
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzanone (HNO): generation, stabilization and detection
Cecilia Mariel
Gallego†
,
Agostina
Mazzeo†
 ,
Paola
Vargas†
,
Sebastián
Suárez
,
Juan
Pellegrino
and
Fabio
Doctorovich
*
,
Paola
Vargas†
,
Sebastián
Suárez
,
Juan
Pellegrino
and
Fabio
Doctorovich
*
Departamento de Química Inorgánica, Analítica, y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, INQUIMAE-CONICET, Ciudad Universitaria, Pab. 2, C1428EHA, Buenos Aires, Argentina. E-mail: doctorovich@qi.fcen.uba.ar
First published on 5th July 2021
Abstract
HNO (nitroxyl, azanone), joined the ‘biologically relevant reactive nitrogen species’ family in the 2000s. Azanone is impossible to store due to its high reactivity and inherent low stability. Consequently, its chemistry and effects are studied using donor compounds, which release this molecule in solution and in the gas phase upon stimulation. Researchers have also tried to stabilize this elusive species and its conjugate base by coordination to metal centers using several ligands, like metalloporphyrins and pincer ligands. Given HNO's high reactivity and short lifetime, several different strategies have been proposed for its detection in chemical and biological systems, such as colorimetric methods, EPR, HPLC, mass spectrometry, fluorescent probes, and electrochemical analysis. These approaches are described and critically compared. Finally, in the last ten years, several advances regarding the possibility of endogenous HNO generation were made; some of them are also revised in the present work.
1. Introduction
Nitric oxide, NO˙, was initially known for being one of the environmentally polluting gasses. Since its identification as an endogenous molecule in humans as the endothelium derived relaxing factor (EDRF),1 countless studies followed to shed light on its important role in biological systems. Nowadays, it is well known, for example, that NO˙ is capable of selectively activating enzymes such as soluble guanylate cyclase (sGC), generating an increase in cyclic guanosine monophosphate (cGMP) levels, leading to cGMP-dependent signalling pathways.2 Azanone (HNO, nitroxyl), the one electron reduced and protonated congener of nitric oxide, also reacts with O2, thiols, hemoproteins, and transition metals. However, their overall chemical reactivity is different. For example, in aqueous solution, HNO dimerizes with a second-order rate constant of approximately 8 × 106 M−1 s−1 to produce hyponitrous acid, which eventually decomposes to produce nitrous oxide and water,3 making HNO detection difficult. There is evidence for very little NO dimerization in the gas state. However, in solution, or inside solid materials, the story might be different. For example, when confined within nanoporous carbon, nitric oxide is found to react completely to form the dimer, (NO)2, even though almost no dimers are present in the bulk gas phase in equilibrium with the pore phase.4 Houk et al. presented theoretical evidence for the role of the NO dimer in reactions of NO with nucleophiles, showing that the concentration of (NO)2 increases in aromatic environments.5 Also, properties predicted by theoretical studies were in excellent agreement with experimental studies based on Infrared and Raman spectroscopy in the gas phase.6HNO and NO˙ also react with each other, with a second-order rate constant of 6 × 106 M−1 s−1.7 Besides, HNO can be converted to NO˙ by relatively mild hydrogen atom abstractors, since the H–NO bond strength is only ∼50 kcal mol−1,8 making it a better hydrogen atom donor than many other biological antioxidants. Indeed, HNO is a potent antioxidant due to its ability to react with oxidizing radicals and generating NO˙,9 which can also react with and quench reactive radical species. Moreover, it has been established that HNO is capable of rapidly reducing a variety of biological oxidants including hemoproteins,10 metalloenzymes (such as superoxide dismutase),11 and flavins12 to give NO˙. In the last ten years, the reactions between HNO and several physiological species were studied.13,14 In all cases, the second-order rate constants were obtained with values ranging between 103 and 105 M−1 s−1.
Historically, direct HNO formation from the chemical reduction of NO˙ seemed unlikely to occur due to the rather negative reduction potential; however, this value is currently under review. Rocha et al. computed E°′ (NO˙, H+/HNO) = −0.161 V versus NHE at pH = 7,15 so, in this context, NO˙ could be reduced by biologically compatible species with reduction potentials between −0.3 and −0.5 V. The more negative redox potential of −0.55 V at pH 7 was derived in 2002 by Shafirovich and Lymar,3 by using ΔfG°(NOaq), the gas phase enthalpy of formation for 1HNO, the tabulated entropy S°(HNOgas) and approximations for free energy of formation of HNO in aqueous solution, ΔfG°(1HNOaq). In 2017 an efficient protocol was derived to obtain the solvation free energy of HNO using three different computational approaches.17 Therefore −0.161 V is probably a more exact value for E°′ (NO˙, H+/HNO).
| HNO | NO˙ | |
|---|---|---|
| Production | NOS byproduct | NOS |
| RSNO decomposition NH2OH oxidation | ||
| NO reduction | ||
| Direct effect – molecular targets | Thiols | Free radicals |
| Metal complexes (specially Fe3+) | Metal complexes (specially Fe2+) | |
| Indirect effects | Formation of oxidative species from reaction with O2 | RNOS formation: NO2, N2O3 and ONOO− |
| Physiological activity | Vasodilation | |
| Neuromodulation and neurotoxic in CNS | ||
| Favours myocardial contraction | Macrophage and immune response activation | |
| Protection against ischemia/reperfusion injuries | Mitochondrial hemeprotein regulation |
Both HNO and NO˙ share many pharmacological effects related to the cardiovascular system,16 and respiratory diseases17 among others. However, the biochemical pathways through which they act are quite different. A comparative summary of their biologically relevant chemisry is shown in Table 1. It is well established that NO˙ maintains normal cardiovascular functions via the activation of the sGC enzyme. In the case of HNO, it emerged as a possible biologically active compound in the mid-1980s due to studies related to cyanamide (H2NCN), a drug used to treat alcoholism.18 In a series of studies, cyanamide was shown to be oxidatively bioactivated (for example, by catalase/H2O2) to generate HNO that, in turn, was responsible for the inhibition of aldehyde dehydrogenase via modification of the cysteine thiolate active site. Further work showed that HNO released via cyanamide also resulted in vasorelaxation in rabbit thoracic aorta.19 Although it is debatable whether HNO is also an endogenously generated EDRF, it has also been proposed that HNO could be an endothelium-derived hyperpolarizing factor (EDHF).20
More recently, HNO has been shown to irreversibly hinder the activity of the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH). This makes HNO a possible anticancer agent, since most solid tumours use glycolysis as their main source of energy. Furthermore, recent studies show that breast cancer cells are affected by the presence of azanone through their interaction with the poly(ADPRibose) polymerase inhibitor (PARP).21 In addition to these biological effects, HNO has also been shown to affect mycobacterium tuberculosis growth by interfering with its general physiological state.22
Another therapeutic application for HNO in the cardiovascular system is its use as a powerful preconditioning agent, which helps to alleviate the negative consequences of an ischemic event and the consequent reperfusion injury. This injury is characterized by blood flow deprivation followed by necrosis and possible heart tissue hypoxia. Animal studies show that a dramatic decrease in infarct size was achieved by pretreating heart tissue with Angeli's salt (an HNO donor); however, HNO has also been shown to increase infarct size if administered during an ischemic event.23
Information on HNO donors that are in clinical trials for heart failure treatments is currently publicly available (Scheme 1).24 Cimlanod (BMS-986231, CXL-1427) represents a second generation donor that delivers HNO through a pH-dependent chemical breakdown when exposed to the neutral pH environment of the bloodstream, and as a consequence, generates positive lusitropic and inotropic effects, as well as vasodilator effects.25 CXL-1020 is a first-generation HNO donor that generates an inactive organic by-product, CXL-105. It was shown to produce beneficial vasodilatory, inotropic, and lusitropic effects in normal and heart failure animal models; however, due to injection-site toxicity, its development was halted.26
 | ||
| Scheme 1 Structures of HNO-donors currently in medical trials. | ||
Finally, a recent discovery on the pharmacological effects of HNO focusing on diabetes-related complications of the cardiovascular system was published.27 Oxidative stress induced by hyperglycemia impairs both endogenous NO˙ generation and its response capacity within the myocardium, which leads to complications for traditional NO˙ therapies. However, the inotropic and lusotropic responses to HNO are enhanced, demonstrating the potential role for therapeutic HNO administration in acute treatment of ischemia and/or heart failure in diabetics.
In this account, the latest achievements on HNO generation, coordination chemistry and detection will be reviewed, with special focus on contributions made by our group.
2. HNO generation
2.1 Solution
Until 2015, although HNO formation from the reaction of NO˙ with alcohols had been considered, for example, using oxidized polyolefins,28 ubiquinol29 and 3,4-dihydroxyphenylacetic acid (DOPAC);30 none of these studies provided its direct, unequivocal detection. Recently, we studied the reaction of NO˙ with aromatic and pseudoaromatic alcohols, the ascorbate anion (AscH−), phenol (PhOH), hydroquinone (HQ), and tyrosine (Y) through several approaches.31 The reaction occurred after mixing NO˙ with ascorbate and aromatic alcohols, but not with aliphatic ones like methanol, D-mannitol, or malic acid. HNO concentration was linearly dependent on both ROH and NO concentrations; the resulting bimolecular constants (keff) are reported in Table 2. Data shows that the diols (hydroquinone and ascorbate) react approximately five-ten times faster than phenols, with ascorbate being the fastest. Alcohol-derived radicals were detected as reaction intermediates by EPR spectroscopy. Additionally, we decided to inspect whether other vitamins bearing phenolic groups, such as α-tocopherol (vitamin E, absorbed and accumulated in humans), or over-the-counter medications, such as acetylsalicylic acid (aspirin) or acetaminophen (paracetamol), can undergo the reaction.32 We found that all three species are capable of producing HNO from NO˙. Aspirin, an aromatic acetyl ester, although initially incapable of reacting with NO˙ due to its protected –OH group, is transformed in the stomach into salicylic acid, whose reactivity towards NO˙ could be demonstrated. Piroxicam also produced HNO after reacting with NO˙.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nitrite ratio obtained for the reactions of NO with reducing agents
:nitrite ratio obtained for the reactions of NO with reducing agents
| Compound | Functional group | k eff (M−1 s−1) | NO2−: N2Ob | Ref. |
|---|---|---|---|---|
| a pH = 7.4, RT, anaerobic, in the presence of DPTA (diethylenetriamine-pentaacetic acid). b Estimated error is (±0.1). c Bovine serum albumin (BSA) is a high molecular weight protein (Mr ≈ 7 × 104) that has only one free thiol group. | ||||
| Benzenethiol | –SH | 110 ± 8 | 1.0 | 40 |
| Cysteine | 25 ± 6 | 1.2 | 40 | |
| BSAc | 0.6 | — | 42 | |
| H2S | 6300 ± 300 | 1.0 | 39 | |
| Ascorbic acid | –OH | 8.1 ± 0.4 | 1.2 | 31 |
| Hydroquinone | 6.0 ± 0.4 | 1.2 | 31 | |
| α-Tocopherol | 3.3 ± 0.4 | — | 32 | |
| Acetaminophen | 1.4 ± 0.6 | — | 32 | |
| Piroxicam | 0.26 ± 0.04 | — | 32 | |
| Isopropylamine | –NH2 | 0.070 ± 0.007 | 1.0 | 41 |
| Diethylamine | –NH | 0.030 ± 0.005 | 1.2 | 41 |
From these results, we proposed that these reactions occur via a proton-coupled nucleophilic attack (PCNA) by the alcohol on NO˙, producing an intermediate RO–N(H)O˙ species, which further decomposes to release HNO (Scheme 2).
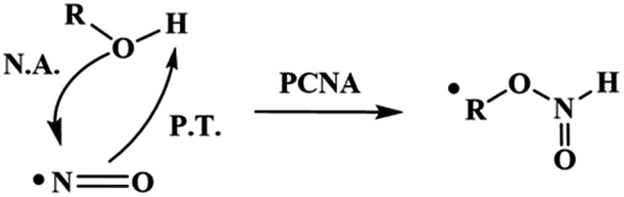 | ||
| Scheme 2 PCNA mechanism. PCNA: proton-coupled nucleophilic attack, P.T.: proton transfer, N.A.: nucleophilic attack. | ||
HNO, in turn, reacts with NO˙ to give nitrite, and with itself to give N2O.33 (eqn (1))
| 2HNO(g) → N2O(g) + H2O(l) | (1) |
The alkoxyl radicals, on the other hand, may react with another equivalent radical, such as tyrosine, which produces dityrosine, or with a second NO˙ molecule to produce an O-nitroso compound, as is the case for diols like ascorbate or hydroquinone.
