
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct C–H amidation of 1,3-azoles: light-mediated, photosensitiser-free vs. thermal†
David T.
Mooney
a,
Heather
McKee
a,
Tabea S.
Batch
a,
Samuel
Drane
b,
Peter R.
Moore
b and
Ai-Lan
Lee
 *a
*a
aInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: A.Lee@hw.ac.uk
bEarly Chemical Development, Pharmaceutical Sciences, R&D BioPharmaceuticals, AstraZeneca, Macclesfield SK10 2NA, UK
First published on 19th August 2024
Abstract
We have developed one thermal and one light-mediated method for direct Minisci-type C–H amidation of 1,3-azoles, which are applicable to thiazoles, benzothiazoles, benzimidazoles, and for the first time, imidazoles. The new visible light-mediated approach can be rendered photosensitiser/photocatalyst-free and likely proceeds via an electron donor–acceptor (EDA) complex, the first direct Minisci-type amidation to do so.
1,3-Azoles are important scaffolds in medicinal chemistry.1 For example, thiazoles, imidazoles and benzimidazoles are included in the top 25 most frequent N-heterocycles in FDA approved drugs,1,2 while benzothiazoles have emerged as an important scaffold in drug design.1 Efficient routes for late-stage C–H functionalisation of these 1,3-azoles would therefore be highly desirable. The Minisci reaction3 could potentially be a powerful method, but 1,3-azoles are more electron-rich and thus more challenging substrates compared to more typically studied electron-poor 6-membered N-heterocycles such as quinolines, pyrazines and pyridines, leading to far fewer reports on 1,3-azoles.3 Azole drugs are notably absent from a recent review of drug molecules that can undergo Minisci-type reactions.4
In particular, the ability to directly C–H amidate 1,3-azoles would be of interest, given the diverse activities of azole-2-carboxamides (e.g.Fig. 1), as well as the paucity of direct methodologies.5 Nevertheless, there are currently only two Minisci-type carboxyamidation studies that are focussed on 1,3-azoles (Scheme 1).6 There are also no reported Minisci amidations on imidazoles, a particularly challenging substrate, despite their prevalence in medicinal chemistry.
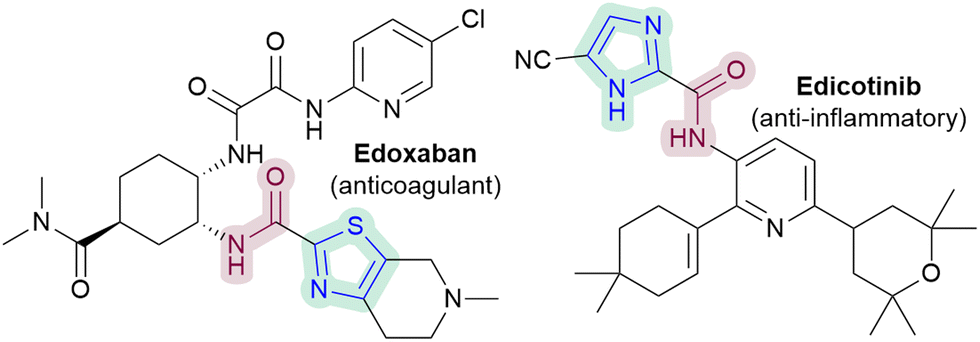 | ||
| Fig. 1 Examples of biologically active 1,3-azole-2-carboxamides. | ||
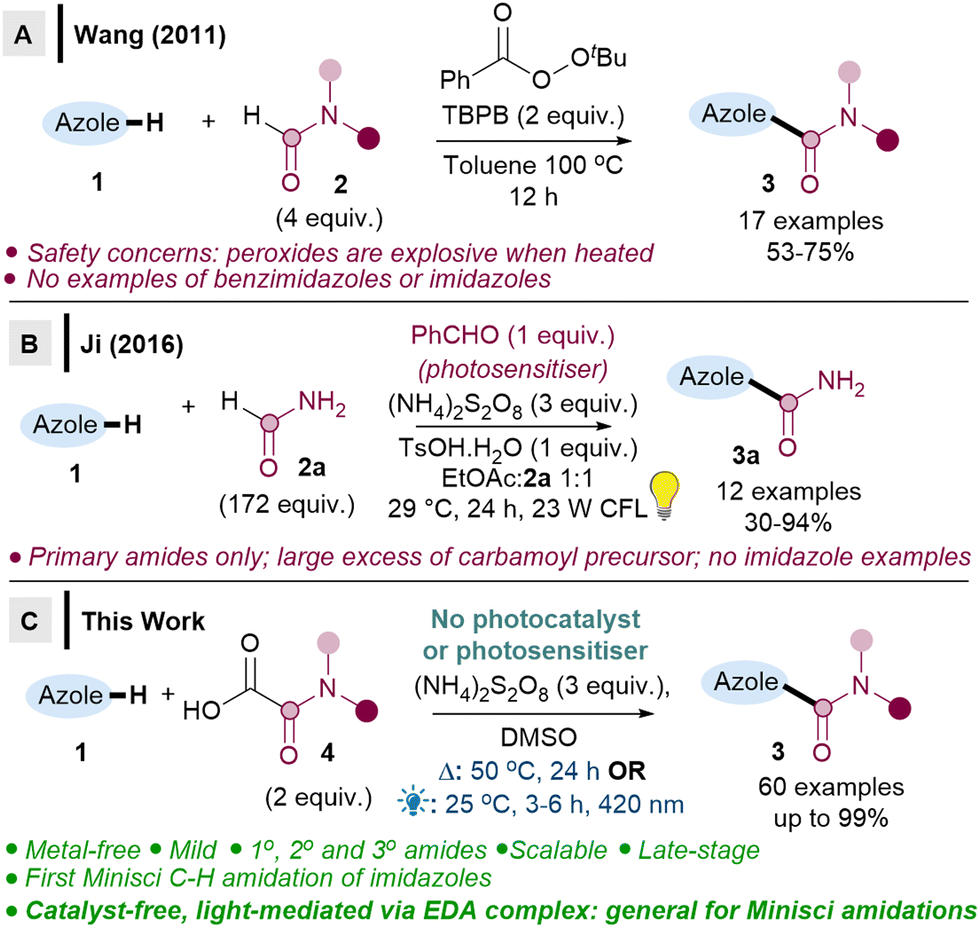 | ||
| Scheme 1 Direct C–H amidations of 1,3-azoles. | ||
In Wang's seminal work, TBPB (flash point 93 °C) is heated at 100 °C, which may have safety implications (Scheme 1A),6a and may also be problematic for late-stage functionalisations. Ji revealed a milder method using a stoichiometric photosensitiser (Scheme 1B),6b but the reaction is limited to primary amidations, and radical precursor 2a is used in large excess (172 equiv.). A handful of other studies have included only 2–3 benzimidazoles or benzothiazoles as part of their wider substrate scope studies, with low to moderate yields (0–59%).7
It is therefore clear that major limitations exist, including: (i) no successful Minisci amidations on imidazoles, (ii) issues with either safety or limited substrate scope, (iii) no late-stage functionalisations. We herein report two metal-free methodologies (one thermal, one light-mediated via EDA) that address all the aforementioned limitations (Scheme 1C). Crucially, we show that imidazoles can be C–H amidated for the first time via a Minisci reaction, along with benzimidazoles and (benzo)thiazoles, the 1,3-azoles which are the most important scaffolds in drug design.1,2 We also demonstrate the first photosensitiser-free, light-mediated direct Minisci amidation, which occurs via EDA complexes.8
We based our initial studies on our persulfate-mediated metal and light-free methodology9 (conditions A) using oxamic acid104a as radical precursor (Table 1). Upon full optimisation studies (see ESI†), it became apparent that concentration was the most crucial variable for achieving good yields (e.g. entries 1–3) and that different optimal concentrations were required for each azole class (e.g. entries 2 vs. 4). The reaction under thermal conditions A occurs just as well in the dark (entry 5), confirming it is not mediated by ambient light.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Cond. | Deviation/notes | Yield (%) | |
| a Yields determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. Conditions B were carried out in a Penn PhD M2 photoreactor at 100% light intensity. Non-anhydrous DMSO used. See ESI for full optimisation studies. b 0.35 M. c No reaction. Ir cat. = Ir(dF(CF3)ppy)2(dtbpy)PF6; Ad = 1-adamantyl. | |||||

|
1 | 1a | A | 0.4 M | 50 |
| 2 | 1a | A | 0.5 M | 70 | |
| 3 | 1a | A | 0.6 M | 15 | |
| 4 | 1b | A | 0.3 M | 82 | |
| 5 | 1b | A | 0.3 M in the dark | 84 | |

|
6 | 1b | —b | Ir cat. 1.5 mol%, 450 nm, 24 h | 85 |
| 7 | 1b | —b | [Mes-Acr]+[ClO4]− 1 mol%, 450 nm, 24 h | 70 | |
| 8 | 1b | —b | 4CzIPN 1 mol% 450 nm, 24 h | 41 | |
| 9 | 1b | Bb | No cat., 450 nm, 24 h | 85 | |
| 10 | 1b | B | None (420 nm, no cat. 3 h) | 85 | |
| 11 | 1b | Bb | In the dark, 3 h | —c | |
| 12 | 1b | Bb | No persulfate, 3 h | —c | |
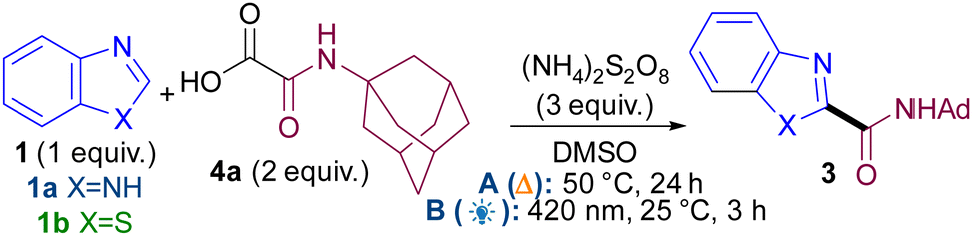
Notably, a second, novel light-mediated reaction was developed (conditions B, see ESI†), in a bid for a milder, room temperature reaction. Initially, various photocatalysts were evaluated (entries 6–8) as they were previously thought to be necessary,7a,b,9a,b,d,11 but to our surprise, the reaction worked well in the absence of photocatalyst (entry 10). In fact, the new photocatalyst-free reaction is also more efficient than thermal conditions A (3 h vs. 24 h). Control reactions confirm that both light (entry 11) and persulfate (entry 12) are necessary under conditions B.
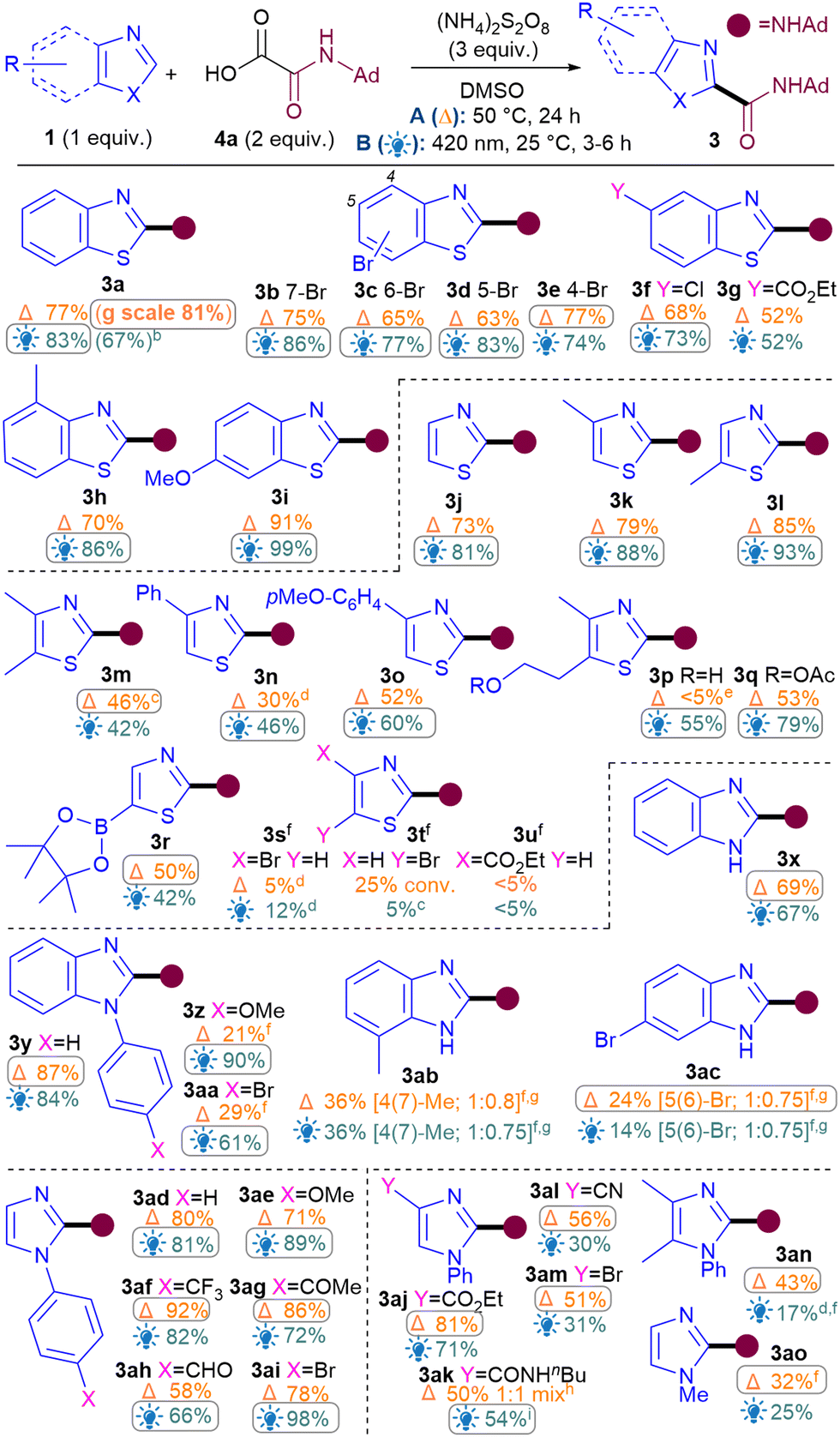
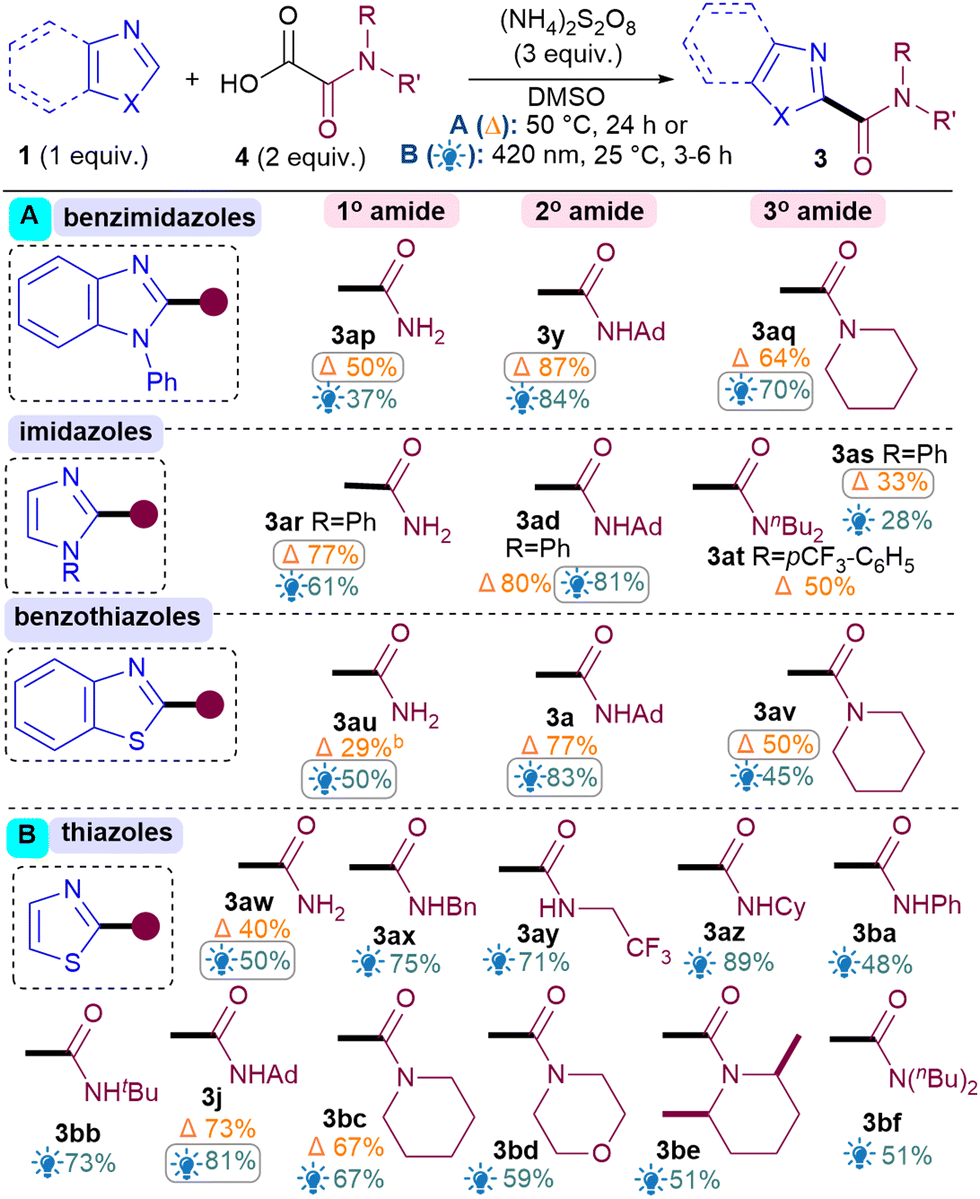
Next, the benzothiazole scope was investigated using both conditions (Table 2). Pleasingly, Br substitutions at 4-,5-,6- and 7-positions were tolerated well under both conditions (3b–3e) as are Cl (3f) and ester groups (3g). Benzothiazoles with electron-donating substituents (3h–i) generally performed better than electron-withdrawing ones (3b–g). A variety of thiazoles also reacted well (3j–3r). Notably, the yields with 3p and 3q exemplify that the milder light-mediated Conditions B can be more functional group tolerant (3p A: <5%; B: 55%). Unlike benzothiazoles, electron-withdrawing substituent on thiazoles caused poor reactivity (3s–3u) but boronic ester was pleasingly tolerated (3r). The reaction was successfully scaled up in batch under both conditions but thermal conditions A (3a, 1 g scale of 1, 81%) performed better than light-mediated conditions B (3a, 5.25 mmol scale of 1, 67%), as expected.12
| a Isolated yields unless otherwise stated. b 5.25 mmol scale of 1 (maximum scale in photoreactor). c 70 °C. d By 1H NMR analysis using 1,3,5-trimethoxybenzene as internal standard. e Complex mixture. f Low yields due to poor conversions. g Major:minor ratio. h Mixture of 3ak and diamidation (3ak′) product. i 1 h. |
|---|
|
Next, the benzimidazole and imidazole scope was investigated (Table 2). Both benzimidazole (3x) and N-Ph-benzimidazoles (3y-aa) performed well. Substituents on the benzimidazole core produced isomers, as expected due to tautomerisation,13 although the conversions were also hampered (3ab–3ac). Pleasingly, phenyl imidazole underwent amidation with good yields (3ad) and various electron-donating (3ae, 3an) as well as withdrawing substituents (3af–am) were tolerated, although an sp3C substituent on the N causes the conversion to drop (3ao).14
For the oxamic acid scope studies, we initially wanted to ascertain whether primary, secondary and tertiary amides are all tolerated for each class of 1,3-azole investigated (Table 3A). For installing primary amides, Conditions A were better for (benz)imidazole (3ap, 3ar) cores while the light-mediated conditions B were better for (benzo)thiazoles (3au, 3aw). Secondary amides were all installed smoothly using both conditions (3y, 3ad, 3a, 3j). Tertiary amides were sluggish with imidazoles (3as, 3at) but installed well on the others (3aq, 3av, 3bc). A more comprehensive oxamic acid scope was also carried out with thiazoles (Table 3B). Conditions B were used in most cases here since they were generally found to produce higher yields with thiazoles (Table 2). Gratifyingly, primary (3aw), secondary (3ax–bb, 3j) and tertiary amides were all successfully installed, including cyclic (3bc–be) and acyclic ones (3bf).
Next, we set out to ascertain why conditions B can occur in the absence of any photocatalyst and a potential explanation is the formation of EDA complexes.15 While EDA complexes have been exploited in Minisci alkylations16 and acylations,17 they have typically involved redox active esters16 or α-keto acids17 as acceptors. There are no examples of EDA induced, light-mediated direct Minisci amidations. UV-vis absorption studies show that apart from 1b, other 1,3-azoles 1 investigated do not absorb at 420 nm and only showed increased absorption, bathochromic shift and colour change for 1 + persulfate (EDA-A), indicating that any general mechanism for 1,3-azoles likely proceeds via EDA-A (Fig. 2 and ESI†). Job's plot analyses18 show maximum absorptions that are not at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (see ESI†), implying that the mechanism does not proceed via a SET from donor to acceptor of the EDA.19 Instead, since the trends in yields are generally similar for both conditions A and B, we propose that the reaction goes via a similar general pathway but differ in the generation of SO4˙− II (Scheme 2). Under conditions A, thermolysis breaks down S2O82− to II.9,20 Photodecomposition of S2O82− requires UV light, but under conditions B, excitation of EDA-A allows for homolytic decomposition of S2O82− at 420 nm.17,21 SET between II and I, followed by decarboxylation22 gives III,23 which then adds to the protonated heterocycle to form IV. H abstraction by II forms product 3. The average quantum yield (Φ) under conditions B is 0.014 (std. dev. 0.0017), implying that a radical chain propagation mechanism does not occur.24 Crucially, we show that photocatalyst-free light-mediated conditions B can be more generally applied to other N-heterocycles beyond azoles, including, phenanthrolines, xanthines, phenanthridines, (iso)quinolines and quinazolines (see ESI†).
:1 ratio (see ESI†), implying that the mechanism does not proceed via a SET from donor to acceptor of the EDA.19 Instead, since the trends in yields are generally similar for both conditions A and B, we propose that the reaction goes via a similar general pathway but differ in the generation of SO4˙− II (Scheme 2). Under conditions A, thermolysis breaks down S2O82− to II.9,20 Photodecomposition of S2O82− requires UV light, but under conditions B, excitation of EDA-A allows for homolytic decomposition of S2O82− at 420 nm.17,21 SET between II and I, followed by decarboxylation22 gives III,23 which then adds to the protonated heterocycle to form IV. H abstraction by II forms product 3. The average quantum yield (Φ) under conditions B is 0.014 (std. dev. 0.0017), implying that a radical chain propagation mechanism does not occur.24 Crucially, we show that photocatalyst-free light-mediated conditions B can be more generally applied to other N-heterocycles beyond azoles, including, phenanthrolines, xanthines, phenanthridines, (iso)quinolines and quinazolines (see ESI†).
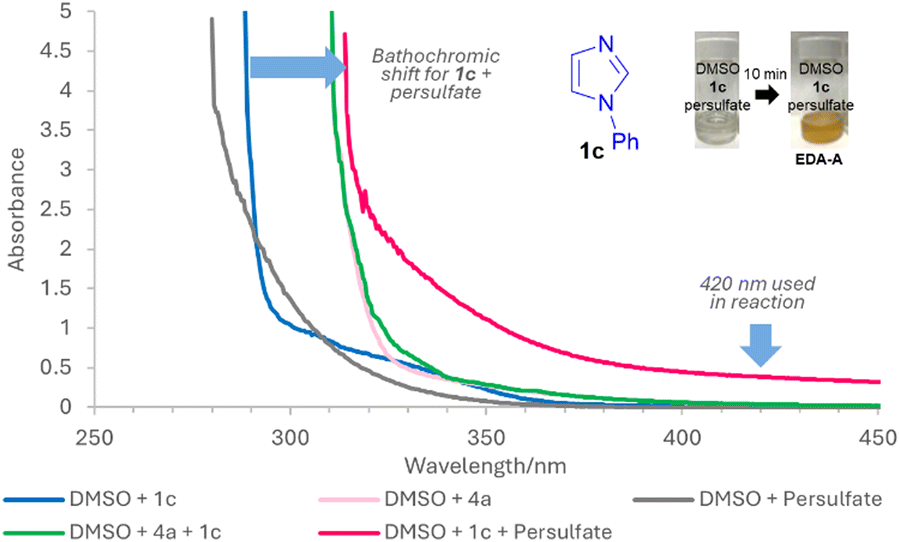 | ||
| Fig. 2 UV-vis absorption studies for phenylimidazole 1c. | ||
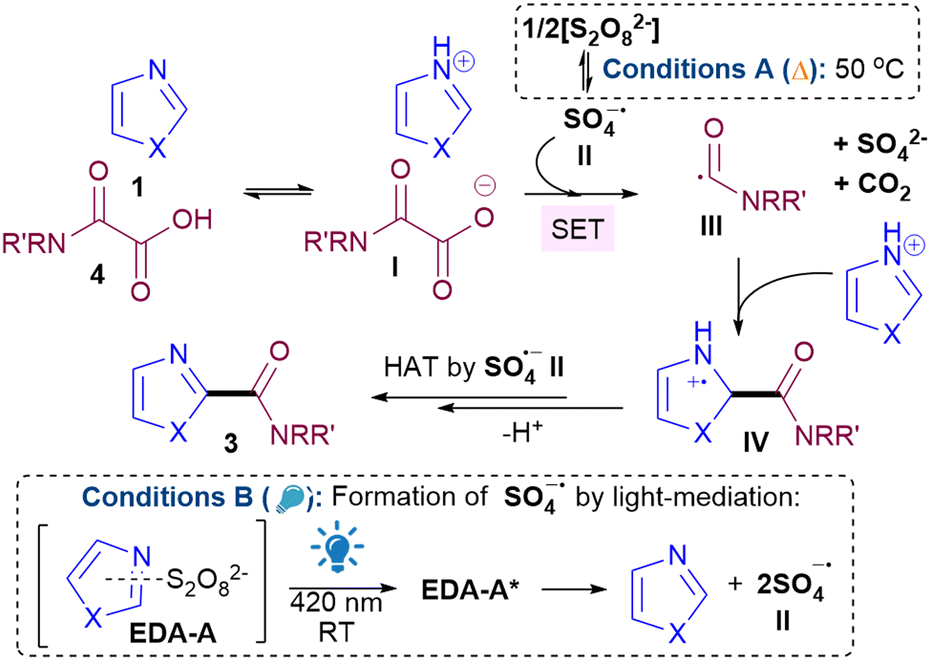 | ||
| Scheme 2 Proposed mechanisms. | ||
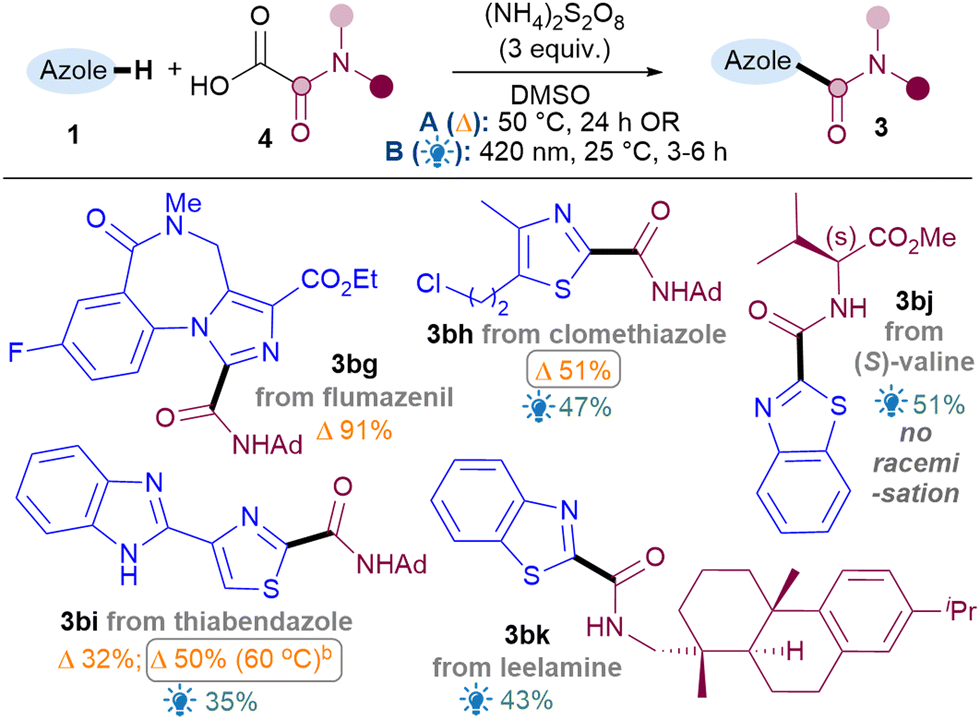
To showcase the application of the methodology, late-stage C–H functionalisations of 1,3-azole drug molecules as well as installation of more complex amides were investigated, initially using the best general conditions ascertained for each class (Table 4). To our delight, amidation of flumazenil occurred in excellent yield (3bg, 91%). Thiazoles clomethiazole (3bh, 51%) and thiabendazole (3bi, 50%) were also amidated smoothly. As expected, thiabendazole initially worked better under conditions B, but the advantage of the thermal reaction is that it could be pushed to 50% 3bi using more forcing conditions. Good functional group tolerance was observed for the chloroalkyl (3bh) and benzimidazole (3bi). More complex amides were also successfully applied, such as amino acid valine (3bj, 51%) and natural product leelamine (3bk, 43%).
In conclusion, we have developed direct Minisci-type C–H amidations of 1,3-azoles, which are amenable to late-stage C–H functionalisations. A wide substrate scope of amides can be directly installed, providing a significant advance on previous methods and facilitating rapid access to a wide range of amidated 1,3-azoles. We also reveal the first photocatalyst-free, light-mediated direct decarboxylative Minisci amidation which occurs via EDA complexes, allowing for more efficient, cheaper and more atom economical C–H amidation reactions.
We thank the Engineering and Physical Sciences Research Council and AstraZeneca for financial support [Industrial CASE PhD studentship to DTM; grant code: EP/V519522/1].
Data availability
Data available at https://doi.org/10.17861/4616a665-9166-4a45-8541-c643e58d7054 and in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Shearer, et al., J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- E. Vitaku, et al., J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS.
- X. Zhang, et al., Green Chem., 2024, 26, 3595–3626 RSC.
- There is only one non-Minisci pathway, using Ir4(CO)12 with isocyanates and silanes at 110 °C: Y. Fukumoto, et al., Synthesis, 2021, 3011–3018 CrossRef CAS.
- (a) T. He, et al., Chem. Commun., 2011, 47, 8946–8948 RSC; (b) Y. Zhang, et al., Chem. Sci., 2016, 7, 2111–2118 RSC.
- (a) X.-L. Lai, et al., Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS; (b) A. H. Jatoi, et al., Chem. Commun., 2019, 55, 466–469 RSC; (c) Z. Zhou, et al., Org. Biomol. Chem., 2021, 19, 2917–2922 RSC; (d) I. Sokolovs and E. Suna, Org. Lett., 2023, 25, 2047–2052 CrossRef CAS; (e) V. S. Bhat and A. Lee, Eur. J. Org. Chem., 2021, 3382–3385 CrossRef CAS.
- Previous reports using 1,4-dihydropyridines and activated N-amidopyridinium salts to form the EDAs are indirect as well as non atom economical: (a) I. Kim, et al., Org. Lett., 2020, 22, 8730–8734 CrossRef CAS PubMed ; for review, see: ; (b) B. T. Matsuo, et al., Chem. Commun., 2022, 58, 8322–8339 RSC.
- (a) D. T. Mooney, et al., J. Org. Chem., 2021, 86, 17282–17293 CrossRef CAS; (b) D. T. Mooney, et al., Org. Lett., 2022, 24, 8008–8013 CrossRef CAS; (c) D. R. Sutherland, et al., Org. Lett., 2018, 20, 6863–6867 CrossRef CAS PubMed; (d) M. T. Westwood, et al., Org. Lett., 2019, 21, 7119–7123 CrossRef CAS.
- I. M. Ogbu, et al., Chem. Commun., 2022, 58, 7593–7607 RSC.
- (a) M. Jouffroy and J. Kong, Chem. – Eur. J., 2019, 25, 2217–2221 CrossRef CAS PubMed; (b) J.-W. Yuan, et al., Org. Biomol. Chem., 2020, 18, 2747–2757 RSC.
- D. Cambié, et al., Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley-Blackwell, Chichester, England, 5 edn, 2010 Search PubMed.
- Unsubstituted imidazole (with no substituent on N) is not a suitable substrate and no product was observed.
- (a) G. E. M. Crisenza, et al., J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS; (b) Z. Yang, et al., Beilstein J. Org. Chem., 2021, 17, 771–799 CrossRef CAS PubMed.
- (a) M. Sharique, et al., Chem. Sci., 2022, 13, 5701–5706 RSC; (b) J. Li, et al., Adv. Synth. Catal., 2022, 364, 802–810 CrossRef CAS; (c) E. de Pedro Beato, et al., J. Am. Chem. Soc., 2021, 143, 12304–12314 CrossRef CAS PubMed; (d) M.-C. Fu, et al., Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (e) J. Li, et al., Adv. Synth. Catal., 2022, 364, 802–810 CrossRef CAS.
- L. Guillemard, et al., Adv. Synth. Catal., 2018, 360, 4184–4190 CrossRef CAS.
- P. MacCarthy, Anal. Chem., 1978, 50, 2165 CrossRef CAS.
- (a) W. Guo, et al., J. Org. Chem., 2019, 84, 14168–14178 CrossRef CAS PubMed; (b) Y. Liu, et al., J. Org. Chem., 2023, 88, 12474–12480 CrossRef CAS.
- (a) E. B. McLean, et al., Org. Lett., 2022, 24, 686–691 CrossRef CAS; (b) D. M. Kitcatt, et al., Chem. Sci., 2023, 14, 9806–9813 RSC.
- (a) S. Saha and A. K. Bagdi, Org. Biomol. Chem., 2022, 20, 3249–3262 RSC; (b) S. Devari and B. A. Shah, Chem. Commun., 2016, 52, 1490–1493 RSC; (c) Y. Xiong, et al., Asian J. Org. Chem., 2020, 9, 292–295 CrossRef CAS.
- J. D. Griffin, et al., J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS.
- (a) C. Raviola, et al., Green Chem., 2019, 21, 748–764 RSC; (b) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full optimisation studies, mechanistic studies, characterisation data and copies of NMR spectra of new compounds. See DOI: https://doi.org/10.1039/d4cc02742f |
| This journal is © The Royal Society of Chemistry 2024 |