
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA robust hollow metal–organic framework with enhanced diffusion for size selective catalysis†
Chunhui
Wu‡
 ,
Xiaowen
Zhao‡
,
Dongxu
Wang
,
Xiaomeng
Si
and
Tao
Li
*
,
Xiaowen
Zhao‡
,
Dongxu
Wang
,
Xiaomeng
Si
and
Tao
Li
*
School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, People's Republic of China. E-mail: litao1@shanghaitech.edu.cn
First published on 24th October 2022
Abstract
Single crystalline (SC) hollow metal–organic frameworks (MOFs) are excellent host materials for molecular and nanoparticle catalysts. However, due to synthetic challenges, chemically robust SC hollow MOFs are rare. This work reports the construction of a defect-free and chemically stable SC hollow MOF, MOF-801(h), through templated growth from a unit cell mismatched core, UiO-66. Under the protection of excess MOF-801 ligand, fumaric acid, the MOF-801 shell was perfectly retained while the isoreticular UiO-66 core was selectively and completely etched away by formic acid. The combination of a large cavity, small aperture and short diffusion length allows the Pt nanoparticle encapsulated composite catalyst, Pt⊂MOF-801(h), to perform size selective hydrogenation of nitro compounds at an accelerated speed. Impressively, the catalyst can undergo concentrated HCl or boiling water treatment while maintaining its crystallinity, morphology, catalytic activity, and size selectivity. In addition, Au nanoparticles encapsulated catalyst, Au⊂MOF-801(h), was used for the size selective nucleophilic addition of HCl to terminal alkynes for the first time, which is a harsh reaction involving high concentrations of a strong acid.
Introduction
Size selective catalysis has found wide spread applications in the petrochemical and chemical industries.1 Currently, commercially available size selective catalysts are limited to a few zeolite molecular sieves that exhibit comparable pore size and pore geometry to a particular reactant or product.2,3 For example, HZSM-5 is one of the most successful size-selective Lewis acid catalysts widely implemented in many industrial processes, including alkylation, isomerization, and disproportionation.4–8 Furthermore, other catalytic active sites such as metal nanoparticles, metal oxides, etc. can also be immobilized in a porous host to exhibit size selectivity.9,10 In this case, the porous host not only functions as a size selective barrier but also can protect the catalyst from thermal sintering or poisoning by small molecules in the surrounding media. In addition, the native active sites carried by the porous host can co-operate with the immobilized catalyst to achieve synergistic catalysis.11–13 To further extend size selective catalysts to more complex reaction systems, the design of new porous materials with a tailor-made aperture is key.Amongst various types of porous materials, metal–organic frameworks (MOFs) are widely acknowledged as a promising supporting substrate for catalysts.14–20 Benefiting from its organic-inorganic hybrid nature, MOFs can be designed either through the metal nodes or the organic struts to achieve sub-angstrom aperture size regulation necessary for size selectivity. More importantly, the synthetic conditions of most MOFs are mild, which enables non-destructive immobilization of fragile catalysts (e.g., enzymes and transition metal complexes).21–23 Nevertheless, the long diffusion pathway through narrow apertures in MOFs inevitably slows down the reaction kinetics. Meanwhile, the pore walls in close proximity to the catalyst surface may also block the active sites, resulting in a further decrease in catalytic performance.24,25
One feasible approach to avoid these limitations is to encapsulate the catalyst in a single crystalline (SC) hollow MOF.26,27 The single crystallinity eliminates the grain boundary thus minimizing defects. The thin shell effectively shortens the diffusion length of reactants while the inner cavity can physically confine catalysts without altering their native active sites. To achieve SC hollow MOF architecture, a sacrificial template is often employed.28 In a typical protocol, the MOF is first grown on the surface of a sacrificial template29–32 which is later removed through chemical etching. The challenge is that the sacrificial template needs to be sufficiently stable to withstand the solvothermal synthetic condition of the MOF shell and labile enough to be later removed without damaging the MOF. For this very reason, currently reported SC hollow MOFs are largely limited to ZIF-8,33,34 MOF-74,35 and UMOM-2,36 all of which are acid labile. Although SC hollow MIL-101(Cr) is considered chemically robust, it is not suitable for size selective catalysis due to its large aperture (∼1.6 nm).37,38 The only exception is SC hollow UiO-66 and its derivatives which were synthesized through harsh chemical etching either by using concentrated nitric acid or by using reactive oxygen species.39 Therefore, developing a chemically robust yet defect-free single crystal hollow MOF capsule to host catalysts is still challenging yet highly sought after.
This work reports the construction of a chemically robust ultra-microporous SC hollow MOF-801 for the encapsulation of Pt nanoparticles (NPs) (Scheme 1). The MOF-801 shell can be directionally grown from the surfaces of Pt NP-containing-UiO-66 single crystals into a complete and uniform shell despite large unit cell mismatch. While MOF-801 and UiO-66 are typically considered to be similar in chemical stability, selective etching of one of them is very difficult. We developed a ligand-protected etching (LPE) strategy to protect the MOF shell by incorporating excess ligand (fumaric acid). While both MOF-801 and UiO-66 can be digested by formic acid, the presence of excess fumaric acid allows MOF-801 to heal simultaneously. This selective etching process leads to the formation of SC hollow MOF-801 particles with Pt NPs encapsulated within. Due to the small aperture size of MOF-801 (∼4.6 Å), the resultant composite catalyst exhibits size selective hydrogenation of different nitro compounds with the reaction kinetics approaching that of the neat Pt NPs. Meanwhile, the ultrasmall aperture can block sterically hindered thiol molecules from poisoning the encapsulated Pt. Most impressively, the hollow MOF-801 can maintain its morphology, catalytic activity and size selectivity even after harsh chemical treatment with concentrated HCl and boiling water. Utilizing the excellent stability of MOF-801, another composite catalyst containing Au NPs was synthesized for the nucleophilic addition reaction of HCl to terminal alkynes. Despite the presence of excess strong acids, the size selective feature was also realized.
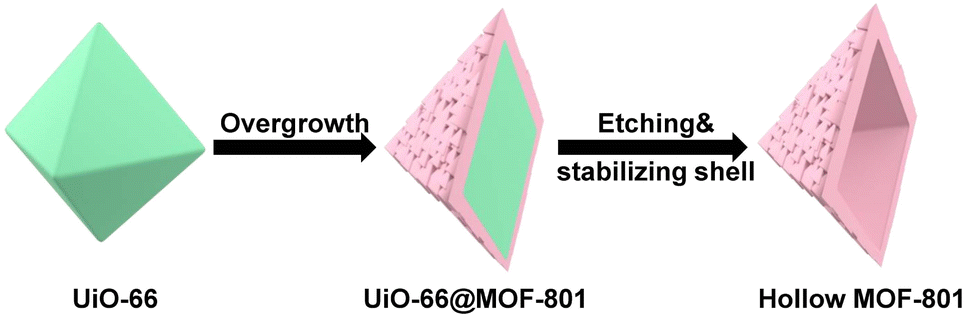 | ||
| Scheme 1 Scheme of the synthesis of hollow MOF-801. | ||
Results and discussion
The high chemical stability of MOF-801 has been widely demonstrated in water sorption, membrane separation and other applications.40–43 This is likely due to its more compact crystal structure compared to that of other structural analogues. This feature along with its ultra-small aperture size (∼4.6 Å)44 render MOF-801 an ideal candidate for size selective catalysis. Nonetheless, unlike the UiO-66 series that can undergo heteroepitaxial growth between various structural analogues,39 MOF-801 does not have sacrificial structural analogues that also exhibit similar unit cell parameters. Fortunately, recent discoveries have shown that facet-selective oriented growth between topologically distinct MOFs is possible even with a high degree of unit cell parameter mismatch.45–47 We hence reason that UiO-66 can serve as a suitable sacrificial template for the construction of SC hollow MOF-801. To start with, monodispersed UiO-66 particles with an average size of 410 ± 30 nm were synthesized according to a previously reported method (Fig. S1†).48 Our previous work shows that rapid nucleation of the MOF shell tends to form a polycrystalline structure whereas slow nucleation favors oriented growth.49 Therefore, in order to obtain a SC MOF-801 shell, various amounts of HCOOH were used to modulate the growth of the MOF-801 shell. First, we regulated the UiO-66/MOF-801 growth solution ratio (theoretical maximum yield of MOF-801 divided by that of UiO-66) from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 to 1:5, 1:10 and 1:15 while maintaining the metal/ligand precursor concentrations at 15 mM with the addition of HCOOH (184 eq. with respect to fumaric acid) as the modulator to synthesize a core–shell MOF, UiO-66@MOF-801. After 2 h of reaction, MOF-801 shells were formed in all four samples. With the growth solution ratio at 1:2.5, the MOF-801 shell was visibly polycrystalline as evidenced by its uneven surface texture. With the increase of the UiO-66/MOF-801 growth solution ratio to 1:5 and 1:10, the MOF-801 shell became denser and smoother (Fig. S2A†). Further increasing the growth solution ratio to 1:15 led to the deposition of free MOF-801 crystallites on the core–shell particle surfaces, which is an indication of homogeneous nucleation of MOF-801 (Fig. S2Aiv†). The powder X-ray diffraction (PXRD) patterns of all samples show a superposition of the characteristic peaks from UiO-66 and MOF-801 suggesting good crystallinity for both phases (Fig. S2B†). Based on the results obtained so far, a growth solution ratio of 1:10 was selected for the following experiment.
:2.5 to 1:5, 1:10 and 1:15 while maintaining the metal/ligand precursor concentrations at 15 mM with the addition of HCOOH (184 eq. with respect to fumaric acid) as the modulator to synthesize a core–shell MOF, UiO-66@MOF-801. After 2 h of reaction, MOF-801 shells were formed in all four samples. With the growth solution ratio at 1:2.5, the MOF-801 shell was visibly polycrystalline as evidenced by its uneven surface texture. With the increase of the UiO-66/MOF-801 growth solution ratio to 1:5 and 1:10, the MOF-801 shell became denser and smoother (Fig. S2A†). Further increasing the growth solution ratio to 1:15 led to the deposition of free MOF-801 crystallites on the core–shell particle surfaces, which is an indication of homogeneous nucleation of MOF-801 (Fig. S2Aiv†). The powder X-ray diffraction (PXRD) patterns of all samples show a superposition of the characteristic peaks from UiO-66 and MOF-801 suggesting good crystallinity for both phases (Fig. S2B†). Based on the results obtained so far, a growth solution ratio of 1:10 was selected for the following experiment.
Next, the amount of HCOOH was varied from 92 to 368 eq. As shown in Fig. 1A and S3,† with increasing concentration of HCOOH, the thickness of the MOF-801 shell increased accordingly from ∼34 nm (92 eq.) to ∼70 nm (276 eq.). However, a smooth shell was only obtained when 92 or 184 eq. HCOOH was used. This phenomenon seems to contradict the previous report that slow nucleation tends to favor well-oriented shell growth.47 One possible explanation is that a high HCOOH concentration resulted in surface etching of UiO-66 which disrupted the oriented growth of MOF-801. Indeed, a visible gap can be observed at two MOF interface when 184 eq. HCOOH was used (Fig. 1Aii). At 276 and 368 eq. of HCOOH, UiO-66 can be mostly etched after the reaction leaving a polycrystalline MOF-801 shell (Fig. 1Aiii andiv). To maintain the single crystallinity of the MOF-801 shell, 92 eq. HCOOH was used from here on.
 | ||
| Fig. 1 (A) TEM images of UiO-66@MOF-801 synthesized with various amounts of formic acid. (B) TEM images of UiO-66@MOF-801 obtained after various reaction times. (C) SEM (i) and TEM (ii) images of MOF-801(h). | ||
To better understand the nucleation and growth process of the MOF-801 shell, we monitored the time dependent morphological evolution of the MOF-801 shell under the optimal synthetic conditions (92 eq. HCOOH and the UiO-66/MOF-801 growth solution ratio is 1:10) using TEM (Fig. 1B). After 30 min, small nuclei of MOF-801 started to appear uniformly on UiO-66 (Fig. 1Bii). Its PXRD pattern confirmed the emergence of a peak at 9.91° (2θ) corresponding to the {200} facet of MOF-801 (Fig. S4†). On extending the growth time to 60 min, the continuous growth of the MOF-801 nuclei interconnected in a continuous and defect-free shell with an average thickness of ∼27 nm is observed (Fig. 1Biii). On further prolonging the growth time to 120 min, the shell thickness increased to ∼34 nm (Fig. 1Biv and S5†). This product is thus denoted as 66@801 from here on.
Our first attempt to remove the UiO-66 template using a DMF solution of HCOOH (4.6 M) led to complete dissolution of 66@801 (Fig. S6†). To preserve the crystallinity of MOF-801 during etching, the ligand of MOF-801, fumaric acid, was added to the etchant to prevent MOF-801 degradation. This very trick made a world of difference as the TEM image revealed the formation of well-defined hollow MOF-801 particles (Fig. 1Cii). The average shell thickness is ∼90 nm which is significantly thicker than the MOF-801 layer on the core–shell form. This is likely because the Zr digested from UiO-66 was utilized for the continuous growth of MOF-801 inside the shell. We hypothesize that the presence of fumaric acid encourages the growth of MOF-801. This will allow the MOF-801 shell to heal while being digested. When the healing rate outcompetes the dissolution rate, the shell is preserved. On the other hand, UiO-66 cannot be healed in the presence of fumaric acid due to mismatched ligand length. Therefore, it will continuously digest in the presence of formic acid. The SEM image showed that MOF-801(h) manifests octahedron-morphology with a smooth surface texture (Fig. 1Ci). The PXRD patterns and Fourier transform-infrared (FT-IR) spectra also confirmed the complete removal of the UiO-66 template and the preservation of the crystallinity of MOF-801 (Fig. 2A and S7†). Furthermore, TGA experiments were performed for UiO-66, MOF-801, 66@801, and MOF-801(h) under an O2 atmosphere. The ZrO2 weight was recorded at 750 °C after an isothermal step. The mass ratio of ZrO2 and dry MOF is indicative of the MOF composition. For pristine UiO-66 and MOF-801, the ZrO2:MOF ratios are 0.456 and 0.533, respectively. The ratio of 66@801 is 0.480, falling in between that of UiO-66 and MOF-801. MOF-801(h) exhibits a ZrO2 to MOF ratio of 0.516 which is close to that of pristine MOF-801 (Fig. S8†). This result suggests that the etching of UiO-66 was complete. However, the discrepancy between 0.516 and 0.533 is likely due to the presence of residual terephthalic acid in the hollow cavity. Based on the TGA curves, we calculated that ∼3.2 wt% of terephthalic acid remains in the hollow MOF-801 structure after the removal of the UiO-66 core. In addition, the 1H NMR spectrum of the digested MOF-801(h) suggested that ∼5.4 wt% terephthalic acid was present in MOF-801(h) (Fig. S9†). This conclusion was further confirmed by the elemental analysis results (Table S1†).
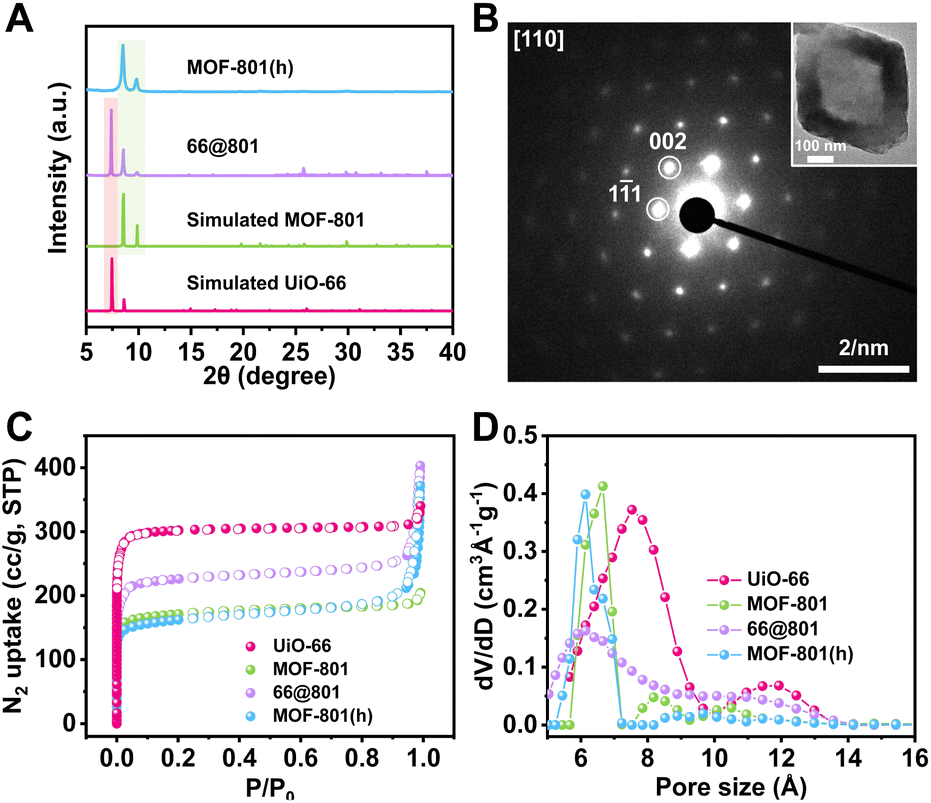 | ||
| Fig. 2 (A) PXRD patterns of 66@801, MOF-801(h), simulated MOF-801 and UiO-66. (B) SAED pattern of a MOF-801(h) particle (TEM image is shown in the inset). (C) N2 adsorption–desorption isotherms at 77 K for UiO-66, MOF-801, 66@801 and MOF-801(h). (D) Pore size distribution profiles of UiO-66, MOF-801, 66@801 and MOF-801(h). | ||
To reveal the crystal orientation of the MOF-801 shell, the selected area electron diffraction (SAED) experiment was conducted on a MOF-801(h) particle. The SAED pattern through the [110] zone axis exhibited a hexagonal array of diffraction spots identical to that of a MOF-801 single crystal suggesting that MOF-801(h) particles are indeed single crystals (Fig. 2B). This observation confirmed that the UiO-66 core directed the growth orientation of MOF-801 crystallites which eventually merged into a single crystalline shell.
The porosity of these MOF samples was investigated using N2 adsorption experiments at 77 K (Fig. 2C). Being the larger analogue, UiO-66 exhibited a BET surface area of 1230 m2 g−1, higher than that of MOF-801 (680 m2 g−1). The BET surface area of 66@801 is 914 m2 g−1, lower than that of UiO-66 but higher than that of MOF-801. The N2 adsorption isotherm of 66@801 also falls in between that of MOF-801 and UiO-66. After etching, MOF-801(h) manifested a similar N2 adsorption isotherm and BET surface area (677 m2 g−1) compared to that of MOF-801 (680 m2 g−1) (Table S2†). Meanwhile, the pore-size distribution analysis also confirmed that both MOF-801(h) and MOF-801 exhibited a 0.6–0.7 nm pore, distinct from the 0.8 nm pore of UiO-66 (Fig. 2D). These results unequivocally prove that MOF-801(h) is highly crystalline.
Encouraged by the positive results, we turned to apply MOF-801(h) as a porous support to host Pt NPs for size selective catalysis. Following a previously reported method,50,51 PVP covered Pt NPs (∼3 nm) (Fig. S10†) were introduced during the synthesis of UiO-66 which led to the inclusion of multiple Pt NPs distributed at the center of UiO-66 crystals (Pt⊂UiO-66) (Fig. 3Ai and S11A†). The Pt NPs loading was identified to be 2.86 wt% using inductively coupled plasma-optical emission spectroscopy (ICP-OES) (Table S3†). By measuring the residual Pt content in the supernatant, the encapsulating efficiency of Pt NPs was calculated to be 92% (Table S4†). Through similar overgrowth and etching processes, Pt⊂MOF-801(h) was obtained (Fig. 3Aiv and S12B†). As shown in the TEM images, Pt NPs remained at the center of the 66@801 core–shell particles (Fig. 3Aiii and S12A†). After etching, Pt NPs were successfully included inside the macroporous cavities of MOF-801(h). The PXRD patterns show that the introduction of Pt NPs has no negative impact on the MOF crystallinity (Fig. S13†).
To demonstrate the size selective catalysis of Pt⊂MOF-801(h), a model reaction, liquid-phase catalytic hydrogenation of nitrobenzene (NB) and 1-nitronaphthalene (NN), was carried out (Fig. S14 and S15†). The reaction condition was slightly modified from that in previously reported studies.49,52–54 Being the smaller nitro compound, NB exhibits a size of 6.6 × 4.6 Å whereas the larger nitro compound, NN exhibits a size of 7.3 × 6.6 Å (Fig. 3Ciii).55 Apart from Pt⊂MOF-801(h), Pt⊂UiO-66 and Pt⊂66@801 were also included to investigate the impact of the pore size and diffusion length on the catalytic hydrogenation performance. To compare hollow versus solid architectures, another catalyst, Pt⊂MOF-801, was prepared (Fig. 3Aii and S11B†). The Pt NP loading was identified to be 3.00 wt% by ICP-OES (Table S3†). The encapsulating efficiency of Pt NPs was calculated to be 89% (Table S4†). While pristine Pt NPs achieved high conversion (>98%) for both NB and NN after 90 min, Pt⊂MOF-801(h) reached 91% conversion for NB but only 12% for the larger compound NN, which demonstrates a clear size selectivity (Fig. 3B). This is expected because the size of NN significantly exceeds the aperture size of MOF-801 (Fig. 3C). The size selectivity of Pt⊂MOF-801(h) is comparable to that of Pt⊂MOF-801 which exhibited 66% NB conversion and 8.5% NN conversion. However, Pt⊂MOF-801(h) out-performs Pt⊂MOF-801 in terms of reaction kinetics due to its thinner shell (∼23 nm) which leads to a shorter diffusion distance (Fig. S11B and S12B†). Pt⊂UiO-66, on the other hand, did not exhibit size selectivity as it showed moderate conversion for both NB (78%) and NN (53%) due to its larger aperture size (∼6.0 Å) (Fig. 3Ci). Meanwhile, Pt⊂66@801 showed a slightly higher NN conversion (26.0%) and lower NB conversion (75%) than Pt⊂MOF-801(h) likely due to the presence of defects on the MOF-801 shell layer which were successfully repaired during the subsequent etching process (Fig. S12A†). In contrast, the catalytic hydrogenation of NB and NN on pristine UiO-66, MOF-801, 66@801 and MOF-801(h) showed negligible catalytic activity (Table S5†). To investigate whether such size selectivity is applicable to mixtures, the catalytic hydrogenation of an NN/NB mixture (n/n = 1/1) on Pt NPs and Pt⊂MOF-801(h) was performed. For Pt NPs, the TON of NN at 10 min was 44 whereas the TON of NB at 10 min was 14 (Fig. 3D). This reactivity difference is due to the electronic effect between the two molecules. The conversion of NB and NN at various reaction time points is shown in Fig. S16.† Interestingly, for Pt⊂MOF-801(h), the TON was much higher (77) for NB than for NN (21) despite its lower reactivity (Fig. 3D). This demonstrates that the size selective catalysis feature of Pt⊂MOF-801(h) can also be applied to mixtures.
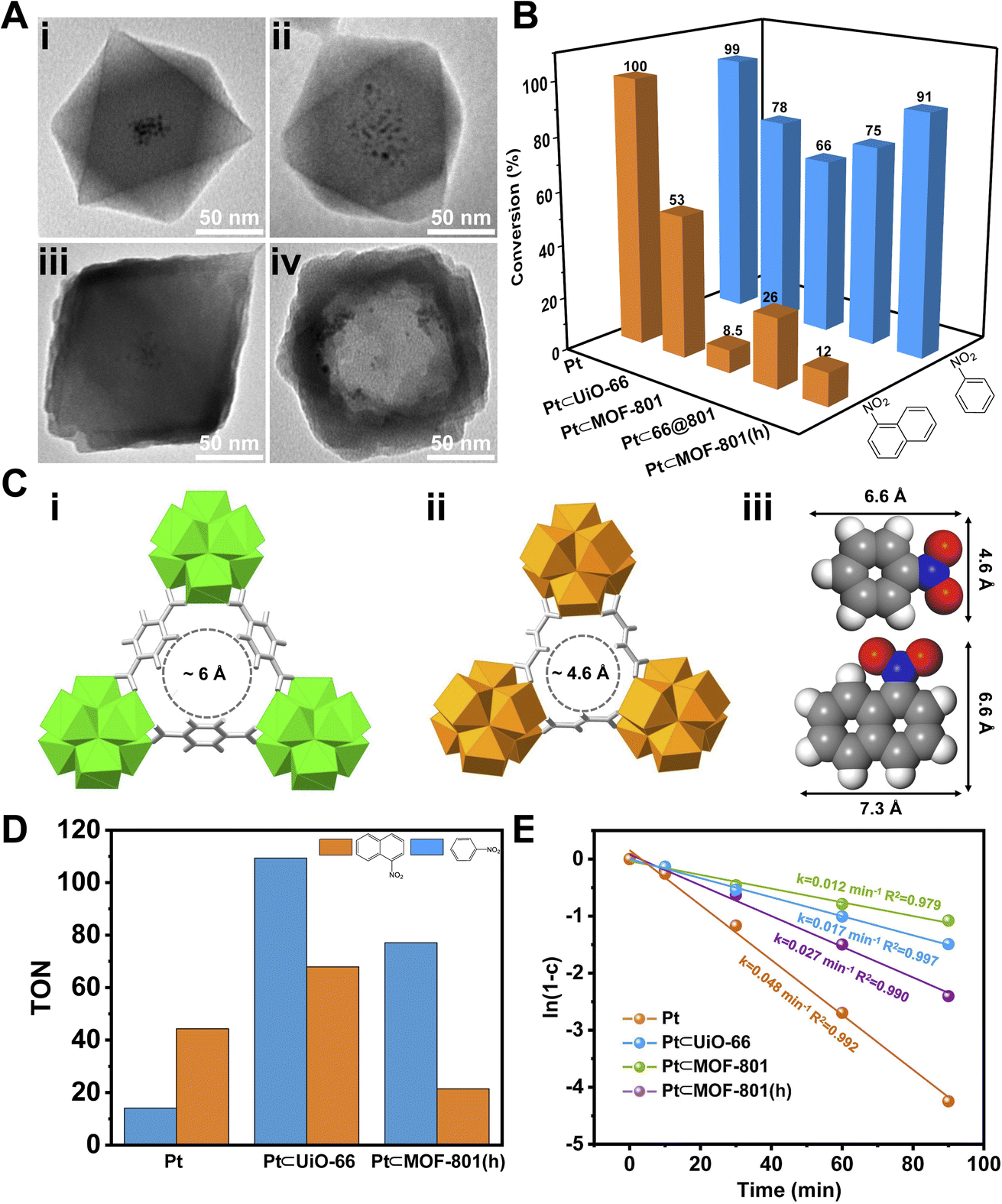 | ||
| Fig. 3 (A) TEM images of Pt⊂UiO-66 (i), Pt⊂MOF-801 (ii), Pt⊂66@801 (iii) and Pt⊂MOF-801(h) (iv). (B) The conversion of nitrobenzene and 1-nitronaphthalene over different catalysts after 90 min of the hydrogenation reaction. (C) Comparison of aperture sizes of UiO-66 (i) and MOF-801 (ii) with the molecular sizes of nitrobenzene and 1-nitronaphthalene (iii). (D) The conversion of NB and NN for a NN/NB mixture over Pt NPs for 10 min, and over Pt⊂UiO-66 and Pt⊂MOF-801(h) for 90 min. The maximum possible turnover number (TON) is 135. (E) The catalytic reaction kinetic curves for various catalysts. The solid line represents the linear fitting curves. k and c are the reaction rate constant and the conversion of the reactant. | ||
To confirm the retention of Pt NPs in the MOF structure after catalysis, the mass ratios of Zr and Pt for Pt⊂UiO-66 and Pt⊂MOF-801 before and after hydrogenation of NB were determined by ICP-OES. The results showed that the mass ratio of Zr and Pt remained the same after catalysis. Moreover, after filtering out Pt⊂UiO-66 and Pt⊂MOF-801, no Pt was found in the supernatants of the reaction mixtures (Table S6†). We further evaluated the catalytic activity of the supernatant from NB hydrogenation over Pt⊂MOF-801 at the 90 min time point. The catalyst was completely removed from the supernatant through centrifugation and filtration. The residual NB was quantified by GC with an external standard and used as the initial concentration. The supernatant was re-exposed to this exact hydrogenation condition for 90 min after which the conversion of the residual NB was found to be only 1.8%. This suggests that no substantial amount of catalytically active species was leached out from the MOF. This result is also consistent with the ICP results (Table S6†).
Next, we sought to investigate the reaction kinetics. To quantitatively compare the reaction kinetics of NB over the four catalysts, we first assumed that the kinetic order in NB equals one (Fig. S17†). The kinetic data were fit to the first-order integrated rate law (eqn S(1)†). The rate constant (k) was then calculated. As shown in Fig. 3E, Pt NPs exhibited the fastest reaction rate (k = 0.048 min−1) among the four catalysts. On the other end of the spectrum, Pt⊂MOF-801 was the slowest (k = 0.012 min−1) of the four catalysts due to its small aperture size as well as long diffusion distance (∼60 nm) (Fig. S11B†). It is also possible that part of the Pt active sites was shielded by the MOF pore walls, thus further decreasing its catalytic activity. For similar reasons, the k value of Pt⊂UiO-66 only reached 0.017 min−1. Despite having a smaller pore size than UiO-66, the rate constant of Pt⊂MOF-801(h) is 0.027 min−1, which is 2.3 times that of Pt⊂MOF-801 (0.012 min−1). The control experiment shows that formic acid treatment has little effect on the catalytic activity of Pt NPs (Fig. S18†). Therefore, it is reasonable to conclude that the increased reaction rate on MOF-801(h) compared to that on solid MOF-801 was due to the shortened diffusion pathway and increased mass transport in the hollow MOF.
Apart from size selective catalysis, the small aperture of Pt⊂MOF-801(h) can also be used to reject molecules that can deactivate the encapsulated catalyst. To demonstrate, Pt⊂MOF-801(h) was incubated with 0.44 mM ethanethiol (ET) or 1-adamantanethiol (AT) in toluene for 10 min before the NB hydrogenation reaction. As shown in Fig. 4A, the catalytic activity of Pt NPs drastically decreased after treatment with thiol. Similarly, the NB conversion on Pt⊂UiO-66 decreased to 26% and 29% after ET and AT treatment, respectively, which suggests that both thiol molecules can diffuse through the aperture of UiO-66. In contrast, while ET can still poison Pt⊂MOF-801, Pt⊂66@801, and Pt⊂MOF-801(h), the larger thiol, AT, has little poisoning effect on these composite catalysts due to the size exclusion by the MOF-801 shell. These results suggest that MOF-801 can serve as a barrier to protect the encapsulated catalyst from poisoning by bulky contaminants in external media. The reusability of Pt⊂MOF-801(h) was evaluated by performing 9 consecutive cycles of the NB hydrogenation reaction. The initial conversion of NB was deliberately set at 45% to better evaluate the change in catalytic activity. As shown in Fig. 4B, the NB conversion over Pt⊂MOF-801(h) slightly decreased from the initial 45% to 33% after 9 catalytic cycles. This result suggests that Pt⊂MOF-801(h) exhibited reasonable reusability. The TEM image, the SEM image and the PXRD pattern of the post-catalysis sample show that the morphology and crystallinity were also well-maintained (Fig. 4C, Eiii and S19†). The Zr/Pt mass ratios in Pt⊂MOF-801(h) before and after recycling 9 times remained the same, which indicates that there was no loss of the Pt catalyst after 9 runs (Table S6†). These evidences suggest that the decrease in catalytic activity may be attributed to MOF-801 pore clogging by the reaction by-products or the sintering of the ultra-small Pt NPs.
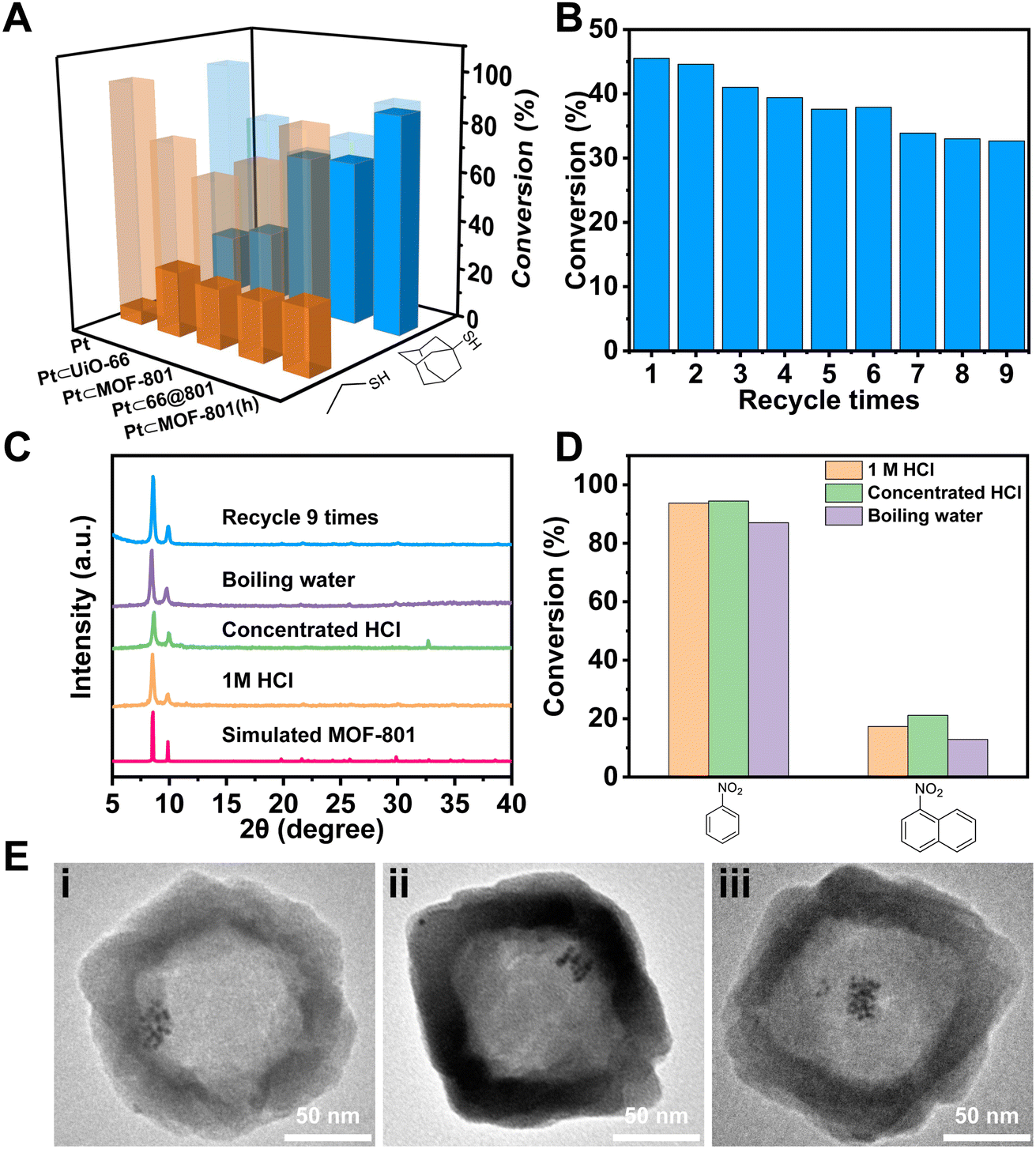 | ||
| Fig. 4 (A) The hydrogenation conversion of nitrobenzene over different catalysts after treatment with ethanethiol (orange) and 1-adamantanethiol (blue). The transparent columns represent nitrobenzene conversion prior to thiol treatment. (B) Recycling performance of Pt⊂MOF-801(h) for the nitrobenzene hydrogenation reaction. (C) PXRD patterns of Pt⊂MOF-801(h) after treatment with boiling water, 1 M and concentrated HCl, and recycling 9 times. (D) The hydrogenation conversion of nitrobenzene and 1-nitronaphthalene over Pt⊂MOF-801(h) after harsh chemical treatment with boiling water, 1 M and concentrated HCl. (E) TEM images of Pt⊂MOF-801(h) after treatment with concentrated HCl (i), boiling water (ii), and 9 catalytic cycles (iii). | ||
Furthermore, the chemical stability of Pt⊂MOF-801(h) was investigated. To test its acid stability, Pt⊂MOF-801(h) was treated with 1 M or concentrated HCl (∼12 M) for 1 h. For hydrothermal stability, Pt⊂MOF-801(h) was boiled in water for 3 h. The PXRD patterns show that these harsh treatments did not lead to a noticeable decrease in MOF-801 crystallinity (Fig. 4C). Moreover, the hollow morphology of Pt⊂MOF-801(h) was retained with no visible defects (Fig. 4Ei and ii and S20†). Then these treated catalysts were used again for hydrogenation of NB and NN. Fig. 4D shows that both the catalytic activity and the size selectivity of Pt⊂MOF-801(h) were fully preserved despite harsh treatment. To the best of our knowledge, this is the first example of a robust hollow MOF composite catalyst that can withstand strong acid and boiling water treatment.
Encouraged by the high chemical stability of MOF-801(h), a series of Au NP-containing composite catalysts were prepared in a similar manner to investigate the possibility of using such hollow MOF catalysts in chemical reactions under harsh reaction conditions. A previous work by Corma et al. demonstrated that Au NPs were a highly efficient catalyst for the nucleophilic addition of HCl to terminal alkynes.56 Therefore, this reaction was selected as a model reaction here due to the involvement of high HCl concentration which highlights the necessity to use an acid stable catalyst. We aim to study whether the hollow MOF-801 shell can enable size selective catalyst between a small alkyne, 4-ethynyltoluene (EL), and a bulky alkyne, 1-ethynylnaphthalene (EN).
For the synthesis, following a previously reported method, Au NPs (∼5 nm) were introduced into UiO-66 and MOF-801 to give Au⊂UiO-66 and Au⊂MOF-801 (Fig. S21†).57 Through similar overgrowth and etching processes, Au⊂66@801 and Au⊂MOF-801(h) were obtained (Fig. 5A and B, S22 and S23†). The Au loading in Au⊂UiO-66 and Au⊂MOF-801 was identified to be 4.62 wt% and 1.67 wt% using ICP-OES (Table S7†). The TEM images show that the Au NPs were successfully confined within the cavity of MOF-801(h) (Fig. 5A and S23A†). The crystallinity of the MOF-801 shell was again well-maintained during the etching process (Fig. 5B).
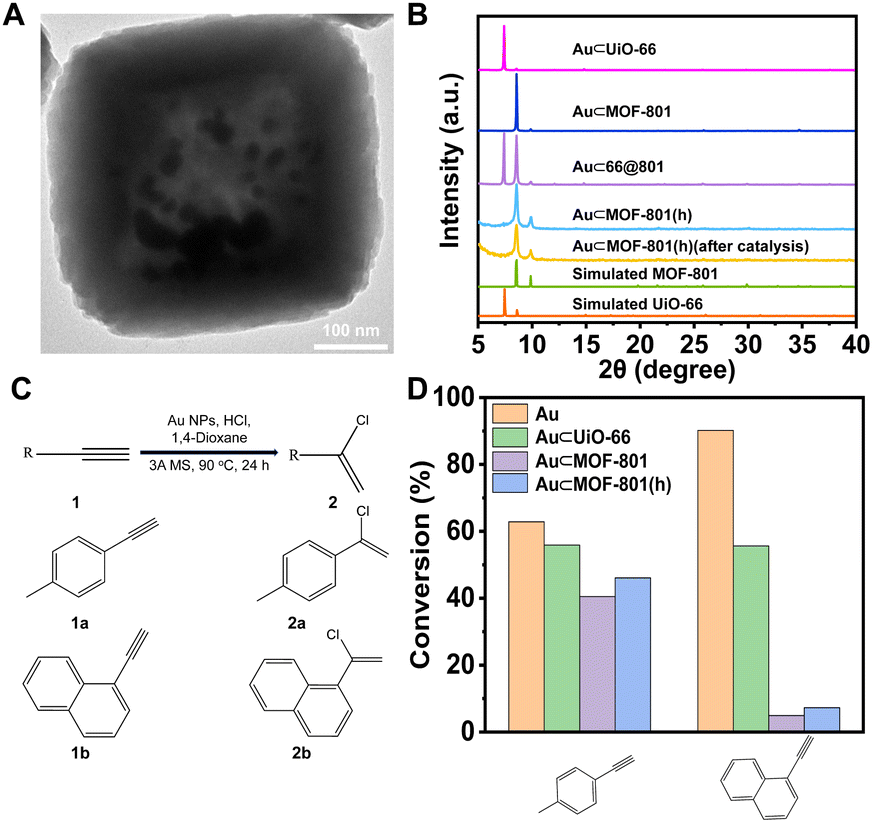 | ||
| Fig. 5 (A) TEM images of Au⊂MOF-801(h). (B) PXRD patterns of Au⊂UiO-66, Au⊂MOF-801, and Au⊂MOF-801 before and after catalysis. (C) Au catalyzed reaction of terminal alkynes with HCl. (D) The conversion of 4-ethynyltoluene and 1-ethynylnaphthalene over different catalysts after 24 h of the reaction with HCl. | ||
The nucleophilic addition of HCl to terminal alkynes was performed according to the reported catalytic procedure (Fig. 5C).56 Apart from Au⊂MOF-801(h), Au⊂UiO-66 and Au⊂MOF-801 were also included to investigate the impact of the pore size and diffusion length on the catalytic performance. While pristine Au NPs (∼13 nm) achieved a high conversion for both EL (63%) and EN (90%) after 24 h reaction (Fig. S24†). Au⊂MOF-801(h) reached 47% conversion for EL but only 7.3% for the larger EN (Fig. 5D). This demonstrates a clear size selectivity due to the MOF-801 shell. Similarly, Au⊂MOF-801 exhibited 40% EL conversion and 4.9% EN conversion. Au⊂UiO-66, on the other hand, did not exhibit size selectivity as it showed moderate conversion for both EL (56%) and EN (55%) due to its larger aperture size (∼6.0 Å) (Fig. 3Ci). These results indicate that Au⊂MOF-801(h) can retain its catalytic performance and size selectivity even in the presence of a strong acid (0.92 M). In addition, the TEM image, the SEM image, the PXRD pattern and BET surface area of the post-catalysis sample showed that the crystallinity and morphology of the catalyst were also fully retained after catalysis (Fig. 5B, S26 and S27†). These results highlight the robust properties of hollow catalysts in practical catalytic applications.
Conclusions
In summary, this work presents the synthesis of a chemically robust SC hollow MOF, MOF-801(h), that can be used to physically confine noble metal NPs for size selective hydrogenation of nitro compounds and nucleophilic addition of HCl to terminal alkynes. Similar to metal–organic layers,58,59 hollow MOFs effectively shorten the diffusion distance of the substrate molecules, thereby increasing reaction kinetics. What's distinctive is that the closed shell architecture creates an ideal environment for the encapsulation of hetero and homogeneous catalysts within. The successful growth of a well-oriented and defect-free MOF-801 shell from a core MOF of a mismatched unit cell substantially expanded the possibility of hollow MOFs that can be constructed from the templated method. In combination with the fact that UiO-66 is capable of hosting a myriad of molecular and nanoparticle catalysts through de novo growth or post-synthetic modifications, this work opens new avenues for exploring size selective catalysis.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
T. L. conceived and developed this study. C. W. and X. Z. performed the experiments and analyzed the data. X. Z. drew the scheme picture. X. S. conducted select area electron diffraction (SAED) experiment. D. W. carried out the ICP-OES measurement. The manuscript was written by C. W. and T. L. with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22075181, T. L.) and the start-up funding from ShanghaiTech University. The authors thank the support from the Analytical Instrumentation Center (# SPST-AIC10112914), SPST, ShanghaiTech University and the Centre for High-resolution Electron Microscopy (CħEM) of SPST at ShanghaiTech University under Grant No. EM02161943. The computing resources were made available through the high-performance computing platform of ShanghaiTech University.Notes and references
- N.-Y. Chen, Shape selective catalysis in industrial applications, CRC press, 1996 Search PubMed.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- J. Weitkamp, Solid State Ionics, 2000, 131, 175–188 CrossRef CAS.
- J. Čejka and B. Wichterlová, Catal. Rev., 2002, 44, 375–421 CrossRef.
- T.-C. Tsai, S.-B. Liu and I. Wang, Appl. Catal., A, 1999, 181, 355–398 CrossRef CAS.
- D. Fraenkel and M. Levy, J. Catal., 1989, 118, 10–21 CrossRef CAS.
- W. W. Kaeding, G. C. Barile and M. M. Wu, Catal. Rev.: Sci. Eng., 1984, 26, 597–612 CrossRef CAS.
- L. Young, S. Butter and W. Kaeding, J. Catal., 1982, 76, 418–432 CrossRef CAS.
- T. Andana, K. G. Rappé, F. Gao, J. Szanyi, X. Pereira-Hernandez and Y. Wang, Appl. Catal., B, 2021, 291, 120054 CrossRef CAS.
- D. Farrusseng and A. Tuel, New J. Chem., 2016, 40, 3933–3949 RSC.
- A. B. Laursen, K. T. Højholt, L. F. Lundegaard, S. B. Simonsen, S. Helveg, F. Schüth, M. Paul, J. D. Grunwaldt, S. Kegnæs and C. H. Christensen, Angew. Chem., 2010, 122, 3582–3585 CrossRef.
- C. Wang, E. Guan, L. Wang, X. Chu, Z. Wu, J. Zhang, Z. Yang, Y. Jiang, L. Zhang and X. Meng, J. Am. Chem. Soc., 2019, 141, 8482–8488 CrossRef CAS PubMed.
- H. J. Cho, D. Kim, J. Li, D. Su and B. Xu, J. Am. Chem. Soc., 2018, 140, 13514–13520 CrossRef CAS PubMed.
- T. Kajiwara, M. Fujii, M. Tsujimoto, K. Kobayashi, M. Higuchi, K. Tanaka and S. Kitagawa, Angew. Chem., 2016, 128, 2747–2750 CrossRef.
- P. Hu, J. V. Morabito and C.-K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed.
- S. Ahn, S. L. Nauert, C. T. Buru, M. Rimoldi, H. Choi, N. M. Schweitzer, J. T. Hupp, O. K. Farha and J. M. Notestein, J. Am. Chem. Soc., 2018, 140, 8535–8543 CrossRef CAS PubMed.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- T. Drake, P. Ji and W. Lin, Acc. Chem. Res., 2018, 51, 2129–2138 CrossRef CAS PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef PubMed.
- W. Zhang, G. Lu, C. Cui, Y. Liu, S. Li, W. Yan, C. Xing, Y. R. Chi, Y. Yang and F. Huo, Adv. Mater., 2014, 26, 4056–4060 CrossRef CAS PubMed.
- F. Meng, S. Zhang, L. Ma, W. Zhang, M. Li, T. Wu, H. Li, T. Zhang, X. Lu and F. Huo, Adv. Mater., 2018, 30, 1803263 CrossRef PubMed.
- D. Liu, J. Wan, G. Pang and Z. Tang, Adv. Mater., 2019, 31, 1803291 CrossRef PubMed.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed.
- T. Qiu, S. Gao, Z. Liang, D. G. Wang, H. Tabassum, R. Zhong and R. Zou, Angew. Chem., Int. Ed., 2021, 60, 17314–17336 CrossRef CAS PubMed.
- J. Ha and H. R. Moon, CrystEngComm, 2021, 23, 2337–2354 RSC.
- D. H. Hong, H. S. Shim, J. Ha and H. R. Moon, Bull. Korean Chem. Soc., 2021, 42, 956–969 CrossRef CAS.
- L. Chai, J. Pan, Y. Hu, J. Qian and M. Hong, Small, 2021, 17, 2100607 CrossRef CAS PubMed.
- C. Liu, J. Wang, J. Wan and C. Yu, Coord. Chem. Rev., 2021, 432, 213743 CrossRef CAS.
- X. Y. Liu, F. Zhang, T. W. Goh, Y. Li, Y. C. Shao, L. Luo, W. Huang, Y. T. Long, L. Y. Chou and C. K. Tsung, Angew. Chem., 2018, 130, 2132–2136 CrossRef.
- C. Rosler, A. Aijaz, S. Turner, M. Filippousi, A. Shahabi, W. Xia, G. Van Tendeloo, M. Muhler and R. A. Fischer, Chem.–Eur. J., 2016, 22, 3304–3311 CrossRef PubMed.
- C. Wu, L.-Y. Chou, L. Long, X. Si, W.-S. Lo, C.-K. Tsung and T. Li, ACS Appl. Mater. Interfaces, 2019, 11, 35820–35826 CrossRef CAS PubMed.
- J. Lee, J. H. Kwak and W. Choe, Nat. Commum., 2017, 8, 1–8 CrossRef PubMed.
- Y. Qin, X. Han, Y. Li, A. Han, W. Liu, H. Xu and J. Liu, ACS Catal., 2020, 10, 5973–5978 CrossRef CAS.
- W. Liu, J. Huang, Q. Yang, S. Wang, X. Sun, W. Zhang, J. Liu and F. Huo, Angew. Chem., Int. Ed., 2017, 56, 5512–5516 CrossRef CAS PubMed.
- L. Luo, W.-S. Lo, X. Si, H. Li, Y. Wu, Y. An, Q. Zhu, L.-Y. Chou, T. Li and C.-K. Tsung, J. Am. Chem. Soc., 2019, 141, 20365–20370 CrossRef CAS PubMed.
- B. Kim, J. Na, H. Lim, Y. Kim, J. Kim and E. Kim, Adv. Funct. Mater., 2019, 29, 1807549 CrossRef.
- C. Wu, K. Zhang, H. Wang, Y. Fan, S. Zhang, S. He, F. Wang, Y. Tao, X. Zhao and Y.-B. Zhang, J. Am. Chem. Soc., 2020, 142, 18503–18512 CrossRef CAS PubMed.
- H. Furukawa, F. Gandara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- J. Zhang, H.-J. Bai, Q. Ren, H.-B. Luo, X.-M. Ren, Z.-F. Tian and S. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 28656–28663 CrossRef CAS PubMed.
- Y. Liu, Z. Chen, G. Liu, Y. Belmabkhout, K. Adil, M. Eddaoudi and W. Koros, Adv. Mater., 2019, 31, 1807513 CrossRef PubMed.
- M. Zhao, J. Chen, B. Chen, X. Zhang, Z. Shi, Z. Liu, Q. Ma, Y. Peng, C. Tan and X.-J. Wu, J. Am. Chem. Soc., 2020, 142, 8953–8961 CrossRef CAS PubMed.
- S. Choi, T. Kim, H. Ji, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2016, 138, 14434–14440 CrossRef CAS PubMed.
- F. Wang, Y. Fan, Y. Ma and T. Li, Cryst. Growth Des., 2021, 21, 4571–4578 CrossRef CAS.
- G. Lu, C. Cui, W. Zhang, Y. Liu and F. Huo, Chem.–Eur. J., 2013, 8, 69–72 CAS.
- F. Wang, S. He, H. Wang, S. Zhang, C. Wu, H. Huang, Y. Pang, C.-K. Tsung and T. Li, Chem. Sci., 2019, 10, 7755–7761 RSC.
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816–5822 CrossRef CAS PubMed.
- F. Wang, H. Wang and T. Li, Nanoscale, 2019, 11, 2121–2125 RSC.
- X. Yan, L. Chen, H. Song, Z. Gao, H. Wei, W. Ren and W. Wang, New J. Chem., 2021, 45, 18268–18276 RSC.
- H. Pan, Y. Peng, X. Lu, J. He, L. He, C. Wang, F. Yue, H. Zhang, D. Zhou and Q. Xia, Mol. Catal., 2020, 485, 110838 CrossRef.
- X. Wang and Y. Li, J. Mol. Catal. A: Chem., 2016, 420, 56–65 CrossRef CAS.
- T. L. Cui, W. Y. Ke, W. B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Angew. Chem., 2016, 128, 9324–9328 CrossRef.
- J. Oliver-Meseguer, A. Doménech-Carbó, M. Boronat, A. Leyva-Pérez and A. Corma, Angew. Chem., 2017, 129, 6535–6539 CrossRef.
- X. Qin, S. He, J. Wu, Y. Fan, F. Wang, S. Zhang, S. Li, L. Luo, Y. Ma and Y. Lee, ACS Cent. Sci., 2020, 6, 247–253 CrossRef CAS PubMed.
- L. Cao and C. Wang, ACS Cent. Sci., 2020, 6, 2149–2158 CrossRef CAS PubMed.
- L. Cao, Z. Lin, F. Peng, W. Wang, R. Huang, C. Wang, J. Yan, J. Liang, Z. Zhang and T. Zhang, Angew. Chem., 2016, 128, 5046–5050 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc02838g |
| ‡ The authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |