
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of EGFRwt/EGFRvIII homo- and hetero-dimers in glioblastoma cells as detected by single molecule localization microscopy†
Kevin
Jahnke‡§
a,
Nina
Struve‡
*b,
Daniel
Hofmann
a,
Martin Julius
Gote¶
a,
Margund
Bach
a,
Malte
Kriegs‡
b and
Michael
Hausmann‡
 *a
*a
aKirchhoff-Institute for Physics, Heidelberg University, Im Neuenheimer Feld 227, 69120 Heidelberg, Germany. E-mail: hausmann@kip.uni-heidelberg.de
bDepartment of Radiotherapy & Radiation Oncology, University Medical Center Hamburg – Eppendorf, Martinistr. 52, 20246 Hamburg, Germany. E-mail: ni.struve@uke.de
First published on 24th July 2024
Abstract
Super-resolution microscopy has been used to show the formation of receptor clusters and adapted lipid organization of cell membranes for many members of the ErbB receptor family. The clustering behaviour depends on the receptor size and shape, possibly ligand binding or expression activity. Using single molecule localization microscopy (SMLM), we also showed this typical clustering for the epidermal growth factor receptor variant III (EGFRvIII) in glioblastoma multiforme (GBM) cells. EGFRvIII is co-expressed with the wild type (EGFRwt) and both receptors are assumed to preferentially form hetero-dimers leading to transactivation and elevated oncogenic EGFR-signalling in GBM cells. Here, we analysed EGFRvIII and EGFRwt co-localization using our already described model system of the glioblastoma cell line DKMG, displaying endogenous EGFRvIII expression. Using EGFRvIII and EGFRwt specific antibodies, EGFR localization and their potential for dimerization in a given membrane cluster were analysed by dual colour SMLM supported by novel approaches of mathematic evaluations including Ripley statistics, persistent homology and similarity algorithms. Surprisingly, cluster analysis, Ripley point-to-point distance statistics for cluster geometry and persistent homology comparing cluster topology, revealed that both EGFRvIII and EGFRwt do primarily not form hetero-dimers but the results support the hypothesis that they tend to form homo-dimers. The ratio of homo-dimers obtained by this calculation was significantly higher (>5σ, standard deviation) than expected from randomly arranged points. In comparison, hetero-dimer formation was only slightly increased. We confirmed these data by immunoprecipitation, which show no co-precipitation of EGFRvIII and EGFRwt. Furthermore, we showed that the topology of the clusters was more similar among the same type than among the different types of receptors. Taken together, these data indicate that EGFRvIII does induce oncogenic signalling by homo-dimerisation and not preferentially by hetero-dimer formation with EGFRwt. These data offer a new perspective on EGFRvIII signalling which will lead to a better understanding of this tumour associated receptor variant in GBM.
Introduction
The ErbB family (ErbB1, ErbB2, ErbB3, ErbB4) of receptor tyrosine kinases (RTK) is a prominent group of plasma membrane receptors, which is highly involved in cell growth and development.1 Several members are also well known to be involved in the development and progression of cancer.2,3 For the activation of ErbB-receptors, dimerization has to be induced by specific ligands like for instance epidermal growth factor (EGF) or transforming growth factor α (TGFα) for ErbB1. ErbB receptors form dimers with the same receptor (homo-dimerization), e.g., ErbB1 with ErbB1, or with a different partner of the ErbB family (hetero-dimerization), e.g., ErbB3 (=Her3) with ErbB2 (=Her2).4 The likelihood of dimerization depends on the ErbB receptor shape and spatial organisation of the receptors in the cellular bi-lipid membrane. Therefore, not only the level of expression of ErbB receptors is of high interest in research and diagnosis but also their spatial organisation and interactions leading to dimerization in the membrane.4,5 Such investigations contribute to a better understanding of mechanisms behind cancer genesis and could help to further improve anti-cancer therapy strategies.6 In particular, enhanced expression of the epidermal growth factor receptor (EGFR = ErbB1) has been accounted for oncogene signalling especially in glioblastomas.7 This has increased interest and research activities on EGFR and its variants in these tumour entities.8–10Glioblastoma multiforme (GBM), a high-grade aggressive glioma with an estimated 5-year survival rate of less than 10%,11,12 is a common and one of the most malignant brain cancers.13 Despite extensive research and many approaches for aggressive treatment,14–16 GBM remains one of the cancers with mostly poor prediction and outcome for the patient, i.e., the majority of patients is dying within 15 months after their diagnosis.17 GBM is treatment-resistant and highly invasive which is believed to be the reason for the high recurrence rates observed after surgery, radiation treatment and chemotherapy.18
About 50% of GBM cases show increased wild type EGFR (EGFRwt) expression19 due to egfr gene amplification which often is accompanied by gene rearrangements. The most common gene mutation leads to the epidermal growth factor receptor variant III (EGFRvIII20 or de2-7EGFR21) frequently expressed in GBM. EGFRvIII lacks the exons 2–7 resulting in a loss of the EGFR's N-terminal extra-cellular domain which is usually involved in ligand binding and dimerization. EGFRvIII has been shown to be an important factor involved in many different cellular mechanisms influencing tumour genesis and treatment response,22 which is, however, under controversial debate. For example, EGFRvIII mediates the sensitivity towards the chemotherapeutic temozolomide by upregulating the DNA mismatch repair which is associated with better survival of EGFRvIII-positive patients.23 While this is true for patients with methylguanine-DNA methyltransferase (MGMT) promotor methylation, the opposite could also be found for other entities.24 EGFRvIII induces replication stress and genomic instability which can lead to increased sensitivity towards irinotecan.25 However, EGFRvIII does not affect radio-sensitivity with and without anti-EGFR treatment.26,27 These studies show on the one hand that EGFRvIII may be a promising biomarker for future GBM therapy but on the other the need for mechanistic insights to better understand EGFR biology and the patients’ response towards GBM therapy.
In contrast to EGFRwt, which is activated by ligand binding and subsequent dimerization, EGFRvIII is thought to be constitutively active because of the loss of EGFR's extra-cellular domain which is usually involved in ligand binding and dimerization.28 The precise mechanisms of this activation and regulated signalling pathways are not completely understood; however, dimerization-independent activity and oncogenic signalling relationships between EGFR and EGFRvIII have been described.29 A selective pressure for EGFRwt expression in EGFRvIII positive cells may be suggested29 which may be due to physical binding of EGFRwt and EGFRvIII.21 Although EGFRvIII is known to be ligand-independent, the addition of EGF ligands resulted in an increased phosphorylation of EGFRvIII on double positive cells suggesting a EGFRwt ligand-dependent cross-phosphorylation of EGFRvIII.29 If EGFRvIII expression can only be observed in the presence of EGFRwt, this raises the intriguing questions as to whether EGFRvIII promotes hetero-dimerization and how these hetero-dimers between a fully intact and a deleted receptor variant should be formed.
EGFRwt is known to be localized in membranous structures such as coated pits.30 In addition, super-resolution imaging has revealed the clustering of EGFRwt.31 However, less is known about the precise localization and arrangement of EGFRvIII. By means of super-resolution single molecule localization microscopy (SMLM) we have shown that EGFRvIII undergoes cluster formation.5 The precise functions and consequences of these spatial arrangements for the dimerization potential, however, have remained elusive. While homo-dimerization was suggested as the predominant mechanism of EGFRvIII in glioblastoma,32 the potential of hetero-dimerization10 or even both possibilities33 could not be excluded. This has motivated further detailed studies of dimerization-correlated receptor arrangements using super-resolution (SMLM) as used in previous studies.4,5
SMLM has been established as an approach for super-resolution light microscopy34 that circumvents the Abbe-Rayleigh boundary conditions of optical resolution35 and enables precise single molecule localization.36 The precision is on the order of a few ten nanometres and even about 10 nm (ref. 36) in 3D conserved cells whereby standard objective lenses and optics are used.37,38 The fundamental concept of localization microscopy is based on optical isolation by different spectral signatures, e.g., by using fluorophores that can be switched between two different spectral states39–41 to achieve a temporal isolation and thus a spatial separation of single signals. This allows the determination of signal positions (spatial coordinates) and their spatial distances even if they are adjacent below the Abbe-Rayleigh resolution limit. All acquired positions of individual fluorescent molecules can then be merged into a coordinate matrix (called “ortematrix”).
This resulting coordinate matrix can be subjected to further data processing actions and especially mathematical algorithms for point pattern analysis without further typical procedures for image processing for quality improvements of the visual image.42 Distances of points can be calculated from the coordinates as Euclidian distances between the coordinates scaled with object size units obtained from the pixel size of the camera and the magnification used. Interpretation tools as basis for biological investigations are algorithms derived from Ripley's statistics based on the point-to-point distance frequency distributions and topological calculations obtained by persistence homology and appropriate similarity measures (see42 and citations therein).
SMLM and other super-resolution microscopy techniques have been successfully applied to study membranes and receptor cluster formation (see for instance4,5,31,43) or chromatin arrangements to distinguish cell types according to their heterochromatin or euchromatin networks, e.g.44 or to study radiation induced DNA damaging and repair, e.g.38,45 Here, we apply SMLM to study the probability for the formation of hetero- and homo-dimers of EGFRwt and EGFRvIII in a well-established GBM cell model.5
Results and discussion
Expression of EGFRwt and EGFRvIII and antibody specificity
The supramolecular organization of EGFRwt and EGFRvIII on membranes was studied with respect to dimerization processes. Two iso-genetic GBM sublines, DKMGvIII − (EGFRvIII − subline) and DKMGvIII + (EGFRvIII + subline), were used.5 Western blot analysis of these cells did not reveal EGFRvIII expression in the EGFRvIII − subline while strong expression in the EGFRvIII + subline was observed. Since we used an antibody which detects both, the wt and the vIII form of EGFR, also an association of EGFRvIII expression with an increase in the expression of EGFRwt was detected (Fig. 1A). Quantifying EGFRvIII expressing cells by flow cytometry using the EGFRvIII specific antibody L8A4, revealed that more than 90% of EGFRvIII positive cells were measured in the EGFRvIII + subline while the EGFRvIII − subline only showed approximately 1% EGFRvIIII expressing cells (Fig. 1B).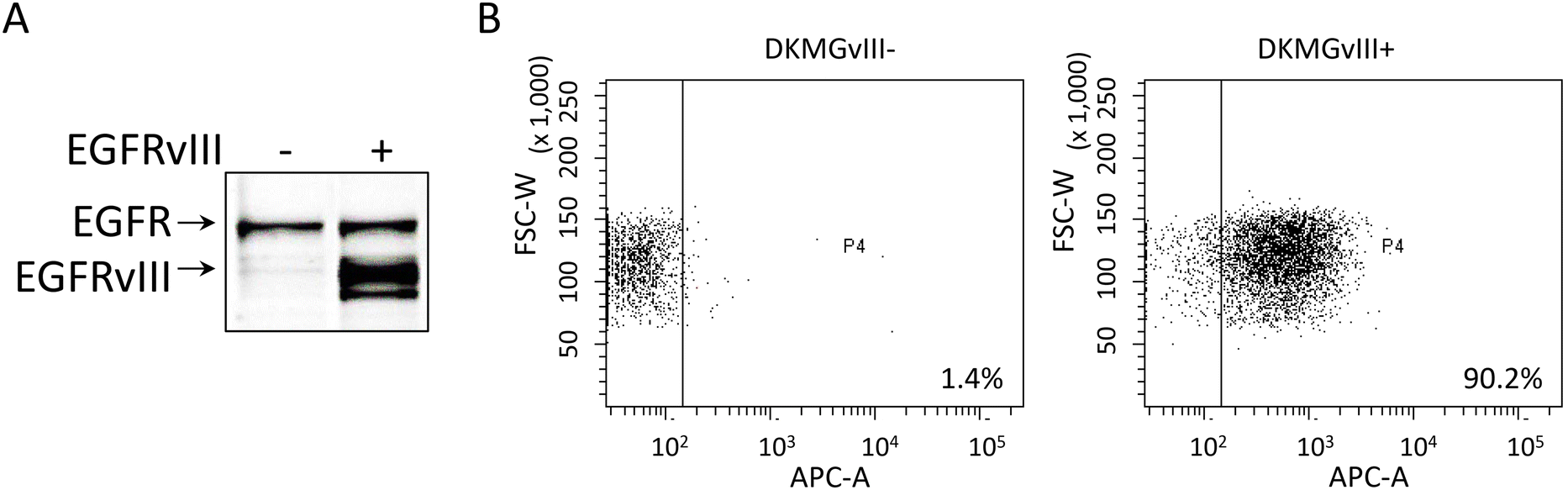 | ||
| Fig. 1 Expression of EGFRwt and EGFRvIII in DKMGvIII− and DKMGvIII + cell lines. (A) Western blot analysis using whole cell lysates and EGFR specific antibodies. (B) Dot blot of flow cytometry analysis using EGFRvIII specific antibody L8A4 and fluorescence labelled secondary antibodies. The percentage of EGFRvIII positive cells is indicated. (FSC-W: foreword scatter; APC-A: EGFRvIII signal; P4: gate for EGFRvIII positive cells). | ||
For the subsequent dimer analysis, antibodies from different species were needed which had to be specific either for EGFRwt or for EGFRvIII. The specificity of the selected antibodies (mouse-derived αEGFR R-1 for EGFRwt and rabbit-derived αEGFR D6T2Q for EGFRvIII) was tested using Chinese hamster ovary cells (CHO) which lack EGFR expression. These cells were transfected by EGFRwt or EGFRvIII encoding vectors and analysed by immunofluorescence microscopy. As shown in Fig. 2, both antibodies were highly specific and showed no cross reactivity.
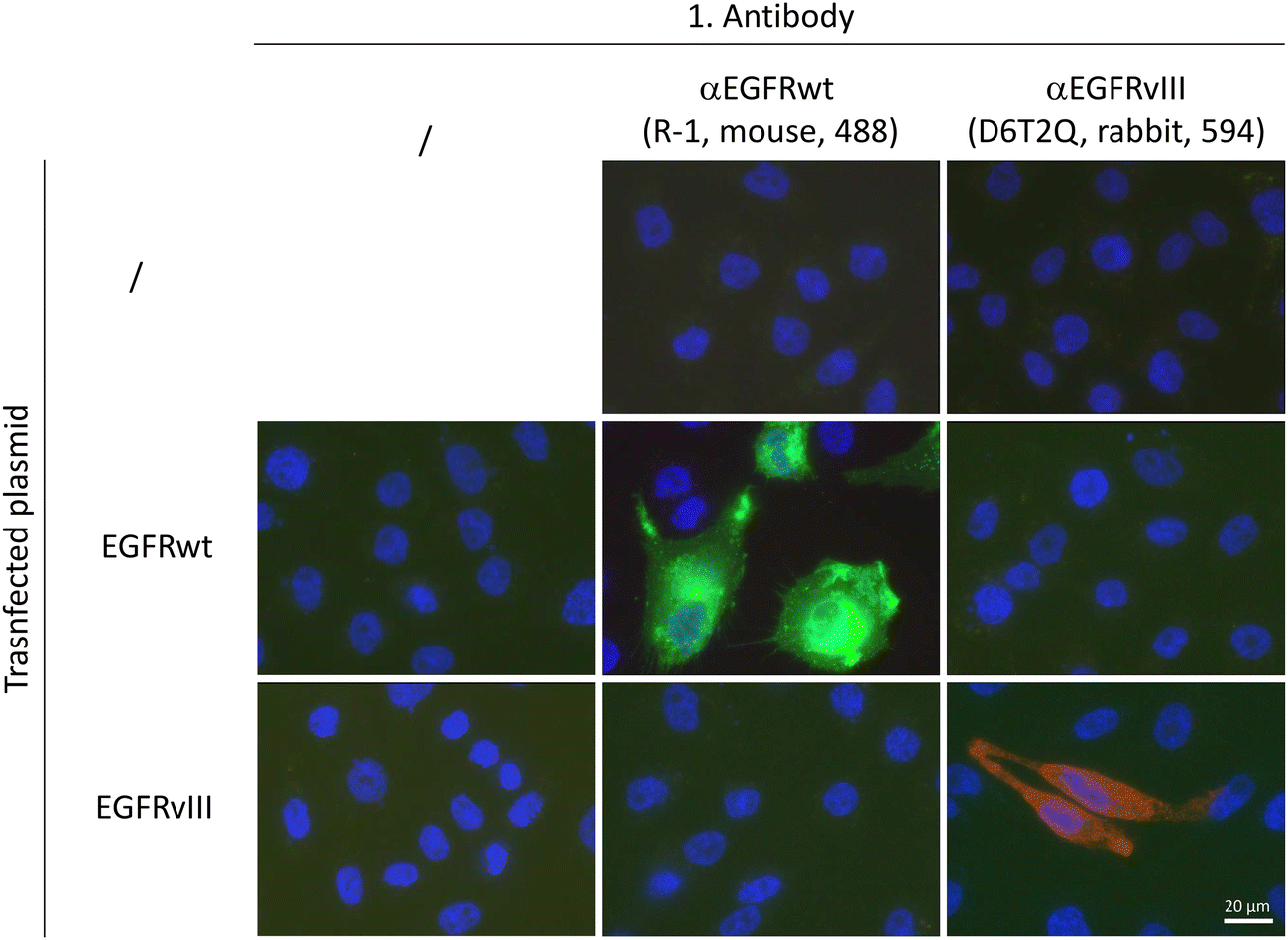 | ||
| Fig. 2 Immunofluorescence imaging of EGFR-non expressing CHO cells transfected with plasmids carrying either DNA for EGFRwt or EGFRvIII receptors. EGFRwt was detected using mouse derived αEGFR R-1 antibody (green), EGFRvIII by using rabbit derived αEGFR D6T2Q antibody (red). The cell nuclei are counterstained by DAPI. | ||
Single molecule localization microscopy (SMLM)
Since the cell periphery and the cell membrane are of particular interest in this work, the detection areas were chosen in such a way that only these regions of the cells were recorded (Fig. 3a and b). In addition, cells were selected that were largely in one plane and had a flat membrane outlet. Finally, equally-sized ROIs (regions of interest) were cut out and subjected to localization microscopy data evaluation. | ||
| Fig. 3 Examples of SMLM images (a) EGFRwt, (b) EGFRvIII, (c) and (d) visualization of the respective cluster formation. Scale bar is 1 μm. The inserts are enlarged by a factor of about 3. | ||
From 8 prepared slides, two labelled ones (slide 1 and slide 5) and one control (labelled by secondary antibodies only) were selected according to the quality required for SMLM (see Experimental chapter). The two completely labelled slides in general showed different absolute numbers of blinking events (Fig. 4a) but the same tendencies in structural formations. Although the data were only obtained from a few slides it has to be considered that all results are obtained from single cell data sets subjected to established, standardized tools of a computer analysis. This ensures a high reproducibility of the evaluation process for each cell. However, the cell line analysed showed a broad variability in the quantitative values as showed by the standard box plot graphics. It might be apposite to ask for reasons of this variability observed. At a first glimpse one might think about preparation effects that on one hand can never be excluded completely. On the other hand, real biological reasons have to be included into the consideration. Recently, Alekseenko et al.46 have pointed out reasons for low reproducibility of quantitative results between individual tumour cells. Each cell that could be subjected to single cell experiments has slightly different conditions in the microenvironment which might lead to little functional differences in the individual cells. This might be negligible in bulk experiments but in single cell measurements it could become visible. Or with other words, the more precisely single cell measurements are performed, the more variations between individual cells may become obvious.
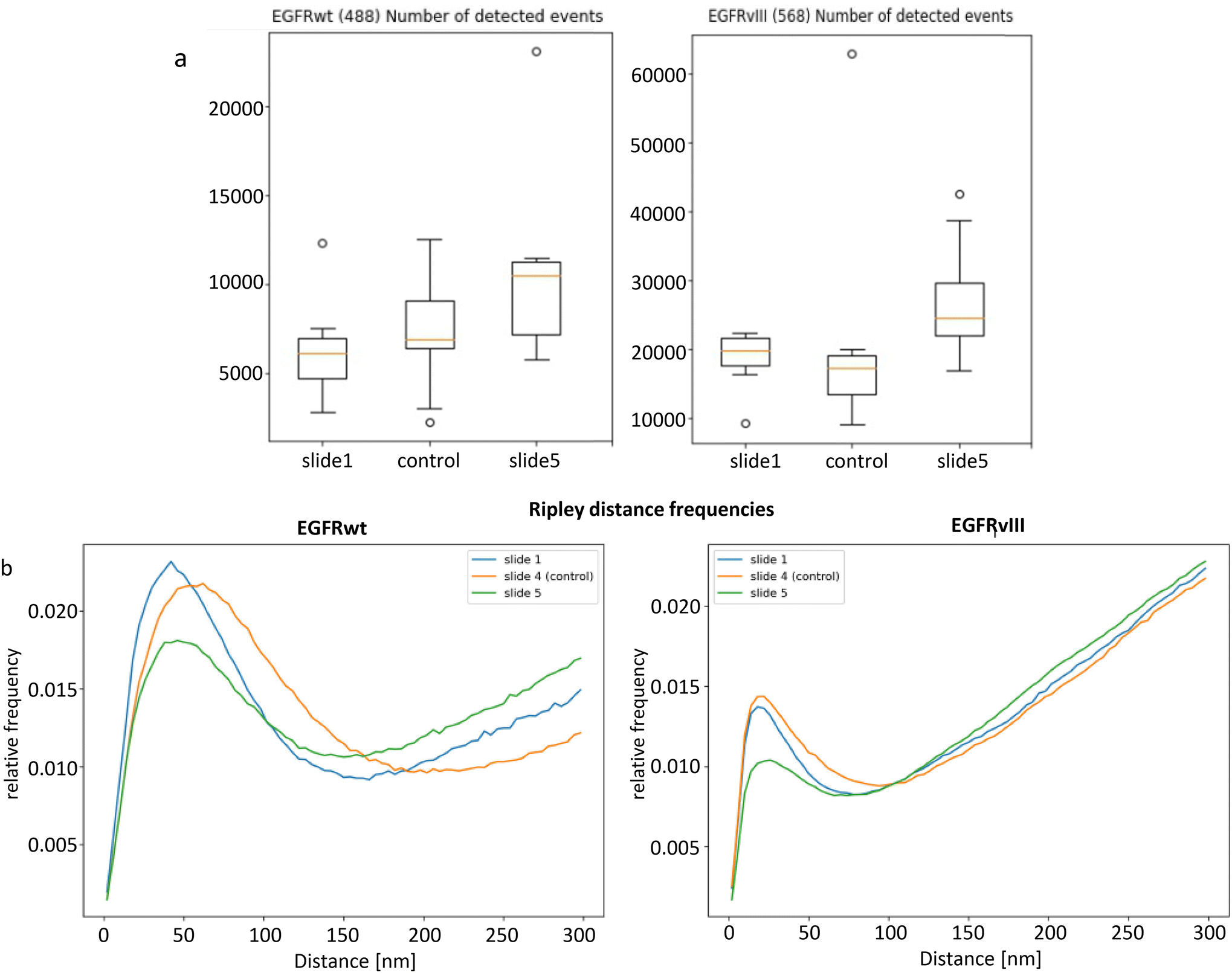 | ||
| Fig. 4 (a) Box plots of the number of detected events for EGFRvIII and EGFRwt (slide 1 and 5) and controls carrying the secondary antibodies only. (b) Ripley frequency histograms of pairwise point distances. The peaks represent the receptor clusters. In case of the controls without specific binding, the peaks are broadened. The linear increase on the right side indicates randomly arranged points around the clusters. [The boxplots show the median point number of the detected signals (red line), the lower and upper quantile (box), and the value range within ±2 standard deviations (line). Additional crosses refer to values that differ more than 3 box lengths from the median.]. | ||
For the number of blinking events, no significant differences were seen between the control group and the slides with the complete colouring. Obviously the secondary antibodies tend to non-specifically attachment on the cell membranes. This seems to be a typical shortcoming of the cell model used as being discussed and elaborated in more detail in Boyd et al.5 Therefore it was necessary also to calculate the frequencies of distances where the two groups could be separated from the controls (see Ripley statistics below). Both groups exhibited signal numbers in the same orders of magnitude in two detection channels; for EGFRwt, the mean detection numbers fluctuated between 7000 and 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, for EGFRvIII the mean detection numbers were much higher, i.e., in the range between 18000 and 25000. The event numbers, however, differed significantly between the 488 nm (EGFRwt) and 568 nm (EGFRvIII) channels. On average, up to twice as many events were found for EGFRvIII compared to EGFRwt. This corresponds to the fact that the deletion mutant is more highly expressed than EGFRwt in the cell line used.22 The higher event number for EGFRvIII can also be seen in the localization image (Fig. 3b). However, the clearly more pronounced fluorophore-rich area outside the cell in the EGFRvIII channel, visible in Fig. 3b on the right, also indicates an elevated background. This suggests that there could be also some background inside the cell. Thus, the increased signal number in EGFRvIII might result from a more pronounced expression and a presumably increased background of non-specifically bound secondary antibodies.
000, for EGFRvIII the mean detection numbers were much higher, i.e., in the range between 18000 and 25000. The event numbers, however, differed significantly between the 488 nm (EGFRwt) and 568 nm (EGFRvIII) channels. On average, up to twice as many events were found for EGFRvIII compared to EGFRwt. This corresponds to the fact that the deletion mutant is more highly expressed than EGFRwt in the cell line used.22 The higher event number for EGFRvIII can also be seen in the localization image (Fig. 3b). However, the clearly more pronounced fluorophore-rich area outside the cell in the EGFRvIII channel, visible in Fig. 3b on the right, also indicates an elevated background. This suggests that there could be also some background inside the cell. Thus, the increased signal number in EGFRvIII might result from a more pronounced expression and a presumably increased background of non-specifically bound secondary antibodies.
The different number of blinking events reasoned the selection of the dyes for the secondary antibodies and that in case of distance measurements between the two receptor types, always the distances from EGRFwt to EGFRvIII were measured.
Statistical evaluation of SMLM data sets
The high background in EGFRvIII was also visible by the linear increase42 in the Ripley47 distributions (Fig. 4b, right). Especially in comparison to EGFRwt (Fig. 4b, left) the background dominated there. The curves show the relative frequencies of the pair-wise point distances. In these curves the peaks on the left indicate clustering while the linear part on the right indicates a more or less random distribution depending on the steepness of the curves. In the cases of the completely labelled specimens (slide 1 and 5), the maxima of the peaks were positioned at the same values (EGFRvIII at about 20 nm; EGFRwt at about 43 nm) and showed the same widths while in both cases the control was shifted to the right with an increased width. The peaks of slide 5 were significantly lower and the linear part was steeper, which was due to the different point numbers of the specimens. These additional signals presumably consisted in a larger part on an amplified background. It is noticeable that peaks of EGFRwt had a broader width than the peaks of EGFRvIII and the control had the widest maxima, which indicates a reduced and less dense clustering.The results of the Ripley analysis were supported by the cluster analysis using DBSCAN with the cluster conditions that a minimum of 5 points must be located within a radius of 60 nm around a single point to form a cluster. In Fig. 3c and d, the results of this clustering are shown for the examples of Fig. 3a and b. On average about four times more EGFRvIII clusters than EGFRwt clusters were obtained (Fig. 5a). While the mean cluster size was only slightly smaller for EGFRvIII (Fig. 5c), EGFRvIII clusters showed a much larger variation in the mean density than the clusters of EGFRwt (Fig. 5b). However, the mean number of points per cluster did not vary between EGFRvIII and EGFRwt (Fig. 5d).
 | ||
| Fig. 5 Box plots (for description see Fig. 4) for different features of the clusters according to the DBScan with a minimum of 5 points within a radius of 60 nm around a point: (a) number of clusters per cell; (b) mean point density in the clusters; (c) mean cluster size; (d) mean number of points per cluster. | ||
Dimerization of EGFRwt and EGFRvIII according to SMLM data sets
Cluster formation seems to be a prerequisite for the dimerization of EGFRs. In the following, we will elucidate whether EGFRwt and EGFRvIII prefer the formation of homo-dimers or hetero-dimers. Since SMLM data only reflect the position of the fluorochromes of the secondary antibodies used, an estimate was calculated for the largest distance of fluorochromes of the antibody construct that would still be compatible with dimerization of the receptors.In the case of EGFR, it is known that the receptors, induced by a ligand via their extracellular domain, form homo- or hetero-dimers with other ErbB receptors.48 Since EGFR has only one transmembrane domain, the dimer spacing would be the distance between the two transmembrane domains. With the help of the crystal structure of an EGFRwt homo-dimer this distance can be estimated to 7.9 nm.49 Both primary antibodies used bind to the extracellular domain around the EGF binding site.50,51 Accordingly, these two were about 8 nm apart.
The primary antibody for EGFRwt has a molecular mass of 150 kDa and the one for EGFRvIII has a molecular mass of 130 kDa. These have an approximate size52 of 14 × 8 × 4 nm3 and have the shape of a Y, which consists of several protein chains. The two primary antibodies were fluorescently labelled by specific secondary antibodies “loaded” with the fluorophores, which were assumed to have about the same spatial extension. The size of the fluorophores was negligibly small since it is limited by a molecular mass of 759 Da for AlexaFluor594 or 721 Da for AlexaFluor488. These secondary antibodies were not free in their binding, as the binding site was located at the lower end of the primary antibody. This also restricted the binding angle. In addition it has to be considered that it is possible that even two or perhaps three secondary antibodies bind to one primary antibody. Unfortunately, neither literature nor the manufacturer could give concrete estimates of the probability of double or triple secondary antibody binding.
Under the first assumption that only one secondary antibody targets one primary antibody, the theoretically maximum possible distance for a dimer was estimated to:
| Dimer distance (max) = 2 × (4 nm + 8 nm + 8 nm) = 40 nm |
The first term (4 nm) originated from the distance between the receptors as obtained in the crystal structure. The binding and expansion of the primary and secondary antibodies determined the second (8 nm) and third (8 nm) terms. The bonds they formed were restricted but not uniform. The minimum dimer distance that can be registered is given by the localization precision of the point detection. This means the precision with which the coordinate of a single point can be determined. Or with other words, a point is detected as a single point as long as it only fluctuates within this region. The localization precision was in the order of 10 nm (see Table S1 in the ESI†). In the estimate the minimum distance was set to 0 nm which means within localization precision only.
The relative frequencies of the next neighbour distances were counted for EGFRwt–EGFRwt, for EGFRvIII–EGFRvIII and for EGFRwt–EGFRvIII. In Fig. 6a–c the results are shown for slide 1. The red curve was calculated as the envelope of the histograms. These histograms were compared to next neighbour distance histograms of a random distribution obtained from the same number of points randomly distributed on the same area as the total membrane section evaluated (Fig. 6d–f).
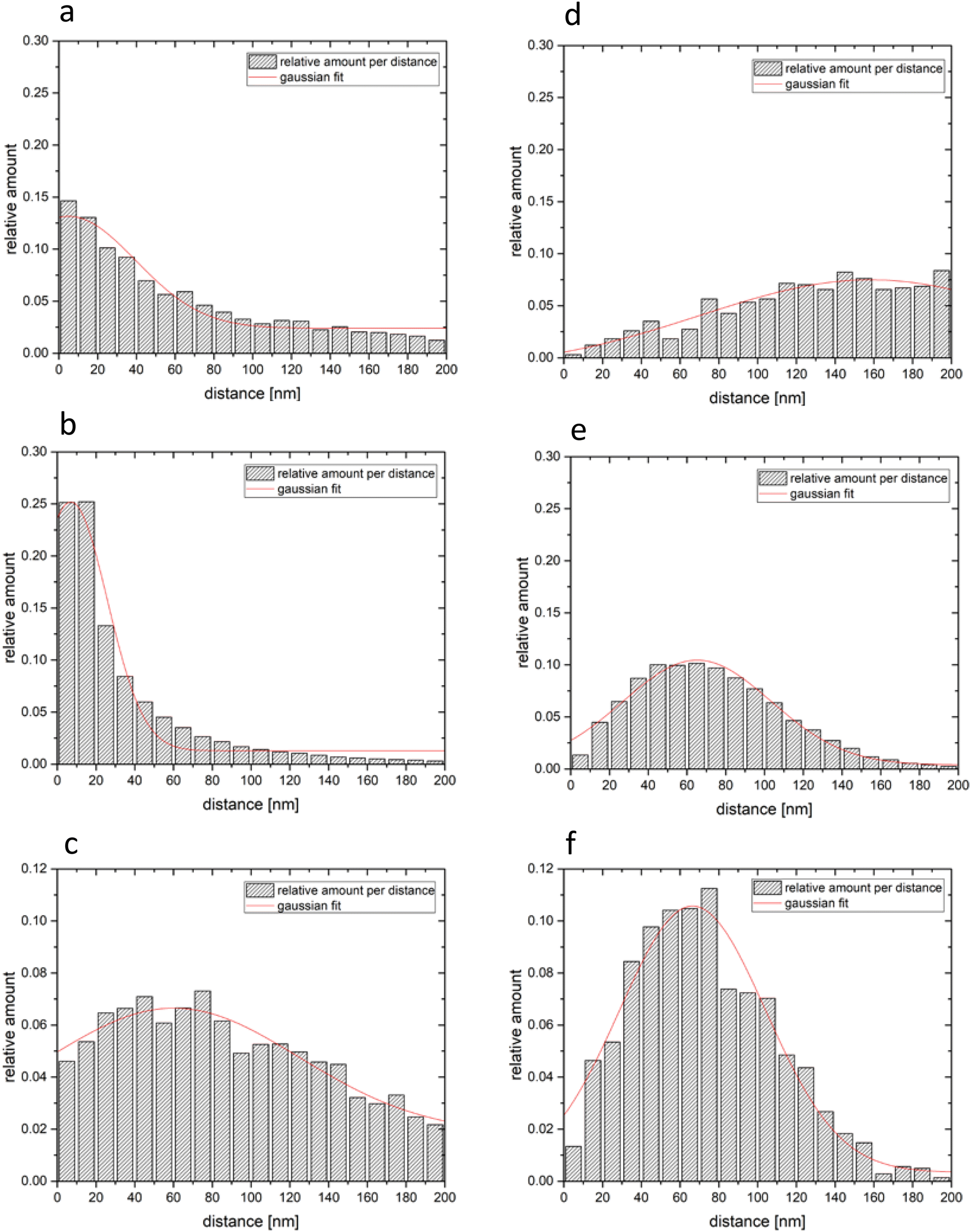 | ||
| Fig. 6 Relative numbers of distances measured between (a) EGFRwt – EGFRwt; (b) EGFRvIII – EGFRvIII; (c) EGFRwt – EGFRvIII. The right column (d), (e) and (f) shows the results for the same number of points randomly distributed. The grey columns show the relative amount of events per distance window of 10 nm. The red lines show the resulting Gaussian fit curve for these relative frequency distributions. | ||
Using the estimated maximum value for dimerization as calculated above, the relative amount of homo-dimers and hetero-dimers were calculated from the frequency distributions of the next neighbour distances. On average 38.04% of the EGFRwt and 69.14% of the EGFRvIII seemed to form homo-dimers according to our distance criteria.
These relative high values of presumptive homo-dimers have to be discussed in more detail. In principle these values are biased by the possibility that two or even three secondary antibodies can bind to one primary antibody. Assuming that for instance two secondary antibodies bind in such a way that their axes are perpendicularly orientated to the axis of the primary antibody, their distance could be estimated to 20 nm (2 × 8 nm for the length of the secondary antibodies + 4 nm for the thickness of the primary antibody). This distance is larger than the localization precision so that the value would contribute to the results of our point distance determination and would be interpreted as a homo-dimer. The frequency of such an antibody situation cannot be determined. However, since the secondary antibodies do not preferentially bind in the described perpendicular arrangement, it could be expected that some of them are orientated in a small angle to the axis of the primary antibody so that the two dyes cannot be separated within the localization precision. Moreover, it cannot be excluded that many primary antibodies carry only one secondary antibody or that a measured distance between two blinking events derive from two secondary antibodies that are attached to two closely adjacent primary antibodies although both primary antibodies carry more than one secondary antibody.
We also analysed the frequencies of distances to the next neighbour, the two next neighbours and the five next neighbours (Fig. S1, ESI†). For both types of receptors the three curves revealed a very similar shape with a maximum at distances below 40 nm. Since the probability that five next neighbours are secondary antibodies of one primary antibody is very low or even tends to zero, we assume in comparison with the data obtained for hetero-dimerization that it seems to be reasonable that a considerable amount of distance pairs in our data set were due to homo-dimerization.
Another point that should be mentioned in the context of the secondary antibodies is the degree of labelling (DOL) of such antibodies which can range from 2–8 dye molecules per antibody. Usually in a biological micro-environment several of these dye molecules quench each other, but in general it cannot be excluded that more than one dye molecule is actively blinking at one secondary antibody. However, if it is assumed that the length of such an antibody is about 8 nm along which the dye molecules can be arranged, the multiple blinking events are below the localization precision (Table S1†) and therefore recorded as one event.
Our data support the hypothesis that EGFRs prefer homo-dimerisation.53 These results obtained for homo-dimerization were significantly different from the results obtained for the same number of randomly distributed points (5.95% or 18.13%, respectively) (Table 1). The difference of these two values of the random distributions corresponds to the different numbers of blinking events in general which we observed for EGFRwt and EGFRvIII.
| Sample | Receptor EGFR | Measurement | Random data | ||
|---|---|---|---|---|---|
| Mean distance [nm] | Amount of dimers [%] | Mean distance [nm] | Amount of dimers [%] | ||
| Slide 1 | wt | 5.37 ± 3.14 | 47.02 | 158.24 ± 11.27 | 5.95 |
| vIII | 7.05 ± 3.05 | 72.1 | 62.2 ± 1.39 | 21.01 | |
| Control | wt | 45.16 ± 4.27 | 34.96 | Not available | |
| vIII | 11.29 ± 1.69 | 74.36 | 74.35 ± 1.80 | 17 | |
| Slide 5 | wt | 31.15 ± 3.83 | 32.15 | Not available | |
| vIII | 15.30 ± 2.27 | 60.97 | 74.37 ± 1.73 | 16.37 | |
| Mean wt | 38.04 ± 6.45 | Significance 8.7σ | 5.95 | ||
| Mean vIII | 69.14 ± 5.85 | Significance 5σ | 18.13 ± 2.05 | ||
In contrast to the results for homo-dimerization, the results obtained for hetero-dimerization of 18.47% did not significantly differ from the result of randomly distributed points (17.06%) within one standard deviation (Table 2). This rather low value for hetero-dimerization might also support the assumption that multiple binding of secondary antibodies is not the major labelling situation.
| Sample | Measurement | Random data | ||
|---|---|---|---|---|
| Mean distance [nm] | Amount of dimers [%] | Mean distance [nm] | Amount of dimers [%] | |
| Slide 1 | 60.18 ± 4.38 | 23.08 | 66.42 ± 1.84 | 19.76 |
| Control | 87.93 ± 3.03 | 17.49 | 73.45 ± 2.17 | 15.63 |
| Slide 5 | 93.48 ± 2.61 | 14.85 | 75.48 ± 1.82 | 15.79 |
| Mean | 18.47 ± 3.43 | Mean | 17.06 ± 1.91 | |
In summary, these calculations and measurements by SMLM indicated that the formation of homo-dimers is considerably preferred to the formation of hetero-dimers for EGFRwt and EGFRvIII. These findings are also supported by bio-physical simulation models (see below) of clustering of proteins integrated into a bi-lipid membrane where clustering of equally sized proteins is preferred which could better support homo-dimerization that hetero-dimerization.54–56
Topological results of SMLM data
Physical models of a lipid bilayer membrane (lipid with one unit hydrophilic head and two or three units hydrophobic tails) with transmembrane receptors incorporated, suggested due to physico-chemical and thermodynamic conditions that the membrane adapts its thickness to the receptor, i.e., the distance of the hydrophobic parts of the lipid layers, to the hydrophobic part of the receptor so that the receptor axis remains approximately perpendicular to the membrane surface.54,55 Due to thermodynamic reasons, this favours the formation of clusters of the receptors of the same type.56 Based on these theoretical approaches, we studied whether the clusters of EGFRwt and EGFRvIII are similarly organized so that they can easily intermingle and the receptors could preferentially form homo- or hetero-dimers. Based on the SMLM data, persistence homology in dimension 0 (components) and 1 (holes) was calculated42 and the results were transferred in a bar-code pattern. In Fig. 7a, typical examples for the bar-code pattern of the holes are presented for EGFRwt and EGFRvIII. Each bar represents the beginning and the end of a hole in a net-like distribution of points.57 The difference of the receptor clusters is obvious. While the holes for EGFRvIII have a short “life-time” (short bars), the holes for EGFRwt persist longer (larger bars). This indicated that the clusters are different in their topology and in principle their geometry and size. This can be reflected by the frequency distribution of the endpoints of the bars which is known to be a characteristic measure in persistent homology.58 In both cases, i.e. for the components and for the holes, these frequency distributions for the clusters differed significantly (Fig. 7b), i.e., the distributions for EGFRvIII are shifted to smaller values compared to EGFRwt, indicating that the clusters of EGFRvIII and EGFRwt are differently organized.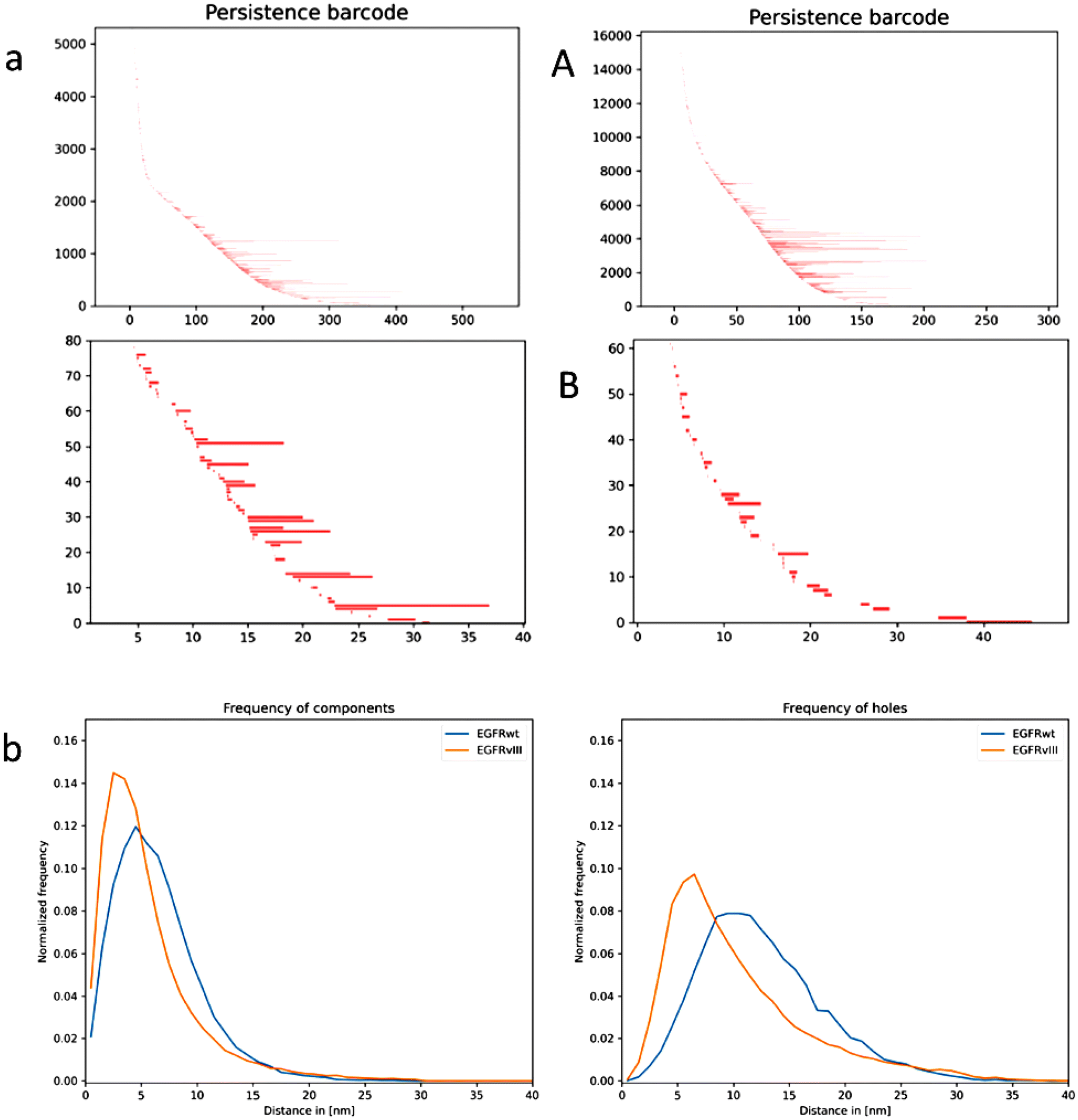 | ||
| Fig. 7 (a) Typical examples of a bar-code distribution for EGFRwt (left) and EGFRvIII (right). The distribution shows the number of bar-codes vs. the length in nm for the holes (dim 1) in persistence homology analysis for (A) the whole cell and (B) the clusters of a cell only. (Note: different scaling). (b) Relative frequency of the positions of endpoints of a bar-code distribution for EGFRwt and EGFRvIII within the clusters identified (see aB). The left histogram represents the components bar-codes; the right one the holes bar-codes. | ||
A well-established measure to compare the barcode patterns of two receptor clusters is the Jaccard index.57,59 Due to normalization of this similarity measure, index values between 0 and 1 can be obtained. A value of 0 means that there is no overlap when the two compared bar sets are superimposed; with a value of 1, the two bar sets are identical.
For a representative data set obtained from slide 1, the Jaccard indices were calculated for the components and for the holes and averaged. The results are depicted in a heatmap representation in Fig. 8a. When comparing EGFRwt with EGFRwt similarity values between 0.420 and 0.445 were obtained. For the comparison of EGFRvIII with EGFRvIII, the similarity values were lower between 0.410 and 0.425. These values were further but only partly decreased in the case of comparing EGFRwt with EGFRvIII (between 0.400 and 0.430). For a better overview, 2nd generation heatmaps59 were calculated in which one pixel represents the mean value of the values of one 1st generation heatmap. These 2nd generation heatmaps are shown in Fig. 8b for the components, the holes and the average of components and holes. In all three heatmaps the comparison of EGFRwt with itself has the highest similarity followed by slightly lower similarities for EGFRvIII with itself. The comparison of EGFRwt with EGFRvIII and vice versa resulted in the lowest similarity values.
 | ||
| Fig. 8 Heatmaps of the averaged Jaccard indizes of clusters in cells (a) show the mean values for the results of the components and holes. In the heatmaps 2nd generation (b) the respective heatmaps of all cells are averaged. The data show the similarity of the EGFRwt and EGFRvIII for themselves and indicate the disimilarity for the two types of receptor clusters. | ||
Binding studies for the receptor dimers
To further analyse the presence of EGFRwt/EGFRvIII dimers, we performed immunoprecipitation studies using whole cell lysates. As shown in Fig. 9, EGFRwt was successfully precipitated using wt specific antibody R-1. As positive controls, whole cell SDS-lysates were used as well as the detection of the EGFR-binding protein Grb2, which was co-precipitated when the αEGFRwt antibody was used but not in the control precipitation. However, we detected no EGFRvIII in the αEGFRwt antibody precipitate which further suggests no significant interaction of EGFRwt and EGFRvIII. | ||
| Fig. 9 Immunoprecipitation of EGFRwt using αEGFR R-1 antibody. EGFR and Grb2 were detected specific using appropriate antibodies by Western blot experiment. (Super.: Supernatant; Precip.: Precipitate). | ||
Experimental
Cell culture
Cultivation of the EGFRvIII + cell model system DKMGvIII + has been described in detail elsewhere.26Briefly, iso-genetic cell lines with a very high amount of EGFRvIII expressing cells (DKMGvIII+) or a very low amount of EGFRvIII expressing cells (DKMGvIII−) were generated as described in.52 The cells were cultured in RPMI (10% heat inactivated FCS, 2 mM L-glutamine, 1 mM sodium pyruvate).EGFR negative Chinese Hamster Ovary (CHO) cells were cultivated as described in ref. 61 using α-minimal essential medium (Gibco) supplemented with 5% fetal calf serum (FCS, Gibco). For EGFRwt and EGFRvIII over-expression, CHO cells were transfected with 2 μg plasmid carrying either DNA for EGFRwt or EGFRvIII receptors using Lipofectamine 2000 according to the manufacture's protocol (Thermo Fisher) and protein expression was analysed after 24 h using immunofluorescence microscopy. All cells were grown at 37 °C, 5% CO2 and 100% humidification. All cells were identified by a short tandem repeat multiplex assay (Applied Biosystems) and were negatively tested for mycoplasma.
Western blot (WB)
Proteins from whole-cell extracts were detected by Western blot according to standard protocols. The Odyssey® CLx Infrared Imaging System (LI-COR Biosciences) was used for signal detection and quantification. Primary antibodies: EGFR (1:1000, rabbit, Cell Signaling, #2232); β-actin (1:20000, mouse, Sigma-Aldrich, #A-2228); Grb2 (1:1000, rabbit, Cell Signaling, 3972). All primary antibodies were diluted in 5% bovine serum albumin (BSA) in PBS supplemented with 0.2% Tween. Secondary anti-mouse and anti-rabbit antibodies were purchased from LI-COR Biosciences.
Immunoprecipitation (IP)
1.2 × 106 cells were grown in a T75 flask for 3 d and were harvested in 1 ml IP-buffer (50 mM HEPES, pH 7.4; 150 mM NaCl; 1.5 mM MgCl2; 1 mM EDTA; 0.1% Triton X-100) by scraping on ice. The lysate was sonicated (10 s) and centrifuged (1000g, 15 min) at 4 °C. The pellet (Pellet sample) and supernatant (Input sample) were separated. The Pellet was lysed using SDS-lysis buffer (200 mM Tris/HCl, pH 8.8; 5 mM EDTA; 0.1% bromophenol blue; 10% sucrose; 3.3% SDS; 2% 2-mercaptoethanol) while protein concentration of the Input was measured by BCA-test. Immunoprecipitation was performed using 100 μL protein A/G-agarose per reaction. For precipitation of EGFRwt 1 μg anti-EGFR antibody R-1 (mouse, Santa Cruz Biotechnologies, #sc-101) was used which epitope maps amino acids 6-273. The antibody was pre-incubated with the protein A/G-agarose for 30 min at RT, and 10% BSA solution (in PBS) was added 1:1. The mixture was incubated for 10 min at RT, centrifuged (1000g), washed once with PBS, centrifuged again and re-suspended in 500 μl IP-lysis buffer. A mock treated sample without antibody was used as control. 100 μl of the agarose mix was incubated with 700 μg protein of the input over night at 4 °C using a rotator. The precipitate was pelleted and the supernatant saved for further analysis (Supernatant). The pellet was washed once with PBS (Wash sample), with wash buffer 1 (0.5 M LiCl, 0.1 M Tris/HCl, pH 7.4) and wash buffer 2 (0.01 M Tris/HCl, pH 7.4) followed by resuspension with 70 μl SDS-lysis buffer for 5 min at 95 °C (precipitate sample).
Specimen preparation and immunofluorescence staining for microscopy and SMLM
For localization imaging as well as standard immunofluorescence microscopy, the cells were cultivated on round cover glasses in a 12-well plate at 37 °C. After 24 h the cells were washed in 2× PBS and fixed in 4% formaldehyde (freshly prepared from paraformaldehyde). After blocking in 1× PBS and 3% BSA for 1 h at RT, the cells were incubated with the rabbit anti αEGFRvIII antibody D6T2Q (concentration 1:500; Cell Signaling Technology, #64952)51 and with the mouse anti αEGFRwt antibody R-1 (concentration 1:1000; Santa Cruz Biotechnologies, #sc-101)50 for 1 h. After washing three times with 1× PBS/0.5% Tween20, the specimens were incubated with the ALEXAfluor®488 labelled secondary goat anti-mouse antibody (concentration 1:1000; Thermo Fisher Scientific, #A-11001)62 and the ALEXAfluor®594 labelled secondary goat anti-rabbit antibody (concentration 1:1000, Thermo Fisher Scientific, #a-11037)63 for SMLM. All primary antibodies were diluted in 5% BSA in PBS supplemented with 0.2% Tween. These dye combinations were chosen in order to label the lower number of receptor signals by the dye with the higher detection efficiency, i.e., the dye which absorption spectrum better fits the illumination wavelength. In some additional control experiments, we also swapped the dyes of the secondary antibodies (data not shown). These experiments, however, revealed no measurable advantage. The fluorescence labelled cells were embedded in ProLongGold® (Thermo Fisher Scientific, Waltham, MA, USA), which was left to polymerize for 24 h in the dark at RT. Finally, the slides were sealed and stored in the dark at 4 °C.
For the data presented here, four replicates of slides with primary and secondary antibodies and four control replicates with secondary antibodies only were prepared. After a first pre-screening those slides which were used for the results presented here were selected by visual inspection and according to the quality standards required for SMLM data acquisition and evaluation. This means that due to blinking background signals outside the cells, the data sets acquired for two of the slides were excluded as being insufficient for further evaluation. Slides No. 1 and No. 5 were subjected to detailed evaluation. The quality selection also excluded two slides of the controls. Since the remaining control slides both showed the same results, they were merged to one control data set for better overview.
Flow cytometry
Flow cytometry was performed as described previously61 using a FACSCanto and FACSDiva software (BD Biosciences). For EGFRvIII detection and quantification anti-EGFRvIII antibody L8A4 (1:1000, mouse, Absolute antibody, #Ab00184-1.4) and Alexa fluor®647 labeled secondary antibody (1:1000, Life Technologies, #A-21235) were used.
3D-microscopy
For 3D-imaging a Zeiss Axioplan 2 fluorescence microscope equipped with an Apotome scanning unit was used. An objective lens 63×/NA 1.4 was applied in combination with appropriate filter settings for the staining used. From the 3D-images maximum projection images were used for further evaluation.Localization microscopy
To study cell receptors on the nanoscale, a single molecule localization microscope (SMLM) was used as described elsewhere.5,37,38 SMLM is based on the principle of reversible photo-bleaching inducing stochastic blinking of the fluorophores.39 This allows for an accurate localization of the single molecule (order of 10 nm).36,42 To distinguish between the two receptors in general two different Coherent Saphire solid state lasers with wavelengths of 488 nm and 568 nm were used for illumination. Within each pathway, the laser intensity can be regulated from few mW to 200 mW. The search for suitable cells was conducted with low intensities whereas for the data acquisition, the intensity was adjusted to be 200 mW in order to provide perfect blinking-conditions. This value corresponds to 10 kW cm−2.A so-called neutral density filter wheel allows the regulation of the lasers intensity with twelve different grey filters of different densities. After the filter wheel, the light beam is expanded and focussed onto the specimen. The emitted light of the fluorophores then is separated from the laser beam by a dichroic and a blocking filter wheel before it is focussed onto the CCD camera (Sensicam QE, PCO, Kehlheim, Germany) which measures the intensity of the incoming light. The CCD camera used, has a size of 1376 × 1040 pixels whereas each pixel covers an area of 6.45 × 6.45 μm2. In combination with the objective 100×/NA 1.4, this yields an effective pixel size of 64.5 × 64.5 nm2.
The selection and in-focus adjustment of cells for data acquisition was accomplished by visual means. Therefore, low intensities were chosen such that the shape of the cells could be seen but nearly no bleaching of the fluorophores occurred. In order to measure only receptors on the membrane of the cell and to irradiate the specimen equally within a confined area, a region of interest (ROI) was chosen for each measurement. These were chosen to be planar membrane extensions of single cells. After the ROIs were determined, a wide-field image and a localization measurement were recorded for each wavelength. To avoid bleaching of the fluorophores, data acquisition always started with the larger wavelength (568 nm before 488 nm). For the localization measurements, 1000 image frames were acquired at a frequency of approximately 7.7 frames per s and with an integration time of 100 ms.
Data acquisition and processing
Typically, 20 to 35 image sets of 1000 frames were recorded for DKMGvIII + cells of each specimen. The obtained localization data was processed with house-made programs written on Python42 or in some cases on MATLAB scripts:36 The positions of the fluorescence molecules of the antibodies were calculated from registration of molecular blinking events according to an algorithm based on subtraction of the brightness values of two successive frames. Dark states over more than two successive frames can also be registered. For fitting a two dimensional Gaussian distribution, a centre of mass calculation was applied as described in Grüll et al.64 A so-called ‘orte-matrix’ was produced, which consists of nine columns: (a) the amplitude of the signal in photoelectrons, (b) the lateral y-coordinate in nm, (c) the lateral x-coordinate in nm, (d and e) the measurement errors for x and y coordinates, (f and g) the standard deviations, (h) the number of photoelectrons in the signal i.e. counts and (i) the number of the image in which the signal is found. To get from pixel space into position space a conversion factor is needed.An important advantage of the orte-matrix is its simplicity and the opportunity to sort out signals with too low intensities. This can be done by a threshold factor, which in this work was set to be 3. This means that all intensities which occur in the orte-matrix have to be at least as big as three times the background intensity. Therefore, autofluorescence of the cell and light emission of molecules different than the used fluorophores become negligible.
In addition, the localization precision of the obtained signals is determined by the brightness of a point. On average it is in the order of 10 nm (Table S1, ESI†), which is above the theoretical limits for SMLM because of the influence of the setup, the fluorophores and the preparation of the cells.
The density of signals on the membrane was calculated by manually cutting the images into defined sized (2 × 2 μm2) squares. This is necessary because in general the membrane was only making up 75% of the acquired ROIs. Therefore, the membrane part of each ROI was subdivided into 20–25 squares of the same size and the density of signals on the membrane thereby calculated.
For the investigation of clustering of receptors, the DBSCAN algorithm was used65 with a cluster definition with a minimum number of five signals within a radius of 60 nm. All the points which fulfill this condition are considered to be in a cluster. This method yielded values for cluster sizes, cluster diameter and distance distributions within and out of clusters.
The coordinate values of the points were subjected to Ripley statistics47 for which the point-to-point distances were measured by distances from each central point to all its peripheral ones. These pairwise distances were summarized in a distance frequency histogram and the envelope function was used to determine point arrangements.42 A homogeneous point-to-point distance distribution leads to a linear curve with a different slope. The diagonal means a random distribution. If the points form clusters, smaller distances are more frequent which results in a peak.
In order to determine the probability of homo- or hetero-dimerization, a custom-written script was used to determine the next, two next, and five next distances for each point. The orte-matrices therefore had to be shift corrected. In this work special significance lies on the next-neighbour distances because they would be possible dimer distances. Because the total number of points of EGFRwt of the cell line DKMGvIII+ is comparably small in contrast to the number of EGFRvIII, the hetero-distances were determined from wild type to the deletion mutant.
As a final processing step Persistent Homology (PH)57,58 analysis was used to explore the topology of the labelled proteins.42 This method aims to find out the significant structures of a point pattern. In order to obtain a pattern that represents these major features, each point is surrounded by a circle with an increasing radius. Here, two properties were considered: (a) the number of “components”, which is the number of elements that are still separated by this process and (b) the number of “holes” of the structures inside the components. These holes are “born” if the circles around the points have formed one closed component with free space inside. In algebraic topology, these properties are called zero- and one-dimensional complexes. These two dimensions can be represented by barcodes. Therefore, a geometric relationship among detected points is obtained by growing circles around each point (starting of bars for dimension 0). Whenever two of these circles mutually embed each-other's center, these centers are connected and they belong to the same component (end of a bar of dimension 0). With increasing radii, more points are connected to one component. Whenever a component forms a closed curve (in its simplest form a triangle) with free space inside, the area of this space is considered as a hole (start of a bar for dimension 1). With reducing the number of components, the number of holes is first increasing and then decreasing again when the holes are closed (end of a bar for dimension 1). The end points values of the bars for the sets of barcodes can be shown in frequency histograms giving information of the structural arrangements of the point pattern. The similarity of the bar codes can be quantified by the Jaccard index.59 The result of this normalized similarity measure is a value between 0 and 1, where 0 means no overlap and 1 the identity of two bars. The Jaccard indizes were calculated for each receptor cluster. All values for one cell were averaged and presented in a heatmap.57 Such heatmaps were computed for components and holes or for the mean values of both. The results obtained by these heatmaps were averaged for each type of receptor and the mean values can be compared by a 2nd generation heatmap.60
Conclusion
It is textbook knowledge that receptors of the ErbB family can form homo-dimers and hetero-dimers which are related to different activities. In glioblastoma cells the ErbB1 (=EGFRwt) occurs together with a mutant (=EGFRvIII).22 Up to now, it is controversially discussed whether EGFRwt and EGFRvIII prefer to form homo- or hetero-dimers.32,33,53 Here, we studied the dimerization behaviour of these two receptors by means of SMLM in an established cell model (DKMG+) that is overexpressing EGFRvIII. Our results supported the hypothesis that EGFRwt and EGFRvIII show a significant preference for homo-dimerization. Hetero-dimerization could not be excluded but it was compatible to random association of the relevant two colour signals. Using modern approaches of mathematical evaluation of SMLM data (Ripley pairwise distance frequency statistics, persistent homology, and comparison of topologies),42 we supported this finding by investigating the characteristics of cluster formation in cell membranes. From the perspective of receptor clustering and receptor cluster topology intermingling of EGFRwt and EGFRvIII clusters, a prerequisite for hetero-dimerization, appears to be less probable since the clusters showed a certain degree of dis-similarity. Although our findings are based on probability assumptions and do not strictly exclude hetero-dimerization, they are supported by other biophysical investigations of membrane lipids that seem to create a lipid composition in nano-domains which depends on the type of receptors incorporated.66 Moreover, our data are supported by the study of Stec et al.32 which postulates a homo-dimerization as the predominant mechanism of EGFRvIII regulation and by Fan et al.29 who failed to show direct interaction of EGFRwt and EGFRvIII, although describing an intensive crosstalk. Due to these data, a ligand induced hetero-dimerization is also unlikely. The fact, that other studies describe hetero-dimerization might be explained by the different cellular system. In contrast to others,67,68 we used human GBM cell lines with endogenous EGFRvIII expression, i.e., the receptor could be investigated in its natural molecular environment. All in all, the data presented here indicate the importance of the spatial organization of molecules in cell membranes for cellular functioning. Modern super-resolution light microscopy techniques in combination with novel evaluation approaches beyond imaging may open an avenue for the better understanding of mechanisms behind molecular organization and cellular functioning.Author contributions
Conceptualization, N. S., M. K. and M. H.; methodology, N. S., M. K. and M. H.; software, M. H.; validation, N. S., M. K., D. H. and M. H.; formal analysis, K. J., N. S., D. H. and M. G.; investigation, K. J., N. S., M. G., M. B.; resources, M. H.; data curation, K. J. and D. H.; writing—original draft preparation, N. S., M. K. and M. H.; writing—review and editing, K. J., N. S., D. H., M. G., M. B., M. K. and M. H.; visualization, K. J., D. H., M. G.; supervision, M. H.; project administration, M. K., M. H.; funding acquisition, M. K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the access to the localization microscope of Christoph Cremer, Institute of Pharmacy and Molecular Biotechnology, Heidelberg University. M. K. was supported by the Wilhelm Sander-Stiftung and the Brigitte und Dr Konstanze Wegener-Stiftung. The financial support to M. H. by the Deutsche Forschungsgemeinschaft within the funding programme “Open Access Publikationskosten” is gratefully acknowledged.References
- G. Carpenter and H. J. Liao, Cold Spring Harbor Perspect. Biol., 2013, 5, 1–17 CrossRef PubMed
.
- T. M. Brand, M. Lida, C. Li and D. L. Wheeler, Discov. Med., 2011, 12, 419–432 Search PubMed
- M. Ono and M. Kuwano, Clin. Cancer Res., 2006, 12, 7242 CrossRef CAS PubMed
- G. Pilarczyk, I. Nesnidal, M. Gunkel, M. Bach, F. Bestvater and M. Hausmann, Int. J. Mol. Sci., 2017, 18, 362 CrossRef PubMed
- P. S. Boyd, N. Struve, M. Bach, J. P. Eberle, M. Gote, F. Schock, C. Cremer, M. Kriegs and M. Hausmann, Nanoscale, 2016, 8, 20037 RSC
- P. Maier, L. Hartmann, F. Wenz and C. Herskind, Int. J. Mol. Sci., 2016, 17, 102 CrossRef PubMed
- B. F. El-Rayes and P. M. LoRusso, Br. J. Cancer, 2004, 91, 418–424 CrossRef CAS PubMed
- D. A. Sabbah, R. Hajjo and K. Sweidan, Curr. Top. Med. Chem., 2020, 20, 1–20 CrossRef PubMed
- Y. Zhang, Pharmacol. Rev., 2023, 75, 1218–1232 CrossRef CAS PubMed
- A. Rutkowska, E. Stoczynska-Fidelus, K. Janik, A. Włodarczyk and P. Rieske, J. Oncol., 2019, 1092587, 1–20 Search PubMed
- R. Stupp, M. E. Hegi, W. P. Mason, M. J. van den Bent, M. J. Taphoorn, R. C. Janzer, S. K. Ludwin, A. Allgeier, B. Fisher, K. Belanger, P. Hau, A. A. Brandes, J. Gijtenbeek, C. Marosi, C. J. Vecht, K. Mokhtari, P. Wesseling, S. Villa, E. Eisenhauer, T. Gorlia, M. Weller, D. Lacombe, J. G. Cairncross and R. O. Mirimanoff, Lancet Oncol., 2009, 10, 459–466 CrossRef CAS PubMed
- C. Adamson, O. O. Kanu, A. I. Mehta, C. Di, N. Lin, A. K. Mattox and D. D. Bigner, Expert Opin. Invest. Drugs, 2009, 18, 1061–1083 CrossRef CAS PubMed
- Q. T. Ostrom, H. Gittleman, G. Truitt, A. Boscia, C. Kruchko and J. S. Barn-holtz-Sloan, Neuro-Oncology, 2018, 20, iv1–iv86 CrossRef PubMed
- F. Hanif, K. Muzafar, K. Perveen, S. M. Malhi and S. U. Simjee, Asian Pac. J. Cancer Prev., 2017, 18, 3–9 Search PubMed
- M. E. Davis, Clin. J. Oncol. Nurs., 2016, 20, S2–S8 CrossRef PubMed
- S. M. Batool, K. Muralidharan, T. Hsia, S. Falotico, A. S. Gamblin, Y. B. Rosenfeld, S. K. Khanna, L. Balaj and B. S. Carter, Clin. Cancer Res., 2022, 28, 4070–4082 CrossRef CAS PubMed
- P. Y. Wen and S. Kesari, N. Engl. J. Med., 2008, 359, 492–507 CrossRef CAS PubMed
- A. Vehlow and N. Cordes, Biochim. Biophys. Acta, 2013, 1836, 236–244 CAS
- A. Del Vecchio, C. P. Giacomini, H. Vogel, K. C. Jensen, T. Florio, A. Merlo, J. R. Pollack and A. J. Wong, Oncogene, 2013, 32, 2670–2681 CrossRef PubMed
- H. K. Gan, A. N. Cvrijevic and T. G. Johns, FEBS J., 2013, 280, 5350–5370 CrossRef CAS PubMed
- R. B. Luwor, H.-J. Zhu, F. Walker, A. A. Vitali, R. M. Parera, A. W. Burgess, A. M. Scott and T. G. Johns, Oncogene, 2004, 23, 6095–6104 CrossRef CAS PubMed
- G. Zadeh, K. P. L. Bhat and K. Aldape, Cancer Cell, 2013, 14, 403 CrossRef PubMed
- N. Struve, Z. A. Binder, L. F. Stead, T. Brend, S. J. Bagley, C. Faulkner, L. Ott, J. Müller-Goebel, A.-S. Weik, K. Hoffer, L. Ktug, T. Riekmann, L. Bußmann, M. Henze, J. J. D. Morrissette, K. M. Kurian, U. Schüller, C. Petersen, K. Rothkamm, D. M. O'Rourke, S. C. Short and M. Kriegs, Oncogene, 2020, 39, 3041–3055 CrossRef CAS PubMed
- Z.-F. Shi, G.-Z. Li, Y. Zhai, C.-Q. Pan, D. Wang, M.-C. Yu, C. Liu, W. Zhang and X.-G. Xu, Genes, 2023, 14, 651 CrossRef CAS PubMed
- N. Struve, K. Hoffer, A. S. Weik, B. Riepen, E. Ka, M. Cetin, J. Burmester, L. Ott, J. Liebing, F. Gatzemeier, J. Müller-Goebel, L. Bußmann, A. Parplys, K. Unger, W. Mansour, U. Schüller, T. Rieckmann, C. Petersen, K. Rothkamm and M. Kriegs, Neuro-Oncology Adv., 2021, 4, 1–14 Search PubMed
- N. Struve, M. Riedel, A. Schulte, T. Riekmann, T. J. Grob, A. Gai, K. Rothkamm, K. Lamszus, C. Petersen, E. Dikomey and M. Kriegs, Oncotarget, 2015, 6, 33867 CrossRef PubMed
- M. Riedel, N. Struve, J. Müller-Goebel, S. Köcher, C. Petersen, E. Dikomey, K. Rothkamm and M. Kriegs, Oncotarget, 2016, 7, 61988 CrossRef PubMed
- J. Wong, M. Ruppert, S. H. Bigner, C. H. Grzeschik, P. A. Humphrey, D. S. Bigner and B. Vogelstein, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2965–2969 CrossRef PubMed
- Q.-W. Fan, C. K. Cheng, W. C. Gustafson, E. Charron, P. Zipperm, R. A. Wong, J. Chen, J. Lau, C. Knobbe-Thomsen, M. Weller, N. Jura, G. Reifenberger, K. M. Shokat and W. A. Weiss, Cancer Cell, 2013, 24, 438–449 CrossRef CAS PubMed
- A. Abulrob, Z. Lu, E. Baumann, D. Vobornik, R. Taylor, D. Stanimirovic and L. J. Johnston, J. Biol. Chem., 2010, 285, 3145–3156 CrossRef CAS PubMed
- J. Gao, Y. Wang, M. Cai, Y. Pan, H. Xu, J. Jiang, H. Ji and H. Wang, Nanoscale, 2015, 7, 2511 RSC
- W. Stec, K. Rosiak, C. Treda, M. Smolarz, J. Peciak, M. Pacholczyk, A. Lenart, D. Grzela, E. Stoczynska-Fidelus and P. Rieske, Oncotarget, 2018, 9, 8560–8572 CrossRef PubMed
- P. C. Pan and R. S. Magge, Int. J. Mol. Sci., 2020, 21, 8471 CrossRef CAS PubMed
- C. Bond, A. N. Santiago-Ruiz, Q. Tang and M. Lakadamyali, Mol. Cell, 2022, 82, 315–332 CrossRef CAS PubMed
- M. Lelek, M. T. Gyparaki, G. Beliu, F. Schueder, J. Griffié, S. Manley, R. Jungmann, M. Sauer, M. Lakadamyali and C. Zimmer, Nat. Rev. Methods Primers, 2021, 1, 39 CrossRef CAS PubMed
- M. Stuhlmüller, J. Schwarz-Finsterle, E. Fey, J. Lux, M. Bach, C. Cremer, K. Hinderhofer, M. Hausmann and G. Hildenbrand, Nanoscale, 2015, 7, 17938–17946 RSC
- M. Eryilmaz, E. Schmitt, M. Krufczik, F. Theda, J.-H. Lee, C. Cremer, F. Bestvater, W. Schaufler, M. Hausmann and G. Hildenbrand, Cancers, 2018, 10, 25 CrossRef PubMed
- M. Hausmann, E. Wagner, J.-H. Lee, G. Schrock, W. Schaufler, M. Krufczik, F. Papenfuß, M. Port, F. Bestvater and H. Scherthan, Nanoscale, 2018, 10, 4320–4331 RSC
- J. Hendrix, C. Flors, P. Dedecker, J. Hofkens and Y. Engelborghs, Biophys. J., 2008, 94, 4103–4113 CrossRef CAS PubMed
- P. Lemmer, M. Gunkel, D. Baddeley, R. Kaufmann, A. Urich, Y. Weiland, J. Reymann, P. Müller, M. Hausmann and C. Cremer, Appl. Phys. B, 2008, 93, 1–12 CrossRef CAS
- P. Lemmer, M. Gunkel, Y. Weiland, P. Müller, D. Baddeley, R. Kaufmann, A. Urich, H. Eipel, R. Amberger, M. Hausmann and C. Cremer, J. Microsc., 2009, 235, 163–171 CrossRef CAS PubMed
- J. Weidner, C. Neitzel, M. Gote, J. Deck, K. Küntzelmann, G. Pilarczyk, M. Falk and M. Hausmann, Comput. Struct. Biotechnol. J., 2023, 21, 2018–2034 CrossRef CAS PubMed
- X. Xu, Y. Wang, W.-S. Choi, X. Suna and R. Godbout, Nanoscale, 2021, 13, 9706–9722 RSC
- J. Erenpreisa, A. Giuliani, K. Yoshikawa, M. Falk, G. Hildenbrand, K. Salmina, T. Freivalds, N. Vainshelbaum, J. Weidner, A. Sievers, G. Pilarczyk and M. Hausmann, Int. J. Mol. Sci., 2023, 24, 2658 CrossRef CAS PubMed
- M. Falk and M. Hausmann, Cancers, 2021, 13, 18 CrossRef CAS PubMed
- I. Alekseenko, L. Kondratyeva, I. Chernov and E. Sverdlov, Int. J. Mol. Sci., 2023, 24, 2796 CrossRef CAS PubMed
- B. D. Ripley, J. R. Stat. Soc. Ser. B (Methodol.), 1977, 39, 172–192 CrossRef
- J. Schlessinger, Cell, 2002, 110, 669–672 CrossRef CAS PubMed
- H. Ogiso, R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J.-H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu and S. Yokoyama, Cell, 2002, 110, 775–787 CrossRef CAS PubMed
- https://www.scbt.com/scbt/product/egfr-antibody-r-1 , Santa Cruz Biotechnology.
- https://www.cellsignal.com/products/primary-antibodies/egf-receptor-viii-d6t2q-xp-rabbit-mab/64952 , Cell Signaling Technology.
- Y. H. Tan, M. Liu, B. Nolting, J. G. Go, J. Gervay-Hague and G.-Y. Liu, ACS Nano, 2008, 2, 2374–2384 CrossRef CAS PubMed
- M. A. Lemmon, Exp. Cell Res., 2009, 315, 638–648 CrossRef CAS PubMed
- G. Guigas and M. Weiss, Biochim. Biophys. Acta, 2016, 1858, 2441–2450 CrossRef CAS PubMed
- U. Schmidt, G. Guigas and M. Weiss, Phys. Rev. Lett., 2008, 101, 128104 CrossRef PubMed
- D. Morozova, M. Weiss and G. Guigas, Soft Matter, 2012, 8, 11905 RSC
- A. Hofmann, M. Krufczik, D. W. Heermann and M. Hausmann, Int. J. Mol. Sci., 2018, 19, 2263 CrossRef PubMed
- G. Máté, A. Hofmann, N. Wenzel and D. W. Heermann, Biochim. Biophys. Acta, 2014, 1838, 1180–1190 CrossRef PubMed
- P. Jaccard, Bull. Soc. Vaudoise Sci. Nat., 1901, 37, 547–579 Search PubMed
- H. Hahn, C. Neitzel, O. Kopečná, D. W. Heermann, M. Falk and M. Hausmann, Cancers, 2021, 13, 5561 CrossRef CAS PubMed
- J. Dahm-Daphi, Int. J. Radiat. Biol., 1996, 69, 615–621 CrossRef CAS PubMed
- https://www.thermofisher.com/antibody/product/goat-anti-mouse-igg-h-l-cross-adsorbed-secondary-antibody-polyclonal/a-11001 , Thermo Fisher Scientific.
- https://www.thermofisher.com/antibody/product/goat-anti-rabbit-igg-h-l-highly-cross-adsorbed-secondary-antibody-polyclonal/a-11037 , Thermo Fisher Scientific.
-
F. Grüll, M. Kirchgessner, R. Kaufmann, M. Hausmann and U. Kebschull, Proc. – 21st Int. Conf. Field Program. Logic Appl, FPL, 2011, pp. 1–5 Search PubMed
- E. Schubert, J. Sander, M. Ester, H. P. Kriegel and X. Xu, ACM Trans. Database Syst., 2017, 42, 1–21 CrossRef
- E. Sezgin, I. Levental, S. Mayor and C. Eggeling, Nat. Rev. Mol. Cell Biol., 2017, 18, 361–374 CrossRef CAS PubMed
- R. B. Luwor, H.-J. Zhu, F. Walker, A. A. Vitali, R. M. Perera, A. W. Burgess, A. M. Scott and T. G. Johns, Oncogene, 2004, 23, 6095–6104 CrossRef CAS PubMed
- L. Li, S. Chakraborty, C.-R. Yang, K. J. Hatanpaa, D. J. Cipher, V. T. Puliyappadamba, A. Rehman, A. J. Jiwani, B. Mickey, C. Madden, J. Raisanen, S. Burma, D. Saha, Z. Wang, S. C. Pingle, S. Kesari, D. A. Boothman and A. A. Habib, Oncogene, 2014, 33, 4253–4264 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01570c |
| ‡ Equal contribution. |
| § Present addresses: School of Engineering and Applied Sciences, Harvard University, 9 Oxford St, 02138 Cambridge, MA, USA. |
| ¶ Present addresses: KU Leuven, Department of Biosystems, Division of Animal and Human Health Engineering, Geel Campus, Kleinhoefstraat 4, 2440 Geel, Belgium. |
| This journal is © The Royal Society of Chemistry 2024 |