
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the role of solvent polarity in mechanochemical Knoevenagel condensation: in situ investigation and isolation of reaction intermediates†
Kerstin
Scheurrell‡
a,
Inês C.
B. Martins‡§
 *a,
Claire
Murray
a and
Franziska
Emmerling
*ab
*a,
Claire
Murray
a and
Franziska
Emmerling
*ab
aFederal Institute for Materials Research and Testing (BAM), Berlin, Germany. E-mail: ines.martins@sund.ku.dk; franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Berlin, Germany
First published on 24th August 2023
Abstract
Mechanochemistry has proven to be a highly effective method for the synthesis of organic compounds. We studied the kinetics of the catalyst-free Knoevenagel reaction between 4-nitrobenzaldehyde and malononitrile, activated and driven by ball milling. The reaction was investigated in the absence of solvents (neat grinding) and in the presence of solvents with different polarities (liquid-assisted grinding). The reaction was monitored using time-resolved in situ Raman spectroscopy and powder X-ray diffraction (PXRD). Our results indicate a direct relationship between solvent polarity and reaction kinetics, with higher solvent polarity leading to faster product (2-(4-nitrobenzylidone)malononitrile) formation. For the first time, we were able to isolate and determine the structure of an intermediate 2-(hydroxy(4-nitrophenyl)methyl)malononitrile based on PXRD data.
Introduction
The Knoevenagel condensation1 is an important carbon–carbon bond-forming reaction. The reaction comprises the condensation between an aldehyde or a ketone with active hydrogen compounds to yield α,β-unsaturated compounds.2,3 The mechanism involves two steps: (i) an aldol addition of an active hydrogen compound to an aldehyde or carbonyl compound and (ii) 1,2-elimination of water or dehydration.4–6 Usually, the Knoevenagel reaction is conducted in solution under homogeneous conditions using weak bases which react with an aldehyde to form the β-hydroxy intermediate.5,7 Protic and aprotic polar solvents, such as EtOH and DMF, favour the kinetics for the first step in the mechanism and aprotic solvents accelerate the reaction in the second step. The intermediate is further converted into the final α,β-unsaturated olefin, after the elimination of water.8In response to the urgent need for environmentally friendly synthetic processes, alternative methods using minimal amounts of bulk catalysts and solvents are under exploration.9 In this context, several modifications of the traditional Knoevenagel reaction have been developed in recent years including the use of heterogenous catalysts (alkali-earth or modified silica),10 water as catalyst and solvent,11,12 and catalyst-free environments using ethanol/water as reaction media.13 For solution-based conditions, the solvent and catalyst greatly affect the kinetics of the reaction, whereas the solid-state reaction profile is influenced by parameters such as the amount of solvent and the mechanical force used.14–19
Kaupp20 and Suzuki21 have shown that an efficient and quantitative Knoevenagel condensation can be achieved in the solid state. In their study, Kaupp et al. documented the first Knoevenagel condensation between 4-nitrobenzaldehyde and malononitrile using a ball mill. They achieved a significant reduction in catalyst concentration compared to the traditional solution-based method.20 These discoveries have led to further studies of solvent-free Knoevenagel condensation reactions, including 4-nitrobenzaldehyde (4-NBA) and malononitrile (MN).22–27 This reaction has been explored in planetary and vibrational ball mills under catalyst-free conditions and in the presence or absence of solvents, showing complete conversions to the 2-(4-nitrobenzylidone)malononitrile (2-NMN) product.25,28
Here, we investigate how solvents of different polarities affect the kinetics and mechanism of the Knoevenagel reaction between 4-NBA and MN using mechanochemical techniques (Fig. 1). To achieve this, we used in situ Raman spectroscopy and PXRD to observe the reaction in the presence of three solvents (liquid-assisted grinding-LAG) with different polarities (octane, ethanol (EtOH) and dimethylformamide (DMF)) as well as in the absence of solvents (neat grinding-NG). By using different solvents, we were able to detect and isolate the reaction intermediate (2-(hydroxyl(4-nitrophenyl)methyl)malononitrile-2-H-NMN), and determine its structure using PXRD data. This tunability of the Knoevenagel reaction kinetics confirms the power of mechanochemistry to simultaneously streamline reaction pathways and minimise the necessary volumes of reactants, such as solvents.
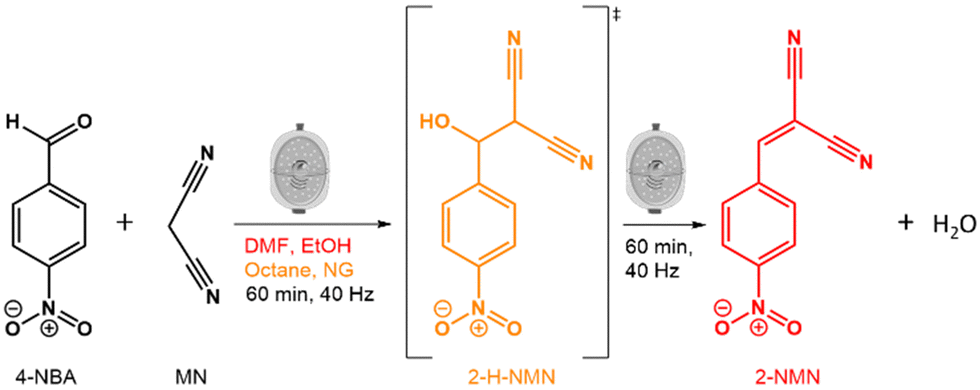 | ||
| Fig. 1 Schematic representation of the Knoevenagel reaction between 4-NBA and MN, highlighting the intermediate 2-H-NMN (in orange) and the resulting product, 2-NMN (in red). | ||
Results and discussion
The Knoevenagel reaction between 4-NBA and MN (Fig. 1) was conducted using a vertical vibrational ball mill. The reactants were milled for 60 minutes at 40 Hz, under two specific conditions: (1) LAG (60 μL, η = 0.1 μL mg−1; η = V (liquid; μL) m−1 (reagents; mg))29 of solvent with different polarities – octane, ethanol (EtOH) and dimethylformamide (DMF); and (2) absence of solvent (NG). The reaction was investigated by time-resolved in situ (TRIS) Raman spectroscopy in tandem with synchrotron PXRD. For reactions under LAG conditions when using high polarity solvents (EtOH and DMF) the Raman data (Fig. 2, top) show that, the reaction progresses towards the formation of the product 2-NMN (high intensity C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching band at 2232 cm−1). This stretching band appears at lower frequency than the MN CN stretch band (2267 cm−1) due to the high π-electrons conjugation between the new C
N stretching band at 2232 cm−1). This stretching band appears at lower frequency than the MN CN stretch band (2267 cm−1) due to the high π-electrons conjugation between the new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the CN group. In addition, the reaction intermediate 2-H-NMN is also detected in very small amounts via the presence of a low intensity CN stretching band at 2260 cm−1.
