
Reprocessable, creep-resistant covalent adaptable networks synthesized using conventional free-radical polymerization conditions with piperidine-based and non-piperidine-based dynamic dialkylamino disulfide chemistry†
Mohammed A.
Bin Rusayyis
 a,
Logan M.
Fenimore‡
b,
Nathan S.
Purwanto‡
a and
John M.
Torkelson
*ab
a,
Logan M.
Fenimore‡
b,
Nathan S.
Purwanto‡
a and
John M.
Torkelson
*ab
aDept. of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: j-torkelson@northwestern.edu
bDept. of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 13th July 2023
Abstract
Conventional cross-linked polymers or thermosets cannot be melt-processed because of their permanent cross-links. Covalent adaptable networks (CANs) with covalent cross-links that are dynamic under a stimulus allow for reprocessing. Disulfide chemistry is widely used in CANs. In contrast to sulfur–sulfur bonds in dialkyl disulfides with a dynamic nature activated by internal or external catalysts, sulfur–sulfur bonds in dialkylamino disulfides have a lower bond dissociation energy, allowing for catalyst-free CANs. However, dialkylamino disulfides have only been applied in CANs synthesized under limited conditions. CANs made by free radical polymerization (FRP) of monomer with dialkylamino disulfide cross-linkers were previously synthesized at or near room temperature, highly unconventional FRP conditions, and the dialkylamino disulfides were based on substituted piperidine rings. Here, we show that these limitations are unnecessary to achieve robust CANs. With BiTEMPS methacrylate, a piperidine-based dialkylamino disulfide cross-linker, FRP of n-hexyl methacrylate with azo initiators leads to CANs with the same cross-link density and excellent reprocessability whether synthesized at room temperature or a conventional FRP temperature, 70 °C. We also synthesized a non-piperidine-based dialkylamino disulfide cross-linker, BiTEBES methacrylate. Using this cross-linker and FRP at 70 °C, we prepared catalyst-free poly(n-hexyl methacrylate) CANs that are reprocessable with full recovery of cross-link density. Their stress relaxation has a temperature dependence that is independent of cross-link density and an activation energy within uncertainty equal to the bond dissociation energy of the dialkylamino disulfide bond. The relaxation distribution breadth is a strong function of cross-link density. CANs made with 5 mol% BiTEBES methacrylate have very low cross-link density and exhibit single-exponential-decay stress relaxation. CANs made with 10 mol% BiTEBES methacrylate or 5 mol% BiTEMPS methacrylate and having factors of 5 to 9 higher cross-link density exhibit substantial relaxation distribution breadth. The BiTEBES-based CANs have excellent creep resistance at 70 °C, like that shown previously for BiTEMPS-based CANs, with creep strains of 0.75% or less after ∼14 h of 3.0 kPa stress. This work demonstrates the versatility of dialkylamino disulfide chemistry and its utility in developing catalyst-free CANs using conventional FRP conditions.
Introduction
Cross-linked polymers, known as thermosets, exhibit improved heat and solvent resistance, excellent dimensional stability, and generally better mechanical properties than analogous non-cross-linked, linear polymers due to their network structure. However, because of their permanent, covalent cross-links, conventionally cross-linked polymers cannot be melt-processed or reshaped into new forms and, therefore, cannot be recycled into high-value products at their end of life. This is why millions of tons per year of used rubber tires, which are cross-linked polymeric materials, are landfilled, incinerated, or lost in the environment.1 Thus, while effective recycling of thermoplastic polymers has, at best, been met with limited success,2–4 the inherent non-recyclability of thermosets presents major environmental and economic challenges.To address these challenges, researchers have recently employed dynamic covalent bonds as cross-links to produce recyclable thermosets. Polymers cross-linked with dynamic bonds are known as covalent adaptable networks (CANs)5,6 or dynamic covalent polymer networks (DCPNs).7 Because of the dynamic nature of their cross-links, CANs may undergo network reconfiguration, making them malleable and recyclable under appropriate conditions. Dynamic chemistries employed in CANs are classified as dissociative or associative. With dissociative dynamic chemistries, covalent bonds break and reform reversibly upon applying a stimulus (e.g., heat) and recombine upon stimulus removal. Dissociative chemistries include Diels–Alder reactions,8–12 hindered urea exchange,13–16 and alkoxyamine chemistry.17–24 With associative dynamic chemistries, covalent bonds rearrange by exchange reactions between functional groups. Examples of associative dynamic chemistry include transesterification25–27 and transamination.28–30 When CANs have exclusively associative dynamic covalent chemistry, they are sometimes called vitrimers.31,32 Some CANs, such as those made from polyurethanes, polyhydroxyurethanes, and polythiourethanes, exhibit both associative and dissociative dynamic chemistries.33–40
Disulfide chemistry is one of the most widely used dynamic chemistries reported in the CANs literature.41–55 The great interest in disulfide chemistry stems in part from the application of sulfur chemistry in the vulcanization of rubber materials used in transportation.31 Although disulfide chemistry has been studied extensively, the dynamic mechanism of sulfur–sulfur bonds in disulfides is complex and incompletely understood. For instance, the dynamic mechanism in disulfides has been reported to depend on the use conditions and substitution patterns of the disulfides.31,56 Traditionally, disulfide chemistry has been characterized as dynamic associative chemistry. However, while an associative exchange pathway is possible via addition/elimination substitution with free thiols,41,44,57 dissociative pathways, including the reduction of the disulfides into thiols that can oxidize again to form disulfides, have also been reported.58–60 As such, disulfide dynamic chemistry is often classified as either dissociative or associative.56 Several studies have indicated that the dynamic disulfide mechanism is based on radical-mediated exchange reactions.42,61–64 The latter point is supported by a recent report which shows that, upon heating, sulfur–sulfur linkages in disulfides generally undergo homolytic dissociation into thiyl radicals that are capable of exchanging with other sulfur-based radicals (Scheme 1).65
 | ||
| Scheme 1 Radical-mediated dynamic exchange mechanism in disulfides. | ||
Although disulfide chemistry is viewed as readily dynamic, the dynamic nature of dialkyl disulfide bonds (RS-SR) often needs to be activated with external or internal catalysts.56,66,67 This is due to the disulfide bonds having a relatively high bond dissociation energy (BDE = 250–300 kJ mol−1).68 As a result, in the absence of catalyst, polymer networks cross-linked with dialkyl disulfide bonds exhibit behavior similar to permanently cross-linked networks as manifested in their inability to relax stress at elevated temperature.69 We also tried to process poly(n-hexyl methacrylate) networks cross-linked with dialkyl disulfide bonds but could not obtain a robust film with good optical properties, consistent with the relatively poor dynamic character of dialkyl disulfide bonds even at higher temperatures.
Interestingly, sulfur–sulfur linkages in dialkylamino disulfides (RNS-SNR) have a reported bond dissociation energy (BDE) of 110–130 kJ mol−1,67,70,71 much lower than the dialkyl disulfide BDE. This significant difference is attributed to the stabilization of the generated thiyl radicals by the lone electron pair on the attached nitrogen atoms.70,72 Because of their relatively low BDE, dialkylamino disulfides are good candidates for dynamic covalent cross-links that can be employed in syntheses of CANs. Nevertheless, the dynamic nature of dialkylamino disulfides remains relatively untapped in the polymer field. Only nine studies have reported CANs based on dialkylamino disulfide cross-links. Notably, the networks reported in these studies were cross-linked with dialkylamino disulfides based on substituted five-membered piperidine rings, with a focus on bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS), the sulfur analog of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) nitroxide radical (see Fig. 1), or a modified version of BiTEMPS.69,72–79 With two exceptions,76,77 including one in which BiTEMPS methacrylate was used with radical-based reactive processing at 160 °C to create polyethylene CANs (which exhibited complete cross-link density recovery after several reprocessing steps),77 the other seven studies made CANs by free-radical polymerization (FRP) of monomer with a BiTEMPS-based cross-linker at unusually low temperatures of room temperature to 30 °C and a room-temperature-activated initiator called V-70.69,72–75,78,79 The unconventional conditions and initiator were selected, at least in part, to minimize the possible participation of disulfide or thiyl radicals in FRP with BiTEMPS-based cross-linkers.69,72–75,78,79 Unfortunately, the unconventional FRP conditions reduce the likelihood of BiTEMPS-based dynamic covalent cross-linkers being adopted for commercial production of CANs by FRP.
 | ||
| Fig. 1 Structures of BiTEMPS and BiTEBES used in this study. | ||
Some of the nine BiTEMPS studies were inspired by the early work of Bennett et al.80 in which the disulfide bond in BiTEMPS was shown to dissociate reversibly upon moderate heating into thiyl (or, specifically, TEMPS˙) radicals. However, given that the relatively low BDE of the disulfide bond in BiTEMPS is mainly attributed to the additional stabilization contributed by the attached nitrogen atom rather than the steric hindrance of the substituted, cyclic piperidine structure, it is reasonable to assume that the alkyl moieties do not primarily influence the BDE of sulfur–sulfur bonds in dialkylamino disulfides. The disulfide BDE of bis(diisopropylamino) disulfide, which is not piperidine-based, is reported to be identical within experimental uncertainty to the BDE of the disulfide bond in BiTEMPS.71 Thus, acyclic, non-piperidine-based dialkylamino disulfides have potential utility in catalyst-free CANs.
