
An N-oxide containing conjugated semiconducting polymer with enhanced electron mobility via direct (hetero)arylation polymerization†
Xiandong
He‡
b,
Feng
Ye‡
a,
Jia-Cheng
Guo
a,
Wenju
Chang
e,
Bingxu
Ma
a,
Riqing
Ding
a,
Sijing
Wang
a,
Yong
Liang
 e,
Dehua
Hu
*bd,
Zi-Hao
Guo
*ac and
Yuguang
Ma
b
e,
Dehua
Hu
*bd,
Zi-Hao
Guo
*ac and
Yuguang
Ma
b
aSouth China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter, South China University of Technology, Guangzhou 510640, China. E-mail: guozihao@scut.edu.cn
bState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P. R. China
cGuangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices, South China University of Technology, Guangzhou, 510640, China
dSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, P. R. China
eState Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China
First published on 22nd March 2023
Abstract
In this paper, an N-oxide building block, 4,4′-dimethyl-[2,2′-bithiazole] 3,3′-dioxide (MeBTzO), was designed and synthesized by oxidation of sp2-N in the aromatic ring. Theoretical calculation results showed that MeBTzO has higher reactivity than its non-oxide sp2-N containing monomer MeBTz in direct (hetero)arylation polymerization (DHAP). Therefore, an N-oxide containing conjugated semiconducting polymer, PDPPMeBTzO, was successfully synthesized via DHAP of MeBTzO with thiophene-flanked diketopyrrolopyrrole (DPP). PDPPMeBTzO possesses a lower lowest unoccupied molecular orbital (LUMO) than the non-oxide analogous polymer PDPPMeBTz, which will facilitate electron injection and transport in organic field-effect transistors (OFETs). As a result, PDPPMeBTzO has obviously enhanced electron transport properties with a higher μe of 0.11 cm2 V−1 s−1 compared to PDPPMeBT with a μe of 7.49 × 10−3 cm2 V−1 s−1. Our strategy demonstrated that introducing the N-oxide group in conjugated polymers has great potential for high performance ambipolar and n-type CSP materials.
Introduction
Conjugated semiconducting polymers (CSPs) have the advantages of low cost, light weight, flexibility, and solution processability and therefore have been widely used in various applications from organic electronics1–4 to bioelectronics.5–8 CSPs with high charge carrier mobility (μ) are essential for device performance, especially for application in organic field-effect transistors (OFETs).1,8–12 To date, great achievements have been made in p-type (hole-transporting) CSPs. Several p-type CSPs with a hole mobility (μh) beyond 10 cm2 V−1 s−1 have been reported,13,14 which is comparable to amorphous silicon.15 Regretfully, less progress has been made for n-type (electron-transporting) CSPs, and the value of electron mobility (μe) for n-type CSPs hardly competes with that for p-type ones. The unbalanced hole and electron transport properties of CSPs limit their comprehensive application in many organic electronic devices. For example, a low-power complementary metal-oxide semiconductor (CMOS)-like organic logic gate needs to combine both p-type and n-type CSP materials with balanced hole and electron transport properties.16,17 Even though some ambipolar (transporting both holes and electrons) CSPs have almost equal hole and electron mobilities,18 they are difficult to apply to electronic devices requiring separated hole and electron transporting channels, e.g. organic thermoelectric generators (OTEGs).19 In this context, more and more efforts have been devoted to boosting the electron transport properties of CSPs.The major challenge in obtaining electron transport properties for CSPs is to realize the low-lying lowest unoccupied molecular orbital (LUMO) that matches the work function of metal electrodes for efficient electron injection. Particularly, CSPs with a LUMO energy level below −4.0 eV are necessary for an air-stable n-type OFET device.11 One promising approach is to introduce electron-withdrawing groups (EWGs) to the polymer backbone,9,10 such as halogen (F or Cl),20–22 cyano (CN),23–25 trifluoromethyl (CF3),26,27 nitrogen (N),28–31 and boron ← nitrogen (B ← N) groups.32–34 Replacing C–H in an aromatic ring with sp2-nitrogen (such as thiazole and pyridine) can effectively lower the LUMO energy level and bring electron transport properties to CSPs.35–38 For example, the diketopyrrolopyrrole (DPP) based conjugated polymer PDPP2T exhibits pure hole-transport properties,39 while 2,2′-bithiazole (BTz) is replaced by 2,2′-bithiophene (BT) to afford the unipolar n-type semiconducting polymer PDPPBTz.40 Similarly, the dithienylthienothio-phenebisimide (TBI) based conjugated polymer PTBI2T only exhibits a hole mobility of 0.06 cm2 V−1 s−1, while PTBI2Tz is an n-type CSP with a μe of 0.05 cm2 V−1 s−1.41 We are interested in these sp2-N containing aromatic structures because they can further oxidise to N-oxide compounds, which can afford even lower LUMO energy levels. As shown in Fig. 1, the density functional theory (DFT) calculation results show that N-oxide pyridine (PyO) and N-oxide 2,2′-bithiazole (BTzO) are more electron-deficient and have lower LUMO than pyridine (Py) and BTz. Recently, Schipper and coworkers reported a series of conjugated polymers containing N-oxide groups,42–44 and the LUMO levels of these polymers are lower than those of non-oxide ones. In this context, building CPSs with the N-oxide group in a conjugated backbone is a potential strategy to achieve electron-transporting CPS materials. However, less attention has been paid to these N-oxide containing conjugated polymers for organic electronic materials.
 | ||
| Fig. 1 Chemical structures, electrostatic potential (ESP) surface, and frontier molecular orbital energy levels for (a) Py and PyO, and (b) BTz and BTzO. Calculations are carried out at the B3LYP/6-311++G** level. | ||
On the other hand, CSPs are usually synthesized by transition-metal-catalyzed coupling reactions that involve organometallic reagents. For example, the widely used Stille reaction for CSPs needs organotin monomers.45 Despite the fact that these conventional coupling reactions provide an effective platform to produce CSPs, it always takes tedious steps to synthesize the corresponding organometallic monomers, not to mention their toxicity. A direct (hetero)arylation reaction can directly form an aryl C–C bond through the C–H bond and C–X bond (X usually is a halide, Br or I), which is more environmentally friendly and atom economical.46 Currently, direct (hetero)arylation polymerization (DHAP) is becoming a valuable “greener” alternative to traditional coupling reactions for the synthesis of CSPs.47–52 The broadly recognized mechanism of direct arylation is a base-assisted, concerted metalation–deprotonation (CMD) pathway.53,54 In this process, the activation barrier of the C–H bond cleavage in the transition state is related to the reactivity and regioselectivity of the C–H bond donating substrate. Theoretical calculation shows that N-oxide derivatives have significantly lower CMD activation barriers of C–H bond cleavage than the original N-heterocycles, which make them easier to polymerize by DHAP.55
Herein, we designed and synthesized a new building block containing the N-oxide group, namely MeBTzO, and polymerized it with thiophene-flanked DPP through DHAP to afford PDPPMeBTzO (P1). Compared to the non-oxide analogous monomer MeBTz, MeBTzO shows higher DHAP reactivity due to its lower CMD activation barrier of C–H bond cleavage induced by the N-oxide group. The photophysical and electrochemical results show that P1 has an obviously lower LUMO energy level and smaller bandgap than PDPPMeBTz (P2) without the N-oxide group. Although DFT calculations reveal that the backbone planarity of P1 is relatively poor, P1 still exhibits excellent n-dominant ambipolar transporting properties (μe/μh = 3.5) with μe = 0.11 cm2 V−1 s−1 and μh = 0.03 cm2 V−1 s−1. At the same time, non-oxide P2 has nearly unipolar hole transport properties with a μh/μe over 17, in which μe is about 0.007 cm2 V−1 s−1 and μh is 0.124 cm2 V−1 s−1. The switched charge carrier transport properties from P2 to P1 is benefited from the lower LUMO energy level of P1 that facilitates electron injection.
Results and discussion
Selection of the N-oxide monomer
The thiazole (Tz) group has been widely used for constructing CSPs with excellent charge carrier transport properties.40,41,56–58 Li and coworkers reported that BTz, a Tz containing aromatic compound, can undergo DHAP to copolymerize with DPP.59 Based on the previous report,55 the N-oxide compound BTzO should have higher reactivity than BTz in DHAP. Therefore, we first conducted theoretical calculations to compare the reactivity of BTz and BTzO. DFT calculation at the B3LYP/TZVP (DZVP for palladium) level is applied to determine the CMD activation barriers of C–H bond cleavage during DHAP for both BTz and BTzO. As shown in Fig. 2, BTz has energy barriers of 24.8 kcal mol−1 and 30.0 kcal mol−1 for the α- and β-positions, respectively. As expected, these barriers are reduced to 23.4 kcal mol−1 and 24.1 kcal mol−1 when BTz was replaced by the corresponding N-oxide BTzO. However, the energy barrier difference between the α- and β-positions in BTzO is only 0.7 kcal mol−1, which will make undesirable β-defects in DHAP. According to Arrhenius's law, the regioselectivity ratio between the α- and β-positions is about 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 115 °C. Hence, a methyl group is introduced at the β-position to avoid the formation of β-defects. Calculation results show that MeBTzO has even smaller CMD energy barriers compared to BTzO (23.0 kcal mol−1vs. 23.4 kcal mol−1) at the α-position. These theoretical calculation results indicate that MeBTzO will exhibit high reactivity in the DHAP. Apart from the theoretical prediction, the selection of MeBTzO in our study is also due to its better solubility. Both BTzO and MeBTzO are synthesized. However, BTzO hardly dissolves in common solvents for DHAP, such as THF, DMF and toluene. At the same time, MeBTzO can be well dissolved in these solvents. Overall, based on the theoretical and experimental results, we employed MeBTzO as an N-oxide monomer to synthesize CPSs by DHAP in this study.
:1 at 115 °C. Hence, a methyl group is introduced at the β-position to avoid the formation of β-defects. Calculation results show that MeBTzO has even smaller CMD energy barriers compared to BTzO (23.0 kcal mol−1vs. 23.4 kcal mol−1) at the α-position. These theoretical calculation results indicate that MeBTzO will exhibit high reactivity in the DHAP. Apart from the theoretical prediction, the selection of MeBTzO in our study is also due to its better solubility. Both BTzO and MeBTzO are synthesized. However, BTzO hardly dissolves in common solvents for DHAP, such as THF, DMF and toluene. At the same time, MeBTzO can be well dissolved in these solvents. Overall, based on the theoretical and experimental results, we employed MeBTzO as an N-oxide monomer to synthesize CPSs by DHAP in this study.
 | ||
| Fig. 2 Gibbs free energy of the CMD transition state associated with the transition state for the C–H bond cleavage. | ||
Synthesis
The synthetic routes to the MeBTzO monomer and the corresponding polymer are presented in Scheme 1. The Yamamoto homo-coupling reaction of 2-bromo-4-methylthiazole is carried out to afford 4,4′-dimethyl-2,2′-bithiazole (MeBTz). After oxidizing with meta-chloroperoxybenzoic acid (m-CPBA), MeBTzO is obtained. It takes only two steps to synthesize the MeBTzO monomer from commercially available starting materials under mild conditions, which is much easier than organometallic reagents. The MeBTzO containing polymer P1 is then synthesized via DHAP between MeBTzO and ThDPP-Br catalyzed by Hermann's catalyst. As a comparison, we also synthesized P2 without N-oxidation under the same DHAP conditions. It is worth noting that the reaction rate between MeBTzO and ThDPP-Br is surprisingly fast, evidenced by the color of the reaction solution that switches from red to deep purple 30 minutes after its setting up (Fig. S4†). In contrast, we did not observe the same color change until 4 hours later in the synthesis of P2. This phenomenon well correlates with the computed CMD barriers. The purification of P1 is conducted by Soxhlet extraction with methanol, acetone and hexane, in order to remove the impurities and low-molecular-weight fractions. However, P2 is completely dissolved in hexane after methanol and acetone extraction. This is due to the low reactivity of MeBTz, thus leading to low-molecular-weight P2. The chloroform fraction of P1 and the hexane fraction of P2 were collected and concentrated, and then precipitated in methanol to afford the final product for further characterization and device fabrication. The chemical structures of all monomers and two polymers were verified by 1H NMR spectroscopy (see details in the ESI†). The molecular weights (Mn) of polymers were characterized by high-temperature gel permeation chromatography (HT-GPC) at 150 °C using 1,2,4-tricholorobenzene (TCB) as the eluent and the details are summarized in Table 1. | ||
| Scheme 1 Synthetic route to (a) MeBTzO, (b) PDPPMeBTzO, and PDPPMeBTz. | ||
| Polymer | M n (kDa)/PDIa | E LUMO [eV] | λ max [nm] | λ onset [nm] |
E
optge [nm] |
E HOMO [eV] |
|---|---|---|---|---|---|---|
| a Determined by GPC using 1,2,4-trichlorobenzene (TCB) as an eluent at 150 °C. b The LUMO energy level estimated from the cyclic voltammetry (CV) measurements. c The absorption maximum for the chloroform solution. d The absorption onset for the film spectrum. e Optical band gap estimated using the equation: Eoptg = 1240/λonset. f The HOMO energy level estimated using the equation: EHOMO = ELUMO − Eoptg. | ||||||
| PDPPMeBTzO | 26.7/2.11 | −3.65 | 767 | 906 | 1.37 | −5.02 |
| PDPPMeBTz | 8.4/1.54 | −3.60 | 670 | 852 | 1.46 | −5.06 |
Photophysical and electrochemical properties
The photophysical properties of P1 and P2 are investigated using ultraviolet–visible (UV–vis) absorption spectra both in dilute solution (1 × 10−5 M in chloroform) and thin film (Fig. 3a and b), and the corresponding data are summarized in Table 1. | ||
| Fig. 3 UV–vis–NIR absorption spectra of PDPPMeBTzO and PDPPMeBTz in (a) chloroform solution and (b) as-thin films. (c) Cyclic voltametric reduction curves of both polymers in a thin film. | ||
In solution, the maximum absorption peak of P1 is at 767 nm with the absorption onset at around 880 nm, while P2 presents an absorption maximum at 670 nm with the absorption onset at around 842 nm. Note that after introducing the steric hindrance methyl group in MeBTz, the absorption of P2 is blue-shifted to the previously reported polymer PDBTz without the methyl group,40 probably due to the relatively poor coplanarity of P2. In contrast, P1 containing the N,N′-dioxide MeBTzO unit with two more steric oxygen atoms does not show further blue-shift in the absorption spectra. This is because oxygen atoms in MeBTzO units could form a chalcogen (S–O) bond,44 which will not disturb the coplanarity of the polymer chain. Furthermore, the electron deficient MeBTzO unit in P1 could lower the LUMO level of the polymer, thus leading to a smaller bandgap compared to P2. From solution to thin film, both polymers show an obvious bathochromic shift and increased 0–0 vibrational peaks, indicating strong interchain aggregation and better coplanarity in the solid state. The optical bandgaps (Eoptg) of P1 and P2 are 1.37 eV and 1.46 eV, respectively, calculated from the absorption onset of both polymers in thin films.
The LUMO energy levels of both polymers were determined by cyclic voltammetry (CV). The experiment was conducted with polymer films on a glassy carbon electrode in an acetonitrile solution containing n-Bu4NPF6 as the supporting electrolyte under nitrogen. Due to their highly electron deficient backbone, both polymers exhibit a relatively reversible doping process (Fig. 3c), suggesting their good electron acceptor properties. The LUMO energy levels are calibrated by using ferrocene as a reference, which are −3.65 eV and −3.60 eV for P1 and P2, respectively. Clearly, the LUMO energy level of P1 is decreased after introducing N,N′-dioxide groups compared to that of P2. The HOMO energy levels of both polymers are calculated with the equation EHOMO = ELUMO − Eoptg, which are −5.02 eV and −5.06 eV for P1 and P2, respectively.
DFT calculation
To further understand the frontier molecular orbital (FMO) energy levels and backbone conformation of both polymers, DFT calculations were performed at the B3LYP/6-31G* level based on the trimer of P1 and P2, and the methyl group was considered instead of long branched side chains in order to save the calculation time. Unexpectedly, P2 has a nearly planar conformation with the steric methyl group. The dihedral angles θ1, θ2 and θ3 are almost 0° (Fig. 4), which are similar to those of polymers without a steric methyl group (Fig. S3†). For P1, the dihedral angles are slightly increased after oxidation, in which θ1, θ2 and θ3 are 0.09°, 5.58° and 0.18°, respectively. Remarkably, the O⋯S distance in P1 is 2.70 Å as calculated, obviously smaller than the sum of the van der Waals radii for O–S (3.37 Å), indicating the formation of a chalcogen bond. The calculated LUMO energy levels have the same trend as in the CV results, in which the trimer of P1 has a lower energy level (−3.21 eV) than that of P2 (−3.11 eV). | ||
| Fig. 4 DFT-derived HOMO/LUMO energy levels and side view representations of polymers (a) PDPPMeBTzO and (b) PDPPMeBTz. | ||
OFET device
The charge carrier transport properties of both polymers were evaluated using OFET devices with a bottom-gate/top-contact (BGTC) architecture. After the screening of solvents for device fabrication, chloroform (CHCl3) gave the best OFET device performance.The polymer solutions (5 mg mL−1) were spin-coated on an octadecyltrichlorosilane (OTS-18) decorated substrate.60 Before evaporating electrodes, the as-spun polymer films were annealed at different temperatures over 30 minutes. This process could possibly increase the crystallinity of the polymer and reduce the concentration of charge traps.61 The very detail of the device fabrication is presented in the Experimental section and the results of OFET devices for both polymers are summarized in Table 2. Both polymers showed ambipolar charge carrier properties, but with different features. As shown in Fig. 5, the absence of the saturation region for P2 in the output curve under positive VDS indicated that it mainly shows hole transport properties, and its hole mobility was about 0.13 cm2 V−1 s−1 with a small deviation at each annealing temperature. At the same time, its electron mobility increased from 1.3 × 10−3 to 7.4 × 10−3 cm2 V−1 s−1 as the annealing temperature increased, which may be attributed to the degradation of electron trapping.62 The μh/μe value of P2 was about 17, indicating that P2 was a p-dominant ambipolar CSP.
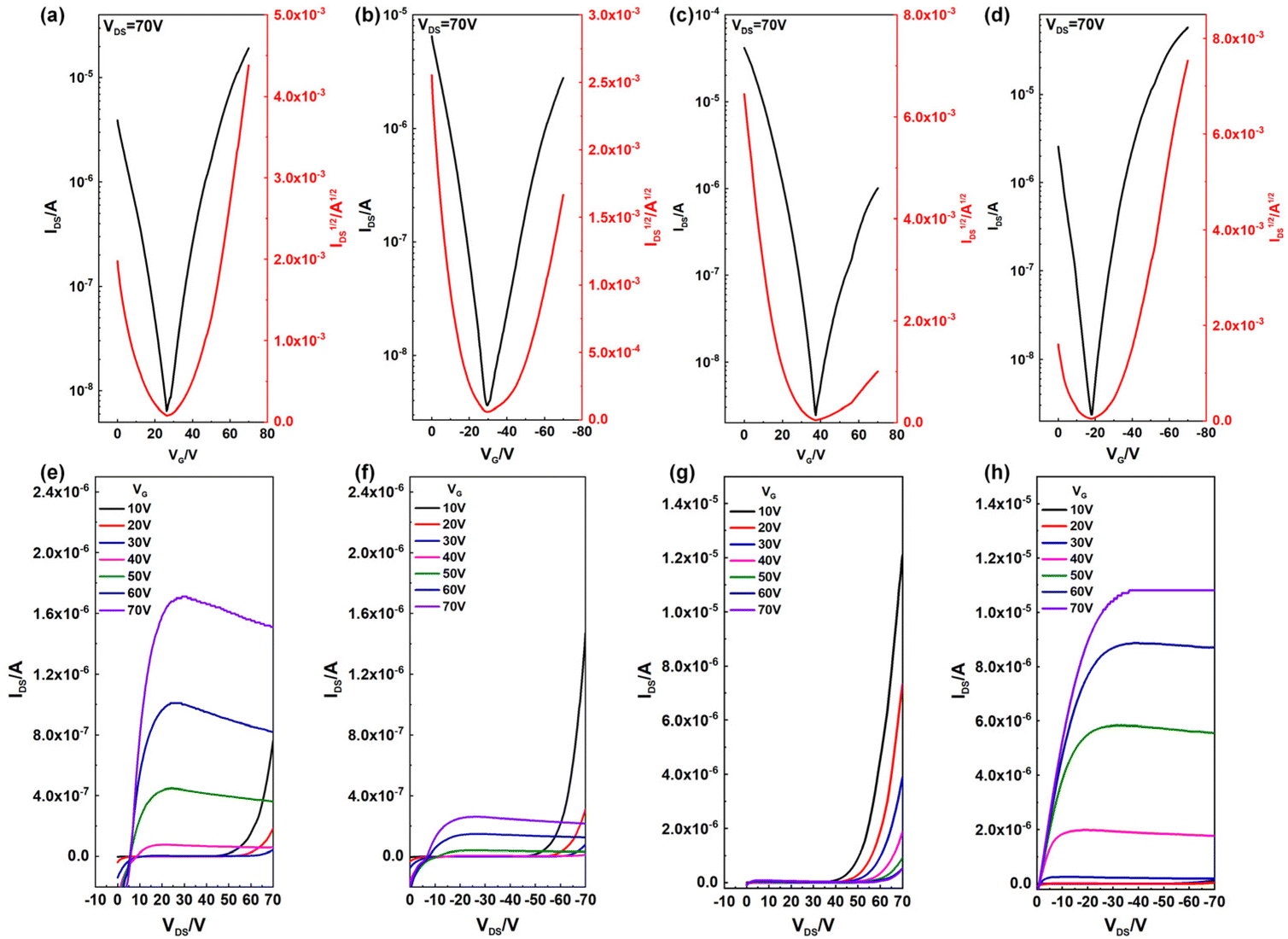 | ||
| Fig. 5 n-Type (a and c) transfer and (e and g) output characteristics of (a and e) PDPPMeBTzO and (c and g) PDPPMeBTz at 250 °C. p-Type (b and d) transfer and (f and h) output characteristics of (b and f) PDPPMeBTzO and (d and h) PDPPMeBTz at 250 °C. | ||
| Polymer | T anneal (°C) | n-Type | p-Type | ||||
|---|---|---|---|---|---|---|---|
| μ e [cm2 V−1 s−1] | I on/Ioffb | V th [V] | μ h [cm2 V−1 s−1] | I on/Ioffb | V th [V] | ||
| a μ e is the average electron mobility calculated from at least five devices. b Current on/off ratio. c Threshold voltage. d μ h is the average hole mobility calculated from at least five devices. | |||||||
| PDPPMeBTzO | 150 | 3.71 × 10−2 | 104 | 50 | 9.80 × 10−3 | 103 | −47 |
| 200 | 5.00 × 10−2 | 104 | 44 | 9.76 × 10−3 | 103 | −45 | |
| 250 | 1.12 × 10−1 | 104 | 44 | 3.19 × 10−2 | 103 | −47 | |
| PDPPMeBTz | 150 | 1.30 × 10−3 | 102 | 40 | 1.28 × 10−1 | 104 | −34 |
| 200 | 7.49 × 10−3 | 102 | 39 | 1.40 × 10−1 | 104 | −38 | |
| 250 | 7.32 × 10−3 | 102 | 45 | 1.24 × 10−1 | 104 | −32 | |
After introducing the N-oxide group in P1, the LUMO energy level decreased from −3.60 eV to −3.65 eV compared to P2. In this way, P1 exhibited more electron transport properties. According to the output curve of P1, the current of the saturation region under positive VDS was bigger than that under negative VDS, which implied the reversion of the dominant charge carrier transporting type, and the average value of μe and μh was 0.112 and 3.19 × 10−2 cm2 V−1 s−1 after annealing at 250 °C, respectively. Correspondingly, the Ion/Ioff of the n-channel in the P1 based OFET and that of the p-channel in the P2 based OFET all reached 104, while Ion/Ioff of the p-channel in the former and that of the n-channel in the latter were relatively low because of the imbalanced ambipolar mobility property of devices.63 In addition, all devices exhibited a considerable threshold voltage of about 30–50 V in absolute value; such a shift originated from charge carrier traps and interface defects.64 In a word, the oxidation strategy makes the main carrier transport type change, while maintaining the same order of magnitude of mobility.
Thin film
To gain deeper insight into the relationship between the device performance and polymer structures, thin film morphologies and microstructures were investigated by atomic force microscopy (AFM) and grazing incidence wide-angle X-ray scattering (GIWAXS). The morphologies of polymer films with different annealing temperatures were characterized by tapping mode AFM. As shown in Fig. 6, P1 and P2 showed obviously different morphological features. P1 had homogeneous thin film morphology with low root-mean-square (RMS) surface roughness, while local aggregations and pinholes were observed for P2 with high RMS surface roughness. | ||
| Fig. 6 AFM height images of the annealed (250 °C) polymer thin films of (a) PDPPMeBTzO and (b) PDPPMeBTz. | ||
This is probably due to the lower molecular weight of P2. Fig. 7, S9 and S10† show the one-dimensional (1D) line-cut profiles and the two-dimensional (2D) GIWAXS patterns of both polymers at three temperatures. For P1, both in-plane (IP) and out-of-plane (OP) profiles presented the (100) diffraction peaks, which represented the lamellar packing, implying their bimodal molecular orientation. For P2, the polymer film exhibited a dominant edge-on orientation, evidenced by only the (100) diffraction peak along the in-plane direction. The d-spacing values of the lamella calculated from the (100) diffraction peaks were 2.59 nm and 2.74 nm for P1 and P2, respectively. Both polymers have weak (010) diffraction peaks and the corresponding π–π distance was 3.87 Å and 3.89 Å for P1 and P2 respectively.
 | ||
| Fig. 7 1D-GIWAXS (a) in-plane and (b) out-of-plane line-cut profiles of the annealed (250 °C) polymer thin films of both polymers. | ||
Conclusions
In summary, we have successfully synthesized two CSPs, PDPPMeBTzO and PDPPMeBTz, using the DHAP method. Through detailed characterization by UV–vis, CV, and DFT calculations, it is found that the oxidation of sp2-N makes PDPPMeBTzO have a low-lying LUMO and smaller bandgap. As a result, the original PDPPMeBTz presents almost unipolar hole transport properties with a μh of about 0.13 cm2 V−1 s−1, while N-oxide PDPPMeBTzO exhibits dominant electron transport properties with a μe of about 0.11 cm2 V−1 s−1. The switched charge carrier properties demonstrate that introducing the N-oxide group in conjugated polymers is an effective way to enhance the electron transport properties. We believe that our strategy will pave a new way to design high performance ambipolar and n-type CSPs.Experimental section
Synthesis of monomers and polymers
The synthetic experimental details are presented in the ESI.†OFET device fabrication
OFETs with bottom-gate and top-contact geometries (BGTC) were used in the present study. All devices were fabricated on a highly doped n++-silicon wafer with a 300 nm thermally grown oxide layer substrate. The wafer serves as a gate electrode and SiO2 acts as a dielectric layer. Prior to the surface treatment of the silicon oxide layer, the wafer was cleaned using deionized water, acetone, and iso-propyl alcohol in an ultrasonic bath and dried in a drying oven. Subsequently, the substrate was plasma treated and transferred to a vacuum oven, adding a small amount of OTS-18 solution to complete the interface modification of the substrate surface under this atmosphere. The polymer was dissolved in chloroform and filtered to obtain a 5 mg mL−1 solution, which was spin-coated (3000 rpm for 30 s) on the substrate to form an active layer under nitrogen conditions. After annealing at different temperatures (150 °C, 200 °C, 250 °C) for 30 min, the sample was transferred to a vacuum evaporation chamber and fixed on a metal shadow mask, and the argentum particles were evaporated to deposit as source and drain electrodes, which have a thickness of about 100 nm. The channel length (L) and width (W) of the device were 50 and 2000 μm, respectively.The film samples for GIWAXS and AFM characterization were also spin-coated on the OTS-treated SiO2 substrate from chloroform solution and annealed at different temperatures.
Characterization of the OFET device
The volt−ampere characteristics of OFETs were tested using the Keithley 2636B semiconductor analyzer. All the measurements were performed in a nitrogen-filled glovebox without illumination. Voltages are applied to the gate and source and drain electrodes, respectively, using the probe station of the semiconductor analyzer. The FET mobilities were calculated from the saturation regime by extracting the slope of the linear range of I1/2DSversus VG plot and using the following equation.where IDS is the drain current and Ci is the capacitance per unit area of the gate dielectric, that is 11.5 nF cm−2 for SiO2. VG and VT are the gate voltage and threshold voltage, respectively. At room temperature, for the measurement of n-type properties, VD was set to 70 V and VG was scanned from 0 to 70 V to obtain the transfer characteristic curve (IDS–VG); in addition, under the increase of the VG from 10 to 70 V with the voltage gradient of 10 V, scanning the VD from 0 to 70 V corresponds to every VG to acquire the output characteristic curve (IDS–VDS). For the measurement of p-type properties, the corresponding voltages were set to equal negative values.

Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. X. H. and F. Y.: investigation, formal analysis, and writing – original draft; J.-C. G., W. C., B. M., R. D. and S. W.: investigation and formal analysis; Y. L.: formal analysis and supervision; D. H and Y. M.: conceptualization, supervision, and funding acquisition; Z.-H. G: conceptualization, supervision, funding acquisition, and writing – review & editing.Conflicts of interest
The authors declare that there are no conflicts.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 22271101), the Natural Science Foundation of Guangdong Province (2020A1515011506), the R&D Program of Guangzhou (202102020993), and the Recruitment Program of Guangdong (2016ZT06C322). D.H. acknowledges the support from the National Key R&D Program of China (2020YFA0714604), the National Natural Science Foundation of China (U20A6002), and the Research and Development Funds for Science and Technology Program of Guangzhou (202007020004). The authors also thank Prof. Xing Jiang (South China University of Technology) for his valuable discussion.References
- S. Yuvaraja, A. Nawaz, Q. Liu, D. Dubal, S. G. Surya, K. N. Salama and P. Sonar, Chem. Soc. Rev., 2020, 49, 3423–3460 RSC.
- P. Kordt, J. J. M. van der Holst, M. Al Helwi, W. Kowalsky, F. May, A. Badinski, C. Lennartz and D. Andrienko, Adv. Funct. Mater., 2015, 25, 1955–1971 CrossRef CAS.
- Y. Li, W. Huang, D. Zhao, L. Wang, Z. Jiao, Q. Huang, P. Wang, M. Sun and G. Yuan, Molecules, 2022, 27, 1800–1830 CrossRef CAS PubMed.
- Q. Fan, W. Su, S. Chen, W. Kim, X. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. Ma, T. Yang, W. Zhuang, Y. Li, Y. Li, T.-S. Kim, L. Hou, C. Yang, H. Yan, D. Yu and E. Wang, Joule, 2020, 4, 658–672 CrossRef CAS.
- A. N. Sokolov, B. C.-K. Tee, C. J. Bettinger, J. B.-H. Tok and Z. Bao, Acc. Chem. Res., 2012, 45, 361–371 CrossRef CAS PubMed.
- S. Wang, J. Y. Oh, J. Xu, H. Tran and Z. Bao, Acc. Chem. Res., 2018, 51, 1033–1045 CrossRef CAS PubMed.
- Y. H. Lee, O. Y. Kweon, H. Kim, J. H. Yoo, S. G. Han and J. H. Oh, J. Mater. Chem. C, 2018, 6, 8569–8612 RSC.
- H. Li, W. Shi, J. Song, H. J. Jang, J. Dailey, J. Yu and H. E. Katz, Chem. Rev., 2019, 119, 3–35 CrossRef CAS PubMed.
- J. Chen, J. Yang, Y. Guo and Y. Liu, Adv. Mater., 2021, 2104325 Search PubMed.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, C. Gao, Z. Ni, X. Zhang, W. Hu and H. Dong, Chem. Soc. Rev., 2023, 52, 1331–1381 RSC.
- D. I. James, S. Wang, W. Ma, S. Hedström, X. Meng, P. Persson, S. Fabiano, X. Crispin, M. R. Andersson, M. Berggren and E. Wang, Adv. Electron. Mater., 2016, 2, 1500313 CrossRef.
- G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y.-Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- X. Liu, Y. Guo, Y. Ma, H. Chen, Z. Mao, H. Wang, G. Yu and Y. Liu, Adv. Mater., 2014, 26, 3631–3636 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2005, 17, 2411–2425 CrossRef CAS.
- J. Lee, A. R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed.
- J. Yang, H. Wang, J. Chen, J. Huang, Y. Jiang, J. Zhang, L. Shi, Y. Sun, Z. Wei, G. Yu, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1606162 CrossRef PubMed.
- S. Cho, J. Lee, M. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910–1916 CrossRef CAS.
- A. Yusuf, Y. Demirci, T. Maras, S. E. Moon, J. Pil-Im, J. H. Kim and S. Ballikaya, ACS Appl. Mater. Interfaces, 2021, 13, 61275–61285 CrossRef CAS PubMed.
- C. J. Mueller, C. R. Singh, M. Fried, S. Huettner and M. Thelakkat, Adv. Funct. Mater., 2015, 25, 2725–2736 CrossRef CAS.
- Y. Sui, Y. Shi, Y. Deng, R. Li, J. Bai, Z. Wang, Y. Dang, Y. Han, N. Kirby, L. Ye and Y. Geng, Macromolecules, 2020, 53, 10147–10154 CrossRef CAS.
- M. Kim, W. T. Park, S. A. Park, C. W. Park, S. U. Ryu, D. H. Lee, Y. Y. Noh and T. Park, Adv. Funct. Mater., 2019, 29, 1805994 CrossRef.
- Y. Sui, Y. Deng, Y. Han, J. Zhang, W. Hu and Y. Geng, J. Mater. Chem. C, 2018, 6, 12896–12903 RSC.
- K. Feng, H. Guo, J. Wang, Y. Shi, Z. Wu, M. Su, X. Zhang, J. H. Son, H. Y. Woo and X. Guo, J. Am. Chem. Soc., 2021, 143, 1539–1552 CrossRef CAS PubMed.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- C. Wei, W. Zhang, J. Huang, H. Li, Y. Zhou and G. Yu, Macromolecules, 2019, 52, 2911–2921 CrossRef CAS.
- P. Deng, Y. Yan, S. D. Wang and Q. Zhang, Chem. Commun., 2012, 48, 2591–2593 RSC.
- Y. Z. Dai, N. Ai, Y. Lu, Y. Q. Zheng, J. H. Dou, K. Shi, T. Lei, J. Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- X. Yan, M. Xiong, J. T. Li, S. Zhang, Z. Ahmad, Y. Lu, Z. Y. Wang, Z. F. Yao, J. Y. Wang, X. Gu and T. Lei, J. Am. Chem. Soc., 2019, 141, 20215–20221 CrossRef CAS PubMed.
- Y. Shi, H. Guo, M. Qin, Y. Wang, J. Zhao, H. Sun, H. Wang, Y. Wang, X. Zhou, A. Facchetti, X. Lu, M. Zhou and X. Guo, Chem. Mater., 2018, 30, 7988–8001 CrossRef CAS.
- Y. Lu, Y. Liu, Y. Z. Dai, C. Y. Yang, H. I. Un, S. W. Liu, K. Shi, J. Y. Wang and J. Pei, Chem. – Asian J., 2017, 12, 302–307 CrossRef CAS PubMed.
- R. Zhao, Y. Min, C. Dou, B. Lin, W. Ma, J. Liu and L. Wang, ACS Appl. Polym. Mater., 2019, 2, 19–25 CrossRef.
- X. Cao, H. Li, J. Hu, H. Tian, Y. Han, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202212979 CAS.
- K. Zhao, Z.-F. Yao, Z.-Y. Wang, J.-C. Zeng, L. Ding, M. Xiong, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2022, 144, 3091–3098 CrossRef CAS PubMed.
- W. Li, Y. An, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2015, 3, 6756–6760 RSC.
- W. Li, W. S. Roelofs, M. Turbiez, M. M. Wienk and R. A. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- Y. Yu, F. Yang, Y. Ji, Y. Wu, A. Zhang, C. Li and W. Li, J. Mater. Chem. C, 2016, 4, 4134–4137 RSC.
- A. Zhang, Q. Wang, R. A. A. Bovee, C. Li, J. Zhang, Y. Zhou, Z. Wei, Y. Li, R. A. J. Janssen, Z. Wang and W. Li, J. Mater. Chem. A, 2016, 4, 7736–7745 RSC.
- Y. Li, P. Sonar, S. P. Singh, M. S. Soh, M. van Meurs and J. Tan, J. Am. Chem. Soc., 2011, 133, 2198–2204 CrossRef CAS PubMed.
- B. Fu, C.-Y. Wang, B. D. Rose, Y. Jiang, M. Chang, P.-H. Chu, Z. Yuan, C. Fuentes-Hernandez, B. Kippelen, J.-L. Brédas, D. M. Collard and E. Reichmanis, Chem. Mater., 2015, 27, 2928–2937 CrossRef CAS.
- M. Saito, I. Osaka, Y. Suda, H. Yoshida and K. Takimiya, Adv. Mater., 2016, 28, 6921–6925 CrossRef CAS PubMed.
- R. A. Mirabal, L. Vanderzwet, S. Abuadas, M. R. Emmett and D. Schipper, Chem. – Eur. J., 2018, 24, 12231–12235 CrossRef CAS PubMed.
- G. S. Sinclair, A. J. Kukor, K. K. G. Imperial and D. J. Schipper, Macromolecules, 2020, 53, 5169–5176 CrossRef CAS.
- G. S. Sinclair, R. C. M. Claridge, A. J. Kukor, W. S. Hopkins and D. J. Schipper, Chem. Sci., 2021, 12, 2304–2312 RSC.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- J. R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupre and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- H. Bohra and M. Wang, J. Mater. Chem. A, 2017, 5, 11550–11571 RSC.
- M. Mooney, A. Nyayachavadi and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 14645–14664 RSC.
- Y. Ran, Y. Guo and Y. Liu, Mater. Horiz., 2020, 7, 1955–1970 RSC.
- S. Yu, F. Liu, J. Yu, S. Zhang, C. Cabanetos, Y. Gao and W. Huang, J. Mater. Chem. C, 2017, 5, 29–40 RSC.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- S. Y. Tang, Q. X. Guo and Y. Fu, Chem. – Eur. J., 2011, 17, 13866–13876 CrossRef CAS PubMed.
- S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS.
- Z. Yuan, B. Fu, S. Thomas, S. Zhang, G. DeLuca, R. Chang, L. Lopez, C. Fares, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2016, 28, 6045–6049 CrossRef CAS.
- C. Buckley, S. Thomas, M. McBride, Z. Yuan, G. Zhang, J.-L. Bredas and E. Reichmanis, Chem. Mater., 2019, 31, 3957–3966 CrossRef CAS.
- Z. Yuan, C. Buckley, S. Thomas, G. Zhang, I. Bargigia, G. Wang, B. Fu, C. Silva, J.-L. Brédas and E. Reichmanis, Macromolecules, 2018, 51, 7320–7328 CrossRef CAS.
- C. Guo, J. Quinn, B. Sun and Y. Li, Polym. Chem., 2016, 7, 4515–4524 RSC.
- Y. Ito, A. A. Virkar, S. Mannsfeld, J. H. Oh, M. Toney, J. Locklin and Z. Bao, J. Am. Chem. Soc., 2009, 131, 9396–9404 CrossRef CAS PubMed.
- M. C. Gwinner, S. Khodabakhsh, H. Giessen and H. Sirringhaus, Chem. Mater., 2009, 21, 4425–4433 CrossRef CAS.
- Z. Chen, M. J. Lee, R. S. Ashraf, Y. Gu, S. Albert-Seifried, M. M. Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed.
- J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, Polym. Chem., 2015, 6, 278–282 RSC.
- L. Bürgi, M. Turbiez, R. Pfeiffer, F. Bienewald, H.-J. Kirner and C. Winnewisser, Adv. Mater., 2008, 20, 2217–2224 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00207a |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |