
Self-assembly of β-cyclodextrin-pillar[5]arene molecules into supramolecular nanoassemblies: morphology control by stimulus responsiveness and host–guest interactions†
Jie
Lu
,
Yingying
Deng
,
Peng
Liu
,
Qingqing
Han
* and
Long Yi
Jin
 *
*
Department of Chemistry, National Demonstration Centre for Experimental Chemistry Education, Yanbian University, Yanji 133002, P. R. China. E-mail: 0000008802@ybu.edu.cn; lyjin@ybu.edu.cn
First published on 2nd February 2023
Abstract
Macrocyclic molecules have attracted considerable attention as new functional materials owing to their unique pore size structure and excellent host–guest properties. With the development of macrocyclic compounds, the properties of mono-modified macrocyclic materials can be improved by incorporating pillar[n]arene or cyclodextrin derivatives through bridge bonds. Herein, we report the self-assembly of amphiphilic di-macrocyclic host molecules (H1–2) based on β-cyclodextrin and pillar[5]arene units linked by azophenyl or biphenyl groups. In a H2O/DMSO (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixed polar solvent, an amphiphile H1 with an azophenyl group self-assembled into unique nanorings and exhibited an obvious photoresponsive colour change. This photochromic behaviour makes H1 suitable for application in carbon paper materials on which arbitrary patterns can be erased and rewritten. The amphiphile H2, with a biphenyl unit, self-assembled into spherical micelles. These differences indicate that various linker units lead to changes in the intermolecular and hydrophilic–hydrophobic interactions. In a CHCl3/DMSO (19:1, v/v) mixed low-polarity solvent, the amphiphile H1 self-assembled into fibrous aggregates, whereas the molecule H2 assembled into unique nanoring aggregates. In this CHCl3/DMSO mixed solvent system, small nanosheet aggregates were formed by the addition of a guest molecule (G) composed of tetraphenylethene and hexanenitrile groups. With prolonged aggregation time, the small sheet aggregates further aggregated into cross-linked nanoribbons and eventually formed large nanosheet aggregates. The data reveal that the morphology of H1–2 can be controlled by tuning the intermolecular interactions of the molecules via the formation of host–guest complexes. Moreover, the polyhydroxy cyclodextrin unit on H1–2 can be strongly adsorbed on the stationary phase in column chromatography via multiple hydrogen bonds, and the singly modified pillar[5]arenes can be successfully separated by host–guest interactions.
:1, v/v) mixed polar solvent, an amphiphile H1 with an azophenyl group self-assembled into unique nanorings and exhibited an obvious photoresponsive colour change. This photochromic behaviour makes H1 suitable for application in carbon paper materials on which arbitrary patterns can be erased and rewritten. The amphiphile H2, with a biphenyl unit, self-assembled into spherical micelles. These differences indicate that various linker units lead to changes in the intermolecular and hydrophilic–hydrophobic interactions. In a CHCl3/DMSO (19:1, v/v) mixed low-polarity solvent, the amphiphile H1 self-assembled into fibrous aggregates, whereas the molecule H2 assembled into unique nanoring aggregates. In this CHCl3/DMSO mixed solvent system, small nanosheet aggregates were formed by the addition of a guest molecule (G) composed of tetraphenylethene and hexanenitrile groups. With prolonged aggregation time, the small sheet aggregates further aggregated into cross-linked nanoribbons and eventually formed large nanosheet aggregates. The data reveal that the morphology of H1–2 can be controlled by tuning the intermolecular interactions of the molecules via the formation of host–guest complexes. Moreover, the polyhydroxy cyclodextrin unit on H1–2 can be strongly adsorbed on the stationary phase in column chromatography via multiple hydrogen bonds, and the singly modified pillar[5]arenes can be successfully separated by host–guest interactions.
Introduction
Macrocyclic molecules, a class of cyclic molecules with specific pore sizes, have attracted the attention of many chemists and materials scientists in recent years as excellent host molecules in host–guest chemistry.1–8 Dimacrocyclic host molecules (DHM), as the name suggests, are a type of double host molecule formed by the covalent bridging of two macrocyclic molecules.9–12 As the host molecules, DHMs not only retain the original host–guest properties of macrocyclic compounds, but also display new properties based on the selection of specific bridging agents. For example, photoresponsive azobenzene can be selected as a bridging agent to confer specific photoresponsive properties to the designed materials.13–19 Different types of macrocyclic molecules can also be selected for bridging to form functional materials.20 This provides macrocyclic molecules with a more specific recognition ability to form host–guest molecules, leading to more selective separation or recognition properties for the fabrication of functional materials.Cyclodextrins (CD), which have a hydrophilic outer cavity and hydrophobic inner cavity, are ideal host molecules, similar to enzymes, and have been widely applied in the fields of catalysis, separation, food, and medicine.21–29 For example, Liu et al. constructed hyaluronidase-responsive polysaccharide supramolecular assemblies using chlorambucil, β-CD with hexylimidazolium units, and adamantyl-grafted hyaluronic acid. These aggregates can specifically target cancer cells and respond to hyaluronidase to induce the release of anticancer drugs. Interestingly, the functional β-CD obtained by hydrolysis can form a stable 1:1 host–guest complex with ATP, which considerably inhibits the hydrolysis of ATP and cuts off the energy supply of cancer cells, thus effectively alleviating the multidrug resistance of cancer cells.30 Pillar[n]arenes formed by methylene bridges linking p-dialkoxy benzene were first reported by Ogoshi in 2008, as a new host family of macrocyclic molecules.31 Although pillar[n]arene molecules have unique columnar structures and specific pore sizes, their poor solubility in water considerably limits their applications in materials science. To overcome these shortcomings, studies have attempted to modify the pillar[n]arene matrix via different methods to change the properties of pillar[n]arenes and expand their application range.32–38 For example, we previously reported the synthesis and self-assembly of an amphiphilic macromolecule composed of pillar[5]arene, biphenyl, and hydrophilic polyethylene glycol monomethyl ether groups, which can detect aliphatic diamines in aqueous solutions.39 Huang et al. linked crown ethers with pillar[5]arenes through a click reaction, and formed a dynamic rotane with guest molecules through a pH response.40 However, there are few examples of pillar[n]arenes and CD linked together via covalent bonds. To the best of our knowledge, only Xin et al. used a click reaction to bridge azide-modified α-CD and pillar[5]arene groups to form hybrid molecules. These molecules can be effectively inserted into a lipid bilayer to form artificial ion channels. As the length of the linker increases, the selectivity of the cation transport changes, where the hybrid molecules with the shortest channel have a specific transmembrane transport preference for K+. These results provide a biomimetic alternative to natural K+ channels.41 To date, there is no report on the synthesis of molecules based on β-CD and pillar[5]arene units connected by a covalent bond, as a linker.
With this in mind, in this study, we synthesised an amphiphilic β-CD-pillar[5]arene molecule H1 (Scheme 1), which is formed by the linkage of β-CD and pillar[5]arenes through azobenzene. Notably, the azobenzene unit, as a linker, leads to the photoisomerization of H1 for the construction of various nanostructural aggregates. To investigate the effect of the linker structure on the morphology of the self-assemblies, we also synthesised a molecule H2 containing biphenyl units (Scheme 1). Moreover, sheet-like supramolecular aggregates are constructed from the host–guest complex formed by H1–2 and the guest molecule G with a tetraphenylethene group, having strong aggregation-induced emission (AIE) properties.
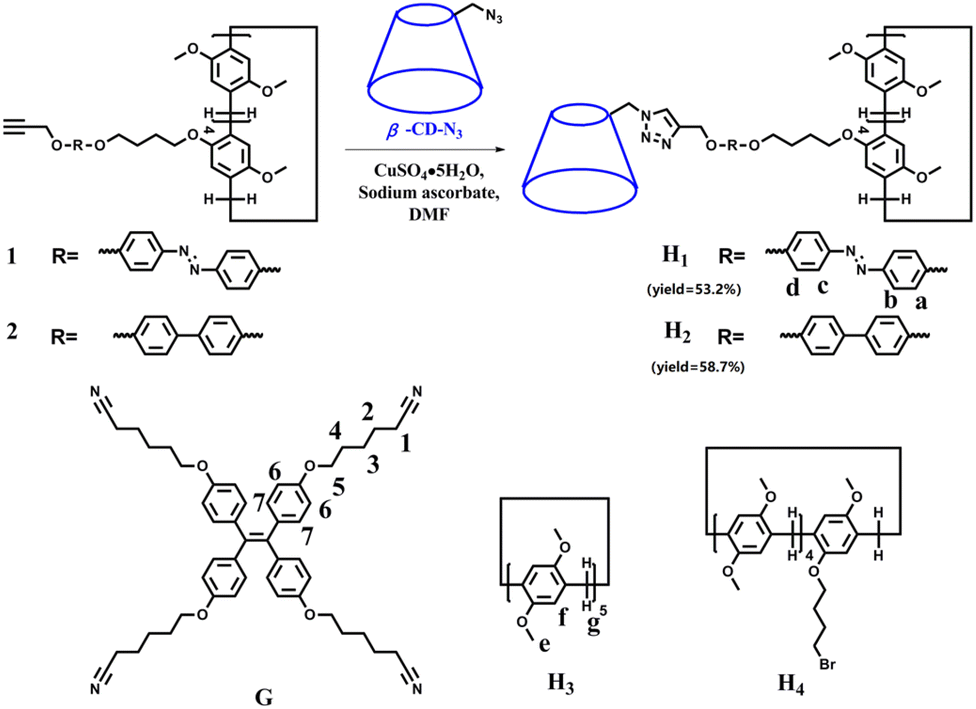 | ||
| Scheme 1 Synthesis of H1–2 and the chemical structures of G, H3, and H4. | ||
Results and discussion
Synthesis of H1 and H2 and the formation of nanoassemblies in H2O/DMSO or CHCl3/DMSO
In recent years, bridged pillar[n]arene derivatives42–44 and bridged cyclodextrin derivatives45–48 have been widely reported for applications in materials science. These molecules are either covalently bridged by two or more pillar[n]arene units or covalently bridged by CD units. However, there are few reports of pillar[n]arenes and cyclodextrins linked together by covalent bonds. Therefore, amphiphilic molecules H1 and H2 were synthesised by the linkage of β-CD and pillar[5]arene groups using azobenzene or biphenyl, respectively. Molecules H1 and H2 were obtained from a click reaction between an azide-modified β-CD and an azobenzene-functionalized pillar[5]arene or a biphenyl-functionalized pillar[5]arene. The structures of H1 and H2 were characterised using 1H-NMR and 13C-NMR spectroscopy (Fig. S27, S28, S29, and S30†), elemental analysis, and MALDI-TOF-MS (Fig. S31 and S32†). Analysis of the experimental data showed that the molecular structure was consistent with that shown in Scheme 1.Molecules H1 and H2 are composed of hydrophilic β-CD and hydrophobic rod segments that contain an azobenzene-functionalized pillar[5]arene or a biphenyl-functionalized pillar[5]arene. The strong amphiphilic characteristics of the β-CD unit enable it to exhibit a strong self-assembly behaviour through hydrophilic/hydrophobic interactions in polar solvents, as well as strong hydrogen bonding interactions induced by the large number of hydroxyl groups on β-CD in low-polarity solvents. Therefore, H2O/DMSO (v/v = 19:1) with high polarity and CHCl3/DMSO (v/v = 19:1) with low polarity were selected to study the aggregation behaviour of the molecules. Fig. 1 and Fig. S33† show the UV-vis and fluorescence (FL) spectra of H1 and H2 in DMSO, H2O/DMSO (v/v = 19:1), and CHCl3/DMSO (v/v = 19:1) solutions, respectively (3.03 × 10−6 mol L−1). Compared with that of H1 in DMSO solution, the UV-vis spectra (Fig. 1a and Fig. S33a†) of H1 exhibited a blue shift in H2O/DMSO and CHCl3/DMSO solutions. This indicates that the hydrophobic rod segment of H1 tends to adopt H-type aggregation.49–51 However, compared with that of H2 in DMSO, the UV-vis spectra (Fig. 1c and Fig. S33c†) of H2 exhibited a red shift in H2O/DMSO solution and a blue shift in CHCl3/DMSO solution. It is proposed that this shift is due to the different dipole moments of the azobenzene and biphenyl units of the H1 and H2 molecules, leading to differences in the strength of the hydrophilic–hydrophobic interactions and steric hindrance of the two molecules. Compared with the molecules in DMSO solution, obvious fluorescence quenching of H1 or H2 was observed in both H2O/DMSO and CHCl3/DMSO solutions. This further indicates the formation of molecular aggregates in the selected solvents. To determine the size of such aggregates, dynamic light scattering (DLS) data were acquired for the H2O/DMSO and CHCl3/DMSO solutions of H1 and H2. Molecules H1 and H2 formed aggregates with diameters of approximately 21 nm and 20 nm, respectively, in H2O/DMSO solution (Fig. S34†), and formed aggregates with diameters of approximately 20 nm and 18 nm, respectively, in CHCl3/DMSO solution.
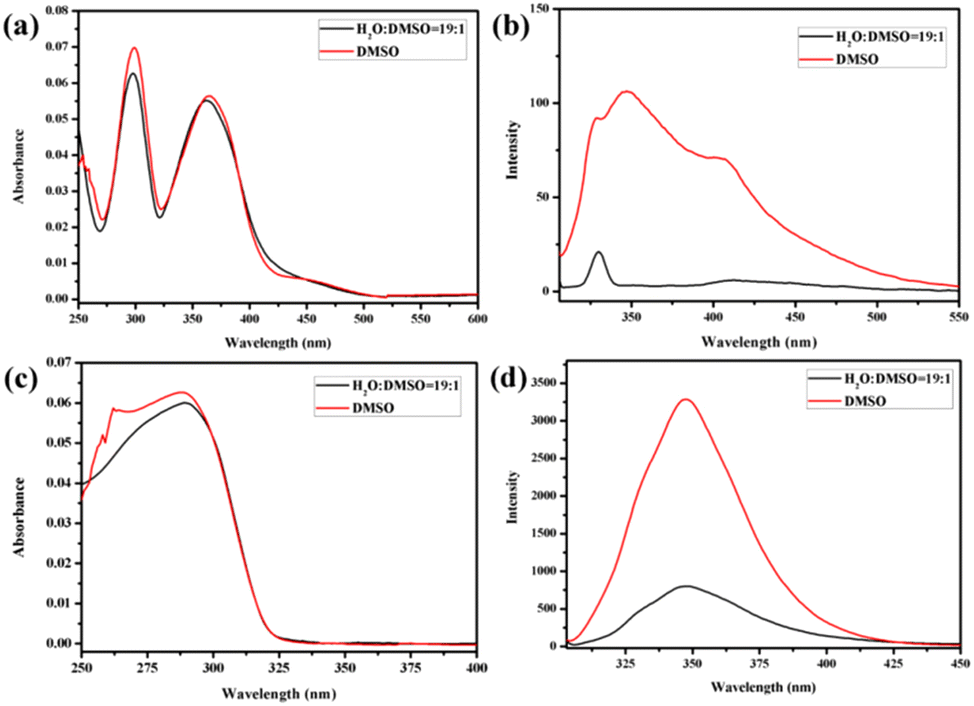 | ||
| Fig. 1 The absorption spectra of (a) H1 and (c) H2, and emission (excited at 298 nm, Ex bandwidth: 2.5 nm; Em bandwidth: 5 nm) spectra of (b) H1 and (d) H2 in DMSO and H2O/DMSO (v/v = 19:1) solutions (3.03 × 10−6 mol L−1). | ||
Atomic force microscopy (AFM) and transmission electron microscopy (TEM) were used to obtain detailed information about the morphology of the aggregates formed by H1 and H2. H1 self-aggregated into nanorings with a uniform diameter and width of ∼7 nm and ∼20 nm (Fig. 2a and b), respectively, in H2O/DMSO (v/v = 19:1) solutions. Additionally, the full spread length of H1 was found to be 3.3 nm (Fig. S35a†) using CPK modelling, which is consistent with the observed half-diameter. This implies that the rigid hydrophobic segments of H1 are arranged in a circular shape surrounded by hydrophilic β-CD units to minimise the surface tension of the molecule in aqueous solution (Fig. 3a). Unlike the structure in H2O/DMSO solution, in CHCl3/DMSO (v/v = 19:1) solution, H1 self-assembled into a nanofibre structure with a diameter of approximately 4.5 nm, which is between one and two times the molecular length when the molecules are fully unfolded. It is proposed that the hydrophilic β-CD units were staggered into rings through hydrogen bonding and surrounded by hydrophobic pillar[5]arene units to minimise the surface tension of the molecules in the CHCl3/DMSO solution (Fig. 3b). This may also be due to the weak hydrophilic–hydrophobic interactions of the molecules and stronger hydrogen bond interactions between the β-CD units in the less polar CHCl3/DMSO solution. For H2 with a biphenyl unit as the linker group, the dipole moment is weaker than that of molecule H1 with azobenzene as the linker unit. Thus, H2 undergoes weaker hydrophilic–hydrophobic interactions in the H2O/DMSO solution with relatively high polarity, leading to assembly into spherical micelles with a diameter of approximately 20 nm, as shown in Fig. 2(d and e). This is considerably longer than the length of 3.1 nm (Fig. S35b†) when the molecule is fully unfolded. Thus, in an aqueous solution, the rigid hydrophobic segments of H2 first aggregate into a nucleus, which is wrapped by hydrophilic β-CD units, and then gradually aggregate to form stable spherical micelles by synergistic driving forces such as hydrogen bonds, π–π stacking, and hydrophobic interactions (Fig. 3c). Compared with the azobenzene units, the biphenyl units with weaker dipole moments undergo weaker π–π stacking interactions in CHCl3/DMSO solution; thus, H2 was assembled into circular aggregates with a uniform diameter and width of ∼5 nm and ∼18 nm (Fig. 2f), respectively. Unlike the assembly arrangement of H1, the hydrophilic β-CD units of H2 are staggered into rings through hydrogen bonding and surrounded by hydrophobic pillar[5]arene units in CHCl3/DMSO solution (Fig. 3d).
 | ||
| Fig. 2 The TEM image of (a) H1 and (d) H2 and AFM image of (b) H1 and (e) H2 obtained from 3.03 × 10−6 mol L−1 H2O/DMSO (v/v = 19:1) solutions. The TEM image of (c) H1 and (f) H2 obtained from 3.03 × 10−6 mol L−1 CHCl3/DMSO (v/v = 19:1) solution. | ||
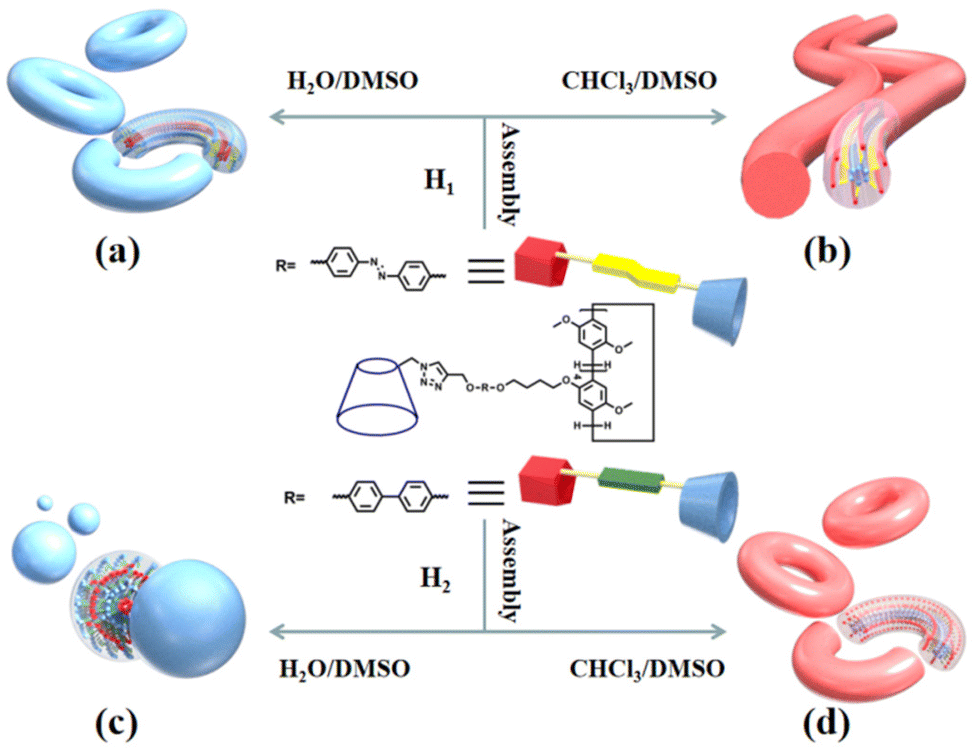 | ||
| Fig. 3 Diagrammatic representation of the self-assembly of molecules H1 (a and b) and H2 (c and d) in H2O/DMSO and CHCl3/DMSO solutions, respectively. | ||
Construction of reversible photoresponsive nanostructures of H1 in aqueous solution
The trans azobenzene derivatives can form cis isomers under ultraviolet (UV) irradiation, which can be restored to the trans form upon irradiation with visible light or by increasing the temperature.52–54 Thus, the photoresponsive properties of H1 in DMSO solution were initially studied by 1H NMR spectral analysis. As shown in Fig. 4, the signals of the protons on the azobenzene of the trans isomer were shifted upfield, whereas those of the protons on the pillar[5]arene units or β-CD units were not shifted when H1 was irradiated with 365 nm UV light for 20 min. This is attributed to the photo-induced transformation of the trans azobenzene units into the cis isomers (cis-H1). Interestingly, the DMSO solution of H1 changed from pale-yellow to an orange-yellow solution of cis-H1 upon irradiation with UV light (Fig. S36†), confirming the occurrence of photoisomerization. UV-vis spectroscopy was used to further examine the photoisomerization of H1 in H2O/DMSO (v/v = 19:1) mixed solution. Before UV irradiation, the H2O/DMSO solution of H1 at 25 °C exhibited two characteristic absorption bands (Fig. 5a) at 298 nm and 362 nm, which are related to the n–π* transitions of the pillar[5]arene units and the π–π* transitions of the trans isomers of the azobenzene moieties, respectively. After irradiation with 365 nm UV light, the n–π* absorption band around 298 nm corresponding to the pillar[5]arene units was almost unchanged; however, a noteworthy decrease in the π–π* absorption band at 362 nm and a new n–π* absorption band at approximately 450 nm were observed, with a clear isosbestic point at 436 nm (Fig. 5a). This indicates trans-to-cis isomerisation of the azobenzene units. When the solution of cis-H1 was irradiated with visible light, the n–π* absorption band at 362 nm gradually increased and the π–π* absorption band at 450 nm gradually decreased (Fig. 5b). This phenomenon became more obvious as the visible-light exposure time increased. A similar phenomenon occurred when the solution of cis-H1 was heated (Fig. 5c), suggesting cis-to-trans isomerisation of the azobenzene units. The trans-to-cis or cis–trans isomerisation was cycled by alternate illumination with 365 nm UV light or visible light (Fig. 5d). Taking advantage of this property of H1, as well as the obvious colour change of H1 shown in Fig. S36,† a H2O/DMSO solution of H1 was applied to ordinary copy paper to fabricate photosensitive erasable writing materials. As shown in Fig. S37,† the copy paper coated with H1 was erasable, and specific pictures and text were rewritten with arbitrary patterns.
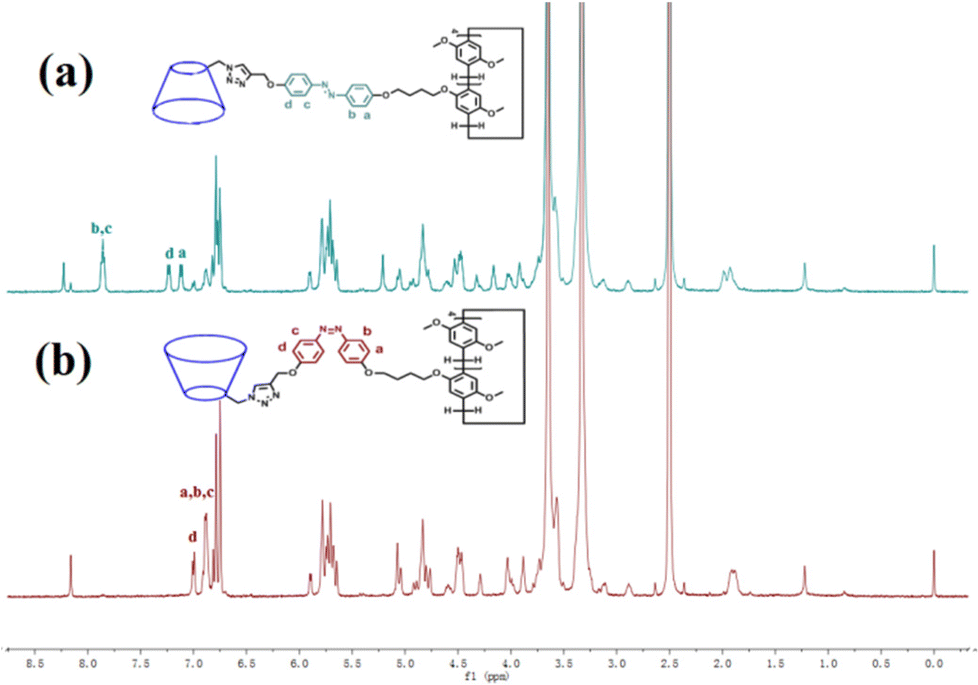 | ||
| Fig. 4 1H NMR spectra (500 MHz, DMSO-d6, 25 °C) of (a) H1 (2.72 × 10−3 mol L−1) and (b) H1 (2.72 × 10−3 mol L−1) after irradiation with 365 nm UV light for 20 min. | ||
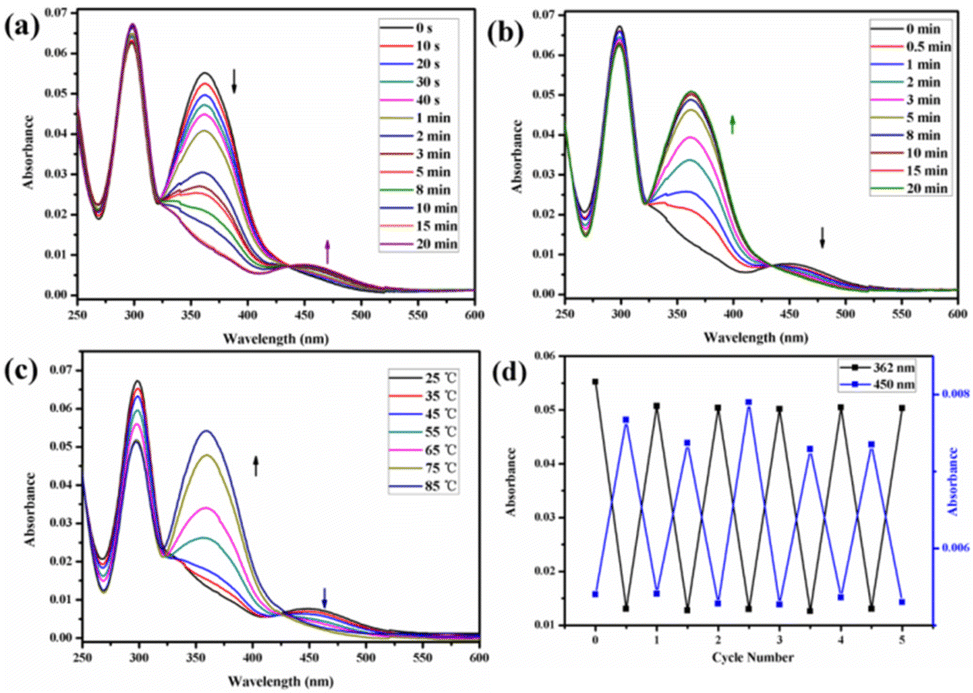 | ||
| Fig. 5 UV-vis spectra of H1 in H2O/DMSO (v/v = 19:1) solution upon exposure to different stimuli: (a) duration of UV irradiation was set to 0–20 min at 365 nm; (b) duration of vis irradiation was set to 0–20 min; (c) different temperatures of H1 in H2O/DMSO (v/v = 19:1) solution after 20 min of UV exposure. (d) Absorbance at wavelengths of 362 nm and 450 nm under alternate UV (365 nm) irradiation for 20 min and vis irradiation for 20 min. | ||
TEM and AFM were used to intuitively show the effect of UV light on the morphology of the molecular aggregates in solution. Fig. 6 shows the TEM and AFM images of cis-H1. The images show annular aggregates with an outer diameter of approximately 38 nm. The size of the aggregates increased compared with that before illumination (Fig. 2a and b) when the H2O/DMSO solution of the dimacrocyclic molecule H1 was irradiated with 365 nm UV light for 20 min. This may result from the synergistic driving force of the molecular arrangement. In this system, the polarity of cis-H1 is larger than that of trans-H1, which enhances the hydrophilic–hydrophobic interactions in H2O/DMSO solution. In contrast, the increased polarity leads to increased intermolecular force to form a larger-scale arrangement.
 | ||
| Fig. 6 (a) Negative-staining TEM images and (b) AFM images of cis-H1 (H1 after irradiation with 365 nm UV light for 20 min) obtained from 3.03 × 10−6 mol L−1 H2O/DMSO (v/v = 19:1) solutions. (c) Enlarged image of part b. | ||
Control of the nanostructures of H1–2 by host–guest complexes
It has been reported that a stable host–guest complex can be formed between alkylnitrile molecules and pillar[5]arene units. Tetraphenyl ethylene, a small organic molecule with AIE properties, has been widely used in advanced materials in recent years owing to its unique luminescence properties.55–57 Based on this, we prepared a guest molecule G containing tetraphenylethylene and four alkylnitrile units that are strongly associated with pillar[5]arene units. The formation of host–guest complexes between H1–2 and G was investigated using 1H NMR and FL spectroscopy. The above results show that H1 and H2 undergo very strong aggregation in chloroform solution. This induces poor solubility of molecules H1 and H2 in the chloroform solution and limits detailed investigation of the host–guest interaction in chloroform. Furthermore, because the aggregation behaviour leads to weak absorbance of H1 and H2 and fluorescence quenching, it is impossible to determine the association constant of the host–guest complex by UV and fluorescence titration. Therefore, H3 was selected as a model for H1–2 to study the host–guest interaction between the pillar[5]arene unit and alkyl nitrile. In the 1H NMR spectrum of the chloroform solution of G and 4 molar equivalents of H3, the signals of the He,f protons of H3 were shifted from 6.75 ppm and 3.64 ppm to 6.79 ppm and 3.66 ppm, respectively, whereas the signals of the alkyl H1–5 protons on molecule G were both shifted upfield (Fig. S38†). The shift suggests the formation of a host–guest complex in chloroform. To further confirm this conjecture, 1H–1H NOESY NMR experiments were carried out. As shown in Fig. 7, the He,f protons of H3 and the H1–5 alkyl protons of G were significantly correlated, which indicates that there is a strong host–guest interaction between molecular H3 and G. To calculate the value of the association, the equilibrium constant was calculated from the FL titration in CDCl3 (Fig. S39a†).58 When the concentration of G was 0.05 mM, the fluorescence intensity gradually increased with an increase in H3. According to the Benesi–Hildebrand equation, the association constant Ka for the interaction of the G and H3 molecules was calculated to be 1.22 × 1017 M−4 (Fig. S39b†). This higher association constant is reasonable because compound G has four recognition sites for complexation. As shown in Fig. S40,† the plot of 1/ΔX vs. 1/[host]4 had a good linear least-squares fit, with a better correlation coefficient compared to that of 1/ΔX vs. 1/[host]n (n = 1, 2, 3), indicating that the stoichiometry of the inclusion complex between H3 and compound G was 4:1. The stoichiometry was also confirmed using the continuous variation method (Job's plot), as shown in Fig. S40.†
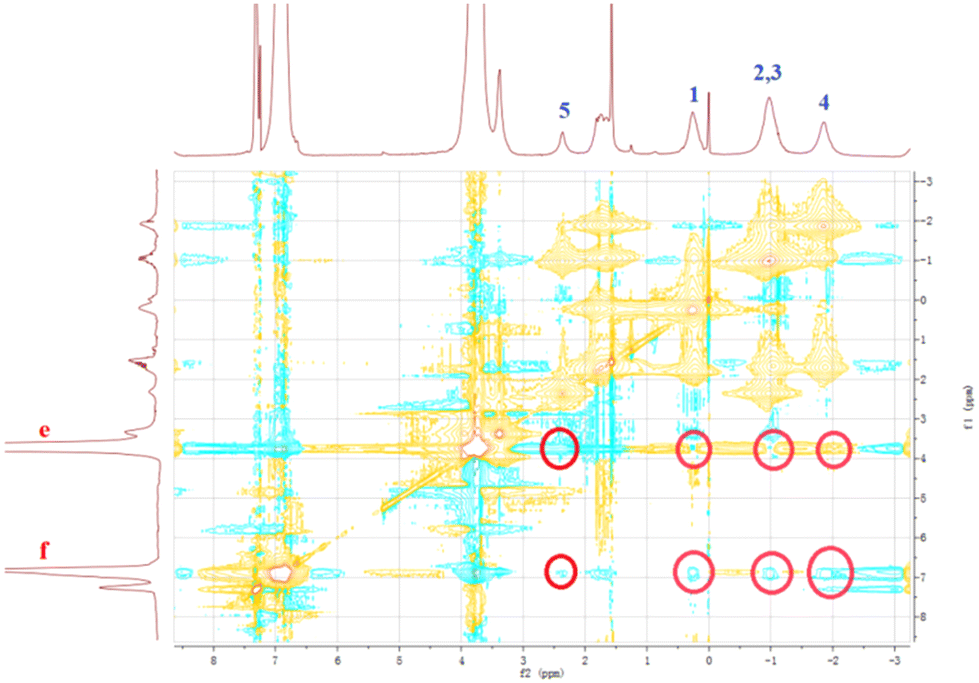 | ||
| Fig. 7
1H–1H NOESY NMR spectrum (500 MHz) of the complex of G with H3 at a 1:4 molar ratio in CDCl3 at 25 °C. | ||
Subsequently, G molecules were added to H1 and H2, and the 1H NMR spectra were recorded to monitor the host–guest interactions in CDCl3:DMSO-d6 (v/v = 1:1) solution. As shown in Fig. 8 and S41,† the signals of the proton on the benzene ring of the pillar[5]arene unit in H1 or H2 were shifted downfield, and the signals of the alkyl H1–5 protons on molecule G were both shifted upfield. The chemical shift values were consistent with those of molecule G in deuterated chloroform solution of H4, indicating that H1 or H2 and the guest molecule G formed complexes through strong host–guest interactions. The signals of the protons on the cyclodextrin unit did not change during this process (Fig. S42†), which further confirmed that a stable 4:1 host–guest complex (H1⊂G or H2⊂G) was formed between the pillar[5]arene unit and the G molecule in CHCl3/DMSO.
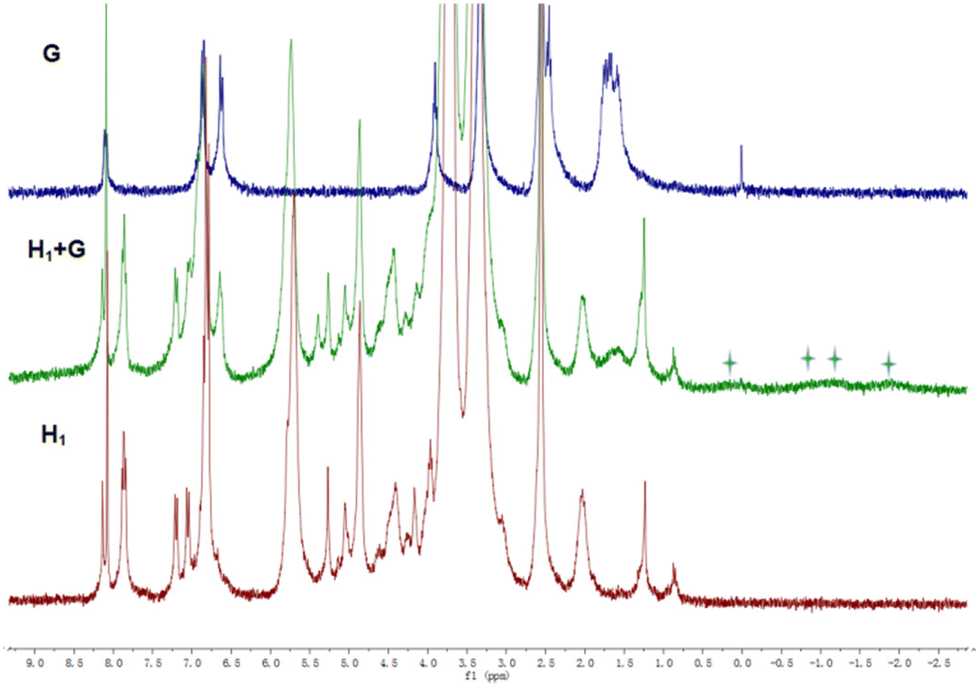 | ||
| Fig. 8
1H NMR spectra of G (blue line), H1 + G (green line), and H1 (red line) in CDCl3:DMSO-d6 (v/v = 1:1) at 25 °C. | ||
Since the guest molecule G itself is an excellent aggregation-induced luminescent material, fluorescence spectroscopy was used to further verify the formation of the host–guest complex. However, as illustrated in Fig. S43(a),† no obvious fluorescence emission was observed when 1 equiv. of the guest molecule G was added to a solution of CHCl3/DMSO (v/v = 19:1) containing 4 equiv. of H1. Interestingly, when the guest molecule G was added to the solution of CHCl3/DMSO (v/v = 19:1) of H2, a strong fluorescence emission peak appeared at 488 nm (Fig. S43b†). This means that H2⊂G exhibits strong AIE, compared to H1⊂G, which is useful for application in materials science. The fluorescence quenching of H1⊂G is attributed to the absorption of the emission of molecule G by the photo-responsive azobenzyl group in the H1 molecule, which suppresses the strong fluorescence of the tetraphenylethene unit, consistent with the reports by other groups.59 To clearly observe such fluorescence changes with the naked eye, 1 equiv. of the guest molecule G was added to 4 equiv. of H1 or H2 in CHCl3/DMSO (v/v = 5:1). Under irradiation with 365 nm ultraviolet light, the solution of G presented a weak light-blue fluorescence (Fig. S44†), whereas H1⊂G in solution appeared orange-yellow without obvious fluorescence. Interestingly, the H2⊂G solution showed bright-white fluorescence when irradiated with UV light, which is consistent with the results of the fluorescence spectroscopy analysis. Notably, the presence of the H1 molecule significantly quenched the fluorescence; thus, this amphiphilic molecule can potentially be used as a sunscreen material.
In order to intuitively demonstrate the influence of the host–guest complex on the aggregate morphology in solution, TEM images of H1⊂G and H2⊂G were acquired. The experimental data are presented in Fig. 9. When the host–guest complex H1⊂G (7.58 × 10−7 mol L−1) was placed in CHCl3/DMSO (v/v = 19:1) solution for 1 h, an irregular circular small sheet structure with a diameter of approximately 100 nm was observed. When the solution was allowed to stand for 4 h, the aggregate changed from an irregular circular sheet structure to a cross-linked nanoribbon aggregate. After standing for 24 h, larger nanosheet aggregates were observed. The height of this lamellar aggregate was approximately 7 nm, as measured by AFM (Fig. S45a and c†), which is similar to the sum of the H1 and G lengths when the molecules are fully extended (Fig. S35c†). Compared to H1, which formed nanofibres in CHCl3/DMSO, the supramolecular H1⊂G molecule composed of rigid pillar[5]arenes and tetraphenylethene through a host–guest interaction undergoes a strong π–π stacking interaction to induce the formation of nanosheet-like aggregates with less curvature than the aggregates of H1 in CHCl3/DMSO solution. In CHCl3/DMSO (v/v = 19:1) solution, tetraphenylethene, azobenzene, and the rigid segment of the pillar[5]arenes units are connected by an alkyl nitrile group arranged in the inner sheet through strong π–π stacking interactions, and are surrounded by four cyclodextrin units at the outer side of the supramolecule (Fig. 10a).
 | ||
| Fig. 9 TEM images of molecule H1⊂G (c/c = 4:1) obtained from CHCl3/DMSO (v/v = 19:1) solutions after (a) 1 h, (b) 4 h and (c) 24 h. TEM images of molecule H2⊂G (c/c = 4:1) obtained from CHCl3/DMSO (v/v = 19:1) solution after (d) 1 h, (e) 4 h and (f) 24 h. | ||
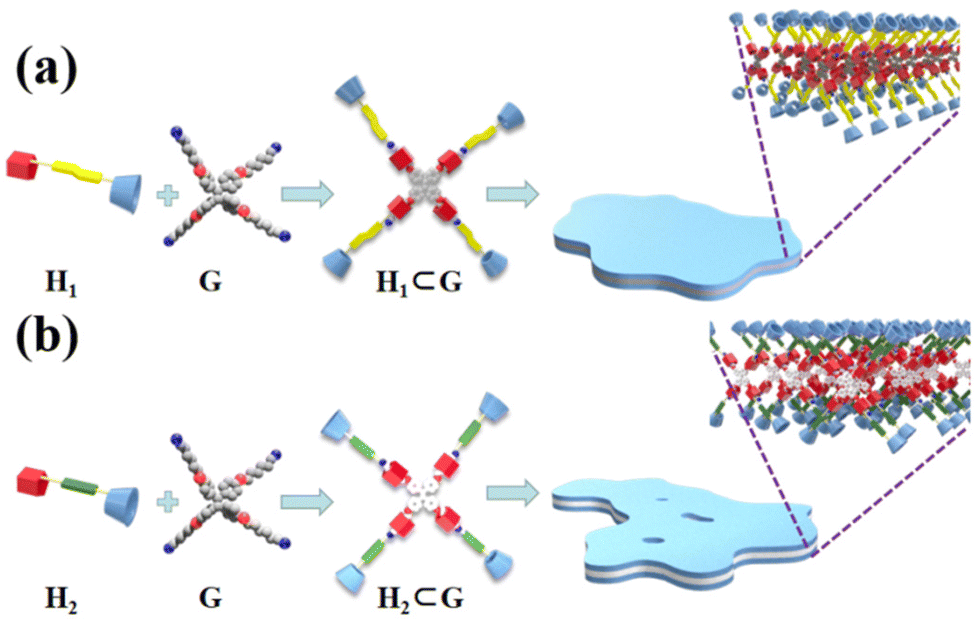 | ||
| Fig. 10 Formation of host–guest complexes (a) H1⊂G and (b) H2⊂G, and self-assembly in CHCl3/DMSO solution, respectively. | ||
Similarly, after immersion of the host–guest complex H2⊂G (7.58 × 10−7 mol L−1) in CHCl3/DMSO (v/v = 19:1) solution for 1 h, small nanosheet-like aggregates with a diameter of approximately 50 nm were obtained, where the size was smaller than that of the aggregates of H1⊂G (Fig. 9d). We speculate that this is because the linker of H1 contains azobenzene units with a larger dipole moment than that of the biphenyl units. The different driving forces of π–π stacking between the two molecules result in slightly different aggregate sizes of the molecules in solution. With an increase in the immersion time to 4 h, cross-linked nanoribbon aggregates were also found (Fig. 9e), and when the immersion time was extended to 24 h, large nanosheet aggregates with a height of ∼7 nm (Fig. 9f and S45b and d†) were observed. The experimental results revealed that various nanostructural aggregates can be constructed by the formation of host–guest supramolecular molecules by precisely tuning the intermolecular interactions (Fig. 10b).
In the synthesis of singly modified pillar[5]arene H4, owing to the excessive addition of p-dimethoxybenzene and the low solubility of the by-product H3, a large amount of the by-product H3 was generated. Since the side-chain bromobutoxy group in H4 can form a host–guest complex60 with H3, separating these molecules by column chromatography presents challenges. Accordingly, we considered the application of host–guest complexes in the separation of H3 and H4. Due to the strong intermolecular hydrogen bonds between the multiple hydroxyl units on cyclodextrin and ordinary column chromatography silica gel or neutral aluminium oxide (N-Al2O3), compounds containing cyclodextrin cannot be separated by ordinary column chromatography using silica gel and N-Al2O3. Taking advantage of this property, H1 or H2 was thoroughly stirred with N-Al2O3 in DMSO as the stationary phase for column chromatography, and then fully flushed with chloroform solution. After standing for 24 h, a mixture of chloroform and petroleum ether (v/v = 1:8) was used to separate the mixture of H3 and H4. After separation using this chromatographic column, relatively pure H4 was obtained. It is proposed that the effective separation of H3 and H4 was achieved due to the side-chain bromobutoxy group in H4, which can form host–guest complexes with the pillar[5]arene unit of H1 or H2. However, for molecule H3, there is only a weak π–π stacking interaction between H3 and H1 or H2. This interaction is much weaker than the host–guest interaction of H1 or H2. Infrared (IR) spectroscopy was used to confirm the strong absorption between H1 or H2 and N-Al2O3 (Fig. S46(a)†). Compared with the separated column material, the strength of the stretching vibration peak of the hydroxyl group of H1 decreased, and the absorption peak moved to a lower wavenumber, indicating that H1 interacts with N-Al2O3 by hydrogen bonding. The stationary phase composed of H2 and N-Al2O3 also showed the same phenomenon in the IR tests (Fig. S46b†). Thin-layer chromatography (TLC) was used to clarify the separation process. As shown in Fig. S47,† when the TLC plate did not contain H1 or H2, the Rf values of H3 and H4 were almost the same; however, when the TLC plate contained H1 or H2, molecules H3 and H4 were separated owing to the host–guest interactions of H1/H2 and H4. We believe that this material may be effective for separating molecules such as alkyl amines and halogenated hydrocarbons, which can form host–guest complexes with H1 or H2.
Experimental section
Molecules 1, 2, H3, H4, β-CD-N3, and G (Scheme 1 and Scheme S1†) were prepared according to previously described methods. All the structural characterisation data are summarised in the ESI (Fig. S1–S32†).Synthesis of H1 and H2
These compounds were synthesised according to the same procedure. The synthesis of H1 is described as a representative example.Conclusions
Amphiphilic bridged β-cyclodextrin-pillar[5]arene molecules H1 and H2 were successfully synthesised, and their self-assembly behaviour was investigated. Molecule H1 contains a photoresponsive azophenyl group, whereas H2 contains a biphenyl group as a linker. In the H2O/DMSO mixed solvent, H1 self-assembled into nanorings, whereas H2 self-assembled into spherical micelles. Similarly, in CHCl3/DMSO, H1 self-assembled into fibrous aggregates, whereas H2 assembled into nanoring aggregates, revealing that a slight modification of the chemical structure of the bridging bond can precisely control the morphology of the nanoaggregates. The effect of UV irradiation on the self-assembly of H1 in H2O/DMSO solution was investigated. The synergistic interaction between π–π stacking and hydrophilic–hydrophobic interactions of H1 in solution led to larger nanoaggregates of cis-H1 than those of trans-H1 in the H2O/DMSO mixed system. Notably, the photoresponsive colour change of H1 enabled its application in carbon paper materials for the fabrication of photosensitive erasable writing materials. Analysis of the self-assembly behaviour of the host–guest complex formed between H1–2 and G in CHCl3/DMSO solution showed that H1 and G form stable host–guest complexes, which assemble into small sheet aggregates under the action of strong hydrogen bonding and π–π stacking. With prolonged assembly time, the small sheet aggregates further aggregate into cross-linked nanoribbon aggregates and finally assemble into large sheet aggregates. Similarly, the host–guest complex between H2 and G eventually aggregates into large sheet-like aggregates. Notably, because the molecule contains polyhydroxyl β-cyclodextrin units, the molecules can be bound to the neutral alumina filler through hydrogen bonding. Interestingly, the molecular filler can be effectively used in the separation of singly modified pillar[5]arenes from the derivatives of pillar[5]arenes. We expect that these functional molecules will be widely applied in the fields of molecular recognition, sensing, and separation.Author contributions
J. Lu: methodology, validation, formal analysis, conducting the experiments, analysis of the experimental results, and writing – original draft. Y. Deng and P. Liu: assisted in the preparation of the molecules. Q. Han: performed the TEM and AFM experiments and analyzed the experimental results. Long Yi Jin: conceptualization, methodology, investigation, resources, supervision, project administration, funding acquisition, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 21961041 and 21562043) and the Higher Education Discipline Innovation Project (D18012).References
- Y. Mei, Q.-W. Zhang, Q. Gu, Z. Liu, X. He and Y. Tian, J. Am. Chem. Soc., 2022, 144, 2351–2359 CrossRef CAS PubMed.
- H. Zhu, L. Shangguan, D. Xia, J. H. Mondal and B. Shi, Nanoscale, 2017, 9, 8913–8917 RSC.
- H. B. Jeon, S. Park, K. R. Ryu, S. K. Ghosh, J. Jung, K. M. Park and J. W. Ha, Chem. Sci., 2021, 12, 7115–7124 RSC.
- Y.-F. Li, Z. Li, Q. Lin and Y.-W. Yang, Nanoscale, 2020, 12, 2180–2200 RSC.
- H. Aramoto, M. Osaki, S. Konishi, C. Ueda, Y. Kobayashi, Y. Takashima, A. Harada and H. Yamaguchi, Chem. Sci., 2020, 11, 4322–4331 RSC.
- T. Xiao, L. Xu, L. Zhou, X.-Q. Sun, C. Lin and L. Wang, J. Mater. Chem. B, 2019, 7, 1526–1540 RSC.
- C. Tu, W. Wu, W. Liang, D. Zhang, W. Xu, S. Wan, W. Lu and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202203541 CrossRef CAS PubMed.
- Y.-G. Jia, M. Zhang and X. X. Zhu, Macromolecules, 2017, 50, 9696–9701 CrossRef CAS.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Sci., 2022, 13, 5951–5956 RSC.
- H. Li, Y. Yang, F. Xu, Z. Duan, R. Li, H. Wen and W. Tian, Org. Chem. Front., 2021, 8, 1117–1124 RSC.
- H. Kitagishi, S. Kurosawa and K. Kano, Chem. – Asian J., 2016, 11, 3213–3219 CrossRef CAS PubMed.
- Y. Liu, Z. Su, S. Jiang, H. Sun, H. Lyu and Z. Xie, Polym. Chem., 2021, 12, 6123–6133 RSC.
- L. Zhang, J. Gu, X. Luo, Z. Tang, Y. Qu, C. Zhang, H. Liu, J. Liu, C. Xie and Z. Wu, Nanoscale, 2022, 14, 976–983 RSC.
- M. Fujiwara, M. Akiyama, M. Hata, K. Shiokawa and R. Nomura, ACS Nano, 2008, 2, 1671–1681 CrossRef CAS PubMed.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- M. Zheng and J. Yuan, Org. Biomol. Chem., 2022, 20, 749–767 RSC.
- A. Abdollahi, H. Roghani-Mamaqani, B. Razavi and M. Salami-Kalajahi, Polym. Chem., 2019, 10, 5686–5720 RSC.
- G. K. Joshi, K. N. Blodgett, B. B. Muhoberac, M. A. Johnson, K. A. Smith and R. Sardar, Nano Lett., 2014, 14, 532–540 CrossRef CAS PubMed.
- T. Ogoshi, K. Yoshikoshi, T. Aoki and T.-a. Yamagishi, Chem. Commun., 2013, 49, 8785–8787 RSC.
- X. Yan, D. Xu, J. Chen, M. Zhang, B. Hu, Y. Yu and F. Huang, Polym. Chem., 2013, 4, 3312–3322 RSC.
- L. Yue, H. Ai, Y. Yang, W. Lu and L. Wu, Chem. Commun., 2013, 49, 9770–9772 RSC.
- W. C. E. Schofield, C. D. Bain and J. P. S. Badyal, Chem. Mater., 2012, 24, 1645–1653 CrossRef CAS.
- F.-F. Shen, Y. Chen, X. Dai, H.-Y. Zhang, B. Zhang, Y. Liu and Y. Liu, Chem. Sci., 2021, 12, 1851–1857 RSC.
- F. Seidi, A. A. Shamsabadi, M. Amini, M. Shabanian and D. Crespy, Polym. Chem., 2019, 10, 3674–3711 RSC.
- S. Datz, B. Illes, D. Gößl, C. v. Schirnding, H. Engelke and T. Bein, Nanoscale, 2018, 10, 16284–16292 RSC.
- B. Zhang, W. Guan, S. Zhang, B. Li and L. Wu, Chem. Commun., 2016, 52, 5308–5311 RSC.
- D. J. DeDora, C. Suhrland, S. Goenka, S. M. Chowdhury, G. Lalwani, L. R. Mujica-Parodi and B. Sitharaman, J. Biomed. Mater. Res., Part B, 2016, 104, 1457–1464 CrossRef CAS PubMed.
- Z. Liu, W. Zhou, J. Li, H. Zhang, X. Dai, Y. Liu and Y. Liu, Chem. Sci., 2020, 11, 4791–4800 RSC.
- L. Zhou and J. Yu, J. Sep. Sci., 2022, 45, 2310–2320 CrossRef CAS PubMed.
- C. Chen, Y. Chen, X. Dai, J. Li, S. Jia, S. Wang and Y. Liu, Chem. Commun., 2021, 57, 2812–2815 RSC.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- T. Ogoshi, T.-a. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- H. Lei, X. Chen, L. Xue, L. Sun, J. Chen, Z. Tan, Z.-G. Zhang, Y. Li and G. Fang, Nanoscale, 2018, 10, 8088–8098 RSC.
- Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Lo and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 8577–8589 CrossRef CAS PubMed.
- T. Ogoshi, K. Maruyama, Y. Sakatsume, T. Kakuta, T.-a. Yamagishi, T. Ichikawa and M. Mizuno, J. Am. Chem. Soc., 2019, 141, 785–789 CrossRef CAS PubMed.
- T. Xiao, W. Zhong, L. Xu, X.-Q. Sun, X.-Y. Hu and L. Wang, Org. Biomol. Chem., 2019, 17, 1336–1350 RSC.
- T. Ogoshi, T. Kakuta and T.-a. Yamagishi, Angew. Chem., Int. Ed., 2019, 58, 2197–2206 CrossRef CAS PubMed.
- T. Ogoshi, R. Shiga and T.-a. Yamagishi, J. Am. Chem. Soc., 2012, 134, 4577–4580 CrossRef CAS PubMed.
- J. Lu, X. Gou, Y. Deng, Y.-R. Pei, Z. Huang and L. Y. Jin, Dyes Pigm., 2022, 199, 110052 CrossRef CAS.
- X. Yan, P. Wei, Z. Li, B. Zheng, S. Dong, F. Huang and Q. Zhou, Chem. Commun., 2013, 49, 2512–2514 RSC.
- P. Xin, H. Kong, Y. Sun, L. Zhao, H. Fang, H. Zhu, T. Jiang, J. Guo, Q. Zhang, W. Dong and C.-P. Chen, Angew. Chem., Int. Ed., 2019, 58, 2779–2784 CrossRef CAS PubMed.
- Y. Cai, X. Yan, S. Wang, Z. Zhu, M. Cen, C. Ou, Q. Zhao, Q. Yan, J. Wang and Y. Yao, Inorg. Chem., 2021, 60, 2883–2887 CrossRef CAS PubMed.
- Y. Wang, M.-Z. Lv, N. Song, Z.-J. Liu, C. Wang and Y.-W. Yang, Macromolecules, 2017, 50, 5759–5766 CrossRef CAS.
- S. Sun, X.-Y. Hu, D. Chen, J. Shi, Y. Dong, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 2224–2229 RSC.
- X. Dai, X. Dong, Z. Liu, G. Liu and Y. Liu, Biomacromolecules, 2020, 21, 5369–5379 CrossRef CAS PubMed.
- Y. Liu, Y. Chen, L. Li, H. Y. Zhang, S. X. Liu and X. D. Guan, J. Org. Chem., 2001, 66, 8518–8527 CrossRef CAS PubMed.
- Y. Liu, G.-S. Chen, L. Li, H.-Y. Zhang, D.-X. Cao and Y.-J. Yuan, J. Med. Chem., 2003, 46, 4634–4637 CrossRef CAS PubMed.
- X. Guan, D. Zhang, T. Jia, Y. Zhang, L. Meng, Q. Jin, H. Ma, D. Lu, S. Lai and Z. Lei, Ind. Eng. Chem. Res., 2017, 56, 3913–3919 CrossRef CAS.
- N. Ye, Y.-r. Pei, Q. Han, M. Lee and L. Y. Jin, Soft Matter, 2021, 17, 6661–6668 RSC.
- S. Yu, R. Shan, G.-Y. Sun, T. Chen, L. Wu and L. Y. Jin, ACS Appl. Mater. Interfaces, 2018, 10, 22529–22536 CrossRef CAS PubMed.
- J. Lu, Y. Deng, K. Zhong, Z. Huang and L. Y. Jin, Polym. Int., 2022, 71, 478–486 CrossRef CAS.
- S. Yu, T. Yang, M. Sui, G.-Y. Sun, T. Chen and L. Y. Jin, Dyes Pigm., 2019, 171, 107694 CrossRef CAS.
- S. Qian, S. Li, W. Xiong, H. Khan, J. Huang and W. Zhang, Polym. Chem., 2019, 10, 5001–5009 RSC.
- A. Adam and G. Haberhauer, J. Am. Chem. Soc., 2017, 139, 9708–9713 CrossRef CAS PubMed.
- G. Zhang, B. Li, Y. Zhou, X. Chen, B. Li, Z.-Y. Lu and L. Wu, Nat. Commun., 2020, 11, 425 CrossRef CAS PubMed.
- S. Wang, Y. Wang, Z. Chen, Y. Lin, L. Weng, K. Han, J. Li, X. Jia and C. Li, Chem. Commun., 2015, 51, 3434–3437 RSC.
- N. Song, D.-X. Chen, Y.-C. Qiu, X.-Y. Yang, B. Xu, W. Tian and Y.-W. Yang, Chem. Commun., 2014, 50, 8231–8234 RSC.
- H. Zhang, K. T. Nguyen, X. Ma, H. Yan, J. Guo, L. Zhu and Y. Zhao, Org. Biomol. Chem., 2013, 11, 2070–2074 RSC.
- R. Arumugaperumal, W.-L. Hua, P. Raghunath, M.-C. Lin and W.-S. Chung, ACS Appl. Mater. Interfaces, 2020, 12, 29650–29660 CrossRef CAS PubMed.
- X. Wang, K. Han, J. Li, X. Jia and C. Li, Polym. Chem., 2013, 4, 3998–4003 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07097a |
| This journal is © The Royal Society of Chemistry 2023 |