C bond and the CN group. In addition, the reaction intermediate 2-H-NMN is also detected in very small amounts via the presence of a low intensity CN stretching band at 2260 cm−1.
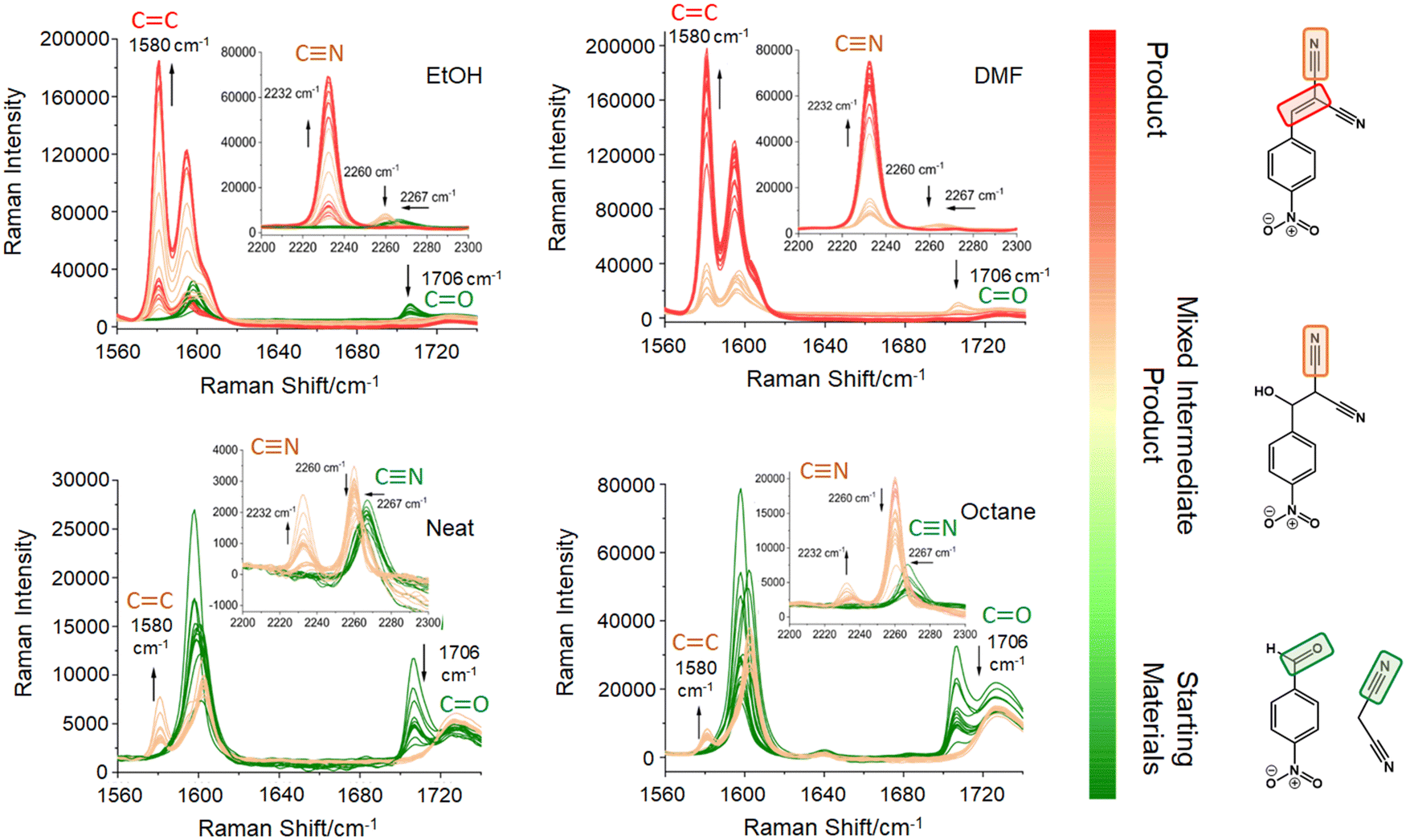 | ||
| Fig. 2 In situ Raman data of the Knoevenagel ball milling reaction between 4-NBA and MN (starting materials) under neat grinding and LAG conditions using octane, EtOH and DMF. Green spectra – reaction induction period containing the educts; beige vibration bands – formation of the intermediate and mixture of the intermediate with the product; red spectra – formation of the product. | ||
Under NG conditions and LAG with non-polar solvents (octane), the formation of the intermediate 2-H-NMN is detected in a higher amount, as observed by the presence of an intense CN stretching band at 2260 cm−1 (Fig. 2, bottom). As the reaction progresses, the main stretching bands of the intermediate and starting materials (CN and CO stretching bands) decrease in intensity. Consequently, the emergence of the product 2-NMN becomes increasingly evident. New CC and CN stretching bands appear at 1580 cm−1 and 2232 cm−1, albeit with low intensities and coexisting with the CN stretching band of the intermediate.
Trapping the Knoevenagel intermediate 2-H-NMN – structure determination
Identification of intermediates in mechanochemical bond forming reactions has occasionally been reported previously.30,31 Here, the intermediate 2-H-NMN was detected during NG and LAG (octane) at 40 Hz, but only in combination with 2-NMN. Therefore, an optimization of the reaction conditions to isolate 2-H-NMN, in pure form, was performed. We investigated an ex situ NG experiment in very mild conditions, using a lower frequency (15 Hz) milling for a period of 60 minutes. The PXRD data presented in Fig. 3a demonstrates the almost pure crystalline phase of the intermediate. Only low intensity reflections of 4-NBA are still present. An additional inspection of both reactivity progress and stability of the intermediate, after stopping the milling process (Fig. 3a and b), show that the reaction continues slowly until complete consumption of the starting materials and formation of the product (4 days). The known melt-mediated reaction during ball milling of 4-NBA and MN may be responsible for this observation.23 Based on previous studies, after approximately 35 minutes of milling and at the same time that the reaction starts, MN melts and the milling jar temperature quickly increases to 35 °C, which is above the melting point of MN (32 °C).23,24 The mixture between molten MN and crystalline 4-NBA, together with the accumulated energy from the mechanical impact of the milling balls into the powder, may help in the reaction progress after stopping the milling process.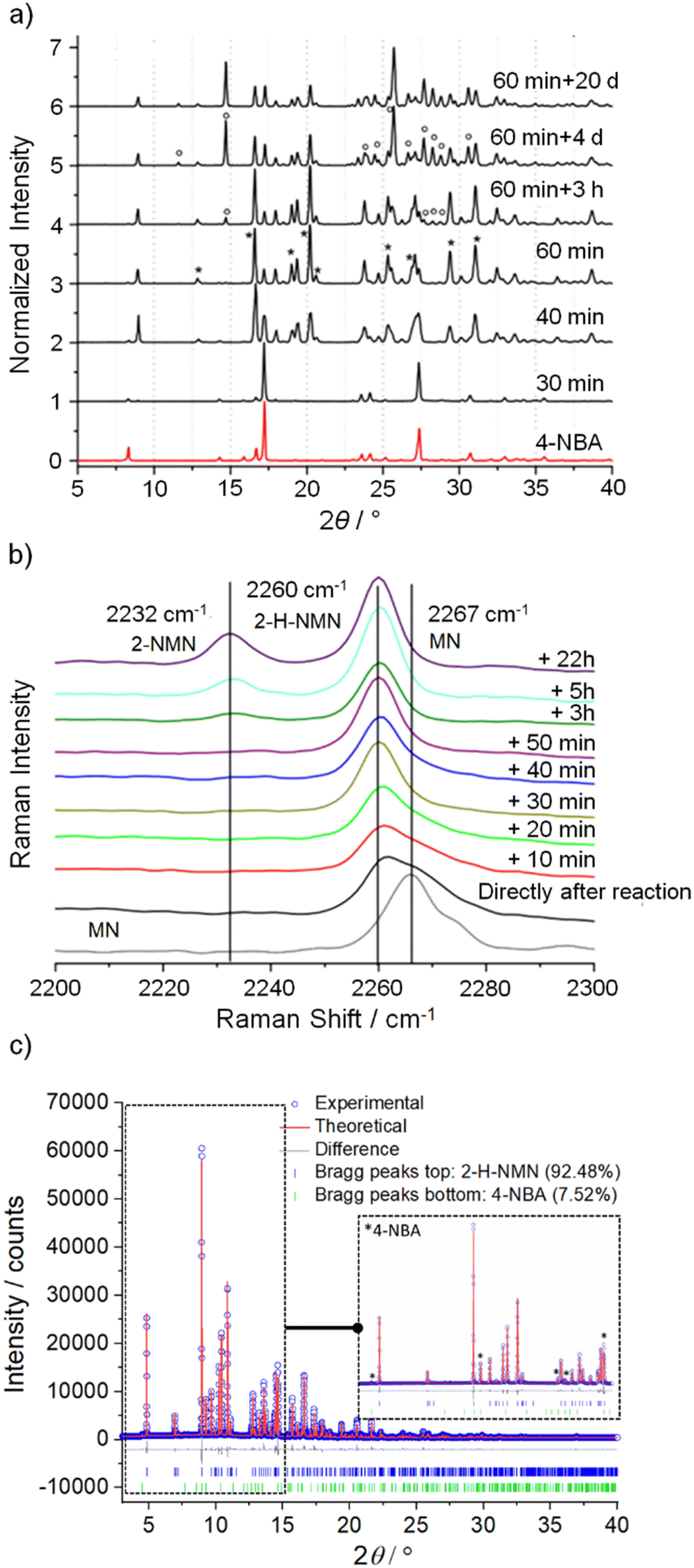 | ||
| Fig. 3 (a) Ex situ PXRD pattern (λ = 1.54056 Å) of 4-NBA and the intermediate/product after milling at 15 Hz for 60 min and after storage at different times (3 h, 4 d and 20 d); (b) Ex situ Raman spectra of the Knoevenagel reaction between 4-NBA and MN under NG conditions (15 Hz, 60 min) during different waiting times after milling; (c) Rietveld refinement plot with quantitative phase analysis of 2-H-NMN and 4-NBA, displaying the measured powder pattern (blue dots, λ = 0.8266 Å), the calculated powder pattern (red solid line), the difference curve (grey), and the reflection positions (Bragg peaks) for each phase. An expansion shows the fitted peaks of 4-NBA. | ||
The powder pattern of 2-H-NMN obtained after 60 minutes of milling was unambiguously indexed as monoclinic unit cell, using both TOPAS32–34 and DICVOL35 in DASH.36 Using the Hoffmann's volume increments,37 the expected molecular volume was 232.03 Å3, corresponding to four molecules per unit cell (Z = 4) for a unit cell volume of 995.90 Å3 (Table 1). The structure was solved by the real-space method and refined using the Rietveld method.
| 2-H-NMN | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Dashed values corresponds to values after background subtraction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chemical formula | C10H7N3O3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formula weight/g mol−1 | 217.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal system | Monoclinic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Space group | P21/c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a/Å | 10.89048(15) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| b/Å | 9.54267(14) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| c/Å | 10.63046(15) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| β/° | 115.6448(4) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| V/Å3 | 995.94(3) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Z | 4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
R
p,a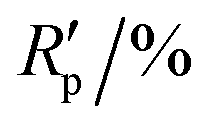 |
4.258, 11.326 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
R
wp,a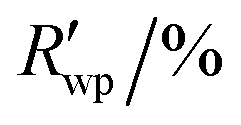 |
5.895, 12.277 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
R
exp,a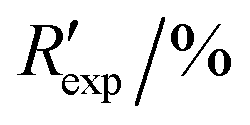 |
3.403, 7.087 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| R Bragg | 3.527 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gof | 1.732 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Quantitative phase analysis was performed using TOPAS,32–34 shown in Fig. 3c, which determined that the phase mixture contains 92.48% of intermediate and 7.52% of 4-NBA.
The crystal structure of the intermediate 2-H-NMN is presented in Fig. 4. The packing structure is based on an intermolecular OH⋯O hydrogen bond interaction, dH⋯O = 1.94(5) Å, enabling the formation of a chain along b. This HB interaction is probably responsible for the stabilization of the intermediate, delaying the formation time of 2-NMN product. Furthermore, there is an extra HB interaction between the acidic hydrogen atom, directly connected to the malononitrile group, and the nitrogen atom of another malononitrile group (CH⋯O, dH⋯N = 2.47(6) Å). These two HB interactions allow the interconnection between intermediate chains along c, forming a R33(19) synthon. An additional π⋯π interaction, dπ⋯π = 3.748(5) Å, supports the packing growing in all directions (Fig. 4).
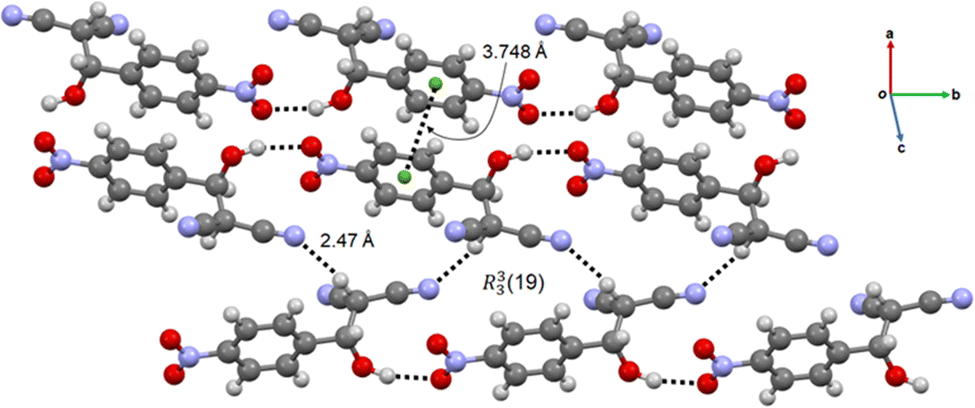 | ||
| Fig. 4 Chain-like packing arrangement of 2-H-NMN evidencing the OH⋯O and CH⋯O HB, the π⋯π interactions and the R33(19) synthon. | ||
Kinetic investigation of the Knoevenagel condensation
For a better understanding of the reaction progress using LAG or NG conditions, we analysed the time-dependent changes in the Raman spectra during the reaction course using the concentration in real time algorithm. A two-point background correction with normalization of the CN stretching bands at 2232 cm−1 (MN) and 2260 cm−1 (2-H-NMN), and CO stretching band at 1706 cm−1 (2-NMN) was used to generate the kinetic curves of the reactions presented in Fig. 5 (for more details, see the Experimental section).
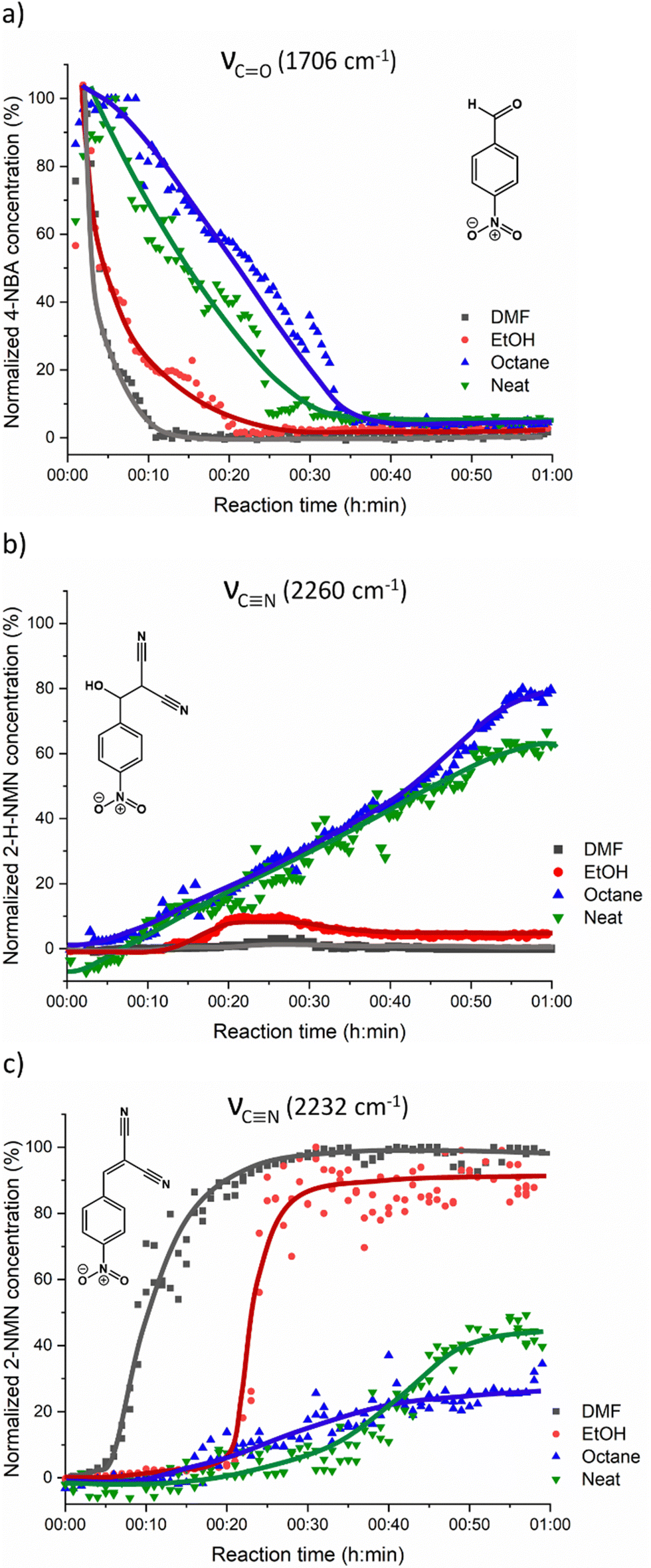 | ||
| Fig. 5 Kinetic plots for the Knoevenagel reaction showing (a) consumption of the 4-NBA starting material and formation of (b) 2-H-NMN intermediate and (c) 2-NMN product. No fitting was performed and the curves in the kinetic data are only a guide to the eye. | ||
Under LAG conditions with EtOH and DMF, the starting material MN is consumed totally after 20 and 30 min of reaction, respectively (Fig. 5a), with complete formation of the product 2-MNM with no intermediate detection (Fig. 5c). For the reaction conducted with DMF, a small induction period of 3 min is observed, during which no product formation is detected. The induction period slightly increases to ca. 5 min when using EtOH as LAG solvent. These observations are supported by in situ PXRD data (Fig. 6a) and are also in accordance with what is expected in solution when using such solvents.13 The aprotic polar solvent DMF favours the reaction kinetics in both steps, while EtOH favours mostly the first step (aldol addition), giving rise to a slightly faster reaction kinetics in the presence of DMF.
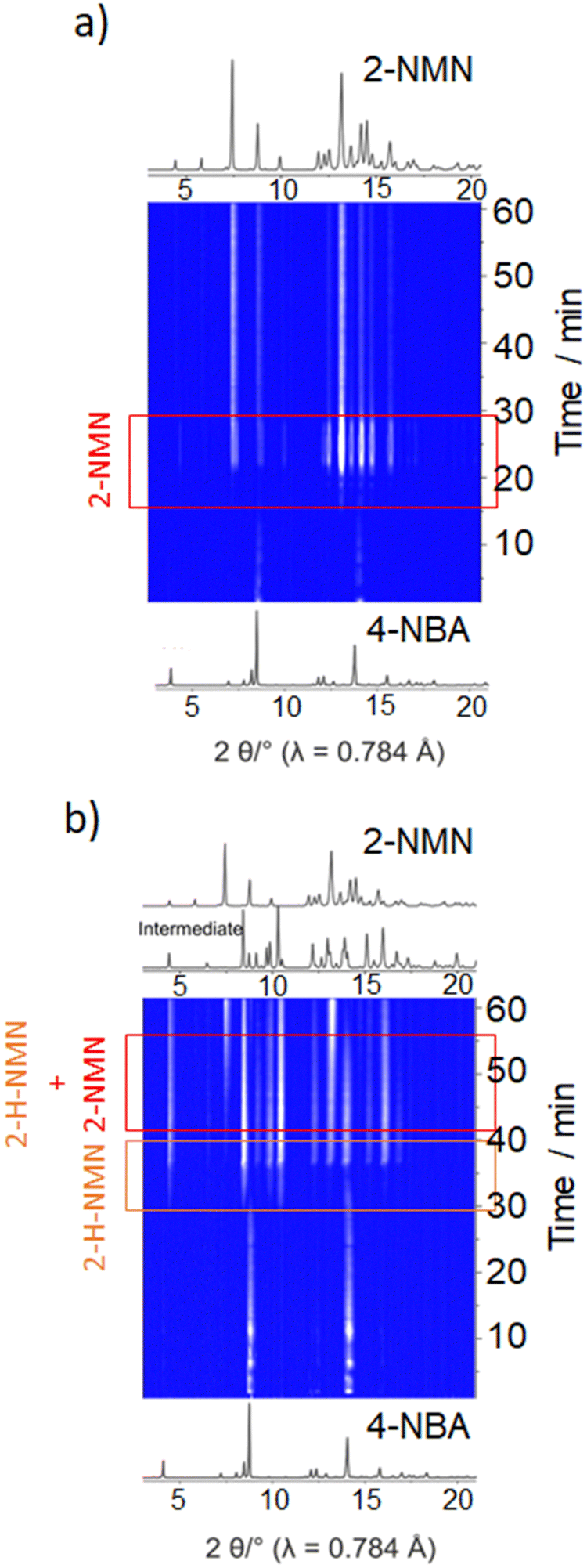 | ||
| Fig. 6 In situ PXRD data of the Knoevenagel reaction between 4-NBA and MN performed by ball milling using (a) EtOH and (b) octane. | ||
Performing the reaction using octane and under NG conditions leads to the formation of the intermediate 2-H-MNM (Fig. 5b) in the amount of 78% and 60%, respectively, after 60 min of reaction. The intermediate 2-H-MNM and the product 2-MNM appear concomitantly after ca. 10 min of induction period. These results are also in accordance with the in situ PXRD data (Fig. 6b). Considering that non-polar solvents do not favour the 1,2-elimination step, it was expected that under LAG with octane and NG conditions, the intermediate would be formed. In both reaction conditions, the kinetic profile is similar, and the product formation is not completed during 60 min of milling.
Previous in situ PXRD and Raman study performed by Haferkamp and Kulla et al23,38 showed that the reaction between MN and 4-NBA using NG at 50 Hz during 60 min lead to a complete conversion to the product 2-NMN without the detection of the intermediate 2-H-NMN. However, it is known that milling frequencies can hugely influence the reaction kinetics.16,39,40 In our in situ studies, we have used a lower milling frequency of 40 Hz. This difference of 10 Hz appears to be sufficient to detect the formation of 2-H-NMN during the same reaction time (60 min).
To complement our in situ studies ex situ experiments were performed. We selected two reaction conditions as examples (LAG in EtOH and octane) and collected PXRD for every single reaction point using Rietveld refinements for quantification (Fig. S2 and S3, ESI†). The kinetic profile of both reactions presented in Fig. 7 are similar to those obtained after in situ data collection (Fig. 5). Using EtOH, a complete conversion to the product 2-MNM is achieved after ca. 25 min whereas when using octane, the reaction is not complete. Residual staring material 4-NBA is still present (ca. 10%) after 60 min of reaction together with ca. 20% of the intermediate 2-H-NMN and ca. 70% of the product 2-MNM.
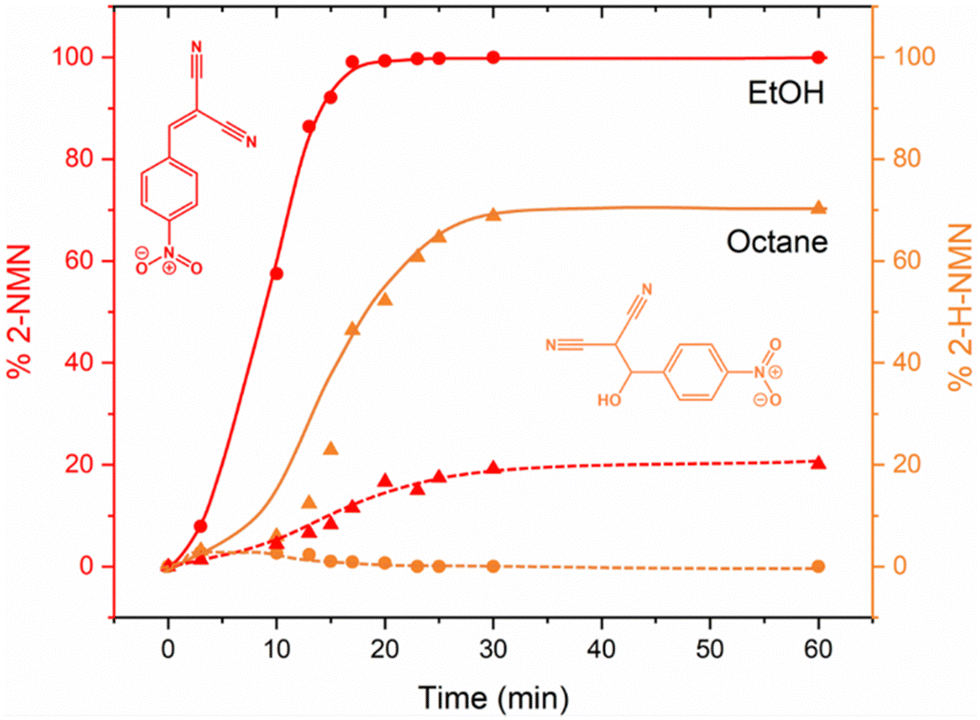 | ||
| Fig. 7 Kinetic plots for the Knoevenagel reaction, obtained by Rietveld refinement, showing the formation of 2-NMN product (red) and 2-H-NMN intermediate (orange) after milling in EtOH (sphere) and octane (triangle). | ||
Experimental section
Chemicals
The chemicals 4-NBA (≥98%, Roth, Germany), MN (99%, ABCR, Germany), hydroquinone (99.5%, Acros, USA), absolute EtOH (99.9%, Th. Geyer, Germany), octane (for synthesis, Merck, Germany) and DMF (p. A., Th. Geyer, Germany) were purchased and used without further purification.Mechanochemical synthesis
The mechanochemical Knoevenagel condensation was studied using both in situ Raman spectroscopy and in situ PXRD. To initiate the reaction, 600 mg of 4-NBA and MN were weighed out in stoichiometric amounts and placed in either a 10 mL Perspex (for the vertical vibrational ball mill (Pulverisette, Fritsch, Germany)) or 25 mL stainless steel milling jar (for the horizontal vibrational ball mill (Retsch MM 400)). A small amount of hydroquinone, approximately 10 mg, was added as a stabilizer along with 60 μL of solvent, which could be either EtOH, octane, or DMF, depending on the LAG requirement. Two milling balls (stainless steel, 10 mm diameter, 4 g) were added to two reaction mixtures. The milling was performed in a vertical vibrational ball mill (Pulverisette, Fritsch, Germany) and also in a horizontal vibrational ball mill (Retsch MM 400) for 60 min at 40 Hz and 15 Hz, respectively. Directly after the reaction, the products were characterized by ex situ PXRD.Powder X-ray diffraction for structural determination
The measurements were performed on a D8 diffractometer (Bruker AXS, Karlsruhe, Germany) in transmission geometry using Cu Kα1 radiation (λ = 1.54106 Å) in a 2θ range from 5 to 65° and a step size of 0.02°.The high-resolution synchrotron PXRD data was collected at beamline P08 (DESY, Research Centre of the Helmholtz Association). The experiments were carried out in a 1 mm capillary at a wavelength of 0.8266 Å and an energy of 14.998 keV using a liquid nitrogen cooled double crystal monochromator Si(111). Scattered intensities were collected with a linear X-ray detector (Mythen 1k; spot size 200 × 1000 μm; detector-sample-distance 875.208 mm).
The powder pattern was indexed using DICVOL35 in DASH,36 and TOPAS,32–34 excluding some peaks from the 4-NBA starting material impurity. Structure solution was achieved by the real-space method, using simulated annealing routine implemented in DASH.36 The starting model of 4-NBA was obtained from the cif file of CSD (Cambridge Structural Database) with the entry YAWCEG47 and modified by removing a double bond and adding two hydrogen atoms and a hydroxyl group. During simulated annealing no restrictions in the degrees of freedom were applied.
Rietveld refinement was carried out using TOPAS32–34 for the full 2θ range. A Pawley fit was first performed in the following refinement steps: scale factor, background, atomic positions, and isotropic displacement parameters were refined.
For the structure refinement, two different isotropic displacement parameters were refined: one for the non-hydrogen (non H) atoms and one for the hydrogen atoms, Biso(H) = 1.2 × Biso(non H). Restraints were applied for all bond lengths, angles, and planar rings/groups. Data collection and structure refinement details are summarized in Table 1. All information about hydrogen bonding distances and angles, obtained using PLATON,41 are presented in Table 2.
| Sym op | D–H⋯A | d(D–H) (Å) | d(H⋯A) (Å) | d(D⋯A) (Å) | DHA (deg) | |
|---|---|---|---|---|---|---|
| 2-H-NMN | x, −1 + y, z | O–H⋯O | 0.97(5) | 1.94(5) | 2.872(8) | 163(5) |
| 1 − x, 1/2 + y, 3/2 − z | C–H⋯N | 0.96(7) | 2.47(6) | 3.377(14) | 158(5) |
A quantitative phase analysis was performed to determine the percentage of 4-NBA and 2-H-MNM present (Fig. 3c and Table 1). The structure cif file (CSD entry KAYSUY) was used in the respective Rietveld refinement.
In situ experiments
The in situ synchrotron XRD investigations were performed at the μSpot beamline (BESSY II, Helmholtz Centre Berlin for Materials and Energy) using a vertical ball mill (Pulverisette, Fritsch, Germany) and a Perspex grinding jar containing 2 balls (10 mm, 4 g) at 40 Hz for 60 min. The experiments were carried out at a wavelength of 0.8266 Å and an energy of 17 keV using a double crystal monochromator Si(111). Scattered intensities were collected with a two-dimensional X-ray detector (Eiger9M, CCD 3072 × 3072). The exposure time was 15 s. The images were processed with DPDAK 1.4.1 followed by a background correction with a Python 3.7.1 script using Spyder from Anaconda.49 For a better comparison with the ex situ PXRD data, the q values were transformed into the diffraction angle 2θ (Cu Kα1).The in situ Raman spectroscopic measurements were performed on a Raman RXN1TM analyser (Kaiser Optical Systems, Lyon, France). A contactless probe with a working distance of 6 cm and a spot size of 1 mm was used. The excitation wavelength of the laser was 785 nm. Raman spectra were recorded with iC Raman V.4.1.915 (Mettler Toledo) at an acquisition time of 5 s and 5 accumulations with a new spectrum every 30 s. The subtraction of jar contribution, baseline correction, and normalization were carried out with a script designed for basic analysis and visualization of in situ Raman monitoring data.42 Acquired spectra were processed a curve-resolution algorithms having the capability to group wavenumber values that change absorbance intensity in the same manner (ConcIRT module, implemented in the iC Raman software).43,44 For each group, the algorithm calculates the associated component spectrum and relative concentration profile. As each new reaction spectrum is acquired, all the reaction spectra are re-analysed and the individual component spectra and profiles are updated. In this way, the calculation results evolve as the reaction progresses and additional components such as intermediates are detected.
Relative concentration profiles were calculated for products, intermediates and starting materials from single Raman bands after a two-point (start and end of the vibration band) background correction.
Conclusions
We explored the catalyst-free mechanochemical Knoevenagel condensation reaction between 4-nitrobenzaldehyde (4-NBA) and malononitrile (MN). We investigated the effect of solvents with different polarities (EtOH, DMF and octane) on the reaction mechanism using time-resolved in situ Raman spectroscopy and PXRD. The use of EtOH and DMF resulted in faster reaction kinetics, leading to complete formation of the product 2-NMN. In contrast, the use of non-polar solvents such as octane or performing the reaction under neat conditions slowed the reaction kinetics and led to the formation of the intermediate 2-H-NMN with incomplete conversion to the product 2-NMN. In addition, by performing ex situ mechanochemical experiments at 15 Hz without solvent, we were able to isolate and determine the structure of the intermediate 2-H-NMN in an almost pure form under mild conditions.Our results show that in situ monitoring of mechanochemical reactions is a powerful tool for detecting reaction intermediates that would otherwise go undetected using conventional approaches. This method enables the facile optimisation of reaction conditions and times by changing parameters such as solvent and milling frequency. This in turn creates opportunities to study these reactions in detail, including following the reaction pathway or isolating important intermediates or by-products under different conditions. The potential to tune chemical activity and product selectivity through mechanochemistry, which directly addresses many environmental sustainability concerns, should therefore not be underestimated.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Funding by the Deusche Forschungsgemeinschaft (DFG, German Research Foundation) - EM 198-12 is gratefully acknowledged. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at Petra III and we would like to thank Dr Florian Bertram for assistance at beamline P08. The authors are grateful to Torvid Feiler, Kevin Linberg and Maria Heilmann for measuring PXRD data at BESSY II and DESY.Notes and references
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2596–2619 CrossRef CAS.
- F. T. Lutz and B. Uwe, in Comprehensive Organic Synthesis, ed. M. T. Barry and F. Ian, Pergamon, Oxford, 1991, pp. 341–394 DOI:10.1016/B978-0-08-052349-1.00033-0.
- M. Ricardo, in Green Chemistry, ed. K. Mazaahir and M. Neeraj Kumar, IntechOpen, Rijeka, 2012, ch. 2 DOI:10.5772/36489.
- K. van Beurden, S. de Koning, D. Molendijk and J. van Schijndel, Green Chem. Lett. Rev., 2020, 13, 349–364 CrossRef CAS.
- E. V. Dalessandro, H. P. Collin, L. G. L. Guimarães, M. S. Valle and J. R. Pliego, Jr., J. Phys. Chem. B, 2017, 121, 5300–5307 CrossRef CAS PubMed.
- R. Devi, P. Begum, P. Bharali and R. C. Deka, ACS Omega, 2018, 3, 7086–7095 CrossRef CAS PubMed.
- E. J. J. Cragoe, C. M. Robb and J. M. Sprague, J. Org. Chem., 1950, 15, 381–390 CrossRef CAS.
- A. C. O. Hann and A. Lapworth, J. Chem. Soc., Trans., 1904, 85, 46–56 RSC.
- Green Synthetic Processes and Procedures, ed.R. Ballini, The Royal Society of Chemistry, 2019 10.1039/9781788016131-00001.
- R. Wirz, D. Ferri and A. Baiker, Langmuir, 2006, 22, 3698–3706 CrossRef CAS PubMed.
- J. P. H. Li, A. A. Adesina, E. M. Kennedy and M. Stockenhuber, Phys. Chem. Chem. Phys., 2017, 19, 26630–26644 RSC.
- X.-L. Zhao, K.-F. Yang, Y.-P. Zhang, J. Zhu and L.-W. Xu, Chin. Chem. Lett., 2014, 25, 1141–1144 CrossRef CAS.
- J. M. G. O. Ferreira, J. B. M. d Resende Filho, P. K. Batista, E. E. S. Teotonio and J. A. Vale, J. Braz. Chem. Soc., 2018, 29(7), 1382–1387 CAS.
- I. C. B. Martins and F. Emmerling, Cryst. Growth Des., 2021, 21, 6961–6970 CrossRef CAS.
- A. A. L. Michalchuk, I. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2019, 21, 2174–2179 RSC.
- A. M. Belenguer, A. A. L. Michalchuk, G. I. Lampronti and J. K. M. Sanders, Beilstein J. Org. Chem., 2019, 15, 1226–1235 CrossRef CAS PubMed.
- A. A. L. Michalchuk, I. A. Tumanov and E. V. Boldyreva, J. Mater. Sci., 2018, 53, 13380–13389 CrossRef CAS PubMed.
- S. Darwish, S.-Q. Wang, D. M. Croker, G. M. Walker and M. J. Zaworotko, ACS Sustainable Chem. Eng., 2019, 7, 19505–19512 CrossRef CAS.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- G. Kaupp, M. R. Naimi-Jamal and J. Schmeyers, Tetrahedron, 2003, 59, 3753–3760 CrossRef CAS.
- S. Wada and H. Suzuki, Tetrahedron Lett., 2003, 44, 399–401 CrossRef CAS.
- D. V. Demchuk, M. N. Elinson and G. I. Nikishin, Mendeleev Commun., 2011, 4, 224–225 CrossRef.
- S. Haferkamp, F. Fischer, W. Kraus and F. Emmerling, Beilstein J. Org. Chem., 2017, 13, 2010–2014 CrossRef CAS PubMed.
- S. Haferkamp, A. Paul, A. A. L. Michalchuk and F. Emmerling, Beilstein J. Org. Chem., 2019, 15, 1141–1148 CrossRef CAS PubMed.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 873–878 RSC.
- R. Trotzki, M. M. Hoffmann and B. Ondruschka, Green Chem., 2008, 10, 767–772 RSC.
- L. Rong, X. Li, H. Wang, D. Shi, S. Tu and Q. Zhuang, Synth. Commun., 2006, 36, 2407–2412 CrossRef CAS.
- C. F. Burmeister, R. Schmidt, K. Jacob, S. Breitung-Faes, A. Stolle and A. Kwade, Chem. Eng. J., 2020, 396, 124578 CrossRef CAS.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- S. Lukin, M. Tireli, I. Lončarić, D. Barišić, P. Šket, D. Vrsaljko, M. di Michiel, J. Plavec, K. Užarević and I. Halasz, Chem. Commun., 2018, 54, 13216–13219 RSC.
- J. G. Hernández, Chem. – Eur. J., 2017, 23, 17157–17165 CrossRef PubMed.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 1991, 24, 987–993 CrossRef CAS.
- K. Shankland, E. Pidcock, J. Van De Streek, W. I. F. David, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910–915 CrossRef.
- D. W. M. Hofmann, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 489–493 CrossRef PubMed.
- H. Kulla, S. Haferkamp, I. Akhmetova, M. Röllig, C. Maierhofer, K. Rademann and F. Emmerling, Angew. Chem., Int. Ed., 2018, 57, 5930–5933 CrossRef CAS PubMed.
- P. A. Julien, I. Malvestiti and T. Friščić, Beilstein J. Org. Chem., 2017, 13, 2160–2168 CrossRef CAS PubMed.
- L. Vugrin, M. Carta, S. Lukin, E. Meštrović, F. Delogu and I. Halasz, Faraday Discuss., 2023, 241, 217–229 RSC.
- A. Spek, J. Appl. Cryst., 2003, 36, 7–13 CrossRef CAS.
- S. Lukin, K. Užarević and I. Halasz, Nat. Protoc., 2021, 16, 3492–3521 CrossRef CAS PubMed.
- D. E. Pivonka, J. M. Chalmers and P. R. Griffiths, Applications of Vibrational Spectroscopy in Pharmaceutical Research and Development, Wiley, 2007 Search PubMed.
- A. Chanda, A. M. Daly, D. A. Foley, M. A. LaPack, S. Mukherjee, J. D. Orr, G. L. Reid III, D. R. Thompson and H. W. Ward, Org. Process Res. Dev., 2015, 19, 63–83 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1997414. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp02883f |
| ‡ Authors equally contributed to the manuscript. |
| § Current address: Department of Pharmacy, University of Copenhagen, Denmark. |
| This journal is © the Owner Societies 2023 |