
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of heteroleptic bis-phosphine bis-NHC iron (0) complexes: a strategy to enhance small molecule activation†
Christian M.
Andre
and
Nathaniel K.
Szymczak
 *
*
Department of Chemistry, University of Michigan, 930 North University Avenue, Ann Arbor, MI 48109, USA. E-mail: nszym@umich.edu
First published on 8th November 2024
Abstract
We report the synthesis of heteroleptic iron complexes supported by both a bis-phosphine ligand (depe) and a bis-NHC ligand. The mixed ligand sets provide access to iron (0) adducts of N2 and CO that are highly activated, in comparison to homoleptic (i.e. Fe(depe)2L) variants. Computational and experimental studies revealed the mixed ligand set distorts the geometric and electronic structure to yield an unusually basic iron. Although protonation occurred at Fe, silylation of the Fe(0)N2 complex afforded a highly activated silyldiazenido [FeNNSiMe3]+ complex.
Low-valent iron complexes are routinely targeted as models for Fe-nitrogenase, given their ability to activate N2‡ as well as other small molecules (CO, CO2, etc.),1 with iron phosphines among the most established (Fig. 1A).2 Of particular note is the Fe0N2 complex of 1,2-bis(diethylphosphino)ethane (depe) because it features a significantly activated N2 ligand and is a catalyst for selective N2 reduction to hydrazine.2a These properties have motivated studies by our group3 and others4 to examine additional design principles, such as those achieved by introducing secondary sphere acids, to further enhance the activation imparted by the Fe0(depe)2 unit to small molecules. While adjusting the primary coordination sphere of the iron centre to increase its electron density is the most common redesign strategy, modification of the phosphine from depe typically§ affords systems that are less donating than Fe(depe)25 and/or have a dramatically different steric profile and binding geometry.2i,j,6 In analogy to phosphines, lower-coordinate iron complexes of monodentate N-heterocyclic carbenes (NHC) are reported to activate N2 to a greater extent (Fig. 1B), a result of their stronger σ-donor ability than phosphines.7
 | ||
| Fig. 1 (A) Fe phosphines demonstrated to activate N2.2c,d,g,6a (B) Fe N2 NHC complexes and the lack of a reported Fe N2 tetracarbene.7b,c (C) Analogy between depe and bis-NHC ligand and the prospect of a heteroleptic complex. | ||
In contrast to the breadth of low-valent chemistry reported with phosphine donors, there are no such examples of 18-electron Fe0 complexes containing four NHC donors.8 To bridge the gap between Fe0 complexes containing four phosphines (known) and those containing four NHCs (unknown), we targeted systems containing two phosphines and two NHCs. Although mixed NHC/phosphine transition metal complexes9 have been reported, few examples contain bis-NHCs/bis-phosphines,9h–k and none involve iron. Assembling two distinct sets of bidentate ligands to form a heteroleptic complex is precedented for tuning a metal's reactivity and/or stability;9j,10 however, this approach has not been employed to improve N2 activation.
Our design strategy employed a methylene-linked bis-NHC ligand, because it is a neutral bidentate ligand with similar steric profile and bite-angles to depe (Fig. 1C).2d,11 We hypothesized that a heteroleptic iron complex of both a bis-phosphine and a bis-NHC ligand would be structurally analogous to the established bis-(bis-phosphine) Fe0(depe)2 system, but better able to promote substrate activation with a more electron-rich iron centre.
We developed a general one-pot metalation/ligand substitution procedure (Scheme 1), to access a mixed NHC/phosphine system. Introduction of 1 equiv. 1 (bis-N-butyl-imidazolium salt) to a suspension of Fe(N(SiMe3)2)2 in CH3CN afforded a clear red solution after 2 h at room temperature. Solvent removal by vacuum followed by washing with THF yielded a deep red solid. This red intermediate exhibited two aromatic 1H NMR singlets at 7.46 and 7.32 ppm without further downfield resonances, consistent with a symmetrically bound bis-NHC tetrakis-MeCN complex (1a) analogous to a previous report.12 To a CH3CN solution of 1a was added 1.0 equiv. depe as a CH3CN solution, which caused a colour change to deep orange. Concentration of this product under vacuum yielded a viscous solution, from which residual depe was removed via pentane extraction. Solvent removal yielded a bright orange diamagnetic complex 2 as a mixture of isomers in 95% yield.
 | ||
| Scheme 1 Synthesis of heteroleptic complexes 2–5. Molecular structure of 4 determined by single crystal XRD. Thermal ellipsoids shown at 50% probability with hydrogen atoms omitted and non-interacting alkyl chains are in wireframe for clarity. | ||
ESI-mass spectrometry of 2 provided a m/z of 567.2671, consistent with [Fe(HCOO)(butylCC)(depe)]+ (m/z = 567.2674),¶ which features a heteroleptic composition of the primary coordination sphere. The 1H NMR spectrum in CH2Cl2 exhibited a set of four strong aromatic singlets (7.53, 7.38, 7.28, & 7.04 ppm) and a set of two weaker singlets (7.58 and 7.24 ppm) that integrate in a net ratio of ∼ 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The 31P NMR spectrum exhibited a pair of doublets (75.87 and 62.93 ppm, JPP = 15Hz) and a smaller singlet (62.56 ppm) that integrate to the same ratio (Fig. S2 and S3, ESI†). This pattern is consistent with a mixture of the cis-heteroleptic isomer (2-cis), where both phosphorus and NHC donors are inequivalent, and the trans heteroleptic isomer (2-trans), where they are equivalent. Following addition of MeCN-d3 to 2, we observed a loss of coordinated MeCN 1H NMR resonances (2.45 and 2.33 ppm) and growth of free MeCN (1.94 ppm). This behavior indicates facile exchange of MeCN ligands and may implicate a ligand exchange pathway for isomerization of 2. The structure of 2-cis was further confirmed by single crystal x-ray diffraction (SC-XRD) studies from crystals grown in CH2Cl2.
:1. The 31P NMR spectrum exhibited a pair of doublets (75.87 and 62.93 ppm, JPP = 15Hz) and a smaller singlet (62.56 ppm) that integrate to the same ratio (Fig. S2 and S3, ESI†). This pattern is consistent with a mixture of the cis-heteroleptic isomer (2-cis), where both phosphorus and NHC donors are inequivalent, and the trans heteroleptic isomer (2-trans), where they are equivalent. Following addition of MeCN-d3 to 2, we observed a loss of coordinated MeCN 1H NMR resonances (2.45 and 2.33 ppm) and growth of free MeCN (1.94 ppm). This behavior indicates facile exchange of MeCN ligands and may implicate a ligand exchange pathway for isomerization of 2. The structure of 2-cis was further confirmed by single crystal x-ray diffraction (SC-XRD) studies from crystals grown in CH2Cl2.
Although 2 was isolated as a mixture of cis and trans isomers, we found that addition of a neutral π-acid, such as CO, afforded a single geometric isomer. Charging a solution of 2 in CH2Cl2 with 30 psig CO caused the colour to slowly fade over 6 h. Evaporation of the solvent afforded a pale-yellow species (3). Its 31P NMR spectrum featured a singlet at 64.6 ppm, while the 1H NMR spectrum exhibited aromatic resonances at 7.57 and 7.17 ppm that integrate 4:3 with a singlet at 2.48 ppm. This result is consistent with a trans geometry (Cs symmetry) containing a single acetonitrile ligand. ESI mass spectrometry of 3 provided a m/z of 295.6467, consistent with [Fe(CO)(MeCN)(butylCC)(depe)]2+ (m/z = 295.6451). IR spectroscopy of 3 revealed a single νCO stretch at 1947 cm−1. These data support assignment of 3 as a heteroleptic complex containing a single carbonyl ligand trans to a coordinated CH3CN. We attribute formation of the trans isomer of 3 to the strong trans donor properties of CO, which favours trans ligands that are weaker donors (MeCN), rather than stronger donors (NHC or phosphine), as would be necessitated by the cis isomer.
Following the synthesis of 2 and 3, we pursued an Fe0 species. Reduction of 2 with excess KC8 followed by pentane extraction/removal afforded a deep red diamagnetic solid (4). This species exhibits a sharp 31P NMR singlet at 90.05 ppm, and two aromatic 1H singlets at 6.44 and 6.35 ppm. Subjecting 3 to analogous reduction conditions and workup yielded a diamagnetic orange solid (5). NMR spectra of 5 similarly exhibit a broad 31P NMR singlet at 95.85 ppm and two 1H singlets at 6.45 and 6.32 ppm. The breadth of these NMR resonances of 4 and 5 vary with temperature (−75 °C to 45 °C), indicating fluxionality in solution.
Both 4 and 5 exhibited more activated π-acceptor ligands than their bis-depe Fe0 analogues: 4 displayed a strong νN2 stretch at 1913 cm−1 (νN2 = 1955 cm−1 for FeN2(depe)2) and 5 exhibited a strong νCO stretch at 1737 cm−1 (νCO = 1800 cm−1 for FeCO(depe)2). The ligand environment imposed on iron was assessed using cyclic voltammetry experiments. Compounds 4 and 5 exhibited reversible events at −2.48 V and −2.06 V (THF, vs. Fe(Cp2)/Fe(Cp)2+), respectively, which we assign as Fe(0/1) redox couples. Relative to the bis-depe analogues, 4 and 5 are significantly more reducing (480 mV and 520 mV more cathodic, respectively2a), further supporting strongly reduced Fe centers.
SC-XRD studies on 4 and 5 reveal structural differences from the analogous Fe(depe)2 complexes. 5 exhibits an intermediate 5-coordinate (τ = 0.4) geometry and 4 has a square pyramidal (τ = 0.02) geometry, which contrast with the reported TBP geometry of FeN2(depe)2 and FeCO(depe)2 (τ = 0.9 for both).7b,13 While the N–N bond distance of 4 is not statistically different from that of FeN2(depe)2, (1.144(2) Å vs. 1.139(13) Å), the C–O bond of 5 is longer than that of FeCO(depe)2 (1.199(3) Å vs. 1.179(8) Å), consistent with its more activated IR stretch. Both the Fe–N2 bond of 4 and the Fe–CO bond of 5 are longer than their Fe(depe)2 analogues (1.8016(16) Å vs. 1.748(8) Å for FeN2; 1.733(6) Å vs. 1.179(8) Å for FeCO), which reflects less multiple bond character between iron and the bound diatoms, despite greater π-backdonation (Scheme 1).
 | ||
| Fig. 2 Frontier orbitals from DFT performed at TPSS def2-TZVP level of theory with SMD solvation model on optimized structure (isosurface values = 0.05). | ||
Geometry optimization of both 4 and 5 using density functional theory (DFT) converged to intermediate structures (τ = 0.4). IR spectra and Fe(0/1) redox potentials for both FeN2(depe)2 and 4 calculated from these DFT optimization studies corroborate solution-phase experimental results (Table S4, ESI†), supporting the intermediate geometry as the solution-phase structure of 4. This optimized structure also reveals an elongated N–N bond in 4 with respect to FeN2(depe)2 (1.142 Å vs. 1.139 Å), indicating increased activation of the N2 ligand. Molecular orbital (MO) calculations for 4 and FeN2(depe)2 illustrate energetic differences between the frontier orbitals (Fig. 2). The HOMO of 4 is localized on the Fe centre, is σ-antibonding with respect to N2 coordination, and higher in energy than that of FeN2(depe)2, which validates elongated Fe–N2 and Fe–CO bonds found in crystal structures of 4 and 5.
The HOMO-1 of 4 is the primary orbital involved in π-backdonation into the N2 π* orbital; however it is −0.475 eV lower than the HOMO. Despite the HOMO-1 being higher in energy in 4 than in FeN2(depe)2, this orbital sits far below the metal-centred HOMO. This electronic structure renders the complex more basic at Fe, and less likely to protonate at N2 than for FeN2(depe)2. We attribute the higher orbital energies of 4 to the greater donor strength of the NHC ligands, while the distortion of its geometric and electronic structure is due to the inherent asymmetry of the heteroleptic ligand environment.
To augment the DFT studies that predict high Fe-basicity of 4, we studied its reactivity with Brønsted acids. Treating 4 with [NH2Ph2][OTf] in THF afforded a rapid color change from red to yellow. This product exhibits a triplet 1H NMR resonance spectrum at −16.7 ppm (2JHP = 60 Hz), a doublet 31P NMR resonance at 81.9 ppm (2JHP = 59 Hz), and an IR absorbance at 2072 cm−1. These data are consistent with prior reports of [FeH(N2)]+ complexes,2a,k as protonation of FeN2(depe)2 with 1 equiv. [NH2Ph2][OTf] afforded trans-[Fe(H)N2(depe)2]+, characterized by a 1H NMR quintet at −18.20 ppm, a 31P doublet at 81.14 ppm (2JHP = 50 Hz), and an IR feature at 2090 cm−1 (νN2). Therefore we assign this product as trans-[FeH(N2)(depe)(butylCC)][OTf] ([6][OTf]) (Fig. 3). Treatment of 4 with a weaker acid, tBuOH, affords 6 quantitatively by NMR spectroscopy, but subjecting FeN2(depe)2 to the same conditions yields <15% protonation. This divergence in reactivity demonstrates the higher basicity of the Fe center predicted by DFT in 4 compared to FeN2(depe)2.
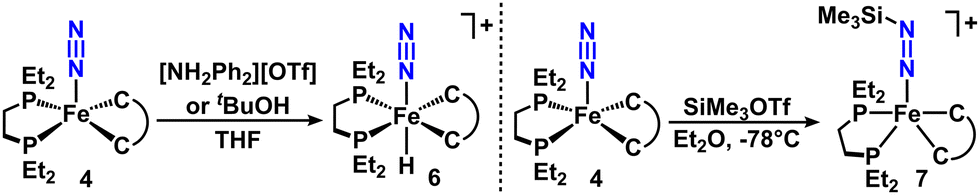 | ||
| Fig. 3 Synthesis of 6 and 7 from 4. | ||
To minimize Fe-centered reactivity, we targeted a more sterically encumbered electrophile, trimethylsilyl triflate (SiMe3OTf), for N2 functionalization, analogous to reported silylation of FeN2(depe)2.14 Addition of 1 eq of SiMe3OTf to an Et2O solution of 4 at −78 °C afforded a deep green precipitate (7) in 75% yield (Fig. 3). This species exhibits a 31P NMR singlet at 85.3 ppm, and two aromatic 1H singlets at 7.68 and 7.16 ppm, which is consistent with a FeII heteroleptic complex. These aromatic features integrated net 4:9 with respect to a 1H singlet at 0.19 ppm, consistent with incorporation of one SiMe3 group. A single 19F NMR resonance at −78.9 ppm is consistent with a free triflate anion in the product, indicating a cationic iron complex. 29Si NMR spectroscopy revealed a singlet at -5.4 ppm, and ATR-IR characterization of 7 exhibited a strong absorbance at 1693 cm−1; both of which are consistent with a silyldiazenide (−NNSiMe3) substituent. These spectra are similar to those of the reported [Fe(N2SiMe3)(depe)2]+ (N2SiMe3 unit = 1H NMR: 0.20 ppm, 29Si NMR: 6.4 ppm, IR: 1732 cm−1).14 In contrast to the reaction of FeN2(depe)2 with SiMe3OTf, which was reported to be reversible, 7 formed cleanly and irreversibly, supporting the increased basicity of the N2 moiety of 4. Compared to other reported iron silyldiazenido complexes, of which most are neutral,1b,15 the IR absorbance suggests that 7 is among the most activated, only surpassed by an anionic example1b from the Holland group, despite assignment of 7 as cationic. While the heteroleptic iron system and Fe(depe)2 demonstrate similar aptitude for N2 silylation, each heteroleptic intermediate (the N2 (4) and silylhydrazide (7) complexes) exhibit substantially more activated N–N bonds at each step.
In conclusion, we have reported the synthesis and characterization of a new class of heteroleptic Fe0 complexes jointly supported by a bis-phosphine and a methylene-linked bis-NHC ligand. The dinitrogen and carbonyl complexes, 4 and 5, are highly reduced, as exemplified by their highly activated νN2/νCO stretches and cathodic redox potentials. SC-XRD and DFT studies reveal that both 4 and 5 adopt an intermediate 5 coordinate geometry (τ = 0.4). Silylation of 4 yields a highly activated silydiazenido complex 7, demonstrating the system's potential for N2 functionalization. This mixed-ligand Fe0 system exhibits properties uncharacteristic of reported Fe0 phosphine or NHC complexes, and the modular access to heteroleptic Fe0N2 compounds presented herein invites further work to tune the primary sphere donors and induce new reactivity at iron.
C. A. performed the experiments. N.K.S managed the project. Both C. A. and N. K. S. designed and analysed experiments and wrote the manuscript.
This work was supported by the NIH (R35GM136360). We thank Fengrui Qu for SCXRD collection of 2-cis, 4, and 5 and the Buss Lab for assistance with inert atmosphere IR spectroscopy.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2-cis, 4, and 5 have been deposited at the CCDC under and can be obtained from https://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef; (b) S. F. McWilliams, D. L. J. Broere, C. J. V. Halliday, S. M. Bhutto, B. Q. Mercado and P. L. Holland, Nature, 2020, 584, 221 CrossRef PubMed; (c) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef; (d) S. Kuriyama, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef PubMed; (e) S. M. Rummelt, H. Zhong, I. Korobkov and P. J. Chirik, J. Am. Chem. Soc., 2018, 140, 11589 CrossRef PubMed.
- (a) P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521 CrossRef PubMed; (b) M. M. Deegan and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 2561 CrossRef PubMed; (c) S. Kuriyama, T. Kato, H. Tanaka, A. Konomi, K. Yoshizawa and Y. Nishibayashi, Bull. Chem. Soc. Jpn., 2022, 95, 683 CrossRef; (d) M. Hirano, M. Akita, T. Morikita, H. Kubo, A. Fukuoka and S. Komiya, Dalton Trans., 1997, 3453 RSC; (e) M. V. Baker and L. D. Field, Organometallics, 1986, 5, 821 CrossRef; (f) H. H. Karsch, Chem. Ber., 1977, 110, 2213 CrossRef; (g) S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105 CrossRef; (h) T. T. Adamson, S. P. Kelley and W. H. Bernskoetter, Organometallics, 2020, 39, 3562 CrossRef; (i) D. J. Schild and J. C. Peters, ACS Catal., 2019, 9, 4286 CrossRef; (j) T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2004, 126, 6252 CrossRef; (k) L. D. Field, N. Hazari and H. L. Li, Inorg. Chem., 2015, 54, 4768 CrossRef; (l) G. J. Leigh and M. Jimenez-Tenorio, J. Am. Chem. Soc., 1991, 113, 5862 CrossRef; (m) A. Hills, D. L. Hughes, M. Jimenez-Tenorio, G. J. Leigh and A. T. Rowley, Dalton Trans., 1993, 3041 RSC.
- J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952 CrossRef PubMed.
- (a) H. Corona, M. Pérez-Jiménez, F. de la Cruz-Martínez, I. Fernández and J. Campos, Angew. Chem., Int. Ed., 2022, 61, e202207581 CrossRef; (b) A. D. Piascik, R. Li, H. J. Wilkinson, J. C. Green and A. E. Ashley, J. Am. Chem. Soc., 2018, 140, 10691 CrossRef PubMed.
- (a) R. A. Cable, M. Green, R. E. Mackenzie, P. L. Timms and T. W. Turney, J. Chem. Soc. Chem. Commun., 1976, 7, 270 RSC; (b) A. M. Tondreau, B. L. Scott and J. M. Boncella, Organometallics, 2016, 35, 1643 CrossRef; (c) J. D. Gilbertson, N. K. Szymczak and D. R. Tyler, J. Am. Chem. Soc., 2005, 127, 10184 CrossRef.
- (a) R. Gilbert-Wilson, L. D. Field, S. B. Colbran and M. M. Bhadbhade, Inorg. Chem., 2013, 52(6), 3043–3053 CrossRef; (b) L. D. Field, H. L. Li, S. J. Dalgarno and R. D. McIntosh, Eur. J. Inorg. Chem., 2019, 2006–2011 CrossRef; (c) F. F. van de Watering, W. Stroek, J. Ivar van der Vlugt, B. de Bruin, W. I. Dzik and J. N. H. Reek, Eur. J. Inorg. Chem., 2018, 1254–1265 CrossRef PubMed; (d) D. E. Prokopchuk, E. S. Wiedner, E. D. Walter and C. V. Popescu, et al. , J. Am. Chem. Soc., 2017, 139(27), 9291–9301 CrossRef PubMed; (e) A. Cavaillé, B. Joyeux, N. Saffon-Merceron, N. Nebra, M. Fustier-Boutignon and N. Mézailles, Chem. Commun., 2018, 54(84), 11953 RSC.
- (a) Y. Fan, J. Cheng, Y. Gao, M. Shi and L. Deng, Chem. Sin., 2018, 76, 445 Search PubMed; (b) Z. Ouyang, J. Cheng, L. Li, X. Bao and L. Deng, Chem. – Eur. J., 2016, 22, 14162 CrossRef PubMed; (c) S. A. Lutz, A. K. Hickey, Y. Gao, C.-H. Chen and J. M. Smith, J. Am. Chem. Soc., 2020, 142, 15527 CrossRef PubMed.
- C. R. Groom, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., 2016, B72, 171 Search PubMed.
- (a) A. Ahmida, F. M. Elmagbari, H. Egold, U. Flörke and G. Henkel, Polyhedron, 2020, 181, 114472 CrossRef; (b) A. Ahmida, F. M. Elmagbari, H. Egold and G. Henkel, Polyhedron, 2021, 198, 115083 CrossRef; (c) P. Ai, A. A. Danopoulos and P. Braunstein, Dalton Trans., 2016, 45, 4771 RSC; (d) B. R. Galan, E. S. Wiedner, M. L. Helm, J. C. Linehan and A. M. Appel, Organometallics, 2014, 33, 2287 CrossRef; (e) P. Nagele, U. Herrlich, F. Rominger and P. Hofmann, Organometallics, 2013, 32, 181 CrossRef; (f) E. Mosaferi, L. Pan, T. Wang, Y. Sun, C. Pranckevicius and D. W. Stephan, Dalton Trans., 2016, 45(4), 1354 RSC; (g) N. Stylianides, N. Tsoreas and A. A. Daopoulos, J. Organomet. Chem., 2005, 690, 5948 CrossRef; (h) X. Liu and W. Chen, Dalton Trans., 2011, 41(2), 599 RSC; (i) O. Bárta, P. Pinter, I. Císařová, T. Strassner and P. Štěpnička, Eur. J. Inorg. Chem., 2020, 575–580 CrossRef; (j) A. L. Ostericher, K. M. Waldie and C. P. Kubiak, ACS Catal., 2018, 8(10), 9596 CrossRef; (k) M. V. Baker, S. K. Brayshaw, B. W. Skelton, A. H. White and C. C. Williams, J. Organomet. Chem., 2005, 690(9), 2312 CrossRef.
- (a) S. Gonell, E. A. Assaf, J. Lloret-Fillol and A. J. M. Miller, ACS Catal., 2021, 11(24), 15212 CrossRef; (b) Y. Liu, K. S. Kjær, L. A. Fredin, P. Chábera and T. Harlang, et al. , Chem. – Eur. J., 2015, 21(9), 3628 CrossRef; (c) K. Witas, S. S. Nair, T. Maisuradze, L. Zedler, H. Schmidt and P. Garcia-Porta, et al. , J. Am. Chem. Soc., 2024, 146(29), 19710 CrossRef.
- S. Meyer, C. M. Orben, S. Demeshko, S. Dechert and F. Meyer, Organometallics, 2011, 30(24), 6692 CrossRef.
- J. Rieb, A. Raba, S. Haslinger, M. Kaspar, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2014, 53, 9598 CrossRef PubMed.
- S. Komiya, M. Akita, A. Yoza, N. Kasuga, A. Fukuoka and Y. Kai, J. Chem. Soc. Chem. Commun., 1993, 9, 787 RSC.
- A. D. Piascik, P. J. Hill, A. D. Crawford, L. R. Doyle, J. C. Green and A. E. Ashley, Chem. Commun., 2017, 53, 7657 RSC.
- (a) Y. Ohki, K. Munakata, Y. Matsuoka, R. Hara, M. Kachi, K. Uchida, M. Tada, R. E. Cramer, W. M. C. Sameera and T. Takayama, et al. , Nature, 2022, 607, 86–90 CrossRef PubMed; (b) M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118 CrossRef PubMed; (c) Y. Lee, N. P. Mankad and J. C. Peters, Nat. Chem., 2010, 2, 558 CrossRef PubMed; (d) J. Rittle and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15898 CrossRef PubMed.
- J. L. Crossland and D. R. Tyler, Coord. Chem. Rev., 2010, 254, 1883 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2368195, 2390974 and 2394085. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05463f |
| ‡ For N2, we use the term “activation” to refer to a lower N–N bond order, assessed structurally, or by IR spectroscopy (νN2).16 |
| § A notable exception is the ligand DMeOPrPE.5c |
| ¶ Formate (HCOO−) is present in the ESI-MS eluent solution. |
| This journal is © The Royal Society of Chemistry 2024 |