
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses and polymerization of monoterpene-based (meth)acrylates: IBO(M)A as a relevant monomer for industrial applications
Franziska
Obermeier
 a,
Dominik
Hense
a,
Paul N.
Stockmann
*b and
Oliver I.
Strube
*a
a,
Dominik
Hense
a,
Paul N.
Stockmann
*b and
Oliver I.
Strube
*a
aInstitute for Chemical Engineering, Universität Innsbruck, Innsbruck, Austria. E-mail: oliver.strube@uibk.ac.at
bFraunhofer IGB, Bio, Electro and Chemocatalysis BioCat, Straubing Branch, Schulgasse 11a, 94315, Straubing, Germany. E-mail: paul.stockmann@igb.fraunhofer.de
First published on 8th March 2024
Abstract
Monoterpenes, a class of natural compounds that occur as secondary plant metabolites, have received increasing attention as promising starting materials for the synthesis of (meth)acrylate monomers. This review addresses the synthesis, polymerization, and diverse applications of monoterpene (meth)acrylates and provides a comprehensive overview of the progress in this field in recent decades. Various synthetic routes and techniques are discussed, highlighting the adaptability of monoterpenes as starting materials. The different material properties, due to the diverse chemical structures, are examined in the context of different polymerization methods. The potential for tailoring polymer properties through the selection of appropriate monomers and polymerization techniques is demonstrated. The wide range of applications for such polymers, including their use in adhesives, coatings, biomedical materials and beyond, will be highlighted. Innovative applications are presented to highlight the practical relevance of these materials in various industries. This review is intended to offer researchers a comprehensive resource on the synthesis, polymerization, and applications of monoterpene (meth)acrylates. It highlights the versatile nature of these compounds, which offer a promising avenue for the development of environmentally friendly and high-performance polymeric materials.
 Franziska Obermeier | Franziska Obermeier studied renewable raw materials at Technical University of Munich and received her Master degree in 2021. Since graduating, she has been working as a research assistant at the University of Innsbruck. Under the supervision of Prof. Dr Strube, she is currently working on her doctoral thesis, in which she investigates the use of terpenes as a raw material towards acrylate monomers for application in coating systems. Her current research interests range from green organic synthesis to material science and chemical engineering topics. |
 Dominik Hense | Dominik Hense studied materials science at the University of Paderborn, where he received his Master degree under supervision of Prof. Dr Strube in 2019. He started his Ph.D. studies in chemistry in the same working group. One year later, together with Prof. Dr Strube, he moved to the University of Innsbruck. Already during his master and Ph.D. studies, he investigated new materials based on biological polymers. In 2023, he completed his Ph.D. and started working as senior scientist at University of Innsbruck. In his research, he continues his work in the field of biobased polymers. The main focus is innovative use of enzymatic processes and protein-based materials. |
 Paul N. Stockmann | Paul N. Stockmann studied chemistry at LMU Munich and graduated in 2015. He joined Prof. Volker Sieber's Chair of Chemistry of Biogenic Resources at TU Munich and received his PhD about the synthesis of monoterpene-based monomers in 2020 in cooperation with Fraunhofer IGB. His interest is the conversion of biogenic molecules to monomers and polymers to increase sustainability and integrate new properties to plastics by utilization of nature-exclusive chemical structures He leads the group Biobased Polymers and Additives at Fraunhofer IGB, Straubing branch. |
 Oliver I. Strube | Oliver I. Strube gained his diploma in chemistry from Clausthal University of Technology in 2008 and completed his doctoral studies with Prof. Dr Schmidt-Naake on modern polymer materials in 2011. In the following, he initiated his independent research on the field of bioinspired materials at Paderborn University. During his career, he has received numerous awards, such as multiple Coatings Chemistry Awards from the German Chemical Society and the Innovation Award of Coatings Science International. After his habilitation in 2020 he became full professor for chemical and materials engineering at University of Innsbruck in 2021. His main fields of research are biobased & bioinspired materials, coatings technology, and circular economy. |
1. Introduction
In recent years, there has been a significant effort to enhance the sustainability of various common products in the chemical industry. This shift is primarily driven by the growing awareness among both customers and business stakeholders. The plastics industry, like many others, has also embraced this trend by focusing on renewable feedstock, recycled materials, and energy-efficient production methods, all guided by the principles of green chemistry.1 To ensure the most complete utilization of resources and to create new raw materials, the use of industrial waste streams, e.g. from pulp and paper or the food industry, is of great interest. One abundant group of natural raw materials that falls into this category is that of terpenes and terpenoids, which are derived from sources like wood waste and citrus peels (Table 1).1 The different monoterpenes can be obtained from the plant parts by steam extraction.| Source | Terpene | |
|---|---|---|
| Turpentine oil (co-product of paper industry) | α- and β-pinene | 330![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| Citrus oil (co-product of orange juice industry) | R-(+)-Limonene | 30000 |
| Mentha arvensis essential oil | (−)-Menthol | 4800 |
| Peppermint essential oil | (−)-Menthol, (−)-menthone | 3200 |
| Cedarwood essential oil | α- and β-cedrene | 2600 |
| Eucalyptus globulus essential oil | 1,8-Cineole | 2070 |
| Litsea cubeba essential oil | Citral | 2000 |
| Clove leaf essential oil | β-Caryophyllene | 2000 |
| Spearmint essential oil | R-(−)-Carvone | 1300 |
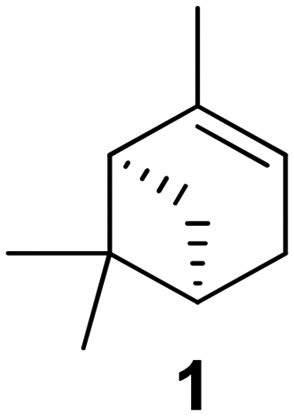
Terpenes are already commonly used as fragrance or therapeutic agents, like the highly available monoterpene (MT) pinene (1,2).2
Due to the presence of double bonds, terpenes can be easily functionalized or – in the case of terpenoids – already include functional groups such as carbonyl or hydroxy groups (Fig. 1).
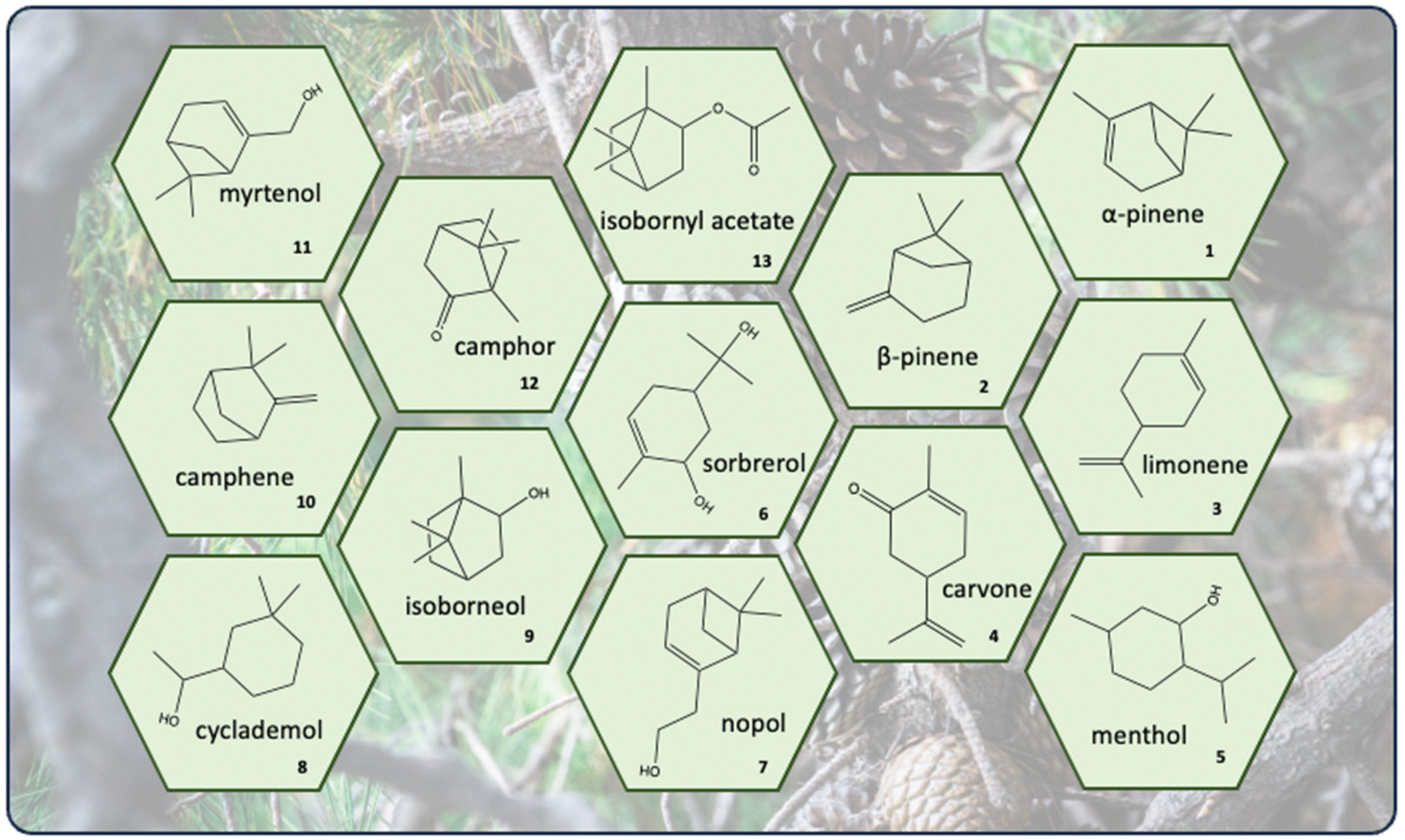 | ||
| Fig. 1 Cyclic terpenes and terpenoids discussed in this review (1–13). | ||
This characteristic makes cyclic and aliphatic (Fig. 2) terpenes, particularly monoterpenes, highly attractive as raw materials for polymers.3,4
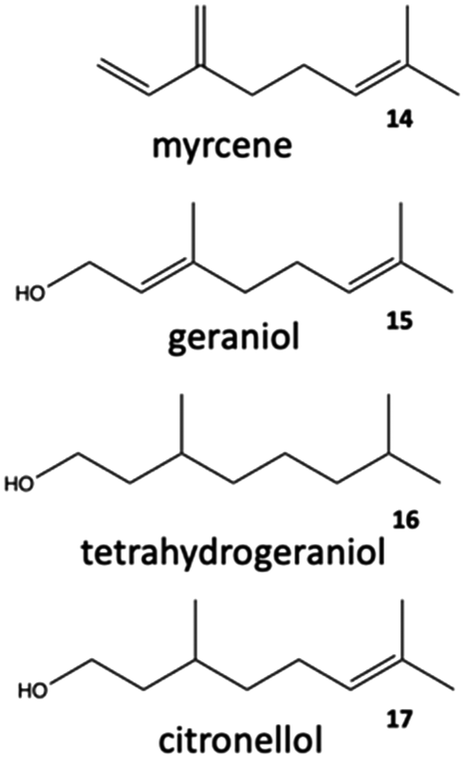 | ||
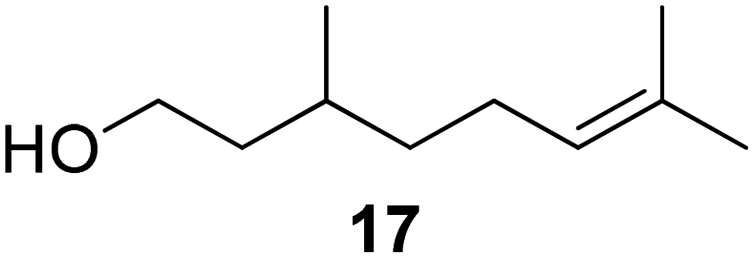
| Fig. 2 Triple unsaturated acyclic monoterpene myrcene (14) and additional aliphatic terpenoids (15–17). | ||
The general adaption of biobased polymers is still quite low (counting for just about 2% of the production volume of petroleum-based plastics in 2018). The main reasons are economically unfavorable differences to oil-based counterparts and necessary substitution of established production processes.5 To be a real alternative, biobased plastics and additives need to be superior in performance and/or price point. Recently, the German agency FNR (Scientific Agency for Biobased Raw Materials) analyzed in a market study of biobased plasticizers, which aspects are most relevant for companies to introduce such materials into the market. These aspects are sustainability, image, market requirement, cost-effectiveness, and political demands.6 To ensure the competitiveness of biobased monomers, biobased plastic products have to be integrated into existing processes. The first step to realize this goal is a good availability of these monomers. Especially the food industry produces huge annual quantities of terpenes and terpenoids as byproducts,7 which means they are readily available worldwide. Using terpene-containing waste streams can therefore lower the transport costs of the raw materials while at the same time reducing the overall amount of waste. However, the partly high price of terpenes, especially if they are not used as a mixture of stereoisomers, should not be overlooked.
To effectively reduce the carbon footprint of plastic products by using terpenes, two steps are necessary: first, if the terpenes do not yet contain functional groups, they must be sustainably modified and then provided with a polymerizable group. Common examples are acrylate or methacrylate groups, which are of great interest due to their easy conversion into poly(meth)acrylates by radical polymerization. This aim can be accomplished, for example, through catalytic8 or enzymatic approaches.9,10 At this point, it should be mentioned that all methods described here are based on fossil based acrylic acid due to the non-commercial availability of bio-based alternatives.
Second, green polymerization methods are an important key factor in reducing energy consumption and minimizing CO2 emissions.11,12 Common strategies also include enzyme-initiated processes13 as well as water-based emulsion polymerization.14 Many properties and, therefore, applications depend on the molecular weight (Mw, Mn) of the polymers. While the selection of suitable monomers enables some control over the functionality and molecular structure of polymers, it does not offer control over chain length and molecular weight distribution. Living polymerization can be used to solve this issue and enable synthesis of more tailor-made polymers.15,16 Using such controlled polymerization techniques17,18 can be employed to prepare polymers with a defined molecular weight based on terpene monomers. However, the synthesis of polymers using controlled polymerization methods is seldom employed on an industrial scale due to their often uneconomical nature. Nevertheless, this strategy holds potential for utilizing renewable raw materials in high-end applications that demand specific polymer properties (Fig. 3).
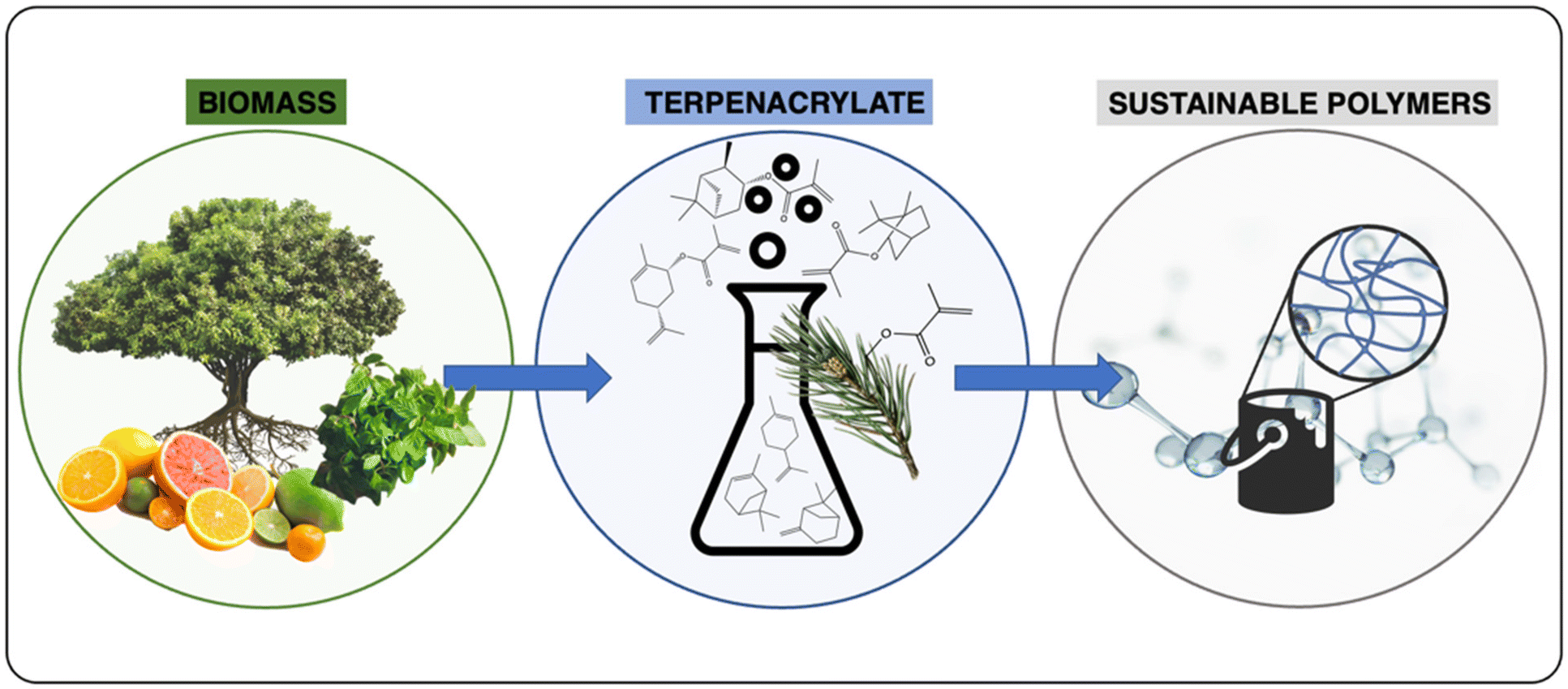 | ||
| Fig. 3 Conceptualization of the extraction of terpenes from biological resources and their conversion to polymers. | ||
The objective of this review is to offer an overview of the diverse (meth)acrylate monomers synthesized from monoterpenes (MT(M)A) and the resultant polymers. Additionally, we will consider the synthesis of terpene polymers through both free radical polymerization and controlled radical polymerization methods. The material properties attained through the distinctive molecular structure of monoterpene building blocks will be highlighted, with a specific emphasis on the commercially available acrylate monomer IBO(M)A (MA9).
2. Monoterpene-based (meth)acrylate synthesis
Homopolymerization of non-functionalized monoterpenes poses challenges, with cationic polymerization often proving most successful,19,20 as well as in the form of living cationic polymerization.21 The utilization of free radical methods for homopolymerization typically leads to low conversion rates and the production of low molar mass oligomers/polymers.22 In this context, pinene monomers (Fig. 1, molecules 1; 2) have been extensively investigated (Fig. 4).19,21,23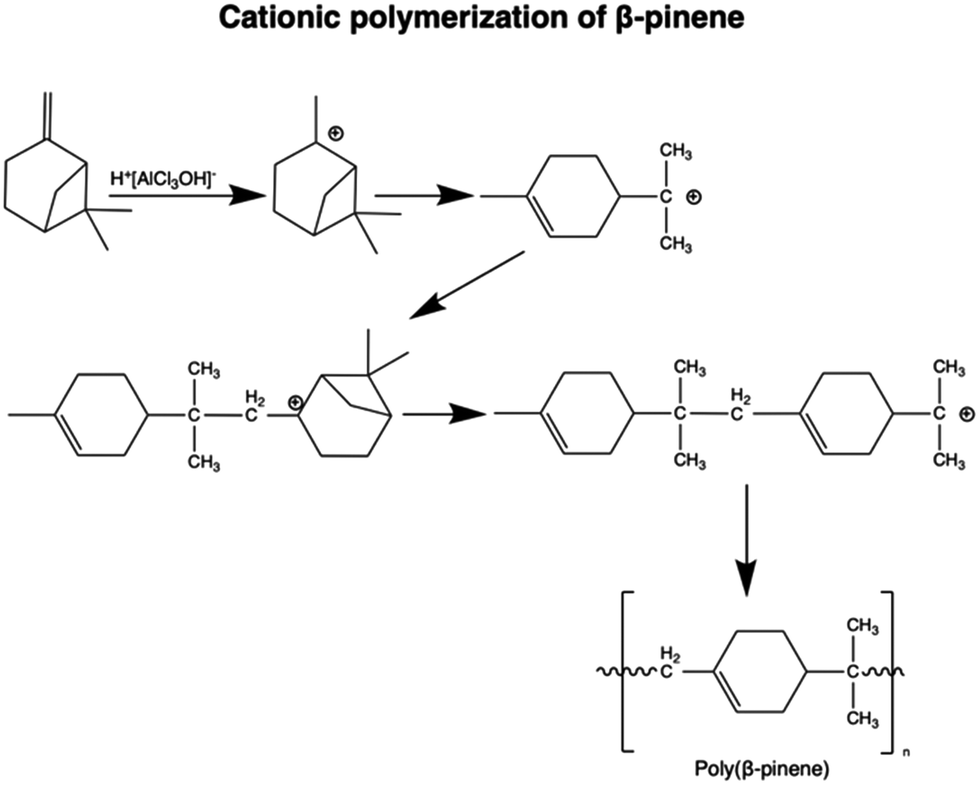 | ||
| Fig. 4 Reaction mechanism for the cationic polymerization of β-pinene with AlCl3 as a catalyst.19 | ||
In general, achieving high conversion rates in free-radical homopolymerization is challenging for most monoterpenes. One possible explanation is the high reactivity of allylic hydrogen atoms in terpene molecules during chain growth. This leads to the preferential reaction of the propagating polymer radical with an allylic hydrogen, resulting in chain transfer to the terpene. Consequently, a new allylic radical forms, leading to the termination of the polymer chain.24 This circumstance complicates the creation of copolymers with high terpene content.25 Various terpenes, including β-pinene (Fig. 1, molecule 2), myrcene (Fig. 2, molecule 14), and limonene (Fig. 1, molecule 3), have been explored for copolymerization in radical polymerization processes, but only low yields have been achieved.26
As discussed earlier, the polymerization of unfunctionalized monoterpenes poses challenges. To address this, functional groups can be introduced to the monoterpenes. The research group led by Meier modified monoterpenes by adding thiol groups. They functionalized limonene and β-pinene and varied the amount of thiol groups by adjusting the feed ratio.27 Monoterpene can also be modified with epoxide groups, like in the case of limonene dioxide. This difunctional monomer can be polymerized with a diamine via polyaddition reaction. However, due to the different reactivity of the endocyclic and exocyclic epoxide group of the limonene dioxide, high conversation cannot be achieved.28
In contrast, (meth-)acrylic groups exhibit high reactivity in polymerization. Numerous monomers derived from biomass (like plant oil, eugenol and isosorbide) containing a (meth)acrylate group have already been investigated.29–31 The integration of (meth-)acrylic groups can be accomplished by esterification of (meth-)acrylic acid and a monoterpene alcohol. The synthesis of several monomers has been reported in the academic literature, including linear, branched, cyclic, and bicyclic (meth-)acrylates derived from primary, secondary, and even tertiary monoterpene-based alcohols (Fig. 5).
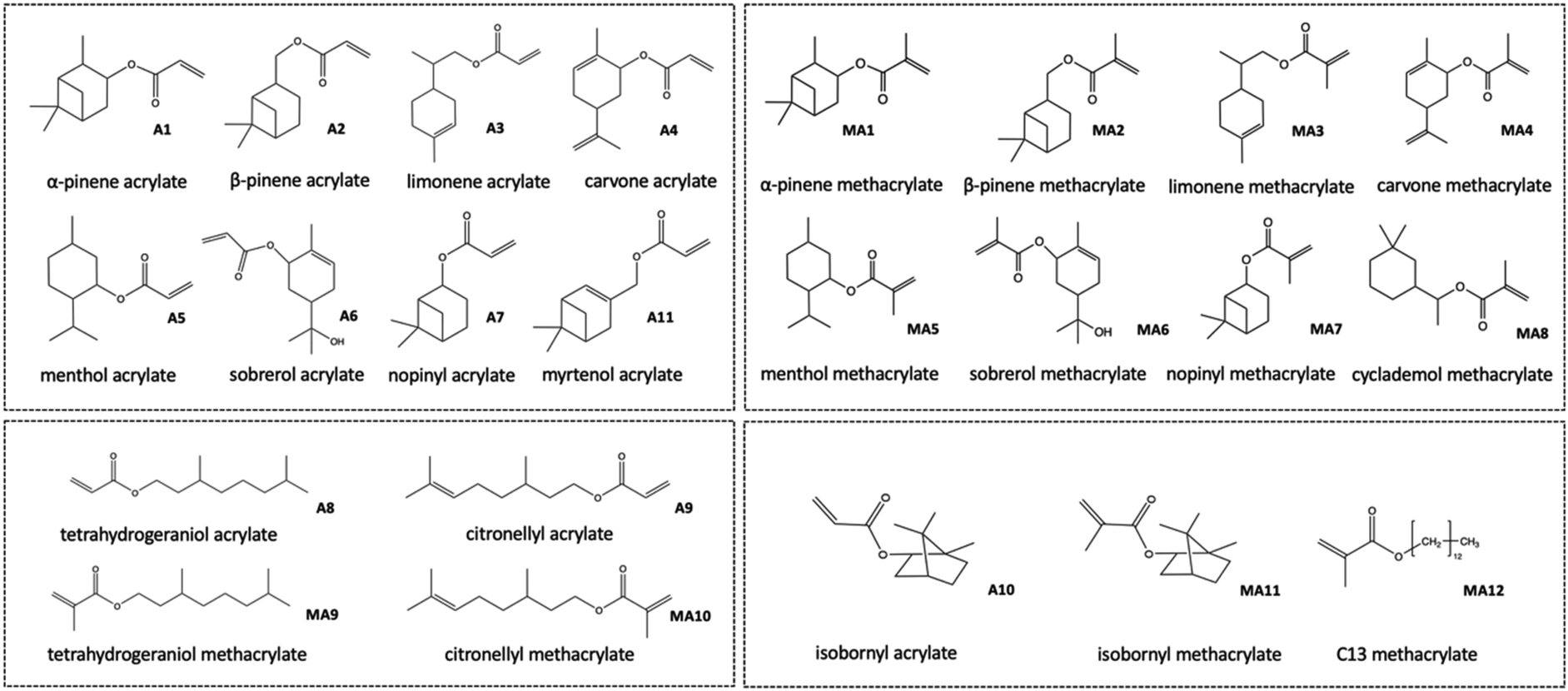 | ||
| Fig. 5 Cyclic monoterpene acrylates (A1–A7, A11) and methacrylates (MA1–MA8); acyclic monoterpene-based acrylates (A16; A17) as well as methacrylates (MA16; MA17) and conventionally available isobornyl acrylate (A9) and methyacrylate (MA9) discussed in this review. | ||
The following chapter deals with different synthesis methods to produce (meth)acrylate monomers based on monoterpenes.
2.1. Synthesis of monoterpene acrylate monomers by esterification of monoterpenoids
Many monoterpenoids – which are monoterpenes containing an oxygen function – feature an accessible hydroxy group, as nature already provided the initial oxidation during biosynthesis. However, these compounds are less abundant, more expensive, and often valuable contents of various industrial applications. Linear, cyclic, and bicyclic monoterpenoids containing primary, secondary, and even tertiary hydroxy groups demonstrated their potential as (meth)acrylate precursor, and their synthesis in academic literature is discussed below.Standard esterification methods often involve acyl chlorides, nitrogen bases, and – often halogenated – organic solvents (Fig. 6).
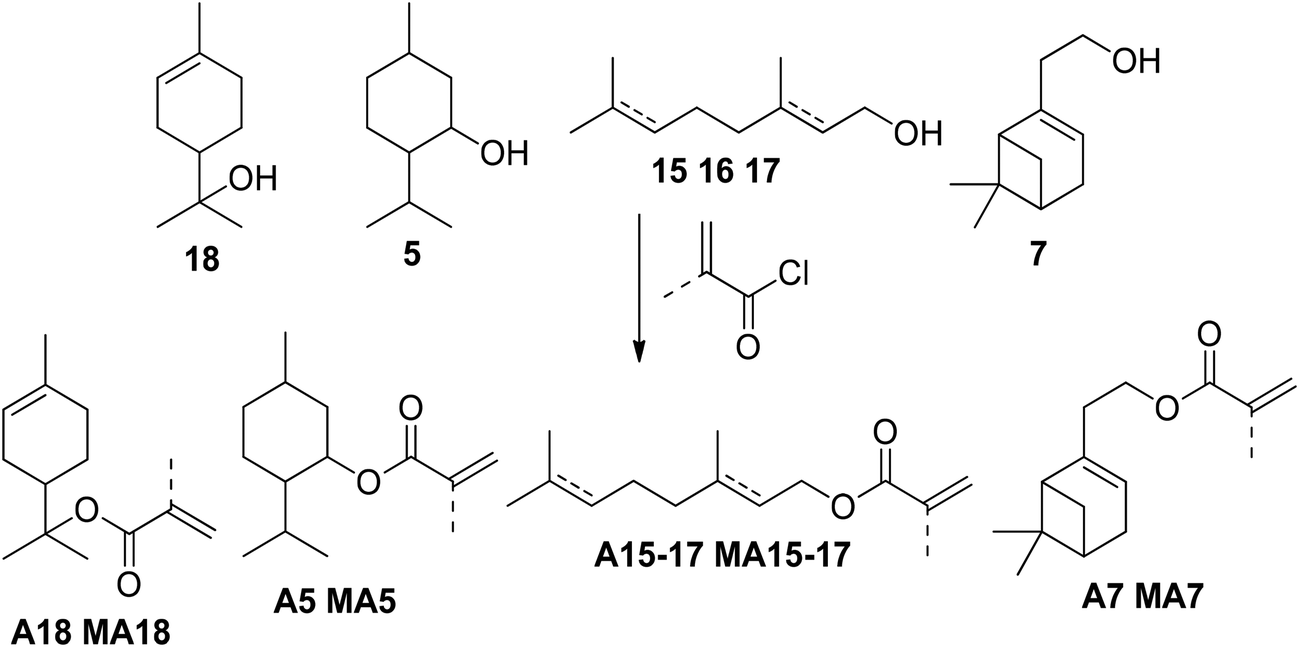 | ||
| Fig. 6 General scheme of the esterification of monoterpene alcohols with (meth)acryloyl chloride. | ||
Altomare et al. published a series optically active copolymers containing menthyl acrylate (molecule A5), using methacryloyl chloride in dry diethyl ether and Et3N as base for its synthesis.32–35 The reaction time was 6 h, and the yield 89%. However, using DCM as a solvent decreased the yield to 56%.36 Similar approaches by Baek et al. achieved a 72% yield for acrylates A5 and A16 in a reaction system of acryl chloride, DCM and TEA, also requiring column chromatography.37,38 Greiner et al. opted for a Steglich esterification for the synthesis of menthyl acrylate (A5) and reached a yield of 81% after chromatographic purification.39 Myrtenol (11) led to the corresponding acrylate A11 with yields of 62% after flash chromatography using a standard method with methacryloyl chloride, TEA, and diethyl ether.40 Some monoterpenes have been converted to azoles and were then evaluated as potential antifungal agents. These azole derivatives required an α,β-unsaturated terpenoid precursor for aza-Michael addition. For the integration of that chemical motif, Akhmedov et al. converted several terpenes, such as geraniol (15) and myrtenol (11), with stochiometric amounts of acryloyl chloride and Hünig-base in chloroform to the corresponding acrylates in small-scale reactions.41 The reaction time was 10 h at room temperature, and purification by column chromatography was required. The yield of myrtenyl acrylate (A11) was 72%.
S. Noppalit et al. used acryloyl chloride and Et3N in DCM for the esterification of tetrahydrogeraniol 16, achieving a yield of 85% without additional purification after passing through basic alumina to remove acidic residues.42 Cuzzucoli Crucitti et al. attempted to synthesize α-terpenyl methacrylate (MA18) by the conversion of α-pinene (1) with methacrylic acid in DCM at 20 °C, and the expected carbon skeleton rearrangement and esterification were supposed to be catalyzed by the strong Lewis acid Fe(OTf)3.43 However, the authors found that the resulting product mixture was composed of IBOMA (MA9, 60%) and bornyl methacrylate (40%). The combined yield was 22% and improved to 35% by application of silica-supported TfOH as a catalyst. In both cases, the initial target molecule MA18 was not detected. A α-terpenyl methacrylate (MA18) was, therefore, produced in a one-pot reaction starting from α-terpineol (18), as depicted in Fig. 7.
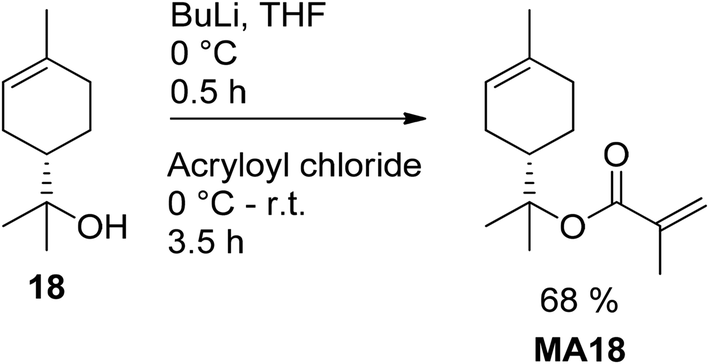 | ||
| Fig. 7 Deprotonation of α-terpineol (18) with BuLi and subsequent esterification. | ||
The scale of this reaction was 100 g per batch, and product purification was achieved through distillation (88–90 °C at pressures below 1 mbar).
The reaction of alcohols with methacryloyl chloride has also been investigated in a continuous flow process.44 DCM served as the solvent, and Et3N acted as the base. The use of ultrasonic sound prevented the reactor from being blocked by the formation of insoluble trimethylamine hydrochloride. Table 2 summarizes the reaction outcomes for α-terpineol and β-citronellol.
| Monoterpene alcohol | Concentration [M] | Temperature [°C] | Reaction time [min] | NMR yield [%] | Isolated yields [%] |
|---|---|---|---|---|---|
|
1.5 | 20 | 1 | 89 | 75 |
| 1.5 | 20 | 5 | 95 | 65 | |
| 1.5 | 20 | 10 | 96 | 80 | |
| 1.5 | 40 | 1 | 90 | — | |
| 1.5 | 40 | 5 | 88 | — | |
|
2.0 | 20 | 1 | 97 | 70 |
Very worth mentioning are the facts that even the tertiary hydroxy group of α-terpineol reacted to the corresponding acrylate, yielding a 97% product at a comparatively high concentration of 2.0 M without the use of strong bases such as butyl lithium and within only one minute of reaction time. The authors also highlight that this system might be suitable for combination with continuous flow polymerization.
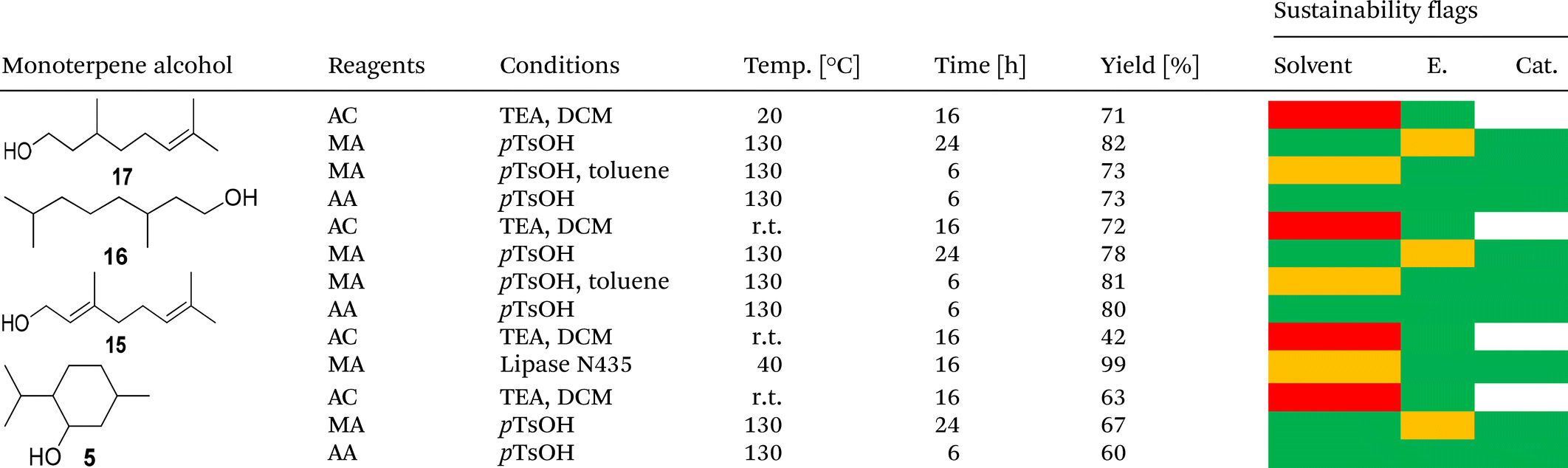
While flow chemistry processes are generally considered valuable tools in green synthesis, the methods mentioned above involve organic solvents, VOCs, or toxic chemicals. Since the sustainability of a chemical compound highly depends on the sustainability of its synthesis, green methods are required for esterification. The CHEM21 green metrics toolkit uses several parameters (renewability, energy, waste, catalysis) to estimate the sustainability of chemical reactions and has proven to be a powerful tool for the sustainability evaluation of four different esterification methods of monoterpene alcohols (Table 3).45
|
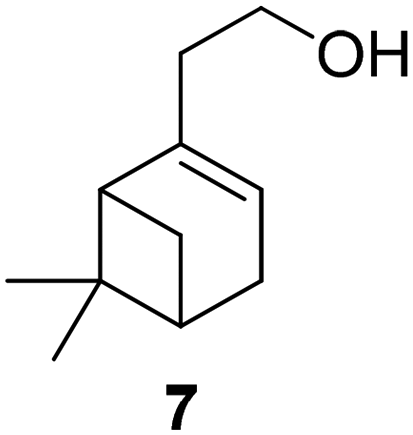
In all cases, a solvent-free, straightforward esterification of the alcohols with methacrylic acid catalyzed by pTsOH was identified as the most sustainable approach. The application of acrylic acid yielded similar results. The exception is geranyl acrylate, as the reaction conditions led to isomerization of the geranyl structure; this might also affect other monoterpene alcohols that easily undergo side reactions, such as Wagner Meerwein rearrangements. pTsOH had previously been employed for the esterification of hydrogenated nopol (7) in 1958, resulting in an 85% yield.46 The ion exchange resin Dowex-50-X8 was used as a heterogeneous sulfonic acid-containing catalyst in the same publication, with a similar yield.
Lipases are well-known catalysts for the esterifications of alcohols. In 2001 Athawale et al. described a enzymatic method for the transesterification of menthol (5) using pseudomonas cepacia lipase (lipase-PS), porcine pancreatic lipase, and candida rugose lipase.47 Methyl methacrylate, vinyl methacrylate, and 2,3-butanedione mono-oxime methacrylate functioned as acylating agents (Fig. 8). Lipase PS showed the best conversion of 98.7% within 24 h. Regarding the solvent, using diisopropyl ether (DIPE) led to a conversion of 97.2% compared to other 61.3% and 18.9% in toluene and chloroform, respectively. In addition, whereas all other solvents led to an ee >96%, chloroform only reached an ee of 68%. Interestingly, the oxime was the most suitable acylating reagent. The authors figured that this is due to methanol being an inferior leaving group during transesterification, and that acetaldehyde, the byproduct of transesterification using vinyl esters, negatively affects the enzyme activity.
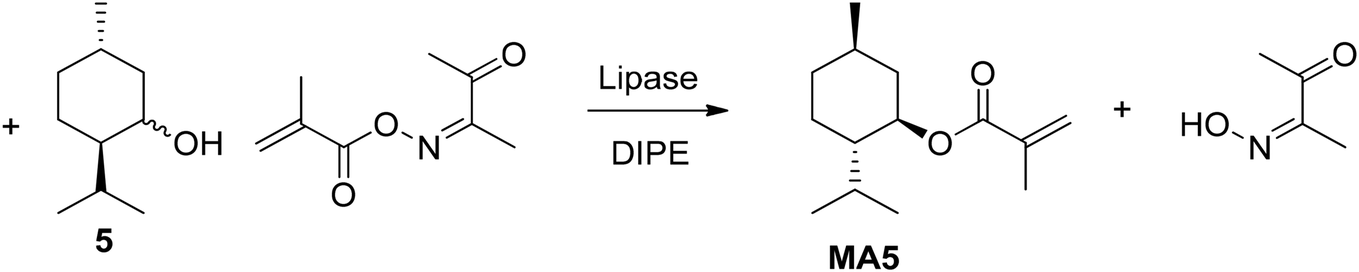 | ||
| Fig. 8 Oxime-functionalized methacrylate as esterification agent suitable for lipase catalysts. | ||
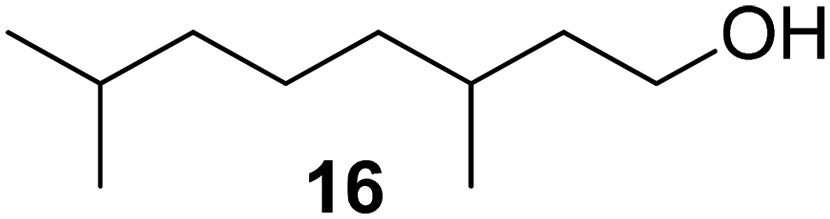
In a follow-up publication, citronellol (17) was investigated using a similar approach, with pseudomonas cepacia lipase exhibiting the highest substrate concentration tolerance (>90% conversion at a concentration of 0.6 M within 16 h).48 To achieve high conversions of citronellol (17), the enzyme concentration was four times higher than that required for menthol (5).
Billon and coworkers thoroughly investigated the influence of microwaves on the transesterification of several MTs catalyzed by Novozyme 435.49 This method is environmentally benign as it is free of metals and halogens and operates without solvents. Table 4 displays the best reaction conditions for acrylic and methacrylic anhydride, respectively.
| Terpenoid | Microwave power [W] | T [°C] | Time (min) | |
|---|---|---|---|---|
| Acrylate | Methacrylate | |||
|
150 | 40 | 30 | 5 |
|
n.a. | 40 | 120 | 120 |
|
150 | 40 | 110 | 110 |
|
150 | 40 | 150 | 120 |
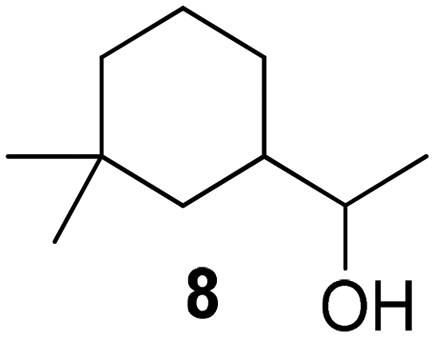
As anticipated, the reaction proceeds considerably faster using anhydrides instead of the corresponding free acids. Furthermore, the reaction time was accelerated by microwaves. The primary alcohols reached full conversion to the corresponding (meth-)acrylates within only 5 to 150 minutes. For unsaturated compounds, microwaves are not suitable, as they induce uncontrolled crosslinking. Another noteworthy aspect is the esterification of the secondary alcohol cyclademol (8), which initially achieved only a conversion of 42% within 24 h. Optimized reaction conditions (40 °C, 150 W) enabled full conversion and a yield of 100% within only 2 h. Equally impactful is the high purity of the produced (meth-)acrylates, which reacted to polymers without previous distillation or column chromatography.
2.2. Synthesis of monoterpene acrylate monomers starting from monoterpenes
To make monoterpenes accessible as monomer feedstock, the integration of reactive groups is a common strategy. For terpene-based (meth)acrylates, the synthesis typically involves two major steps: first, the integration of a hydroxy group, and second, its subsequent esterification with—often activated—(meth)acrylic acid. Alcohol synthesis is achievable while preserving the molecular structure of the monoterpene or accompanied by rearrangements of the carbon skeleton, often resulting in terpenoid structures that also occur in nature.As previously mentioned, the conversion of monoterpenes to (meth)acrylates requires double bond oxidation. One of the most comprehensive studies in this regard, published by S. M. Howdle and co-workers in 2016, describes the oxidation of α-pinene (1), β-pinene (2), and limonene (3) by hydroboration.50 BH3SMe2 served as the boron source for conversion of the pinenes 1 and 2, while 9-BBN was suitable for limonene. While the oxidation of pinenes 1 and 2 was stereo-selective, two evenly distributed isomers emerged from limonene, as the external double bond is accessible from both ring sides. In addition, the treatment of carvone (4) with LiAlH4 gave the corresponding alcohol in quantitative yield. The alcohols were then transformed into the respective (meth-)acrylates through treatment with (meth-)acryloyl chloride or (meth-)acrylic acid in combination with propanephosphonic acid anhydride (T3P®), respectively (Table 5). In all cases, the alcohols and the final products underwent purification through chromatographic techniques or crystallization.
50
| Monoterpene | Alcohol synthesis [method] | Monoterpene alcohol [yield %] | Esterification [method] | (Meth-)acrylates [yield/total yield %] |
|---|---|---|---|---|
|
1 |
|
A |
|
| Acrylate: 55/48 | ||||
| Methacrylate: 63/55 | ||||
| B | Acrylate: 99/86 | |||
|
1 |
|
A |
|
| Acrylate: 66/65 | ||||
| Methacrylate: 47/46 | ||||
| B | Acrylate: 99/98 | |||
|
2 |
|
A |
|
| Acrylate: 75/62 | ||||
| Methacrylate: 64/52 | ||||
| B | Acrylate: 96/79 | |||
|
3 |
|
A |
|
| Acrylate: 91/90 | ||||
| Methacrylate: 73/72 | ||||
| B | Acrylate: 99/98 |
The authors also established an alternative oxidation via oxidative acrylation of β-pinene (2). This reaction was catalyzed by Pd(OAc)2, with additional benzoquinone (BQ) serving as an oxidant, ligand for palladium, and also inhibits polymerization during the synthesis The reaction proceeded without the need for additional solvent, yielding 82% (both isomers, Fig. 9).
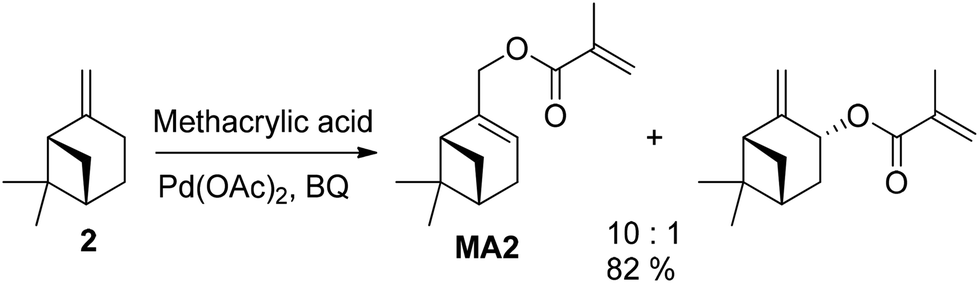 | ||
| Fig. 9 Methacrylation of β-pinene (2) catalyzed by palladium acetate with high regio-stereomeric selectivity for the direct synthesis of acrylates without prior double bond oxidation. | ||
This method holds the potential for greater environmental friendliness compared to the hydroboration pathway, provided that the catalyst and benzoquinone can be isolated and reused. In all cases, both the alcohols and the final products underwent purification through chromatography or crystallization before polymerization.
Another common method for the oxidation of double bonds involves the formation of epoxides, typically using peroxy acids. Cutajar et al. synthesized two new acrylates (Fig. 10) derived from pinene (1) via sobrerol (6) in three and four reaction steps, respectively. These acrylates may find utility as consolidants for archaeological wood.51–53 Regarding their chemical structure, it is noteworthy that the two sobrerol acrylate diastereoisomers carry free hydroxyl groups for further modification or cross-linking. Fig. 10 illustrates the reaction pathway.51,52,54
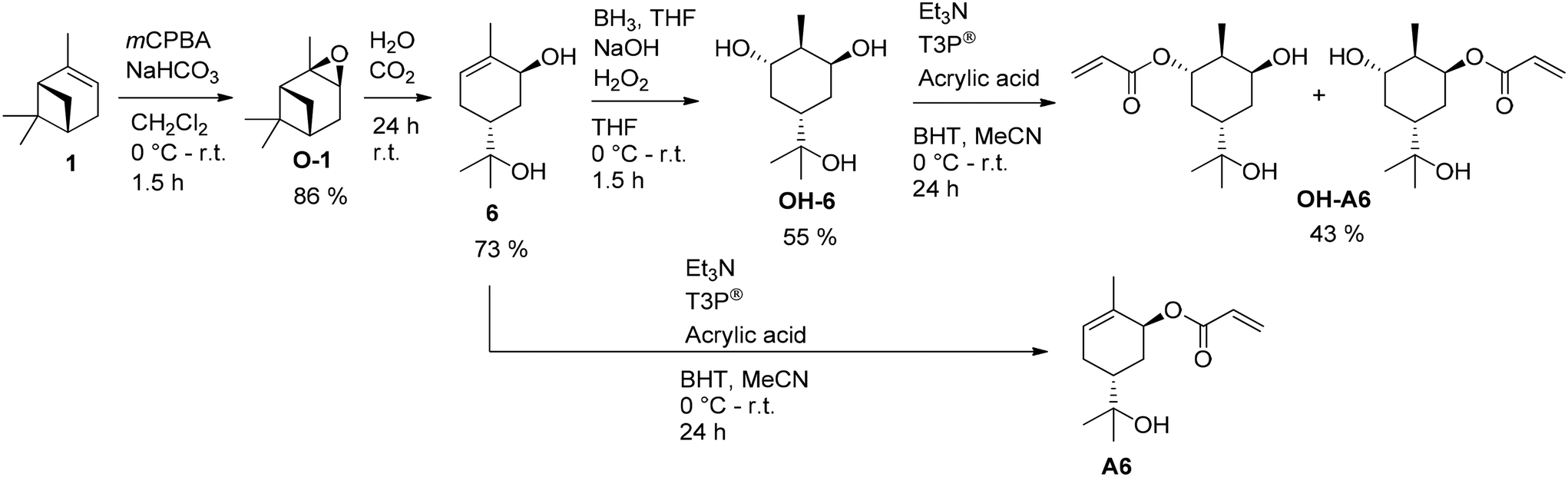 | ||
| Fig. 10 Synthesis pathway to sobreroyl acrylate (A6) starting from α-pinene (1). | ||
The initial step involves the epoxidation of 1 with meta-chloroperoxybenzoic acid (mCPBA) in dichloromethane. Sobrerol (6) can be synthesized from α-pinene oxide (O-1) by stirring in acidic aqueous emulsion, for example at pH 5 caused by continuous introduction of CO2 to the emulsion. The secondary hydroxy group reacted with acrylic acid catalyzed by propanephosphonic acid anhydride (T3P®) to the corresponding acryl ester A6.52 Alternatively, the newly formed double bond was oxidized by hydroboration in anti-Markovnikov position, resulting in the triol OH-6 that is esterified to acrylate OH-A6 accordingly in a total yield of 15%.51 A. C. Serra et al. used a similar method to produce sobrerol (6) in a yield of 62% as colorless crystals (Fig. 11).55 Sorbreol (6) and acryloyl chloride gave acrylate A6 in a yield of 72% in THF as solvent and TEA as a base within 48 h at 50 °C. Alternatively, methacrylate MA6 was accessible in a yield of 84% by using methacrylic anhydride and DMAP.
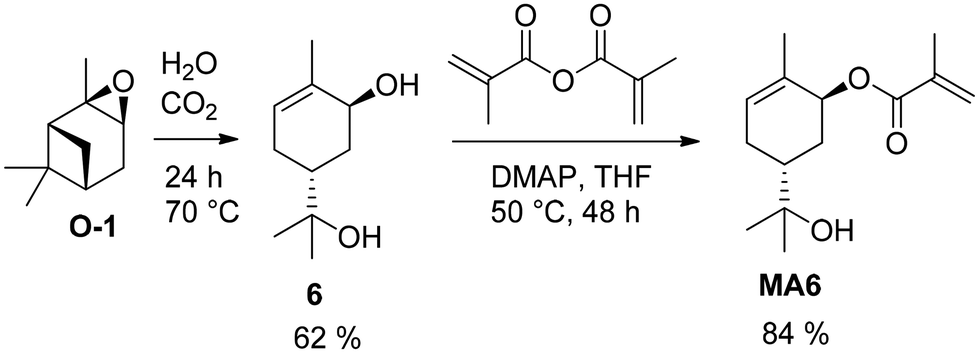 | ||
| Fig. 11 Acid-catalyzed rearrangement of α-pinene oxide to sorbreol (6) and conversion with methacryl anhydride. | ||
Sobrerol (6) is also directly available from α-pinene (1) in the presence of oxygen in aqueous media through oxidation catalyzed by mutated Cytochrome P450 from Bacillus megaterium.56
Limonene (3), one of the most abundant monoterpenes, has also been transformed into new (meth)acrylates in a multi-step synthesis. M. A. R. Meier and co-workers introduced a new set of limonene-based acrylates containing an amide bond.57 The synthesis strategy comprised the conversion of the external double bond to an aldehyde followed by a Passerini three-component reaction, as depicted in Fig. 12.
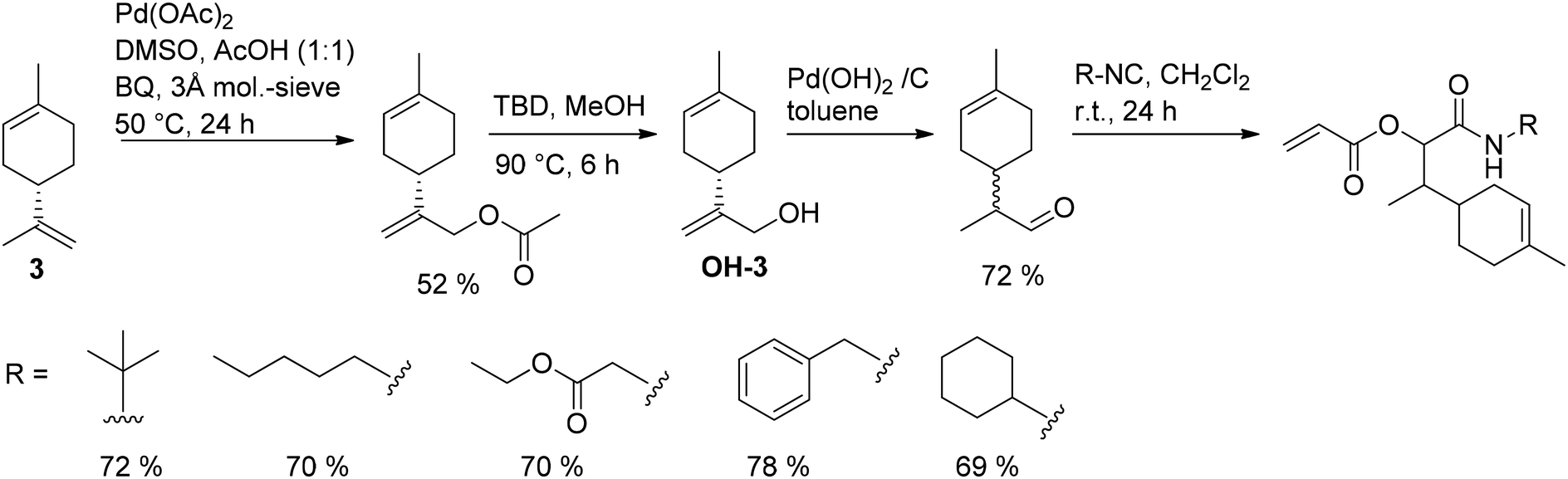 | ||
| Fig. 12 Conversion of limonene (3) to acrylates containing an amide bond. | ||
The oxidative acetoxylation of the external double bond reached a yield of 52%. Complete purification of the desired acetate was not possible. However, the side products did not interfere with the subsequent ester cleavage and isomerization of the limonene allyl alcohol to the corresponding aldehyde (yield: 72% after column chromatography). Finally, limonene aldehyde, acrylic acid, and various isocyanates reacted to novel acryl esters of α-acyloxy amides in yields of up to 78%.
Utilizing both double bonds, Mühlhaupt and co-workers converted the diepoxide of limonene (2O-2) to limonene-based dimethacrylate (LDMA) by direct reaction with methacrylic acid.58 In addition to the expected regioisomers, oligomers formed as a side product due to nucleophilic ring-opening by the emerging hydroxy groups as displayed in Fig. 14.
The reaction conditions and several catalysts were investigated in addition to the auto-catalytic activity of methacrylic acid. Higher reaction temperatures and catalysts in general increased the reaction rate, whereas the type of the applied catalyst was less relevant. Triphenylphosphine (TPP), 1,2-dimethylimidazol (DMI), and benzyltriethylammonium chloride (BTAC) also reduced the formation of oligomers as they predominantly catalyzed the addition of methacrylic acid to the epoxide functions as clarified by comparison of the residual methacrylic acid contents (Table 6).
| Catalyst type 2 wt% | MA/2O-2 [mol mol−1] | T [°C] | Full epoxy conversion [h] | Residual MA [mol%] |
|---|---|---|---|---|
| — | 2.1 | 80 | 24 (89% conv.) | 60 |
| TPP | 2.1 | 80 | 13 | 34 |
| DMI | 2.1 | 80 | 13 | 44 |
| — | 2.1 | 100 | 13 | 60 |
| TPP | 2.1 | 100 | 8 | 35 |
| DMI | 2.1 | 100 | 6 | 42 |
| BTAC | 2.1 | 100 | 6 | 42 |
| BTAC | 2.1 | 120 | 2 | 43 |
| DMI | 1.7 | 100 | 8 | 28 |
| DMI | 1.3 | 100 | 13 | 5 |
L. A. Berglund et al. employed the mono-epoxide of limonene in a solvent-free ring-opening acrylation process without the need for an additional catalyst The reaction was conducted at 75 °C in the presence of 4-methoxyphenol as a radical inhibitor.59 The yield reached 85% after three hours. The resulting limonene-based acrylate possesses three distinct functional groups: the acrylate for polymerization, an external alkene suitable for cross-linking or other chemical modifications, and a hydroxyl group acting as a (sterically hindered) nucleophile. This hydroxyl group also influenced the compound's polarity and compatibility, as illustrated in (Fig. 13).
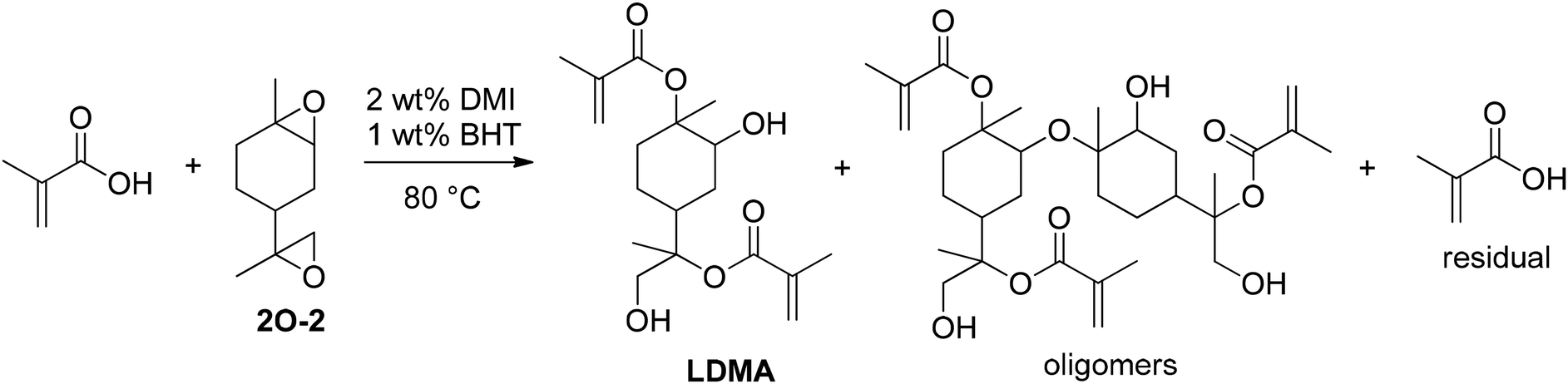 | ||
| Fig. 13 Addition of methacrylic acid to limonene diepoxide (2O-2) and formation of oligomeric side products. | ||
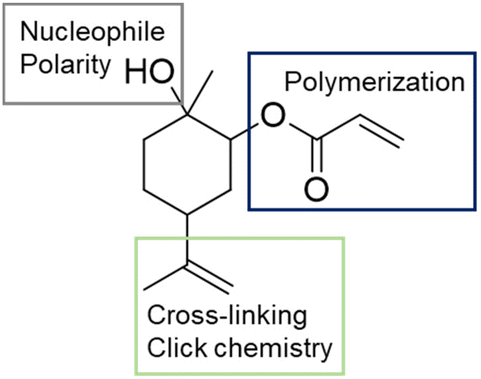 | ||
| Fig. 14 Addition product of acrylic acid to limonene epoxide (O-6) and description of the function of the functional groups. | ||
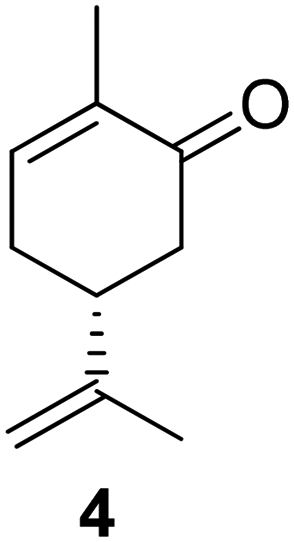
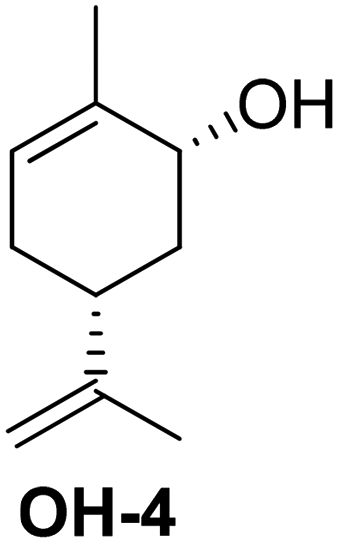
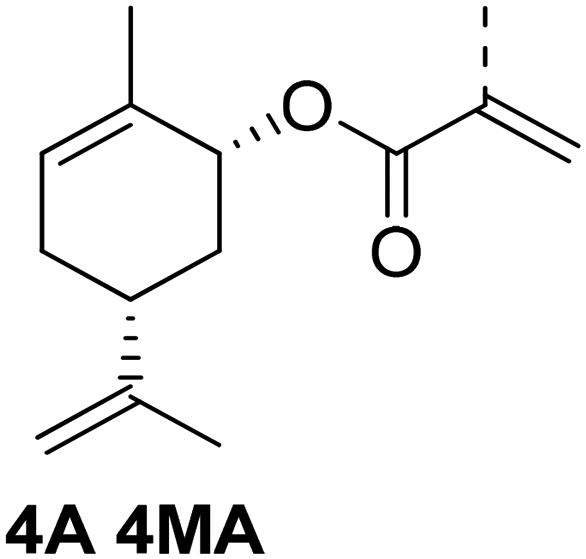
Another strategy to obtaining a multifunctional monoterpene acrylate was introduced by S. M. Howdle and co-workers in 2020.60 The carvone-based acrylate (A4), derived from the reduction of carvone (4) using LiAlH4, followed by subsequent esterification,50 incoporates two additional double bonds (Fig. 15).
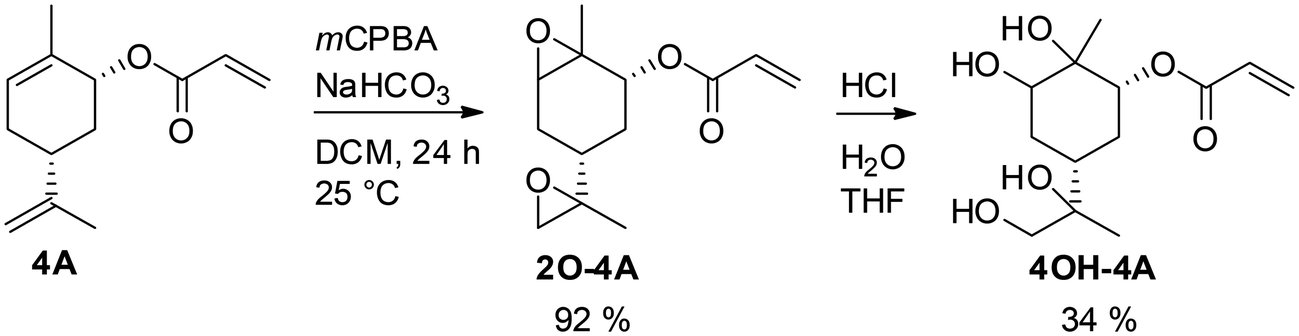 | ||
| Fig. 15 Selective epoxidation of an internal and an external double bond in the presence of the electron deficient acrylic double bond and epoxide hydrolysis to a biobased tetraol acrylate. | ||
Both double bonds are selectively accessible for epoxidation with mCPBA. The electron-deficient double bond of the acrylate motif remains unreactive under these conditions. However, the epoxidation process was not stereo-selective, as the authors noted the formation of several isomers, as detected by NMR. The use of mCPBA resulted in an excellent yield of 92%, while oxone® only yielded 64%. Due to the overlapping proton signals of these isomers in 1H-NMR, quantifying each isomer was not feasible. Treatment of the diepoxide acrylate with HCl solution in THF for 48 hours at room temperature led to ring opening and the formation of carvone-based tetraol acrylates, with a yield of 34%.
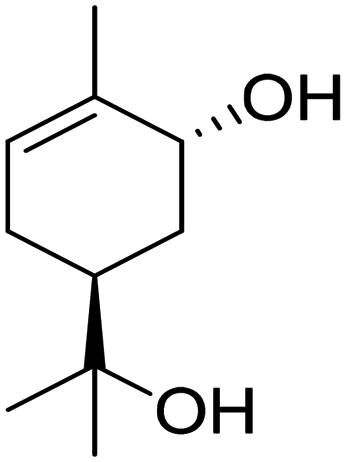
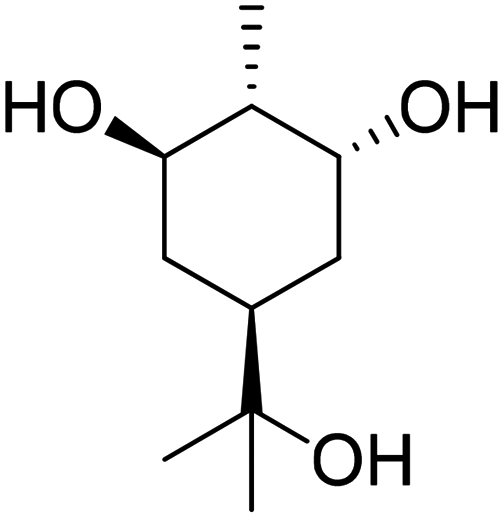
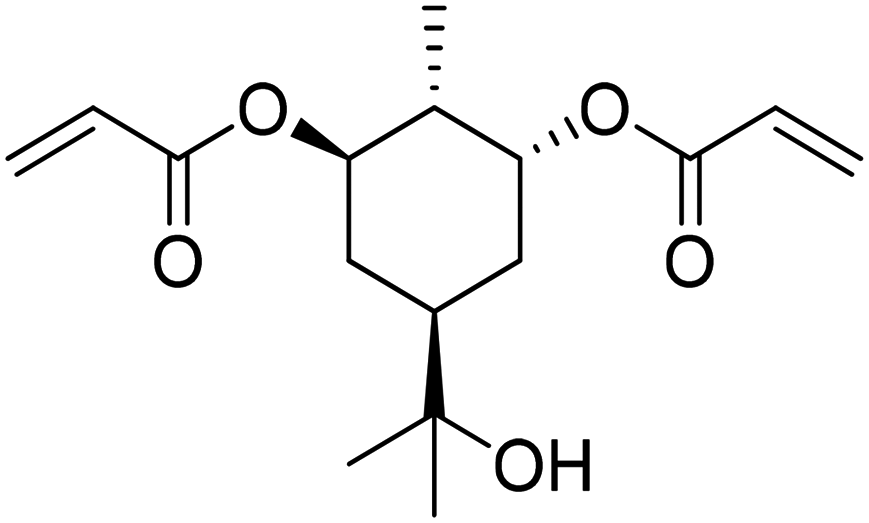
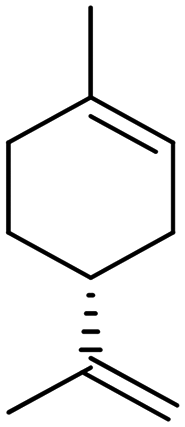
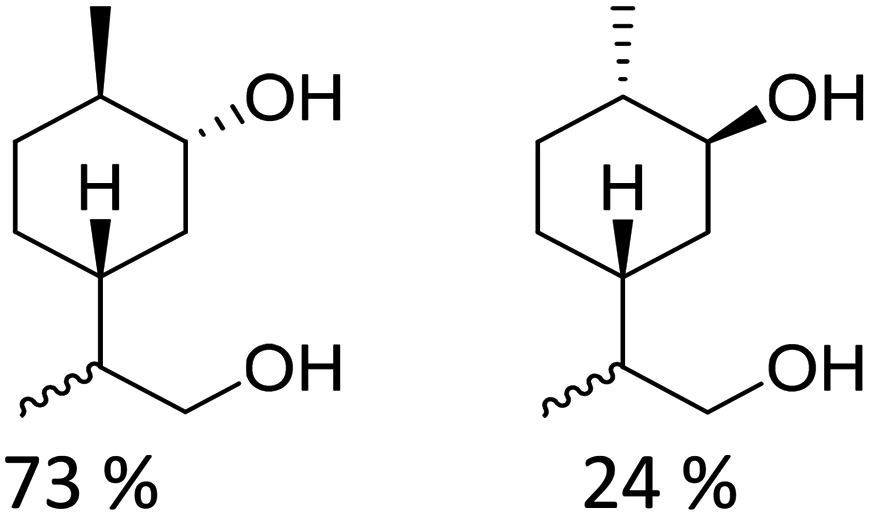
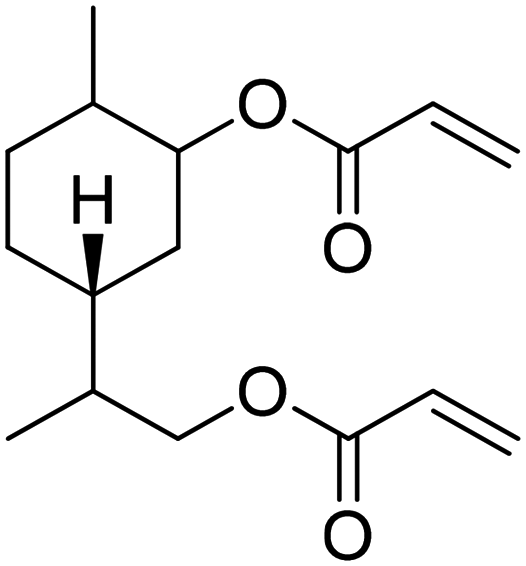
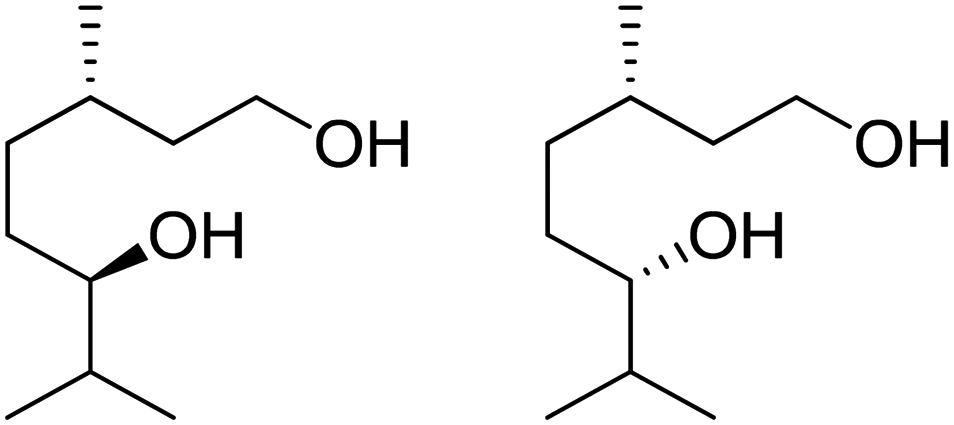
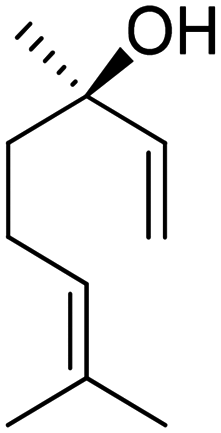
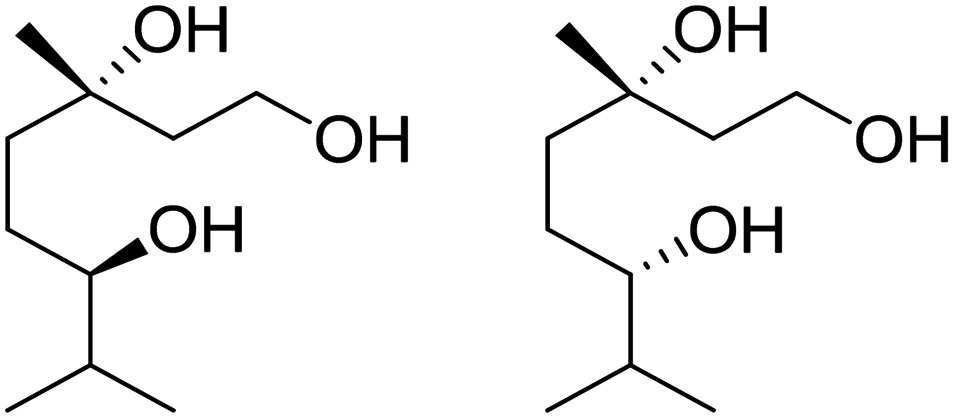
Recently, a “toolbox” for the oxidation of monoterpenes to diols and triols followed by their conversion to diacrylates gave way to a more generalized synthesis approach.61 Hydroboration under argon in dry THF was the reaction of choice for the oxidation of the double bonds, and the yields varied between 25% and 97% after crystallization of chromatographic purification. The esterification of the polyols with acrylic acid was accomplished using a combination of propanephosphonic acid anhydride (T3P®), Et3N, MeCN as a solvent, and small amounts of BHT to prevent polymerization. After work-up, the volatiles were removed by airflow to prevent polymerization and distillation was avoided. Table 7 summarizes the results.
| Monoterpene | Polyol [yield ] | Diacrylate [total yield] |
|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
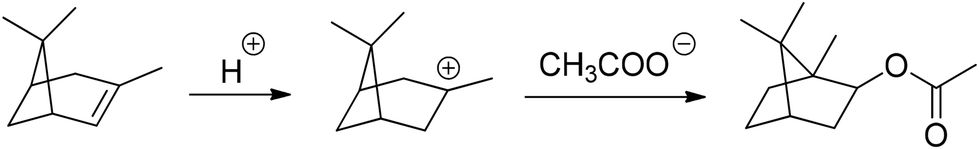 | ||
| Fig. 16 Synthesis of isobornyl acetate (13) by the conversion of α-pinene (1) with acetic acid. | ||
Alternatively, camphene (10) is an often used starting molecule for IBO(M)A (A9, MA9) synthesis. The following section offers a brief overview of the methods applied in IBO(M)A (A9, MA9) production, including scientific and patent literature. These methods can be categorized by their general synthesis strategy: the (trans-)esterification of isoborneol (9) or isobornyl acetate (13) and the direct conversion of camphene (10) or α-pinene (1) and (meth-)acrylic acid.
2.2.1.1. Synthesis of isobornyl (meth)acrylate (IBO(M)A) by (trans-)esterification. The general esterification of biobased alcohols with (meth-)acrylic acid or activated derivatives such as acryloyl chloride or acrylic anhydride by homogeneous, heterogeneous, or enzymatic catalysis is well described in the literature, and we figure that many of these methods can also be applied to isoborneol (9).62,63 Some of these methods are also described in patents. As an exampIe, K. Ishihara and M. Hatano described several alkali and alkaline earth metal phenolates as general catalysts for esterification of alcohols with MA in 2020.64 Sodium 2,6-di-tert-butyl-4-methylphenoxide was used as catalyst for the synthesis of IBOMA (MA9) as displayed in Fig. 17.
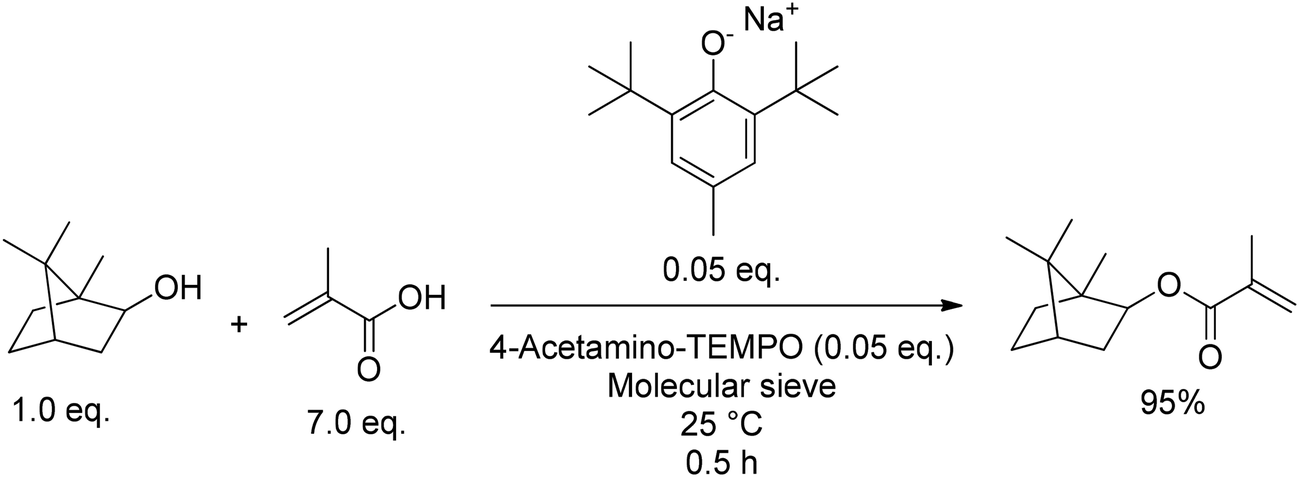 | ||
| Fig. 17 Direct esterification of isoborneol (9) with a sodium phenoxide catalyst. | ||
This method yielded 95% of IBOMA (MA9) in only 30 min at 25 °C but was only tested in small scale and required polymerization inhibitors such as 4-acetylamino-2,2,6,6-tetramethylpiperidin-1-oxyl (4-acetamino-TEMPO) as well as purification by column chromatography. Furthermore, moisture or water formed during the reaction can decrease the effectiveness of this catalysis, and therefore application of molecular sieve 5 Å is mandatory. These catalysts, including potassium, magnesium, lithium, and sodium phenolates were the topic in a detailed follow-up publication by the same authors.64 Already in 2018, the group had introduced a metal-free esterification protocol, using tetramethylammonium methyl carbonates as catalysts.65 The catalytically active ammonium species is formed in situ under release of MeOH and CO2 as illustrated in Fig. 18. Application of [Me4N]+[OCO2Me]− gave a yield of 93% for IBOMA (MA9) from the esterification of isoborneol (9) with MMA at 70 °C for 16 h in the presence of molecular sieve 5 Å.
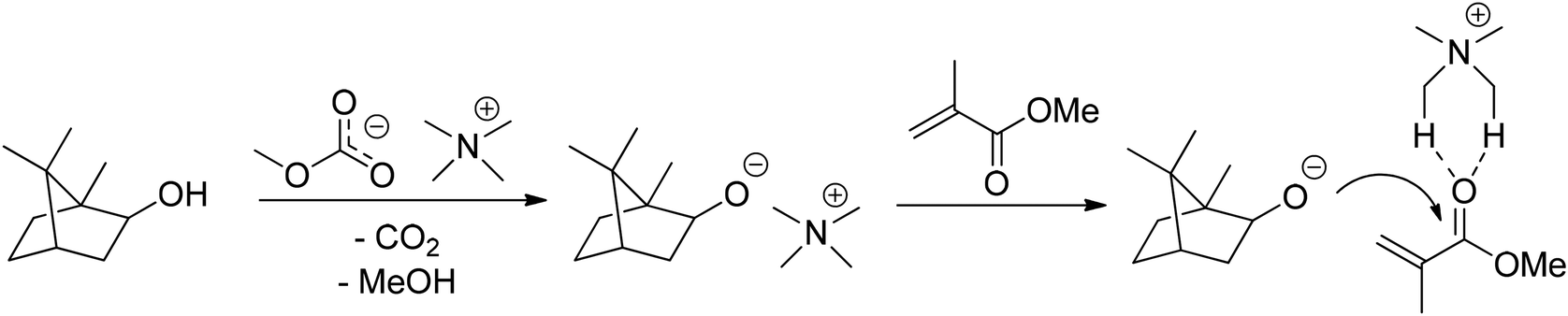 | ||
| Fig. 18 Proposed mechanism of the formation of the active ammonium species and attraction of the ester group by acidic α-H during transesterification. | ||
D. Xu et al. reported a straightforward approach in 2020.66 Methacrylic acid or acrylic anhydride formed IBO(M)A (A9, MA9) with isoborneol (9) in toluene at reflux temperature catalyzed by p-toluene sulfonic acid or sulfuric acid (0.25 w%) in a batch reaction within 6 h. After washing with an aqueous solution of NaOH, drying, and vacuum distillation, IBOMA (MA9) and IBOA (A9) reached a yield of more than 97%.
J. Knebel and D. Saal stated67 that catalysts such as Ca(OH)2,68 alkali carbonates,69 mixed catalysts of organyl tin oxides and organyl tin halides,70 lead-based catalysts,71 sulfuric acid and carboxylic acids,72 and sulfuric acid with BF3 are suitable catalysts for the esterification of isoborneol (9) with methacrylic acid, achieving high purity and good yields. However, they also observed that these catalysts do not catalyze the transesterification of isobornyl acetate (13) and a (meth-)acrylic ester to IBO(M)A (A9, MA9) and that this transesterification would require either complex mixed catalysts or an extensive amount of polymerization inhibitors. Therefore, they suggested a one-pot method with a catalyst consisting of CaO and LiOH, converting isobornyl acetate and the nucleophilic alcohol methanol to isoborneol (9) and low-boiling methyl acetate. Methyl acetate is separated by distillation during the reaction, shifting the reaction equilibrium to isoborneol (9). In a second step, MMA and low amounts of polymerization inhibitors such as hydrochinons or derivatives of TEMPO are added to the reaction mixture, and the esterification to IBOMA (MA9) is catalyzed by the same CaO/LiOH catalyst (Fig. 19).
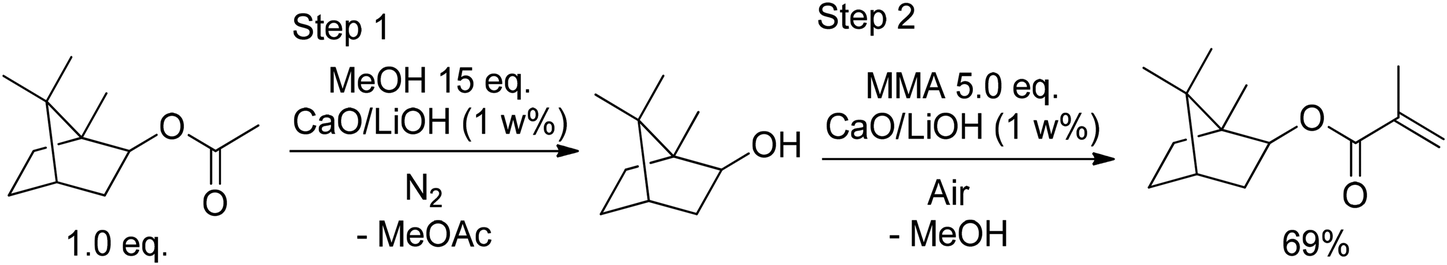 | ||
| Fig. 19 Deacetylation of isobornyl acrylate (13) and esterification with MMA catalyst by a CaO/LiOH mixed catalyst. | ||
The first step is achieved within 7 h at 57–65 °C and the second step within 6 h at 64–100 °C. MeOH, MMA, and methyl acetate are recovered. The CaO/LiOH catalyst is recovered by pressure filtration. Depending on the exact reaction conditions, the yield is 70–81% with a purity of 94–97% and only up to 200 ppm of polymerization inhibitor remain in the product.
M. Miller et al. reported an example for the transesterification of isobornyl acetate (13) with MMA without the occurrence of isoborneol (9) as intermediate in 1999. Here, alkali metal alkoxides were used as catalyst and bromide salts as polymerization inhibitors. Lithium bromide was active as catalyst and inhibitor and can easily be separated from the product by distillation or washing with an aqueous solution of 15% NaCl and 2% NaHCO3. The yield of this reaction was 97.5% with a purity of 99.1% after vacuum distillation. The water occurring during esterification was distilled of as an azeotrope with the solvent cyclohexane.73
2.2.1.2. Synthesis of isobornyl (meth)acrylate (IBO(M)A) by direct conversion of camphene or α-pinene. The conversion of camphene (10) and (meth-)acrylic acid is well described in patent literature. In 1963, B.M. Newman reported that the synthesis of IBOA (A9) and IBOMA (MA9) can be achieved using sulfuric acid or boron trifluoride (3.0%) within 2 h at 50 °C.74N,N′-Diphenyl-p-phenylene was used as polymerization inhibitor (0.08%). Removal of the catalyst by washing with basic solution and subsequent distillation in the presence of another 0.25% of polymerization inhibitor gave IBOMA (MA9) in a yield of 83%. Application of the strongly acidic cation exchange resin amberlyst 15 in the presence of the polymerization inhibitor hydrochinone at 40 °C led to yields of close to 90% and a purity of 98% for IBOMA (MA9) and IBOA (A9) after distillation of the crude reaction oil.75 J. Knebel describes these methods as difficult because of their long reaction times (for acid ion exchange resins), corrosion, and the need of catalyst neutralization, washing and extraction in the case of soluble catalysts, causing costs and environmental issues. He therefore suggested phosphomolybdic acid as a suitable catalyst – also for a continuous production process – for IBO(M)A (A9, MA9) synthesis.76 However, the yield was only about 50°% and the presence of polymerization inhibitors is mandatory during the synthesis and the work-up by distillation. The catalyst precipitates after addition of concentrated potassium hydroxide solution, enabling filtration as an easy way for catalyst separation. In 1996, J-M. Paul used a reactor system that included the pre-mixing of camphene (10), IBO(M)A (A9, MA9) and polymerization inhibitors such as hydrochinone methyl ether (250 ppm).77 The mixture then circulated through a cartridge loaded with amberlyst 15. This method reduced the contact time from 60 to about two minutes compared to other continuous processes with amberlyst 15.78 The product mixture contained 81% IBOMA (MA9), 8–9% methacrylic acid, and camphene (10) (that could be reused), and only 1.5% of oligomer/polymer side products. The purity of IBOMA (MA9) was 99.8% after distillation. More recently, a method by Osaka Organic Chemical Industry comes to similar results using amberlyst 1579 and in academic literature, this catalyst is also used.80 A. Heidekum et al. investigated the addition of carboxylic acids to cyclic olefins and compared amberlyst 15 and Nafion/silica composite (SAC 13) catalysts for the synthesis of IBOA (A9).81 They found that both types of catalysts are suitable and that the reaction temperatures can be surprisingly low (Table 8).
| Entry | Catalyst | Temperature [°C] | Conversion [%] | Selectivity [%] |
|---|---|---|---|---|
| 1 | SAC 13 | 60 | 94 | 92 |
| 2 | Amberlyst 15 | 60 | 93 | 85 |
| 3 | SAC 13 | 20 | 94 | 87 |
| 4 | Amberlyst 15 | 20 | 93 | 89 |
A. Riondel synthesized the superacidic Zr-based catalyst ZrSA15 and used it in IBOMA (MA9) synthesis (Fig. 20). This catalyst was obtained by calcination of pre-mixed Zr(OH)4 and (NH4)2SO4 (6:1) at 650 °C.82
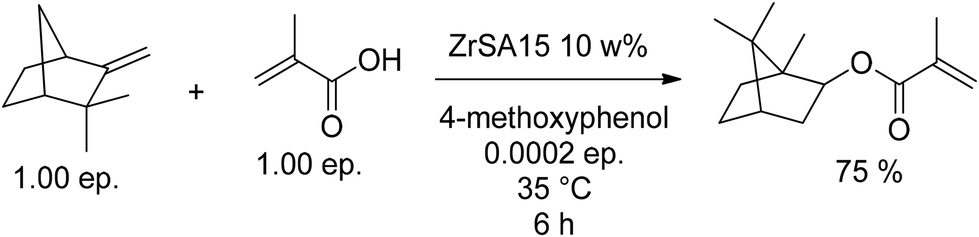 | ||
| Fig. 20 Rearrangement of camphene (10) and esterification with MA catalyzed by a Zr-based catalyst. | ||
This method led to a comparably low conversion of only 75%, but the selectivity towards IBOA (A9) was over 98%. In 2000, J. Knebel et al. described aqueous sulfuric acid (65–85 w%) in the presence of a mixture of polymerization inhibitors as a cheap and effective catalyst for IBO(M)A (A9, MA9) synthesis.83 The inhibitor mixtures consisted of TEMPO-derivatives, hydrochinone ethers, phenols substituted with bulky alkyl groups, and triazine derivatives. Chelating agents such as ethylenedinitrilotetraacetic acid disodium salt dihydrate prevented iron cations, either as impurity of the sulfuric acid or leaching from the stainless steel reactors, to decrease the reaction efficiency. The conversion after 4.5 h at 70 °C was only about 70%, but the selectivity of the reaction was almost 98%. Furthermore – after addition of 2,6-di-tert-butyl-α-dimethylamino-p-cresol to avoid polymerization – it was possible to separate camphene and methacrylic acid for reuse as well as IBOMA (MA9) by distillation directly from the mixture without a significant amount of polymeric side product. A control experiment clarified that the application of concentrated sulfuric acid leads to polymerization of IBOMA (MA9) during the distillation.
In 2010, an application filed by Nanjing Forestry University introduced activated carbon supporting tin tetrachloride as catalyst for the isomerization of unrefined crude α-pinene (1) and esterification with methacrylic acid.84 The reaction time was 8 h at 40–60 °C and afforded the presence of phenothiazine as polymerization inhibitor. As already described for other methods, no washing was required as the addition of a base inactivated the catalyst, enabling distillation of the reaction mixture to afford IBOMA (MA9) in a total yield of 84%. Another invention of J. Knebel et al. for the former Evonik Röhm GmbH is the application of montmorillonite as a heterogeneous catalyst, preferably in a continuously stirred tank reactor.85 Montmorillonite is a phyllosilicate with the chemical formula (Al1,67Mg0,33)[(OH)2Si4O10]·Na0,33(H2O)4 that occurs naturally. Table 9 shows the application of commercially available montmorillonite K10 in the conversion of camphene (10) with methacrylic acid. The catalyst was recycled by filtration and reused for all experiments.
| Entry | Unreacted MA [%] | Unreacted camphene [%] | IBOMA [%] | Camphene dimer [%] | Isoborneol [%] |
|---|---|---|---|---|---|
| 1 | 10.9 | 21.3 | 61.3 | 4.6 | 0.5 |
| 2 | 10.8 | 22.1 | 61.4 | 3.9 | 0.6 |
| 3 | 10.9 | 22.5 | 61.9 | 3.2 | 0.5 |
| 4 | 10.8 | 23.1 | 61.8 | 2.8 | 0.4 |
| 5 | 11.2 | 25.0 | 60.3 | 2.0 | 0.2 |
| 6 | 11.0 | 24.1 | 61.3 | 2.1 | 0.4 |
The reusability of the catalyst is underlined by very low differences of the reaction outcomes, and surprisingly the formation of the camphene (10) dimer as a side product is reduced by over 50% after six runs. The Chinese company Guangdong Lanyang Science & Technology and the South China Agricultural University investigated UV-light in combination with several catalysts for the synthesis of IBO(M)A (A9, MA9) from camphene (10) and (meth)acrylic acid as depicted in Table 10.86
| Entry | UV light [W] | Catalyst | Reaction time [min] | Product | Yield [%] |
|---|---|---|---|---|---|
| 1 | 100 | Phosphotungstic acid/silica gel HPW/SiSO2 | 10 | IBOMA | 91.2 |
| 2 | 200 | Triarylsulfonium hexafluoroantimonate salts (UVI6976 by Dow Chemical Company) | 20 | IBOA | 92.4 |
| 3 | 400 | p-Toluene sulfonic acid | 10 | IBOA | 91.6 |
| 4 | 500 | Bis(4-dodecylphenyl)iodonium hexaflurorantimonate | 20 | IBOMA | 92.0 |
| 5 | 300 | Amberlyst 15 | 30 | IBOA | 92.8 |
The reaction times are very short compared to other methods, polymerization inhibitors prevented side reactions, and direct distillation from the crude reaction mixture gave the pure products without previous separation of the catalysts.
3. Polymerization of terpene acrylates
Poly(methyl methacrylate) (PMMA) is a crucial polymer for numerous applications owing to its advantageous properties, including thermal stability,87 transparency,88 and high glass transition temperature (Tg) of 105 °C.87,89 Additionally, PMMA is known for its excellent mechanical properties, good weather resistance, chemical resistance, and outstanding optical characteristics.89 Suitable for a large variety of applications, polyacrylate resins are indispensable in clear coatings in the automotive industry, in many optical devices, and physically drying paints.Given the versatility of polyacrylate resins, it is highly desirable to explore the replacement of commonly used petrochemical-based monomers with functionalized terpenes. The subsequent sections provide an overview of the approaches that have been investigated to achieve this goal.
3.1. Free radical polymerization
Numerous monomers derived from biomass, like plant oil, eugenol and isosorbide, containing a (meth)acrylate group, have already been investigated29–31 but only a few are based on monoterpenes.In 2013, S. M. Howdle et al. investigated polymerization various terpene-based acrylates (A) and methacrylates (MA) using radical polymerization. By employing different amounts of the chain transfer agent (CTA) dodecyl mercaptan (DDM), they obtained a range of polymers with diverse properties and molecular weights. The summarized results of this study are presented in Table 11.
| Monomer | Polymer structure | [DDM] (wt%) | Conversion (%) | M n (g mol−1) | M w/Mn | T g (°C) |
|---|---|---|---|---|---|---|
| A1 | PA1 | 5.0 | 95 | 4800 | 1.34 | 12 |
| A1 | PA1 | 1.0 | 97 | 15100 |
2.04 | 63 |
| A1 | PA1 | 0.5 | 99 | 23600 |
2.20 | 71 |
| MA1 | PMA1 | 5.0 | 99 | 7900 | 1.41 | 85 |
| MA1 | PMA1 | 1.0 | 99 | 17400 |
1.71 | 140 |
| MA1 | PMA1 | 0.5 | 99 | 22200 |
1.85 | 142 |
The study demonstrated the correlation between molecular weight and glass transition temperature and highlighted the distinctions between acrylate and methacrylate with respect to Tg. Poly(α-pinene MA) (PMA1) exhibited a notably high glass transition temperature, reaching up to 142 °C, while maintaining transparency. The observed difference in the glass transition temperature of poly(α-pinene acrylate) (PA1) and poly(α-pinene methacrylate) (PMA1) was attributed to steric hindrance caused by the methacrylate group within the polymer chain. This inhibits segment rotation and increases the stiffness of the chain, resulting in a higher Tg compared to the acrylates. However, there is no description of the DSC measurement method used to obtain information about Tg. Since the detected value of Tg depends on parameters such as the heating rate, the prehistory of the polymer, and the measuring method itself, it is challenging to compare these results with others presented in this review. The different homopolymer structures can be observed in Fig. 21.
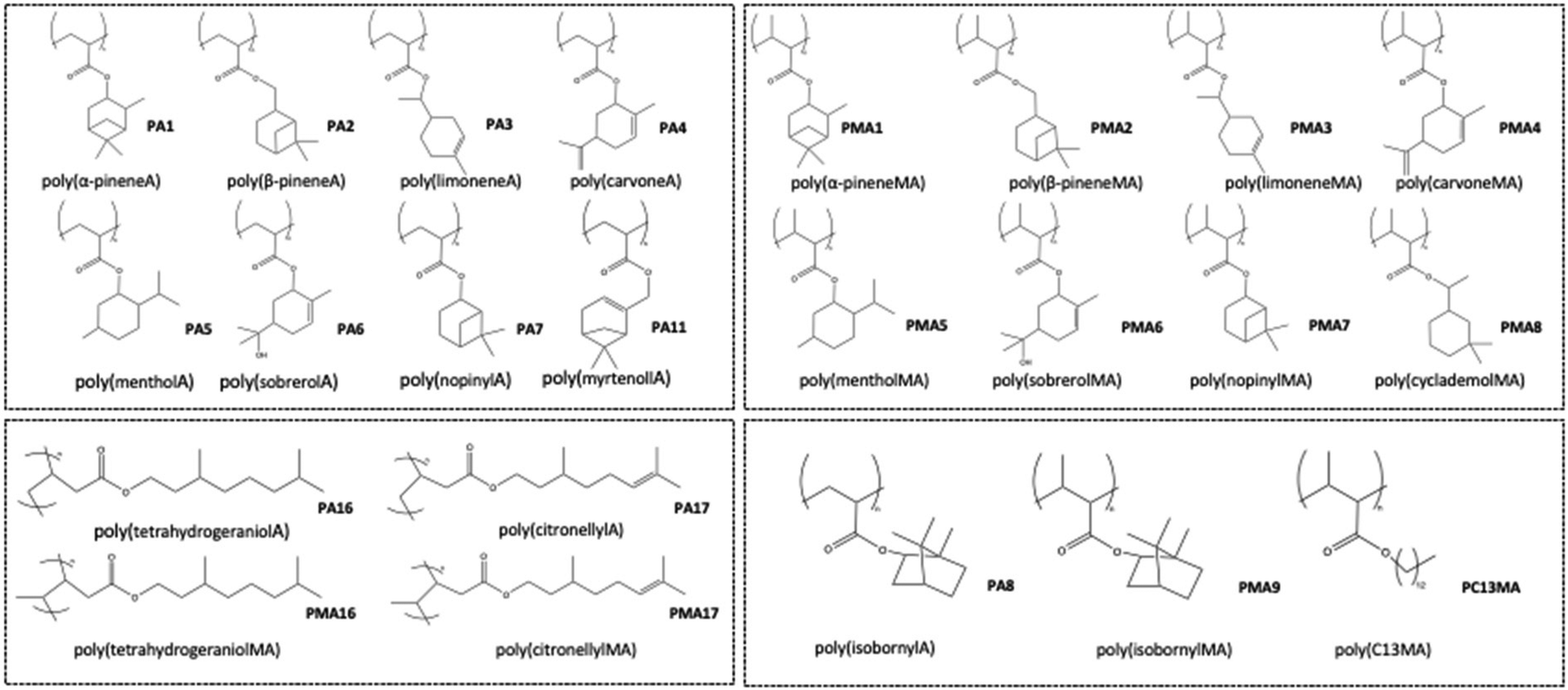 | ||
| Fig. 21 Polyacrylates based on cyclic monoterpene (PA1–PA7; PA11) and polymethacrylates (PMA1–PMA8); acyclic monoterpene based polyacrylates (PA16; PA17) as well as polymethacrylates (PMA16; PMA17) and polymers from conventionally available acrylate (PA8; PMA9; PC13MA) and methacrylate. | ||
While the pinene-based polymers showed high conversions, only a small fraction of carvone acrylate (A4) could be polymerized. Increasing the temperature from 60 to 110 °C increased the degree of conversion but led to gelation. Polymerization of carvone-based methacrylate (MA4), however, resulted in high yields and no crosslinking or gelation. Both reactions were followed by FTIR microscopy. This analysis showed the conversion of only one double bond for methacrylate, whereas both double bonds reacted in the case of carvone acrylate. Therefore, they showed that crosslinking arises from the reactivity of the exocyclic olefin. The research group explained this behavior with the preferred attack of the initiator at the exocyclic olefin to form the thermodynamically favored tertiary radical. At higher temperatures, the acrylic double bond was involved in the reaction, leading to crosslinked gels. In the case of the methacrylate, the initiator attacks preferably on the methacrylic double bond, which leads to linear polymers. Due to the consumption of only one reactive double bond of the carvone methacrylate, the remaining double bond increases the attractiveness of the monomer for biobased polymeric coatings. The obtained polymers can be further crosslinked with commercial crosslinkers for use in different coating applications.50
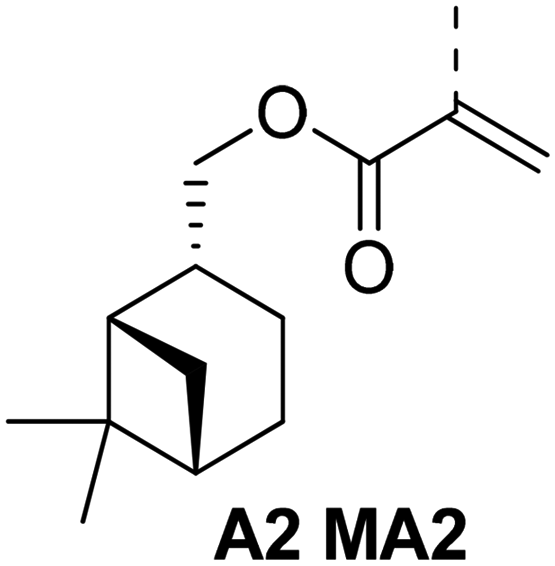
The same research group designed new monomers based on nopinyl (meth)acrylate (A7, MA7) due to the correlation between Tg and the distance of the (meth)acrylate group to the bridging unit in the terpene. The corresponding structural formulas are shown in Fig. 22. They predicted a higher Tg for nopinyl MA, due to the absence of one carbon atom between the cyclic structure and the acrylic double bond in contrast to β-pinene (meth)acrylate (A2, MA2). They reported a very high Tg of around 170 °C for the poly(nopinyl methacrylate) homopolymer measured by DMA.90 Several other publications about polymers based on pinene have been published in recent years.43,52,53,61,91
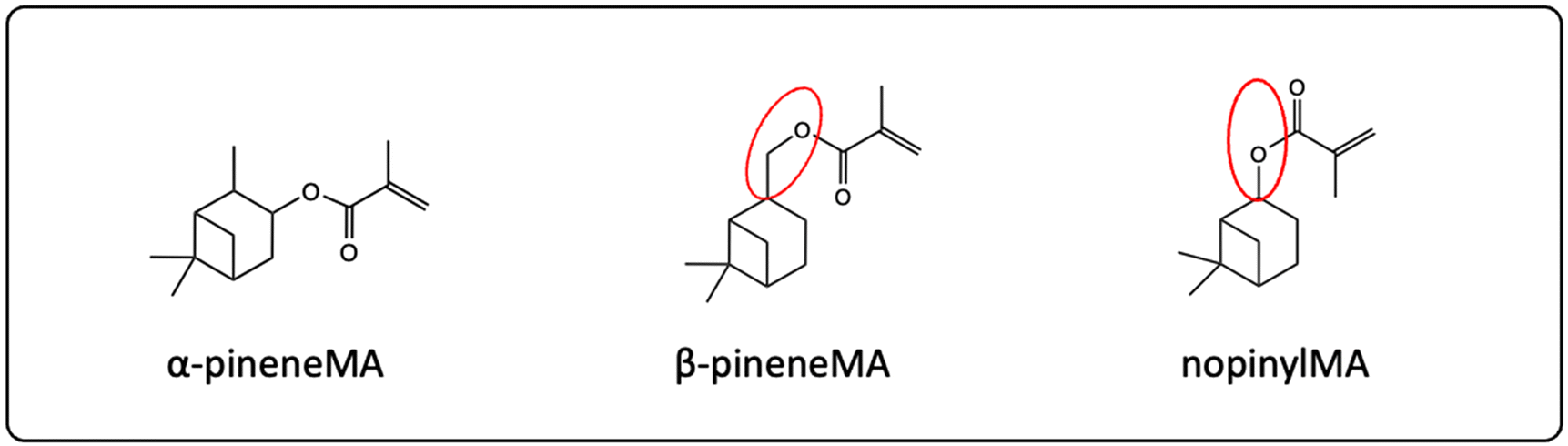 | ||
| Fig. 22 Chemical structure of α-pineneMA, β-pineneMA, and nopinylMA. In case of nopinylMA, the bridging unit is closer to the (meth)acrylate functionality, which results in less flexibility of the terpene group. | ||
The group of Billon investigated the synthesis of terpenoid based (meth)acrylic monomers by using an enzymatic synthesis pathway. They used the terpenoids tetrahydrogeraniol (16), citronellol (17), cyclademol (8), and nopol (7) to create the corresponding (meth)acrylate monomers. They used free radical polymerization technique to obtain homopolymers. Unfortunately, they performed the polymerization only for the acrylate monomers PA7, PA8, PA16 and PA17 and not for the methacrylate monomers. Neither did they analyze a glass transition temperature of the homopolymers, which makes it difficult to classify the possible applications of the monomers.49
In 2020, Mülhaupt et al. introduced the use of limonene-based dimethacrylate for 3D printing application. They demonstrated the potentiol to substitute bisphenol A glycidyl methacrylate (BisGMA) with this monomer, offering the additional advantage of significantly lower viscosity (5–117 Pa s) compared to BisGMA (560 Pa s).58
By Min and colleagues investigated the free radical polymerization of menthol methacrylate (MA5) using benzoyl peroxide as the initiator, both in solution and in bulk. With this method, a homopolymer (PMA5) and a copolymer were synthesized with Tg of 83 °C and 113 °C, respectively.36 Another group prepared a copolymer through AIBN-initiated free radical polymerization, containing trans-4-methacryloxyazobenzene and menthol acrylate (MA5).32 In 2015, menthol acrylate was used for the formulation of transparent acrylic pressure-sensitive adhesives (PSAs). The polymerization was carried out in bulk under UV irradiation obtaining polymers with a Mw between 998–1410 kg mol−1.37 Two years later, UV-cured transparent PSAs were prepared using menthol acrylate (MA5) and tetrahydrogeraniol (A16). The research group investigated adhesive performance, viscoelastic properties, and optical properties of these transparent acrylic PSAs.38
Another study details the conversion of an acrylate based on sobrerol (6) via free radical polymerization. They obtained sobrerol acrylate (A6) achieved a Mw of (4.3 ± 0.2) kDa, measured by sedimentation equilibrium method.51
The group led by Droesbeke investigated terpenoid-based (meth)acrylates (A5, A6, A17, MA9, MA17) in combination with acrylic acid and MMA as copolymers via emulsion polymerization. The objective was, once again, to use these polymers as pressure-sensitive adhesives. They claimed to produce 100% biobased and waterborne polymers. However, it is important to note that they obtained the acrylate building block from (meth)acrylic acid, and therefore, the process cannot be considered fully biobased for large-scale applications.92
In another emulsion polymerization, myrcene was combined with methacrylate monomers of varying chain lengths (stearyl, lauryl, and butyl). They descripted the effect of the side chain length on the polymerization reaction. Additionally, they investigated the Tg and the morphology of the copolymers.93
The aliphatic methacrylate citronellylMA (MA17) was utilized for the chemical modification of potato starch to produce amphiphilic materials. Grafting resulted in altered morphology, polarity, solubility, chemical stability, etc., showcasing the potential of terpene acrylates for enhancing the performance of copolymers.94
In summary, all the (meth)acrylates synthesized from different monoterpenes can be converted into homopolymers (Table 12) and copolymers by the free radical polymerization method. The conversion and the molar mass depend on the respective experimental conditions. The Tg of the individual homopolymers depends both on the molecular structure itself and, to a certain extent, on the molar mass.
| Polymer structure | Conversion (%) | M n (g mol−1) | T g (°C) | Ref. |
|---|---|---|---|---|
| PA1 | 95 | 4800 | 12 | 50 |
| PA1 | 97 | 15100 |
63 | 50 |
| PA1 | 99 | 23600 |
71 | 50 |
| PMA1 | 99 | 7900 | 85 | 50 |
| PMA1 | 99 | 17400 |
140 | 50 |
| PMA1 | 99 | 22200 |
142 | 50 |
| PA3 | 88 | 17000 |
−5 | 50 |
| PA4 | 60 | 10600 |
5 | 50 |
| PMA4 | 99 | 26500 |
117 | 50 |
| PMA5 | — | 124500 |
83 | 36 |
| PMA6 | ||||
| PA7 | 86 | 97400 |
74 | 90 |
| 77 | 211000 |
49 | ||
| PMA7 | 91 | 48000 |
170 | 90 |
| PA16 | 80 | 215000 |
— | 49 |
| 99 | 278000 |
−46 | 42 | |
| PA17 | 78 | 205000 |
— | 49 |
In summary, the chemical structure of terpene (meth)acrylates is versatile, allowing for the production of alternatives with high biobased content for various commercial acrylates. Combining different terpene-based acrylics offers the ability to tailor the mechanical properties of the resulting polymer. While this principle is already applied in the design of “classic” polyacrylates, the presented studies demonstrate that monomers derived from renewable sources enable the tuning of polyacrylate properties in a similar manner.
3.2. Controlled radical polymerization
To obtain a high degree of control over the physical and mechanical polymer properties, the polymerization technique must be properly selected in addition to the monomers. Radical polymerization is suitable for a wide range of vinyl monomers with different functional groups, as well as many solvents. It is used to manufacture around 45% of synthetic polymers worldwide.95 However, this approach is not suited for the synthesis of polymers with specific molecular mass and low polydispersity. To overcome this limitation, controlled radical polymerization (CRP), also called reversible deactivating radical polymerization (RDRP), can be used. The advantages of CRP methods are their good control over composition, topology, dispersity, and end-group functionality.96 In contrast to living polymerization, it can be readily done in dispersed aqueous media and can be used for a broad range of monomers.97The most common methods for controlled radical polymerization are the atom transfer radical polymerization (ATRP),98,99 the reversible addition–fragmentation chain transfer (RAFT) polymerization,100 and the nitroxide-mediated polymerization (NMP).97
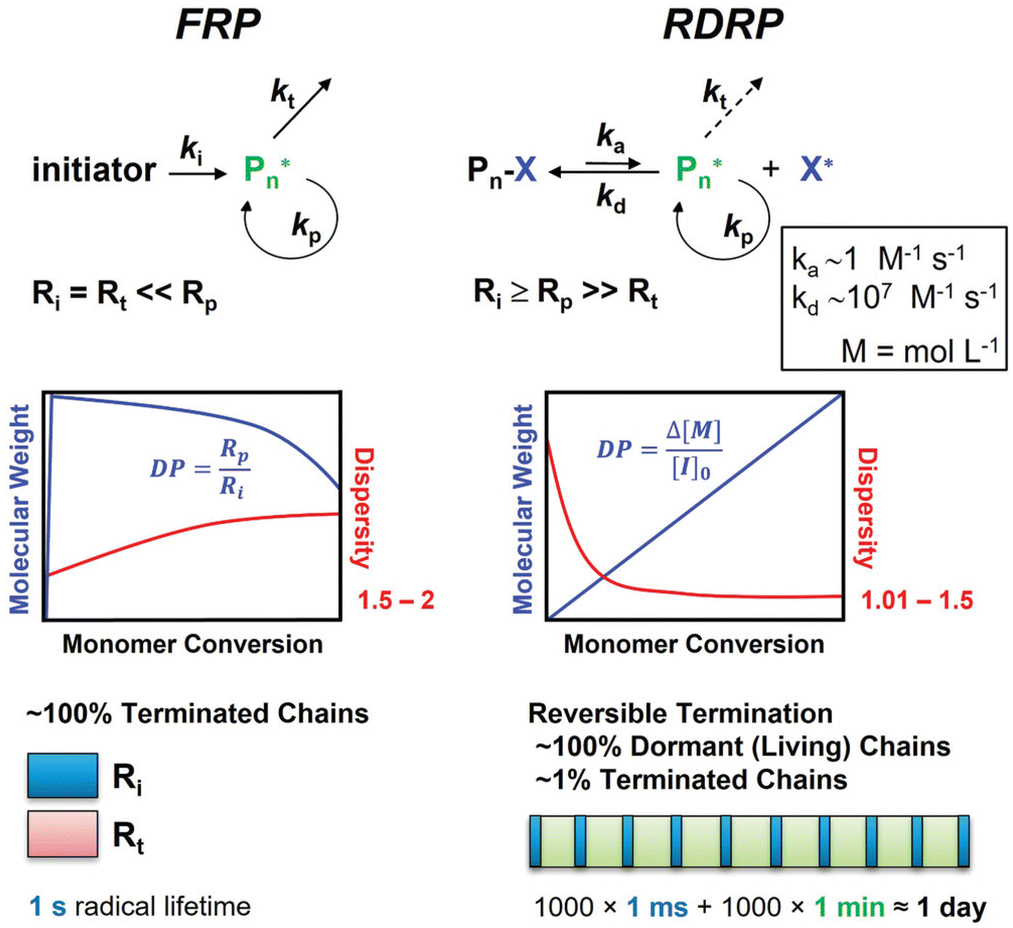 | ||
| Fig. 23 Free radical polymerization and reversible deactivation radical polymerization. Ri, Rt, and Rp are representing the rate of initiation, termination, and propagation; ki, kt, and kp are the corresponding rate constants; ka and kd are the radical activation and deactivation rate constants. DP is the degree of polymerization, M is the monomer, and I the initiator.96 (Figure: Sylwia Dworakowska, Francesca Lorandi, Adam Gorczyńskif, Matyjaszewski Krzyszto, https://onlinelibrary.wiley.com/doi/full/10.1002/advs.202106076, CC BY 4.0.) | ||
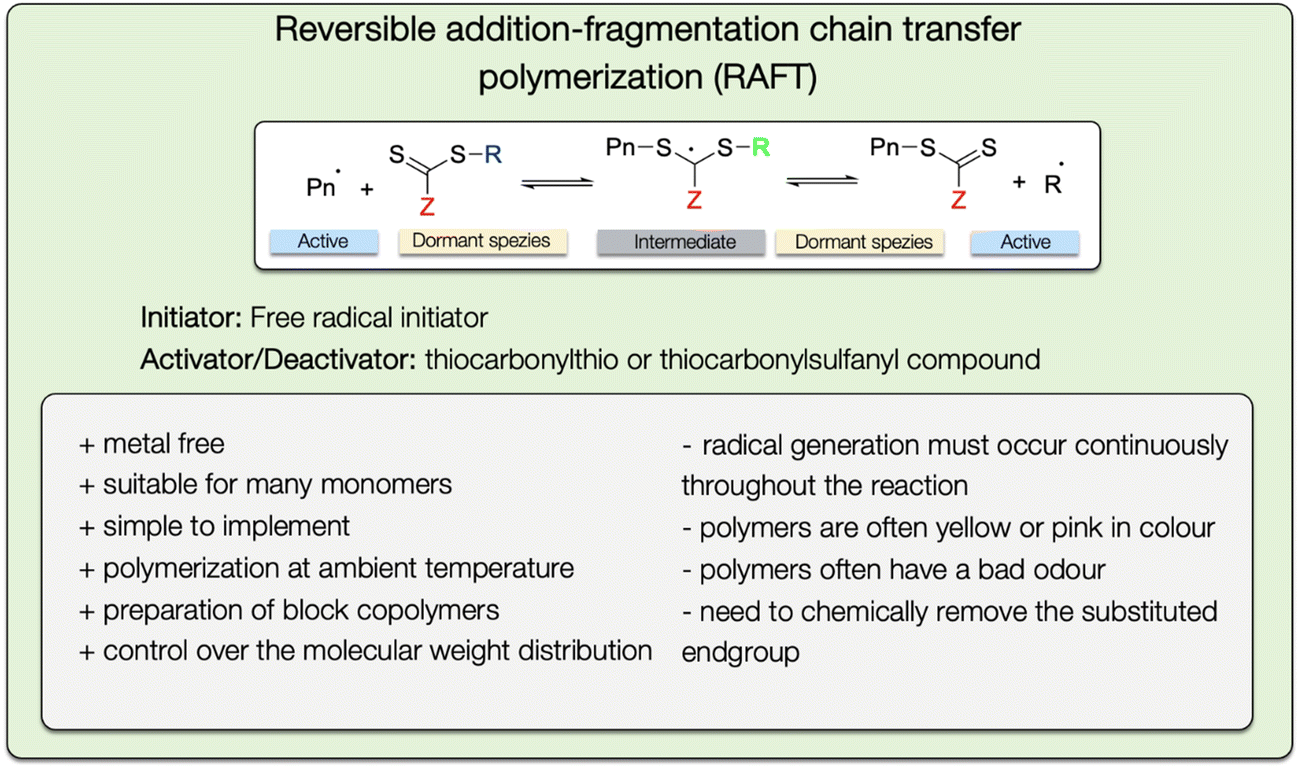 | ||
| Fig. 24 General reaction mechanism of the RAFT polymerization method with a list of advantages and disadvantages. | ||
The RAFT technique can be used for production of block copolymers, which are however not as pure as the ones created via ATRP (see section 3.2.3). Because of the continuously generated radicals in case of the RAFT method, high-order blocks are contaminated by a small amount of homopolymer.100
An important advantage of the RAFT technique compared to other CRP is, that it is well suited for implementation in industrial processes due to the similarity to free radical polymerization (Fig. 24). In both polymerization methods, the same chemicals are used with the addition of the RAFT agent. However, the high cost and the need to chemically remove the substituted end group created via the RAFT reagent are still obstructions.
Recently, the working group of Howdle investigated the polymerization of α-pinene methacrylate (MA1), β-pinene methacrylate (MA2), and limonene acrylate (A3) via RAFT method to obtain di- and multiblock copolymer architectures. At first, they examined the glass transition temperature of the homopolymers with a value of −3 °C for the poly(limonene acrylate) (PA3) and up to 168 °C for poly(α-pinene methacrylate) (PMA1). Additionally, they investigated the synthesis of hard–soft block copolymers, i.e. ABA triblock copolymers of poly(α-pinene methacrylate) (MA1) and poly(butyl acrylate). Both showed promising adhesive properties comparable to that observed for the commercial material. Another triblock copolymer was reported, containing poly(limonene acrylate) (PA3) as soft blocks.102
In 2019, Noppalit and coworkers synthesized a tetrahydrogeraniol acrylate (A16), which was polymerized via controlled radical polymerization. The resulting polymer had a relatively low Tg of −46 °C. Therefore, it could be used as a replacement for low Tg acrylic monomers, such as the widely used n-butyl acrylate (Tg −54 °C).89 The use of poly(tetrahydrogeraniol acrylate) (PA16) as an elastomeric building block in triblock copolymers with poly(styrene) using RAFT polymerization method. They observed a phase separation of the soft PA16 (Tg of −22 °C) and the hard poly(styrene) phase (Tg of 110 °C), followed by AFM (Fig. 25).42
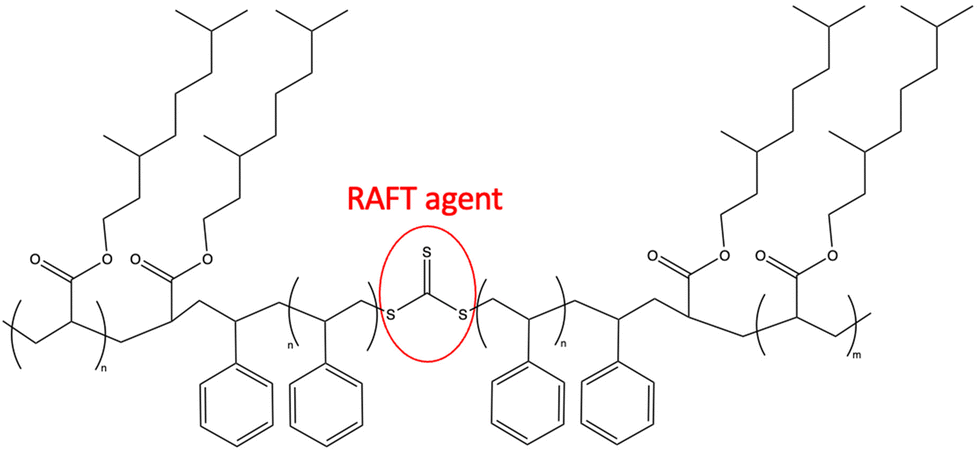 | ||
| Fig. 25 Use of bifunctional RAFT agent to build up the ABA triblock copolymer poly(tetrahydrogeraniol acrylate)-poly(styrene)-b-poly(tetrahydrogeraniol). | ||
In the same year, they also published an article about pressure-sensitive adhesives based on tetrahydrogeraniol acrylate (A16) and cyclademol methacrylate (MA8) triblock copolymers. They used a RAFT miniemulsion polymerization technique to achieve a waterborne process. The resulting poly(tetrahydrogeraniol acrylate)-b-(cyclademo methacrylate)-b-(tetrahydrogeraniol acrylate) copolymers had a high molecular weight of Mn ≈ 105500 g mol−1 and Đ ≈ 1.6. They observed nanophase segregation by AFM and rheological measurements. The copolymer shows good mechanical properties and adhesive performances compared to copolymers partially based on styrene.103
The research group of E. Malmström56 compared free radical polymerization, RAFT, and ATRP for the terpenoid-based sorbrerol methacrylate (MA6). Additionally, they investigated an enzymatic pathway using horseradish peroxidase-mediated free radical polymerization. Peroxidases like horseradish peroxidase (HRP) have been used as bio-initiators, whereby some other radical-building enzymes are known as well (alcohol oxidase, xanthine oxidase, laccases). HRP is oxidized by H2O2 and builds its two catalytically active forms. The initiator monomer is oxidized by the active form of the enzyme, creating two radicals, and returns to its native state afterwards. A great advantage is, that the only byproduct of this reaction is water (Fig. 26).56
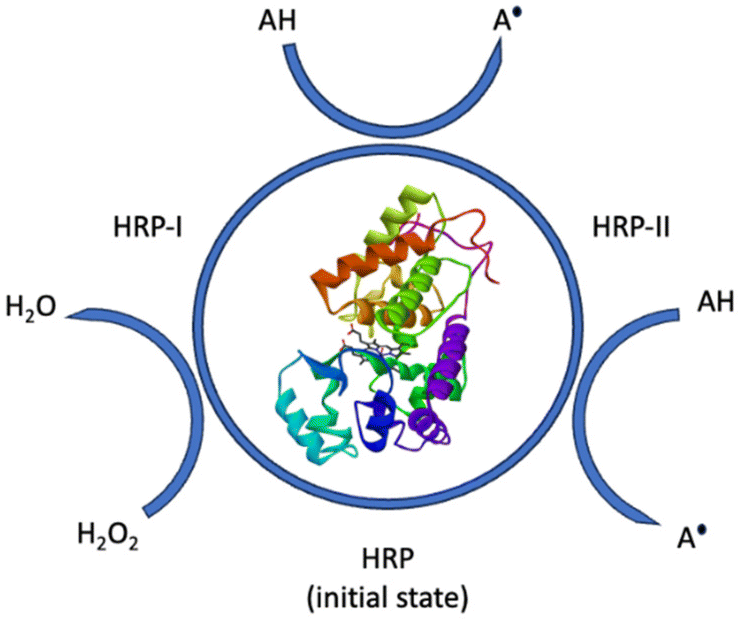 | ||
| Fig. 26 Simplified catalytic mechanism of peroxidase-catalyzed radical formation. | ||
The polymers (PMA3) show in all cases a high Tg between 116 and 155 °C for the homopolymers, and a control of the molar mass ratio could be obtained for the RDRP methods. Surprisingly, only small differences in the molar mass led to a large difference the glass transition temperature. For the homopolymer created by RAFT polymerization, the research group reported a Tg of 116 °C and Mn = 7200 g mol−1. The ratio of monomer:RAFT agent:initiator was 50:1:0.2. On the other hand, the homopolymer obtained via free radical polymerization (ratio monomer:initiator = 250:1) showed a Tg of 154 °C with a much higher number average molar mass of Mn = 48200 g mol−1. The difference in Tg of 38 °C can be compared with another study dealing with α-pinene methacrylate homopolymers.50 Here, the Tg of two polymers with an number average molecular weight of 7900 g mol−1 and 22200 g mol−1 differed by 57 °C, which is significantly higher than the previously discussed example.
Further insights into the polymerization of α-pinene methacrylate were obtained by preparation of coatings based on PMA3. In this case, additional crosslinking of the remaining double bond using thiol-ene click chemistry with UV curing was carried out. Additionally, thermal crosslinking of the tertiary alcohol was achieved by adding hexamethoxymethylmelamine, as well as a catalyst. They monitored the conversion of the double bond with FT Raman spectroscopy. In the second case, the reaction of the hydroxy group was studied via FTIR. Finally, the crosslinked films were tested regarding their solubility in THF (shaking for 24 h), which proved their insolubility. However, no further information about the mechanical properties of the polymer films were reported.56 As mentioned before, the research group of Howdle investigated the use of α-pinene, β-pinene, limonene, and carvone for methacrylate monomer synthesis. They showed that the obtained monomers can be readily polymerized via free radical polymerization and can additionally be used for reversible deactivation radical polymerization methods. A block copolymer with poly(α-pinene methacrylate) (PMA1) as hard blocks and poly(limonene acrylate) (PA3) functioning as soft blocks were successfully synthesized using RAFT polymerization.102
A few years later, they investigated the RAFT polymerization of THGMA (MA16) and the novel monomer nopinyl methacrylate (MA7), resulting in hard–soft–hard ABA triblock copolymers. They used 2-cyano-2-propyl dodecyl trithiocarbonate as an RAFT agent and reached a conversation of up to 96% in 24 h with Mn = 10800 g mol−1 and a polydispersity of 1.14 for the homopolymer poly(nopinyl methacrylate).
A special case of block copolymers are triblock copolymers, which are also accessible via RAFT polymerization. Preparation of triblock copolymers requires a difunctional RAFT agent, which has to be synthesized first. In one example, a macro-RAFT agent based on poly(MA4) was used. In the next step, the macro-RAFT agent was polymerized with (nopinyl methacrylate) MA7 to obtain the triblock copolymers (Fig. 27). A polymer with a low polydispersity index of 1.10 and an number-average molar mass of 16100 g mol−1 was achieved with a conversion of up to 63%.90
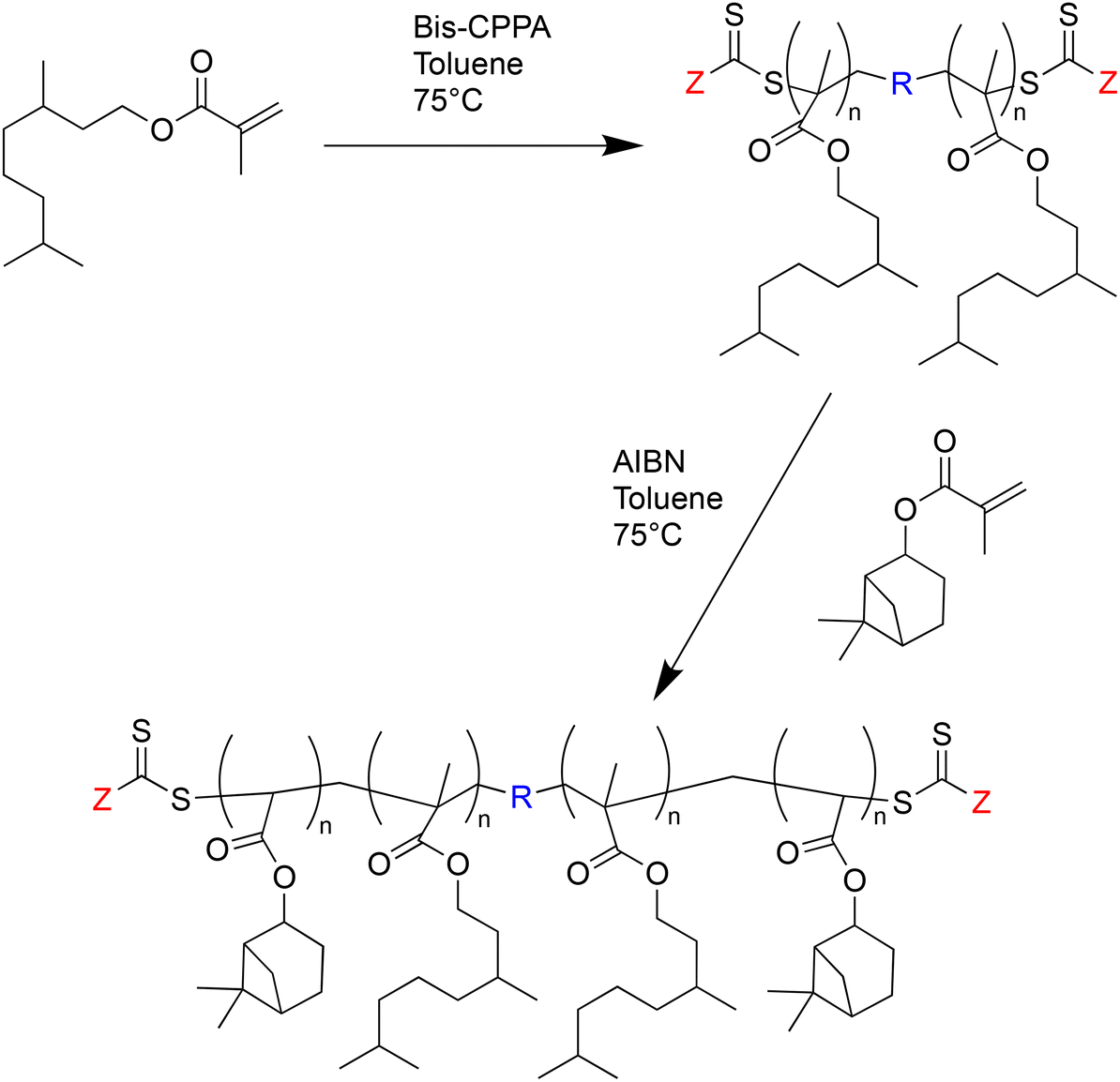 | ||
| Fig. 27 Synthesis of PNPMA-b-PTHGMA-b-PNPMA triblock copolymers using macro-RAFT agent. | ||
In another study, tetrahydrogeraniol acrylate (A16) was used as a soft block in combination with polystyrene as a hard block to form a thermoplastic elastomer.42
Another interesting attempt was made to synthesize polycarvon acrylate diepoxide for use as a monomer in a one-step electrospinning process to create epoxy functional surface fibers. The group of Montanari blended the polymer with polyvinylidene fluoride in solution and create fibers with an epoxy functional surface.104
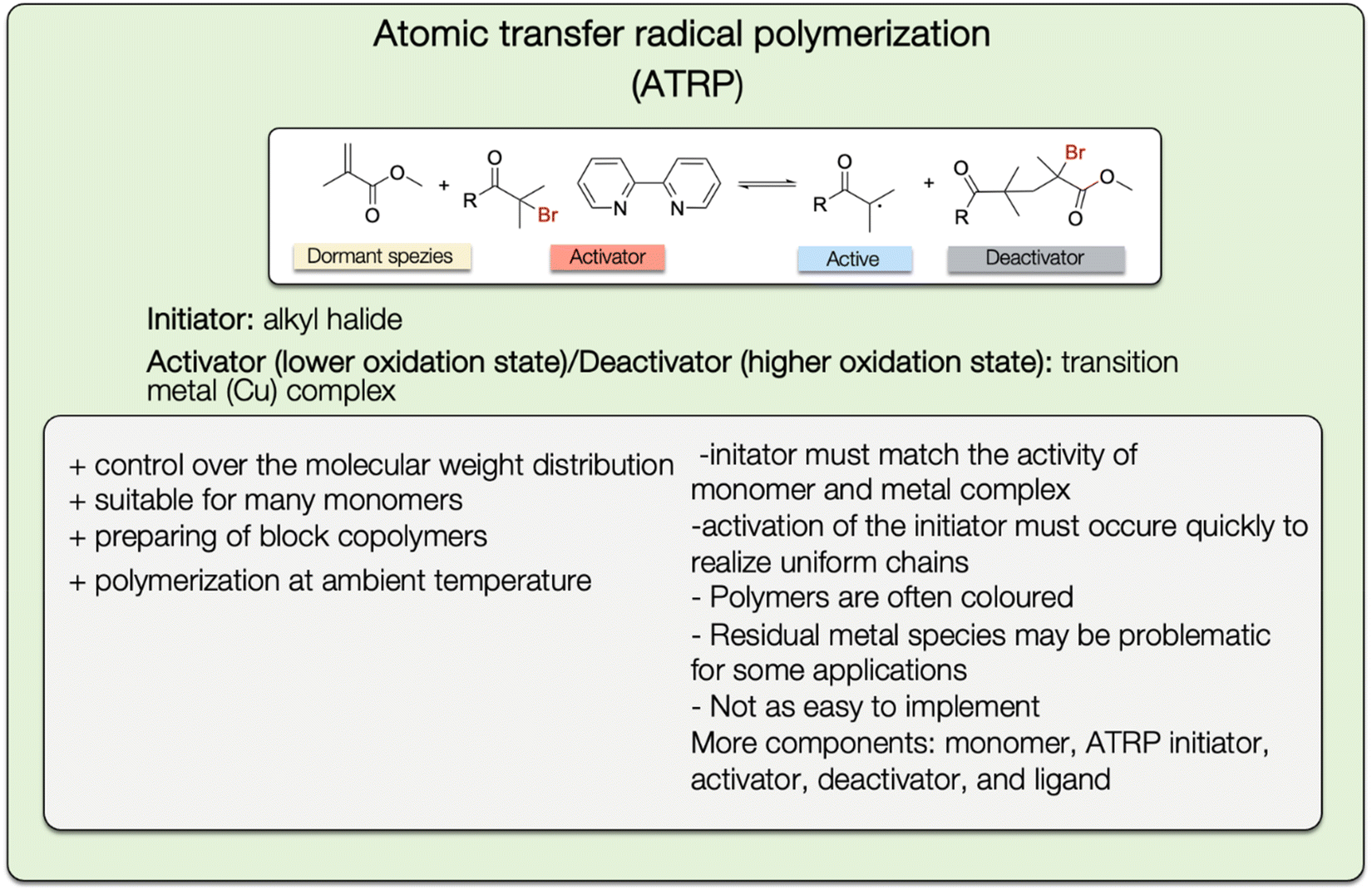 | ||
| Fig. 28 General reaction mechanism of the ATRP polymerization method with a list of advantages and disadvantages. | ||
In 2011, the polymerization of L-menthyl methacrylate (MA5) using ATRP was explored, employing bis[2-(2-bromoisobutyrooxy)ethyl]disulfide as the ATRP agent.39
Maric et al.105 published the use of an ATRP technique for the copolymerization of the biobased methacrylate IBOMA (MA9) and C13MA (similar to lauryl methacrylate) with ethylene glycol dicyclopentenyl ether methacrylate (EGDEMA). Specifically, they utilized ARGET ATRP, a method where the activator is continuously regenerated through electron transfer. The notable advantage of this approach is the minimal catalyst requirement, in the order of a few parts per million (ppm). Consequently, issues such as odor and coloration associated with the catalyst could be significantly mitigated. With this method, they synthesized both statistical poly(EGDEMA-stat-C13MA) copolymers and poly(C13MA-b-EGDEMA) block copolymers. To enhance the rigidity of the polymers, IBOMA was copolymerized with EGDEMA, and the resulting product was cured through thiol–ene clicking of the pendant double bonds (Fig. 29).106
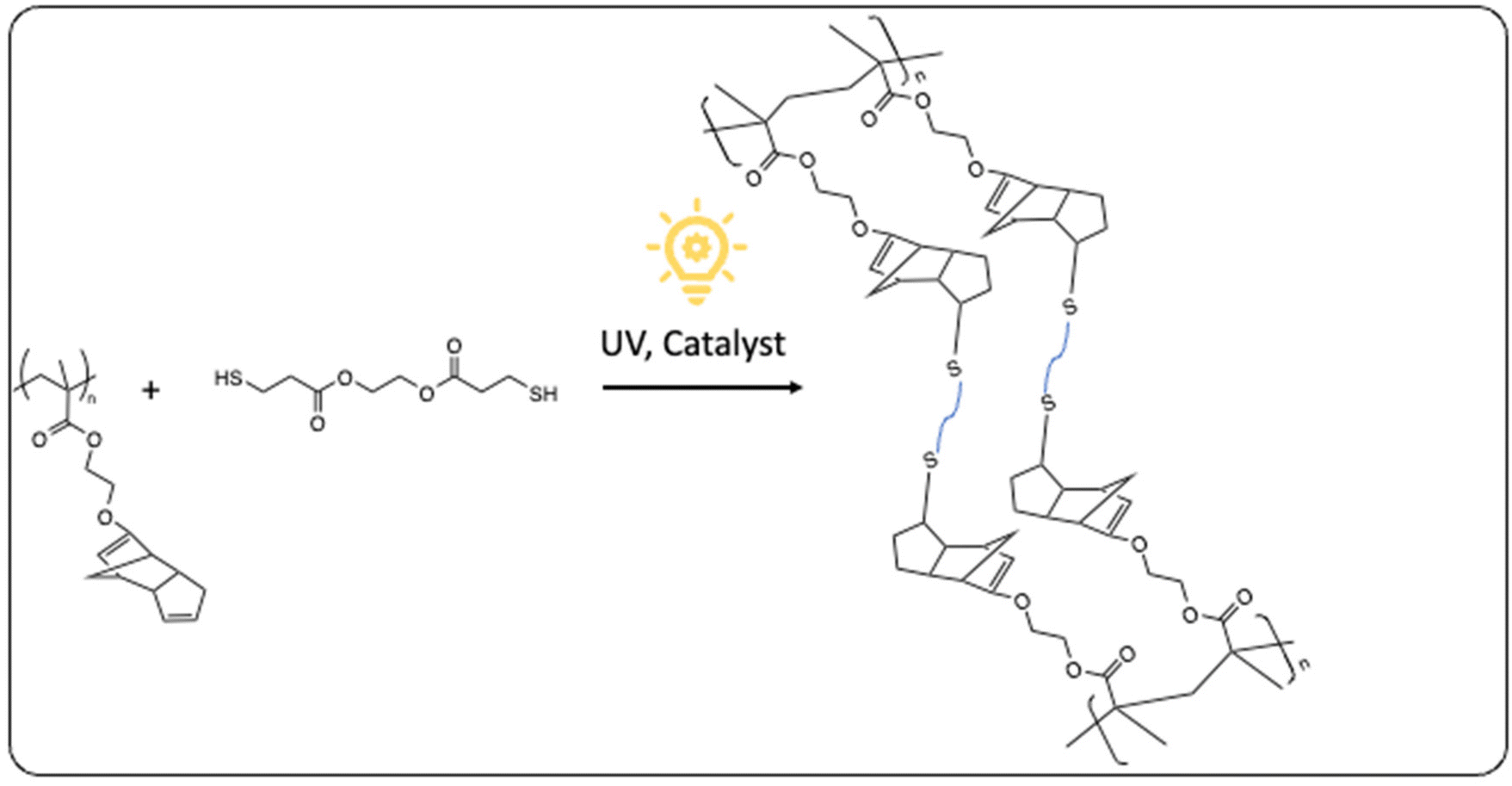 | ||
| Fig. 29 Curing of copolymerized IBOMA/EGDEMA by photoinitiated thiol–ene clicking of the free double bonds of EGDEMA. | ||
Liu and Mishra used an ATRP strategy for polymerization of menthol acrylate (A5). They investigated various ligands and achieved a conversion rate for PA5 of 78.4% within 0.5 h (Mn = 14000 g mol−1; Mw/Mn = 1.11) using tris[2-(dimethylamino)ethyl]amine (Me6TREN) as the ligand. Additionally, they successfully synthesized diblock copolymers with methyl acrylate.107
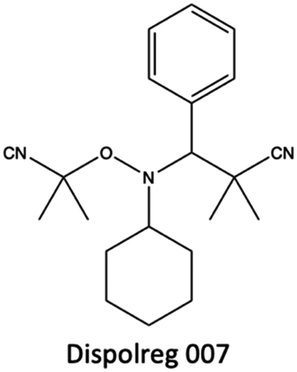 | ||
| Fig. 30 Chemical structure of the alkoxiamine Dispolreg 007. | ||
Three years later, the same research group explored the synthesis of block copolymers using two terpenoid-based monomers, cyclademol MA (MA8) and tetrahydrogeraniol MA (MA16), in a miniemulsion with Dispolreg 007. Their goal was to address the limitations associated with other controlled living polymerization methods, such as the use of toxic solvents, expensive control agents, and the need for intermediate purification. They successfully achieved the synthesis of a poly(MA16)-b-poly(MA8) diblock copolymer, achieving a conversion of 98% for both the MA16 and MA8 monomers. The synthesis duration was 8 hours at 97 °C. The resulting poly(MA16)-b-poly(MA8) exhibited molecular weights (Mn) of 124000 g mol−1 and 77000 g mol−1, with low polydispersity values of 1.35 and 1.62, respectively.110 Nitroxide-mediated polymerization of methacrylate is reported in emulsion,111–113 miniemulsion,114–116 micro-emulsion,117,118 and suspension.119 Certainly, a notable advantage of Nitroxide-Mediated Polymerization (NMP) is that the removal of the control agent is not necessary, as the utilized nitroxides do not impact the color or odor of the polymers (Fig. 31). This characteristic is a key factor contributing to the suitability of NMP for industrial applications.97 As mentioned in the previous sections, the RAFT and ATRP technique are triggered by sulfur-based chain transfer agents or metallic ligands, respectively. Consequently, the products have to undergo exhausting post-polymerization treatments to remove residual catalysts or other reagents.101
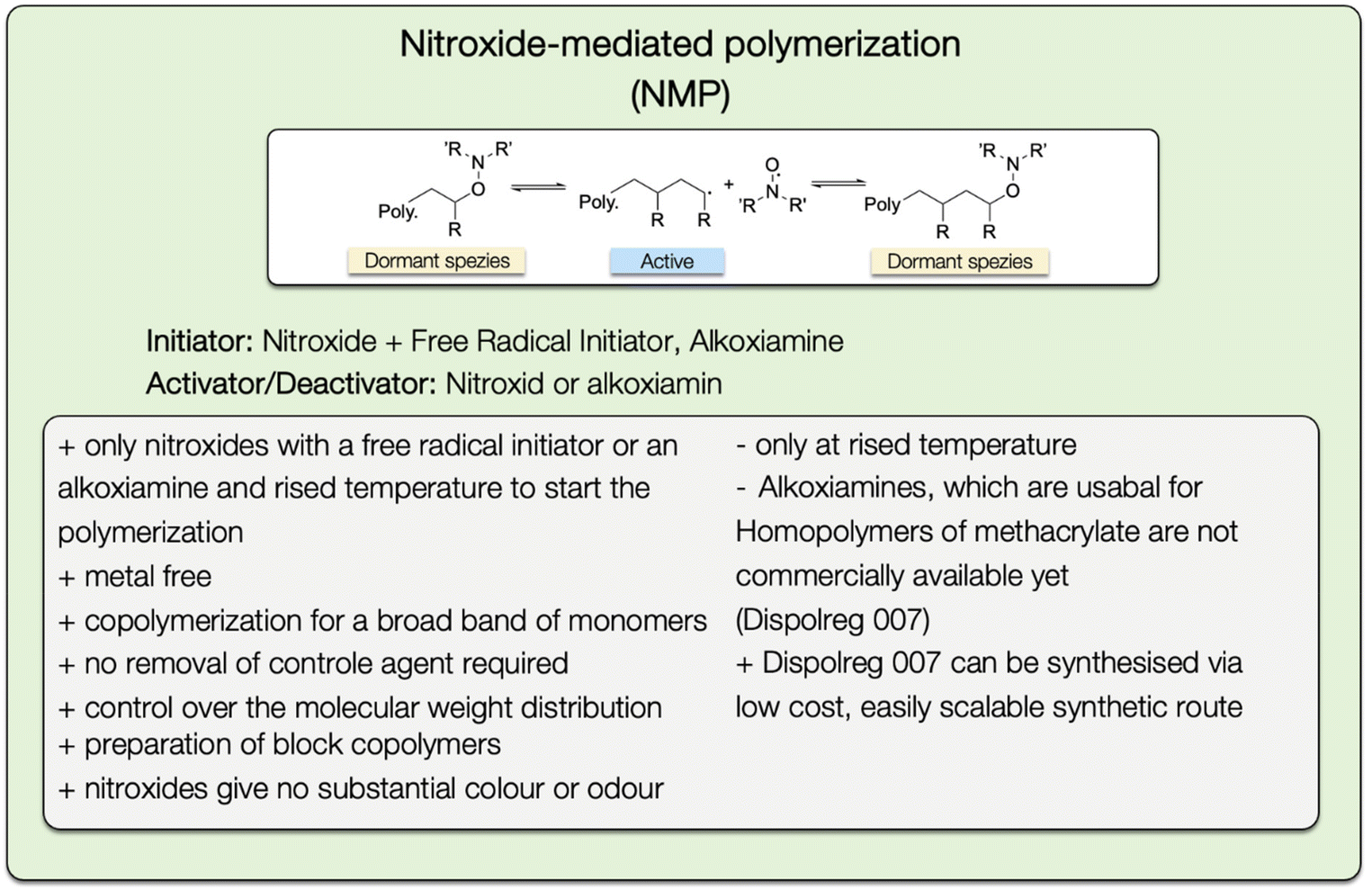 | ||
| Fig. 31 General reaction mechanism of the NMP polymerization method with a list of advantages and disadvantages. | ||
Despite many new developments, only a few products in the field of adhesives, coatings, and additive markets are produced with a controlled radical polymerization process.120,121 In order to obtain biobased polymers with tailored properties, controlled polymerization of terpene acrylates bears high potential for further research. Progress on this field will most likely lead to even more breakthroughs to finally establish this material class in our everyday lives. The most prominent example, how this can succeed, is polymerization of the terpene acrylate IBOMA.
3.3. Polymerization of IBO(M)A
Isobornyl methacrylate (IBOMA, MA11) serves as an example of a monomer already employed in industrial applications. Polyacrylates with high chemically reactive grades and low molecular weights, typically around 20000 g mol−1, are utilized in the production of two-component lacquers. These lacquers can be crosslinked with melamine resins or isocyanates. Polymers with molecular weights exceeding 100000 g mol−1 find applications in high-solid paints, where the elevated molecular weight in polymer dispersions ensures rapid drying of the film.89 Controlled radical polymerization techniques provide a means to attain tailor-made polymers with specific molar masses (and polydispersities) suited for a desired application.
In 2018, a research group investigated the polymerization of IBOMA (MA11) with nitroxide-mediated polymerization technique.122 They used different experimental conditions to produce IBOMA (MA11)/acrylonitrile (AN) copolymers and a poly(IBOMA-stat-AN)-b-poly(TDMA-stat-AN) block copolymer in toluene solution. An optimal temperature of 100 °C was determined for a sufficiently high reaction rate and good control of the polymerization. Thereby, different monomer ratios were investigated to adjust the Tg of the resins. The AN was used as a controlling co-monomer and two different nitroxide initiators were considered. The initiator BlocBuilder-MA™(BB) and the prepared NHS-BB (2-methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl)-aminoxy]-N-pro-pionyloxysuccinimide) showed advantages like lower activation energy and higher dissociation rate constants122 in comparison to first-generation TEMPO-based initiators.123 The new initiators allow controlled polymerization of different acrylates at lower temperatures.124
In 2020, Tajbakhsh and co-workers106 also reported the homopolymerization and statistical copolymerization IBOMA. They synthesized the polymer via nitroxide-mediated miniemulsion polymerization in dispersed aqueous media, without any controlling comonomer. Once again, Dispolreg 007 served as initiator. Operating at the optimum polymerization temperature of 90 °C, the homopolymerization of IBOMA (MA11) resulted in a relatively narrow molecular weight distribution (1.46 < Đ < 1.58) and a conversion rate of up to 83% within 2 hours. Additionally, they investigated the statistical copolymerization of IBOMA (MA11) and C13MA (MA12). A schematic representation of this reaction is shown in Fig. 32. The researchers were able to prepare copolymers in emulsion with tunable glass transition temperatures in aqueous solvents (−52 °C < Tg < 123 °C) and in organic solvent (−40 °C < Tg < 169 °C).
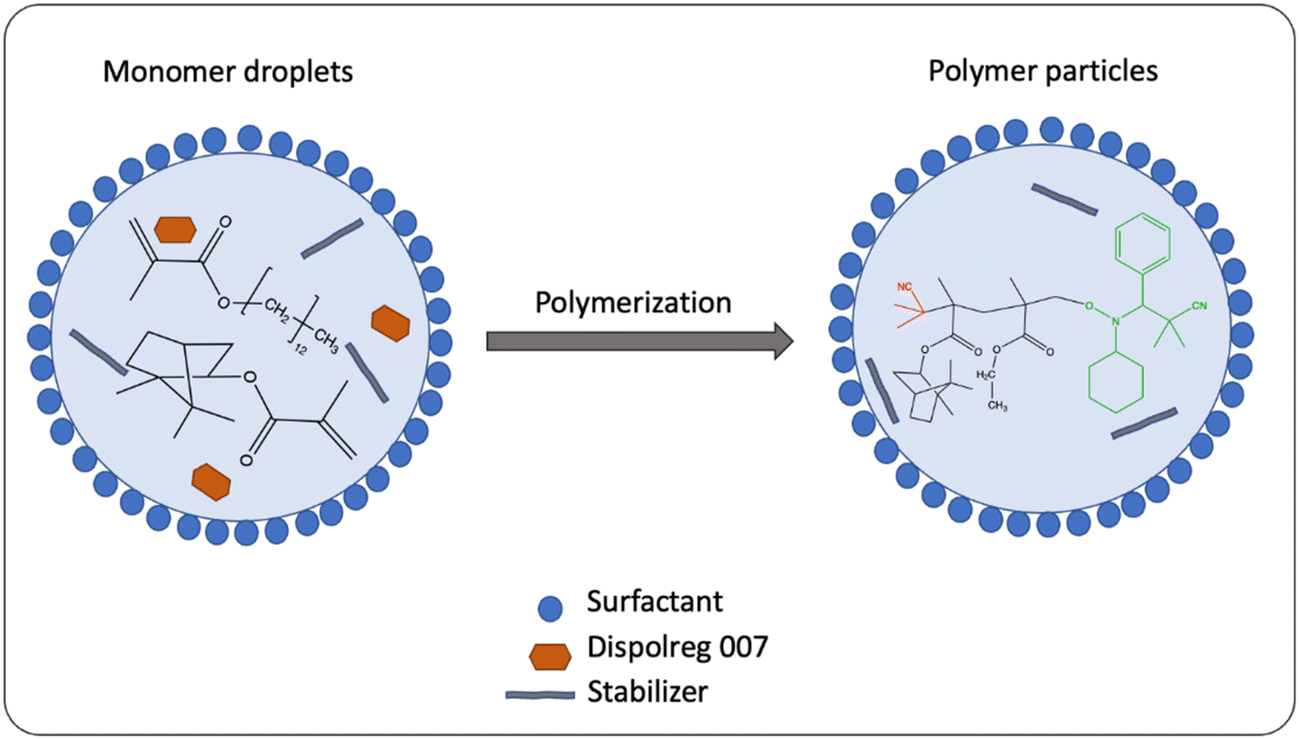 | ||
| Fig. 32 Emulsion polymerization of IBOMA and C13MA with Dispolreg 007. | ||
They achieved a conversion for resins created in emulsion of up to 92.7%, with a molecular weight of Mn = 68000 g mol−1, and a polydispersity ranging from 1.62 to 1.72. Consequently, they demonstrated the feasibility of conducting NMP of IBOMA (MA11) and C13MA (MA12) in dispersed aqueous media. The high glass transition temperature (Tg) of IBOMA (MA11) homopolymers makes it an intriguing comonomer for various fields of application.
4. Structure–property relationships and possible applications of terpene-based (meth-)acrylate, with a focus on isobornyl (meth)acrylate (IBO(M)A)
Isobornyl (meth)acrylate (IBO(M)A, A9, MA9) features a hydrophobic, bulky, bicyclic isobornyl group linked to the methacrylate group through an ester bond (Fig. 33) The relatively large volume of the side group can enhance the dimensional stability of copolymers after thermal processing.125,126 Moreover, it contributes to high glass transition temperature values ranging between 140 and 195 °C for poly(IBOMA).125–127 In comparison, Tg of poly(IBOA), is approximately 94 °C. This difference arises from the additional methyl group in IBOMA (MA9), leading to increased steric hindrance of polymer chains.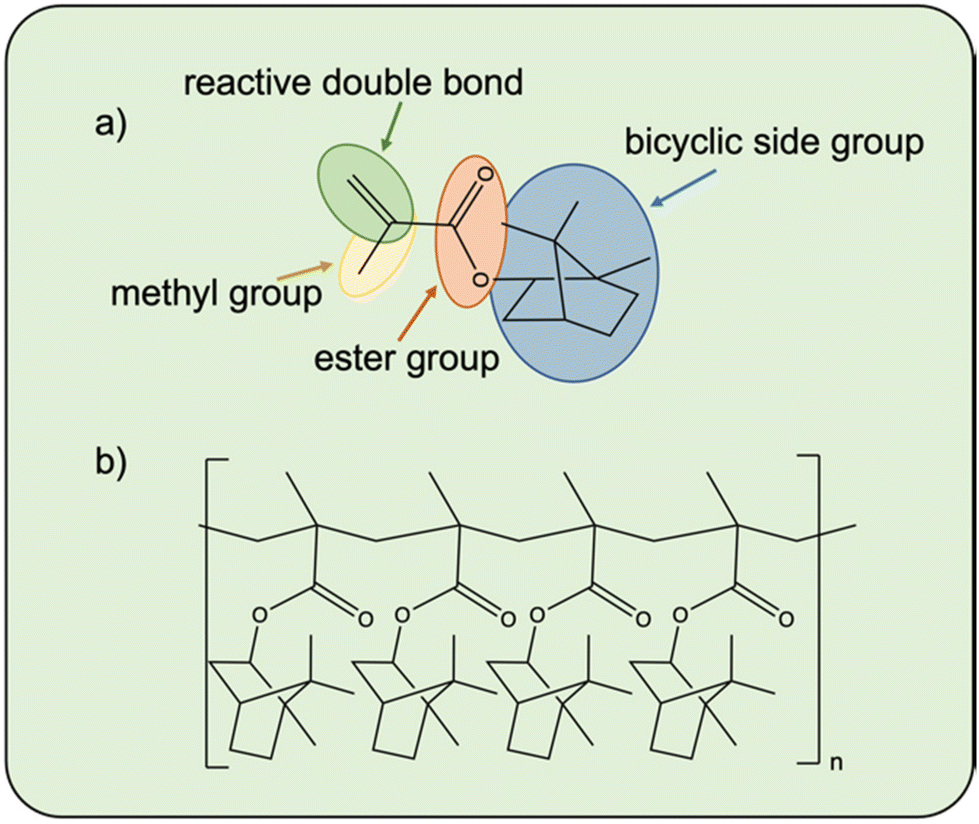 | ||
| Fig. 33 (a) Monomer structure of IBOMA with highlighted parts of the molecule, (b) polymerized IBOMA. | ||
The tendency for water adsorption is diminished in IBO(M)A (A9, MA9) compared to typical poly(meth)acrylates, owing to its relatively non-polar nature. This characteristic results in a rapid drying of polymer films. Additionally, the hardness and flexibility of the polymer can be adjusted to a certain extent.89 These structure–property relationships play a crucial role in determining the overall properties of the polymer and can be controlled by selecting the specific monoterpene building block.
4.1. Using structure–property relationships of terpene-based polyacrylates for development of coatings
The number of carbon atoms between the ring structure and the acrylate group affects the plasticization point of the polymer as well as the mechanical properties, solubility, and compatibility with other resins.128 Howdle et al.90 mentioned this effect for the terpene-based monomers α-pinene (MA1) and β-pinene methacrylate (MA2), with a higher Tg in the case of α-pineneMA (MA1). They explained this by the smaller distance between the bridging unit of the bicyclic ring and the acrylate functionality, which also leads to less distance between the side group and polymer backbone.By combining IBO(M)A (A9, MA9) with other monomers, properties like hardness, flexibility, and viscosity can be easily tailored.122,125,129 As already mentioned, the introduction of polar groups offers further possibilities to increase the glass transition temperature, since the association of the polymer chains with each other is increased. Resins with low polarity offer several further advantages, being especially suited application behavior, good wetting of different surfaces, and better spraying properties. All these properties allow to create homogeneous films.89
To ensure effective film formation, it is desirable for the resin's viscosity to be low. Typically, the viscosity of polymethacrylate solutions is higher compared to polyacrylates due to the steric hindrance introduced by methyl groups. However, poly(IBO(M)A) is an important exception from this rule: it shows low solution viscosity but a high glass transition temperature. Generally, monomers with cycloaliphatic side chains tend to show lower solution viscosity than those with short (aliphatic) side chains or aromatic structures. This characteristic makes such polymers promising candidates for use in high-solid paints89 or as biobased reactive diluent. Regarding the latter, there are some reports describing the use of IBO(M)A as reactive diluent The motivation behind these studies is evidently the increasing demand for lacquers with reduced volatile organic compounds (VOCs). Since IBO(M)A is a comparatively large molecule, it evaporates very slowly (see also section 4). In contrast to common reactive diluents, the polymerization of IBO(M)A (A9, MA9) is characterized by very low shrinkage.130
Another advantage of IBO(M)A (A9, MA9) is that the cycloaliphatic side group is transparent to UV light and possesses a high refractive index. Unlike aromatic building blocks, they are less prone to yellowing. Aromatic monomers, such as styrene, can undergo oxidation of the free double bond when exposed to heat and UV light, leading to the formation of free radicals. This process can result in the degradation of the polymer chain and the formation of yellow decomposition products.131 In general, the more tightly the polymer chains can be arranged together, the higher is the refractive index due to the higher density. Aromatic molecules containing conjugated π-bonds show high polarizability and therefore an increased refractive index (Table 13).132 Also monomers with a compact molecular structure like bicyclic rings, show a high refractive index. Compared to other polymers with high Tg like the aromatic polystyrene, poly(terpene acrylates) additionally exhibit reduced susceptibility to UV degradation.133 Due to the compact molecular structure and the increased molar refraction, they have a high reflection index and are consequently well suited for optical polymers.89
| Monomer | Styrene | MMA | Cyclohexylacrylate | IBOMA |
|---|---|---|---|---|
| Birefringence n20D | 1.5480 | 1.4160 | 1.5028134 | 1.4790 |
So far, we have discussed how IBO(M)A, in particular, can customize resin properties through its structure–property relationships. In coatings industry, another common strategy to create coatings with desired properties is by cross-linking of the polymer chains. Optimal crosslinking can lead to excellent flexibility, mechanical strength, and chemical and solvent resistance.89
Thermally curable polymer resins can be obtained by copolymerization of functional acrylic monomers that contain, for example, hydroxy groups, amide groups, or epoxy groups (Fig. 34). It is important to note that both film properties and chemical reactivity are influenced by the type and number of functional groups. Monomers with electrophilic or nucleophilic groups, such as hydroxy groups (–OH) or amino groups (–NH2), exhibit increased reactivity. Additionally, carboxyl groups can have a catalytic effect, leading to an accelerated reaction rate.89 Many acrylic polymers incorporate a hydroxy-functional (meth)acrylate as one monomer. These resins find significant application, particularly in automotive clear coats.89
 | ||
| Fig. 34 (Meth)acrylate monomers with different functional groups. | ||
Coatings cured thermally from these monomers exhibit improved resistance against cracking in weatherability tests. The susceptibility to cracking is closely related to the glass transition temperature, with the trend being most pronounced for monomers containing an aromatic sidechain. Clear coat films, crafted from acrylic monomers with a high Tg but devoid of aromatic compounds, demonstrate a reduced tendency to crack.89
As mentioned in section 2.2, Howdle et al. investigated the monoterpene carvone for synthesizing multifunctional (meth)acrylate monomers. They designed a carvone acrylate monomer with two additional epoxy groups and, in a subsequent step, obtained four hydroxy groups through hydrolysis (Fig. 15). The diepoxide monomer underwent polymerization via free radical polymerization, resulting in a polycarvone acrylate di-epoxide with a wide range of molecular weights achieved by adjusting the reaction conditions (Mn = 3000–32000 g mol−1). No uncontrolled crosslinking reactions occurred due to the epoxy groups during the polymerization of the acrylate group, as confirmed by solubility tests in different solvents. The DSC characterization of the final polymer showed a Tg at 64 °C for the entire range of molecular weights (except for Mn = 2800; Tg at 50 °C). This renders it an ideal biobased alternative for glycidyl methacrylate.91
Another method of curing polymers involves the use of radiation, typically UV light. To prepare reactive acrylic systems for this purpose, (meth)acrylates can be copolymerized with olefins, and the resulting product can be crosslinked using UV light. However, these monomers tend to harden at ambient temperatures, making them more challenging to handle. Terpene-based acrylates like carvone (meth)acrylate or limonene (meth)acrylate carry a reactive double bond, enabling them to be crosslinked via thiol–ene click chemistry during UV curing.50
In summary, polymers based on IBO(M)A offer excellent mechanical properties such as hardness, resistance, and sufficient flexibility. This combination makes such polymers promising for a wide range of applications (Fig. 35).
 | ||
| Fig. 35 Replacing fossil-based acrylates with bio-based alternatives. Terpene acrylates have the potential to achieve the same or even better material properties like hardness due to high glass transition; low shrinkage; reduced VOC; high refractive index; high UV transmittance; low solution viscosity. | ||
To fully substitute fossil-based acrylates with monoterpene-based acrylates, the development of acrylates with different functionalities is crucial. This enables petroleum-based acrylates to be replaced by terpene-based acrylates without restricting the range of applications.
4.2. Isobornyl (meth)acrylate (IBO(M)A) as alternative to styrene
To maximize the benefits of polyacrylates, several monomers are combined to create resins with advanced, tailored properties. These even have the potential to compete with commercial polymer resins, as the example of IBOMA shows:The most important monomer to increase the refractive index of a polymer is styrene. Renowned for its rigid structure, wide availability, dissolution capacity, low viscosity, cost-effectiveness, and high reactivity, styrene is crucial in achieving excellent mechanical properties.135 Additionally, it is known to facilitate rapid physical drying of the resulting films.
Finally, the styrene-containing polymers usually have a high Tg,89 however also depends on the amount and type of the further comonomers. Although styrene exhibits highly beneficial properties, it is also known as human carcinogen as classified by the USA's Department of Health and Human Services through the National Toxicology Program.135
Moreover, it is listed as a hazardous air pollutant by the environmental protection agency. Due to its high volatility, using styrene in polymer production requires careful handling and suited safety precautions.136 Another disadvantage of styrene-containing resins is their photocatalytic degradation, which results in a yellowing of the polymer.137
To replace styrene, current research considers especially bicyclic terpene-based acrylates since they also exhibit suited hardness, and thermal stability. Due to their lower volatility, they have fewer volatile organic compounds (VOCs) compared to styrene.
The research group of Cousinet et al.138 investigated IBO(M)A (A9, MA9) as an appropriate substitute for styrene in unsaturated polyester resins. They mentioned the high volatility of styrene and MMA, which evaporate completely in less than 1 h at room temperature. In contrast, IBOMA (MA9) showed a weight loss of only 12 wt% after 15 h, which makes it ideal for the use as harmless reactive diluent.
4.3. Antibacterial adhesion
The formation of biofilms by bacteria, algae, and other marine organisms poses a significant challenge for the marine transport industry. The attachment of these organisms leads to increased fouling, resulting in higher roughness and reduced maneuverability of ships. The most straightforward approach to prevent marine fouling on surfaces is using anti-fouling coatings, which traditionally consisted of tributyltin or copper-based materials. However, the release of these substances not only deters biofilm-forming organisms but also causes harm to other marine organisms and the ecological environment. To safeguard the marine habitat, these substances were prohibited by the International Marine Organization in 2008.139 To address the need for an anti-fouling coating with high mechanical and chemical stability and low toxicity, the borneol-based acrylate IBOMA comes into play. The chiral effects and hydrophobic interactions of the chiral bicyclic monoterpene borneol are well-known for their antibacterial properties.140 Due to the universal antibacterial effect of the borneol building block and its high rigidity, Wang and coworkers tested this compound for use in anti-fouling coatings. They explored the combination of IBOMA with fluorine monomers and synthesized a copolymer, poly(methyl methacrylate-co-ethyl acrylate-co-hexafluorobutyl methacrylate-co-isobornyl methacrylate) (PBAF), using the free radical polymerization technique. The coatings exhibited a remarkable resistance of 98.2% and 92.3% against the bacteria Escherichia coli and Staphylococcus aureus, respectively. This superior antibacterial activity of isoborneol acrylate was evident when compared to the investigated poly(methyl methacrylate-co-ethyl acrylate) copolymer.141 In another approach, borneol acrylate was employed to create copolymers with PMMA to enhance the antibacterial performance of implants.142The research group of Tan developed a diblock copolymer, poly[(N-3,4-dihydroxyphenthyl acrylamide)-b-(borneolacrylate)] synthesized via reversible addition-fragmentation chain transfer polymerization. The coating incorporates dopamine as a mussel-inspired adhesive and borneol acrylate as an antibacterial component. Characterized by atomic force microscopy (AFM), the coating exhibited a homogeneous and smooth morphology. Furthermore, it demonstrated robust adhesion to various substrates such as silica, alumina, silicon, and stainless steel, coupled with excellent broad-spectrum antibacterial activity.143
In 2020, Zhang et al. synthesized a copolymer of IBOMA, triisopropylsilylacrylate and n-butyl methacrylate. They selected IBOMA for its universal antibacterial capabilities and demonstrated the potential of the copolymer as an antifouling coating.144 However, care must be taken to ensure that no residual monomers diffuse from the coating film which could harm aquatic organisms. Monoterpenes, however, offer great potential for use in antifouling coatings and should therefore be investigated further.
4.4. Medical applications
As mentioned earlier, monoterpenes are highly suitable for use in optical polymers. They exhibit a compact molecular structure, increased molar refraction, and a high refractive index. For optical applications in medicine, such as contact lenses, the ideal polymers should not only have a high refractive index but also offer exceptional flexibility. Due to the complex side-groups of common high refractive index polymers, they all possess a high Tg.133 Bicyclic terpene acrylates like IBO(M)A (A9, MA9) provide a solution, allowing the production of transparent and highly glossy polymer films with both high flexibility and a high Tg.137 Polymers containing IBO(M)A also exhibit antibacterial effects, which can be advantageous for applications involving contact lenses or other medical devices.143In another study, poly(IBOMA-co-BA) copolymer was synthesized by free radical polymerization. The incorporation of the difunctional crosslinker 4,4′-isopropylidenediphenol dimethacrylate was introduced to produce crosslinked polymer films. The resulting polymer films exhibited good transparency, excellent chemical resistance, refractive indices ranging from 1.47 to 1.54, and very low birefringence. These properties make it very attractive for fabrication of optical devices.145
For the application as a dental restorative material, a nanogel of IBOMA and the di-functional crosslinker ethoxylated bisphenol-A dimethacrylate was synthesized. These components were chosen to form a resin matrix with minimized polymerization shrinkage and stress, along with enhanced resistance to degradation, making it suitable for use as an intraocular lens in postoperative treatments.146
In 2023, IBOMA was used to increase hydrophobicity of antibiotic-loaded intraocular lenses used for postoperative treatment. The bulky structure of IBOMA resulted in hindered drug release, attributed to the effect of steric hindrance obstructing the diffusion pathway.146
4.5. Pressure sensitive adhesives
IBOMA (MA9) was evaluated to replace traditionally used petroleum based MMA as a hard monomer for use in an acrylic latex pressure sensitive adhesive (PSA). Literature reports indicate that a PSA containing IBOMA exhibited a high glass transition temperature, superior water resistance compared to its MMA counterpart, and also demonstrated higher gel content.147For the use as UV-curable transparent acrylic pressure sensitive adhesives, the monoterpene acrylates menthyl acrylate (A5) and tetrahydrogeraniol acrylate (A16) were investigated by Hwang and coworkers. They assessed the adhesion performance and environmental reliability of the synthesized acrylic PSAs by varying the molar ratio of A5 and A16. In combination with 2-hydroxyethyl acrylate, they obtained PSAs with high transparency (>92%) and stable weatherability.148
In a recent publication, Droesbeke et al. investigated tetrahydrogeranyl acrylate (A16), menthyl methacrylate (MA5), citronellyl methacrylate (MA17) and isobornyl methacrylate (MA9) as potential monomers for PSA materials. They aimed to create renewable alternatives for both soft and hard monomers in acrylic PSAs, using IBOMA as the hard component. The peak probe adhesive stresses ranged from 200 to 600 kPa for different compositions, confirming promising values for PSAs.149
Zang et al. reported the positive effect of IBOMA on the PSA performance, as it increases the spacing between the chains. This results in improved wetting and, consequently, enhanced adhesion properties.30
5. Discussion and conclusion
Terpene-based monomers, particularly Isobornyl Acrylate (IBO(M)A), have gained attention for their potential as sustainable alternatives in synthetic polymer production due to their widespread availability. Various publications have explored the conversion of monoterpenes (MT) into (meth-)acrylates and poly(meth-)acrylates. IBO(M)A has demonstrated successful integration into industrial (meth-)acrylate chemistry, offering a renewable and eco-friendly option.150 The introduction of a hydroxy group into the monoterpenes (MT) carbon framework is a crucial step, often accomplished through chemical transformations such as epoxidation with mCPBA, Pd-catalysis, or hydroboration. While some protocols involve toxic and unsustainable chemicals for (trans-)esterifications, there is potential in environmentally friendly methods, including enzymatic catalysis. However, the literature lacks examples of enzymatically catalyzed (trans-)esterification on a kg scale or higher, likely due to the associated high costs. Furthermore, many approaches employ purification methods, particularly column chromatography, that are not industrially relevant. Developing and scaling green synthesis methods is essential to harness the environmental benefits of renewable MTs. Additionally, (meth-)acrylic acid, a key component, is not currently sourced from renewables, as the production costs do not compete with the established oxidation of propene. In 2021, Fouilloux and Thomas reviewed the production and polymerization of biobased acrylates, presenting various synthetic pathways for acrylic acid and methacrylic acid. Methods mentioned for synthesizing MAA include decarboxylation of citric acid, itaconic acid, or dehydration of 2-hydroxyisobutyric acid (2-HIBA). However, these methods are still in the research state, with biobased MAA less advanced than AA.151The potential of monoterpenes in acrylate chemistry is not only the use of renewable resources, but also the creation of a new monomer feedstock that can integrate novel properties to polymers and formulations due to their great structural diversity and chirality. However, the exploration of these properties is in its early stages, with most reports focusing on the general polymerization behavior rather than in-depth investigations into polymer properties or performance in applications – although the few exceptions underline that the work is worth the effort (section 4).
Finally, although MTs are relatively abundant for a non-polymeric, high purity bio-resource and are accessible from industrial residues,7 their annual production volume is still low from the perspective of the plastic industry. Therefore, the production of terpenes by biotechnological methods via direct conversion of CO2 with different cyanobacteria or similar abundant resources is required to bring terpene-based (meth)acrylates to the next level.152–154
Acrylates are important polymers used in a wide range of applications, including the automotive industry, the production of coatings and adhesives, the electronics industry, packaging industry as well as for medical applications like medical devices or implants.
One already used MT based methacrylate is isobornyl methacrylate (IBOMA) (MA9), which is prepared from isoborneol (9).155 The amount of biobased carbon in terpene based (meth)acrylate monomers is already very high as 71% for IBOMA, and can even be raised to 100% if bio-based methacrylic acid is be accessible at industrial scale.138
By supporting research and development, as well as providing incentives for companies and consumers through improved material performance, terpene acrylates could help reduce dependence on non-renewable resources and promote a sustainable economy.
Conflicts of interest
There are no conflicts of interest to declare.References
- W. Schwab, C. Fuchs and F. C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- A. C. Rivas da Silva, P. M. Lopes, M. M. Barros de Azevedo, D. C. Machado Costa, C. S. Alviano and D. S. Alviano, Molecules, 2012, 17, 6305–6316 CrossRef PubMed.
- K. Loos, Biocatalysis in Polymer Chemistry, Wiley-VCH Verlag, Groningen, 2010 Search PubMed.
- M. N. Belgacem and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, Elsevier, Oxford, 1st edn, 2008 Search PubMed.
- R. Chinthapalli, P. Skoczinski, M. Carus, W. Baltus, D. de Guzman, H. Käb, A. Raschka and J. Ravenstijn, Ind. Biotechnol., 2019, 15, 237–241 CrossRef.
- K. Schmidt, P. Schmidt, M. Lang and A. Rusam, Einsatz und Potential biobasiereter Additive in Kunststoffen, 2020 Search PubMed.
- I. John, K. Muthukumar and A. Arunagiri, Int. J. Green Energy, 2017, 14, 599–612 CrossRef CAS.
- Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed.
- L. O. Wiemann, C. Faltl and V. Sieber, Z. Naturforsch., B: Chem. Sci., 2012, 67, 1056–1060 CrossRef CAS.
- M. Hofer, H. Strittmatter and V. Sieber, ChemCatChem, 2013, 5, 3351–3357 CrossRef CAS.
- R. T. Mathers and S. P. Lewis, in Green Polymerization Methods, ed. R. T. Mathers and M. A. R. Meier, 2011, pp. 89–128 Search PubMed.
- K. Schröder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- K. J. Rodriguez, M. M. Pellizzoni, R. J. Chadwick, C. Guo and N. Bruns, Enzyme-initiated free radical polymerizations of vinyl monomers using horseradish peroxidase, Elsevier Inc., 2019 Search PubMed.
- M. Kohri, Polym. J., 2014, 46, 373–380 CrossRef CAS.
- J. Smid, M. Van Beylen and T. E. Hogen-Esch, Prog. Polym. Sci., 2006, 31, 1041–1067 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- K. Matyjaszewski, Prog. Chem., 2001, 22, 2079–2088 Search PubMed.
- M. S. Masami Kamigaito and T. Ando, Chem. Rev., 2001, 101, 3689–3745 CrossRef.
- W. J. Roberts and A. R. Day, Am. Chem. Soc., 1949, 72, 1226–1230 CrossRef.
- M. K. K. Satoh and H. Sugiyama, Green Chem., 2006, 8, 878–882 RSC.
- K. Satho, A. Nakahara, K. Mukunoki, H. Sugiyama, H. Saito and M. Kamigaito, Polym. Chem., 2014, 5, 3222 RSC.
- A. M. Ramos, L. S. Lobo and J. M. Bordado, Macromol. Symp., 1998, 127, 43–50 CrossRef CAS.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- S. Ren, E. Trevino and M. A. Dubé, Macromol. React. Eng., 2015, 9, 339–349 CrossRef CAS.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed.
- M. Firdaus, L. Montero De Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- A. Mija, E. Louisy, S. Lachegur, V. Khodyrieva, P. Martinaux, S. Olivero and V. Michelet, Green Chem., 2021, 23, 9855 RSC.
- Z. Demchuk, A. Kohut, S. Voronov and A. Voronov, ACS Sustainable Chem. Eng., 2018, 6, 2780–2786 CrossRef CAS.
- S. Molina-Gutiérrez, V. Ladmiral, R. Bongiovanni, S. Caillol and P. Lacroix-Desmazes, Ind. Eng. Chem. Res., 2019, 58, 21155–21164 CrossRef.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2015, 3, 662–667 CrossRef CAS.
- A. Altomare, C. Carlini, F. Ciardelli and R. Solaro, Polym. J., 1988, 20, 801–809 CrossRef CAS.
- A. Altomare, C. Carlini, M. Panattoni and R. Solaro, Macromolecules, 1984, 17, 2207–2212 CrossRef CAS.
- A. Altomare, C. Carlini, F. Ciardelli, R. Solaro and N. Rosato, J. Polym. Sci., Polym. Chem. Ed., 1984, 22, 1267–1280 CrossRef CAS.
- A. Altomare, C. Carlini and R. Solaro, Polymer, 1982, 23, 1355–1360 CrossRef CAS.
- E. H. Min, K. H. Wong, E. Setijadi, F. Ladouceur, M. Straton and A. Argyros, Polym. Chem., 2011, 2, 2045–2051 RSC.
- S. S. Baek and S. H. Hwang, Polym. Bull., 2016, 73, 687–701 CrossRef CAS.
- S. S. Baek and S. H. Hwang, Eur. Polym. J., 2017, 92, 97–104 CrossRef CAS.
- K. Gries, K. Bubel, M. Wohlfahrt, S. Agarwal, U. Koert and A. Greiner, Macromol. Chem. Phys., 2011, 212, 2551–2557 CrossRef CAS.
- B. Lepoittevin, X. Wang, J. P. Baltaze, H. Liu, J. M. Herry, M. N. Bellon-Fontaine and P. Roger, Eur. Polym. J., 2011, 47, 1842–1851 CrossRef CAS.
- A. Akhmedov, R. Gamirov, Y. Panina, E. Sokolova, Y. Leonteva, E. Tarasova, R. Potekhina, I. Fitsev, D. Shurpik and I. Stoikov, Org. Biomol. Chem., 2023, 21, 4863–4873 RSC.
- S. Noppalit, A. Simula, N. Ballard, X. Callies, J. M. Asua and L. Billon, Biomacromolecules, 2019, 20, 2241–2251 CrossRef CAS.
- V. Cuzzucoli Crucitti, A. Ilchev, J. C. Moore, H. R. Fowler, J. F. Dubern, O. Sanni, X. Xue, B. K. Husband, A. A. Dundas, S. Smith, J. L. Wildman, V. Taresco, P. Williams, M. R. Alexander, S. M. Howdle, R. D. Wildman, R. A. Stockman and D. J. Irvine, Biomacromolecules, 2023, 24, 576–591 CrossRef CAS PubMed.
- J. Salaklang, V. Maes, M. Conradi, R. Dams and T. Junkers, React. Chem. Eng., 2018, 3, 41–47 RSC.
- M. A. Droesbeke and F. E. Du Prez, ACS Sustainable Chem. Eng., 2019, 7, 11633–11639 CrossRef CAS.
- R. J. Marvel, C. S. Schwen, R. Hobson and R. W. Coleman, J. Polym. Sci., 1958, 33, 27–37 CrossRef.
- V. Athawale, N. Manjrekar and M. Athawale, J. Mol. Catal. B: Enzym., 2001, 16, 169–173 CrossRef CAS.
- V. Athawale, N. Manjrekar and M. Athawale, Biotechnol. Prog., 2003, 19, 298–302 CrossRef CAS PubMed.
- T. Castagnet, G. Aguirre, J. M. Asua and L. Billon, ACS Sustainable Chem. Eng., 2020, 8, 7503–7512 CrossRef CAS.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC.
- M. Cutajar, F. Andriulo, M. R. Thomsett, J. C. Moore, B. Couturaud, S. M. Howdle, R. A. Stockman and S. E. Harding, Sci. Rep., 2021, 11, 1–12 CrossRef PubMed.
- M. Cutajar, F. Machado, V. Cuzzucoli Crucitti, S. Braovac, R. A. Stockman, S. M. Howdle and S. E. Harding, Sci. Rep., 2022, 12, 1–14 CrossRef PubMed.
- M. Cutajar, S. Braovac, R. A. Stockman, S. M. Howdle and S. E. Harding, Sci. Rep., 2023, 13, 1–13 CrossRef PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- M. S. Lima, C. S. M. F. Costa, J. F. J. Coelho, A. C. Fonseca and A. C. Serra, Green Chem., 2018, 20, 4880–4890 RSC.
- A. Stamm, M. Tengdelius, B. Schmidt, J. Engström, P. O. Syrén, L. Fogelström and E. Malmström, Green Chem., 2019, 21, 2720–2731 RSC.
- M. von Czapiewski, K. Gugau, L. Todorovic and M. A. R. Meier, Eur. Polym. J., 2016, 83, 359–366 CrossRef CAS.
- V. Schimpf, A. Asmacher, A. Fuchs, K. Stoll, B. Bruchmann and R. Mülhaupt, Macromol. Mater. Eng., 2020, 305, 1–9 CrossRef.
- C. Montanari, Y. Ogawa, P. Olsén and L. A. Berglund, Adv. Sci., 2021, 8, 2100559 CrossRef CAS PubMed.
- U. Montanari, V. Taresco, A. Liguori, C. Gualandi and S. M. Howdle, Polymer International, 2021, 70, 499–505 CrossRef CAS.
- H. R. Fowler, D. M. O'Brien, D. J. Keddie, C. Alexander, D. J. Irvine, R. A. Stockman, S. M. Howdle and V. Taresco, Macromol. Chem. Phys., 2023, 46, 1–6 Search PubMed.
- C. Veith, F. Diot-Néant, S. A. Miller and F. Allais, Polym. Chem., 2020, 11, 7452–7470 RSC.
- H. Fouilloux and C. M. Thomas, Macromol. Rapid Commun., 2021, 42 Search PubMed.
- K. Ishihara and M. Hatano, Catalyst for synthesizing carboxylic acid ester, and method for producing carboxylic acid ester, JP2021137728A 63, 2021.
- M. Hatano, Y. Tabata, Y. Yoshida, K. Toh, K. Yamashita, Y. Ogura and K. Ishihara, Green Chem., 2018, 20, 1193–1198 RSC.
- D. Xu, Y. Shaoshan, F. Tao, Z. Xiaolan, C. Peng, D. Gu, M. Z. and X. Congying, Preparation method of isobornyl acrylate, CN Pat., CN112094188A, 2020.
- J. Knebel and D. Saal, EP Pat 1067110B1, 1999.
- J. Knebel and W. Spalt, Progress for the preparation of alkoxypolyglycol (meth) acrylates, DE19602035A1, 1997.
- K. Joachim and O. Thomas, Carbohydrate (meth)acrylate preparation by ester exchange without solvent, DE19545870, 1997.
- J. D. Knebel, M. D. Pfirmann and T. Ohl, Process for the transesterification of (meth) acrylic acid esters, DE4401132A1, 1995.
- J. D. Knebel, Nitrogen heterocycle-substd. (meth)acrylate esters(s), DE4301673A1, 1994.
- G. Wolfgang, H. Heinz-Jürgen, S. Günter and D. S. Hermann-Josef, Method for continuosly establishing methacrylic acid, DE3146191A1, 1983.
- M. T. Miller, N. Pondicherry, E. L. Trapasso and A. V. De Sande, DE69914642T2, 1999.
- N. M. Bortnick, Process of making esters of acrylic and methacrylic acids, US Pat., 3087962A, 1980 Search PubMed.
- M. D. Pfirmann and J. D. Knebel, Verfahren zur Herstellung von Isobornyl(meth)acrylat, DE4316004A1, 1994.
- J. D. Knebel and M. D. Bader, Verfahren zur Herstellung von Isobornyl(meth)acrylate, DE4316004A1, 1994.
- J.-M. Paul and G. Desire, Process for the preparation of isobornyle (meth) acrylate, EP0718271A1, 1994.
- Y. Hiraga and T. Morikawa, Preparation of unsaturated monocarboxylic acid isobornyl ester, JPS5849337A, 1981.
- M. Matsuura, R. Nadano and T. Furumata, Isobornyl (meth)acrylate-containing composition and method for production thereof, WO2019193925A1, 2019.
- X. Xu, L. Chen, J. Guo, X. Cao and S. Wang, R. Soc. Chem., 2017, 7, 30439–30445 CAS.
- A. Heidekum, M. A. Harmer and W. F. Hoelderich, J. Catal., 1999, 181, 217–222 CrossRef CAS.
- A. Riondel, Process for the preparation of isobornyl (meth)acrylate, NBER Working Papers, US5719314A, 1996 Search PubMed.
- J. Knebel and M. Bader, Production of isobornyl (meth)acrylate, used in lacquer binder production, uses aqueous sulfuric acid and inhibitor in reaction of camphene with (meth)acrylic acid, DE 19920796A1, 2000.
- W. Jianxin, L. Haojin, X. Xiaowei, W. Baisong and W. Shifa, Method for catalytically synthesizing isobornyl methacrylate by activated carbon supported stannic chloride, CN101863763A, 2010.
- D. K. Joachim, T. Schütz, G. Gräff and T. Ohl, Process for the preparation of bicyclic or tricyclic (meth) acrylates, DE102011089504A1, 2013.
- Y. Teng, L. Yingling, X. Yaliang, L. Xiaoping and Y. Z. Hong, Preparation method of isobornyl (meth) acrylate of biological origin, CN112142593, 2020.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- T. L. Tsai, C. C. Lin, G. L. Guo and T. C. Chu, Ind. Eng. Chem. Res., 2008, 47, 2554–2560 CrossRef CAS.
- B. B. Mathur, News Bull. Indian Dent. Assoc., 1973, 4, 16–19 CAS.
- R. L. Atkinson, M. Elsmore, S. Smith, M. Reynolds-Green, P. D. Topham, D. T. W. Toolan, M. J. Derry, O. Monaghan, V. Taresco, D. J. Irvine, R. A. Stockman, D. S. A. De Focatiis and S. M. Howdle, Eur. Polym. J., 2022, 179, 111567 CrossRef CAS.
- U. Montanari, V. Taresco, A. Liguori, C. Gualandi and S. M. Howdle, Polym. Int., 2021, 70, 499–505 CrossRef CAS.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Green Chem., 2020, 22, 4561–4569 RSC.
- P. Sarkar and A. K. Bhowmick, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2639–2649 CrossRef CAS.
- M. Worzakowska, J. Polym. Environ., 2018, 26, 1613–1624 CrossRef CAS.
- W. H. Binder, Macromol. Rapid Commun., 2019, 40, 1–7 CrossRef PubMed.
- S. Dworakowska, F. Lorandi, A. Gorczyński and K. Matyjaszewski, Adv. Sci., 2022, 9, 1–39 Search PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365–398 CrossRef CAS.
- R. L. Atkinson, O. R. Monaghan, M. T. Elsmore, P. D. Topham, D. T. W. Toolan, M. J. Derry, V. Taresco, R. A. Stockman, D. S. A. De Focatiis, D. J. Irvine and S. M. Howdle, Polym. Chem., 2021, 12, 3177–3189 RSC.
- S. Noppalit, A. Simula, L. Billon and J. M. Asua, ACS Sustainable Chem. Eng., 2019, 7, 17990–17998 CrossRef CAS.
- U. Montanari, D. Cocchi, T. M. Brugo, A. Pollicino, V. Taresco, M. Romero Fernandez, J. C. Moore, D. Sagnelli, F. Paradisi, A. Zucchelli, S. M. Howdle and C. Gualandi, Polymer, 2021, 13, 1804–1822 CAS.
- M. Alrefai, V. Meola and M. Maric, J. Polym. Sci., 2022, 1–15 Search PubMed.
- S. Tajbakhsh, F. Hajiali and M. Maric, Ind. Eng. Chem. Res., 2020, 59, 8921–8936 CrossRef CAS.
- S. Liu and M. K. Mishra, Macromolecules, 2007, 40, 867–871 CrossRef CAS.
- N. Ballard, M. Aguirre, A. Simula, A. Agirre, J. R. Leiza, J. M. Asua and S. Van Es, ACS Macro Lett., 2016, 5, 1019–1022 CrossRef CAS PubMed.
- A. Simula, F. Ruipérez, N. Ballard, J. R. Leiza, S. Van Es and J. M. Asua, Polym. Chem., 2019, 10, 106–113 RSC.
- S. Noppalit, A. Simula, L. Billon and J. M. Asua, Polym. Chem., 2020, 11, 1151–1160 RSC.
- J. W. Ma, J. A. Smith, K. B. McAuley, M. F. Cunningham, B. Keoshkerian and M. K. Georges, Chem. Eng. Sci., 2003, 58, 1163–1176 CrossRef CAS.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2005, 38, 9963–9973 CrossRef CAS.
- E. Groison, S. Brusseau, F. D'Agosto, S. Magnet, R. Inoubli, L. Couvreur and B. Charleux, ACS Macro Lett., 2012, 1, 47–51 CrossRef CAS PubMed.
- N. Ballard, M. Aguirre, A. Simula, J. R. Leiza, S. Van Es and J. M. Asua, Polym. Chem., 2017, 8, 1628–1635 RSC.
- Y. Guo and P. B. Zetterlund, Macromol. Rapid Commun., 2011, 32, 1669–1675 CrossRef CAS PubMed.
- E. Mehravar, A. Agirre, N. Ballard, S. Van Es, A. Arbe, J. R. Leiza and J. M. Asua, Macromolecules, 2018, 51, 9740–9748 CrossRef CAS.
- P. B. Zetterlund, J. Wakamatsu and M. Okubo, Macromolecules, 2009, 42, 6944–6952 CrossRef CAS.
- W. S. J. Li and M. F. Cunningham, Polym. Chem., 2014, 5, 3804–3816 RSC.
- N. Ballard, M. Aguirre, A. Simula, J. R. Leiza, S. van Es and J. M. Asua, Chem. Eng. J., 2017, 316, 655–662 CrossRef CAS.
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC.
- M. Destarac, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- F. Hajiali, A. Métafiot, L. Benitez-Ek, L. Alloune and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2422–2436 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485–5492 CrossRef CAS.
- B. Lessard and M. Marić, Macromolecules, 2008, 41, 7870–7880 CrossRef CAS.
- B. Zhang, Y. Ma, D. Chen, J. Xu and W. Yang, J. Appl. Polym. Sci., 2013, 129, 113–120 CrossRef CAS.
- S. Il Park, S. I. Lee, S. J. Hong and K. Y. Cho, Macromol. Res., 2007, 15, 418–423 CrossRef.
- L. J. F. Nikos Hadjichristidis, J. Mays and W. Ferry, Polym. Phys., 1984, 22, 1745–1751 CrossRef.
- A. Goldschmidt and H.-J. Streitberger, BASF Handbuch Lackiertechnik, 2002 Search PubMed.
- J. M. Yu, P. Dubois and R. Jérôme, Macromolecules, 1996, 29, 7316–7322 CrossRef CAS.
- Rohm and Haas Co., DE 2622022A1, 1976.
- J. Qu, J. Cheng, Z. Wang, X. Han and M. Zhao, Opt. Mater., 2014, 36, 804–808 CrossRef CAS.
- Y. C. Xu Fei and Y. Shi, Appl. Phys. A: Mater. Sci. Process., 2010, 100, 409–414 CrossRef.
- Z. Ahmad, in Encyclopedia of High Performance Silicones, 2014, pp. 65–74 Search PubMed.
- C. Dams, PhD thesis, University of Marburg, 2019.
- M. Malik, V. Choudhary and I. K. Varma, J. Macromol. Sci., Polym. Rev., 2000, 40, 139–165 CrossRef.
- D. Mandrioli and E. K. Silbergeld, Environ. Health Perspect., 2016, 124, 6–11 CrossRef PubMed.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 1–32 CrossRef CAS PubMed.
- S. Cousinet, A. Ghadban, E. Fleury, F. Lortie, J. P. Pascault and D. Portinha, Eur. Polym. J., 2015, 67, 539–550 CrossRef CAS.
- J. Bannister, M. Sievers, F. Bush and N. Bloecher, Biofouling, 2019, 35, 631–648 CrossRef CAS PubMed.
- L. Luo, G. Li, D. Luan, Q. Yuan, Y. Wei and X. Wang, Appl. Mater. Interfaces, 2014, 6, 9371–19377 Search PubMed.
- F. Song, L. Zhang, R. Chen, Q. Liu, J. Liu, J. Yu, P. Liu, J. Duan and J. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 33417–33426 CrossRef CAS PubMed.
- X. Sun, Z. Qian, L. Luo, Q. Yuan, X. Guo, L. Tao, Y. Wei and X. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 28522–28528 CrossRef CAS PubMed.
- Y. T. Xin Wang, S. Jing, Y. Liu and S. Liu, Polymer, 2017, 116, 314–323 CrossRef.
- J. Hu, B. Sun, H. Zhang, A. Lu, H. Zhang and H. Zhang, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- M. Qu, J. Cheng, J. Wang, Z. Han and X. Zhao, Opt. Mater., 2014, 30, 1252–1262 Search PubMed.
- M. Li, J. W. Xu, J. Li, W. Wang, C. Luo, H. Han, Z. K. Xu and K. Yao, Bioact. Mater., 2023, 20, 271–285 CAS.
- C. Fang, X. Zhu, Y. Cao, X. Xu, S. Wang and X. Dong, Int. J. Adhes. Adhes., 2020, 100, 1–9 CrossRef.
- S.-H. H. Seung-Suk Baek, Eur. Polym. J., 2017, 92, 97–104 CrossRef.
- M. A. Droesbeke, A. Simula, J. M. Asua and F. E. Du Prez, Green Chem., 2020, 22, 4561–4569 RSC.
- M. Peplow, Nature, 2016, 536, 266–268 CrossRef CAS PubMed.
- H. Fouilloux and C. M. Thomas, Macromol. Rapid Commun., 2021, 42, 2000530 CrossRef CAS PubMed.
- M. Li, B. Long, S. Y. Dai, J. W. Golden, X. Wang and J. S. Yuan, BioDesign Res., 2022, 2022, 9897425 CrossRef PubMed.
- T. Krieg, A. Sydow, S. Faust, I. Huth and D. Holtmann, Angew. Chem., Int. Ed., 2018, 57, 1879–1882 CrossRef CAS PubMed.
- H. J. Lee, J. Lee, S. M. Lee, Y. Um, Y. Kim, S. J. Sim, J. Il Choi and H. M. Woo, J. Agric. Food Chem., 2017, 65, 10424–10428 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, in Chemicals from Biomass, Pacific Northwest National Laboratory, Springfild, 2004, pp. 1–76 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |