
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoscale control over water-film thickness using temperature modulation: tuning mineral carbonation reactivity†
Christopher J.
Thompson
 *a,
Sebastian T.
Mergelsberg
b,
Bavan P.
Rajan
b,
Paul F.
Martin
a,
Sebastien N.
Kerisit
b and
John S.
Loring
*b
*a,
Sebastian T.
Mergelsberg
b,
Bavan P.
Rajan
b,
Paul F.
Martin
a,
Sebastien N.
Kerisit
b and
John S.
Loring
*b
aEnergy and Environment Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: chris.thompson@pnnl.gov
bPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: john.loring@pnnl.gov
First published on 6th March 2024
Abstract
Induced temperature gradients within a high-pressure reactor were used to assess how the thickness of nanoscale water films affect the carbonation of forsterite, Mg2SiO4, in water-saturated supercritical carbon dioxide (scCO2). Water monolayer (ML) coverages on the mineral surface were estimated by in situ infrared spectroscopy and were shown to increase nearly exponentially from approximately 5 to 170 ML when the forsterite region was cooled from +0.5 to −0.3 °C relative to a nearby pool of excess water. Carbonation rates and extents were strongly dependent on the water film thickness, with the highest rates and complete mineral conversion occurring within 40 h under bulk-like water coverages. Reaction passivation, possibly due to a reduction in reactive surface area, was observed for thinner water films ranging from 5–53 ML. Temperature modulation can be an effective technique for investigating basalt reactivity over a range of water coverages in humidified scCO2.
Environmental significanceInjecting CO2 into basaltic geologic reservoirs where carbon mineralization can occur is a promising approach to confront global warming. Deep within the reservoir, water can dissolve in the CO2 to form a variably hydrated supercritical fluid (scCO2). Silicate minerals exposed to this fluid become covered in Å-to-nm thick water films, where the minerals react with CO2 to form metal carbonates. Here, a temperature modulation approach was used to control the thickness of nanoscale water films on forsterite (Mg2SiO4) in water-saturated scCO2. This method enables investigation of mineral carbonation reactivity from thin-film to bulk-like water conditions. Our results demonstrate an extreme influence of temperature on water film thickness and highlight the difficulties of studying mineral carbonation under water-saturated scCO2 conditions. |
In 2022, fossil fuel combustion and industrial processes released 36.8 Gt of CO2 into the atmosphere, which is the highest amount recorded in a single year.1 Geologic carbon sequestration (GCS) can help mitigate these emissions by injecting CO2 captured from fossil fuel plants into geologic reservoirs.2 Basalt formations are attractive locations for permanent CO2 storage because they are rich in Mg-, Ca-, and Fe-bearing silicate minerals that can dissolve and transform to metal carbonates.2 Injected CO2 can be dissolved in water3 or it can be a mostly anhydrous high-pressure fluid.4–6 Advantages of the latter include 1) more injected CO2 per unit volume, 2) reduced corrosion of steel pipes and wellbore casings, 3) lower viscosity, and 4) potentially less plugging of pores at the point of injection because metal silicates will not transform to solid carbonates without water.6–8 In an anhydrous CO2 injection scenario, pore water will be displaced, and the CO2 will reside as a variably humidified supercritical fluid (scCO2) with water content that can range from anhydrous near the injection well to fully saturated further into the reservoir.4,5,9
Under humidified scCO2 conditions, the metal silicate minerals in basalts become covered by Å-to-nm thick water films.10–12 Within these films, carbonic acid from the reaction of CO2 and water dissolves the minerals, and carbonate minerals and silica precipitate. Metal silicate dissolution and carbonate precipitation in thin water films are highly dependent on water film thickness,8,10,11,13 which is dictated in part by the relative humidity (RH) of the scCO2. Water film environments are distinct from bulk water because they have extremely low water-volume-to-mineral-surface-area ratios,11,14 are highly-surface structured with low dielectric constants,15–19 and have diffusive mass transport limitations. A fundamental understanding of the reactivity of metal silicate minerals towards carbonation in nanoscale water films is needed to better predict and optimize GCS in basaltic reservoirs.
Soon after GCS injection operations have ceased, the scCO2 in the reservoir may become fully water saturated as pore water flows back towards the injection well. Because of the importance of this condition, several previous studies have investigated metal silicate reactivity in scCO2 humidified to 100% RH by the presence of a separate liquid water phase.20–25 However, in most of these investigations, the extent to which water partitions between the scCO2 and the mineral surface was not directly measured.
In fully water-saturated scCO2, localized differences in temperature can affect water partitioning. This could have a significant impact in laboratory-based mineral carbonation studies, where temperature could be non-uniform due to equipment imperfections. Ideally, uniform temperatures are desirable because this reduces experimental complexity and more closely mimics the conditions in a geologic reservoir. However, intentionally induced temperature gradients could also be used as a tool to control the amount of water on the mineral surface. This would enable investigation of mineral carbonation reactivity over a range of adsorbed water conditions—from nanoscale water films to bulk-like water. Both ends of the spectrum of water conditions have previously been well studied, but little is known about the transition between the two.
The primary goal of this study was to investigate metal silicate carbonation in water-saturated scCO2 over a range of adsorbed water thicknesses. We used a model nanosized basalt mineral, forsterite (Mg2SiO4), in scCO2 at 50 °C, 90 bar and 100% RH and monitored the water coverages and reactivity using in situ infrared spectroscopy. A secondary goal was to demonstrate how induced temperature gradients in a laboratory apparatus can be used to control the amount of water on the mineral surface in these types of studies. We show that by establishing <1 °C differences between separate regions of a high-pressure reactor, we were able to span nearly two orders of magnitude in water monolayer coverages.
IR spectroscopic titrations were carried out to monitor the carbonation of forsterite at 100% RH with excess liquid water present. In this technique, water is added stepwise to a CO2-pressurized cell equipped with both transmission and attenuated total reflection (ATR) IR optics. Transmission IR is used to measure the concentration of water dissolved in the scCO2. ATR IR is used to measure the spectrum of the forsterite, which is coated as a several-microns-thick layer on the ATR internal reflection element (IRE). The cell is jacketed for temperature control using a circulating bath. For this study, we equipped the mirror assembly attached to the IRE mount with cartridge heaters so that the temperature of the forsterite on the IRE could change independent of the rest of the cell. Temperatures were measured in several locations throughout the cell as a function of dwell percentage applied to the cartridge heaters. The estimated precision of the temperature measurements was ±0.02 °C. Key locations were the forsterite-coated surface of the IRE and the region in the bottom of the cell where liquid water pooled when added above 100% RH conditions. We focused on the temperature difference between these locations because the pool is where the excess liquid water resided, and the IRE is where the ATR IR measurements were made.
Six time-dependent IR titration experiments (TDep_CO2_1 to TDep_CO2_6; see Table S2†) were conducted at 100% RH in scCO2 where the dwell percentage of the cartridge heaters was adjusted so that the temperature difference between the liquid water and the IRE where the forsterite was located varied between −0.29 °C (TDep_CO2_1; cartridge heaters off; forsterite was cooler) to 0.51 °C (TDep_CO2_6; forsterite was warmer). Selected ATR IR spectra are shown in Fig. S3 through S8† from time dependent experiments TDep_CO2_1 to TDep_CO2_6, respectively. The spectra from each experiment demonstrate time-dependent changes due to water adsorption, forsterite dissolution, amorphous silicate precipitation, and metal carbonate precipitation. Water adsorption will be discussed here; a detailed interpretation of the spectra in Fig. S3 through S8† concerning the carbonate phases that precipitated and spectral changes due to forsterite dissolution and amorphous silica precipitation is in the ESI.†
The amount of water adsorbed on forsterite was measured for each time-dependent experiment using the following method. Independent experiments were carried out to determine a correlation between the integrated absorbance of the ATR IR OH stretching band of adsorbed water and the coverage of water measured using transmission IR results (Ads_CO2_1 to Ads_CO2_5). The details of these experiments are in the ESI† (see also Fig. S9†). This correlation was then used to extrapolate the coverage of adsorbed water from the integrated absorbance of the ATR IR OH stretching band of water in the time-dependent experiments after 100% fluid RH was achieved (estimated to be after 10 h; see Fig. S10†). Fig. 1 shows this coverage of water as a function of the temperature difference between where titrated liquid water pooled and the surface of the IRE where the forsterite was located for each time-dependent experiment. The water coverage was about 5 ML when the forsterite was warmer (0.2–0.5 °C) than the liquid water. As the forsterite became cooler than the liquid water, the water coverage demonstrates a near-exponential increase. Our best estimate for the amount of water adsorbed on forsterite in scCO2 at 100% RH when there is no temperature difference is 21 ± 1 ML. Interestingly, the forsterite in wet scN2 at this same condition has a lower water coverage of 6.3 ± 0.2 ML. The higher coverage in scCO2 is likely because forsterite solubility is greater in scCO2, and dissolved Mg2+ and HCO3− are hygroscopic.
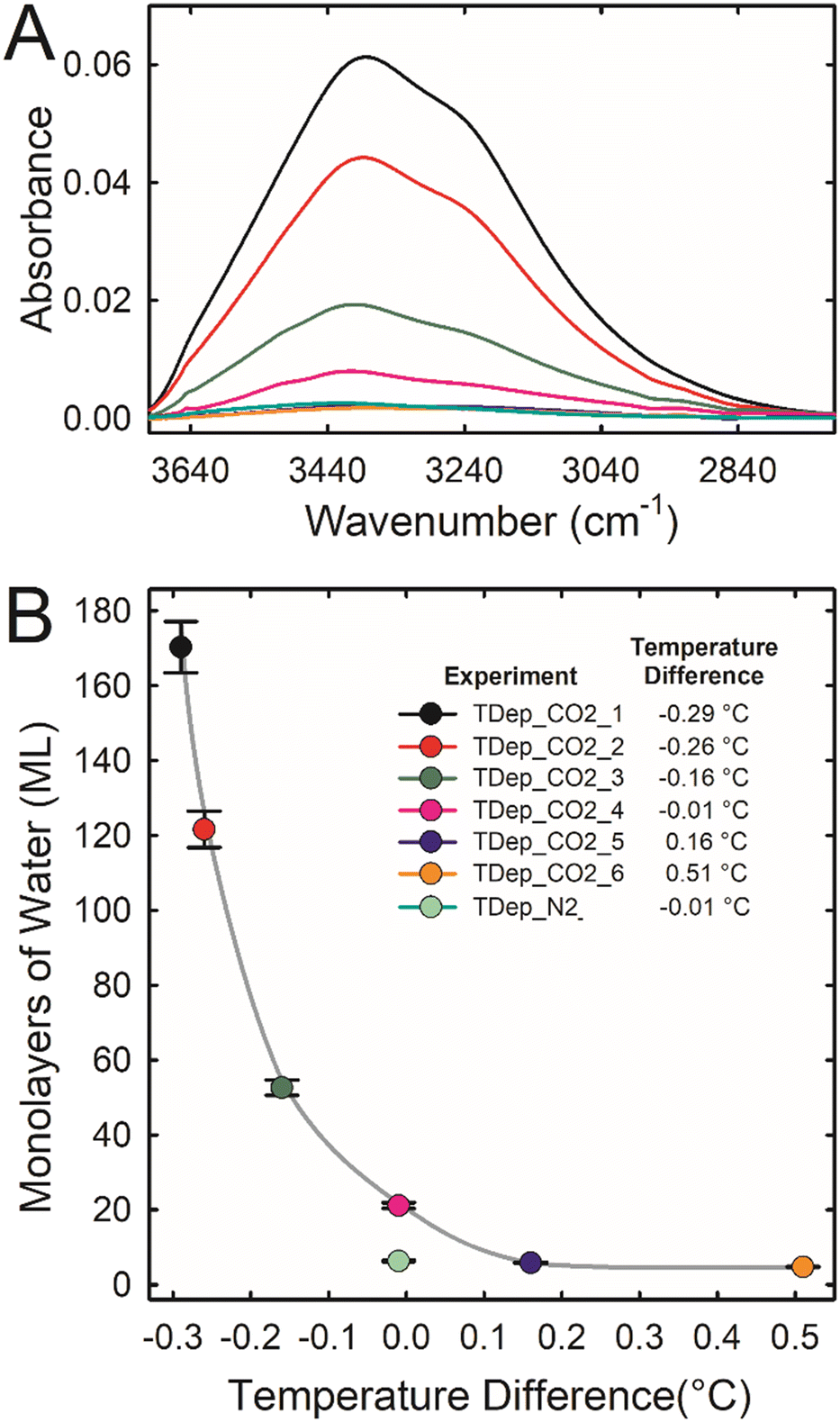 | ||
| Fig. 1 (A) ATR IR spectra in the OH stretching region after 10 h of reaction when 100% fluid RH was achieved (see Fig. S10†) for experiments TDep_CO2_1 through TDep_CO2_6 where forsterite was reacted for a total of 120 hours in scCO2 at 50 °C, 90 bar, and 100% RH with excess water present. Each experiment was conducted with a unique temperature difference between two regions in the sample cell: the IRE surface where the forsterite was located and a nearby water pool area (see ESI† for details). A negative temperature difference indicates that the IRE surface was cooler than the nearby water pool. (B) The estimated average water film thickness for experiments TDep_CO2_1 through TDep_CO2_6. The lines between the data points are to help visualize trends. Data are also shown for experiment TDep_N2 carried out in scN2. | ||
The precise temperature-induced control over water layer thickness in scCO2 is likely due to dissolution of forsterite and formation of a hygroscopic thin water film, rich in Mg2+ and HCO3−. This is supported by fitting a hygroscopic water layer growth model from κ – Köhler theory to the data (see ESI†). Using estimates of water saturation, density, and interfacial tension, we fit a hygroscopicity term (κ) to the data. Results suggest that at positive temperature offsets, κ ≈ 0.01, similar to values reported for insoluble silica materials (e.g., silica dust).26,27 At the two largest negative temperature offsets—and thus highest water supersaturation near the forsterite surface—κ ≈ 0.1, which corresponds to values reported for carbonate dust, i.e., nano- to micron-sized sparingly soluble carbonate particles.27,28 The two measurements at intermediate temperature offsets also correspond to intermediate values of κ, suggesting a hygroscopicity between that of a hydrated silicate surface and a hydrated carbonate surface. These observations from the hygroscopicity model also agree with the differences in reactivity reported in Fig. 2. The most reactive samples exhibit hygroscopicity equivalent to a surface layer rich in solvated Mg2+ and HCO3−. The least reactive samples likely build up a silica-rich surface that is not nearly as hygroscopic. This observation is consistent with the experiment in scN2, where no significant forsterite dissolution is expected and the hygroscopicity would remain low.
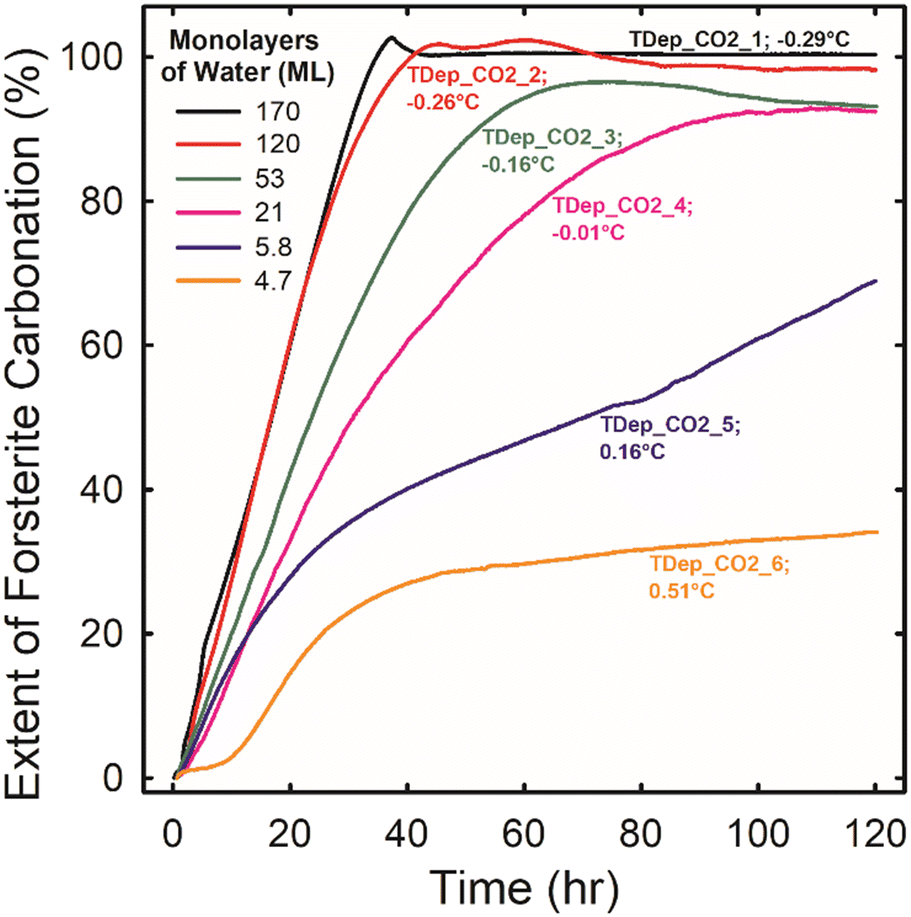 | ||
| Fig. 2 The extent of forsterite carbonation as a function of time during experiments TDep_CO2_1 through TDep_CO2_6 (see Table S2†) where forsterite was reacted for a total of 120 hours in scCO2 at 50 °C, 90 bar, and 100% RH with excess water present. The reaction extent was estimated based on the decrease in the SiO stretching band of forsterite as described in the ESI.† The temperature difference between where titrated liquid water pools and the surface of the IRE where the forsterite was located is listed after each experiment label. A negative temperature difference indicates that the IRE surface was cooler than the water pool. More negative temperature differences resulted in thicker water films on the forsterite, which have been estimated for each experiment (see Fig. 1) and are listed in the legend. | ||
Small temperature differences lead to a wide range of film thicknesses which, in turn, cause significant changes in carbonation reactivity. Fig. 2 is a plot of the extent of forsterite carbonation versus time for experiments TDep_CO2_1 through TDep_CO2_6. We calculated the extent of carbonation based on the decrease in the integrated absorbance of the SiO stretching band of forsterite due to the mineral's dissolution. This is believed to be valid because forsterite dissolution is strongly coupled to carbonate precipitation under low water conditions. Additionally, estimating the reaction extent based on dissolution is expected to be more reliable than following the growth of metal carbonates directly because the absorption coefficients of the various carbonate products (i.e., bicarbonate, hydromagnesite, magnesite, and AMC) are not the same. The analysis to determine the extent of carbonation using the forsterite SiO stretching bands is described in the ESI.† Variations of less than 5% in the reaction extent are likely systematic errors due to spectroscopic baseline changes and the overlapping bands of water and carbonate.
Carbonation rates and extents declined with decreasing water coverage. Rates were fastest for TDep_CO2_1 and TDep_CO2_2 when forsterite was covered with bulk-like water coverages of 170 ± 7 and 121 ± 5 ML of water. Forsterite carbonated at a constant rate in these experiments and was completely consumed in less than two days. However, rates became progressively slower with increasing time in experiment TDep_CO2_3 at a water coverage of 53 ± 2 ML. The slowing of the reaction could be due to a reduction of reactive surface area by the accumulation of reaction products. In fact, we showed in a recent study that a Mg-depleted silica-rich layer that is just a few nm in thickness on reacted forsterite particles can cause a passivation effect under humidified scCO2 conditions.29 Passivation was apparent even in experiment TDep_CO2_4 at a water coverage of 21 ± 1 ML when there was the smallest temperature difference between the liquid water and the IRE where the forsterite is located. The carbonation rate was fastest and relatively constant up to about 30 h but then gradually slowed by a factor of 2 from 30 to 80 h, and there was unreacted forsterite present even after 5 days. The impact of reaction passivation was greatest for experiment TDep_CO2_6 at a water coverage of 4.7 ± 1 ML. The carbonation rate was relatively rapid between 8 and 24 h of reaction, but then slowed by a factor of 15 from 42 to 120 h. A detailed study aimed at elucidating the mechanism by which Mg-depleted silica-rich layers slow forsterite carbonation in humidified scCO2 is ongoing.
This study has furthered an understanding of metal silicate carbonation reactivity in scCO2 at 100% RH with excess liquid water present. Water film thickness is a key controlling parameter that, in our apparatus, could be altered by an order of magnitude with less than a 0.5 °C temperature difference between the metal silicate and nearby liquid water (Fig. 1). Small temperature differences also control the carbonation rate, extent of carbonation, and whether a passivation effect is observed (Fig. 2). Under uniform temperature conditions, our best estimate for the water film thickness on forsterite nanoparticles in fully water-saturated scCO2 at 50 °C and 90 bar is 21 ± 1 ML. Our results highlight the difficulties of working at 100% RH with excess liquid water present. We speculate that unintended bulk-like water conditions have occurred in many previous, laboratory-based studies of mineral carbonation in scCO2 due to the high sensitivity of fully water saturated systems to very small temperature changes (Fig. 1B). However, if temperature differences between the mineral and the liquid water are carefully controlled, this provides the opportunity to investigate metal silicate carbonation reactivity at water film thickness of 10's to 100's of ML. In other words, by modulating temperature, we can study carbonation at water coverages that have not been previously investigated and construct a more complete picture of basalt reactivity in humidified scCO2.
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division through its Geosciences program at Pacific Northwest National Laboratory (FWP 56674). Some analyses were performed at the Environmental Molecular Sciences Laboratory, a user facility sponsored by the DOE's Office of Biological and Environmental Research and located at PNNL.
Conflicts of interest
There are no conflicts to declare.Notes and references
- IEA, CO2 Emissions in 2022, IEA, Paris, 2023 Search PubMed.
- A. Raza, G. Glatz, R. Gholami, M. Mahmoud and S. Alafnan, Earth-Sci. Rev., 2022, 229, 104036 CrossRef CAS.
- J. M. Matter, M. Stute, S. Ó. Snæbjörnsdottir, E. H. Oelkers, S. R. Gislason, E. S. Aradottir, B. Sigfusson, I. Gunnarsson, H. Sigurdardottir, E. Gunnlaugsson, G. Axelsson, H. A. Alfredsson, D. Wolff-Boenisch, K. Mesfin, D. Fernandez de la Reguera Taya, J. Hall, K. Dideriksen and W. S. Broecker, Science, 2016, 352, 1312–1314 CrossRef CAS PubMed.
- B. P. McGrail, H. T. Schaef, F. A. Spane, J. B. Cliff, O. Qafoku, J. A. Horner, C. J. Thompson, A. T. Owen and C. E. Sullivan, Environ. Sci. Technol. Lett., 2017, 4, 6–10 CrossRef CAS.
- B. P. McGrail, H. T. Schaef, F. A. Spane, J. A. Horner, A. T. Owen, J. B. Cliff, O. Qafoku, C. J. Thompson and E. C. Sullivan, Energy Procedia, 2017, 114, 5783–5790 CrossRef CAS.
- B. P. McGrail, F. A. Spane, J. E. Amonette, C. R. Thompson and C. F. Brown, Energy Procedia, 2014, 63, 2939–2948 CrossRef CAS.
- B. P. McGrail, H. T. Schaef, V. A. Glezakou, L. X. Dang and A. T. Owen, Energy Procedia, 2009, 1, 3415–3419 CrossRef CAS.
- E. Placencia-Gómez, S. N. Kerisit, H. S. Mehta, O. Qafoku, C. J. Thompson, T. R. Graham, E. S. Ilton and J. S. Loring, Environ. Sci. Technol., 2020, 54, 6888–6899 CrossRef PubMed.
- S. Krevor, M. J. Blunt, S. M. Benson, C. H. Pentland, C. Reynolds, A. Al-Menhali and B. Niu, Int. J. Greenhouse Gas Control, 2015, 40, 221–237 CrossRef CAS.
- J. S. Loring, C. J. Thompson, Z. Wang, A. G. Joly, D. S. Sklarew, H. T. Schaef, E. S. Ilton, K. M. Rosso and A. R. Felmy, Environ. Sci. Technol., 2011, 45, 6204–6210 CrossRef CAS PubMed.
- S. N. Kerisit, S. T. Mergelsberg, C. J. Thompson, S. K. White and J. S. Loring, Environ. Sci. Technol., 2021, 55, 12539–12548 CrossRef CAS PubMed.
- Q. R. S. Miller, D. A. Dixon, S. D. Burton, E. D. Walter, D. W. Hoyt, A. S. McNeill, J. D. Moon, K. S. Thanthiriwatte, E. S. Ilton, O. Qafoku, C. J. Thompson, H. T. Schaef, K. M. Rosso and J. S. Loring, J. Phys. Chem. C, 2019, 123, 12871–12885 CrossRef CAS.
- J. S. Loring, J. Chen, P. Bénézeth, O. Qafoku, E. S. Ilton, N. M. Washton, C. J. Thompson, P. F. Martin, B. P. McGrail, K. M. Rosso, A. R. Felmy and H. T. Schaef, Langmuir, 2015, 31, 7533–7543 CrossRef CAS PubMed.
- S. T. Mergelsberg, S. N. Kerisit, E. S. Ilton, O. Qafoku, C. J. Thompson and J. S. Loring, Chem. Commun., 2020, 56, 12154–12157 RSC.
- A. W. Knight, N. G. Kalugin, E. Coker and A. G. Ilgen, Sci. Rep., 2019, 9, 8246 CrossRef.
- S. Le Caër, S. Pin, S. Esnouf, Q. Raffy, J. P. Renault, J. B. Brubach, G. Creff and P. Roy, Phys. Chem. Chem. Phys., 2011, 13, 17658–17666 RSC.
- O. Teschke, G. Ceotto and E. F. de Souza, Phys. Rev. E, 2001, 64, 011605 CrossRef CAS PubMed.
- S. Senapati and A. Chandra, J. Phys. Chem. B, 2001, 105, 5106–5109 CrossRef CAS.
- L. Fumagalli, A. Esfandiar, R. Fabregas, S. Hu, P. Ares, A. Janardanan, Q. Yang, B. Radha, T. Taniguchi, K. Watanabe, G. Gomila, K. S. Novoselov and A. K. Geim, Science, 2018, 360, 1339 CrossRef CAS PubMed.
- Q. R. S. Miller, J. P. Kaszuba, H. T. Schaef, M. E. Bowden, B. P. McGrail and K. M. Rosso, Chem. Commun., 2019, 55, 6835–6837 RSC.
- B. Aguila, L. Hardee, H. T. Schaef, S. Zare, M. J. Abdolhosseini Qomi, J. V. Crum, J. E. Holliman Jr, E. T. Rodriguez, L. M. Anovitz, K. M. Rosso and Q. R. S. Miller, Environ. Sci.: Nano, 2023, 10, 2672–2684 RSC.
- M. J. Abdolhosseini Qomi, Q. R. S. Miller, S. Zare, H. T. Schaef, J. P. Kaszuba and K. M. Rosso, Nat. Rev. Chem., 2022, 6, 598–613 CrossRef CAS PubMed.
- Y. E. Chai, Q. R. S. Miller, H. T. Schaef, D. Barpaga, R. Bakhshoodeh, M. Bodor, T. Van Gerven and R. M. Santos, J. Supercrit. Fluids, 2021, 171, 105191 CrossRef CAS.
- Q. R. S. Miller, C. J. Thompson, J. S. Loring, C. F. Windisch, M. E. Bowden, D. W. Hoyt, J. Z. Hu, B. W. Arey, K. M. Rosso and H. T. Schaef, Int. J. Greenhouse Gas Control, 2013, 15, 104–118 CrossRef CAS.
- H. T. Schaef, Q. R. S. Miller, C. J. Thompson, J. S. Loring, M. S. Bowden, B. W. Arey, B. P. McGrail and K. M. Rosso, Energy Procedia, 2013, 37, 5892–5896 CrossRef CAS.
- H. Herich, T. Tritscher, A. Wiacek, M. Gysel, E. Weingartner, U. Lohmann, U. Baltensperger and D. J. Cziczo, Phys. Chem. Chem. Phys., 2009, 11, 7804–7809 RSC.
- M. Tang, D. J. Cziczo and V. H. Grassian, Chem. Rev., 2016, 116, 4205–4259 CrossRef CAS PubMed.
- J. T. Kelly, C. C. Chuang and A. S. Wexler, Atmos. Environ., 2007, 41, 2904–2916 CrossRef CAS.
- S. T. Mergelsberg, B. P. Rajan, B. A. Legg, L. Kovarik, S. D. Burton, G. M. Bowers, M. E. Bowden, O. Qafoku, C. J. Thompson, S. N. Kerisit, E. S. Ilton and J. S. Loring, Environ. Sci. Technol. Lett., 2023, 10, 98–104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and materials, Tables S1–S3 and Fig. S1–S11. See DOI: https://doi.org/10.1039/d3en00884c |
| This journal is © The Royal Society of Chemistry 2024 |