
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorinated polymer zwitterions on gold nanoparticles: patterned catalyst surfaces guide interfacial transport and electrochemical CO2 reduction†
Qiang
Luo
 a,
Joseph
Tapia
e,
Le
Zhou
d,
Chung-Hao
Liu
bf,
Maham
Liaqat
a,
Hanyi
Duan
b,
Zhefei
Yang
d,
Mu-Ping
Nieh
bc,
Todd
Emrick
*d,
Peng
Bai
*e and
Jie
He
*ab
a,
Joseph
Tapia
e,
Le
Zhou
d,
Chung-Hao
Liu
bf,
Maham
Liaqat
a,
Hanyi
Duan
b,
Zhefei
Yang
d,
Mu-Ping
Nieh
bc,
Todd
Emrick
*d,
Peng
Bai
*e and
Jie
He
*ab
aDepartment of Chemistry, University of Connecticut, Storrs, CT 06269, USA. E-mail: jie.he@uconn.edu
bPolymer Program, Institute of Materials Science, University of Connecticut, Storrs, CT 06269, USA
cDepartment of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA
dPolymer Science and Engineering Department, Conte Center for Polymer Research University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: tsemrick@mail.pse.umass.edu
eDepartment of Chemical Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA. E-mail: pengbai@umass.edu
fNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 29th July 2024
Abstract
We report the use of fluorinated polymer zwitterions to build hybrid systems for efficient CO2 electroreduction. The unique combination of hydrophilic phosphorylcholine and hydrophobic fluorinated moieties in these polymers creates a fractal structure with mixed branched cylinders on the surface of gold nanoparticles (AuNPs). In the presence of these polymers, the CO faradaic efficiency improves by 50–80% in the range of −0.7 V to −0.9 V. The fractal structures have a domain size of ∼3 nm, showing enhanced mass transfer kinetics of CO2 approaching the catalyst surfaces without limiting ion diffusion. The phase-separated hydrophilic and hydrophobic domains offer separated channeling to water and CO2, as confirmed by attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and molecule dynamic (MD) simulations. H2O molecules permeate extensively into the polymer layer that adsorbs on zwitterions, forming continuous chains, while CO2 molecules strongly associate with the fluorinated tails of fluorinated polyzwitterions, with oxygen facing the positively charged amine groups. Overall, this coupling of zwitterion and fluorocarbon in a polymer material creates new opportunities for defining microenvironments of metallic nanocatalysts in hybrid structures.
Introduction
The sustainable conversion of CO2 into valuable chemicals and fuels has attracted considerable attention as a means to reduce CO2 emission.1–4 Electroreduction of CO2 in an aqueous electrolyte stands out as a potential solution to achieve CO2 cycling in the form of C1 and C2 fuels, making use of electricity generated from renewable sources.5–8 CO2 electroreduction, however, relies on water as a proton source to drive this chemical transformation.9 The catalyst-electrolyte-reactant (solid–liquid) interface is strongly hydrated during electrolysis.10,11 At this interface, an undesirable competitive proton reduction, known as the hydrogen evolution reaction (HER), occurs at a similar thermodynamic potential.12 Hydrogen production during electroreduction of CO2 therefore lowers the selectivity or faradaic efficiency (FE), the efficiency associated with utilizing electricity to carry out CO2 transformation.13 Happening at the interface of nanocatalysts, the electroreduction of CO2 is a complex process that is affected by various microenvironmental characteristics. These variables may greatly impact the efficiency, selectivity, and overall performance of the process.14 One straightforward solution is to control the localized proton/CO2 concentrations at the solid–liquid interface.10,11,15 A higher localized pH, e.g., in gas-flow cells or porous catalysts, can lower the proton concentrations while at the same time, it can promote the selectivity for CO2 electroreduction.16–23 Notably, mesoporous Au nanostructures with an inverse opal structure could increase the selectivity for CO2 electroreduction by suppressing HER through diffusional limitations of protons imposed by a porous electrode.24 Alternatively, by tuning the cation species in electrolytes, the strong diploe field created by the hydrated metal cations near the electrode can dramatically lower the adsorption energy of *CO2 and *CO.25 For example, Cs+ ions increase the activity of Ag electrodes by 2.4 times, from 59.1% to 80.3%, relative to Li+ ions.26Modifying nanoparticle catalysts with surface ligands represents an attainable solution to addressing this unmet challenge at the solid–liquid interface.17,27–32 The key feature of surface-bound ligands is to create a microenvironment that contributes surface gating, i.e., mass transporting of reactants and products.11,33–35 Such ligands are well-known to control surface hydrophobicity of nanocatalysts.36–39 By adding hydrophobic surface ligands, surface hydrophobicity has been demonstrated to change the product selectivity on Au,33,34,40 Ag41,42 and Cu.11,43,44 On Cu dendrites, hydrophobic 1-octadecanethiol (ODT) dramatically changed the metal surface from hydrophilic to superhydrophobic with a water contact angle of 153°.11 Consequently, the hydrophobic pocket captured gaseous CO2 and favored its reduction with a selectivity ∼90% for CO2 reduction while suppressing HER reaction below 10% at −1.6 V vs. reversible hydrogen electrode (RHE, the same hereafter).11 Unfortunately, this hydrophobicity also lowers the overall activity of Cu because of limited surface wetting (ion diffusion) by the electrolyte. Alternatively, ligands that balance surface hydrophobicity and hydrophilicity create a more dynamic interface without limiting molecular diffusion, with additionally balanced accessibility of CO2 and water, being key for harmonizing catalyst reactivity without compromising activity.18,45
Here we describe a method to control the microenvironment of nanoscale catalysts using polymer zwitterions that balance mass transport of CO2 and protons under CO2 electroreduction in an aqueous electrolyte (Scheme 1). The extreme hydrophilicity of zwitterions, such as poly(2-methacryloyloxyethyl phosphoryl choline) (polyMPC, P1), with inner-salt phosphorylcholine (PC) side chains are offset by the integration of fluorocarbon groups into choline phosphate-based polymers P2 and P3.46 Using these fluorozwitterion copolymers, the CO FE on gold nanoparticles (AuNPs, ∼3 nm) improved from ∼50% to 80%, with perfluorinated P3 in the range of −0.7 V to −0.9 V, while the partial current density for HER was suppressed by ∼50%. The phase-separated hydrophilic zwitterion domains and hydrophobic fluorocarbon domains offer channels for water and CO2, as confirmed by attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and molecule dynamic (MD) simulations. Further analysis reveals that water molecules permeate extensively into the polymer zwitterion channels, forming continuous chains with 1.9 hydrogen bonds per water molecule. Similarly, CO2 molecules strongly associate with the fluorinated tails of P3, with oxygen atoms facing the ammonium groups. Therefore, the desired amphiphilic interfaces built by the combination of perfluorinated alkanes and hydrophilic phosphorylcholine groups provide a means by which to control mass transport at the complex interface of catalyst-electrolyte-reactant, a promising feature for designing new, efficient catalytic systems for CO2 electroreduction.