Here, we address two important issues regarding the use of dialkylamino disulfide cross-linkers in developing CANs. First, we demonstrate that robust CANs may be synthesized by FRP using a conventional FRP temperature, 70 °C,81 and conventional free-radical initiators, azobisisobutyronitrile (AIBN) and benzoyl peroxide (BPO).81 With AIBN, we used conventional FRP to produce robust CANs with the piperidine-based BiTEMPS methacrylate, resulting in the same cross-link densities within experimental uncertainty as those CANs previously produced using room-temperature FRP72,73 and exhibiting the same complete cross-link density recovery after multiple reprocessing steps. By obtaining the same cross-link density within experimental uncertainty and excellent reprocessability independent of FRP temperature, we show that any impact on cross-link density of our as-polymerized and recycled CANs resulting from the participation of BiTEMPS-based disulfide or thiyl radicals is immeasurably small. We further showed the stability of BiTEMPS at our polymerization conditions by small-molecule model studies. Second, we extend our approach to non-piperidine-based dialkylamino disulfide cross-linkers by synthesizing bis(tert-butyl-3-ethylamino methacrylate) disulfide (BiTEBES methacrylate). We show the utility of BiTEBES methacrylate as a dynamic cross-linker in the conventional FRP of catalyst-free, reprocessable networks that can be recycled multiple times with complete retention of cross-link density within experimental uncertainty. Based on the temperature dependence of their stress relaxation response, we show that the bond dissociation of the dialkylamino disulfides in the BiTEBES cross-links dominates the stress relaxation response in the BiTEBES-based CANs. We also show that poly(n-hexyl methacrylate) CANs cross-linked at very low levels with 5 mol% BiTEBES methacrylate exhibit single-exponential-decay stress relaxation response in the temperature range of 120–150 °C and excellent long-term creep resistance at 70 °C. When we increase the cross-link density in BiTEBES-based or BiTEMPS-based CANs by factors of 5 to 9, the stress relaxation exhibits a substantial breadth of relaxation times, and average stress relaxation times increase by as much as a factor of 3 to 3.5.
Experimental
Materials
All chemicals are commercially available and used as received unless otherwise stated. Petroleum ether (anhydrous), 2-(tert-butylamino)ethyl methacrylate (TBEM, 97%), 2,2,6,6-tetramethylpiperidine (99%), sulfur monochloride (98%), n-hexyl methacrylate (HMA, 98%), azobisisobutyronitrile (AIBN, 98%), Luperox A98 (benzoyl peroxide, BPO, 98%), sodium acetate (anhydrous, 99%), sodium sulfate (Na2SO4, anhydrous, 99.5%), N,N-dimethylacetamide (DMAc, anhydrous, 99.8%), N,N-dimethylformamide (DMF, anhydrous, 99.8%), hexane (mixture of isomers, 98.5%), acetonitrile (99.9%), toluene (99.9%), and chloroform-d (99.8 atom% D) were from Sigma-Aldrich. 2,2,6,6-Tetramethyl-4-piperidyl methacrylate (TMPM) was from TCI America. Dichloromethane (Certified ACS) and methanol (99.9%) were from Fisher. V-70 azo initiator was obtained from FUJIFILM Wako Chemicals. n-Hexyl methacrylate monomer was de-inhibited using inhibitor remover (Sigma Aldrich, 311340) in the presence of calcium hydride (Sigma Aldrich, 90%). AIBN was recrystallized from methanol. Petroleum ether, DMAc, and DMF were dried over 4 Å molecular sieves for at least 72 h before use.Synthesis of bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS methacrylate)
The synthesis followed the protocols described previously in ref. 72, 73, and 77.Synthesis of bis(tert-butyl-3-ethylamino methacrylate) disulfide (BiTEBES methacrylate)
2-(tert-Butylamino)ethyl methacrylate (9.81 g, 52.94 mmol) and pre-dried petroleum ether (55 mL) were placed in a beaker and stirred at room temperature for 15 min, after which the mixture was cooled in an acetonitrile/dry ice bath at −40 °C. Sulfur monochloride (1.81 g, 13.43 mmol) mixed with pre-dried petroleum ether (2 mL) was added dropwise to the cooled mixture with vigorous stirring. Once the sulfur monochloride solution was added, the reaction mixture was allowed to stir at −40 °C for 2 h, followed by stirring at room temperature for 30 min. The obtained wet white solid was dried under vacuum at 80 °C for 48 h to obtain the cross-linker as a cream-colored solid (3.75 g, 65%; melting point: 105 °C). 1H NMR (500 MHz, CDCl3) δ 6.28 (s, 2H), 5.63 (s, 2H), 4.65 (t, J = 6.1 Hz, 4H), 3.23 (t, J = 6.1 Hz, 4H), 1.96 (s, 6H), 1.50 (s, 18H). 13C NMR (126 MHz, CDCl3) δ 166.95, 135.40, 127.14, 59.85, 57.54, 40.62, 25.92, 18.34.Synthesis of 1,3-bis(2,2,6,6-tetramethylpiperidin-1-yl)trisulfane (BiTEMPS-SM)
This compound was synthesized using a modification of a reported procedure.76 Under nitrogen atmosphere, sodium acetate (3.0 g, 36 mmol) and 2,2,6,6-tetramethylpiperidine (5.0 g, 36 mmol) were dissolved in dry DMF (20 mL). The resulting solution was cooled to 0 °C, and sulfur monochloride (1.6 g, 12 mmol, distilled prior to use) was added dropwise. After this addition, the mixture was stirred for 1 h and poured into cold water. The crude precipitate was collected by vacuum filtration, dissolved in hexane, and washed with deionized water (3 × 50 mL) and saturated brine (3 × 50 mL). The organic layer was collected and dried with Na2SO4. The solvent was evaporated under reduced pressure to give a yellow oil. This crude product was then purified by silica gel column chromatography (petroleum ether, 100%, Rf = 0.5) to yield an off-white solid, BiTEMPS-SM (0.77 g, 26%). 1H NMR (500 MHz, CDCl3) δ 1.70 (m, 2H), 1.56 (ddd, J = 13.0, 6.4, 4.3 Hz, 4H), 1.52–1.44 (m, 6H), 1.42–1.36 (m, 20H), 1.15 (s, 4H) for BiTEMPS-SM. Hi-Res MS (ESI): m/z found [M − H+] for C18H36N2S3+ 377.05 (calcd 377.20).Small-molecule study
BiTEMPS-SM (48.5 mg, 0.129 mmol) and DMAc (0.5 mL) were each added to three 8 mL scintillation vials. V-70 (8.0 mg, 0.026 mmol) was added to vial 1, and AIBN (4.2 mg, 0.026 mmol) was added to vial 2; vial 3 was left as is. Vials 1 and 2 were degassed with nitrogen (N2) for 5 min. For 24 h, vial 1 was stirred at room temperature, and vials 2 and 3 were stirred at 70 °C. Subsequently, 20 μL of each vial were diluted in 20 mL of acetonitrile, and ESI-MS spectra were collected on each of the resulting solutions. These spectra were compared to an ESI-MS spectrum of vial 3 taken prior to the 24 h stirring to observe changes in the mass spectrum of pure BiTEMPS-SM (377.05 m/z). Each of these solutions were also separated by thin-layer chromatography (TLC, silica gel, petroleum ether, 100%), and resulting spots were identified under UV light exposure (254 nm) and KMnO4 staining. Spots were compared across vials relative to the TLC signature of pure BiTEMPS-SM.Electrospray ionization mass spectrometry (ESI-MS)
ESI-MS was performed using a Bruker Amazon SL equipped with a quadrupole ion trap mass analyzer. Samples were prepared to be ∼0.1 mg mL−1 in acetonitrile and were filtered through Whatman PTFE membrane filters with 0.2 μm pore sizes.Network syntheses
In a typical synthesis, HMA monomer, BiTEMPS methacrylate or BiTEBES methacrylate (used without further purification), and initiator (V-70, AIBN, or BPO) were added to a 20 mL scintillation vial. DMAc (∼1.2 mL DMAc per g of HMA) was added to facilitate dissolving the cross-linker, BiTEMPS methacrylate or BiTEBES methacrylate, in HMA. The cross-linker (BiTEMPS methacrylate or BiTEBES methacrylate) was added in either 5 mol% or 10 mol% concentration with respect to the total amount of monomer and cross-linker. The concentration of the initiator was 1.0 mol% with respect to the total amount of monomer and cross-linker. The solution was stirred at room temperature for 20 min. Once the cross-linker dissolved completely in the monomer, the solution was bubbled with N2 gas for 5 min. Samples containing AIBN and BPO were reacted at 70 °C, and samples containing V-70 were reacted at room temperature. N2 gas was allowed to flow continuously into the vials during polymerization. Gelation was achieved within 4 h, and the polymerization was allowed to continue overnight for complete conversion. The obtained network was cut into small pieces, washed with DCM/methanol mixtures, and then dried in a vacuum oven for 24 h at 60 °C for the BiTEBES-based network and 80 °C for the BiTEMPS-based network.Fourier transform infrared (FTIR) spectroscopy
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy was performed using a Bruker Tensor 37 FTIR spectrophotometer equipped with a diamond/ZnSe attachment. Sixteen scans were collected at room temperature over the 4000 to 600 cm−1 range at 4 cm−1 resolution.Nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectroscopy and proton-decoupled 13C NMR spectroscopy of the synthesized cross-linkers (BiTEMPS methacrylate and BiTEBES methacrylate) were done at room temperature using a Bruker Avance III 500 MHz NMR spectrometer. Deuterated chloroform (CDCl3) was used as a solvent, and the spectra were reported relative to tetramethylsilane.Molding and reprocessing of networks
After drying each synthesized network, it was cut into millimeter-sized pieces and processed using a PHI press (Model 0230C-X1). Unless otherwise noted, the network was molded into films (used for most characterizations) or discs (used for creep characterization) at 130 °C for 1.0 h using a 10-ton ram force.Differential scanning calorimetry (DSC)
The melting temperature (Tm) of the newly synthesized cross-linker BiTEBES methacrylate and the glass transition temperatures (Tgs) of as-synthesized and molded networks were obtained by DSC using a Mettler Toledo DSC822e. The peak Tm value of the cross-linker was determined from the endothermic peak of the first heating cycle (heating rate 10 °C min−1). To determine the network Tgs, samples were annealed at −50 °C for 5 min followed by heating to 80 °C at a heating rate of 10 °C min−1. The samples were then cooled again to −50 °C (cooling rate −10 °C min−1) and then heated to 80 °C at a rate of 10 °C min−1. The Tg values were obtained from the heating ramp of the second heating cycle using the 1/2 ΔCp method.Swelling
Swelling tests were performed at room temperature by placing the networks into 20 mL scintillation vials filled with toluene. The mixtures were homogenized thoroughly by vortex mixing and left to swell for 3 days. Swollen samples were dried in a vacuum oven at 80 °C for at least 48 h to obtain gel content. To confirm gel contents obtained by this method, a Soxhlet extraction was performed in toluene at 130 °C for 18 h on a single sample of BiTEMPS-based (5 mol%) PHMA network. A 1st-molded network piece was massed (∼15 mg) and placed into a Growing Labs cellulose Soxhlet extraction thimble. After extraction with boiling toluene, the insoluble network fraction was dried in a vacuum oven at 80 °C for 48 h. A gel content of 97% was obtained, which is within experimental uncertainty of the previously reported gel content for BiTEMPS-based (5 mol%) PHMA networks.72,73Thermogravimetric analysis (TGA)
Thermogravimetric analysis was done using a Mettler Toledo TGA/DSC3+. The change in sample weight was recorded as a function of temperature at a 20 °C min−1 heating rate from 25 °C to 500 °C under a nitrogen atmosphere.Dynamic mechanical analysis (DMA)
Thermomechanical properties of the molded networks were characterized using a TA Instruments RSA-G2 Solids Analyzer where the storage modulus (E′), the loss modulus (E′′), and the damping ratio (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of the networks were measured as functions of temperature under a nitrogen atmosphere. The instrument was operated in tension mode at a frequency of 1 Hz and 0.03% oscillatory strain. All data were obtained upon heating the rectangular specimens within the range −55 °C to 225 °C with a heating rate of 3 °C min−1. Three measurements were performed for most samples.
δ) of the networks were measured as functions of temperature under a nitrogen atmosphere. The instrument was operated in tension mode at a frequency of 1 Hz and 0.03% oscillatory strain. All data were obtained upon heating the rectangular specimens within the range −55 °C to 225 °C with a heating rate of 3 °C min−1. Three measurements were performed for most samples.
Stress relaxation
Stress relaxation of the 1st-mold network samples was characterized using a TA Instruments RSA-G2 Solids Analyzer. Rectangular specimens measuring ∼1 mm in thickness and ∼3 mm in width were mounted and allowed to equilibrate at the desired temperature for 10 min. Once thermal equilibrium was reached, each sample was subjected to an instantaneous 5% tensile strain, which was maintained throughout the test. The stress relaxation modulus was measured until it had relaxed to 10–30% of its initial value.Creep
Shear creep experiments at 3.0 kPa stress were performed on 1st-molded disk (∼2 mm-thick) samples using an Anton-Paar MCR 302 rheometer with 25 mm parallel-plate fixtures. Samples were equilibrated at the test temperature for 10 min before starting the experiment. Each test was carried out for 50000 s.
Results and discussion
Comparison of CANs made with BiTEMPS methacrylate by free radical polymerization at 70 °C and at room temperature
We and others have previously reported the synthesis of addition-type BiTEMPS-based networks, including poly(n-hexyl methacrylate), via FRP at or near room temperature using the low-temperature initiator, V-70.69,72–75,78,79 In those studies, elevated polymerization temperatures were avoided to minimize or prevent any potential issues associated with the possible generation of thiyl radicals via the dissociation of the disulfide bonds in BiTEMPS during the polymerization reaction.69,72–75,78,79 However, these concerns were not supported by any experimental work that shows specific interference of the thiyl radicals with the polymerization reaction. (Indeed, Fenimore et al.77 grafted BiTEMPS methacrylate to produce polyethylene CANs by reactive mixing at 160 °C and reprocessed those CANs at 160 °C, showing robust CAN formation with complete recovery of cross-link density after multiple reprocessing steps and demonstrating no measurable impact of thiyl radicals on CAN reprocessability.77) Also, given the high cost of the low-temperature V-70 initiator, the use of a conventional and cost-effective initiator would be desired for commercial-scale applications.Here, we have examined the synthesis of BiTEMPS-based CANs using a conventional FRP temperature, 70 °C, and a conventional free-radical initiator, AIBN. Specifically, we polymerized n-hexyl methacrylate (HMA) with 5 mol% and 10 mol% BiTEMPS methacrylate using DMAc as solvent. The polymerization mixtures gelled within 2 h, and the FRPs were allowed to continue overnight for completion. The obtained gels were broken into small pieces, washed with DCM/methanol mixtures, and dried in a vacuum oven to obtain BiTEMPS-based PHMA CANs. These CANs were insoluble in toluene, a good solvent for linear poly(n-hexyl methacrylate), confirming the cross-linked nature of the synthesized networks. Specifically, the CAN synthesized with 5 mol% BiTEMPS methacrylate had a Tg of 18 °C as characterized by DSC (Fig. S1†). This value is substantially higher than the Tg exhibited by linear PHMA (Tg = −6 °C),73 consistent with the cross-linked nature of the obtained CAN. Similar results were previously obtained for PHMA networks synthesized with 5 mol% BiTEMPS methacrylate at or near room temperature using V-70 initiator.69,72,73 Previous reports have shown that BiTEMPS exhibits extremely limited dissociation at 70–80 °C;69,72,76 thus, even if very small amounts of thiyl radicals are present during the FRP, the presence of these radicals does not prevent the polymerization reaction or the formation of a cross-linked polymer. We also note that we attempted to polymerize BiTEMPS methacrylate with HMA at 70 °C without the use of initiator. However, the mixture remained a liquid, and no recoverable solid was formed.
To investigate the impact of using AIBN and the 70 °C conventional FRP temperature on the processability of BiTEMPS-based networks, small pieces of these PHMA networks were hot pressed at 130 °C using a 10-ton ram force, which are the same processing conditions we employed previously for PHMA networks containing 5 mol% BiTEMPS methacrylate and polymerized at room temperature using V-70 initiator.72,73 After 1 h of hot pressing, a uniform, well-consolidated, and robust film was obtained (1st mold sample). The film was cut into small pieces and reprocessed using the same processing conditions to obtain the 2nd mold sample. The 2nd mold samples were also cut into small pieces and molded into a film (3rd mold sample) using the same processing conditions (see Fig. S2† for representative images of the reprocessed networks). 1st mold, 2nd mold, and 3rd mold samples were all observed to swell in toluene, confirming their cross-linked natures after processing.
The three molded samples were characterized by dynamic mechanical analysis (DMA). Fig. 2 shows the temperature (T)-dependent DMA properties, including E′ and damping ratio (tanδ), of reprocessed network samples. The E′ curves of all CANs display a quasi-rubbery plateau well above their Tgs, further confirming the cross-linked character of these dynamic covalent networks. In the quasi-rubbery plateau, there is a slight decrease with increasing T, consistent with the dissociative nature of the BiTEMPS-based dynamic covalent cross-linker.72,73,77 The tanδ peak T, often taken as a “shifted” Tg value,82 remained unchanged after multiple reprocessing steps, indicating that the properties of the CANs remain constant with multiple recycles. Importantly, similar to previous findings on PHMA networks synthesized with 5 mol% BiTEMPS methacrylate at room T using V-70 initiator,72,73 the three molded samples of the analogous PHMA network synthesized with AIBN showed identical E′ values (within experimental uncertainty) in the rubbery plateau region at 120 °C (1.72 ± 0.11 MPa, 1.87 ± 0.25 MPa, 1.81 ± 0.12 MPa for the 1st, 2nd and 3rd mold samples, respectively). In addition, the PHMA network synthesized with 10 mol% BiTEMPS methacrylate at 70 °C with AIBN exhibited an expectedly larger E′ value in the rubbery plateau region at 120 °C of 7.65 ± 0.15 MPa, a factor of 4.4 higher than that of the PHMA network synthesized with 5 mol% BiTEMPS methacrylate. The 2nd molded sample of this PHMA network with 10 mol% BiTEMPS methacrylate shows an identical E′ value within experimental uncertainty (7.57 ± 0.13 MPa) of its 1st molded sample (Fig. 2b). According to Flory's ideal rubber elasticity theory, which indicates that the rubbery plateau modulus is proportional to effective cross-link density,83 our results show the complete recovery of cross-link density, within uncertainty, after multiple recycling steps. Our results illustrate that the use of conventional FRP conditions with AIBN has no apparent adverse effect on being able to produce BiTEMPS-based PHMA CANs or the recovery of properties after reprocessing, highlighting the robustness of BiTEMPS chemistry and expanding the utility of BiTEMPS-based cross-linkers.
 | ||
| Fig. 2 Temperature-dependent dynamic mechanical responses as functions of molding steps for a PHMA CAN synthesized via FRP at 70 °C with (a) 5 mol% and (b) 10 mol% BiTEMPS methacrylate. | ||
Notably, the PHMA CANs made with 5 mol% BiTEMPS methacrylate by FRP at 70 °C with AIBN exhibit E′ values in the rubbery plateau region at 120 °C that are identical within experimental uncertainty to the values obtained for PHMA CANs made with 5 mol% BiTEMPS methacrylate by FRP at room T and with V-70 initiator.72 The room-T synthesized PHMA CANs had E′ values at 120 °C of 1.70 ± 0.16 MPa, 1.68 ± 0.10 MPa, 1.73 ± 0.04 MPa for the 1st, 2nd and 3rd mold samples, respectively. This excellent agreement in quasi-rubbery plateau E′ values and thus effective cross-link densities between the BiTEMPS-based CANs synthesized at room T and at 70 °C indicates that any impact of thiyl radicals generated during the 70 °C FRP with these initiators is so small as to leave both cross-link density and recovery of cross-link density after multiple reprocessing steps unaffected within experimental uncertainty.
We also synthesized a PHMA CAN with 5 mol% BiTEMPS methacrylate at 70 °C using 1 mol% of a peroxide initiator, BPO. The obtained network was sticky yet could be molded into a healed film using the same processing conditions employed for the networks synthesized with azo initiators. DMA characterization (Fig. S3†) revealed a rubbery plateau E′ value at 120 °C of 0.53 MPa, notably lower than the values of analogous networks synthesized with 5 mol% BiTEMPS methacrylate and azo initiators. The tanδ peak T of 4 °C is also lower than that of analogous PHMA networks. These results indicate that the use of BPO instead of AIBN as initiator leads to BiTEMPS-based PHMA networks with a lower cross-link density. It is possible that some oxidation of sulfur atoms in the BiTEMPS-based cross-linker by the peroxide initiator contribute to this lower cross-link density. Also, given that the half-life of BPO is 16.3 h at 70 °C,84 it is possible that some reduction of cross-link density results from a lesser completion of the polymerization, even after 24 h of reaction. Future studies are warranted to investigate the effects of peroxide initiators in conventional FRP of dialkylamino-disulfide-based CANs.
After demonstrating that thiyl radicals generated during the dissociation of BiTEMPS moieties play a negligible role in FRPs of monomer and the (re)processing of CANs, we further sought to demonstrate the stability of BiTEMPS at polymerization conditions using small-molecule studies. We first synthesized a BiTEMPS-based small molecule, BiTEMPS-SM, by reacting 2,2,6,6-tetramethylpiperidine with sulfur monochloride (S2Cl2) using a modified literature procedure (Fig. 3a and S4†).76 We isolated the trisulfide analogue of the BiTEMPS moiety, a common side product of reactions involving S2Cl2,75,77 by column chromatography for the purposes of this study. The reversible dissociation of BiTEMPS-SM generates both thiyl and dithiyl radicals,75 allowing for our investigation of the activity of both kinds of radicals at polymerization conditions. We made three solutions of BiTEMPS-SM in DMAc of equivalent concentration to that of BiTEMPS moieties during polymerizations of HMA in DMAc (0.26 mol L−1). We subjected these respective solutions (vials 1–3) to the following sets of conditions: room temperature with V-70 initiator (0.052 mol L−1), 70 °C with AIBN initiator (0.052 mol L−1), and 70 °C without initiator. Considering that we observe the gelation of PHMA CANs in all FRP cases in under 4 h, we quenched any activity in the vials after 4 h by exposing them to oxygen and diluting 20 μL of the contents in 20 mL of acetonitrile for mass spectrometry.
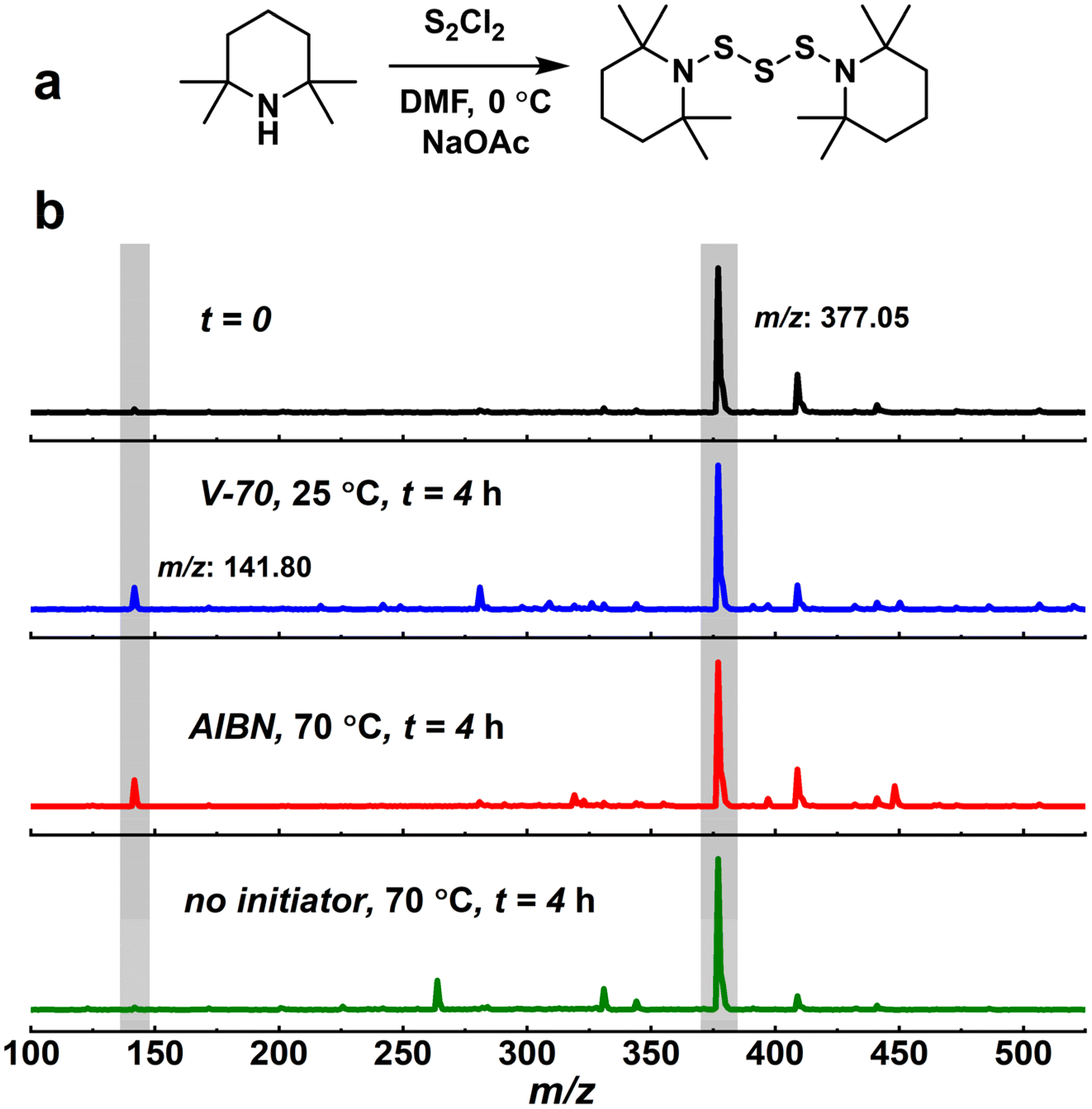 | ||
| Fig. 3 (a) Synthesis of BiTEMPS-SM. (b) ESI-MS spectra of (top to bottom) BiTEMPS-SM, BiTEMPS-SM with V-70 at 25 °C after 4 h (vial 1), BiTEMPS-SM with AIBN at 70 °C after 4 h (vial 2), and BiTEMPS-SM without initiator at 70 °C after 4 h (vial 3). | ||
We collected ESI-MS spectra of the vials after exposure to the emulated polymerization conditions and compared these spectra to an ESI-MS spectrum of BiTEMPS-SM prior to exposure to the prescribed conditions (Fig. 3b). Pure BiTEMPS-SM exhibits a peak in its ESI-MS spectrum at 377.05 m/z, which corresponds to BiTEMPS-SM with a proton (calcd 377.20 m/z). We observed little change in the ESI-MS spectra of vials 1–3 relative to that of pure BiTEMPS-SM; the 377.05 m/z peak remains strong in intensity in all cases. A new, low-intensity peak at 141.80 m/z emerged in the ESI-MS spectra of vials 1 and 2 that corresponds to the BiTEMPS-SM precursor, 2,2,6,6-tetramethylpiperidine (calcd 141.15 m/z). This suggests that BiTEMPS-SM may be sensitive in small amounts to both heat and azo radical exposure over long periods of time in the absence of polymerization sites like carbon–carbon double bonds. Further, in an effort to identify chemical changes to BiTEMPS-SM after exposure to these conditions, we separated the contents of the vials by thin-layer chromatography (TLC) on silica gel with 100% petroleum ether as the mobile phase (Fig. S5†). We observed no difference in the TLC signatures of the vials relative to pure BiTEMPS-SM. We also did not observe any other indication that reactions had occurred during the small-molecule studies such as color changes. Thus, with these investigations, we have further demonstrated the robustness and stability of the BiTEMPS moiety (capable of generating both thiyl and dithiyl radicals) after exposure to emulated FRP conditions beyond previously exhibiting their lack of activity in FRPs themselves.
Polymer networks made with BiTEBES methacrylate by FRP at 70 °C
The dynamic cross-linker bis(tert-butyl-3-ethylamino methacrylate) disulfide, or BiTEBES methacrylate (Fig. 4), was first synthesized by reacting 2-(tert-butylamino)ethyl methacrylate (TBEM), a commercially available yellowish liquid monomer, with sulfur monochloride (S2Cl2) at cold temperature (−40 °C) in the presence of petroleum ether as solvent (Fig. 4a). In this reaction, the nitrogen atoms of the amine groups in TBEM act as nucleophilic reagents that attack the sulfur atoms in S2Cl2. The reaction is completed in less than 3 h, after which a wet, white solid is obtained (Fig. S6a†). After drying under vacuum at 80 °C, we obtained a cream-colored solid (Fig. S6b†). Without further purification, the dried solid was characterized by FTIR spectroscopy (Fig. S7†). It is well known that secondary amines, such as TBEM, show a weak, often broad, N–H stretch peak in the 3300–3400 cm−1 range, consistent with our observation for the starting monomer, TBEM (Fig. S8†). However, in the same figure, the synthesized BiTEBES methacrylate does not show any peak in this range, indicating that a complete reaction within uncertainty between the amine groups in TBEM with S2Cl2 has been achieved successfully. We confirmed the identity of the obtained product by 1H NMR and 13C NMR spectroscopies (Fig. S9 and S10†). | ||
| Fig. 4 (a) Synthesis of the dynamic cross-linker, BiTEBES methacrylate. (b) Synthesis and (re)processing of dynamic BiTEBES-based PHMA networks. | ||
The synthesized cross-linker was also characterized by DSC. As shown in Fig. S11,† an endothermic peak is observed at 105 °C (1st heating cycle), corresponding to the melting of the cross-linker BiTEBES methacrylate. The cross-linker does not recrystallize upon cooling from the melt state as neither a crystallization transition was observed in the DSC thermogram upon cooling nor a melting transition was observed upon reheating (i.e., 2nd heating cycle) the sample again after cooling to room T. In addition to the melting peak, the 1st heating DSC curve shows an exothermic peak at 197 °C. This peak is likely associated with exothermic decomposition of the cross-linker and is consistent with thermogravimetric analysis as the sample shows a decomposition T (Td, 5% weight loss temperature) at 180 °C (Fig. S12†). The decomposition T is nearly 20 °C lower than for BiTEMPS methacrylate (Fig. S12†).
To investigate the utility of BiTEBES methacrylate as a dynamic cross-linker, we synthesized poly(n-hexyl methacrylate) networks with 5 mol% BiTEBES methacrylate by FRP at 70 °C using AIBN (Fig. 4b) and BPO as initiators as well as at room temperature using V-70 as initiator. We also synthesized a PHMA network with 10 mol% BiTEBES methacrylate by FRP at 70 °C using AIBN. Like BiTEMPS-based dynamic cross-linkers,69,72,73 we observed that BiTEBES methacrylate was insoluble in HMA. Thus, we added N,N-dimethylacetamide (DMAc), which facilitated the dissolution of BiTEBES methacrylate in HMA, to the reaction mixtures (Fig. S13†). It is important to note that the polymerizations were performed without any external or internal catalysts; hence, the networks are catalyst-free. The polymerization mixtures gelled within 2 h, and the FRPs proceeded overnight. The obtained transparent gels were washed with methanol/dichloromethane mixtures before being dried in a vacuum oven at 60 °C to remove solvent and unreacted materials. The dried gels swelled in toluene, a good solvent for linear PHMA, indicating that cross-linked materials were obtained. Specifically, the gel content values were 94 ± 1% and 97 ± 2% for the AIBN-initiated networks synthesized with 5 mol% (PHMA-5) and 10 mol% (PHMA-10) BiTEBES methacrylate, respectively. The network Tgs were 10 °C and 24 °C for PHMA-5 and PHMA-10, respectively, as measured by DSC. These temperatures are significantly higher than the Tg of linear PHMA (−6 °C).73 Cross-linked polymers tend to exhibit higher Tgs than their analogous linear polymers.85,86 We and others have reported similar observations for PHMA networks based on other dynamic cross-linkers.13,69,72,73
To test their processability, we broke the synthesized networks into small pieces and hot-pressed them at 130 °C for 1 h using a 10-ton ram force. As shown in Fig. 4b, the networks can be molded under these processing conditions into transparent films (thickness ~1 mm) and discs (thickness ~2 mm). Small pieces of the processed films were immersed and homogenized in 20 mL of toluene at room temperature for 3 days. The samples swelled in the solvent with no apparent disintegration, indicating that covalently cross-linked networks were reformed during processing.
We also performed dynamic mechanical analysis on PHMA-5 and PHMA-10 CANs. Fig. 5 shows the tensile storage modulus (E′) and damping ratio (tanδ = E′′/E′) curves of PHMA-5 and PHMA-10 networks as functions of T. As shown in Fig. 5a, E′ curves of both PHMA-5 and PHMA-10 CANs exhibit quasi-rubbery plateaus above ∼100 °C, further confirming the cross-linked nature of the processed networks. In the rubbery plateau regime, E′ values decrease slightly with increasing T, consistent with the dissociative nature of the CANs. Similar observations have been noted for BiTEMPS-based networks and other dissociative dynamic polymer networks.13,72,77 The PHMA-5 network exhibited a quasi-rubbery plateau E′ value at 120 °C of 0.19 ± 0.02 MPa. This value is similar to the E′ value at 120 °C of the V-70-initiated PHMA CAN synthesized with 5 mol% BiTEBES methacrylate (0.28 ± 0.02 MPa, Fig. S14†). It is worth noting that we were unable to characterize by DMA the BPO-initiated PHMA CAN synthesized with 5 mol% BiTEBES methacrylate, as the film was too sticky to be handled after compression molding. As with the BPO-initiated PHMA CAN with 5 mol% BiTEMPS methacrylate, this may be due to oxidation or the incomplete polymerization after 24 h (vide supra). However, pieces of the BPO-initiated BiTEBES-based network swelled in toluene, confirming its cross-linked nature.
 | ||
| Fig. 5 (a) Tensile storage modulus (E′) and (b) damping ratio (tanδ = E′′/E′) of BiTEBES-based PHMA-5 and PHMA-10 networks as functions of temperature. | ||
At 120 °C, well into the quasi-rubbery plateau region, the E′ value of the AIBN-initiated PHMA-10 network is 1.03 MPa, a factor of 5.4 higher than that of the PHMA-5 network. Thus, invoking Flory's ideal rubber elasticity theory,83 the PHMA-10 network has a factor of at least 5 greater effective cross-link density than PHMA-5. (The fact that the ratio of effective cross-link density exceeds the factor of 2 difference in the BiTEBES methacrylate level used to synthesize the networks arises because the fraction of cross-links that do not effectively percolate across a sample, and thus do not contribute to elastomeric response, increases with decreasing cross-linker level.) We observe that the cross-link density, and thus other associated properties, can be easily tuned by simply varying the concentration of BiTEBES methacrylate used to synthesize the network.
Dynamic polymer networks of a dissociative nature exhibit a gel-to-sol transition or terminal flow at high T facilitated by the dissociation of the dynamic cross-links. As shown in Fig. 5a, PHMA-5 and PHMA-10 CANs exhibit terminal flow as sufficient levels of the BiTEBES cross-links undergo dissociation with gel-to-sol transition temperature (Tgel-to-sol) values of ∼176 °C and ∼192 °C for PHMA-5 and PHMA-10, respectively. Similar to the results we reported earlier for BiTEMPS-based poly(n-hexyl methacrylate) networks,72 we see that the Tgel-to-sol value increases with increasing level of cross-linker concentration used in the CAN synthesis.
Fig. 5b depicts tanδ curves of PHMA-5 and PHMA-10 CANs as functions of T. Both networks exhibit somewhat broad tanδ peaks. The PHMA-5 network showed a broader peak than PHMA-10, consistent with a less homogenous distribution of cross-links. The PHMA-5 and PHMA-10 networks exhibit tanδ peaks at 41 °C and 57 °C, respectively. (The T at which tanδ is a maximum is sometimes considered to be a “shifted” Tg value.82) Importantly, PHMA-5 has a higher tanδ peak value than PHMA-10, slightly above 1.00, consistent with the more viscous or liquid-like behavior of PHMA-5 compared to the more heavily cross-linked PHMA-10 network, which is expected to show less liquid-like and more elastic behavior.
Given the terminal flow behavior of the PHMA-5 network at ∼176 °C, we undertook a test to process the as-synthesized PHMA-5 network at 180 °C for 5 min by hot pressing small pieces of the network under a 10-ton ram force. As shown in Fig. S15,† a uniform film can be obtained with only a 5 min processing time. To assess the degree of cross-linking of the processed film, we took a small piece of the film and immersed it in toluene at room temperature. After three days, we measured the gel content to be ∼44%, in contrast to the ∼94% gel content in the as-synthesized sample. The DMA response also confirmed this significant reduction in the gel content. Fig. S16† shows that the PHMA-5 film processed at 180 °C for 5 min does not exhibit a rubbery plateau that is present the case of the film processed at 130 °C for 1 h. These results indicate that processing conditions play a key role in determining the properties of molded samples of dynamic networks. Processing at 130 °C for 1 h achieves robust network properties whereas processing even for only 5 min at 180 °C, the BiTEBES methacrylate decomposition T as measured by TGA, does not.
To investigate the recyclability of the synthesized BiTEBES-based PHMA networks, we reprocessed the PHMA-5 networks twice by taking the 1st mold film sample processed at 130 °C for 1 h and cutting it into small pieces that were hot pressed at the same processing conditions to obtain the 2nd mold samples. The 2nd mold sample was then cut into small pieces and molded into the 3rd mold sample (Fig. S17†). The DSC-measured Tgs of the (re)processed networks were the same as that of the as-synthesized sample (Fig. S18†). Within experimental uncertainty, the (re)processed samples also had the same gel content as the as-synthesized PHMA-5 network (Table 1), indicating that (re)processing at these conditions had no adverse impact on the degree of cross-linking of these dynamic covalent networks. Fig. 6 shows the DMA response of 1st mold, 2nd mold, and 3rd mold PHMA-5 samples. Each molded sample displays a quasi-rubbery plateau in E′ at temperatures well above Tg, a characteristic of cross-linked polymer networks. As shown in Table 1, we observed no change in the tanδ peak T within experimental error after two recycling steps. Notably, within uncertainty, the three (re)processed PHMA-5 samples exhibited identical E′ values in the rubbery plateau region. We also reprocessed the V-70-initiated PHMA CAN synthesized with 5 mol% BiTEBES methacrylate into a 2nd mold (Fig. S19†); this network exhibited identical E′ values in its rubbery plateau region relative to its 1st mold (Fig. S14†). Thus, these results demonstrate full recovery of effective cross-link density, within uncertainty, of BiTEBES-based PHMA CANs after multiple recycling steps.
 | ||
| Fig. 6 Dynamic mechanical responses of BiTEBES-based PHMA-5 network as a function of molding steps. | ||
We also studied the stress relaxation behavior of PHMA-5 and PHMA-10 CANs at 120–150 °C under a constant tensile strain of 5%. As shown in Fig. 7, both networks undergo stress relaxation at elevated T, demonstrating the ability to relax external stress, an important feature of dynamic covalent networks. This stress relaxation, which is enabled by the dissociation of the dialkylamino disulfide bonds of the BiTEBES cross-links present in the networks, becomes faster with increasing T, consistent with greater dissociation of the dynamic cross-links at higher T. In the research literature associated with stress relaxation of CANs, the relaxation time has often been taken as the time when the stress (or modulus) relaxes to 1/e (∼37%) of its original value. However, this method of determining the relaxation time applies only when stress relaxation is known to follow the Maxwell model27,72,87 with a single-exponential decay. While this approach has been adopted in many analyses of stress relaxation in the CANs literature,15,28,49,51,52,54,56,87–90 it may not be applicable, as, to the best of our knowledge, all previous CANs specifically evaluated for breadth of relaxation response have exhibited responses consistent with a distribution of stress relaxation modes.6,13,35,56,72,79,87,91,92 To the best of our knowledge, there has been no explicit previous demonstration of single-exponential decay response in stress relaxation studies of CANs.
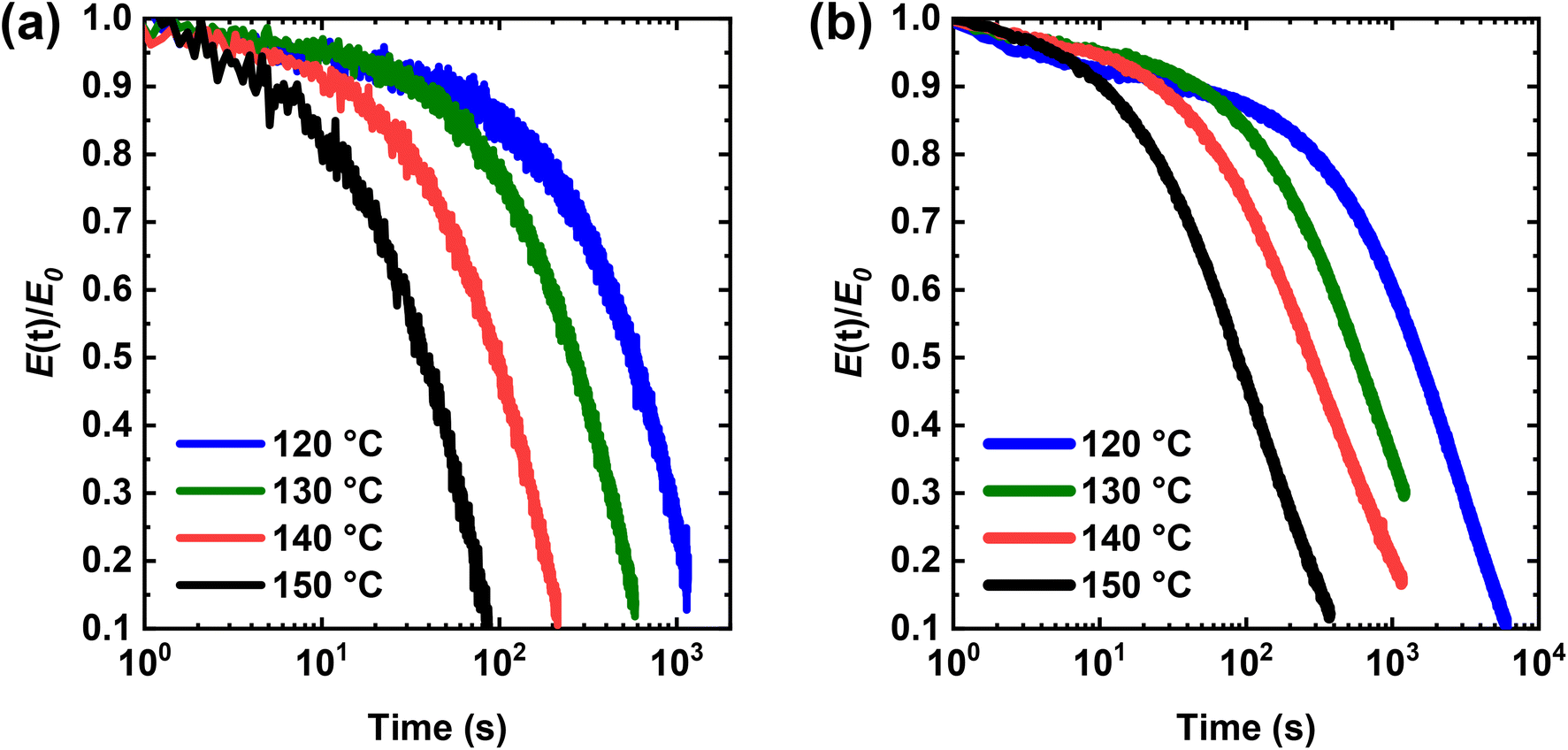 | ||
| Fig. 7 Stress relaxation curves of BiTEBES-based (a) PHMA-5 and (b) PHMA-10 networks at 120–150 °C. | ||
To account for all modes of relaxation, we used the Kohlrausch–Williams–Watts (KWW) stretched exponential decay function to fit the stress relaxation data:13,35,56,72,92–97
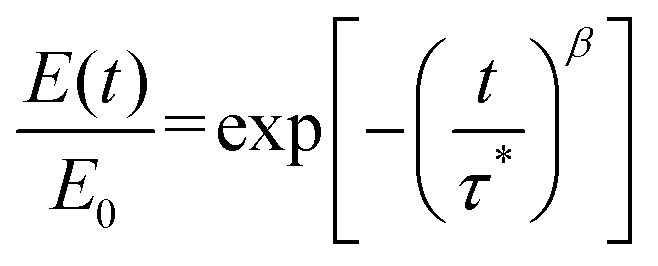 | (1) |
 | (2) |
We fitted our relaxation data to the KWW function and obtained the parameters shown in Table 2. At all temperatures tested, PHMA-5 shows significantly faster stress relaxation (i.e., smaller τ* or <τ>) than PHMA-10, which is expected as PHMA-10 contains a higher concentration of percolated cross-links and thus takes more time to decross-link sufficiently to relax stress. Importantly, within experimental uncertainty, PHMA-5 exhibits a single stress relaxation mode (β = 1.00) at 120–150 °C. Therefore, the stress relaxation in PHMA-5 follows the Maxwell model27,72,87,94 with T-dependent single relaxation times (τ), reported in Table 2. However, in PHMA-10, the resulting network exhibits substantial breadth in stress relaxation response, with β values falling in the range of 0.69–0.79. Additionally, τ values at a given T increase by more than a factor of 3 in PHMA-10 relative to PHMA-5, consistent with the much higher (factor of 5) cross-link density in PHMA-10. Similar qualitative trends in relaxation time and relaxation distribution breadth were observed in AIBN-initiated, BiTEMPS-based PHMA CANs, where, at 130 °C, the PHMA network with 5 mol% BiTEMPS methacrylate displays more rapid stress relaxation and a smaller breadth in its relaxation distribution (<τ> of 835 s and β value of 0.67) than that with 10 mol% BiTEMPS methacrylate (<τ> of 4520 s and β value of 0.46) (see Fig. S20 and Table S1†). These results on BiTEMPS- and BiTEBES-based cross-linkers indicate that the dissociative cross-link density not only influences the rate of stress relaxation in CANs, but also has a significant impact on the modes and distribution of relaxation in these materials. Interestingly, our past stress relaxation study of PHMA CANs made with 5 mol% BiTEMPS cross-linker (and having a cross-link density exceeding that of the BiTEBES-based PHMA-10 CAN) revealed that <τ> = 650 s at 130 °C,72 460 s shorter than <τ> for the BiTEBES-based PHMA-10 CAN at the same T. Thus, both cross-link density and the dynamic covalent cross-linker species have important roles to play in stress relaxation. Importantly, when the density of dissociative covalent cross-links is sufficiently low in CANs, single-exponential decay stress relaxation can occur.
| Sample | PHMA-5 | PHMA-10 | |||||
|---|---|---|---|---|---|---|---|
| T (°C) | τ* = τ (s) | β | R 2 | τ* (s) | β | <τ> (s) | R 2 |
| 120 | 777 | 1.00 | 0.986 | 2365 | 0.78 | 2720 | 0.996 |
| 130 | 355 | 1.00 | 0.989 | 948 | 0.77 | 1110 | 0.999 |
| 140 | 129 | 1.00 | 0.985 | 493 | 0.69 | 632 | 0.998 |
| 150 | 50 | 1.00 | 0.981 | 145 | 0.79 | 166 | 0.999 |
To determine the apparent activation energy (Ea,τ) associated with stress relaxation of the BiTEBES-based networks, the relaxation time (τ) of PHMA-5 and average relaxation time (<τ>) of PHMA-10 were analyzed via an Arrhenius plot in the T range of 120–150 °C. As shown in Fig. 8, the stress relaxation responses of PHMA-5 and PHMA-10 exhibit Arrhenius T dependences despite their dissociative nature. We note that an Arrhenius T dependence of stress relaxation time had been thought to be a characteristic of vitrimers (i.e., associative networks).25,31,87 However, many dissociative networks have been reported to exhibit stress relaxation that follows the Arrhenius relationship.13,64,69,72,87–89,98–100 This outcome is unsurprising given that most stress-relaxation characterization of CANs has been at temperatures nearly 100 °C or more above the network Tg. If network relaxation is expected to follow the Williams–Landel–Ferry (WLF)101 or Vogel–Fulcher–Tammann (VFT)102T dependence, such a T dependence typically exhibits strong non-Arrhenius behavior at Tg < T < Tg + 50 °C. However, at T ≥ Tg + 100 °C, the WLF or VFT T dependence typically becomes Arrhenius-like over ranges of several tens of degrees.87,94
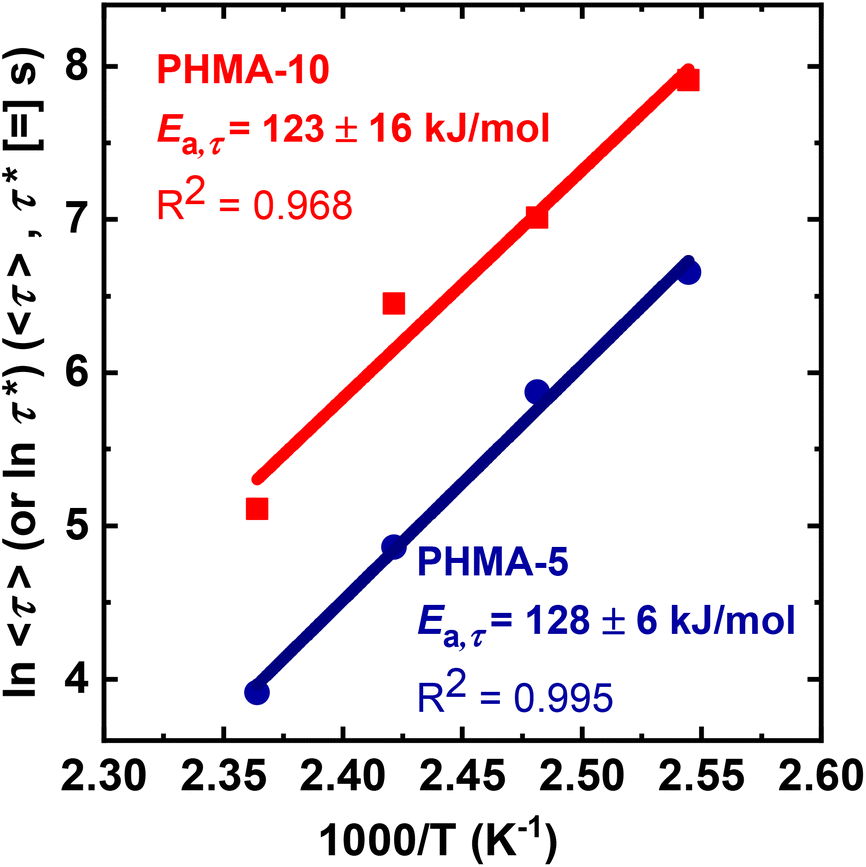 | ||
| Fig. 8 Determination of apparent Arrhenius activation energies of stress relaxation for BiTEBES-based PHMA-5 and PHMA-10 networks. | ||
The Ea,τ values for PHMA-5 and PHMA-10 networks are 128 ± 6 kJ mol−1 and 123 ± 16 kJ mol−1, respectively, identical within experimental uncertainty. Thus, even though the average stress relaxation time increases substantially in going from PHMA-5 to PHMA-10, the T dependence of stress relaxation is independent of cross-link density. Similar observations on the T dependence of stress relaxation have been noted for dissociative CANs based on Diels–Alder chemistry.88 Importantly, the Ea,τ values for PHMA-5 and PHMA-10 networks fall within the range of the BDE of the sulfur-sulfur bond in dialkylamino disulfides (BDE = 110–130 kJ mol−1).67,70,71 These results indicate that the dynamic chemistry of the BiTEBES cross-links dominates the T dependence of the stress relaxation response of the BiTEBES-based networks. Related outcomes were previously noted for PHMA networks cross-linked with the dissociative cross-linker BiTEMPS methacrylate.72,75
Unlike conventional networks cross-linked with permanent bonds, some CANs have been reported to be highly susceptible to creep at elevated T due to the dynamic nature of their cross-links.31 Creep is continuous, time-dependent deformation under a constant load or stress.103 To investigate the creep resistance of BiTEBES-based CANs, we characterized the responses of PHMA-5 and PHMA-10 networks under constant shear stress of 3.0 kPa at 70 °C and 90 °C, well above their Tgs (Fig. 9). Under constant stress, the networks show initial elastic deformation followed by a viscous, time-dependent response. The elastic deformation decreases with increasing cross-link density (i.e., the less cross-linked PHMA-5 shows higher instantaneous elastic strain compared to the more cross-linked PHMA-10), consistent with the higher modulus, E (or E′), and higher resistance to elastic deformation of PHMA-10. As shown in Fig. 9a, PHMA-5 and PHMA-10 exhibit excellent long-term creep resistance at 70 °C as the networks exhibit creep strains of 0.75% and 0.44%, respectively, after ∼14 h of continuous stress. (Here, we calculate creep strain as the difference between the strain at t = 50000 s and t = 1800 s to account only for pure creep and excluding any delayed elastic deformation.)35,72,104,105 These creep strain values are extremely low and comparable to creep strain values associated with permanently cross-linked static networks.104–106
 | ||
| Fig. 9 Creep curves of BiTEBES-based PHMA-5 and PHMA-10 networks at (a) 70 °C and (b) 90 °C. | ||
When T is increased to 90 °C (Fig. 9b), PHMA-5 shows significant creep strain (∼7.1%), whereas PHMA-10 exhibited a much smaller creep strain (∼1.4%) after being subjected to a continuous 3.0 kPa stress for ∼14 h. The much greater creep strain for PHMA-5 is due to its much lower cross-link density relative to PHMA-10. At very low percolated cross-link density, loss of a small fraction of cross-links due to dissociation can result in a loss of locally percolated cross-linking and thus creep; at higher cross-link density, the loss of a similar fraction of cross-links due to dissociation can leave the network with a sufficiently high local cross-link density to strongly suppress creep. Thus, these results are consistent with the notion that the creep response of dissociative CANs may be improved by increasing the concentration of the dynamic cross-links, even if the cross-links are expected to dissociate at the same rate at a given T. Our results indicate that BiTEBES-based dialkylamino disulfide cross-links can be effective in suppressing long-term creep at elevated T up to 70 °C in lightly cross-linked PHMA CANs and up to 90 °C in more highly cross-linked PHMA CANs.
On a related note, we also studied the elevated-T creep behavior of PHMA networks made using 5 mol% BiTEMPS cross-linker and having cross-link density exceeding even that of the PHMA-10 CAN made here with BiTEBES cross-linker. We observed that the BiTEMPS-based PHMA CAN exhibited excellent creep resistance at temperatures as high as 90 °C.72 The creep response was found to be dominated by the dynamic chemistry of the BiTEMPS cross-links present in this CAN.72 We also reported similar results in BiTEMPS-based polyethylene CANs.77
Comparison of PHMA CANs made with BiTEBES methacrylate to those made with BiTEMPS methacrylate
It is important to compare the E′ values and thus cross-link densities of PHMA CANs made with 5 mol% BiTEBES methacrylate and 5 mol% BiTEMPS methacrylate. As reported in ref. 72 and here, our 5 mol% BiTEMPS-based PHMA CANs exhibited E′ = ∼1.7 MPa at 120 °C, a factor of 9 higher than our 5 mol% BiTEBES-based CANs. Consistent with these results, the BiTEMPS-based PHMA networks exhibited Tgel-to-sol = ∼234 °C,72 roughly 40 °C higher than the BiTEBES-based PHMA networks. Such dramatic difference in Tgel-to-sol in BiTEBES-based and BiTEMPS-based networks may be attributed at least in part to the stability of the cross-linkers themselves, with BiTEBES methacrylate having a Td5% nearly 20 °C lower than that of BiTEMPS methacrylate (vide infra). Thus, other things being equal, BiTEBES methacrylate is less effective than BiTEMPS methacrylate in contributing to effective cross-links in CANs made by FRP at 70 °C.The underlying cause for this difference is undetermined at present. However, BiTEBES-based cross-linkers have hydrogen atoms on carbon atoms adjacent to the nitrogen atoms whereas BiTEMPS-based cross-linkers do not. We acknowledge that nitroxyl radicals with hydrogen atoms on carbon atoms adjacent to nitrogen atoms, e.g., di-tert-butyl nitroxide, are less stable than nitroxyl radicals lacking alpha hydrogens, e.g., TEMPO.107 Although the situation is less clear for analogous thiyl radicals, they seem to be less thermally stable.70 Our data on thermal decomposition T shows that BiTEBES methacrylate has Td = 180 °C, nearly 20 °C lower than that of BiTEMPS methacrylate (Fig. S12†). However, the Td values exceed the FRP T by more than 100 °C and the (re)processing T by 50–70 °C; we make robust CANs by FRP, albeit with lower cross-link density with the BiTEBES-based cross-linker, that exhibit excellent recovery of cross-link density after several reprocessing steps. Thus, whatever the underlying cause for the difference in cross-link density in BiTEBES-based and BiTEMPS-based CANs synthesized under otherwise identical conditions, excellent reprocessability with complete cross-link density recovery is obtained in both systems at the (re)molding conditions employed here.
As the main point of our study concerns the synthesis at conventional FRP conditions and reprocessing of CANs made with dialkylamino disulfide cross-linkers, we defer further study of the underlying cause of the difference in cross-link density. Nevertheless, we do not discount any possible involvement of disulfides or thiyl radicals in the FRP69 or that homolytic substitution reactions of carbon-centered radicals with dialkylamino disulfides could lead to the presence of different dynamic cross-links70 or permanent carbon–carbon cross-links. However, if present, such effects are sufficiently small that they do not, within experimental uncertainty, change the facts that CANs are produced and that excellent reprocessability with complete recovery of cross-link density is obtained using appropriate processing conditions.
Conclusions
We demonstrated for the first time the use of conventional free-radical polymerization conditions (reaction at 70 °C with the common initiator AIBN) in synthesizing addition-type CANs using dialkylamino disulfide cross-linkers. A comparison of BiTEMPS-based poly(n-hexyl methacrylate) CANs made under identical conditions except for FRP temperature (room temperature vs. 70 °C) and initiator species (V-70 vs. AIBN) revealed that the same cross-link density within experimental uncertainty was achieved in both CANs. Furthermore, excellent reprocessability was obtained in both CANs with complete cross-link density recovery after several reprocessing steps. Thus, it is not necessary to employ unconventional FRP conditions to obtain robust, reprocessable CANs with dialkylamino disulfide cross-linkers.We also showed for the first time the utility of non-piperidine-based dialkylamino disulfide chemistry in the synthesis of catalyst-free, recyclable polymer networks. We developed a simple method to synthesize a non-piperidine-based dialkylamino disulfide dynamic cross-linker, BiTEBES methacrylate. We demonstrated the utility of BiTEBES methacrylate as a dynamic cross-linker by incorporating it into catalyst-free polymer networks using n-hexyl methacrylate as the base monomer for the network. We showed that the network cross-link density and associated properties could be easily controlled by varying the cross-linker concentration. We illustrated the importance of processing conditions and their impact on the properties of dynamic polymer networks. For example, both BiTEBES-based and BiTEMPS-based networks can be (re)processed multiple times at 130 °C for 1 h with full recovery of cross-link density after recycling. Based on the calculated activation energy (Ea,τ) associated with their stress relaxations, we have showed that the relaxation mechanism in BiTEBES-based networks is dominated by the dynamic chemistry of the BiTEBES cross-links, regardless of the concentration of cross-links. Notably, the breadth of the stress relaxation distribution is a function of tested cross-link density. The network made with 5 mol% BiTEBES methacrylate and a very low cross-link density exhibits single-exponential-decay stress relaxation, something that has not been previously demonstrated explicitly with CANs. In contrast, the network with 10 mol% BiTEBES methacrylate and a factor of 5 higher cross-link density exhibits substantial breadth in its relaxation distribution. We also characterized the creep response of the synthesized BiTEBES-based polymethacrylate networks using a constant 3.0 kPa shear stress. The BiTEBES-based network materials exhibited excellent long-term creep resistance at 70 °C with very low creep strains (≤0.75%) after 14 h of 3.0 kPa shear stress. This study highlights the versatility of dialkylamino disulfide chemistry and demonstrates its broad utility and effectiveness in synthesizing high-performance reprocessable networks.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support of Northwestern University via discretionary funds associated with a Walter P. Murphy Professorship (J. M. T.) and the support from an NSF Graduate Research Fellowship (L. M. F.). This work made use of the MatCI Facility at Northwestern University, which receives support from the MRSEC Program (NSF DMR-1720139) of the Materials Research Center at Northwestern University.References
- N. G. S. Silva, L. Cortat, E. J. O. Teixeira, L. Baptista, D. Orlando and D. R. Mulinari, Waste Manage., 2023, 155, 220–229 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- K. Ragaert, S. Huysveld, G. Vyncke, S. Hubo, L. Veelaert, J. Dewulf and E. Du Bois, Resour., Conserv. Recycl., 2020, 155, 104646 CrossRef.
- K. Khait and J. M. Torkelson, Polym.-Plast. Technol. Eng., 1999, 38, 445–457 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, ACS Macro Lett., 2022, 11, 568–574 CrossRef CAS PubMed.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- L. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064–4065 CrossRef CAS PubMed.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- F. Wang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2012, 22, 13076–13084 RSC.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- C. E. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- Y. Jia, Y. Matt, Q. An, I. Wessely, H. Mutlu, P. Theato, S. Bräse, A. Llevot and M. Tsotsalas, Polym. Chem., 2020, 11, 2502–2510 RSC.
- L. Li, X. Chen, K. Jin, M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2021, 54, 1452–1464 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- L. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- X. Chen, S. Hu, L. Li and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 2093–2101 CrossRef CAS.
- S. Hu, X. Chen, M. A. Bin Rusayyis, N. S. Purwanto and J. M. Torkelson, Polymer, 2022, 252, 124971 CrossRef CAS.
- X. Chen, L. Li and J. M. Torkelson, Macromol. Chem. Phys., 2019, 220, 1900083 CrossRef.
- N. S. Purwanto, Y. Chen, T. Wang and J. M. Torkelson, Polymer, 2023, 272, 125858 CrossRef CAS.
- F. Elizalde, R. H. Aguirresarobe, A. Gonzalez and H. Sardon, Polym. Chem., 2020, 11, 5386–5396 RSC.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, M. A. Hillmyer and W. R. Dichtel, J. Appl. Polym. Sci., 2017, 134, 44984 CrossRef.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- A. Carmine, Y. Domoto, N. Sakai and S. Matile, Chem. – Eur. J., 2013, 19, 11558–11563 CrossRef CAS PubMed.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabañero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- L. Imbernon, E. K. Oikonomou, S. Norvez and L. Leibler, Polym. Chem., 2015, 6, 4271–4278 RSC.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315–4325 RSC.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- I. Azcune and I. Odriozola, Eur. Polym. J., 2016, 84, 147–160 CrossRef CAS.
- L. Zhang, L. Chen and S. J. Rowan, Macromol. Chem. Phys., 2017, 218, 1600320 CrossRef.
- D. J. Fortman, R. L. Snyder, D. T. Sheppard and W. R. Dichtel, ACS Macro Lett., 2018, 7, 1226–1231 CrossRef CAS PubMed.
- M. Zhang, F. Zhao, W. Xin and Y. Luo, ChemistrySelect, 2020, 5, 4608–4618 CrossRef CAS.
- Q. Liu, Y. Liu, H. Zheng, C. Li, Y. Zhang and Q. Zhang, J. Polym. Sci., 2020, 58, 1092–1104 CrossRef CAS.
- L. Li, X. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 4658–4665 CrossRef CAS.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014 CrossRef CAS.
- G. C. Tesoro and V. Sastri, J. Appl. Polym. Sci., 1990, 39, 1425–1437 CrossRef CAS.
- V. R. Sastri and G. C. Tesoro, J. Appl. Polym. Sci., 1990, 39, 1439–1457 CrossRef CAS.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- A. V. Tobolsky, W. J. MacKnight and M. Takahashi, J. Phys. Chem., 1964, 68, 787–790 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS PubMed.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583 RSC.
- Y. Liu, Z. Tang, D. Wang, S. Wu and B. Guo, J. Mater. Chem. A, 2019, 7, 26867–26876 RSC.
- D. Sakamaki, S. Ghosh and S. Seki, Mater. Chem. Front., 2019, 3, 2270–2282 RSC.
- S. Sunner, Acta Chem. Scand., 1955, 9, 837–846 CrossRef CAS.
- A. Takahashi, R. Goseki, K. Ito and H. Otsuka, ACS Macro Lett., 2017, 6, 1280–1284 CrossRef CAS PubMed.
- W. C. Danen and D. D. Newkirk, J. Am. Chem. Soc., 1976, 98, 516–520 CrossRef CAS.
- B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1976, 98, 520–523 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 12, 2760–2771 RSC.
- M. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2020, 53, 8367–8373 CrossRef CAS.
- H. Yokochi, R. Takashima, D. Aoki and H. Otsuka, Polym. Chem., 2020, 11, 3557–3563 RSC.
- M. Aiba, T.-a. Koizumi, M. Futamura, K. Okamoto, M. Yamanaka, Y. Ishigaki, M. Oda, C. Ooka, A. Tsuruoka, A. Takahashi and H. Otsuka, ACS Appl. Polym. Mater., 2020, 2, 4054–4061 CrossRef CAS.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS PubMed.
- L. M. Fenimore, B. Chen and J. M. Torkelson, J. Mater. Chem. A, 2022, 10, 24726–24745 RSC.
- A. Tsuruoka, A. Takahashi, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2020, 59, 4294–4298 CrossRef CAS PubMed.
- M. Aiba, T.-a. Koizumi, K. Okamoto, M. Yamanaka, M. Futamura, Y. Ishigaki, M. Oda, C. Ooka, A. Takahashi and H. Otsuka, Mater. Adv., 2021, 2, 7709–7714 RSC.
- J. E. Bennett, H. Sieper and P. Tavs, Tetrahedron, 1967, 23, 1697–1699 CrossRef CAS.
- G. A. O'Neil and J. M. Torkelson, Macromolecules, 1999, 32, 411–422 CrossRef.
- K. P. Menard, Dynamic mechanical analysis: A practical introduction, CRC Press, 2008 Search PubMed.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- M. Chanda, Introduction to polymer science and chemistry: A problem-solving approach, CRC Press, 2013 Search PubMed.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- K. Jin and J. M. Torkelson, Macromolecules, 2016, 49, 5092–5103 CrossRef CAS.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- X. Kuang, G. Liu, X. Dong and D. Wang, Mater. Chem. Front., 2017, 1, 111–118 RSC.
- Y. Jia, H. Ying, Y. Zhang, H. He and J. Cheng, Macromol. Chem. Phys., 2019, 220, 1900148 CrossRef.
- S. Zhao, D. Wang and T. P. Russell, ACS Sustainable Chem. Eng., 2021, 9, 11091–11099 CrossRef CAS.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2019, 11, 2398–2407 CrossRef CAS PubMed.
- J. C. Hooker and J. M. Torkelson, Macromolecules, 1995, 28, 7683–7692 CrossRef CAS.
- A. Dhinojwala, J. C. Hooker and J. M. Torkelson, J. Non-Cryst. Solids, 1994, 172–174, 286–296 CrossRef CAS.
- A. Dhinojwala, G. K. Wong and J. M. Torkelson, J. Chem. Phys., 1994, 100, 6046–6054 CrossRef CAS.
- R. Kohlrausch, Ann. Phys., 1847, 148, 353–405 CrossRef.
- G. Williams and D. C. Watts, J. Chem. Soc., Faraday Trans., 1970, 66, 80–85 RSC.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- L. Chen, L. Zhang, P. J. Griffin and S. J. Rowan, Macromol. Chem. Phys., 2020, 221, 1900440 CrossRef CAS.
- D. Pratchayanan, J.-C. Yang, C. L. Lewis, N. Thoppey and M. Anthamatten, J. Rheol., 2017, 61, 1359–1367 CrossRef CAS.
- M. L. Williams, R. F. Landel and J. D. Ferry, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- L. H. Sperling, Introduction to physical polymer science, John Wiley & Sons, Ltd., Hoboken, New Jersey, United States, 4th edn, 2006 Search PubMed.
- K. Watanabe, Rubber Chem. Technol., 1962, 35, 182–199 CrossRef.
- D. J. Plazek, J. Polym. Sci., Part A-2, 1966, 4, 745–763 CrossRef CAS.
- L. E. Nielsen, J. Macromol. Sci., Part C, 1969, 3, 69–103 CrossRef CAS.
- A. Nilsen and R. Braslau, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 697–717 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Images of cross-linker before and after drying; NMR & FTIR spectra of BiTEBES methacrylate; DSC thermogram of BiTEBES methacrylate; TGA curve of BiTEBES methacrylate; solubility of BiTEBES methacrylate in HMA monomers; image of PHMA-5 film molded at 180 °C for 5 min; DMA results for PHMA-5 film molded at 180 °C for 5 min; DSC thermograms as-synthesized and (re)processed polymethacrylate networks. See DOI: https://doi.org/10.1039/d3py00498h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |