
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoluminescence and electrochemiluminescence of thermally activated delayed fluorescence (TADF) emitters containing diphenylphosphine chalcogenide-substituted carbazole donors†‡
Shiv
Kumar§
a,
Pauline
Tourneur§
b,
Jonathan R.
Adsetts§
c,
Michael Y.
Wong
 a,
Pachaiyappan
Rajamalli
a,
Dongyang
Chen
a,
Roberto
Lazzaroni
bd,
Pascal
Viville
d,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Yoann
Olivier
*e,
Jérôme
Cornil
*b,
Zhifeng
Ding
*c and
Eli
Zysman-Colman
*a
a,
Pachaiyappan
Rajamalli
a,
Dongyang
Chen
a,
Roberto
Lazzaroni
bd,
Pascal
Viville
d,
David B.
Cordes
a,
Alexandra M. Z.
Slawin
a,
Yoann
Olivier
*e,
Jérôme
Cornil
*b,
Zhifeng
Ding
*c and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: http://www.zysman-colman.com
bLaboratory for Chemistry of Novel Materials, University of Mons, Mons, Belgium. E-mail: Jerome.Cornil@umons.ac.be; Web: http://morris.umons.ac.be/
cDepartment of Chemistry, The University of Western Ontario, London, Ontario N6A 3K7, Canada. E-mail: zfding@uwo.ca; Web: https://publish.uwo.ca/~zfding/
dMateria Nova, Materials R&D center, Mons, Belgium. E-mail: Pascal.Viville@materianova.be; Web: http://www.materianova.be/
eUnité de Chimie Physique Théorique et Structurale (UCPTS) & Laboratoire de Physique du Solide (LPS), Namur Institute of Structured Matter (NISM), University of Namur, Namur, Belgium. E-mail: yoann.olivier@unamur.be
First published on 8th February 2022
Abstract
Aiming to develop efficient blue-emitting thermally activated delayed fluorescence (TADF) compounds, we have designed and synthesized derivatives of the well-known sky-blue emitter 2CzPN that contain electron-accepting phosphine chalcogenide groups to stabilize the HOMO level relative to the pristine compound, thus increasing the HOMO–LUMO gap and blue-shifting the emission wavelength. By cyclic voltammetry, photophysical data and quantum-chemical calculations, it was found that polar solvents and matrices validated the proposed concept, but these trends were not recovered in non-polar media. The suitability of these 2CzPN derivatives in polar matrices for optoelectronic applications was explored with electrochemiluminescence (ECL) by measuring emission delays, radical stability, emission stabilities, emission efficiencies and emission spectra. Some of the 2CzPN derivatives showed an unprecedented delayed onset of the ECL, and delayed rising time to the ECL maximum, as well as long ECL emission decay. All of these mentioned delay times suggest that these luminophores primarily emit via organic long-persistent electrochemiluminescence (OLECL) mechanisms. The derivatization of the donor groups of the emitters affected both the radical stability and the predominant emission mechanism, providing important insight into their potential as emitters in solid-state electroluminescent devices.
1. Introduction
Developing strategies to harvest triplet states in organic light-emitting diodes (OLEDs) based on small molecules has been the focus of considerable effort because, according to spin statistics, they constitute 75% of the excitons generated in the device.1 While phosphorescent heavy-metal-based organometallic complexes have successfully achieved 100% internal quantum efficiency (IQE) thanks to their large spin–orbit coupling (SOC) and hence rapid intersystem crossing (ISC) rates, the use of scarce elements such as iridium and platinum is a detracting feature that will ultimately impact large-scale application of OLED technology.2 Moreover, the inherent instability of blue-emitting heavy metal phosphorescent OLEDs has meant this class of materials is a non-starter in commercial devices.3Materials showing thermally activated delayed fluorescence (TADF), which typically take the form of donor–acceptor organic compounds, have attracted tremendous recent attention as they are capable of efficiently harvesting triplet excitons.4–8 For the TADF mechanism to be operational, there must exist a suitably small splitting energy (ΔEST) between the lowest excited singlet (S1) and triplet (T1) states. When this criterion is met, efficient reverse intersystem crossing (RISC) processes become feasible such that non-emissive triplet excitons are thermally up-converted to emissive singlet excitons.5,9–12 In order to maintain a small ΔEST, the exchange integral between the HOMO and LUMO must be minimized,13 and is commonly achieved by the spatial separation of these orbitals.8,12,14 In general, there are three main molecular designs that respond to this criterion: (1) compounds with twisted conformations between donor and acceptor moieties that emit via an intramolecular charge-transfer state (ICT);10 (2) compounds that promote through-space charge-transfer interactions between adjacent donor and acceptor units (TSCT);15 and (3) compounds that are p- and/or n-doped nanographenes that emit via multi-resonance TADF (MR-TADF).16 Each TADF emitter design principle has its own advantages and disadvantages in terms of emission color, color purity, photoluminescence quantum yield (ΦPL), and RISC efficiency, the latter of which is correlated strongly with device stability. For example, ICT- and TSCT-based TADF emitters have both been reported to be highly emissive; their color is easily tunable but their broad emission profiles lead to poor color purity.15,17,18 In contrast, MR-TADF emitters exhibit excellent color purity and ΦPL but generally possess much lower RISC rates.19 Regardless of emitter design, the stability of TADF OLEDs remains sub-optimal.6
In the pantheon of ICT emitter design, the majority of the research focus has been devoted to new acceptors while the selection of donor moieties has remained rather limited; with carbazole,10,20,21 triphenylamine,22–24 acridan,25–27 phenoxazine,28–31 phenothiazine32,33 and dihydrophenazine34,35 accounting for more that ca. 98% of all reported donors. This library of donors has been enriched through their chemical substitution that serves to tune the photophysical properties of the emitter. The majority of examples revolve around substituted carbazole derivatives and can be broadly categorized into two families: (i) substituents that enhance the electron-donating strength of carbazole and promote a red-shift in the emission, and; (ii) substituents that reduce the electron-donating strength of the carbazole and promote a blue-shift in the emission.36 Electron-rich carbazole derivatives used within TADF emitter design include 3,6-dimethylcarbazole,37 3,6-dimethoxycarbazole,38 3,6-di-t-butylcarbazole,37,38 fluorene-fused carbazole,39 benzofuran-fused carbazole,40 donor-dendronized carbazole41,42 benzothiophene-fused carbazole,43 and paracyclophane-fused carbazole.44 In contrast, 3,6-diphenylcarbazole,10 9-phenylcarbazole,45 α-/β-/δ-carboline,46–50 indolocarbazole,51 diphenylphosphine oxide-substituted carbazole derivatives,52 triaryl and diarylboronate-substituted carbazole derivatives,53 and cyanocarbazoles54,55 represent examples of electron-poor carbazole derivatives reported in the TADF literature. In the context of blue TADF emitters employing functionalized carbazole donors, Adachi et al. have reported several examples of blue TADF emitters. In 2012, Adachi et al. reported the first example of a deep blue emitter, DTC-DPS (λPL = 423 nm, ΦPL = 80%, ΔEST = 0.32 eV in 10 wt% in DPEPO), based on 3,6-di-(tert-butyl)carbazole as the donor and diphenylsulfone as the acceptor. The OLED employing this emitter showed a maximum external quantum efficiency (EQEmax) of 9.9% and CIE coordinates of (0.15, 0.17).56 In a following report, an improved EQEmax of 14.5% and CIE coordinates of (0.16, 0.16) were reported for the emitter, DMOC-DPS (λPL = 455 nm, ΦPL = 80%, ΔEST = 0.21 eV in 10 wt% in DPEPO) in which 3,6-di-(tert-butyl)carbazole (DTC) was replaced by 3,6-dimethoxycarbozole (DMOC) as the donor.38 In another example, a sky-blue emitter BDPCC-TRZ (λPL = 480 nm, ΦPL = 100%, ΔEST = 0.11 eV in 6 wt% in DPEPO), composed of bis(3,6-diphenylcarbazolyl)carbazole (BDPCC) as the donor and triphenyl-1,3,5-triazine (TRZ) as the acceptor, was reported. The OLED with this emitter achieved an EQEmax of 20.6% with λEL at 487 nm and CIE coordinates of (0.19, 0.25).57 Lee et al. reported 5,11-di(9H-carbazol-9-yl)indolo[3,2,1-jk]carbazole-2-carbonitrile (CNICCz, λPL = 445 nm, ΦPL = 46%, ΔEST = 0.27 eV) as a deep blue emitter, with a CN-modified indolocarbazole as the acceptor core and carbazole as the donor. Here, deep-blue OLEDs [λEL = 449 nm, CIE: (0.15, 0.08)] were realized with an EQEmax of 12.4%.58
In the seminal paper published in 2012, Adachi et al. reported one of the earliest sky-blue TADF emitters, 4,5-dicarbazolylphthalonitrile (2CzPN, λPL = 473 nm, ΦPL = 47% in toluene).10 An OLED device employing this emitter produced a sky-blue emission (λEL = 475 nm in 5 wt% PPT doped emitting layer) with an EQEmax of 8.0%. An improved OLED device performance (λEL = 483 nm, CIE: (0.17, 0.30), EQEmax = 13.6%) was achieved when the same emitter was used in a mCP host (λPL = 477 nm, ΦPL = 89% in 6 wt% mCP doped film, ΔEST = 0.09 eV).59 By optimizing the device architecture, Sun et al. were able to achieve an EQEmax of 21.8% [CIE: (0.17, 0.27), λEL = 480 nm] while using the same emitter in the OLED, by adopting a mixed co-host system (mCP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PO15 = 1:1). However, such a high EQE was only achieved at a very low current density of ca. 0.01 mA cm−2, and the device suffered from severe efficiency roll-off at high current density.58 In pursuit of a deeper blue color using the same general structural motif as 2CzPN, Gyeong et al. reported α-CbPN (λPL = 445 nm, ΦPL = 37%, ΔEST =0.28 eV in 20 wt% mCP doped film) and δ-CbPN (λPL = 480 nm, ΦPL = 93%, ΔEST =0.13 eV in 20 wt% mCP doped film), employing α/δ-carboline donors in lieu of carbazole.60 A maximum EQE of 22.5% for a sky-blue OLED with λEL = 486 nm and CIE: (0.19, 0.34) was achieved using 20 wt% δ-CbPN in mCP as the light-emitting layer. The same group also reported a triazine-based emitter 5,5-(2-(9H-carbazol-9-yl)-5-(4,6-diphenyl-1,3,5-triazin-2-yl)-1,3-phenylene)bis(5H-pyrido[3,2-b]indole) (CzDCbTrz) containing a δ-carboline donor, and produced an OLED with an EQEmax of 23.4% and λEL = 471 nm with CIE: 0.16, 0.19 using 6 wt% emitter in 2,8-bis(diphenylphosphineoxide)-dibenzofuran (DBFPO) host. The chemical structures of blue TADF emitters discussed above are shown in Fig. 1.
:PO15 = 1:1). However, such a high EQE was only achieved at a very low current density of ca. 0.01 mA cm−2, and the device suffered from severe efficiency roll-off at high current density.58 In pursuit of a deeper blue color using the same general structural motif as 2CzPN, Gyeong et al. reported α-CbPN (λPL = 445 nm, ΦPL = 37%, ΔEST =0.28 eV in 20 wt% mCP doped film) and δ-CbPN (λPL = 480 nm, ΦPL = 93%, ΔEST =0.13 eV in 20 wt% mCP doped film), employing α/δ-carboline donors in lieu of carbazole.60 A maximum EQE of 22.5% for a sky-blue OLED with λEL = 486 nm and CIE: (0.19, 0.34) was achieved using 20 wt% δ-CbPN in mCP as the light-emitting layer. The same group also reported a triazine-based emitter 5,5-(2-(9H-carbazol-9-yl)-5-(4,6-diphenyl-1,3,5-triazin-2-yl)-1,3-phenylene)bis(5H-pyrido[3,2-b]indole) (CzDCbTrz) containing a δ-carboline donor, and produced an OLED with an EQEmax of 23.4% and λEL = 471 nm with CIE: 0.16, 0.19 using 6 wt% emitter in 2,8-bis(diphenylphosphineoxide)-dibenzofuran (DBFPO) host. The chemical structures of blue TADF emitters discussed above are shown in Fig. 1.
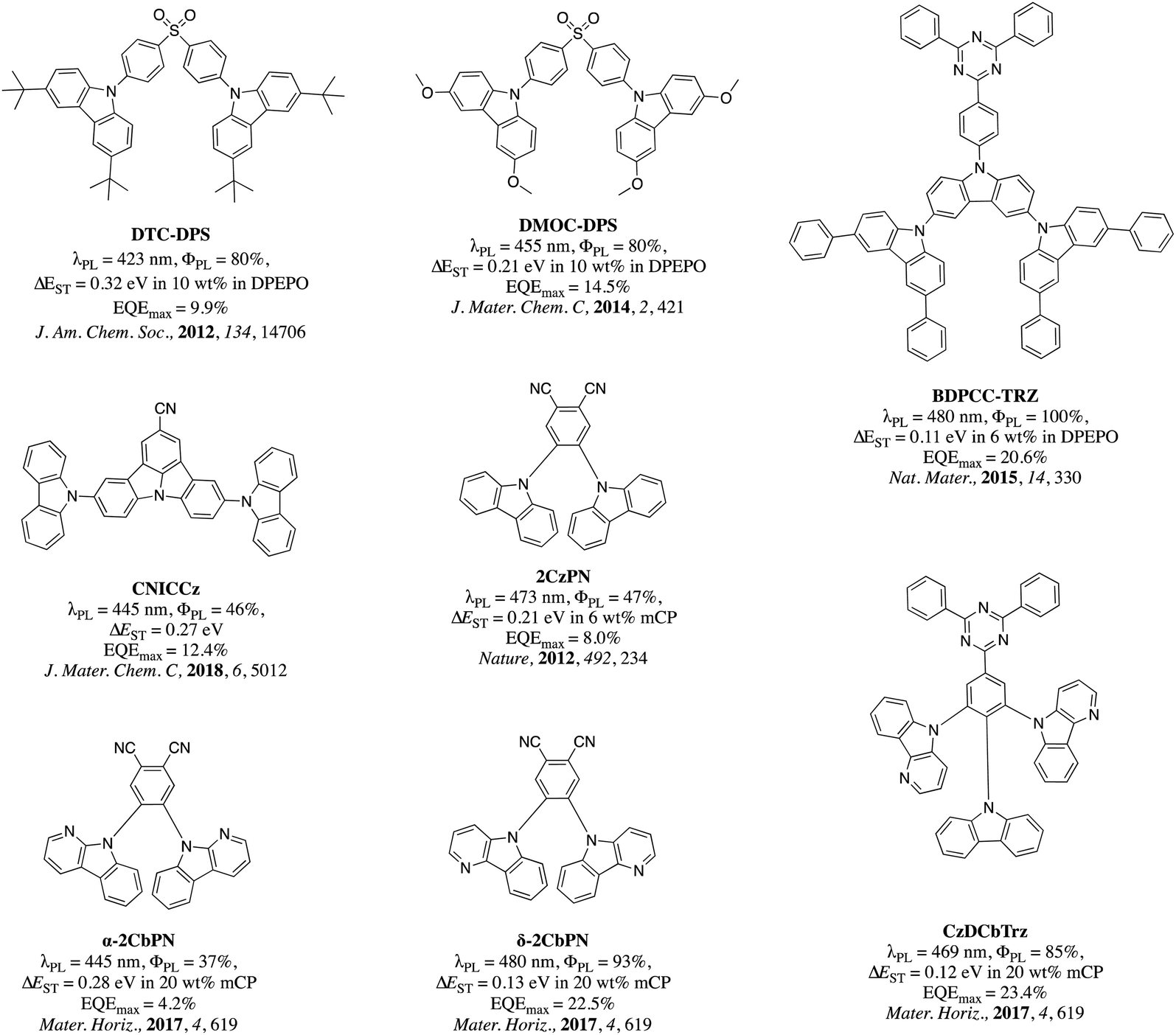 | ||
| Fig. 1 Chemical structures of discussed blue TADF emitters. | ||
Electrochemiluminescence (ECL) or electrogenerated chemiluminescence is emitted when electron transfer between electrochemically-generated radicals results in an exciton formation in the proximity of a working electrode, which relaxes radiatively.61–64 In fact, this combination of charge recombination, radical generation, exciplex formation,65 and possibilities of host–guest systems,66 TADF,67 triplet–triplet annihilation68 and phosphorescent emissions via ECL mirrors photophysical processes that can occur in OLEDs. Furthermore, high luminophore concentrations, and film electrodes61 in ECL studies can mimic bulk and interfacial molecular conditions in OLEDs, allowing predictions of molecular performance before full device manufacturing. ECL finds additional applications in discovering luminophore film enhancement phenomena,69–71 and in commercial applications for antigen sensing.72 Since the early studies on ECL in the 1960s,73–75 various types of emissive compounds have been studied.76 However, the design of commercial ECL luminophores has been mainly limited to noble-metal complexes such as those of Ru, due to their efficient and electrochemically-stable conversion of excitons to light, regardless of the total spin. Analogously to their popular use as emitters in OLEDs, TADF molecules are likewise expected to give high ECL efficiency. Despite the thousands of reports of TADF emitters used in OLEDs, there are only a small number of examples of TADF compounds employed in ECL studies. Imato et al. studied the annihilation ECL properties of carbazole-decorated phthalonitrile based TADF emitters in both DCM and MeCN.67 Of the compounds investigated, the ECL emission maxima, λECL, for 2CzPN were modestly red-shifted (λECL = 550 and 585 nm in DCM and MeCN, respectively) compared to the photoluminescence maxima, λPL, (λPL = 533 and 560 nm in DCM and MeCN, respectively) regardless of solvent. Further, the low ΦPL (ΦPL = 34 and 10% in DCM and MeCN, respectively) translated into low relative ECL efficiencies, ΦECL, of 90 and 22%, respectively, in DCM and MeCN, in comparison to [Ru(bpy)3]2+ which showed ΦECL = 100% under the same conditions. No discussion concerning the kinetics of exciton decay was provided. Most recently, Niu et al. reported the first example of a solid-state ECL study of a TADF polymer containing a backbone acridan-based donor and pendant triazine-containing acceptor, PCzAPT10.77 The polymer showed significantly enhanced ΦECL of 194% relative to [Ru(bpy)3]2+ when using tri-n-propylamine (TPA) as a co-reactant, compared to the ΦPL of 21%. The λECL was also significantly red-shifted, at 587 nm, compared to the λPL (542 nm) for a neat film of PCzAPT10 immersed in 0.1 M TBAP:MeCN electrolyte solution. Furthermore, Niu et al. made water-soluble 4CzIPN nanoparticles which achieved a relative ΦECL = 0.7%, marking the first water-soluble TADF nanoparticle tested.78
Using the same 2CzPN scaffold as a starting point, we hypothesized that adding strong electron-withdrawing group such as phosphine-chalcogenides onto the carbazole donor moieties would promote the desired blue-shift in the emission. In this regard, we designed and synthesized three 2CzPN derivatives and thoroughly investigated their physical, photophysical, electrochemical properties, further supporting these optoelectronic studies with density functional theory (DFT) calculations (Scheme 1). ECL was then used to gauge the suitability of these derivatives for optoelectronic applications in the desirable polar media. Assessing the potential of the three 2CzPN derivatives as OLED materials was possible without the time-intensive and costly task of constructing full OLEDs, since ECL provides a tool to simulate charge imbalances, judge relative stabilities of electrically generated radical cations and anions (holes and electrons, respectively), evaluate relative emission efficiencies and understand emission mechanisms of luminophores.67,79,80 Although our reference compound, 2CzPN is a well-studied TADF emitter in the literature, we have redetermined its optoelectronic properties under the same conditions as the three new emitters, as well as calculating its properties, to allow the most accurate comparison.
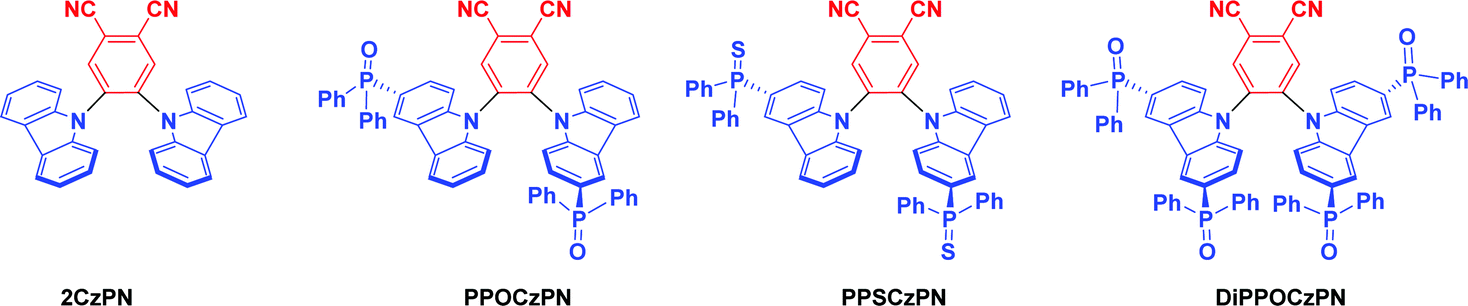 | ||
| Scheme 1 Chemical structure of 2CzPN and the three phosphine chalcogenide derivatives. | ||
2. Experimental section
2.1 General synthetic procedures
All commercially available chemicals and reagent grade solvents were used as received. 3-Bromocarbazole, 3,6-dibromocarbazole, N-TBDMS-3-bromocarbazole and N-TBDMS-3,6-dibromocarbazole were prepared according to the literature.81 Air-sensitive reactions were performed using standard Schlenk techniques under a nitrogen atmosphere. Anhydrous THF was obtained from a solvent purification system. Flash column chromatography was carried out using silica gel (60 Å, 40–63 μm). Analytical thin-layer-chromatography (TLC) was performed using silica plates with aluminium backings (250 μm with F-254 indicator), and were visualized using a 254/365 nm UV lamp. 1H, 13C and 31P NMR spectra in CDCl3 or DMSO-d6 were recorded on an NMR spectrometer (400 MHz for 1H, 101 MHz for 13C and 162 MHz for 31P). The NMR signal is described as follows: s = singlet, d = doublet, dd = doublet of doublets, td = triplet of doublets, ddd = doublet of doublets of doublets, and m = multiplets. Melting points were measured using open-ended capillaries on Electrothermal Mel-Temp® melting point apparatus and are uncorrected. High-resolution mass spectrometry (HRMS) was performed by the EPSRC National Mass Spectrometry Service Centre (NMSSC), Swansea University. Elemental analyses were performed by Mr Stephen Boyer, London Metropolitan University. High performance liquid chromatography (HPLC) analysis was conducted on a Shimadzu Prominence Modular HPLC system. HPLC traces were performed using an ACE Excel 2 C18 analytical (3 × 150 mm) column. Gas chromatography mass spectrometry (GCMS) analysis was carried out on a Shimadzu GCMS-QP2010 SE instrument, an advanced standard gas chromatograph mass spectrometer coupled with automated AOC-5000 sample injection system using a Shimadzu-SH RTX®-1 column (fused silica) (length = 25 m, thickness = 0.25 mm, oven temp.: 40 °C to 250 °C; detector: MS; detection temp.: 250 °C; Carrier gas: helium).ECL was measured by a photomultiplier tube (R928 PMT, Hamamatsu Photonics, Japan) held at −750 V. The voltage signal from the PMT was transduced by a Keithley ammeter (6487, Keithley Instruments, Cleveland, OH) into so-called photocurrent measured in nanoamperes (nA).
The spectroscopic measurement methods were reported elsewhere,84 with a spectrograph (Acton 2300i, Princeton Instruments Inc., Trenton NJ) coupled with a CCD camera (Model DV420-BV, Andor Technology, UK) cooled to −55 °C.
The relative efficiency of the ECL emission was determined by finding the charge input and the ECL output for the specific experimental setup and comparing these values to the commercial standard of ECL emitter systems, [Ru(bpy)3]2+ for annihilation systems and [Ru(bpy)3]2+/BPO or [Ru(bpy)3]2+/TPrA for co-reactant systems, by the following equation:
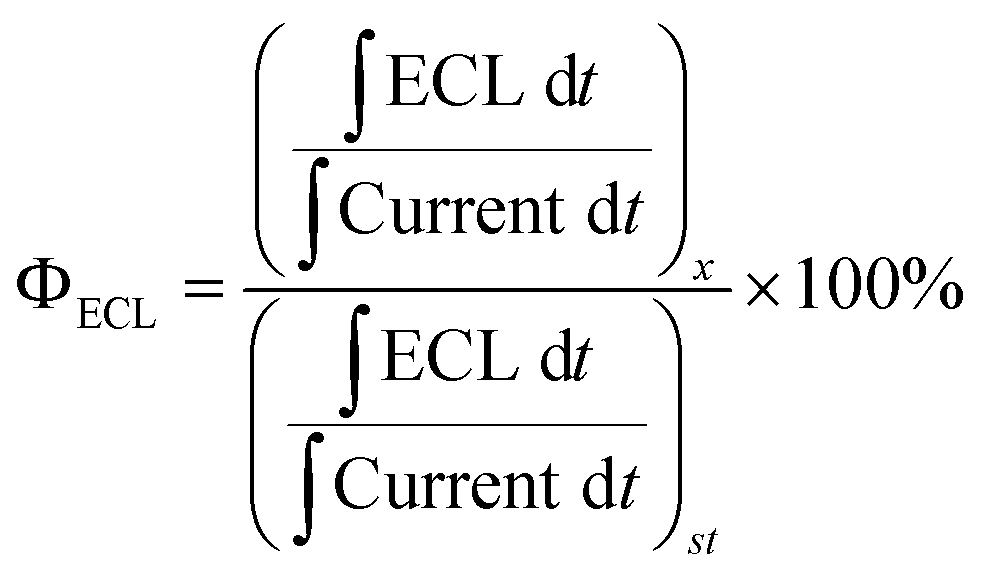 | (1) |
Reaction enthalpies were calculated by the following equation:85
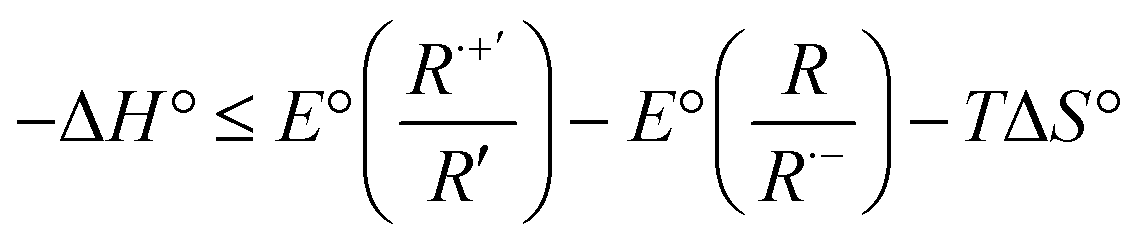 | (2) |
3. Results and discussion
3.1 Synthesis and characterization
The synthesis of the phosphine chalcogenide-modified 2CzPN emitters is shown in Scheme 2. Nitrogen-protected precursors 9-(tert-butyldimethylsilyl)-3-bromo-9H-carbazole, TBDMS-BrCz and 9-(tert-butyldimethylsilyl)-3,6-dibromo-9H-carbazole, TBDMS-DBrCz were prepared from their respective bromo- or dibromo-9H-carbazole derivatives.81 The key intermediates, the phosphine chalcogenide-substituted carbazoles (PPOCz, PPSCz, DiPPOCz), were prepared by lithiation of TBDMS-BrCz and TBDMS-DBrCz followed by diphenylphosphination (TBDMS-PPCz and TBDMS-DiPPCz), and finally oxidation/sulfuration. Removal of the TBDMS group afforded the desired phosphine chalcogenide-substituted carbazole derivatives in moderate-to-excellent yield (40–95%). The respective emitters PPOCzPN, PPSCzPN and DiPPOCzPN were likewise obtained in moderate-to-excellent yield (52–96%) through a nucleophilic aromatic substitution reaction of 4,5-difluorophthalonitrile with PPOCz, PPSCz or DiPPOCz. All the emitters were characterized by 1H, 13C, and 31P NMR spectroscopy and high-resolution mass spectrometry (HRMS), and their purity was demonstrated by HPLC and elemental analysis. The emitters PPOCzPN and PPSCzPN were each found to exist as a mixture of rotamers at room temperature in a ratio of 58:42 and 70:30, respectively, which was observed by both HPLC and 31P{1H} NMR spectroscopy (Fig. S1, ESI‡). This was further substantiated by obtaining single crystals of both rotamers for PPSCzPN.
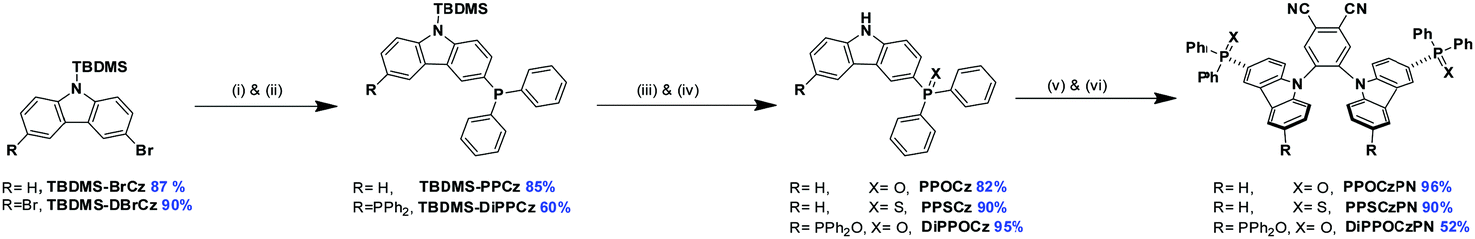 | ||
| Scheme 2 Synthetic routes for the synthesis of PPXCzPN and DiPPOCzPN. Reaction conditions: (i) n-BuLi (1.6M), THF, −78 °C, 30 min, (ii) PPh2Cl, −78 °C – rt, 30 min, (iii) H2O2 or S8, THF, rt, 18 h, (iv) TBAF (1.0 M), THF, rt, 1 h, (v) NaH, THF, 30 min, (vi) 4,5-difluorophthalonitrile, rt, 6 h. | ||
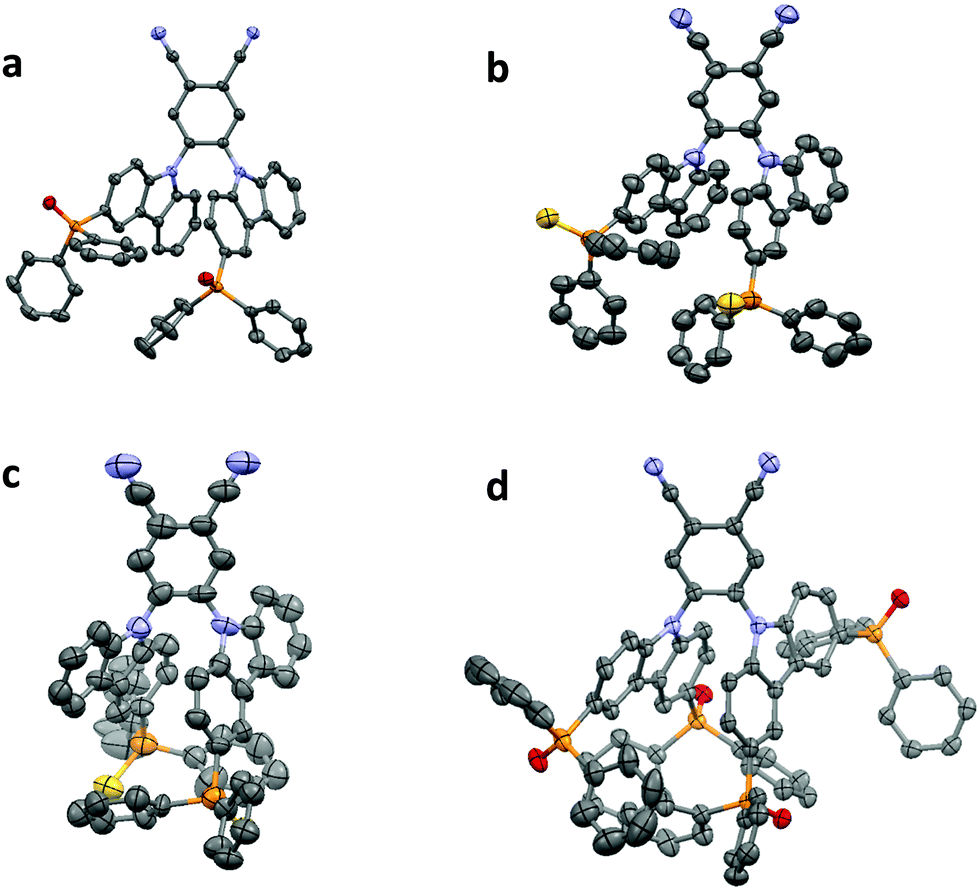 | ||
| Fig. 2 Thermal ellipsoid plots of the crystal structures of (a) PPOCzPN, (b) PPSCzPN-1, (c) PPSCzPN-2 and (d) DiPPOCzPN. Ellipsoids are drawn at the 50% probability level and hydrogen atoms and solvent molecules are omitted for clarity. | ||
The four structures show a number of intermolecular interactions. In both PPOCzPN and DiPPOCzPN a three-dimensional network of weak C–H⋯O and C–H⋯N hydrogen bonded molecules is formed [PPOCzPN H⋯A distances 2.39–2.60 Å, C⋯A separations 3.120(2)–3.529(3) Å; DiPPOCzPN H⋯A distances 2.26–2.54 Å, C⋯A separations 3.018(3)–3.383(3) Å] (where A is the acceptor atom). The networks are reinforced by C–H⋯π interactions in both structures [PPOCzPN H⋯centroid distances 2.79–2.90 Å, C⋯centroid separations 3.549(3)–3.719(2) Å; DiPPOCzPN H⋯centroid distances 2.74 Å, C⋯centroid separations 3.546(3) Å], π–π interactions in PPOCzPN [centroid⋯centroid distances 3.7287(9) Å], and by conventional hydrogen bonds to the water solvent molecule in DiPPOCzPN [H⋯O distances 1.83(3) and 1.87(4) Å, O⋯O separations 2.811(3) and 2.858(4) Å].
Both PPSCzPN structures show a different pattern of interactions where weak interactions of specific types give rise to multiple one-dimensional chain structures, these being linked into two-dimensional sheets when they are combined. In PPSCzPN-1, C–H⋯π interactions [H⋯centroid distances 2.70 and 2.89 Å, C⋯centroid separations 3.623(12) and 3.799(15) Å] form chains along the ac-diagonal axis, and weak C–H⋯N hydrogen bonds [H⋯N distances 2.45 Å, C⋯N separations 3.322(16) Å] form chains along the b-axis. The combination of these leads to sheets in the (1 0 −1) plane. In PPSCzPN-2, π⋯π interactions [centroid⋯centroid distances 3.668(10)–3.803(10) Å] form chains along the b-axis, and C–H⋯π interactions [H⋯centroid distances 2.78 Å, C⋯centroid separations 3.63(3) Å] form chains along the bc-diagonal axis. The combination of these leads to sheets in the (1 0 0) plane, which are further reinforced by weak C–H⋯S hydrogen bonds [H⋯S distances 2.71 Å, C⋯S separations 3.56(2) Å]. The chloroform solvent molecules in PPSCzPN-2 are held within the lattice by weak hydrogen bonds [H⋯N distances 2.54 Å, C⋯N separations 3.41(4) Å; H⋯S distances 2.62 Å, C⋯S separations 3.59(2) Å].
None of these structures show a matching pattern of interactions to that shown in the structure of 2CzPN, where multiple sets of C–H⋯π interactions form sheets in the (0 0 1) plane, which are reinforced by weak C–H⋯N hydrogen bonds.86 This is closest to what is seen in PPSCzPN-1, in that sheets are formed by combinations of C–H⋯π and C–H⋯N interactions; however, in 2CzPN, the C–H⋯π interactions form sheets directly, whereas in PPSCzPN-1 both the C–H⋯π interactions and the weak hydrogen bonds are required to form the sheets.
3.2 Electrochemical and photophysical properties
The three emitters PPOCzPN, PPSCzPN and DiPPOCzPN show irreversible oxidation waves and reversible reduction waves in their cyclic voltammograms (CV), recorded in DCM with 0.1 M tetrabutylammonium perchlorate (TBAP) (red curves in Fig. 3a–c). The values of the HOMO and LUMO energy levels were estimated from the peaks observed in the CV and verified from the differential pulse voltammograms (Fig. S36, ESI‡). The HOMO/LUMO levels of the reference compound 2CzPN, as well as PPOCzPN, PPSCzPN and DiPPOCzPN are determined to be −6.28/−3.50, −6.39/−3.56, −6.56/−3.48 and −6.86/−3.56 eV, respectively. As expected, it is easier to reduce and harder to oxidize DiPPOCzPN than PPOCzPN due to the presence of the second electron-withdrawing diphenylphosphine oxide moiety on each carbazole. PPOCzPN is the easiest to oxidize and has the least stable HOMO. Similar electrochemical trends were seen in MeCN (Fig. S37, ESI‡).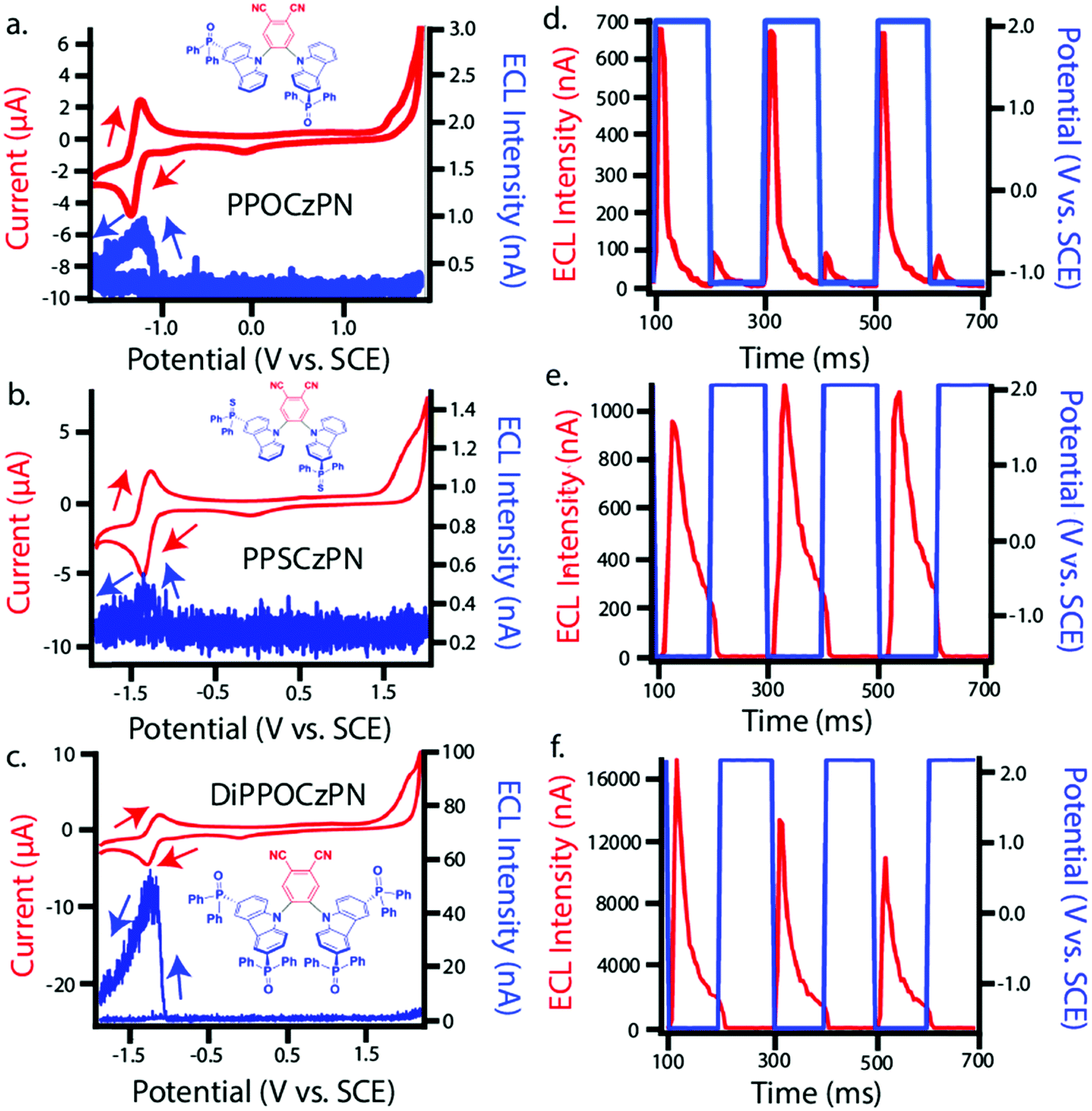 | ||
| Fig. 3 (a–c) CVs (red) along with ECL–voltage curves (blue) and (d–f) ECL-time curves (red) along with voltage-time curves (blue) during potential pulsing at a pulsing frequency of 10 Hz for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively. Scans were recorded in DCM. CVs were all performed at a scan rate of 0.1 V s−1 and the second cycle is shown in every case. Pulsing experiments were carried out between 0.1 V above the first oxidation and below the first reduction potentials of the compounds. | ||
The optical absorption study of PPOCzPN, PPSCzPN, DiPPOCzPN and the reference emitter 2CzPN was carried out in acetonitrile (polar) and toluene (nonpolar) solvents. Table 1 summarizes the photophysical and electrochemical properties of PPOCzPN, PPSCzPN, DiPPOCzPN and the reference emitter 2CzPN in polar solutions and doped films in PMMA (polymethyl methacrylate) as a polar host matrix, leading to the following observations. The phosphine chalcogenide-substituted emitters exhibited similar UV-visible absorption spectra to that of 2CzPN with three major bands observed at around 265 nm, 325 nm, and 360 nm in both solvents (Fig. 4a and d). The high-energy band near 265 nm is attributed to a LE transition of the carbazole moiety (see Fig. S40, ESI‡) while the lowest energy bands near 325 nm and 360 nm are ascribed to ICT transitions in view of their low intensity.59 The ICT bands of the phosphine chalcogenide-substituted emitters are modestly blue-shifted by 380–1020 cm−1 (30–65 nm) compared to the reference emitter 2CzPN due to the presence of the electron-withdrawing substituents. The presence of the second diphenylphosphine oxide group in DiPPOCzPN produced both a more intense absorption of the π–π* band at around 270 nm and the most blue-shifted ICT band. The absorption spectra did not appreciably change between toluene and acetonitrile (see Fig. 4a and d).
| Emitter | MeCN | DCM | 10 wt% doped PMMA film | DCM | ||||
|---|---|---|---|---|---|---|---|---|
| λ abs /nm, (ε × 10−4 M−1 cm−1) | λ PL /nm | Φ PL /% | λ PL /nm | Φ PL /% | E ox/V (HOMOf/eV) | E red/V (LUMOf/eV) | ΔEg/eV | |
| a ICT band measured in MeCN at 298 K. b In degassed DCM (λexc = 358 nm). c Solution ΦPL values were determined by the relative method4 using 0.5 M quinine sulfate in H2SO4 (aq) as the reference (ΦPL: 54.6%).87 d 10 wt% doped PMMA thin films (λexc = 360 nm). e Absolute ΦPL values in 10 wt% doped PMMA thin films under N2. f In DCM with 0.1 M [nBu4N]ClO4 as the supporting electrolyte and SCE as the reference unless otherwise noted.88 The HOMO and LUMO energies were calculated using the relation EHOMO/ELUMO = −(Eox/Eredvs. Fc/Fc+ + 4.8) eV, where the Eox and Ered are anodic and cathodic formal potentials, respectively, obtained from CV.89 g ΔE = |EHOMO − ELUMO|. h Values from ref 77. i Determined in MeCN. | ||||||||
| PPOCzPN | 356 (1.24) | 498 | 51 | 465 | 56 | 1.59 (−5.99) | −1.24 (−3.16) | 2.83 |
| PPSCzPN | 358 (1.49) | 501 | 47 | 465 | 59 | 1.76 (−6.16) | −1.32 (−3.08) | 3.08 |
| DiPPOCzPN | 351 (1.51) | 465 | 61 | 428 | 28 | 2.06 (−6.46) | −1.24 (−3.16) | 3.29 |
| 2CzPN | 366 (1.33) | 515 | 29 | 494 | 77 | 1.48 (−5.88) | −1.30 (−3.10) | 2.78 |
| 2CzPN | 364 (1.14) | 532 | 29 | 492 | 76 | 1.50 (−5.90)i | −1.42 (−3.00)i | 2.90i |
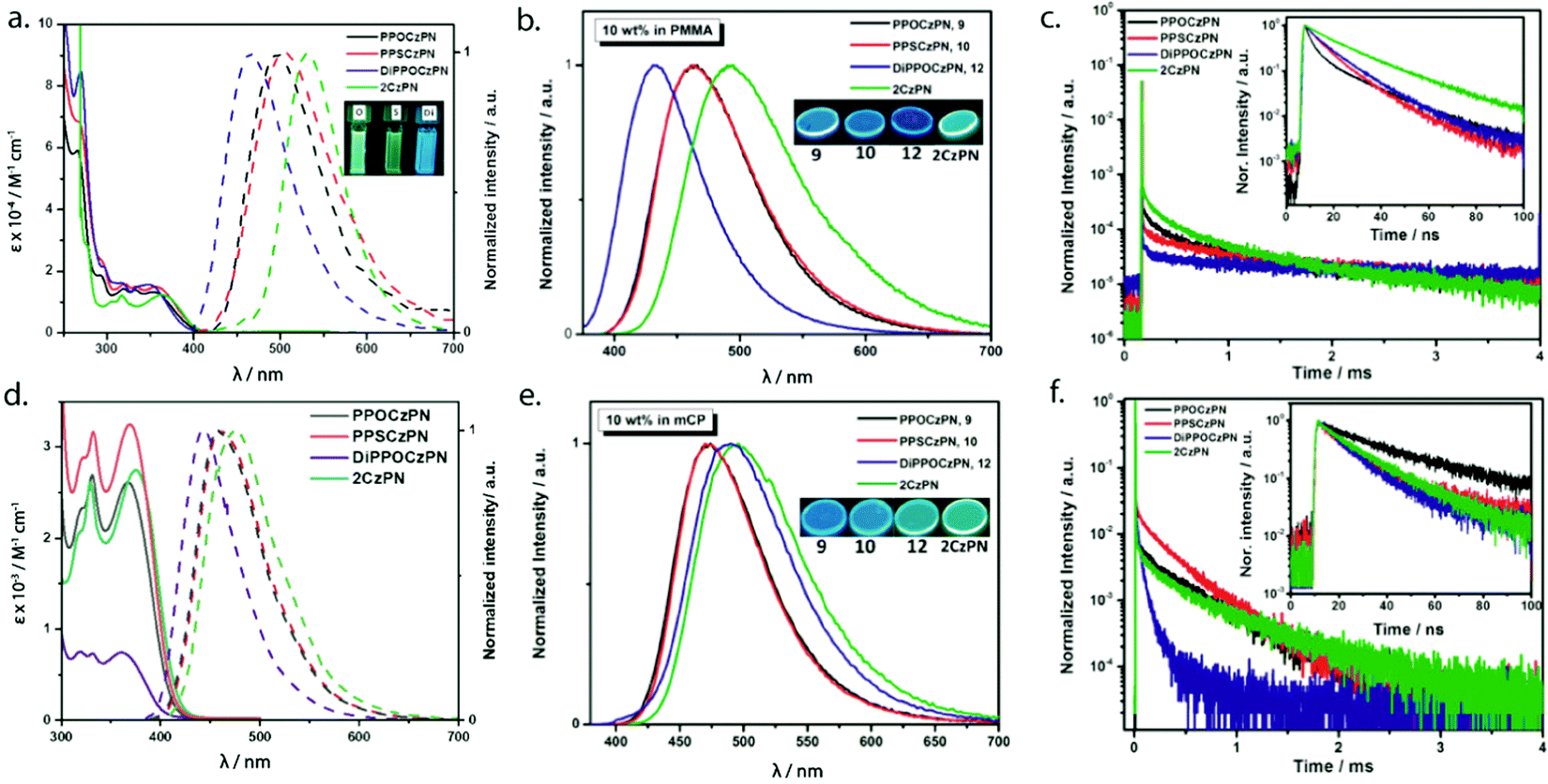 | ||
| Fig. 4 (a) Absorption (solid line) and emission (dashed line) spectra of PPOCzPN, PPSCzPN, DiPPOCzPN and 2CzPN recorded at room temperature in MeCN and DCM solution (λexc = 358 nm), respectively. (b) Steady-state photoluminescence spectra of 10 wt% PMMA films of PPOCzPN, PPSCzPN, DiPPOCzPN, 2CzPN (λexc = 358 nm). (c) Time-resolved photoluminescence decay profiles of 10 wt% PMMA films of PPOCzPN, PPSCzPN, DiPPOCzPN, 2CzPN (λexc = 378 nm). (d) Absorption and emission spectra of PPOCzPN, PPSCzPN, DiPPOCzPN and 2CzPN recorded at room temperature in toluene solution (λexc = 370 nm). (e) Steady-state photoluminescence spectra of 10 wt% mCP films of PPOCzPN, PPSCzPN, DiPPOCzPN, 2CzPN (λexc = 358 nm). (f) Time-resolved photoluminescence decay profiles of 10 wt% mCP films of PPOCzPN, PPSCzPN, DiPPOCzPN, 2CzPN (λexc = 378 nm). | ||
The photoluminescence properties of PPOCzPN, PPSCzPN, DiPPOCzPN and 2CzPN were investigated in DCM and toluene, as neat films and as 10 wt% doped PMMA and mCP (1,3-bis(N-carbazolyl)benzene) films, the latter matrix identified as a suitably high triplet-energy host that had previously been used with 2CzPN. The photophysical properties are summarized in Tables 1 and 2 and the emission spectra and decay profiles of the emitters doped in PMMA and mCP thin films are shown in Fig. 4c and f. In DCM, the emission maxima shift to progressively higher energy across the series from 2CzPN to PPOCzPN, PPSCzPN, and DiPPOCzPN, at 532 nm, 498 nm, 501 nm, and 465 nm, respectively, reflecting the weakening of the donor. On the other hand, owing to its lower polarity, the emission maxima of these emitters are further blue-shifted in toluene compared to DCM, a reflection of their CT character. In 10 wt% doped PMMA films, the λPL values are all blue-shifted compared to those in DCM solution at 492 nm, 465 nm, 465 nm, and 428 nm, due the less polar nature of this medium (Fig. 4b). In 10 wt% doped mCP films, the λPL values are surprisingly red-shifted and clustered closer to each other at 496 nm, 474 nm, 474 nm, and 490 nm for 2CzPN, PPOCzPN, PPSCzPN, and DiPPOCzPN, respectively, in full consistency with the trends observed in toluene (Fig. 4d). The photoluminescence quantum yields, ΦPL, in DCM of PPOCzPN, PPSCzPN and DiPPOCzPN (47–61%) are higher than that of 2CzPN (29%).86 The ΦPL values of the emitters are somewhat lower in PMMA doped films (ranging from 28–59%), and are lower than that of 2CzPN (76%) in the same medium. In mCP, the ΦPL is recovered with values of 72%, 73% and 56% for PPOCzPN, PPSCzPN and DiPPOCzPN, while that of 2CzPN is very similar to that seen in PMMA (77%). A slightly higher ΦPL (89%) has been reported for 6 wt% 2CzPN in co-evaporated mCP thin film.59
| Emitter | Toluene | 10 wt% doped mCP film | Neat film | |||||
|---|---|---|---|---|---|---|---|---|
| λ abs /nm | λ PL /nm | ΔESTg/eV | λ PL /nm | Φ PL /% | ΔESTg/eV | λ PL /nm | Φ PL /% | |
| a ICT band measured in toluene at 298 K. b In toluene (λexc = 370 nm). c In 10 wt% doped mCP thin films. d Absolute quantum yield in 10 wt% doped mCP thin films. e In neat thin films. f Absolute quantum yield in neat thin films. g Measured as the energy difference between the prompt fluorescence onset (at 77 K, λexc = 378 nm, time delay = 1 ns, integration time: 1–100 ns) and the phosphorescence onset (at 77 K, λexc = 378 nm, time delay = 1 ms, integration time = 1–10 ms). h Values from ref. 86. i Values from ref. 59, measured in 6 wt% doped mCP film, ΔEST was estimated from the fluorescence (300 K) and phosphorescence (5 K) peaks. NA = not available. | ||||||||
| PPOCzPN | 321, 331, 367 | 460 | 0.20 | 474 | 72 | 0.19 | 501 | 54 |
| PPSCzPN | 321, 332, 369 | 460 | 0.42 | 474 | 73 | 0.24 | 524 | 26 |
| DiPPOCzPN | 319, 332, 361 | 443 | 0.43 | 490 | 56 | 0.15 | 466 | 38 |
| 2CzPN | 317, 329, 375 | 476 | 0.36 | 496 | 77 | 0.23 | 516 | 33 |
| 2CzPN | 317, 329, 375h | 484h | 0.31h | 477i | 89i | 0.09i | NA | NA |
Fig. 4c shows the time-resolved decay traces of the emitters in a PMMA matrix. The emission decay consisted of both prompt (τp) nanosecond and delayed (τd) microsecond fluorescence components. The prompt fluorescence decays of PPOCzPN, PPSCzPN and DiPPOCzPN were found to be bi-exponential with lifetimes, τp, ranging from 13–20 ns, similar to that of 2CzPN (τp = 8, 21 ns). The delayed fluorescence decay of the three emitters is tri-exponential in nature with average lifetime of 393 μs, 416 μs, and 513 μs for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively, significantly longer than that of 2CzPN (τd = 270 μs). Fig. 4f shows the emission decays in mCP, which are similar to the decay profiles in the PMMA matrix. In this medium, the τp ranges from 8–37 ns, longer than those measured in PMMA, and also longer than that of 2CzPN (τp = 9, 23 ns). The τd values, on the other hand, for PPOCzPN, PPSCzPN and DiPPOCzPN in 10 wt% mCP thin films were found to be 210 μs, 206 μs, and 54 μs, respectively, which are significantly shorter than those in PMMA and are also shorter than that of 2CzPN (τd = 277 μs). An average τd of 273 μs has been reported previously for 6 wt% 2CzPN in mCP thin film.59 The excited state lifetime of the emitters doped in PMMA and mCP films are summarized in Table 3. Altogether, in contrast to 2CzPN, the present data for PPOCzPN, PPSCzPN and DiPPOCzPN emitters clearly demonstrate the high sensitivity of the emitter decay dynamics and emission energy towards the nature of the host medium.
| Emitter | 10 wt% doped in PMMA films | 10 wt% doped in mCP films | ||
|---|---|---|---|---|
| τ p (%)/ns; τd (%)/μs | τ p (%)/ns; τd (%)/μs | S1/eV | T1/eV | |
| S1 = singlet state energy obtained from the onset of prompt fluorescence spectra measured at 77 K (λexc = 360 nm, delay time: 1 ns, time window: 1–100 ns). T1 = triplet state energy obtained from the onset of the phosphorescence spectra measured at 77 K (λexc = 360 nm, delay time: 1 ms, time window: 1–10 ms). | ||||
| PPOCzPN | 6.1 (43), 20.0 (57); 9 (3), 146 (22), 1024 (75) | 10.0 (12), 37.2 (88); 20 (4), 90 (18), 522 (78) | 2.96 | 2.77 |
| PPSCzPN | 5.8 (44), 13.6 (56); 5 (0.5), 174 (10), 1361 (90) | 8.6, (44), 31.6 (56); 23 (4), 175 (33), 422 (62) | 2.97 | 2.73 |
| DiPPOCzPN | 6.7 (40), 14.1 (60); 5 (2), 69 (6), 1176 (92) | 8.1 (58), 26.4 (42); 0.5 (76), 28 (18), 136 (6) | 2.96 | 2.81 |
| 2CzPN | 8.3 (15), 21.1 (85); 6 (4), 124 (22), 680 (72) | 9.9 (29), 23.2 (71); 7 (3), 134 (23), 690 (73) | 2.90 | 2.67 |
Temperature-dependent time-resolved emission decay measurements in 10 wt% doped mCP thin films are shown in Fig. S39 (ESI‡). There is a clear thermal activation of the delayed fluorescence that is consistent with compounds emitting via a TADF mechanism, despite the different trends in comparison to the PMMA matrix.
The singlet–triplet energy gaps (ΔEST) were determined from the difference in energy of the onset of the prompt fluorescence and phosphorescence spectra in 10 wt% doped mCP films measured at 77 K (Fig. 5). The ΔEST values for PPOCzPN, PPSCzPN, DiPPOCzPN and 2CzPN were found to be 0.19 eV, 0.24 eV, 0.15 eV and 0.23 eV, respectively. The experimentally determined ΔEST value for 2CzPN in 10 wt% doped mCP film is in good agreement with the reported value of 0.21 eV in a 6 wt% doped mCP thin film.59,86 The smallest ΔEST value (0.15 eV for DiPPOCzPN) is consistent with the observed τd, value of 54 μs, which is the shortest among these emitters. It is noteworthy that the TD-DFT calculated ΔEST values in the gas phase are consistent with those experimentally determined in the apolar matrix, vide infra, Table 4. The ΔEST values determined for these chalcogenide-substituted 2CzPN derivatives are of similar magnitude to literature 2CzPN derivatives such as α-2CbPN (ΔEST = 0.28 eV) and δ-2CbPN (ΔEST = 0.13 eV) in 20 wt% mCP thin film, where α/δ-carboline was used as carbazole replacement to blue-shift the emission.60
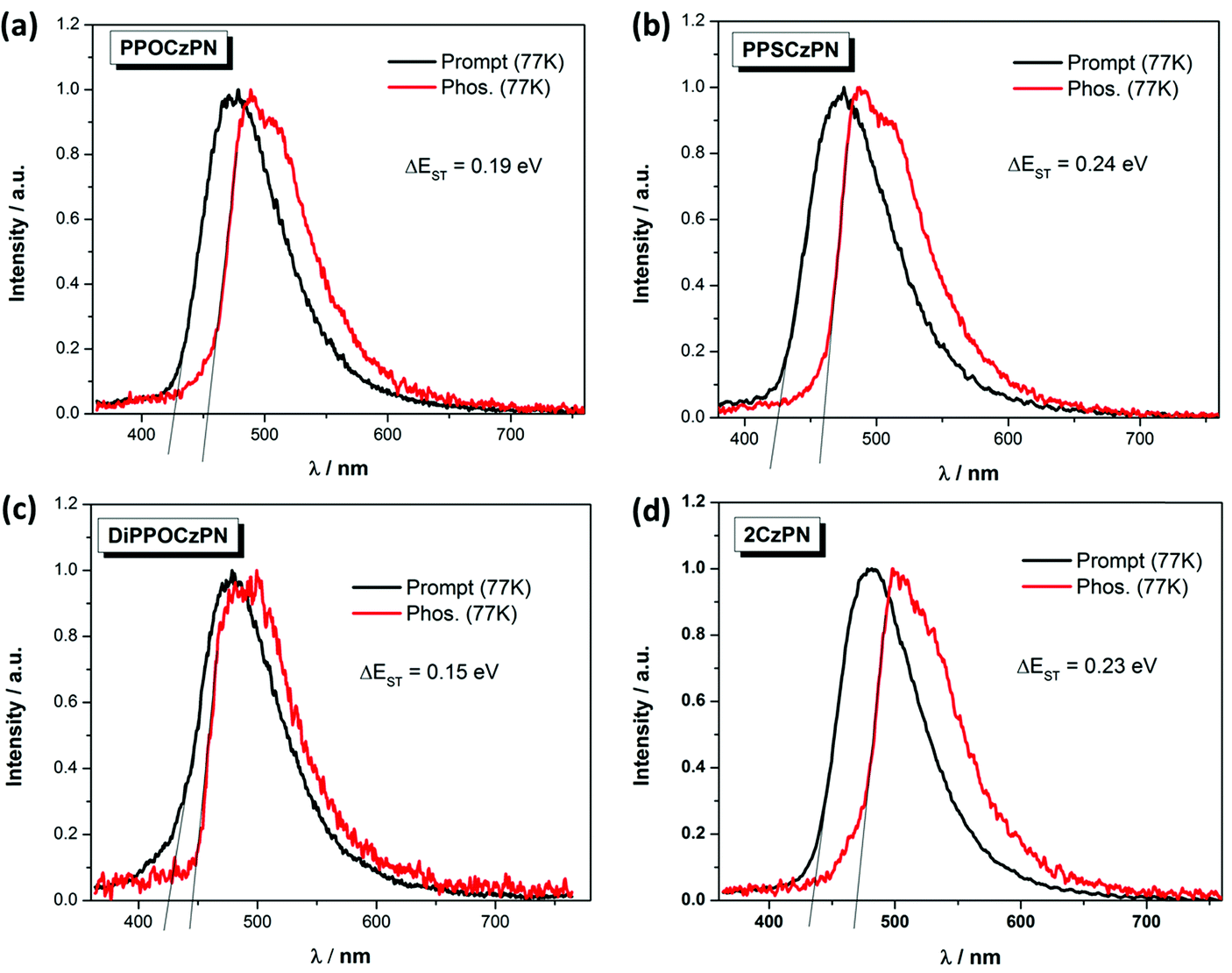 | ||
| Fig. 5 Prompt fluorescence (at 77 K) and phosphorescence spectra (at 77 K) of (a) PPOCzPN, (b) PPSCzPN, (c) DiPPOCzPN, and (d) 2CzPN, in 10 wt% doped mCP thin films (λexc = 360 nm). | ||
| In gas phase | In MeCN | |||||
|---|---|---|---|---|---|---|
| HOMO/eV | LUMO/eV | ΔE/eV | HOMO/eV | LUMO/eV | ΔE/eV | |
| PPOCzPN | −6.05 | −2.52 | 3.53 | −6.06 | −2.48 | 3.58 |
| PPSCzPN | −5.98 | −2.58 | 3.40 | −6.13 | −2.50 | 3.63 |
| DiPPOCzPN | −6.42 | −2.73 | 3.69 | −6.37 | −2.57 | 3.80 |
| 2CzPN | −6.11 | −2.41 | 3.70 | −6.04 | −2.39 | 3.65 |
3.3 Theoretical modelling
To better understand the differences between the PL behaviors observed in the polar DCM solvent or PMMA matrix versus the apolar toluene solvent or mCP matrix, our study has been complemented by quantum-chemical calculations. In line with our previous theoretical studies on TADF emitters,86,90 we have first optimized the ground-state geometry of the 2CzPN and its three derivatives in the gas phase at the Density Functional Theory (DFT) level using the PBE0 functional91 and a 6-31G(d,p) basis set.92 In a second step, we have analyzed the nature of the frontier electronic levels of the compounds. Finally, we have described the lowest singlet and triplet excited states of the emitters by performing TD-DFT calculations within the Tamm–Dancoff approximation,93 employing the same functional and basis set.In the case of the isolated PPOCzPN and PPSCzPN compounds, we have obtained two different low-energy conformers varying by the amplitude of the torsion angles ϕ between the carbazole and phthalonitrile units (see Fig. S40, ESI‡). For PPOCzPN, rotamer 2 with ϕ1 = 134.3° and ϕ2 =-69.2° is more stable by 0.29 eV compared to the other rotamer with ϕ1 = −64.8° and ϕ2 = −65.9°; in the case of PPSCzPN, there is a much larger difference of 1.33 eV between the most stable rotamer (with ϕ1 = 133.2° and ϕ2 = −69.5°) compared to the second rotamer (with ϕ1 = −65.8° and ϕ2 = −64.6°). In view of the large energy difference between the two rotamers (compared to the thermal energy, kT), their co-existence in solution most probably arises from their synthesis, rather than from thermal conversion following synthesis. We will discuss hereafter the properties of the most stable rotamer; for sake of completeness, the very similar properties computed for the second rotamer in the gas phase are collected in the ESI‡ (Fig. S40, S41 and Tables S2, S4). In contrast, we only found a single stable conformer for 2CzPN and DiPPOCzPN with the same torsion angles for both carbazole substituents (ϕ1 = ϕ2 = 58.9° for 2CzPN and ϕ1 = ϕ2 = 65.7° for DiPPOCzPN).
Table 4 collects the HOMO and LUMO energies of the four emitters calculated in the gas phase as well as in acetonitrile by modelling the solvent as a dielectric continuum using the Polarizable Continuum Model (PCM),94 as implemented in Gaussian 16-A03;95 in the latter case, the geometry is fully reoptimized within the PCM model, although this hardly changes the gas phase geometry. In acetonitrile, the HOMO is progressively stabilized going from 2CzPN to PPOCzPN to PPSCzPN and DiPPOCzPN, which is generally consistent with the trends observed in the experimental electrochemistry data in DCM (Table 1) and acetonitrile (Fig. S36, ESI‡). On the other hand, the calculated LUMO energies vary to a lesser extent both according to the calculations and electrochemical measurements, as expected by the fact that the LUMO is localized on the phthalonitrile acceptor while the varying substituents are added on the donor fragment. Strikingly, the trends appear to be quite different when computing the frontier orbital energies in the gas phase, which is expected to reflect the properties in a non-polar medium such as toluene or the mCP matrix. The major difference is that the HOMO is not stabilized but in fact slightly destabilized when going from 2CzPN to PPOCzPN or PPSCzPN, although it is stabilized going from 2CzPN to DiPPOCzPN.
The shape and energies of the orbitals in acetonitrile are displayed in Fig. 6 (and Fig. S41, ESI,‡ for the less stable rotamers of PPOCzPN and PPSCzPN). As expected, the HOMOs are mainly localized on the carbazole moieties, and the LUMOs are strongly localized on the phthalonitrile. In the monosubstituted compounds, the HOMO displays a small amount of electron density on the phosphine chalcogenide units, thus limiting the role played by mesomeric effects in defining the actual HOMO energies. The HOMO level is both the most delocalized in DiPPOCzPN and the most stabilized, which is also evidenced by its electrochemistry (Fig. 3 and Fig. S36, ESI‡). The shift of the HOMO among the compounds is a subtle interplay mostly between the inductively electron-withdrawing effects associated with the phosphine chalcogenide groups and electrostatic interactions between the polar phosphine chalcogenide and cyano moieties, which are expected to be further tuned by changes in the medium polarity. Altogether, the HOMO–LUMO gap in the gas phase is red shifted by 0.2–0.3 eV going from 2CzPN to PPOCzPN and PPSCzPN while it remains unchanged for DiPPOCzPN. In contrast, the electronic bandgaps of 2CzPN, PPOCzPN, PPSCzPN are quite similar in MeCN and larger by ∼0.2 eV for DiPPOCzPN, thus demonstrating the strong impact of the medium polarity on the electronic structure of the compounds and by extension on the charge-transfer state energies, as evidenced at the experimental level.
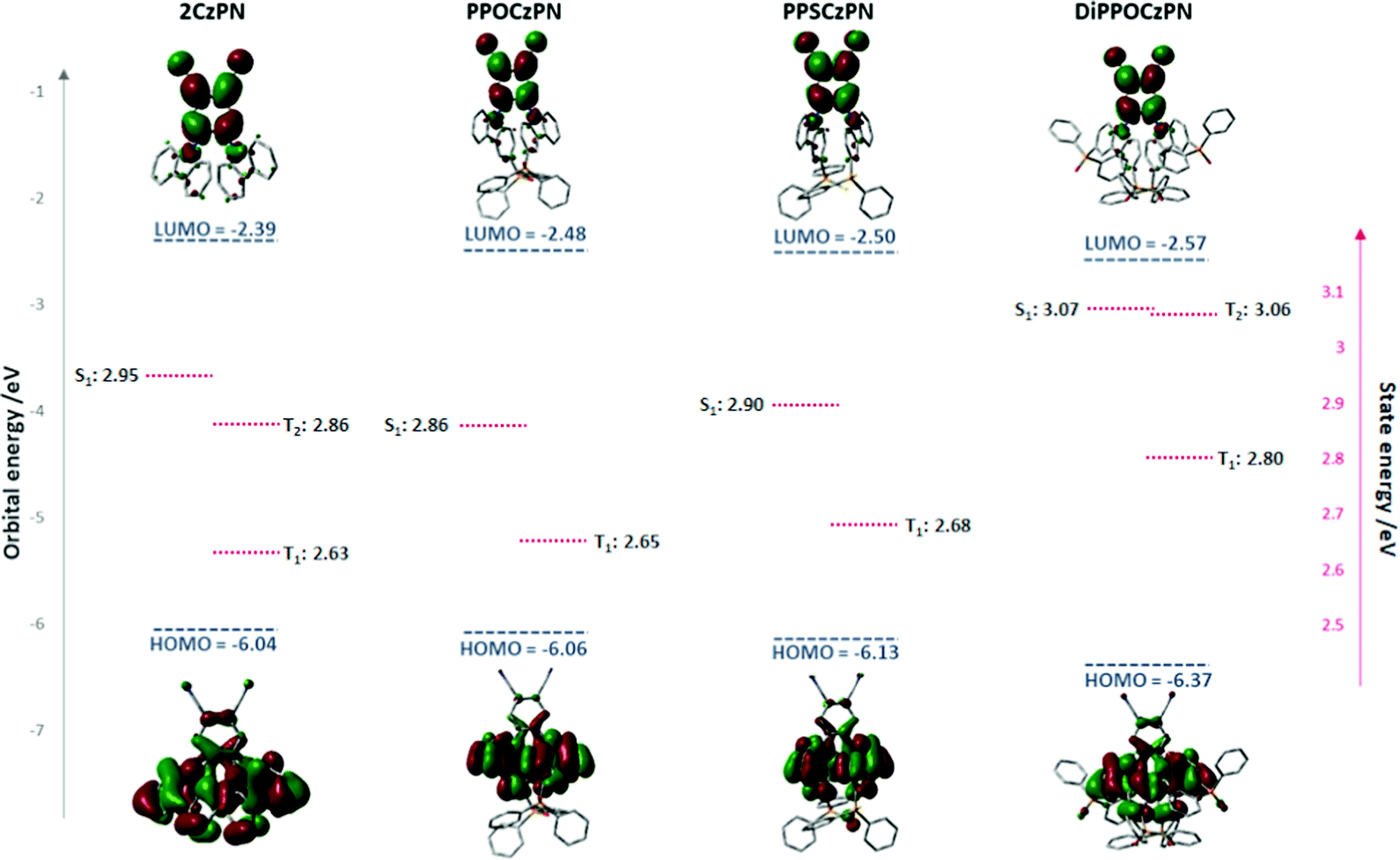 | ||
| Fig. 6 HOMO and LUMO energies of the four compounds in their most stable geometries together with the energy of the lowest singlet and triplet excited states, as calculated in acetonitrile. | ||
When coupled to a time-dependent (TD) formalism, DFT calculations can also give access to the excited-state photophysical properties of the emitters. We will describe below the excited state energies and associated oscillator strength (f) calculated in acetonitrile as well as the energy difference between T1 and S1 (Table 5) for vertical excitation processes (i.e. in the ground-state geometry). We will not address here at the theoretical level the changes in the emission properties as a function of the medium polarity evidenced experimentally, since a proper analysis would require a time-consuming optimization of the S1 state (both in gas phase and in a polar medium described with PCM) which is often impeded by convergence problems and might require the use of another functional.91PPOCzPN and PPSCzPN exhibit a slightly red-shifted S1 state compared to 2CzPN (blue-shifted in the case of DiPPOCzPN), accompanied by a reduction in the oscillator strength, as well as slightly smaller ΔEST values. The calculated ΔEST values of the emitters in the toluene are all around or above 0.2 eV, in good consistency with those experimentally obtained in toluene (Table 2); there is a very good quantitative agreement for 2CzPN (0.31 eV in theory versus 0.36 eV in toluene). These energies are marginally affected when performing the calculations in toluene or in acetonitrile (Table S4, ESI‡). Strikingly, the experimental ΔEST values are systematically reduced in the mCP matrix, most likely due in part to changes in the conformations of the emitters in these thin films. Both 2CzPN and DiPPOCzPN have a second triplet excited state T2 that lies below S1, thus providing a conduit to facilitate reverse intersystem crossing process.77,96 The T1 and S1 excited states are predominantly characterized by a HOMO–LUMO transition and hence both display a significant ICT character.
| Compound | States | Energy/eV | f | Main component of the excitation | Φ s |
|---|---|---|---|---|---|
| PPOCzPN | T1 | 2.65 | HOMO → LUMO (76.5%) | 0.60 | |
| S1 | 2.86 | 0.028 | HOMO → LUMO (97.7%) | 0.35 | |
| ΔEST | 0.21 | ||||
| PPSCzPN | T1 | 2.68 | HOMO → LUMO (80.5%) | 0.61 | |
| S1 | 2.90 | 0.032 | HOMO → LUMO (97.9%) | 0.37 | |
| ΔEST | 0.22 | ||||
| DiPPOCzPN | T1 | 2.80 | HOMO → LUMO (82.1%) | 0.66 | |
| T2 | 3.06 | 0.50 | |||
| S1 | 3.07 | 0.045 | HOMO → LUMO (98.3%) | 0.39 | |
| ΔEST | 0.27 | ||||
| 2CzPN | T1 | 2.63 | HOMO → LUMO (87.8%) | 0.66 | |
| T2 | 2.86 | 0.53 | |||
| S1 | 2.95 | 0.100 | HOMO → LUMO (98.4%) | 0.43 | |
| ΔEST | 0.32 | ||||
The absorption spectra simulated in the gas phase (Fig. S43, ESI‡) predict similar energy for the lowest absorption peaks (around 350 nm and 400 nm) for the four compounds; the same trends are also observed for the experimental spectra recorded both in MeCN and toluene (Fig. 4a and d). Note that the lowest absorption peak around 400 nm is systematically associated with higher-lying excited states while the lowest excited state S1 gives rise to the shoulder on the low energy side. This state is mostly described by a HOMO to LUMO excitation. Analysis of the hole and electron densities (see typical plots for the gas phase in Fig. S42, ESI‡) obtained within the attachment/detachment formalism points to a similar charge transfer character in the S1 state of the four compounds in acetonitrile or in gas phase (Table 5); the overlap index ϕS, ranging from 1 for a pure locally excited state (LE) to 0 for a pure charge-transfer (CT) state, is around 0.4 in all cases, i.e., an indication of a hybrid state with significant LE and CT character. The T1 state exhibits systematically a higher ϕS index pointing to a more local excited state. This change in character between the S1 and T1 (T2) states ensures non-vanishing spin–orbit coupling based on El Sayed's rules and opens up the RISC channel.
3.4 Electrochemiluminescence via the Annihilation pathway
ECL is luminescence generated when electrochemically-generated radical cations (hole) and radical anions (electron) undergo an electron transfer to create an excited state (exciton), which can release its energy as light (Fig. 7a). The behavior of this luminescence can describe radical stability, reactivity and general emission mechanisms of luminophores at concentrations that are typically greater than that in solutions used in photoluminescence studies. Radical cations and anions are produced in solution when the species are oxidized and reduced, respectively in the vicinity of the same working electrode. If these two species collide, an electron transfer can happen from the HOMO (or LUMO) of a radical anion to the HOMO (or LUMO) of a radical cation, potentially creating an exciton that can subsequently emit light in the form of ECL. This mechanism is not limited to the generation of triplets as illustrated in Fig. 7a, but also can form systematically singlets in the case of an annihilation mechanism, in an analogous manner to exciton generation in an organic light-emitting diode. We note that it is unlikely for radicals to undergo further redox reactions at an electrode surface to create excited state species as described in the literature.97 In fact, the electrode surface might quench the radical or excited species.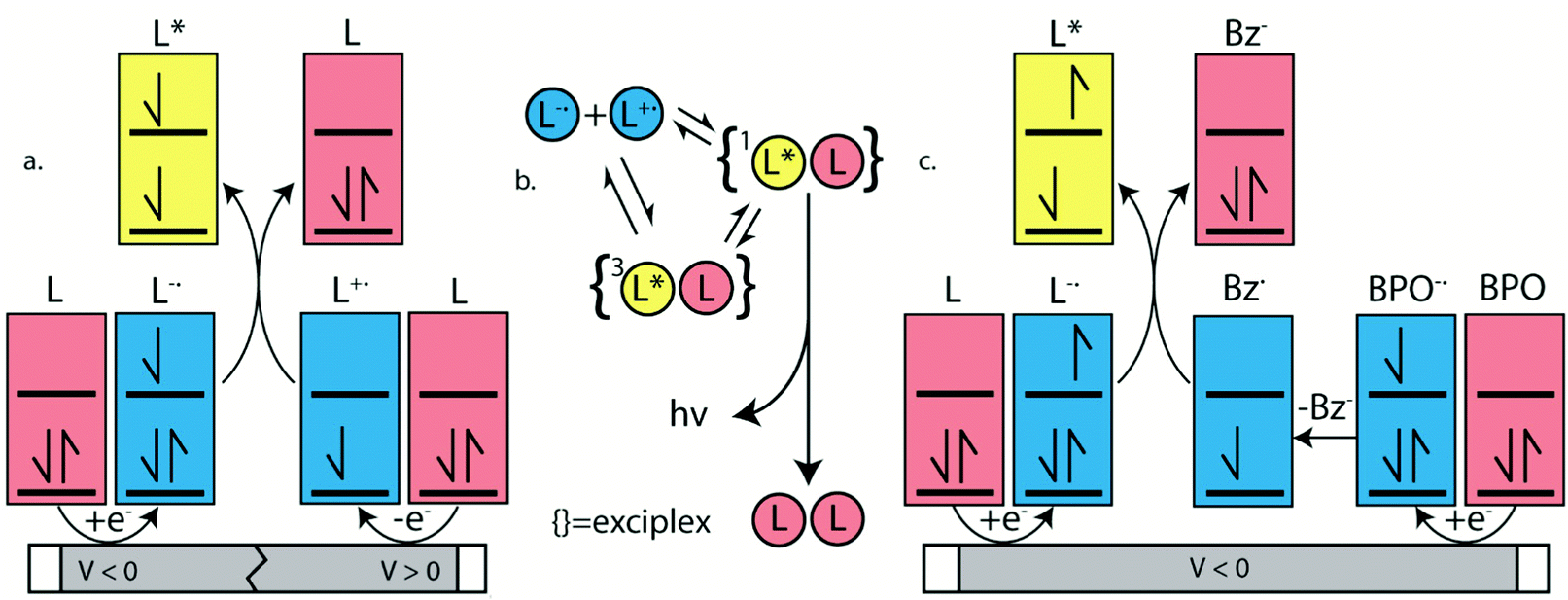 | ||
| Fig. 7 (a) Annihilation and (c) benzoyl peroxide (BPO) co-reactant mechanisms for ECL emission. L, benzoic acid (Bz) and BPO stand for luminophore, benzoate and benzoyl peroxide, respectively. This scheme is illustrative and not energy accurate. Both triplet and singlet energy states can be accessed from either co-reactant or annihilation mechanisms. (b) Diagram illustrating excimer formation and the equilibrium established between anionic and cationic radicals. | ||
To electrogenerate an excited state, the electronic gap of the redox reactions (EEg) must be greater than the photoluminescence emission peak energy (Eg) as defined by eqn (2). For an ECL annihilation system, R′ and R are the same species. However, R′ can become a radical while R remains the luminophore in an oxidative-reduction ECL co-reactant system or R can become an oxidant while R′ remains a luminophore in a reductive-oxidation ECL co-reactant system. Using eqn (2), the electronic gap for PPOCzPN, PPSCzPN and DiPPOCzPN in DCM are measured to be 2.73, 2.98 and 3.19 eV, respectively, from the redox potentials in differential pulse voltammograms (Fig. S35, ESI‡). From the prompt fluorescence spectra in DCM in Fig. 5, the optical gap for PPOCzPN, PPSCzPN and DiPPOCzPN are 2.49, 2.47 and 2.66 eV, respectively. As expected, the energy provided by the redox reactions for all compounds is sufficient to generate both singlet and triplet excited states. The blue curves in Fig. 3a–c demonstrate ECL generated during scan cycles between redox reaction potentials for PPOCzPN, PPSCzPN and DiPPOCzPN. DiPPOCzPN shows a maximum ECL intensity of 60 nA, while PPOCzPN and PPSCzPN display weak emissions up to 1 nA. This indicates that the radical cation is sufficiently stable to survive in solution until the radical anion is created, and the two species collide. DiPPOCzPN˙+ is the most stable radical ion because it produced the highest ECL emission in the annihilation pathway. It also appears that the radical anion is not as stable as the radical cation for all species, because no ECL is seen upon first reduction of the species followed by compound oxidation, even after multiple cycles. The irreversibility of the oxidation reaction shown in CVs might be caused by the potential window limit of the DCM electrolyte solution as can be seen from the CVs in MeCN, Fig. S37 (ESI‡).
Fig. 3d–f shows ECL-time curves during pulsing between 0.1 V more anodic than the oxidation and 0.1 V more cathodic than reduction peak potentials, respectively. PPOCzPN, PPSCzPN and DiPPOCzPN yielded annihilation ECL maxima at 700, 1100 and 17500 nA, respectively. In pulsing experiments, the time between the generation of both radical species in solution is shorter than that in potential cycling, allowing the lifetime of the radical not to be a significant issue and the annihilation ECL to be enhanced. The pulsing annihilation efficiency was determined by eqn (1) where x and st stand for the luminophore and [Ru(bpy)3]2+ under the same conditions, respectively. These ECL efficiencies were determined to be 1, 1 and 10% relative to [Ru(bpy)3]2+, for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively (Table 6). Interestingly, 2CzPN showed no emissions in the annihilation pathway as seen in Fig. S38a and b (ESI‡). DiPPOCzPN continued to have the highest ECL maxima and also had the most efficient emission; interestingly, the structurally similar PPOCzPN had the lowest efficiency. Most emission was observed when a negative potential was applied, further providing evidence for the higher stability of the radical cation over the radical anion. In fact, PPOCzPN is the only species to display emission when a positive potential was applied or when radical cations were actively being produced in pulsing experiments (Fig. 3d–f), indicating a relatively high radical anion reactivity, or a relatively lower radical cation reactivity than the other compounds. For optoelectronic applications, ECL efficiency, ECL maxima, radical stability and radical reactivity are all important parameters to consider. Inequalities between radical cation and radical anion stabilities and reactivities can cause excess charge buildup in OLEDs, leading to fast device degradation. High ECL maxima and efficiencies indicate efficient charge transfers as well as limited non-radiative decay pathways. Therefore, PPOCzPN appears desirable for optoelectronic applications because of relatively more stable and reactive radical ion species, although it has low ECL maxima and efficiency.
| PPOCzPN | PPSCzPN | DiPPOCzPN | [Ru(bpy)3]2+ | |
|---|---|---|---|---|
| a Electronic gaps (EEg) were obtained using eqn (2) and the differential pulse voltammograms of the compounds in Fig. S36. b These values are averages from at least 40 pulses with individual conditions described in Fig. 3. c Obtained from eqn (1) relative to [Ru(bpy)3]2+ at the same concentration, electrolyte and concentration of co-reactant if applicable. d Obtained from eqn (2) but with substituting E°(R˙+′/R′) with the oxidative power of BPO from ref. 98. | ||||
| EEg (eV)a | 2.73 | 2.98 | 3.19 | 2.55 |
| Pulsing ECLonset delay (ms)b | 7 | 14 | 11 | 0 |
| Pulsing ECLmax delay (ms)b | 11 | 19 | 24 | 4 |
| Pulsing ECL efficiency (%)c | 1 | 1 | 10 | 100 |
| Max pulsing ECL (nA) | 700 | 1150 | 17500 |
70000 |
| Annihilation ECL λmax (nm) | 520 | 500 | 475 | 620 |
| EEg BPO (eV)d | 3.01 | 3.48 | 3.39 | 3.04 |
| BPO ECL efficiency (%)c | 17 | 6 | 1 | 100 |
| Max BPO ECL (nA) | 3200 | 1280 | 80 | 26500 |
| BPO ECL λmax (nm) | 520 | 500 | 545 | 620 |
After several pulses, the emission intensity of DiPPOCzPN decreases by over 75%, whereas PPOCzPN and PPSCzPN remain relatively stable over the same time (Fig. S45, ESI‡). It appears that DiPPOCzPN may form side products during the annihilation pulsed ECL experiment that influence the generation of excited species. This fast decay may also be caused by a charge imbalance where negatively or positively charged species that accumulate over time interfere with the exciton states, similar to exciton–polaron quenching of excited states.99
We first considered phosphorescence as the origin of these long ECL onset times. For time comparisons of diffusion properties and emissions, the well-studied commercial phosphorescent ECL emitter [Ru(bpy)3]2+,100–103 was tested under the same conditions (Fig. S47, ESI‡) and showed no observed delay (47 pulses measured). A well-studied organic compound with a known diffusion coefficient is diphenylanthracene (DPA) (DACN = 0.20 × 10−6 cm2 s−1),104 which shows an order of magnitude difference between its diffusion coefficient and that of [Ru(bpy)3]2+ (DACN = 5.8 × 10−6 cm2 s−1).105 If we assume that DPA and these 2CzPN derivatives have similar magnitudes of diffusion coefficient in DCM, from a comparison of their initial emission delays (7 and 0 ms for PPOCzPN and [Ru(bpy)3]2+, respectively), differences in diffusion rates can be ruled out as a cause of emission delays in the 2CzPN derivatives. We believe diffusion delays are on the time scale of microseconds and cannot explain the observed time delay shown in Fig. 3d–f.
We next considered triplet–triplet annihilation as the origin for the long ECL onset times. A TTA mechanism has been used to explain millisecond photoluminescence lifetimes for phenanthrene and may explain the delay of the onset of the ECL seen here.106 Also, the competition for triplet exciton depopulation by TTA, RISC and non-radiative pathways has been studied by Grüne et al. in a m-MTDATA:3TPYMB exciplex TADF OLED to estimate that TTA was responsible for 50% of triplet depopulation, which effectively extended the emission decay time.107 However, Grüne et al. excited their sample for 4 ms at a specific current density and did not see any delay in the onset of emission. TTA has been observed in carbazolyl phthalonitrile derivatives where there is an observed blue-shifting in the solid-state PL as well as serious efficiency roll-off in the OLEDs,108,109 but to the best of our knowledge it has not been demonstrated before by ECL in 2CzPN derivatives. Mechanistically, TTA can be simplified to two triplet excitons (T1) combining to form a singlet exciton (Sn) seen in Fig. 7b. The system in the Sn state then relaxes to the S1 state by internal conversion according to Kasha's law, which can then emit light. TTA is also a common tetramolecular process in ECL where the radical cation of N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD˙+) and the radical anion of DPA (DPA˙−) reside as a well-studied purely TTA-emitting ECL system.68,98,110,111 This TMPD˙+/DPA˙− system can only generate T1 from charge transfer reactions and not S1 as dictated by eqn (2). However, this TTA mechanism has never been accompanied by an observed induction time either in ECL or OLED prior to observed emission.
To the best of our knowledge, there has only been one study of emission delay after excitation in an ECL system, where the ECL of DPA was found to have a delay on the order of 100 μs.112 This delay was attributed to uncompensated solution resistance. In comparison, our experiment also has a platinum working electrode with the same electrolyte (TBAP) at a similar concentration, but the delay we determined is a few orders of magnitude larger than what Rosenmund et al. observed. Thus, neither uncompensated solution resistance, nor triplet–triplet annihilation (TTA), nor phosphorescence is responsible and that this ECL emission delay is currently unexplained in the literature.
Interestingly, an organic long-persistent photoluminescence (OLPL) phenomenon was recently discovered by Kabe et al. in a system comprising a phosphine oxide-containing TADF acceptor (2,8-bis(diphenylphosphoryl)dibenzo-[b,d]thiophene) (PPT)) and N,N,N′,N′-tettramethylbenzidine (TMB) as a donor molecule.113 More recently, this OLPL system has been used in an OLED to achieve organic long-persistent electroluminescence (OLEL).114,115 Unfortunately, solution and solid state emission delays of the system were not investigated on time scales smaller than 1 s, thereby limiting comparisons with the 2CzPN derivatives. However, OLPL and OLEL are both emitted from an exciplex state and therefore the emission was found to be both concentration dependent and layer-thickness dependent. This exciplex emission was also found to have thermal activation processes and thermoluminescence in films.115 Li et al. reported that the TADF emitter, CzPhAP, did not show any delay in the onset of emission but did show 900 s of persistent emission after 60 s of laser excitation, when cast in a PMMA film.116 These unusually long emission times were ascribed to slow charge recombination rates of the luminophore in the thick film, leading to organic long-persistent photoluminescence (OLPL) because of an established equilibrium between exciplex formation with radicals and the subsequent radical disassociation. We thus contend that the delay in the onset of the ECL emission seen in Fig. 3d–f may be best explained by a heretofore previously unreported organic long-persistent electrochemiluminescence (OLECL), analogous to the works of Kabe et al. We hypothesize that an equilibrium exists between the emissive excimer and the dissociated constituents and that the slow association kinetics could cause this delay as illustrated in Fig. 7b.
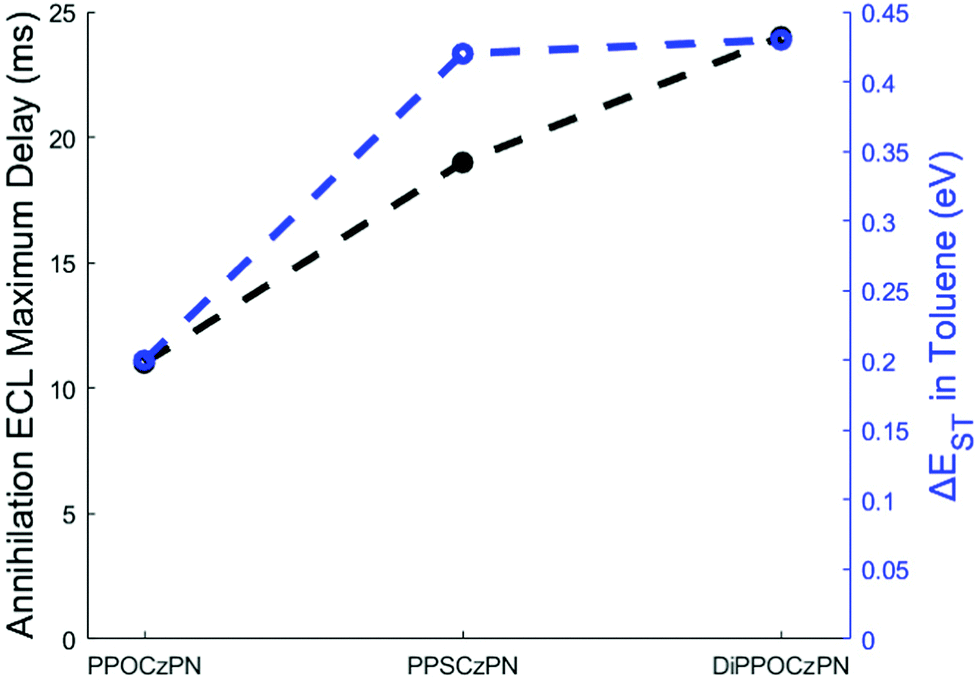 | ||
| Fig. 8 A relationship between the ΔEST in toluene and the annihilation ECL maximum delay of the 2CzPN derivatives in this study. | ||
3.5 ECL of BPO Co-reactant systems
Benzoyl peroxide (BPO) was added as a co-reactant to the electrochemical systems described in Fig. 3 and the potential was scanned in the cathodic direction. This co-reactant pathway is described in Fig. 7c. Around −0.45 V vs. SCE, BPO is reduced and ultimately decomposes into a benzoate radical (Fig. 7c). When PPOCzPN, PPSCzPN and DiPPOCzPN are subsequently reduced to form PPOCzPN˙−, PPSCzPN˙− and DiPPOCzPN˙−, the benzoate radical produced earlier can abstract an electron from the HOMOs of the reduced TADF compounds to produce excited states PPOCzPN*, PPSCzPN* and DiPPOCzPN*, respectively. These excited-state compounds then emit ECL through the so-called co-reactant pathway. Fig. 9a–c show the reduction waves corresponding to the reduction of BPO (red traces) and the corresponding photocurrent generated (blue traces). ECL intensity maxima of 3000, 1280 and 80 nA were measured for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively. PPOCzPN showed the strongest ECL response, which was almost three-fold stronger than that of PPSCzPN and 38 times that of DiPPOCzPN. From previous theoretical and experimental studies,98 benzoate radicals have a redox potential of 1.5 V, which is sufficient to oxidize the three TADF compounds in this study, and for this co-reactant ECL pathway to provide more energy than the annihilation ECL pathway. The difference in emission intensities compared to the annihilation pathway is likely due to this available energy increase.The BPO co-reactant ECL efficiencies for PPOCzPN, PPSCzPN and DiPPOCzPN were determined to be 17, 6 and 1%, respectively, using eqn (1) (Table 6). Interestingly, this ECL efficiency difference between PPOCzPN and DiPPOCzPN resembles the difference in ΦPL for the emitters in a PMMA film (56 to 28%) more than it does the ΦPL difference in DCM (51 to 61%). This indicates that the ECL co-reactant pathway for DiPPOCzPN has an additional non-radiative decay pathway. In ECL co-reactant systems, the lifetime of the radical poses less of an issue than in annihilation experiments, since radical reactants are generated almost simultaneously, typically allowing ECL enhancement in solution. Now that radical stability is less of a factor, PPOCzPN˙− appears to have a higher reactivity than DiPPOCzPN˙− and PPSCzPN˙− towards BPO. A higher anion reactivity for PPOCzPN was noted during pulsing experiments (Fig. 3d–f) and appears to have been observed again in the BPO experiments. With increased available energy, it appears that PPOCzPN is the strongest emitter and maintains its position as the best candidate for optoelectronics.
3.6 ECL spectroscopy
Fig. 9d–f show spooling ECL spectroscopy performed on all co-reactant systems described herein. The ECL spooling spectra add a new data dimension of wavelength to the same spectra seen in Fig. 9a–c. The BPO co-reactant systems show a light green emission for all species with maximum emission CIE coordinates of (0.29,0.40), (0.31,0.42) and (0.33,0.38) for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively. In contrast to the annihilation pathway and also observed in the PL study, the DiPPOCzPN/BPO co-reactant system has the most red-shifted emission spectrum (Table 4 and Fig. 9f), rather than the most blue-shifted. This large red-shift and relative drop in efficiency indicate that DiPPOCzPN may suffer from exciplex formation under these conditions. Exciplexes are heterodimeric species created by intermolecular interactions between a hole on the donor moiety of one molecule and the electron on the acceptor moiety of a second molecule.117 Exciplexes typically have smaller HOMO–LUMO gaps than either of the constituent components, thereby resulting in an observed red-shifted emission. Exciplex formation in OLEDs with luminophores substituted by diphenyl phosphine oxide substituents have been reported by Tourneur et al., who harnessed this exciplex formation to produce emissions from both intramolecular and intermolecular charge-transfer states yielding white light OLEDs.118 The exciplex identified during DiPPOCzPN/BPO co-reactant analysis appears not to be affected by an increasing concentration of reduced species in the vicinity of the electrode, since as the cyclic voltammogram progresses, no shift in emission maxima is seen (insets of Fig. 9d–f with expansions of the insets in Fig. S48, ESI‡). Additionally, there was no identified exciplex formation during the annihilation studies with DiPPOCzPN. The most probable explanation for the observed red-shifted emission is due to the exciplex between DiPPOCzPN˙− and Bz˙+. Newcomb et al. has identified the necessity to screen potential TADF OLED emitters for exciplex formation, as the presence of exciplexes was shown to lower device lifetime.119 Masui et al. also identified large EQE roll-off in OLEDs caused by charge imbalance in the emissive layer that are exacerbated at higher current densities.109,120 The exciplex formation in these compounds is characteristic of compounds that also exhibit OLPL, thereby supporting OLECL emission as the dominant mechanism. Interestingly, the longest pulsing ECLonset delay belonged to DiPPOCzPN, which was the only compound to form a very evident exciplex during BPO co-reactant analysis. Interestingly, 2CzPN showed emissions centered at 550 nm, resembling the emissions seen in the DiPPOCzPN/BPO coreactant studies in Fig. S38c and d (ESI‡). This center wavelength is also 50 nm red-shifted from the 2CzPN PL studies likely due to a similar exciplex formation.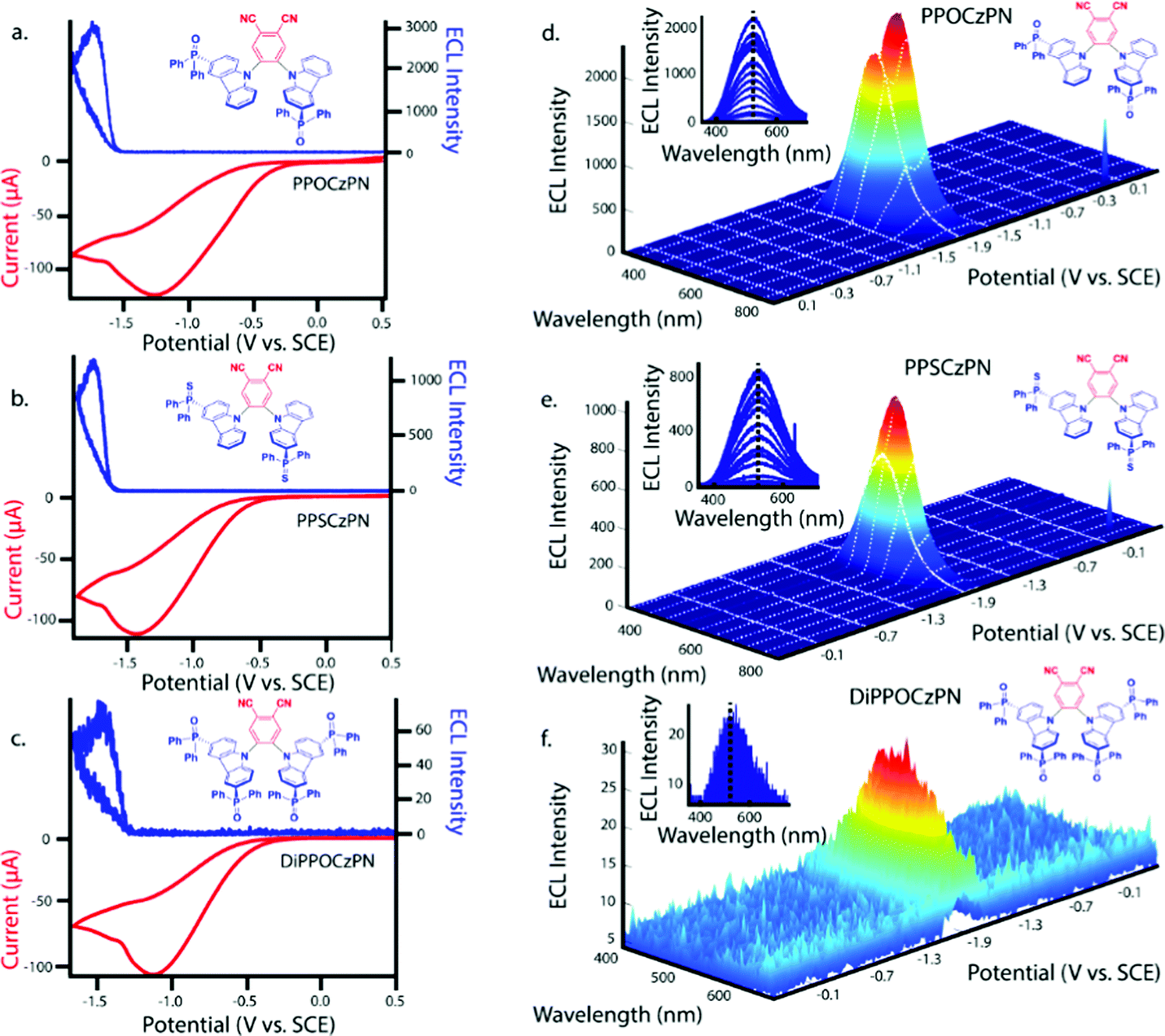 | ||
| Fig. 9 (a–c) CVs (red) along with ECL–voltage curves (blue) during potential pulsing at a pulsing frequency of 10 Hz for PPOCzPN, PPSCzPN and DiPPOCzPN, respectively, all with 10 mM BPO was added as a co-reactant. (d–f) Spooling ECL spectroscopy of the corresponding systems described in Figures a–c. Insets of Figures d–f represent respective stacked spooling ECL spectra. The purple color of these insets was arbitrarily chosen but Fig. S48 (ESI‡) displays the observable color of each recorded spectrum. | ||
In a similar manner to the BPO co-reactant, 5 mM tripropylamine (TPrA) was added as an alternative co-reactant (Fig. S49 and S50, ESI‡). A thorough discussion of the TPrA co-reactant systems follows Fig. S49 (ESI‡) in the ESI.‡ Under these conditions, the PPSCzPN/TPrA system behaved in the exact same manner as the DiPPOCzPN/BPO system, likely indicating a similar exciplex formation. There were no excimers identified in the PPSCzPN annihilation mechanism, indicating that the interactions between PPSCzPN˙+ and NPr3˙ may be responsible for the exciplex formation. The PPSCzPN/TPrA system also seems to be concentration-dependent where an increasing overpotential that creates more PPSCzPN˙+ in the vicinity of the electrode gradually causes a red-shift. The compounds with the longest onset to ECL emission, longest time to achieve a maximum ECL intensity, longest ECL decay time, largest ΔEST and weakest ECL efficiencies likely form exciplexes under co-reactant conditions. These correlations and relationships all support the putative OLECL emission mechanism.
In Fig. 10, the normalized accumulation spectra of all ECL processes discussed so far are overlapped, with ECL peak wavelengths summarized in Table 6. A slight red-shift is seen for all ECL emissions relative to the PL emissions, which is likely due to the more concentrated solutions used in the ECL measurements that result in increased self-absorption of the higher energy components of the emission spectra in the ECL studies. The PL and ECL spectra for PPOCzPN do not shift, indicating a common excited state accessed in all processes. However, there is a large red-shift observed for both the DiPPOCzPN/BPO co-reactant pathway and for the PPSCzPN/TPrA co-reactant pathway. These red-shifts in the ECL spectra typically indicate exciplex formation.
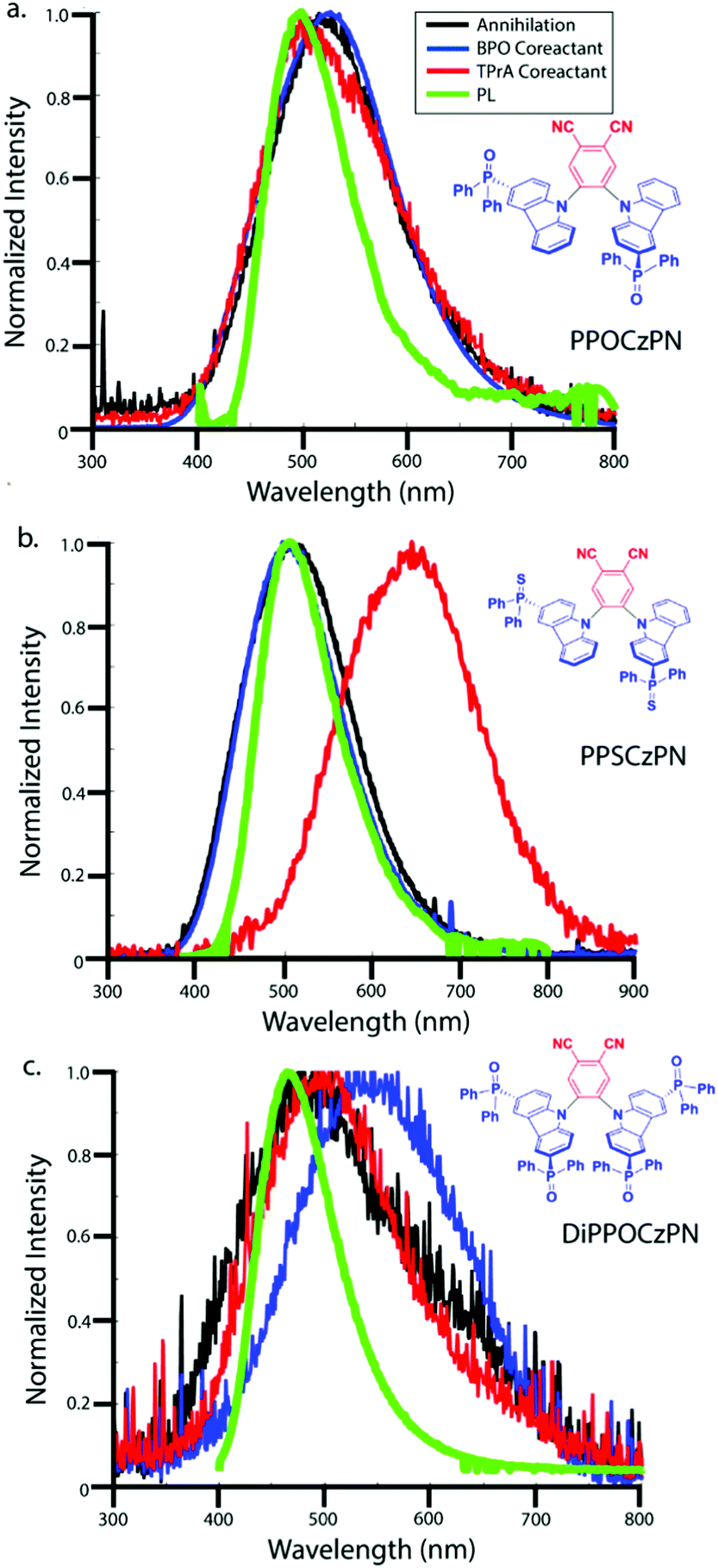 | ||
| Fig. 10 Accumulated ECL spectra during different light generation methods. The annihilation ECL, BPO co-reactant ECL, TPrA co-reactant ECL and photoluminescence emissions are colored in black, blue, red and green traces for (a) PPOCzPN, (b) PPSCzPN and (c) DiPPOCzPN, respectively. | ||
4. Conclusions
Herein, we detailed the synthesis and characterization of three new 2CzPN derivatives with phosphine chalcogenide oxide groups substituted on the donor moiety. This strategy successfully stabilized the HOMO level of the donor, confirmed by cyclic voltammetry. By extension, the lowest-lying charge-transfer state was shifted to higher energy, thereby promoting a deeper blue emission, confirmed by photoluminescence studies. Surprisingly, the same trends are not recovered when measuring emissions in apolar media, thus highlighting solvatochromic effects on the TADF properties of the 2CzPN derivatives. Quantum-chemical calculations support these solvatochromic observations. Electrochemiluminescence studies in the desirable polar medium revealed a heretofore undocumented emission mechanism under electrical excitation in solution. Delayed ECL onset times relative to common ECL emitters in annihilation pathway revealed that organic long-persistent emission was operational and that the delay in the onset times correlated with the ΔEST of the emitters. PPOCzPN, which has the smallest ΔEST, yielded the strongest, fastest, and most efficient ECL emissions when using BPO co-reactant. Thus, this compound should be the most suitable for optoelectronic applications after considering radical stability, emission stability, emission efficiency and emission intensity. Studies on ECL spooling spectroscopy and accumulated ECL spectroscopy identified that an exciplex of DiPPOCzPN forms while in the presence of radical anions. Analogous to the OLPL mechanism advanced by Kabe et al. the slow kinetics of exciplex formation and subsequent emission are likely responsible for the OLECL in this case. This work reveals a heretofore unknown ECL emission mechanism, OLECL, that manifests in both very long onset of ECL emission and long ECL lifetimes. Our study also provides significantly more detailed insight into the ECL of TADF compounds, which can be used to guide and screen TADF compound suitability for optoelectronic applications such as OLEDs.Author contributions
S. K., M. Y. W., and E. Z.-C. designed and synthesized the materials. S. K., P. R. and P. T. carried out the photophysical study. J. R. A and Z. D. performed the electrochemical and ECL studies. D. B. C. and A. M. Z. S. solved the structure of the single crystals. P. T., Y. O., J. C., R. L., and P. V. carried out the theoretical modeling. S. K., E. Z.-C., J. R. A., Z. D., P. T., Y. O., J. C. wrote the manuscript. Y. O., J. C., Z. D. and E. Z.-C. supervised the project.Abbreviations
| TADF | Thermally activated delayed fluorescence; |
| ECL | Electrochemiluminescence |
| 2CzPN | 4,5-Dicarbazolylphthalonitrile |
| BPO | Benzoyl peroxide |
| TPrA | Tripropylamine |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| OLPL | Organic long-persistent photoluminescence |
| OLECL | Organic long-persistent electrochemiluminescence |
| TTA | Triplet–triplet annihilation |
| RISC | Reverse intersystem crossing |
| LE | Locally excited |
| CT | Charge transfer |
| DFT | Density functional theory |
| CV | Cyclic voltammetry |
| DPV | Differential pulse voltammetry. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Sai Kiran Rajendran for providing PL and TCSPC data of 2CzPN in neat film. SK acknowledges the financial support from European Union's Horizon 2020 research and innovation programme under Marie Skłodowska Curie Individual Fellowship (MCIF; Agreement No. 748430-THF-OLED). P. R acknowledges support from a Marie Skłodowska-Curie Individual Fellowship (No. 749557). We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University for analytical services. The work has been supported in Mons by European Union through the Interreg V initiative France-Wallonie-Vlaanderen project LUMINOPTEX and the Belgian National Fund for Scientific Research (FRS-FNRS). Computational resources were provided by the Consortium des Équipements de Calcul Intensif (CÉCI) funded by F. R. S.-FNRS under Grant 2.5020.11. J. C. is an FNRS research director. Y. O. acknowledges funding by the Fonds de la Recherche Scientifique-FNRS under Grant no F.4534.21 (MIS-IMAGINE). We acknowledge the research support from Natural Sciences and Engineering Research Council Canada (NSERC, DG RGPIN-2013-201697, DG RGPIN-2018-06556, and SPG STPGP-2016-493924), Canada Foundation of Innovation, Ontario Innovation Trust (CFI/OIT, 9040) and Western University. J. R. A. appreciates the Ontario graduate scholarships (2018–2022). EZ-C is a Royal Society Leverhulme Trust Senior Research fellow (SRF\R1\201089). For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license.References
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Management of Singlet and Triplet Excitons for Efficient White Organic Light-Emitting Devices, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, Nearly 100% internal phosphorescence efficiency in an organic light-emitting device, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS.
- K. P. Klubek, C. W. Tang and L. J. Rothberg, Investigation of Blue Phosphorescent Organic Light-Emitting Diode Host and Dopant Stability, Org. Electron., 2014, 15, 1312–1316 CrossRef CAS.
- T. Huang, W. Jiang and L. Duan, Recent Progress in Solution Processable TADF Materials for Organic Light-Emitting Diodes, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- X.-K. Chen, D. Kim and J.-L. Brédas, Thermally Activated Delayed Fluorescence (TADF) Path Toward Efficient Electroluminescence in Purely Organic Materials: Molecular Level Insight, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- S. K. Jeon, H. L. Lee, K. S. Yook and J. Y. Lee, Recent Progress of the Lifetime of Organic Light-Emitting Diodes Based on Thermally Activated Delayed Fluorescent Material, Adv. Mater., 2019, 31, 1803524 CrossRef PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, Recent Breakthroughs in Thermally Activated Delayed Fluorescence Organic Light Emitting Diodes Containing Non-Doped Emitting Layers, J. Mater. Chem. C, 2019, 7, 2172–2198 RSC.
- P. Data and Y. Takeda, Recent Advancements in and the Future of Organic Emitters: TADF- and RTP-Active Multifunctional Organic Materials, Chem. – Asian J., 2019, 14, 1613–1636 CrossRef CAS PubMed.
- Y. Olivier, J. C. Sancho-Garcia, L. Muccioli, G. D’Avino and D. Beljonne, Computational Design of Thermally Activated Delayed Fluorescence Materials: The Challenges Ahead, J. Phys. Chem. Lett., 2018, 9, 6149–6163 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- P. de Silva, C. A. Kim, T. Zhu and T. Van Voorhis, Extracting Design Principles for Efficient Thermally Activated Delayed Fluorescence (TADF) from a Simple Four-State Model, Chem. Mater., 2019, 31, 6995–7006 CrossRef CAS.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Rational Design of TADF Polymers Using a Donor–Acceptor Monomer with Enhanced TADF Efficiency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, White Organic Light-Emitting Diodes With Fluorescent Tube Efficiency, Nature, 2009, 459, 234–238 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- E. Spuling, N. Sharma, I. D. W. Samuel, E. Zysman-Colman and S. Bräse, (Deep) Blue Through-Space Conjugated TADF Emitters Based on [2.2]Paracyclophanes, Chem. Commun., 2018, 54, 9278–9281 RSC.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO–LUMO Separation by the Multiple Resonance Effect, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- M. Auffray, D. H. Kim, J. U. Kim, F. Bencheikh, D. Kreher, Q. Zhang, A. D'Aléo, J.-C. Ribierre, F. Mathevet and C. Adachi, Dithia[3.3]paracyclophane Core: A Versatile Platform for Triplet State Fine-Tuning and Through-Space TADF Emission, Chem. – Asian J., 2019, 14, 1921–1925 CrossRef CAS PubMed.
- J. Lee, N. Aizawa and T. Yasuda, Molecular Engineering of Phosphacycle-Based Thermally Activated Delayed Fluorescence Materials for Deep-Blue OLEDs, J. Mater. Chem. C, 2018, 6, 3578–3583 RSC.
- X. Liang, Z.-P. Yan, H.-B. Han, Z.-G. Wu, Y.-X. Zheng, H. Meng, J.-L. Zuo and W. Huang, Peripheral Amplification of Multi-Resonance Induced Thermally Activated Delayed Fluorescence for Highly Efficient OLEDs, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed.
- Y. J. Cho, S. K. Jeon, S.-S. Lee, E. Yu and J. Y. Lee, Donor Interlocked Molecular Design for Fluorescence-like Narrow Emission in Deep Blue Thermally Activated Delayed Fluorescent Emitters, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- L. Mei, J. Hu, X. Cao, F. Wang, C. Zheng, Y. Tao, X. Zhang and W. Huang, The Inductive-Effect of Electron Withdrawing Trifluoromethyl for Thermally Activated Delayed Fluorescence: Tunable Emission from Tetra- to Penta-Carbazole in Solution Processed Blue OLEDs, Chem. Commun., 2015, 51, 13024–13027 RSC.
- H. Wang, L. Xie, Q. Peng, L. Meng, Y. Wang, Y. Yi and P. Wang, Novel Thermally Activated Delayed Fluorescence Materials–Thioxanthone Derivatives and Their Applications for Highly Efficient OLEDs, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Aggregation-Induced Delayed Fluorescence Based on Donor/Acceptor-Tethered Janus Carborane Triads: Unique Photophysical Properties of Nondoped OLEDs, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS.
- H. F. Higginbotham, P. Pander, R. Rybakiewicz, M. K. Etherington, S. Maniam, M. Zagorska, A. Pron, A. P. Monkman and P. Data, Triphenylamine Disubstituted Naphthalene Diimide: Elucidation of Excited States Involved in TADF and Application in Near-Infrared Organic Light Emitting Diodes, J. Mater. Chem. C, 2018, 6, 8219–8225 RSC.
- G. Xie, X. Li, D. Chen, Z. Wang, X. Cai, D. Chen, Y. Li, K. Liu, Y. Cao and S.-J. Su, Evaporation- and Solution-Process-Feasible Highly Efficient Thianthrene-9,9′,10,10′-Tetraoxide-Based Thermally Activated Delayed Fluorescence Emitters with Reduced Efficiency Roll-Off, Adv. Mater., 2016, 28, 181–187 CrossRef CAS PubMed.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Sky-Blue Organic Light Emitting Diode with 37% External Quantum Efficiency Using Thermally Activated Delayed Fluorescence from Spiroacridine-Triazine Hybrid, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- T. Takahashi, K. Shizu, T. Yasuda, K. Togashi and C. Adachi, Donor–Acceptor-Structured 1,4-Diazatriphenylene Derivatives Exhibiting Thermally Activated Delayed Fluorescence: Design and Synthesis, Photophysical Properties and OLED Characteristics, Sci. Technol. Adv. Mater., 2014, 15, 034202 CrossRef PubMed.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, Twisted Intramolecular Charge Transfer State for Long-Wavelength Thermally Activated Delayed Fluorescence, Chem. Mater., 2013, 25, 3766–3771 CrossRef CAS.
- J. Lee, K. Shizu, H. Tanaka, H. Nomura, T. Yasuda and C. Adachi, Oxadiazole- and Triazole-Based Highly-Efficient Thermally Activated Delayed Fluorescence Emitters for Organic Light-Emitting Diodes, J. Mater. Chem. C, 2013, 1, 4599–4604 RSC.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Multi-dipolar Chromophores Featuring Phosphine Oxide as Joint Acceptor: A New Strategy toward High-Efficiency Blue Thermally Activated Delayed Fluorescence Dyes, Chem. Mater., 2016, 28, 5667–5679 CrossRef CAS.
- D.-Y. Chen, W. Liu, C.-J. Zheng, K. Wang, F. Li, S. L. Tao, X.-M. Ou and X.-H. Zhang, Isomeric Thermally Activated Delayed Fluorescence Emitters for Color Purity-Improved Emission in Organic Light-Emitting Devices, ACS Appl. Mater. Interfaces, 2016, 8, 16791–16798 CrossRef CAS PubMed.
- N. Aizawa, C.-J. Tsou, I. S. Park and T. Yasuda, Aggregation-Induced Delayed Fluorescence From Phenothiazine-Containing Donor–Acceptor Molecules for High-Efficiency Non-Doped Organic Light-Emitting Diodes, Polym. J., 2017, 49, 197–202 CrossRef CAS.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, An Organic Molecule with Asymmetric Structure Exhibiting Aggregation-Induced Emission, Delayed Fluorescence, and Mechanoluminescence, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- D. R. Lee, B. S. Kim, C. W. Lee, Y. Im, K. S. Yook, S.-H. Hwang and J. Y. Lee, Above 30% External Quantum Efficiency in Green Delayed Fluorescent Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2015, 7, 9625–9629 CrossRef CAS PubMed.
- S. K. Jeon and J. Y. Lee, Highly Efficient Exciplex Organic Light-Emitting Diodes by Exciplex Dispersion in the Thermally Activated Delayed Fluorescence Host, Org. Electron., 2020, 76, 105477 CrossRef CAS.
- B. Wex and B. R. Kaafarani, Perspective on Carbazole-Based Organic Compounds as Emitters and Hosts in TADF Applications, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- Y. J. Cho, K. S. Yook and J. Y. Lee, High Efficiency in a Solution-Processed Thermally Activated Delayed-Fluorescence Device Using a Delayed-Fluorescence Emitting Material with Improved Solubility, Adv. Mater., 2014, 26, 6642–6646 CrossRef CAS PubMed.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, High-Efficiency Deep-Blue Organic Light-Emitting Diodes Based on a Thermally Activated Delayed Fluorescence Emitter, J. Mater. Chem. C, 2014, 2, 421–424 RSC.
- G. H. Kim, R. Lampande, M. J. Park, H. W. Bae, J. H. Kong, J. H. Kwon, J. H. Park, Y. W. Park and C. E. Song, Highly Efficient Bipolar Host Materials with Indenocarbazole and Pyrimidine Moieties for Phosphorescent Green Light-Emitting Diodes, J. Phys. Chem. C, 2014, 118, 28757–28763 CrossRef CAS.
- D. R. Lee, J. M. Choi, C. W. Lee and J. Y. Lee, Ideal Molecular Design of Blue Thermally Activated Delayed Fluorescent Emitter for High Efficiency, Small Singlet–Triplet Energy Splitting, Low Efficiency Roll-Off, and Long Lifetime, ACS Appl. Mater. Interfaces, 2016, 8, 23190–23196 CrossRef CAS PubMed.
- K. Sun, Y. Sun, T. Huang, J. Luo, W. Jiang and Y. Sun, Design Strategy of Yellow Thermally Activated Delayed Fluorescent Dendrimers and Their Highly Efficient Non-Doped Solution-Processed OLEDs with Low Driving Voltage, Org. Electron., 2017, 42, 123–130 CrossRef CAS.
- K. Albrecht, K. Matsuoka, K. Fujita and K. Yamamoto, Carbazole Dendrimers as Solution-Processable Thermally Activated Delayed-Fluorescence Materials, Angew. Chem., Int. Ed., 2015, 54, 5677–5682 CrossRef CAS PubMed.
- D. R. Lee, S.-H. Hwang, S. K. Jeon, C. W. Lee and J. Y. Lee, Benzofurocarbazole and Benzothienocarbazole as Donors for Improved Quantum Efficiency in Blue Thermally Activated Delayed Fluorescent Devices, Chem. Commun., 2015, 51, 8105–8107 RSC.
- N. Sharma, E. Spuling, C. M. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Bräse, I. D. W. Samuel and E. Zysman-Colman, Turn on of Sky-Blue Thermally Activated Delayed Fluorescence and Circularly Polarized Luminescence (CPL) via Increased Torsion by a Bulky Carbazolophane Donor, Chem. Sci., 2019, 10, 6689–6696 RSC.
- B. Huang, W. Jiang, Y. Y. Liu, Y. A. Zhang, Y. P. Yang, Y. Dai, X. X. Ban, H. G. Xu and Y. M. Sun, Thermally Activated Delayed Fluorescence Materials Based on Carbazole/Sulfone, Adv. Mater. Res., 2014, 1044–1045, 158–163 CAS.
- Y. Im and J. Y. Lee, Effect of the Position of Nitrogen in Pyridoindole on Photophysical Properties and Device Performances of α-, β-, γ-Carboline Based High Triplet Energy Host Materials for deep Blue Devices, Chem. Commun., 2013, 49, 5948–5950 RSC.
- J.-W. Jun, K.-M. Lee, O. Y. Kim, J. Y. Lee and S.-H. Hwang, Synthesis of a Dibenzothiophene/Carboline/Carbazole Hybrid Bipolar Host Material for Green Phosphorescent OLEDs, Synth. Met., 2016, 213, 7–11 CrossRef CAS.
- S. Y. Byeon, S. K. Jeon, S.-H. Hwang and J. Y. Lee, Carbazole-Carboline Core as a Backbone Structure of High Triplet Energy Host Materials, Dyes Pigm., 2015, 120, 258–264 CrossRef CAS.
- Z. Zhang, J. Xie, Z. Wang, B. Shen, H. Wang, M. Li, J. Zhang and J. Cao, Manipulation of Electron Deficiency of δ-Carboline Derivatives as Bipolar Hosts for Blue Phosphorescent Organic Light-Emitting Diodes with High Efficiency at 1000 cd m−2, J. Mater. Chem. C, 2016, 4, 4226–4235 RSC.
- C. W. Lee and J. Y. Lee, Above 30% External Quantum Efficiency in Blue Phosphorescent Organic Light-Emitting Diodes Using Pyrido[2,3-b]indole Derivatives as Host Materials, Adv. Mater., 2013, 25, 5450–5454 CrossRef CAS PubMed.
- C.-C. Lai, M.-J. Huang, H.-H. Chou, C.-Y. Liao, P. Rajamalli and C.-H. Cheng, m-Indolocarbazole Derivative as a Universal Host Material for RGB and White Phosphorescent OLEDs, Adv. Funct. Mater., 2015, 25, 5548–5556 CrossRef CAS.
- K. S. Yook and J. Y. Lee, Bipolar Host Materials for Organic Light-Emitting Diodes, Chem. Rec., 2016, 16, 159–172 CrossRef CAS PubMed.
- M.-S. Lin, L.-C. Chi, H.-W. Chang, Y.-H. Huang, K.-C. Tien, C.-C. Chen, C.-H. Chang, C.-C. Wu, A. Chaskar, S.-H. Chou, H.-C. Ting, K.-T. Wong, Y.-H. Liu and Y. Chi, A Diarylborane-Substituted Carbazole as a Universal Bipolar Host Material for Highly Efficient Electrophosphorescence Devices, J. Mater. Chem., 2012, 22, 870–876 RSC.
- M.-S. Lin, S.-J. Yang, H.-W. Chang, Y.-H. Huang, Y.-T. Tsai, C.-C. Wu, S.-H. Chou, E. Mondal and K.-T. Wong, Incorporation of a CN Group into mCP: A New Bipolar Host Material for Highly Efficient Blue and White Electrophosphorescent Devices, J. Mater. Chem., 2012, 22, 16114–16120 RSC.
- C. S. Oh, J. M. Choi and J. Y. Lee, Chemical Bond Stabilization and Exciton Management by CN Modified Host Material for Improved Efficiency and Lifetime in Blue Phosphorescent Organic Light-Emitting Diodes, Adv. Opt. Mater., 2016, 4, 1281–1287 CrossRef CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Highly Efficient Blue Electroluminescence Based on Thermally Activated Delayed Fluorescence, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- M. Colella, A. Danos and A. P. Monkman, Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS PubMed.
- K. Masui, H. Nakanotani and C. Adachi, Analysis of Exciton Annihilation in High-Efficiency Sky-Blue Organic Light-Emitting Diodes With Thermally Activated Delayed Fluorescence, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- J. R. Adsetts, R. Zhang, L. Yang, K. Chu, J. M. Wong, D. A. Love and Z. Ding, Efficient White Electrochemiluminescent Emission From Carbon Quantum Dot Films, Front. Chem., 2020, 8, 580022 CrossRef CAS PubMed.
- K. Chu, J. R. Adsetts, S. He, Z. Zhan, L. Yang, J. M. Wong, D. A. Love and Z. Ding, Electrogenerated Chemiluminescence and Electroluminescence of N-Doped Graphene Quantum Dots Fabricated from an Electrochemical Exfoliation Process in Nitrogen-Containing Electrolytes, Chem. – Eur. J., 2020, 26, 15892–15900 CrossRef CAS PubMed.
- J. R. Adsetts, S. Hoesterey, C. Gao, D. A. Love and Z. Ding, Electrochemiluminescence and Photoluminescence of Carbon Quantum Dots Controlled by Aggregation-Induced Emission, Aggregation-Caused Quenching and Interfacial Reactions, Langmuir, 2020, 36, 14432–14442 CrossRef CAS PubMed.
- K. Chu, J. R. Adsetts, C. Moore and Z. Ding, Spooling Electroluminescence Spectroscopy of Ru(bpy)32+ Light-Emitting Electrochemical Cells with an Atomic Layer Deposited Zinc Oxide Electron-Transporting/Hole-Blocking Interlayer, ACS Appl. Electron. Mater., 2020, 2, 3825–3830 CrossRef CAS.
- E. W. Grabner and E. Brauer, Electrochemiluminescence of Energy-Deficient Systems II. The Role of Excimer Formation in the Pure Perylene System, Ber. Bunsen-Ges. Phys. Chem., 1971, 76, 111–114 Search PubMed.
- L. Yang, B. Zhang, L. Fu, K. Fu and G. Zou, Efficient and Monochromatic Electrochemiluminescence of Aqueous-Soluble Au Nanoclusters via Host-Guest Recognition, Angew. Chem., Int. Ed., 2019, 58, 6901–6905 CrossRef CAS PubMed.
- R. Ishimatsu, S. Matsunami, T. Kasahara, J. Mizuno, T. Edura, C. Adachi, K. Nakano and T. Imato, Electrogenerated Chemiluminescence of Donor-Acceptor Molecules with Thermally Activated Delayed Fluorescence, Angew. Chem., Int. Ed., 2014, 53, 6993–6996 CrossRef CAS PubMed.
- S. W. Feldberg, A Possible Method for Distinguishing between Triplet–Triplet Annihilation and Direct Singlet Formation in Electrogenerated Chemiluminescence, J. Phys. Chem., 1966, 70, 3928–3930 CrossRef CAS.
- L. Yang, D. Koo, J. Wu, J. M. Wong, T. Day, R. Zhang, H. Kolongoda, K. Liu, J. Wang, Z. Ding and B. L. Pagenkopf, Benzosiloles with Crystallization-Induced Emission Enhancement of Electrochemiluminescence: Synthesis, Electrochemistry, and Crystallography, Chemistry, 2020, 26, 11715–11721 CrossRef CAS PubMed.
- J. R. Adsetts and Z. Ding, Film Electrochemiluminescence Controlled by Interfacial Reactions Along with Aggregation-, Matrix-Coordination-, and Crystallization-Induced Emissions, ChemPlusChem, 2021, 86, 155–165 CrossRef CAS PubMed.
- J. M. Wong, R. Zhang, P. Xie, L. Yang, M. Zhang, R. Zhou, R. Wang, Y. Shen, B. Yang, H. B. Wang and Z. Ding, Revealing Crystallization-Induced Blue-Shift Emission of a Di-Boron Complex by Enhanced Photoluminescence and Electrochemiluminescence, Angew. Chem., Int. Ed., 2020, 59, 17461–17466 CrossRef CAS PubMed.
- D. Ege, W. G. Becker and A. J. Bard, Electrogenerated Chemiluminescent Determination of Ru(bpy)32+ at Low Levels, Anal. Chem., 1984, 56, 2413–2417 CrossRef CAS PubMed.
- E. A. Chandross and F. I. Sonntag, A Novel Chemiluminescent Electron-Transfer Reaction, J. Am. Chem. Soc., 1964, 86, 3179–3180 CrossRef CAS.
- D. M. Hercules, Chemiluminescence Resulting From Electrochemically Generated Species, Science, 1964, 145, 808–809 CrossRef CAS PubMed.
- K. S. V. Santhanam and A. J. Bard, Chemiluminescence of Electrogenerated 9,10-Diphenylathracene Anion Radical, J. Am. Chem. Soc., 1965, 87, 139–140 CrossRef CAS.
- Z. Liu, W. Qi and G. Xu, Recent Advances in Electrochemiluminescence, Chem. Soc. Rev., 2015, 44, 3117–3142 RSC.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Revealing the Spin–Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- Z. Zeng, P. Huang, Y. Kong, L. Tong, B. Zhang, Y. Luo, L. Chen, Y. Zhang, D. Han and L. Niu, Nanoencapsulation Strategy: Enabling Electrochemiluminescence of Thermally Activated Delayed Fluorescence (TADF) Emitters in Aqueous Media, Chem. Commun., 2021, 57, 5262–5265 RSC.
- R. Ishimatsu, T. Edura, C. Adachi, K. Nakano and T. Imato, Photophysical Properties and Efficient, Stable, Electrogenerated Chemiluminescence of Donor–Acceptor Molecules Exhibiting Thermal Spin Upconversion, Chem. – Eur. J., 2016, 22, 4889–4898 CrossRef CAS PubMed.
- A. Kapturkiewicz, Electrochemical Generation of Excited Intramolecular Charge-Transfer States, ChemElectroChem, 2017, 4, 1604–1638 CrossRef CAS.
- M. Y. Wong, G. J. Hedley, G. Xie, L. S. Kölln, I. D. W. Samuel, A. Pertegás, H. J. Bolink and E. Zysman-Colman, Light-Emitting Electrochemical Cells and Solution-Processed Organic Light-Emitting Diodes Using Small Molecule Organic Thermally Activated Delayed Fluorescence Emitters, Chem. Mater., 2015, 27, 6535–6542 CrossRef CAS.
- J. N. Demas and G. A. Crosby, The Measurement of Photoluminescence Quantum Yields. A Review, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- W. H. Melhuish, QUANTUM EFFICIENCIES OF FLUORESCENCE OF ORGANIC SUBSTANCES: EFFECT OF SOLVENT AND CONCENTRATION OF THE FLUORESCENT SOLUTE1, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- J. Adsetts, R. Zhang, L. Yang, K. Chu, J. Wong, D. A. Love and Z. Ding, Efficient White Electrochemiluminescent Emission from Carbon Quantum Dot Films, Front. Chem., 2020, 8, 865–878 Search PubMed.
- L. R. Faulkner, H. Tachikawa and A. J. Bard, Electrogenerated Chemiluminescence. VII. The Influence of an External Magnetic Field on Luminescence Intensity, J. Am. Chem. Soc., 1972, 94, 691–699 CrossRef CAS.
- M. Y. Wong, S. Krotkus, G. Copley, W. Li, C. Murawski, D. Hall, G. J. Hedley, M. Jaricot, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, D. Beljonne, L. Muccioli, M. Moral, J.-C. Sancho-Garcia, M. C. Gather, I. D. W. Samuel and E. Zysman-Colman, Deep-Blue Oxadiazole-Containing Thermally Activated Delayed Fluorescence Emitters for Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2018, 10, 33360–33372 CrossRef CAS PubMed.
- W. H. Melhuish, Quantum Efficiencies of Fluorescence of Organic Substances: Effect of Solvent and Concentration of the Fluorescent Solute, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- M. Moral, L. Muccioli, W. J. Son, Y. Olivier and J. C. Sancho-García, Theoretical Rationalization of the Singlet–Triplet Gap in OLEDs Materials: Impact of Charge-Transfer Character, J. Chem. Theory Comput., 2015, 11, 168–177 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods Without Adjustable Parameters: The PBE0 Model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- S. Hirata and M. Head-Gordon, Time-Dependent Density Functional Theory Within the Tamm–Dancoff Approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- Y. Geng, A. D'Aleo, K. Inada, L.-S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Donor–σ–Acceptor Motifs: Thermally Activated Delayed Fluorescence Emitters with Dual Upconversion, Angew. Chem., Int. Ed., 2017, 56, 16536–16540 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02 Search PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Critical Role of Intermediate Electronic States for Spin-Flip Processes in Charge-Transfer-Type Organic Molecules with Multiple Donors and Acceptors, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- O. V. Klymenko, I. Svir and C. Amatore, A New Approach for the Simulation of Electrochemiluminescence (ECL), ChemPhysChem, 2013, 14, 2237–2250 CrossRef CAS PubMed.
- E. A. Chandross and F. I. Sonntag, Chemiluminescent Electron-Transfer Reactions of Radical Anions, J. Am. Chem. Soc., 1966, 88, 1089–1096 CrossRef CAS.
- R. Coehoorn, P. A. Bobbert and H. van Eersel, Förster-Type Triplet-Polaron Quenching in Disordered Organic Semiconductors, Phys. Rev. B, 2017, 96, 184203 CrossRef.
- N. E. Tokel-Takvoryan, R. E. Hemingway and A. J. Bard, Electrogenerated Chemiluminescence. XIII. Electrochemical and Electrogenerated Chemiluminescence Studies of Ruthenium Chelates, J. Am. Chem. Soc., 1973, 95, 6582–6589 CrossRef CAS.
- N. E. Tokel and A. J. Bard, Electrogenerated Chemiluminescence. IX. Electrochemistry and emission From Systems Containing Tris(2,2'-bipyridine)ruthenium(II) Dichloride, J. Am. Chem. Soc., 1972, 94, 2862–2863 CrossRef CAS.
- W. L. Wallace and A. J. Bard, Electrogenerated Chemiluminescence. 35. Temperature Dependence of the ECL Efficiency of Ru(bpy)32+ in Acetonitrile and Evidence for Very High Excited State Yields from Electron Transfer Reactions, J. Phys. Chem., 1979, 83, 1350–1357 CrossRef CAS.
- F. E. Lytle and D. M. Hercules, Chemiluminescence From The Reduction of Aromatic Amine Cations and Ruthenium(III) Chelates, Photochem. Photobiol., 1970, 13, 123–133 CrossRef.
- H. Saiki, K. Takami and T. Tominaga, Diffusion of Porphyrins and Quinones in Organic Solvents, Phys. Chem. Chem. Phys., 1998, 1, 303–306 Search PubMed.
- G. J. Barbante, C. F. Hogan, D. J. Wilson, N. A. Lewcenko, F. M. Pfeffer, N. W. Barnett and P. S. Francis, Simultaneous Control of Spectroscopic and Electrochemical Properties in Functionalised Electrochemiluminescent Tris(2,2'-bipyridine)ruthenium(II) Complexes, Analyst, 2011, 136, 1329–1338 RSC.
- C. A. Parker and C. G. Hatchard, Delayed Fluorescence From Solutions of Anthracene and Phenanthrene, Proc. R. Soc. London, Ser. A, 1962, 269, 574–584 Search PubMed.
- J. Grüne, N. Bunzmann, M. Meinecke, V. Dyakonov and A. Sperlich, Kinetic Modeling of Transient Electroluminescence Reveals TTA as an Efficiency-Limiting Process in Exciplex-Based TADF OLEDs, J. Phys. Chem. C, 2020, 124, 25667–25674 CrossRef.
- A. Niwa, S. Haseyama, T. Kobayashi, T. Nagase, K. Goushi, C. Adachi and H. Naito, Triplet-Triplet Annihilation in a Thermally Activated Delayed Fluorescence Emitter Lightly Doped in a Host, Appl. Phys. Lett., 2018, 113, 083301–083305 CrossRef.
- K. Masui, H. Nakanotani and C. Adachi, Analysis of Exciton Annihilation in High-Efficiency Sky Blue Organic Light-Emitting Diodes with Thermally Activated Delayed Fluorescence, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- W. Miao, Electrogenerated Chemiluminescence and Its Biorelated Applications, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- M. J. Cormier, D. M. Hercules and J. Lee, Chemiluminescence and Bioluminescence, Springer, Boston MA, 1973 Search PubMed.
- J. Rosenmund and K. Doblhofer, The Effects of Uncompensated Solution Resistance and Rate of the Homogenous Electron Transfer Reaction on Electrochemiluminescence Transients, J. Electroanal. Chem., 1995, 396, 77–83 CrossRef.
- R. Kabe and C. Adachi, Organic Long Persistent Luminescence, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- S. Tan, K. Jinnai, R. Kabe and C. Adachi, Long-Persistent Luminescence From an Exciplex-Based Organic Light-Emitting Diode, Adv. Mater., 2021, e2008844 CrossRef PubMed.
- K. Jinnai, N. Nishimura, C. Adachi and R. Kabe, Thermally Activated Processes in an Organic Long-Persistent Luminescence System, Nanoscale, 2021, 13, 8412–8417 RSC.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Organic Long-Persistent Luminescence from a Thermally Activated Delayed Fluorescence Compound, Adv. Mater., 2020, 32, e2003911 CrossRef PubMed.
- J. Cornil, D. A. dos Santos, X. Crispin, R. Silbey and J. L. Bredas, Influence of Interchain Interactions on the Absorption and Luminescence of Conjugated Oligomers and Polymers: A Quantum-Chemical Characterization, J. Am. Chem. Soc., 1998, 120, 1289–1299 CrossRef CAS.
- P. Tourneur, F. Lucas, C. Quinton, Y. Olivier, R. Lazzaroni, P. Viville, J. Cornil and C. Poriel, White-Light Electroluminescence From a Layer Incorporating a Single Fully-Organic Spiro Compound with Phosphine Oxide Substituents, J. Mater. Chem. C, 2020, 8, 14462–14468 RSC.
- R. Newcomb, J. S. Bangsund, K. W. Hershey, D. C. K. Rathwell, H. Y. Na, J. H. Jeon, P. Trefonas and R. J. Holmes, Role of Host Excimer Formation in the Degradation of Organic Light-Emitting Devices, Appl. Phys. Lett., 2020, 116, 063302–0633028 CrossRef CAS.
- T. T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Thermally Activated Delayed Fluorescence Emitters for Deep Blue Organic Light Emitting Diodes: A Review of Recent Advances, Appl. Sci., 2018, 8, 494–517 CrossRef.
Footnotes |
| † The research data supporting this publication can be accessed at https://doi.org/10.17630/bce6c07c-64b9-4798-9a7b-8eef47d3124e. |
| ‡ Electronic supplementary information (ESI) available: Experimental section, general synthetic procedure, photophysical measurements, electrochemical measurements, theoretical modelling, synthesis, chemical characterization, X-ray crystallography, additional electrochemical experiments and electrochemiluminescence. CCDC 2107137–2107141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc05696d |
| § Equally contributed. |
| This journal is © The Royal Society of Chemistry 2022 |