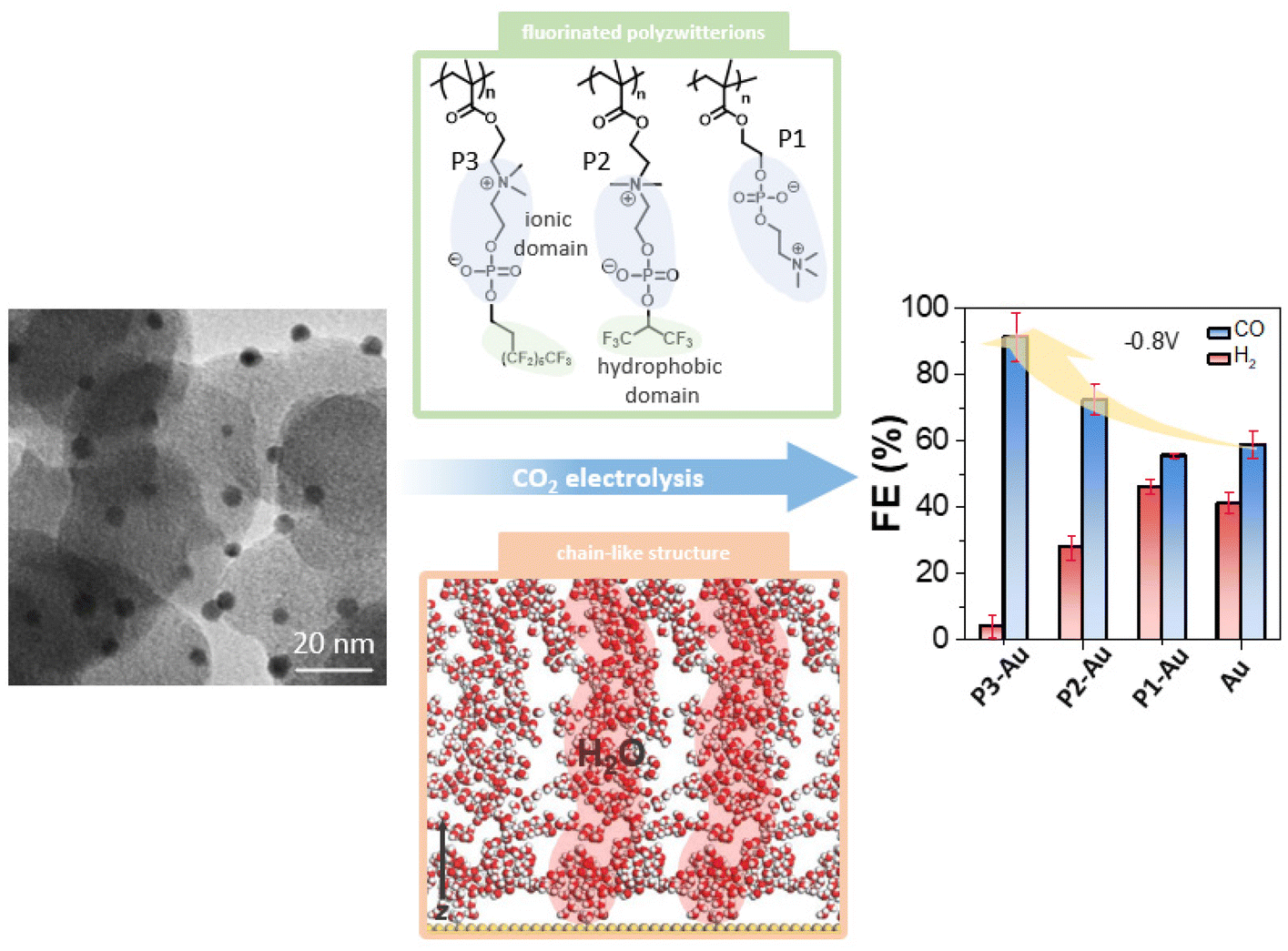 | ||
| Scheme 1 Scheme illustrating the use of fluorinated polymer zwitterions to control the interfacial microenvironment of AuNPs with enhanced selectivity for CO2 reduction. | ||
Results and discussion
Polymer zwitterions P1–P3 were prepared by reversible addition–fragmentation chain-transfer (RAFT) polymerization, using methods described previously.46 P1 and P2 were isolated following their precipitation in diethyl ether, dialysis against deionized water, and lyophilization. P3 was purified by precipitation into a mixture of acetone and diethyl ether, then acetone, and then diethyl ether, followed by drying under vacuum. The number-average molecular weight of polymers was 16.3 kg mol−1 for P1 (dispersity Đ 1.1), 21.5 kg mol−1 for P2 (Đ 1.25) and 43.9 kg mol−1 for P3 (Đ 1.23), respectively (Fig. S1†). AuNPs (an average diameter of 3.2 nm, Scheme 1) were synthesized by direct chemical reduction of HAuCl4 with NaBH4. Before surface modification, AuNPs were first physically adsorbed onto activated carbon (Printex U, ∼50 nm nanospheres). The loading ratio of AuNPs on carbon (Au/C) was about 2.5 wt%, also confirmed by scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX) (see ESI for synthetic details, Fig. S4†). For ligand modification, Au/C catalysts were mixed with polymer zwitterions in trifluoroethanol, which is a good solvent for the polymers. The trithioester chain-ends of P1–P3 serves as a ligand for grafting to AuNP surface.47 The typical surface modification procedure was carried out over 12 h to promote high surface grafting and the resultant Au/C catalysts were washed thoroughly with centrifugation to remove unbound polymers. The surface modification was confirmed by Fourier-transform infrared spectroscopy (FT-IR) (with pure Au supported on a glass slide see Fig. S2†). After surface grafting, the characteristic vibrational frequency of the polymers with Au/C was compared to that of the free polymers. Taking P1 as an example, the characteristic peaks of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1229 cm−1, P–O at 1065 cm−1, and P–O–C at 1482 cm−1 with quaternary ammonium head-group (–N+(CH3)3) at 971 cm−1 was seen, indicating successful modification of P1 on the Au nanocatalysts.48,49 Similarly, typical vibrational peaks of –CF2– and –CF3 at 1100–1300 cm−1 were observed for Au/C catalysts modified with P2 and P3.50 We used thermogravimetric analysis to analyse the grafting density of polymer zwitterions on AuNPs after surface modification process. The ‘grafting to’ method showed a 15.7% weight loss for P1, while those were 14.0%, 16.4% for P2 and P3, respectively (Fig. S3†). As a sequence, P1 gave a slightly higher grafting density of 0.76 chains nm−2; while the grafting density was ∼0.30 chains nm−2 for P2 and P3. Those results are typical for polymer-grafted AuNPs prepared through the grafting-to approach.51–53
O at 1229 cm−1, P–O at 1065 cm−1, and P–O–C at 1482 cm−1 with quaternary ammonium head-group (–N+(CH3)3) at 971 cm−1 was seen, indicating successful modification of P1 on the Au nanocatalysts.48,49 Similarly, typical vibrational peaks of –CF2– and –CF3 at 1100–1300 cm−1 were observed for Au/C catalysts modified with P2 and P3.50 We used thermogravimetric analysis to analyse the grafting density of polymer zwitterions on AuNPs after surface modification process. The ‘grafting to’ method showed a 15.7% weight loss for P1, while those were 14.0%, 16.4% for P2 and P3, respectively (Fig. S3†). As a sequence, P1 gave a slightly higher grafting density of 0.76 chains nm−2; while the grafting density was ∼0.30 chains nm−2 for P2 and P3. Those results are typical for polymer-grafted AuNPs prepared through the grafting-to approach.51–53
The electrochemically active surface area (ECSA) of AuNPs was measured by cyclic voltammetry (CV) to assess the impact of polymer zwitterions modification on the accessibility of Au nanocatalysts at electrode–electrolyte interfaces. The CVs were collected in a N2-saturated 0.5 M H2SO4 environment at a scan rate of 100 mV s−1 (Fig. 1a). As we can see, during the cathodic scan, a reduction peak appeared at ∼1.15 V vs. RHE corresponding to the electrochemical reduction of the surface oxygenated monolayer, correlated with the ECSA of AuNPs (Tables S1–S4†). As shown in Fig. 1b, Au/C catalyst in the absence of ligands has an ECSA of 2.8 ± 0.06 cm2 μg−1. With P1, there is a marginal ECSA change of 2.7 ± 0.03 cm2 μg−1. Hydrophobic polyzwitterions with fluorinated moieties cause a reduction of surface accessibility of Au. For Au-P2/C and Au-P3/C, the ECSA reduced to 2.1 ± 0.01 and 1.9 ± 0.01 cm2 μg−1, respectively, attributed to fluorinated moieties blocking contact of ions for electrochemical redox. Linear sweep voltammograms (LSVs) in CO2-saturated 0.1 M KHCO3 are given in Fig. 1c and d, where the current density was normalized to geometric electrode surface area and ECSA, respectively. In both cases, polymer zwitterions improved the total current density of Au/C as compared to that without ligands. This enhancement was more pronounced when normalized to ECSAs, as evidenced in Fig. 1d.
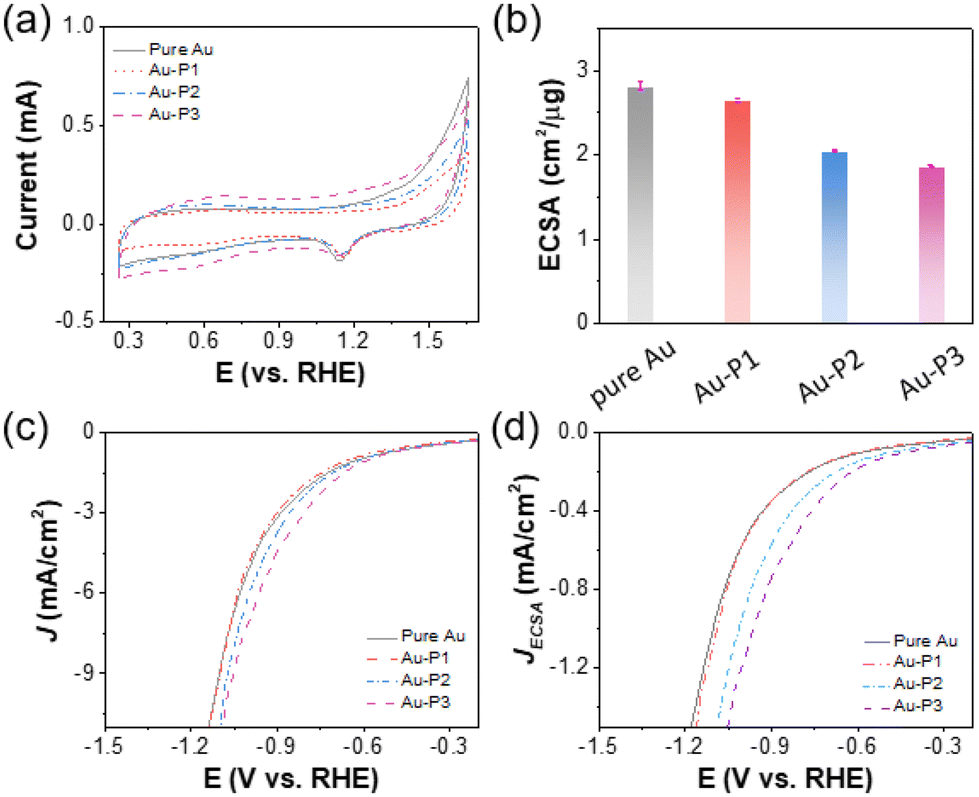 | ||
| Fig. 1 (a) CV analysis of Au/C modified with fluorinated polymer zwitterions recorded under a scan rate of 100 mV s−1 in N2-saturated 0.5 M H2SO4. (b) ECSAs of AuNPs modified with different fluorinated polymer zwitterions. LSV curves measured under a scan rate of 10 mV s−1 in CO2-saturated 0.1 M KHCO3 normalized to (c) geometric area of electrodes and (d) ECSA of AuNPs. | ||
A more quantitative CO2 electroreduction analysis is presented in Fig. 2. Three parameters, including CO FE, the partial current density (JCO and JH2), and the product ratio (nCO/nH2) were employed to compare the electrocatalytic performance of AuNPs with different polyzwitterions as well as citrate-capped Au nanocatalysts as a control. To measure the product of CO2 electroreduction, a customized H-cell separated using a Nafion membrane was used for all electrocatalytic experiments. In the absence of polyzwitterions, Au/C has low selectivity for CO2vs. CO, with CO FE ranging of 54%–60% in the potential window of −0.7 to −0.9 V, given their size effects with corner and edge sites favoring HER. Those results agreed well with the literature.54,55Fig. 2c plots the potential dependent of partial current density (JCO and JH2) calculated out from the corresponding FE and overall current density from LSVs. An accelerated kinetics for CO2 electroreduction was seen under more negative applied bias upon work electrode. Specifically, JCO reached −1.7 mA cm−2 with JH2 of −1.4 mA cm−2 at −0.9 V. In the potential window of −0.7 to −0.9 V, unmodified Au/C exhibited a CO to H2 product ratio around 1.4 (Fig. 2e).
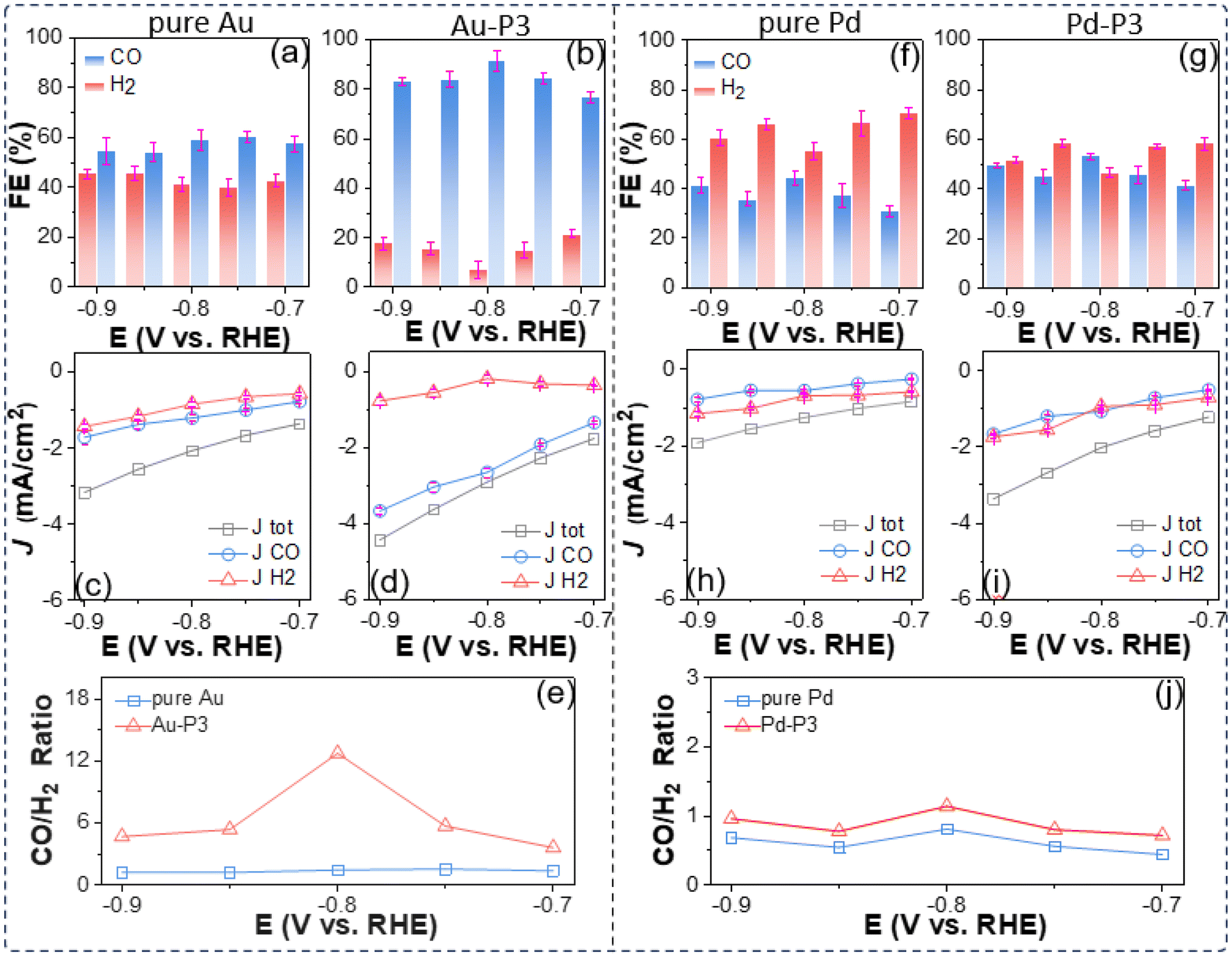 | ||
| Fig. 2 Electrocatalytic performance for CO2 reduction: FE and (partial) current density using Au (a and c), Au-P3 (b and d) and the corresponding products ratio (e). FE and (partial) current density using pure Pd (f and h), Pd-P3 (g and i) and the corresponding products ratio (j). A carbon rod was used as a counter electrode and all measurements were carried out in CO2 saturated 0.1 M KHCO3. | ||
The use of polymer zwitterions resulted in AuNPs exhibiting enhanced selectivity for CO over H2; in the presence of P3, the CO FE increased to above 80% (Fig. 2b), representing a 20–30% improvement in comparison with unmodified Au/C and a notable enhancement in JCO, outperforming other Au-based nanocatalysts (Table S5†) for CO2 electroreduction.54,56–59 For instance, at −0.9 V, Au-P3/C achieved a JCO of 3.7 ± 0.08 mA cm−2 (Fig. 2d), approximately 2.6 times higher than that of for Au/C. Fluorinated polymer zwitterions also suppressed the activity to HER. The JH2 values were in the range from 0.21 ± 0.10 to 0.79 ± 0.12 mA cm−2 for Au-P3 from −0.7 V to −0.9 V, amounting to approximately 50% decrease to produce hydrogen relative to pure AuNPs. The product ratio of CO/H2 increased to around 5. At −0.8 V, Au-P3/C exhibited a CO/H2 ratio of 12.7, as a 9-fold selectivity increase for CO2 reduction compared to Au/C (Fig. 2e). The embedded fluorinated groups into polyzwitterions are critical for selectivity enhancement. In case of hydrophilic P1, nCO/nH2 dropped to ∼1.2, comparable to that of unmodified Au/C (Fig. S5†). In P2, with an hexafluoro isopropyl group, the decrease of fluorocarbon content correlated with a reduction in CO/H2 ratio to 3.5 at −0.9 V (Fig. S5†). Those results suggest that the presence of the fluorinated domains is essential to create a microenvironment that favors reducing CO2 over protons. Hydrophilic P1 onto Au/C nanocatalysts had a negligible effect on the overall current density (Jtot) or JCO. Adding fluorinated zwitterionic polymers introduced an elevation of JCO, indicating that the amphiphilic interfacial property of fluorinated zwitterionic polymers drives such enhancement.
To validate whether such enhancement resulted from the polyzwitterions, additional control experiments were conducted on a commercially available Pd/C (5 wt% Pd loading). With P3, fluorinated polyzwitterions resulted in an increase of about 10% in the selectivity of Pd/C catalyst to CO2 reduction within the potential range of −0.7 V to −0.9 V (Fig. 2f and g and Table S6†). Likewise, hydrophobic polyzwitterions enhanced the activity of Pd/C compared to the one without modification (Fig. S6†). Additionally, JCO approached 1.67 ± 0.03 mA cm−2 at −0.9 V, around 2.1 times larger than that of Pd/C (Fig. 2h and i). At −0.9 V, commercial Pd/C is more selective to HER and the FE of CO was 41%; after modification, Pd-P3 had a CO FE of 50%. Similarly, a slightly higher product ratio around 1 for Pd-P3 was observed (Fig. 2j).
In situ ATR-SEIRAS spectroscopy was further used to study how fluorinated polymer zwitterions would change the solid–liquid interface during electrocatalysis. The SEIRAS spectroscopy was conducted on a gold film electrode in the potential range of 0 to −0.9 V with linear scanning voltammetry at a scan rate of 5 mV s−1. Unbiased gold film electrode was used as a background. Any positive absorption peaks (peak up) signify the formation or adsorption of molecular species on the surface, e.g., reaction intermediates during CO2 electroreduction. Conversely, the downward peaks (peak down) are assigned to desorbed species. Fig. 3a shows the recorded LSV curve in the range of 0 to −0.9 V of pure Au electrode. The corresponding ATR-SEIRAS spectra are presented in Fig. 3b. As cathodic bias increasing, two peaks were seen: a well-defined peak at 1640 cm−1, representing O–H bending (δO–H) and a broad peak centered at 3300–3600 cm−1 for O–H stretching (νO–H).60,61 In addition, the peak intensity study was carried out along with the change of applied bias (Fig. 3e). The O–H stretching peak intensity increase rapidly from −0.3 V, indicating the H-down orientation of adsorbed water molecules on the surface of work electrode. The peak centered around 3400 cm−1. With the increase of cathodic bias, the O–H stretching peak had a red-shift forward lower frequency centered at 3350 cm−1, indicating the dynamic process of water absorbed onto the gold surface.60 At around −0.8 V, the peak intensity plateaued, suggesting the formation of densely packed water molecules on the surface. Those adsorbed water molecules show weak hydrogen bonding as approaching the Au surface.
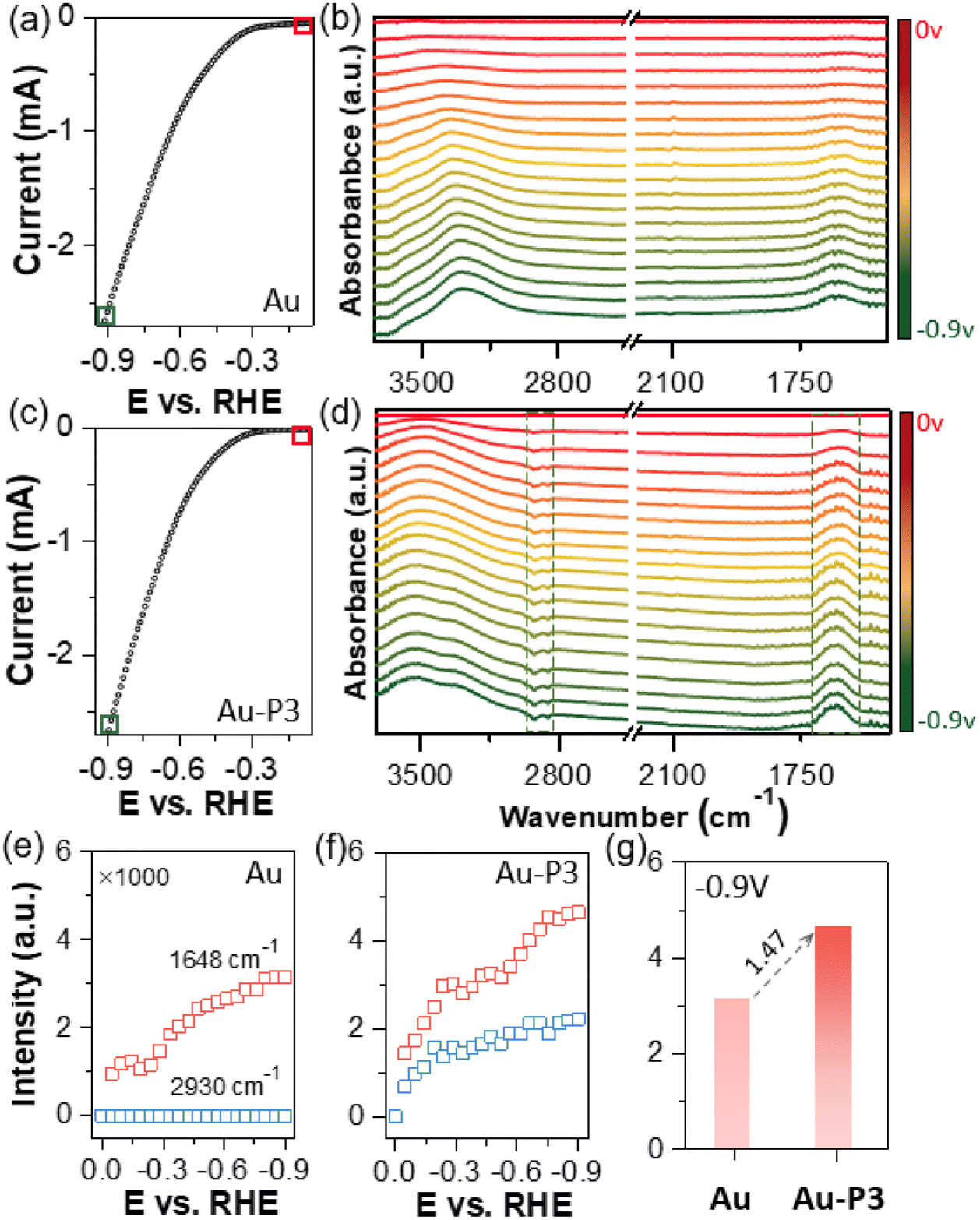 | ||
| Fig. 3 In situ ATR-SEIRAS study of Au electrodes modified with different fluorinated polymer zwitterions. LSV scans in CO2 saturated 0.1 M KHCO3 and in situ ATR-SEIRAS spectra obtained in the range from 0 to −0.9 V for Au (a and b) and Au-P3 (c and d). Peak intensity study for AuNPs (e) and Au-P3 (f) with various reductive potentials. (g) O–H bending model at 1648 cm−1 under −0.9 V. | ||
In the presence of P3, the ATR-SEIRAS spectra displayed noticeable differences. Firstly, the O–H stretching and bending peaks appeared more pronounced under identical potentials (Fig. 3g). The O–H stretching peak also broadened but it centered at 3520 cm−1, suggesting that water molecules did not form extended hydrogen bonding.62 With applied cathodic bias, the O–H stretching peak expanded with two shoulders at ca. 3520 and 3320 cm−1 (Fig. 3c and d). The lower frequency peak nearly overlapped with the O–H stretching peak on pure Au surface, assigned to water molecules with intermolecular hydrogen bonding.63,64 The new peak appeared at around 3520 cm−1 were likely attributed to less hydrogen bonded water. This was also confirmed by Au-P1 and Au-P2. In the presence of polymer zwitterions, a similar O–H stretching peak above 3500 cm−1 was observed during electrocatalysis (Fig. S7†). Those water molecules likely reside in zwitterion domains, where choline phosphate groups form strong ion-dipole interaction with water (i.e., hydration). Lastly, with ligands modified Au electrodes as references, the downward peaks at 3000–2800 cm−1 were observed along with cathodic applied bias for Au-P3, confirmed from unmodified Au under identical condition with no signals. These peaks are assigned to the C–H stretching of polymer backbones and side chains (see details in Fig. S2†), indicating the decay or the orientation away of modified ligands from the electrode surfaces during electrocatalysis (Fig. 3f).
The wettability of polyzwitterions modified surfaces were examined to investigate the interfacial properties where the electrocatalysis exclusively occurred. As studied by previous literature,65 fluorocarbon polymers took on appreciable hydrophilicity while small CAs of P1 and P2 are reasonable due to ionic phosphorylcholine. The change of surface hydrophobicity aligned with the electrocatalytic performance of CO2 reduction, including CO partial current density (JCO) and CO FE. This was confirmed from ligands modification of P1 and P2 with less hydrophobicity showing a lower CO FE and smaller JCO (Fig. S5†), as compared to P3. Furthermore, small-angle X-ray scattering (SAXS) was used to investigate the internal structures of fluorinated polyzwitterions. A distinctive peak at q = 0.21 Å−1, corresponding to a D-spacing 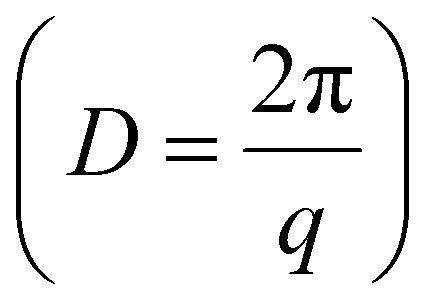 of 3.0 nm, was observed because of the phase separation from hydrophilic and electrostatic interactions of choline phosphate (CP) groups (Fig. S8†). The scattering pattern of P3 can be fitted by a combined cylindrical and lamellar model, indicating the presence of branched cylinders and unstacking lamellae in its fractal morphology. The molecular dynamic (MD) simulation outcome demonstrates the regular separation of P3 with a d-spacing of 2.9 nm (Fig. 4a), consistent with the spacing derived from the corresponding peak in the SAXS data of P3. The phase separation of ionic and fluorinated moieties into two domains would have very different affinity to substrates, e.g., proton and CO2. Together with our SEIRAS results, we would deduce that ionic domains of zwitterions favor the interaction with water, rather than hydrophobic perfluorinated domains.
of 3.0 nm, was observed because of the phase separation from hydrophilic and electrostatic interactions of choline phosphate (CP) groups (Fig. S8†). The scattering pattern of P3 can be fitted by a combined cylindrical and lamellar model, indicating the presence of branched cylinders and unstacking lamellae in its fractal morphology. The molecular dynamic (MD) simulation outcome demonstrates the regular separation of P3 with a d-spacing of 2.9 nm (Fig. 4a), consistent with the spacing derived from the corresponding peak in the SAXS data of P3. The phase separation of ionic and fluorinated moieties into two domains would have very different affinity to substrates, e.g., proton and CO2. Together with our SEIRAS results, we would deduce that ionic domains of zwitterions favor the interaction with water, rather than hydrophobic perfluorinated domains.
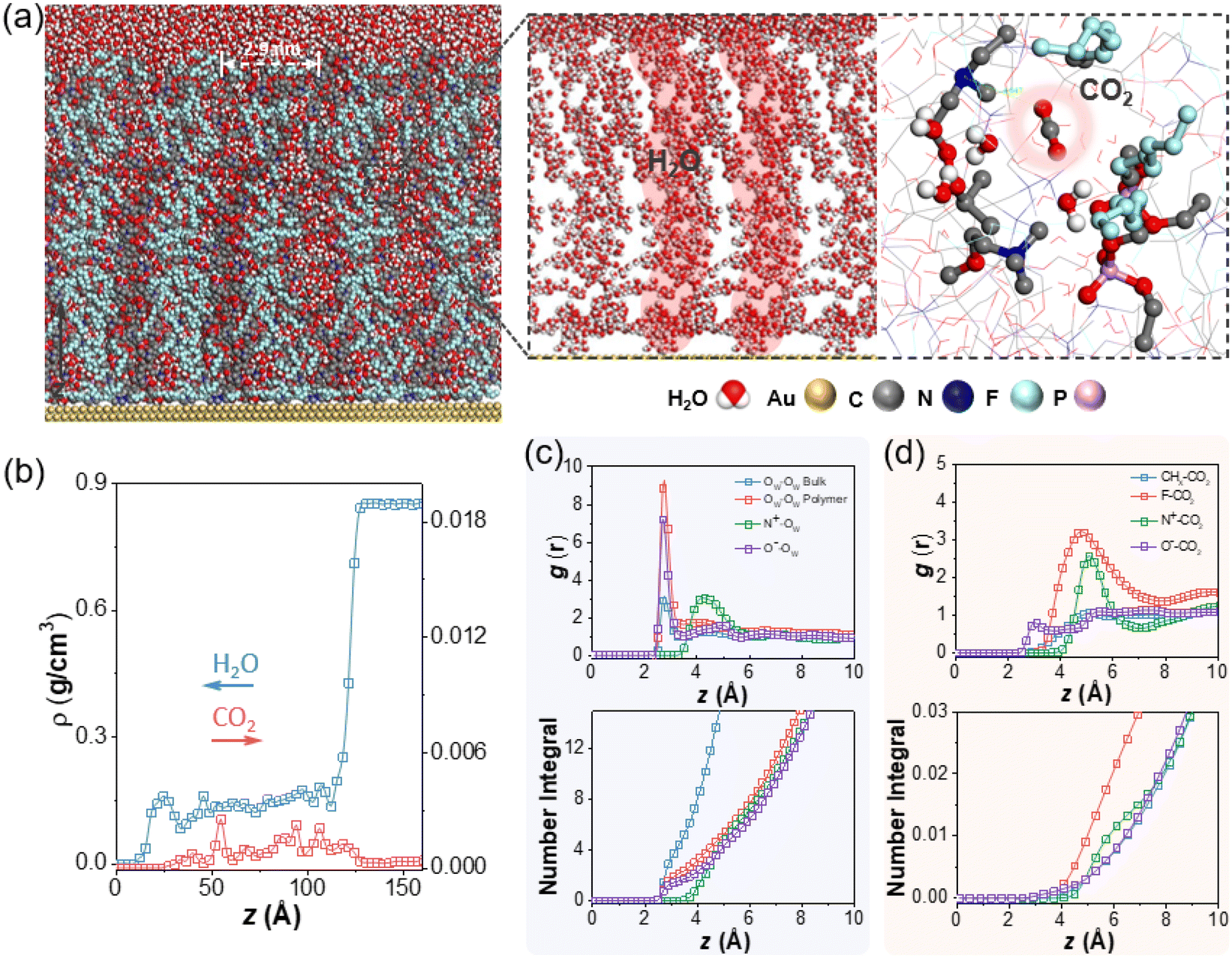 | ||
| Fig. 4 (a) Snapshot of a representative system configuration (left) and zoomed in area of water distribution and solvated CO2 molecule (right). (b) Density profiles as a function of z coordinates, with water and CO2 shown in blue and red. (c) Radial distribution functions and number integrals for O–O of water molecules (OW–OW) at z > 12.76 nm (bulk region), OW–OW at z < 10.95 nm (polymer layer), N+–OW between P3 and water, O−–OW between the phosphorous oxygen in P3 and water. (d) Radial distribution functions and number integrals for nonpolar CHx–CO2 between polymer backbone and methyl branches and CO2, CFx–CO2 between fluorinated tails of the polymers and CO2, N+–CO2 between P3 and CO2, O−–CO2 between the phosphorous oxygen in P3 and CO2. | ||
The effect of zwitterionic ligands on the local solvation environment around AuNPs was investigated using MD simulations. As shown in Fig. 4a, a gold slab was functionalized with P3 ligands at a surface coverage of 0.29 chains per nm2, with each P3 chain consisting of 50 repeating units and the entire system solvated in water with a bulk water layer thickness of ∼ 4.8 nm (Fig. 4b, z > 12.8 nm). Compared to our previous study of AuNPs with hydrophobic polystyrene (Au-PS),33 the fluorinated zwitterionic P3 similarly resulted in an increased CO2 concentration in the polymer layer (Fig. 4b). Examining the radial distribution functions (RDFs) for CO2 (Fig. 4d) and representative snapshots (Fig. 4a), we found that CO2 molecules most strongly associate with the fluorinated tails of the polymer, with its oxygen atom pointing towards the positively charged amine groups. Similar results are also confirmed with various CO2 concentrations (Fig. S9†). However, unlike the case of hydrophobic ligands creating pockets of water clusters, water permeates into the P3 polymer layer more extensively to form continuous chains that adsorb preferentially on the zwitterions (Fig. 4c). The number integrals in Fig. 4c show that the first (r < 0.35 nm) and second (0.35 < r < 0.57 nm) solvation shells around phosphate anion contain 1.8 and 4 water molecules, and the loose structure around the ammonium cation atom (r < 0.63 nm) contains 8 water molecules. Similar continuous chains are also confirmed with different CO2 concentrations (Fig. S10†). To further contrast the water structures induced by the different polymer ligands, we performed a hydrogen-bonding analysis and calculated the tetrahedral order parameter. Here, two water molecules are considered to be connected by a hydrogen bond when rOO < 0.33 nm and ∠O–H⋯O > 113.58°.66 Using this criterion, bulk water forms ∼3.9 hydrogen bonds per molecule, corresponding to an extended tetrahedral network. By contrast, water molecules in the P3 layer form 1.9 hydrogen bonds by average, consistent with a chain-like structure, while water molecules in polystyrene have 3.1 hydrogen bonds, indicating a more localized cluster-like structure (Fig. S11†). To calculate the tetrahedral order parameter, q, for a water molecule in the polymer layer, its four nearest-neighbor water molecules were considered, regardless of whether they are hydrogen-bonded. The order parameter ranges from −3 to 1 (for a perfect tetrahedron). q reaches 0.6 for bulk water at room temperature, compared to q = 0.20 for the polystyrene ligand, while it is −0.17 in the P3 polymer region, indicating a substantially less ordered 3D structure.
To quantitatively assess how fluorinated polymer zwitterions influence the accessibility of Au related to ions and CO2, we further measured the interfacial mass transfer kinetics during electrocatalysis using three distinct molecular probes. Fig. 5a and b display the reversible redox CVs of the anionic probe, K3Fe(CN)6, on Au/C and Au-P3 electrode with scan rates ranging from 80 to 120 mV s−1. A clear linear relationship between peak current (Ip) and the square root of the scan rate (Fig. 5c) enabled examination of the diffusion coefficient of Fe(CN)63− using the Randles–Ševčík equation. The diffusion coefficient (D0) of ferricyanide anions is 1.7 × 10−6 cm2 s−1 in the absence of the polymer zwitterions. With polymer zwitterions, the D0 values exhibited negligible shifts: 1.7 × 10−6 cm2 s−1 for Au-P1, 1.4 × 10−6 cm2 s−1 for Au-P2, and 1.9 × 10−6 cm2 s−1 for Au-P3, respectively (Fig. S12† and Fig. 5d). Polyzwitterions did not slow down the diffusion of ferricyanide anions. Similarly, the diffusion coefficient of the cationic probe, Ru(NH3)63+, showed a similar trend (Fig. S13†). As Fe(CN)63− and Ru(NH3)63+ are significantly larger than proton and water, it is reasonable to assume that polyzwitterions would not block the diffusion of protons. The presence of ionic domains in polymer zwitterions would, in some cases, increase the diffusion of ions. For example, for large molecular probes e.g., ferrocene carboxylic acid (Fc-COOH) with slightly more hydrophobicity, the diffusion coefficient increased approximately 20% and 50% in the presence of P2 and P3 (Fig. 5d and Fig. S14†) as compared to pure Au. This enhancement could be attributed to the affinity of the probe for the hydrophobic fluorocarbon-rich domain. To gain insight into the diffusion kinetics of CO2 during electrocatalysis, electrochemical impedance spectroscopy (EIS) was used to analyze its diffusion at −0.7 V through a frequency range from 100 kHz to 1 Hz (Fig. S15†). The diffusion coefficient of CO2 approached to 1.3 and 2.5 × 10−9 cm2 s−1 for Au-P2 and Au-P3 with hydrophobic domains, respectively, approximately 2- and 4-fold increase compared to the unmodified Au/C. Even though the EIS measurement could not distinguish CO2 diffusion from proton contribution, the proton reduction contribution was not less significant for Au catalysts modified by P3.
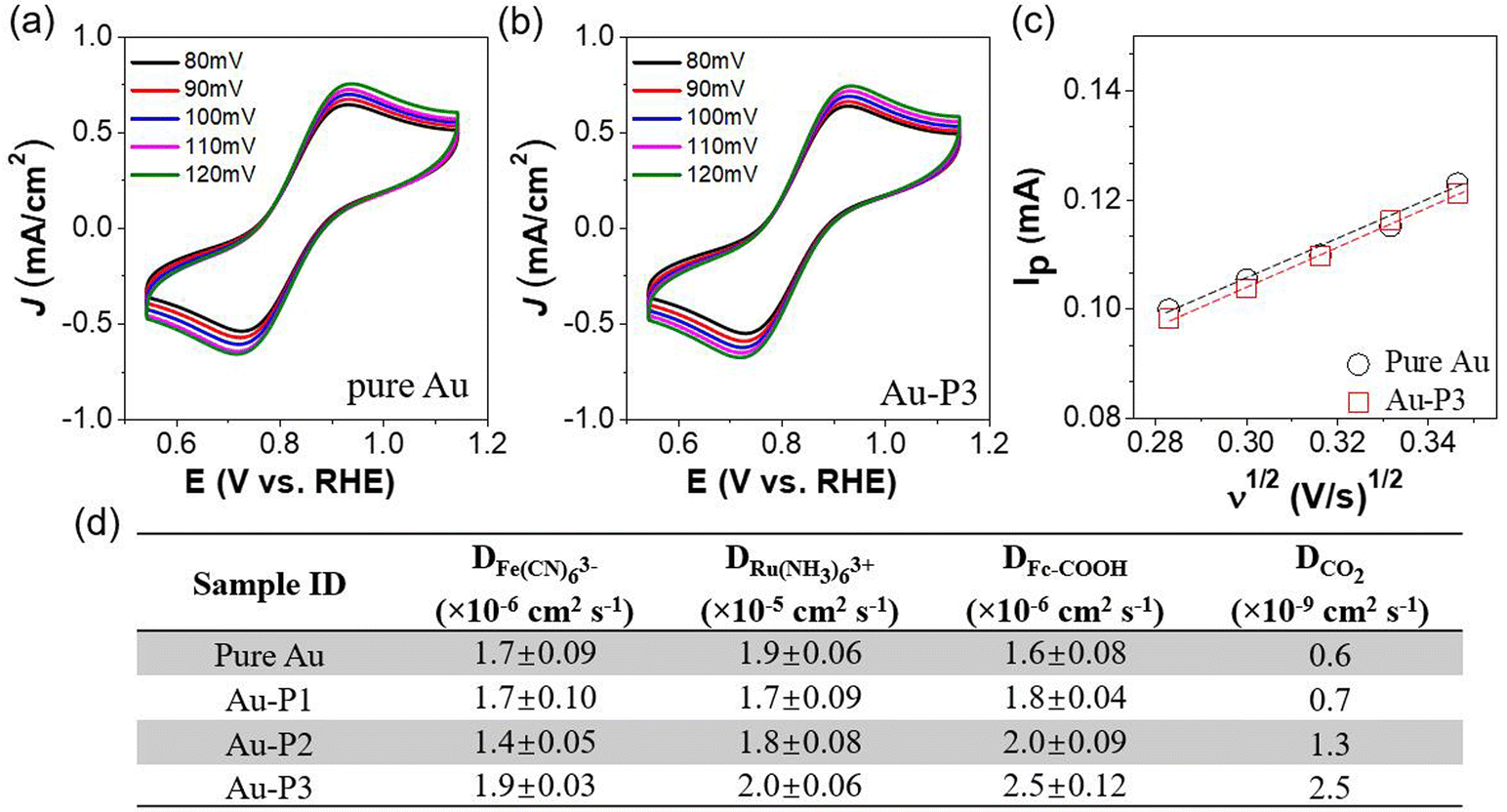 | ||
| Fig. 5 Mass transport properties of all catalysts. (a) CV scans of AuNPs and (b) Au-P3 measured with 5 mM K3Fe(CN)6 under various scan rates. (c) Linear relationship between peak current (Ip) and the square root of scan rates for pure Au and Au-P3. (d) Summary of diffusion coefficient (D) using various probe molecules including Fe(CN)63−, Ru(NH3)63+, Fc-COOH, and CO2. | ||
The diffusion of CO2 across polymer ligands was also analyzed by MD simulations, revealing the nanosecond time scale, CO2 motion is sub-diffusive (Fig. S16†) and exhibits a strong anisotropy in the Au-P3 system. If we were to compute an “effective” diffusivity based on the Einstein relationship, the overall diffusion coefficient of CO2 would be 2 × 10−6 cm2 s−1 while that along the z-axis would be 8 × 10−6 cm2 s−1. This anisotropy was not observed in the Au-polystyrene simulations in which both diffusion coefficients were around 2 × 10−6 cm2 s−1. Although these effective diffusivities are three orders of magnitude larger than those measured by the EIS experiments, the increase of CO2 diffusion is dependent on the hydrophobic perfluorinated moieties of P3. We speculate that the difference between experiments and simulation might be due to a combination of two factors. On one hand, the simulations did not account for chemical interactions and interconversion (e.g., carbonate/carbamate formation) between CO2 and water/zwitterions, which may inhibit CO2 diffusion. On the other hand, charge transfer reactions of CO2 may not be fast enough for the EIS measurement to determine true CO2 diffusivity. Based on the results above, the hydrophobicity from fluorinated subunits likely establishes a localized microenvironment that facilitates transport kinetics and enriches CO2 near the Au surface.
Conclusions
In summary, this study presents a method for controlling the microenvironment of nanocatalysts by balancing the mass transport of CO2 and protons during CO2 electroreduction through fluorinated polyzwitterions. The combination of zwitterions with fluorinated moieties, notably poly(PFO-MCP, P3), provides an effective way to enhance selectivity in CO2 electroreduction. CO FE increased from ca. 60% to 80% for perfluorinated P3 modified AuNPs in the range of −0.7 V to −0.9 V, that is higher than those of small Au nanocatalysts favoring hydrogen product. Furthermore, the partial current density for HER, JH2, is reduced by approximately 50%. The product ratio analysis indicates that the desired interface improved the CO/H2 ratio as high as 12.7, 9-fold higher than pure Au. As confirmed by ATR-SEIRAS, MD simulations, SAXS and diffusion estimation, the phase-separated hydrophilic ionic domains and fluorinated domains offered separated channels to regulate reactants, e.g., CO2 and water. The fluorinated tail acts to adsorb CO2 molecules; simultaneously, water molecules permeate extensively into the polymer layer on zwitterions, forming continuous chain-like structures with 1.9 hydrogen bonds per molecule in comparison to bulk water with 3.9 hydrogen bonds forming an extended tetrahedral network. Our results highlighted the critical role of localized microenvironments from bioinspired design of surface ligands with enhanced selectivity forward CO2 reduction.Author contributions
J. H. and T. E. conceived the concept. Q. L. and M. L. carried out the electrocatalysis experiments, electrochemistry spectroscopic study and analysis. P. B. and J. T. set out the MD simulation process. L. Z. and T. E. designed and synthesized polymer ligands. M-P. N, C-H. L, and H. D. did the SAXS test and analysis. Z. Y. did the surface contact angle analysis. J. H. and Q. L. took the lead in writing the manuscript. All authors here discussed the results and provide suggestions for manuscript.Data availibility
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
JH is grateful for the financial support from the National Science Foundation (CHE 2102245 on CO2 reduction). PB acknowledges the support from the National Science Foundation (CBET 2144360 on simulation). TE acknowledges support for the synthesis of polymer zwitterions from the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering (DE-SC0008876). The TEM images were obtained at the Biosciences Electron Microscopy Facility at the University of Connecticut. This work was also partially supported by the Green Emulsions Micelles and Surfactants (GEMS) Center of the University of Connecticut. The LiX beamline is part of the Center for BioMolecular Structure (CBMS), which is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). LiX also received additional support from NIH Grant S10 OD012331. As part of NSLS-II, a national user facility at Brookhaven National Laboratory, work performed at the CBMS is supported in part by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Program under contract number DE-SC0012704. The computer simulations used resources of the National Energy Research Scientific Computing Center (NERSC) through allocation ERCAP0028612 and of the Advanced Cyberinfrastructure Coordination Ecosystem (ACCESS) through allocation CTS190069.References
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustain. Chem., 2019, 16, 47–56 CrossRef.
- Y. Zhang, X. Wang, S. Zheng, B. Yang, Z. Li, J. Lu, Q. Zhang, N. M. Adli, L. Lei, G. Wu and Y. Hou, Adv. Funct. Mater., 2021, 31, 2104377 CrossRef CAS.
- B. Chen, B. Li, Z. Tian, W. Liu, W. Liu, W. Sun, K. Wang, L. Chen and J. Jiang, Adv. Energy Mater., 2021, 11, 2102152 CrossRef CAS.
- T. Wang, Q. Zhang, K. Lian, G. Qi, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, J. Colloid Interface Sci., 2024, 655, 176–186 CrossRef CAS PubMed.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- A. Razmjoo, L. Gakenia Kaigutha, M. A. Vaziri Rad, M. Marzband, A. Davarpanah and M. Denai, Renewable Energy, 2021, 164, 46–57 CrossRef CAS.
- Z. Yin, G. T. R. Palmore and S. Sun, Trends Chem., 2019, 1, 739–750 CrossRef CAS.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- R. Shi, J. Guo, X. Zhang, G. I. N. Waterhouse, Z. Han, Y. Zhao, L. Shang, C. Zhou, L. Jiang and T. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- Y. Chen, R. K. Miao, C. Yu, D. Sinton, K. Xie and E. H. Sargent, Matter, 2024, 7, 25–37 CrossRef CAS.
