; Y. Chen, S. Dong, X. Xu, X. Liu and X. Feng, Bimetallic Rhodium(II)/Indium(III) Relay Catalysis for Tandem Insertion/Asymmetric Claisen Rearrangement, Angew. Chem., Int. Ed., 2018, 57, 16554–16558 CrossRef PubMed; J.-B. Xi, M.-L. Ma and W. Hu, Rh2(OAc)4 and InCl3 co-catalyzed diastereoselective trapping of carbamate ammonium ylides with aldehydes for the synthesis of β-hydroxyl-α-amino acid derivatives, Tetrahedron, 2016, 72, 579–583 CrossRef.
- E. V. Grachova and G. Linti, Reactivity of InCp* Towards Transition Metal Carbonyl Clusters: Synthesis and Structural Characterization of the Rh6(CO)16−x(InCp*)x Mixed-Metal Cluster Compounds, x = 1–2, Eur. J. Inorg. Chem., 2007, 3561–3564 CrossRef CAS.
- A. V. Protchenko, J. Urbano, J. A. B. Abdalla, J. Campos, D. Vidovic, A. D. Schwarz, M. P. Blake, P. Mountford, C. Jones and S. Aldridge, Electronic Delocalization in Two and Three Dimensions: Differential Aggregation in Indium “Metalloid” Clusters, Angew. Chem., Int. Ed., 2017, 56, 15098–15102 CrossRef CAS PubMed.
- F. P. Gabbai, S.-C. Chung, A. Schier, S. Krüger, N. Rösch and H. Schmidbaur, A Novel Anionic Gold-Indium Cluster Compound: Synthesis and Molecular and Electronic Structure, Inorg. Chem., 1997, 36, 5699–5705 CrossRef PubMed.
- C.-C. Lin, G. Kong, H. Cho and B. R. Whittlesey, Preparation of [K][In{Fe2(CO)8}2](THF) and Fe2{μ-InCl(THF)}2(CO)8 (THF = Tetrahydrofuran), Inorg. Chem., 1993, 32, 2705–2710 CrossRef CAS; W. Uhl, S. U. Keimling, M. Pohlmann, S. Pohl, W. Saak, W. Hiller and M. Neumayer, Organometallic Iron Indium Derivatives Prepared by the Reaction of Carbonyliron Compounds with the Alkylindium(I) Derivative In4[C(SiMe3)3]4, Inorg. Chem., 1997, 36, 5478–5482 CrossRef.
- F. Demartin, M. C. Iapalucci and G. Longoni, Synthesis and Characterization of Novel Types of Adducts of Nickel Carbonyl Clusters with Indium Halides: X-ray Structures of [NEt4]3[Ni6(μ3-InBr3)(η2-μ6-In2Br5)(CO)11]·Me2CO, [NEt4]4[Ni6(η2-μ6-In2Br5)2(CO)10]·Me2CO, and [NEt4]4[Ni12(μ6-In)(η2-μ6-In2Br4OH)2(CO)22], Inorg. Chem., 1993, 32, 5536–5543 CrossRef CAS.
- T. Steinke, C. Gemel, M. Winter and R. A. Fischer, The Clusters [Ma(ECp*)b] (M=Pd, Pt; E=Al, Ga, In): Structures, Fluxionality, and Ligand Exchange Reactions, Chem. – Eur. J., 2005, 11, 1636–1646 CrossRef CAS.
- D. Collini, F. Fabrizi De Biani, S. Fedi, C. Femoni, F. Kaswalder, M. C. Iapalucci, G. Longoni, C. Tiozzo, S. Zacchini and P. Zanello, Synthesis and Electrochemistry of New Rh-Centered and Conjuncto Rhodium Carbonyl Clusters. X-ray Structure of [NEt4]3[Rh15(CO)27], [NEt4]3[Rh15(CO)25(MeCN)2]·2MeCN, and [NEt4]3[Rh17(CO)37], Inorg. Chem., 2007, 46, 7971–7981 CrossRef CAS PubMed.
- A. Fumagalli, T. F. Koetzle, F. Takusagawa, P. Chini, S. Martinengo and B. T. Heaton, Isolation and characterization of penta-.mu.2-carbonyl-dodecarbonylpentarhodate(1-): a key product in rhodium carbonyl chemistry, J. Am. Chem. Soc., 1980, 102, 1740–1742 CrossRef CAS.
- P. Chini and S. Martinengo, Synthesis of rhodium carbonyl compounds at atmospheric pressure. II. Synthesis and properties of the derivatives of the [Rh12(CO)30]2− anion, Inorg. Chim. Acta, 1969, 3, 299–302 CrossRef CAS.
- S. Martinengo, A. Fumagalli and P. Chini, Rhodium carbonyl cluster chemistry. synthesis and chemical characterization of the anion [Rh6(CO)14]4−, J. Organomet. Chem., 1985, 284, 275–279 CrossRef CAS.
- P. R. Raithby, The growth of higher nuclearity carbonyl clusters of ruthenium and osmium, J. Organomet. Chem., 2024, 1005, 122979 CrossRef CAS; B. Berti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Functionalization, Modification, and Transformation of Platinum Chini Clusters, Eur. J. Inorg. Chem., 2018, 3285–3296 CrossRef.
- A. Fumagalli, T. F. Koetzle, F. Takusagawa, P. Chini, S. Martinengo and B. T. Heaton, J. Am. Chem. Soc., 1980, 102, 1740–1742 CrossRef CAS.
- S. F. A. Kettle, E. Diana, R. Rossetti and P. L. Stanghellini, Why Are the Terminal ν(CO) Infrared Spectra of Metal Cluster Carbonyls So Often So Simple?, J. Am. Chem. Soc., 1997, 119, 8228–8231 CrossRef CAS.
- E. V. Grachova, P. Jutzi, B. Neumann, L. O. Schebaum, H. G. Stammler and S. P. Tunik, Unusual selective substitution of triply bridging carbonylligands for GaCp* in Rh6(CO)16. Synthesis and structural characterization of the Rh6(μ3-CO)4−x(μ3-GaCp*)x(CO)12 clusters, x = 1–4, J. Chem. Soc., Dalton Trans., 2002, 302–304 RSC.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, The Role of Gold in Transition Metal Carbonyl Clusters, Coord. Chem. Rev., 2018, 355, 27–38 CrossRef CAS.
- M. Krejčik, M. Daněk and F. Hartl, J. Electroanal. Chem. Interfacial Electrochem., 1991, 317, 179 CrossRef.
- J. D. Roth, G. J. Lewis, L. K. Safford, X. Jiang, L. F. Dahl and M. J. Weaver, J. Am. Chem. Soc., 1992, 114, 6159–6169 CrossRef CAS.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Shape maps and polyhedral interconversion paths in transition metal chemistry, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Topological analysis of the metal-metal bond: A tutorial review, Coord. Chem. Rev., 2017, 345, 150–162 CrossRef CAS.
- B. Berti, C. Cesari, C. Femoni, T. Funaioli, M. C. Iapalucci and S. Zacchini, Redox active Ni-Pd carbonyl alloy nanoclusters: syntheses, molecular structures and electrochemistry of [Ni22−xPd20+x(CO)48]6− (x = 0.62), [Ni29−xPd6+x(CO)42]6− (x = 0.09) and [Ni29+xPd6−x(CO)42]6− (x = 0.27), Dalton Trans., 2020, 49, 5513–5522 RSC.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e01493 Search PubMed; S. Grimme, C. Bannwarth and P. Shushkov, J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. A. Hansen, Consistent structures and interactions by density functional theory with small atomic orbital basis sets, J. Chem. Phys., 2015, 143, 054107 CrossRef CAS; A. A. Otlyotov, A. D. Moshchenkov, L. Cavallo and Y. Minenkov, 16OSTM10: a new open-shell transition metal conformational energy database to challenge contemporary semiempirical and force field methods, Phys. Chem. Chem. Phys., 2022, 24, 17314–17322 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–330568 RSC; F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- H. Kruse and S. Grimme, A geometrical correction for the interand intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems, J. Chem. Phys., 2012, 136, 154101 CrossRef CAS PubMed; S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef; S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- F. Neese, Software update: The ORCA program system - Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1616 Search PubMed.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for empirical absorption correction, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELX 2014/7, Program for crystal structure determination, University of Göttingen, Germany, 2014 Search PubMed.
- E. Keller, SCHAKAL99, University of Freiburg, Germany, 1999 Search PubMed.
- C. Femoni, M. C. Iapalucci, F. Kaswalder, G. Longoni and S. Zacchini, The possible role of metal carbonyl clusters in nanoscience and nanotechnologies, Coord. Chem. Rev., 2006, 250, 1580–1604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of 1–3; ESI-MS spectra of 1–3; EDS results for 1; IR spectra related to CV experiments on 1; carbonyl regions of the simulated IR spectra for 1; crystallographic data and bond lengths of 1–3; solid state packing views of 1, 2 and 3. CCDC 2067535, 2067536, 2067538 and 2067539. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr02922d |
| This journal is © The Royal Society of Chemistry 2024 |