- X. Han, T. Mou, S. Liu, M. Ji, Q. Gao, Q. He, H. Xin and H. Zhu, Nanoscale Horiz., 2022, 7, 508–514 RSC.
- T. Wei, S. Zhang, Q. Liu, Y. Qiu, J. Luo and X. Liu, Acta Phys.-Chim. Sin., 2023, 39, 202207026 Search PubMed.
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z.-J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- A. S. Varela, Curr. Opin. Green Sustain. Chem., 2020, 26, 100371 CrossRef.
- C. Chen, H. Jin, P. Wang, X. Sun, M. Jaroniec, Y. Zheng and S. Z. Qiao, Chem. Soc. Rev., 2024, 53, 2022–2055 RSC.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- Z. Luo, Z. Yin, J. Yu, Y. Yan, B. Hu, R. Nie, A. F. Kolln, X. Wu, R. K. Behera, M. Chen, L. Zhou, F. Liu, B. Wang, W. Huang, S. Zhang and L. Qi, Small, 2022, 18, 2107799 CrossRef CAS PubMed.
- J. Y. T. Kim, C. Sellers, S. Hao, T. P. Senftle and H. Wang, Nat. Catal., 2023, 6, 1115–1124 CrossRef CAS.
- D. Raciti, T. Braun, B. M. Tackett, H. Xu, M. Cruz, B. J. Wiley and T. P. Moffat, ACS Catal., 2021, 11, 11945–11959 CrossRef CAS.
- X. Zhu, C. Tian, H. Wu, Y. He, L. He, H. Wang, X. Zhuang, H. Liu, C. Xia and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 43588–43594 CrossRef CAS PubMed.
- J. Ding, T. Wei, T. Hou, W. Liu, Q. Liu, H. Zhang, J. Luo and X. Liu, Nanoscale, 2024, 16, 10628–10636 RSC.
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 14834–14837 CrossRef CAS PubMed.
- X. Yang, H. Ding, S. Li, S. Zheng, J.-F. Li and F. Pan, J. Am. Chem. Soc., 2024, 146, 5532–5542 CrossRef CAS PubMed.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS PubMed.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS PubMed.
- X. Wei, G. Johnson, Y. Ye, M. Cui, S.-W. Yu, Y. Ran, J. Cai, Z. Liu, X. Chen, W. Gao, P. J. L. Bean, W. Zhang, T. Y. Zhao, F. A. Perras, E. J. Crumlin, X. Zhang, R. J. Davis, Z. Wu and S. Zhang, J. Am. Chem. Soc., 2023, 145, 14298–14306 CrossRef CAS PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883–17905 RSC.
- Q. Luo, H. Duan, M. C. McLaughlin, K. Wei, J. Tapia, J. A. Adewuyi, S. Shuster, M. Liaqat, S. L. Suib, G. Ung, P. Bai, S. Sun and J. He, Chem. Sci., 2023, 14, 9664–9677 RSC.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834–15840 CrossRef CAS PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
- K. Shi, Z. Ren, Z. Meng and X. Feng, ChemCatChem, 2023, e202301308 Search PubMed.
- X. Han, Q. Gao, Z. Yan, M. Ji, C. Long and H. Zhu, Nanoscale, 2021, 13, 1515–1528 RSC.
- Z. Wei, C.-H. Liu, Q. Luo, S. Thanneeru, A. M. Angeles-Boza, M.-P. Nieh and J. He, Mater. Chem. Front., 2023, 7, 2038–2048 RSC.
- Z. Wei and J. He, in Hairy Nanoparticles, 2023, pp. 375–400, DOI:10.1002/9783527835874.ch10.
- Y. Zhao, C. Wang, Y. Liu, D. R. MacFarlane and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1801400 CrossRef.
- D. Kim, S. Yu, F. Zheng, I. Roh, Y. Li, S. Louisia, Z. Qi, G. A. Somorjai, H. Frei, L.-W. Wang and P. Yang, Nat. Energy, 2020, 5, 1032–1042 CrossRef CAS.
- Y. Ma, J. Wang, J. Yu, J. Zhou, X. Zhou, H. Li, Z. He, H. Long, Y. Wang, P. Lu, J. Yin, H. Sun, Z. Zhang and Z. Fan, Matter, 2021, 4, 888–926 CrossRef CAS.
- Q. Chang, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, N. S. Marinkovic, A. A. Park, S. Kattel and J. G. Chen, JACS Au, 2022, 2, 214–222 CrossRef CAS PubMed.
- J. Bai, W. Wang and J. Liu, Chemistry, 2023, 29, e202302461 CrossRef CAS PubMed.
- K. Wei, H. Guan, Q. Luo, J. He and S. Sun, Nanoscale, 2022, 14, 11869–11891 RSC.
- L. Zhou, A. Triozzi, M. Figueiredo and T. Emrick, ACS Macro Lett., 2021, 10, 1204–1209 CrossRef CAS PubMed.
- B. Ebeling and P. Vana, Macromolecules, 2013, 46, 4862–4871 CrossRef CAS.
- L. Jiang, W. Zhu, H. Qian, C. Wang, Y. Chen and P. Liu, J. Mater. Chem. B, 2019, 7, 5078–5088 RSC.
- L. Zhu, Q. Jin, J. Xu, J. Ji and J. Shen, J. Appl. Polym. Sci., 2009, 113, 351–357 CrossRef CAS.
- I. C. P. Rodrigues, L. F. Woigt, K. D. Pereira, A. D. Luchessi, É. S. N. Lopes, T. J. Webster and L. P. Gabriel, J. Mater. Res. Technol., 2020, 9, 7777–7785 CrossRef CAS.
- H. Duan, Y. Lin and J. He, in World Scientific Reference on Plasmonic Nanomaterials, World Scientific, 2021, vol. 22, pp. 409–432 Search PubMed.
- Z. Wei, M. Kayceety, A. Price, K. Wei, Q. Luo, S. Thanneeru, S. Sun and J. He, ACS Appl. Mater. Interfaces, 2022, 14, 55227–55237 CrossRef CAS PubMed.
- L. Zhang, Z. Wei, M. Meng, G. Ung and J. He, J. Mater. Chem. A, 2020, 8, 15900–15908 RSC.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- D.-R. Yang, L. Liu, Q. Zhang, Y. Shi, Y. Zhou, C. Liu, F.-B. Wang and X.-H. Xia, Sci. Bull., 2020, 65, 796–802 CrossRef CAS PubMed.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D.-e. Jiang, S. Sun and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS PubMed.
- D. R. Kauffman, D. Alfonso, C. Matranga, H. Qian and R. Jin, J. Am. Chem. Soc., 2012, 134, 10237–10243 CrossRef CAS PubMed.
- X. Cai, G. Li, W. Hu and Y. Zhu, ACS Catal., 2022, 12, 10638–10653 CrossRef CAS.
- M. Dunwell, Y. Yan and B. Xu, Surf. Sci., 2016, 650, 51–56 CrossRef CAS.
- S. Zhu, T. Li, W.-B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- H. Wang, J. C. Wagner, W. Chen, C. Wang and W. Xiong, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 23385–23392 CrossRef CAS PubMed.
- K. Kong, A.-Z. Li, Y. Wang, Q. Shi, J. Li, K. Ji and H. Duan, Nat. Commun., 2023, 14, 6925 CrossRef CAS PubMed.
- Y.-H. Wang, S. Zheng, W.-M. Yang, R.-Y. Zhou, Q.-F. He, P. Radjenovic, J.-C. Dong, S. Li, J. Zheng, Z.-L. Yang, G. Attard, F. Pan, Z.-Q. Tian and J.-F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- L. Zhou, Z. Yang, J. N. Pagaduan and T. Emrick, Polym. Chem., 2023, 14, 32–36 RSC.
- J. R. Errington and P. G. Debenedetti, Nature, 2001, 409, 318–321 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01484g |
| This journal is © The Royal Society of Chemistry 2024 |