Another question that arises regarding HNO generation is whether its production from the reaction of NO˙ with dihydrogen sulfide (H2S) and thiols is possible. H2S is a small gasotransmitter molecule, which has been shown to have cardioprotective effects on its own.34,35
We observed that HNO formation is first order in both reactants, H2S and NO. The reaction between these compounds could proceed via a direct one-electron transfer, promoting the reduction of NO˙ to HNO or, alternatively, HS− could attack NO in a proton-coupled nucleophilic attack to form HSNO˙-, as reported for the reaction of NO with alcohols.36 This contrasts with the studies performed by Cortese–Krott, who proposed that the main product of the direct reaction was SSNO−, evidenced by an absorption maximum at 412 nm.37 Recently, however, via the transnitrosation reaction (RSNO + R′SH ⇄ R′SNO + RSH), Marcolongo et al. observed that an absorption band at 412 nm reaches its full development one minute after mixing the RSNO/HS− reagents. Simultaneously, the appearance of NO˙ is observed after the initial RSNO reagent has already disappeared. From these data and the evidence that {(H)SNO} has a half-life of six seconds, they concluded that {(H)SNO} is the first intermediate in the transnitrosation reaction, and a precursor for SSNO−.38 Both the development of the absorption band at 412 nm following the reaction between H2S and NO˙, which depends on HS− concentration, and the delayed release of NO˙ are consistent with SSNO− formation. For more detail and discussion on the reactivity and interconversion of these nitrogen and sulfur species, the reader is referred to more specific works.38,39
On the other hand, previous studies showed that NO˙ can also react with thiols, leading to the formation of disulfides, N2O, and, finally, N2.30 In this context, we approached the reaction of NO˙ with thiols the same way as we did for alcohols.40 For this analysis, the reactions of NO˙ with 1-hexanethiol (R6-SH), cysteine (Cys), benzenethiol (Ph-SH), and benzeneselenol (Ph-SeH), were evaluated. The results revealed that HNO production rate varies in the following order: Ph-SeH > Ph-SH ≫ R6-SH > Cys. We also confirmed that the reaction is first order in both reagents. Cys and R6-SH, both aliphatic thiols, display similar reactivity, with effective rate constants approximately four times smaller than those observed for Ph-SH and Ph-SeH. This is because aromatics allow better stabilization of the unpaired spin.
Finally, in a similar way, we studied the reaction between alkylamines and NO˙.41 However, the obtained keff values for HNO production were between twenty and two hundred fifty times smaller than those obtained for other moderate NO˙ reducing agents (at pH 7.4). In all cases, the results presented in Table 2 show that N2O and NO2− are produced in approximately a 1:1 ratio, as expected considering the reaction between HNO and NO˙ (2):
| 2NO˙ + HNO → N2O and NO2− + N2O + H+ | (2) |
2.2 HNO donors
Azanone is impossible to store due to its high reactivity and inherent low stability. In consequence, its chemistry and effects are studied using HNO donor compounds, which release this molecule upon certain stimulation (chemical, photochemical, or thermal). For example, Angeli's Salt (Na2N2O3) releases HNO (and nitrite) at acidic pH (Scheme 3a), while there are many base-catalyzed donors, like Piloty's acid derivatives and pyrazolone-based compounds such as HAPY-1 (4-(N-hydroxylamino)-4-(acetyl-O-methyoxyoxime)-N-phenyl-3-methylpyrazolone) (Scheme 3b and c respectively).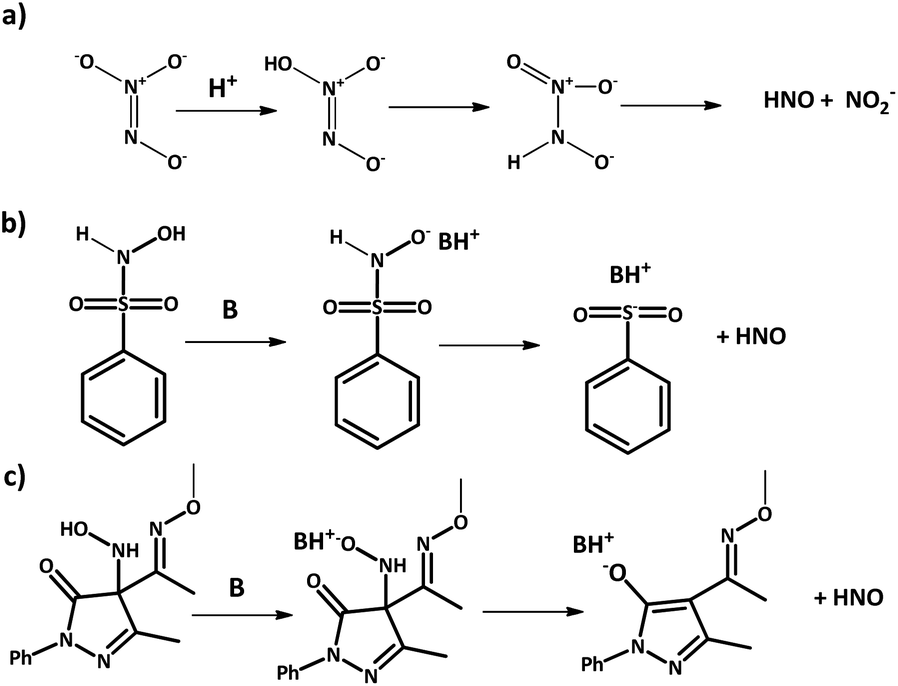 | ||
| Scheme 3 HNO generation from donors. (a) Angeli's salt; (b) Piloty's acid; (c) HAPY-1. | ||
In 2017, we showed that HNO release can be photochemically induced by irradiating Piloty's acid solutions with visible light in the presence of a Ru-bpy complex as an actuator.43 When the Ru complex is irradiated with visible light, amine ligands are released, increasing the pH of the solution. This, in turn, activates HNO release from Piloty's acid derivatives as discussed above. Light-driven azanone release had been previously reported using N-alkoxysulfonamide donors and Xe light irradiation.44 In this case, HNO was released due to an uncaging mechanism and not because of a pH shift. Other examples for photochemical HNO release have also been reported based on the same family of donors,45,46 caged Piloty's acid,47 and even {FeNO}6 complexes with a pendant thiol moiety.48 We would like to remind the reader that {MNO}n is the Enemark–Feltham notation, used to avoid assigning oxidation states in transition-metal nitric oxide (NO) complexes. The exponent “n” counts the total number of metal (d) and NO (π*) electrons.49
2.3 Gas phase
In a work in progress, we describe a new procedure for HNO generation in the gas phase, which consists of the heterogeneous phase reaction of a base-catalyzed solid HNO donor, (Piloty's acid derivatives and HAPY-1), with a gaseous base, such as ammonia.50 In this process, which does not need liquid phases or extreme experimental conditions in contrast to previous methods,51–55 HNO forms after the gaseous base reaches the solid surface and deprotonates the Piloty's acid derivative to give the sulfinite salt, in concordance with the decomposition mechanism in solution. At pressures around 1 bar and room temperature, HNO mainly dimerizes to yield water and N2O (eqn (1)), whose FT-IR signal can be detected once it has left the solid's surface. HNO generation could also be assessed by two different mass spectrometry methods.Kinetic measurements suggest that HNO production rate is affected by diffusion processes inside the solid structure, including both the penetration of the gaseous base and the escaping of the formed HNO into the gas phase. Although the overall kinetic process is somewhat complex, a rapid initial growth is observed during the first seconds, followed by a slower, linear growth. The first process is considered to be mainly dependent on gas-phase diffusion and mixing processes as well as the gaseous base's adsorption rate. Once an equilibrium base concentration has been reached over the solid's surface, N2O concentration begins to grow linearly until the available sites on the surface begin to become scarce. Surprisingly, the kinetic constant associated to the first process, 7 × 10−2 s−1, is similar to that obtained for HNO generation in solution from Piloty's acid derivatives at basic pH (4.4 × 10−2 s−1),56 and both could be fitted to a first-order regime. The reaction appears to be mostly a superficial process, with little diffusion of the gaseous base into the solid particles, since the N2O production yield was calculated as approximately 25% via FT-IR and NMR spectroscopic measurements, and so most of the solid remains unreactive. The synthesis and use of nanoparticulated Piloty's acid derivatives could therefore cause a dramatic increase in HNO production yield.
This economic and straightforward method for azanone generation in the gas phase may allow deepening the study of HNO gas-phase chemistry, and analyze, for example, its reactivity towards oxygen, which has remained an issue of certain controversy. In solution, while peroxynitrite has been proposed as a product for this reaction by some authors, others claim that NO˙ and the hydroperoxide radical are formed instead.57,58 The high reactivity of either possible species prevents more detailed studies, and so this new clean generation method in the gas phase might be of much use.59 Apart from probably allowing new reactivity studies, the controlled generation of HNO can give rise to a new drug delivery scheme for novel treatments, in which azanone may be inhaled instead of administered through donors in solution. In order to achieve these promising applications, of course, an appropriate system is to be constructed, in which the degree of dimerization can be controlled, oxidation is avoided, and, most importantly, the excess gaseous base is properly retained by using selective membranes or acid traps.60–63 Nitric oxide, for example, has already been successfully used as an inhaled therapeutic agent to treat cardiac as well as respiratory conditions, to the point of reducing the need for assisted respiration in some cases.64–70
2.4 Endogenous generation
The endogenous generation of NO˙ has been well established, since nitric oxide synthase (NOS), using NADH and dioxygen, generates NO˙ and citrulline from the amino acid L-arginine.71 Under hypoxic conditions, NO˙ can also be generated by nitrite reduction mediated by Cu or Fe metalloenzymes.72 In contrast, the endogenous generation of HNO remains a key question to be answered, in order to understand its possible biological functions. NO˙ reduction by alcohols, thiols, amines, and H2S were in fact shown to generate HNO, as discussed in Section 2.1. Other studies suggest that in the absence of the biopterin cofactor, NOS generates HNO from arginine.73,74Additionally, in vitro studies demonstrated that the reaction of nitrosothiols (RSNOs) with excess thiols promotes the formation of disulphide and HNO,75,76 while the reaction between RSNOs and ascorbate produces dehydroascorbate and HNO.77 The generation of HNO by the action of certain enzymes has also been proposed. For example, Fe or Mn superoxide dismutase (SOD) enzymes produces azanone from NO˙,78 while bacterial cytochrome C nitrite reductase catalyses the six-electron reduction of nitrite to ammonium via an HNO intermediate, coordinated to an heme centre.79,80 Moreover, some heme enzymes in the nitrogen cycle, such as Cyt P450nor, are proposed to function via HNO intermediates,81
In an interdisciplinary approach, taking advantage of selective methods for HNO detection under in vitro and intracellular conditions, we suggested that H2S could transform endogenous NO˙ into HNO in sensory neurons. These results were surprisingly identical to those observed in stimulation with HNO, observing a clear specific activation of the sensory channel of TRPA1 chemoreceptors through the formation of disulfide bonds in N-terminal cysteines, which activate the HNO-TRPA1-CGRP cascade (TRPA1: transient receptor potential channel A1; CGRP: calcitonin gene-related peptide).82 The endogenous formation of HNO could be observed using a fluorescent probe, which also showed that HNO production was inhibited by NO˙ synthase inhibitors and/or cystathionine beta-synthase (CBS) inhibitors.
Another possible source of endogenous HNO is based on the oxidation of different nitrogen containing species mediated by several hemoproteins, which are capable of stabilizing oxo-ferryl species, such as peroxidases.83–85 For example, we confirmed that myoglobin produces HNO via the peroxidation of hydroxylamine with excellent catalytic activity.86
Another proposal for the production of HNO in vivo results from the enzymatic activity of nitric oxide synthases (NOS) under particular conditions such as the absence of its biopterin cofactor.74 Other non-enzymatic pathways include reactions between biologically relevant molecules with NO˙, such as those named above, which lead to the formation of HNO and suggest its possible endogenous formation. However, this has not been definitively confirmed due to difficulties in unequivocal detection of HNO. In a collaborative work currently in progress, we determined the generation of HNO in human platelet-rich plasma using the electrochemical HNO sensor developed by our group. The results show the effective formation of HNO in platelets, after stimulating them with the addition of agonists such as adenosine diphosphate (ADP) and a synthetic peptide agonist of the PAR-1 thrombin receptor called PAR1-AP.87,88 Currently, HNO detection in this biological system is under study, using a selective phosphine-based fluorescent probe.89
3. Stabilization by metalloporphyrins and other ligands
3.1 Metalloporphyrins
Given the high reactivity of free HNO, researchers have tried to stabilize azanone and its conjugate base, NO−, by coordination to a metal center using a variety of ligands. In particular, heme proteins and synthetic porphyrins have been consistently explored due to the special biological relevance of such biomimetic complexes, which may resemble reactive intermediates in the reaction mechanisms of certain enzymes of the nitrogen cycle. Moreover, the interaction of HNO with heme active sites is closely related to the physiological effects exhorted by this gasotransmitter.Iron porphyrin derivatives are the clear protagonists, since the electronic properties of this metal center allows the formation of {FeNO}6, {FeNO}7 and {Fe(H)NO}8 complexes which can be interpreted as Fe(II)–NO+, Fe(II)–NO˙ or Fe(II)–NO−/HNO, and therefore there is a rich redox reactivity to be exploited.90 In contrast, manganese porphyrins only produce {MnNO}6 complexes which are not reduced to the {MnNO}7/8 forms, whereas cobalt porphyrins form uniquely {CoNO}8 species, and oxidation occurs on the porphyrin ring.91 Attempts to obtain the {CoHNO}8 derivatives, however, result in porphyrin ring protonation.92 Nevertheless, although {CoHNO}8 species have not been stabilized, the cationic porphyrin [Co(II)TMPyP]4+ has been shown to effectively catalyze the reduction of nitrite to ammonia, and so the formation of a HNO complex could be proposed as an intermediate.93
{Fe(H)NO}8 complexes might be prepared from their nitrosyl derivatives via chemical or electrochemical reduction, or via hydride attack on the {FeNO}6 species. The pioneering works of Kadish provided the first evidence for {FeNO}8 complexes in organic media, via spectroelectrochemical studies using TPP (meso-tetraphenyl porphyrin) and OEP (β-octaethyl porphyrin);94,95 they were later obtained by Ryan and coworkers via chemical reduction in THF, which enabled their FT-IR characterization.96,97 More recently, they also succeeded in obtaining the crystal structure of [Fe(II)(OEP)(NO)]−.98 In the presence of weak acids, evidence for the formation of Fe(II)(OEP)(HNO) (Fig. 1a) was also obtained,99 and this compound resulted stable for hours with excess phenol acting as the proton source.100 Abucayon and coworkers could prepare the hexacoordinated Fe(II)(OEP)(HNO)(5-MeIm) by hydride attack on the ferric nitrosyl, and characterize it via1H NMR at low temperature.101 Interestingly, in fact, this strategy had been previously exploited successfully for the preparation of the first HNO porphyrin complex, which featured Ru as the metal center and could also be characterized by FT-IR and 1H NMR, showing greater stability than its iron analogue.102
 | ||
| Fig. 1 Fe(II)NO− and Fe(II)HNO porphyrin complexes. | ||
Using the perhalogenated, electron poor porphyrin TFPPBr8 (2,3,7,8,12,13,17,18-octabromo-5,10,15,20-tetrakis-pentafluorophenyl)porphyrin to prevent oxidation, the first heme-model {FeNO}8 complex was isolated in our group via chemical reduction of the corresponding {FeNO}7 derivative.103 [Fe(II)TFPPBr8NO]− (Fig. 1b) resulted indefinitely stable in anoxic solution, and could be characterized via UV-Vis, FT-IR and 15N NMR spectroscopies, with this last technique being used for the first time for this kind of compounds. In a more recent work by Hu and Li, even its crystal structure (along with that of the {FeNO}7 derivative) could be elucidated.104 DFT calculations assigned its electronic structure to be intermediate between Fe(II)–NO− and Fe(I)–NO˙, as opposed to non-heme, predominantly Fe(II)–NO− complexes,105 as also proposed by Lehnert and coworkers.106 Despite this complex's remarkable stability, the protonated {FeHNO}8 adduct could not be observed, as it immediately yielded the {FeNO}7 precursor after acid addition. Reaction (3) with hydrogen production, previously proposed by Choi et al.,96 was therefore considered as a plausible decomposition mechanism in this case.
| FeII(P)HNO + H+ → FeII(P)NO˙ + 1/2H2 | (3) |
This electron-withdrawing platform also allowed for the exploration of a second reduction process, which was found to occur on the porphyrin ring to give a species best described as [{FeNO}7(TFPPBr8)4−]2−.107 This electronic configuration would account for the unexpected observed positive shift of ν(NO), that was predicted by DFT calculations, for intermediate- and high-spin states.
Using a sterically hindered bis-picket fence porphyrin, (3,5-Me-BAFP), Lehnert and coworkers could obtain the first HNO ferrous-heme model complex, which, additionally, resulted of great stability with a lifetime of several hours (Fig. 1c).106 Notably, their results supported the bimolecular decomposition pathway for {FeHNO}8 complexes, shown in eqn (4), since it is expected for this reaction to be disfavoured by the presence of the bulky substituents hindering the coordination site.
| 2FeII(P)HNO → 2FeII(P)NO + H2 | (4) |
In aqueous media, Lin and Farmer were first successful in stabilizing the HNO adduct of myoglobin, and obtaining its Raman, 15N and 1H NMR, and X-ray absorption spectra.108,109 This extensive characterization could be accomplished due to the remarkable long lifetime of this complex, which extended over weeks. Other globin-HNO adducts could also be prepared, evidencing the protective nature of the proteic environment.110
Almost two decades later, the {FeNO}7 derivative of the broadly studied, anionic water-soluble porphyrin TPPS (meso-tetraphenylsulphonate porphyrin) could be isolated in our laboratory.111 More interestingly, the first {FeNO}8 and {FeHNO}8 derivatives of a water-soluble model could be generated via chemical reduction, starting from carefully degassed solutions of the previously isolated Fe(II)–NO˙ complex and recrystallized sodium dithionite as the reducing agent.112 [Fe(II)TPPS(HNO)]4− (Fig. 1c) could be readily obtained at a UV-Vis scale at pH ≤ 9, as evidenced by the marked spectral changes, which in turn were consistent with early studies involving flash photolysis reductions of [Fe(II)TPPSNO˙]4− generated in situ.113 At more basic pH values, a subtle spectral change was observed after reduction, which suggested the formation of the deprotonated species, [Fe(II)TPPS(NO−)]5−; this could be properly confirmed via acid-base interconversion experiments. Excitingly, as both species could be identified, the pKa value for coordinated HNO could be determined as 9.7 via electrochemical as well as UV-Vis studies, by monitoring the pH dependence of the reduction potential of [Fe(II)TPPSNO˙]4−. The reduction potential for {FeNO}7/{FeNO}8 was estimated as −0.885 vs. Ag/AgCl, while at pH 6, ERED for {FeNO}7, H+/{FeHNO}8 was measured as −0.655 V vs. Ag/AgCl, in agreement with previous results obtained by Meyer and coworkers using [Fe(II)TPPSNO˙]4− prepared in situ.114 Due to the relative stabilization of the NO− moiety upon coordination, the pKa value of 9.7 is expectedly lower than the reported value for free HNO.3 Moreover, the estimated value is in agreement with the one reported for a Ru(II)–HNO complex,115 and falls in the 8–10 range proposed for Fe(II)OEP(HNO).100 It is important to note that this is the first pKa value obtained for a ferrous porphyrin biomimetic complex, suggesting that heme-HNO complexes might exist in the protonated form under physiological conditions.
Although both reduced species eventually reoxidize to the {FeNO}7 precursor, the protonated [Fe(II)TPPS(HNO)]4− complex appears to decay faster, as soon as it is formed, following a first order regime. Notably, these results do not agree with the previously proposed bimolecular reaction shown in eqn (4). The observed unimolecular constant, k = (0.017 ± 0.003) s−1, did not change significantly under a variety of experimental conditions, including pH, although a primary kinetic isotopic effect was observed when the reaction was conducted in D2O suggesting a rate-limiting step involving H–NO bond breaking. Furthermore, the addition of the H˙ abstracting radical TEMPO˙ to a cuvette containing freshly formed [Fe(II)TPPS(HNO)]4− resulted in immediate and complete conversion to the {FeNO}7 species, further evidencing the relative weakness of the H–NO bond. DFT calculations using the Fe(II)TPP(HNO) model suggested a feasible limiting step involving the migration of the H atom in the HNO moiety to the most proximal meso carbon in the porphyrin ligand. As a result, a phlorin radical intermediate is formed, only 2.4 kcal mol−1 more energetic than the starting azanone complex (Fig. 2).
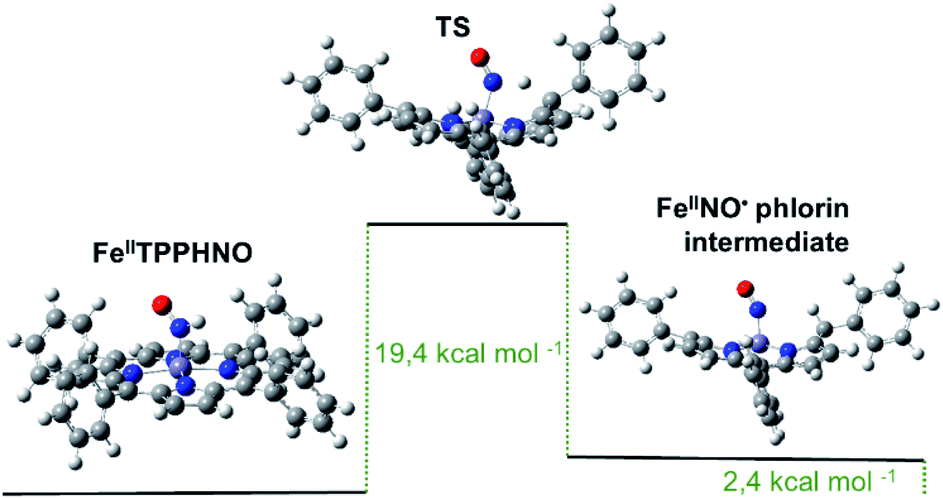 | ||
| Fig. 2 Optimized structures and calculated energy differences for Fe(II)TPP(HNO), the final phlorin intermediate and transition state. | ||
Additionally, the calculated activation energy for this process fell in the range of the experimentally estimated value. Similar proton-coupled electron transfers for other porphyrins have also been reported in recent works, in which phlorin intermediates were proposed and even detected.116,117 The obtained results altogether suggest a first-order decomposition mechanism, new for a {FeHNO}8 species, involving homolytic H–NO cleavage and phlorin radical formation as the rate-limiting step.
Although new information on {FeHNO}8 and {FeNO}8 complexes is disclosed every year, these species remain very elusive, and the fundamental reasons for their instability are not yet completely understood. The bimolecular reaction (4) might be a main decomposition pathway in organic media, while its relevance in aqueous media is not apparent from our results. In this case, although steric protection must also be considered, as shown by the fairly inert globin HNO adducts, hydrogen bonding apparently plays a crucial role in stabilizing the HNO moiety, as has been shown in theoretical studies for heme-protein systems.118,119 This is evidenced by the stability of the water soluble complex [Fe(II)(CN)5(HNO)]3−,120 and the relatively long lifetime of [Fe(II)TPPS(HNO)]4−, a compound based on an unhindered porphyrin in water, as compared to organic-soluble counterparts. The importance of hydrogen bonding has also been recently highlighted by Rahman and Ryan, where they report the existence of an equilibrium between the protonated Fe(II)(OEP)(HNO) complex and the hydrogen bonded Fe(II)(OEP)(NO−)⋯H–O–Ph complex in the presence of phenols.121 The electronic properties and saturation of the porphyrin ring also play determining roles in the reactivity of FeNO porphyrinates, which could even be responsible for the diverse reaction products yielded by different heme-based nitrite reductases, as reported recently by Amanullah and Dey.122 Evidently, further research should be pursued in all these directions in order to disclose the rich bioinorganic chemistry performed by iron porphyrin nitrosyl and azanone derivatives, which remain a very intriguing field.
3.2 Pincer ligand complexes
There have been no reports thus far of HNO stabilization by complexes with pincer ligands, although some reports of HNO release by these type of nitrosyl species do suggest the existence of intermediate HNO adducts. On the one hand, Caulton and coworkers report of the HNO release from the reaction between an osmium polihydride complex Os(PNP)(H)3 (Fig. 3a) and nitric oxide constitutes of a thorough example.123 In this reaction, as depicted in eqn (5), the loss of H2 is a key step which enabled the formation of an intermediate dinitrosyl hydride species (detected by FTIR and XRD) which then undergoes an insertion to form bound HNO. This last species, however, could not be detected, since HNO is reportedly released faster than the adduct is formed.| Os(PNP)(H)3 + 2NO˙ → Os(PNP)(NO) + HNO + H2 | (5) |
 | ||
| Fig. 3 Some structures of pincer (a and b) and other multidentate–ligand (c–e) complexes relevant to HNO stabilization. | ||
On the other hand, in our laboratory, hydride attack on a {RhNO}8 Rh(I)PCPNO+ complex124 produced the reduced {RhNO}9 Rh(I)PCPNO˙ species,125 also suggesting the intermediacy of an HNO complex with H2 production.126
NO− stabilization by complexes with pincer scaffolds is far better known. Several structural analyses of this kind of species exist in bibliography,124,127–133 along with reports of their catalytic activity.134–136 Within our group, several rhodium-based pincer ligand systems have been studied (the most relevant are highlighted in Fig. 3b). For such cases the formal assignation of oxidation states is not direct, and, through the use of different techniques, we have concluded that the combined analysis of the ν(NO) FTIR signal along with the XRD-determined value of the Rh–N–O angle is sufficient to identify NO− configurations.124 A PCN ligand system proved particularly prone to yield Rh(III)NO−, even leading to the stabilization of adducts with the weakly-coordinating anions PF6− and BF4−.137 Most of our studies on pincer nitrosyls focus on the exploration of their rich redox chemistry.125,137
3.3 Other complexes
There have been several studies of HNO/NO− stabilization for complexes based in multidentate ligands that cannot be formally classified neither as pincers nor porphyrins, albeit similar in structure. Much the same as for iron porphyrins, FeNO− electronic structures electronic structures are predominant for non-heme iron systems.138 Worth mentioning are cyclam-based structures105,139 and some other N-tetracoordinate chelate structures140–142 which have been found to stabilize NO− and to release HNO under certain conditions, suggesting the existence of an intermediate M–N(H)O species. Harrop and coworkers have reported a {FeNO}8 complex with potential HNO-donor ability (Fig. 3c),142 which yielded the reductive nitrosylation product of metmyoglobin upon exposure to it and showed inhibition for this same reaction in the presence of glutathione. However, the exact nature of the reactive species could not be verified, and mechanistic details have not yet been reported. In a subsequent work, they also studied the potential of another complex, this time of cobalt with a different type of N-tetracoordinate ligand (Fig. 3d), as an HNO-donor.141 In this last case, HNO could be trapped both by metmyoglobin and a manganese porphyrin, and could also be indirectly detected by an N2O FTIR signal (see Section 4.1) when the {CoNO}9 species was made to react in water. A CoII–HNO intermediate was putatively proposed to be involved in the release of HNO but could not be isolated. Interestingly, a dinitrosyl species {Co(NO)2}10 was also detected in these reactions and was proved to release HNO as well.On the subject of HNO stabilization, Hess and coworkers' study of a Ru complex with a pentacoordinate ligand is most remarkable, since it is one of the few systems where an HNO adduct could be fully characterized (Fig. 3e).143 This adduct was obtained by the reaction of [Ru (pybuS4)(NO)]+ with NaBH4 in methanol at 0 °C, and was characterized by 1H and 13C NMR, FTIR, mass spectrometry and XRD. Complex [Ru (pybuS4)(HNO)] was found to be highly reactive. It could not be deprotonated back to its NO− precursor with Brønsted bases such as triethylamine, butyllithium or lithium methoxide. However, reaction with Brønsted acids (H2PO3−, HBr and triflic acid) did yield [Ru (pybuS4)(NO)]+. This reaction was described as a 2e−/1H+ oxidation and is probably coupled to H2 formation.
Also of relevance are Slep and coworkers' works based on a family of ruthenium complexes with N-chelating ligands, which notably coordinate HNO.115,144,145 Moreover, the HNO coordinated species were stable enough for their pKa for HNO/NO− conversion to be experimentally obtained, as aforementioned, and clear correlations between HNO acidity and the electronic parameters of the coordination sphere could be established.144,145
4. HNO detection methods
4.1 Indirect detection methods
The detection of HNO is a challenging matter due to the molecule's inherent instability towards dimerization (eqn (1)) and its overall short lifetime. Historically, this tendency towards dimerization was exploited as a means to detect HNO through N2O by CG-MS and FT-IR,146–148 but later evidence of other azanone-independent mechanisms that could also lead to release of N2O proved this method to be less reliable.149,150 Some of the first detection methods that were devised as an alternative were mainly based on HNO trapping with different scavengers and monitoring by either UV-spectrophotometry or EPR. Thus, investigation on HNO trapping became extremely relevant.One of the most studied HNO traps in bibliography are metalloporphyrins, and our laboratory group has conducted broad research on this subject. Early reports of HNO trapping by these species included both hemoproteins10,108,151–153 as well as isolated porphyrins.154 These last type of heme-model systems are a powerful tool, since they essentially retain the reactivity of the hemoprotein active site but allow for a simpler manipulation and reactivity monitoring. In initial works, the trapping kinetics of Fe and Mn porphyrins were reported and it was established that, whereas Fe porphyrins reacted with azanone yielding the same product as for reductive nitrosylation with excess NO˙, Mn porphyrins had a much slower reaction -if any- with NO˙, therefore being capable of HNO/NO˙ discrimination.155–157 This was further investigated in a separate work where the trapping kinetics of several porphyrins was studied. The work led to the conclusion that manganese-porphyrins with the most negative potentials were the most suitable traps for azanone detection, since they enabled a pathway through free HNO, instead of proceeding via a direct donor–porphyrin interaction, together with a large Soret band shift.158 Other relevant studies were focused on improving the stability of the final trapping product by protecting it from air-induced oxidation, which represented the main limitation of the method. Our group worked on the insertion of the Mn porphyrin in a protein environment,159 whereas Shoenfisch and co-workers developed a gel-encapsulated Mn porphyrin system, both with great results.160 However, these colorimetric methods bear the disadvantage of monitoring a wavelength where most biological compounds typically absorb, and so interfere in the detection of azanone for in vivo analysis.
Thiols are also optimal for HNO trapping, partly because they have fast kinetics for its reaction with this molecule.161 It has long been established that the reaction of azanone with thiols leads to the formation of RS(O)NH2, which are very specific products that can be exploited as markers, even enabling HNO/NO˙ discrimination.151,162–166 Although these species have been successfully monitored through NMR,167 their detection by HPLC-UV is more useful due to its higher inherent sensitivity.163,166,168 It must be noted, however, that purification is required for UV detection of the marker.
Another successful trapping system, albeit far less frequently reported for its use in colorimetric detection, are phosphines. Most relevant is King and co-workers’ study on several phosphine-based traps which includes one of the few reports of an indirect colorimetric method that has enabled quantification of HNO in vitro.169
As far as EPR-monitoring is concerned, nitronyl nitroxides have been demonstrated to work as efficient paramagnetic probes capable of HNO/NO˙ discrimination.170,171 In particular, their encapsulation in liposomes has been proposed as a means to improve indirect HNO detection.172 A great advantage of this last method is that it could be suitable for biological systems, since no interference is expected and the reactants are protected from decomposition by the liposome matrix. However, no further experiments have been carried out to date on this matter. Some other works using iron dithiocarbamates as traps had also been previously reported, but their ability to discriminate between HNO and NO˙ is limited.173,174
Although these indirect methods have allowed a better understanding of the reactivity of the elusive HNO species, they have some significant flaws, mostly concerning their applicability on biological samples (for a summary, see Table 3).
| Detection | Trapping system | Advantages | Disadvantages |
|---|---|---|---|
| UV-Vis | Mn-porphyrin in a protein matrix159 | + Air stable | − Limited application in biological samples due to UV Soret band interference |
| + Good sensitivity (low nM level) | |||
| + HNO/NO discrimination | |||
| UV-Vis | Mn-porphyrin in a xerogel160 | + Air stable | − Limited application in biological samples due to UV Soret band interference |
| + Good sensitivity (low nM level) | − Cannot be reused due to gel aging | ||
| + HNO/NO discrimination | |||
| HPLC-UV | Thiol (glutathione)166 | + HNO/NO discrimination | − Requires previous purification |
| + Good sensitivity (low μM level) | − Formation of GSSG may interfere | ||
| + Can be applied to biological samples | |||
| UV-Vis | Phosphine carbamates169 | + Good sensitivity (low μM level) | − Limited application in biological samples due to phosphine hydrolysis |
| + HNO/NO discrimination | |||
| EPR | Liposome-encapsulated nitronyl nitroxides172 | + Potential compatibility with biological samples (no experiments have been carried out to date) | − Requires special and expensive equipment, which is not always readily available |
| + HNO/NO discrimination |
4.2 Direct detection methods: electrochemistry and mass spectrometry
As mentioned before, indirect methods for azanone detection are based on the detection of a certain reaction product formed due to HNO decomposition. However, direct methods for HNO detection have also been developed, which rely on mass spectrometry and electrochemistry.In 2011 HNO detection by MIMS was achieved by Toscano and coworkers, by adapting a specific sensor for nitric oxide.177,178 This method allowed the detection of HNO generated from azanone donors by the observation of the fragmented ion (NO+), whose presence was evidenced by a large peak at m/z 30. However, it was not always possible to unequivocally observe the signal at m/z 31. This signal can emerge from HNO generation or from naturally abundant 15NO˙ formed from N2O fragmentation. N2O control experiments were performed, and the m/z 30 to m/z 31 ratio was found to be 255:1. Therefore, if the observed ratio is smaller, it can be considered as evidence for direct HNO production. HNO generation rate produced via reaction of solid donors with gaseous ammonia (but not its yield) was found to be dependent on base injection rate. The ion corresponding to the dimerization product, N2O+, could also be observed at m/z 44. Although a detection limit of approximately 50 nM could be reached, additional experiments, such as HNO trapping, are necessary to corroborate the origin of the m/z 30 signal, since both NO˙ and fragmentations from N2O parent ions could also contribute to this signal. Recently, another method was validated for HNO detection which uses a high-pressure (1–1000 mbar) radiofrequency mass spectrometer. Due to a higher temperature of operation and a different setup, HNO dimerization was less favoured in this procedure, giving a very clear signal at m/z 31 due to HNO generation.179
On the other hand, as mentioned earlier, the reactivity of HNO towards synthetic metalloporphyrins containing divalent transition metals has been intensely studied, and so different colorimetric detection techniques mentioned above have been derived. In particular, Co(II) porphyrins, explored as isoelectronic models of protoheme oxygenases,180 have a high reactivity towards NO˙(g) to give Co(III)(Por)NO− in a few minutes.181 Correspondingly, the same complex is obtained when Co(III) porphyrins react directly with azanone; in contrast, the reaction of Co(III) with NO˙(g), gives the unstable Co(III)(Por)NO˙.182,183 This critical difference in reactivity, added to the ease and efficiency with which thiol derivative porphyrins can be covalently attached to metal electrodes (e.g., gold, silver),184,185 have allowed the use of cobalt(II) 5,10,15,20-tetrakis[3-(p-acetylthiopropoxy)phenyl]porphyrin [Co(P)], as a tool for the electrochemical discrimination between HNO and NO˙.186 In this context, our group has developed a specific time-resolved electrochemical sensor for HNO working at the nanomolar level, which is inert towards other biocompatible reactive oxygen and nitrogen oxide species (ROS and RNOS).187
This sensor operates at a fixed potential of 0.8 V vs. Ag°/AgCl, where [Co(P)] is stable and no current is observed. In the presence of HNO, Co(III)(P)NO− is produced, which oxidizes to Co(III)(P)NO˙ under these conditions. The resulting complex is unstable and releases NO˙, regenerating the original porphyrin and allowing the cycle to restart (Scheme 4). The current intensity obtained in the experiment is proportional to the amount of HNO present. This method is capable of quantitatively and selectively detecting HNO in real-time, with a linear response in the range 1–1000 nM.187,188
The HNO selective time-resolved electrochemical sensor has been used to verify the production of HNO from NO˙ mediated by H2S,189 ascorbate, tyrosine, and other alcohols.31 In addition, it provided clear evidence of the reaction of NO˙ with certain biological aromatic alcohols obtained from food, such as vitamin E, or used as over-the-counter medications, such as aspirin,32 and allowed to understand the kinetics behind the reactions of NO˙ with thiols to give HNO.40 A recent study confirmed HNO production by the myoglobin-mediated oxidation of hydroxylamine using this selective sensor.86
Compatibility of this sensor with biological media has also been evaluated: for example, HNO detection was achieved after the addition of ascorbate to immunostimulated macrophages.31
 | ||
| Scheme 4 Reactions Involved in the amperometrical detection of HNO by Co(P). | ||
In 2010, Lippard et al. developed the first copper(II)-based fluorescent probe, Cu(II)(BOT1)11 (BOT1 = BODIPY-triazole 1) for HNO detection (λem = 526 nm, ϕ = 0.12). One year later, Yao et al. reported another probe, coumarin-based Cu(II)(COT1)190 this time bearing a green-emitting chromophore (λem = 499 nm, ϕ = 0.63). Although these probes are also sensible to cysteine and ascorbate, the intracellular levels of these reductants in biological media are not enough to generate a fluorescent response comparable to those observed in the presence of HNO. A similar probe, Cu(II)(COET),191 based on a 7-diethylamine-coumarin fluorophore with a slightly modified chelating site was reported by Yao et al. in 2012. In parallel, Lippard and collaborators designed the benzoresorufin based Cu(II)(BRNO1–3) probes for detection of both HNO and NO˙.192 A few years later, the Cu(II)(HCD) probe based on N-(1H-1,2,3-triazole-4-yl)methyl-N,N-di (2-pyridylmethyl)amine was reported by Xing et al. to sense HNO and/or H2S depending on the probe's microenvironment, making it particularly interesting.193 Finally, a few fluorescent probes for HNO detection emitting in the infrared region were developed, such as dihydroxanthene-based Cu(II)(DHX1) (λem= 715 nm, ϕ = 0.05) whose emission spectrum does not overlap with other blue or green-emitting probes.194 This represents a substantial advantage, since they allow the generation of multi-coloured images and the simultaneous monitoring of two or more analytes inside the cell.195 In the recent years, many other Cu-based fluorescent probes for HNO detection have been developed, which have helped to understand the photophysical properties of these systems.196,197
Nitroxide compounds, which are organic compounds bearing an unpaired electron in an NO˙ motif, can also act as fluorogenic species, since they can act as quenchers when bound to a fluorophore. Nitroxides can be oxidized to the oxoammonium cations and are easily reduced by several compounds, including HNO, to yield the corresponding hydroxylamine. For instance, the reaction between TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxyl radical) and HNO gives TEMPOL-H and NO˙ via hydrogen abstraction from the HNO moiety.164 This method was used to design HNO detection probes which are unreactive towards NO˙.198
Phosphine-based probes have proven to possess greater selectivity and sensitivity than Cu(II) probes. In this case, the reductive ligation of HNO produces the release of a fluorescent chromophore.199–201 Recently developed phosphine based probes have also been able to image HNO in tumors,202 and simultaneously in the Golgi apparatus and mitochondria.203 Finally, a thiol based probe takes advantage of the special reactivity of HNO towards these groups, allowing its detection with a metal-free structure.204 For more detail on the structures and mechanism of action of these probes, the reader is referred to a recent review which summarizes the detection of HNO and other gasotransmitters using these fluorescent tools.205
In summary, the biological compatibility shown by fluorescent probes and electrochemical sensors give them an advantage for performing in vivo measurements, while MIMS requires the use of complementary techniques to ensure the unequivocal detection of HNO. On the other hand, the fact that most of the fluorogenic probes are based on the one-electron reducing nature of HNO can lead to possible interferences from biological reducing agents such as thiols or ascorbate. In contrast, the externally applied potential in the electrochemical sensor avoids this inconvenience, since the metal centre oxidation state is fixed as Co(III). HNO detection methods are summarised in Fig. 4.
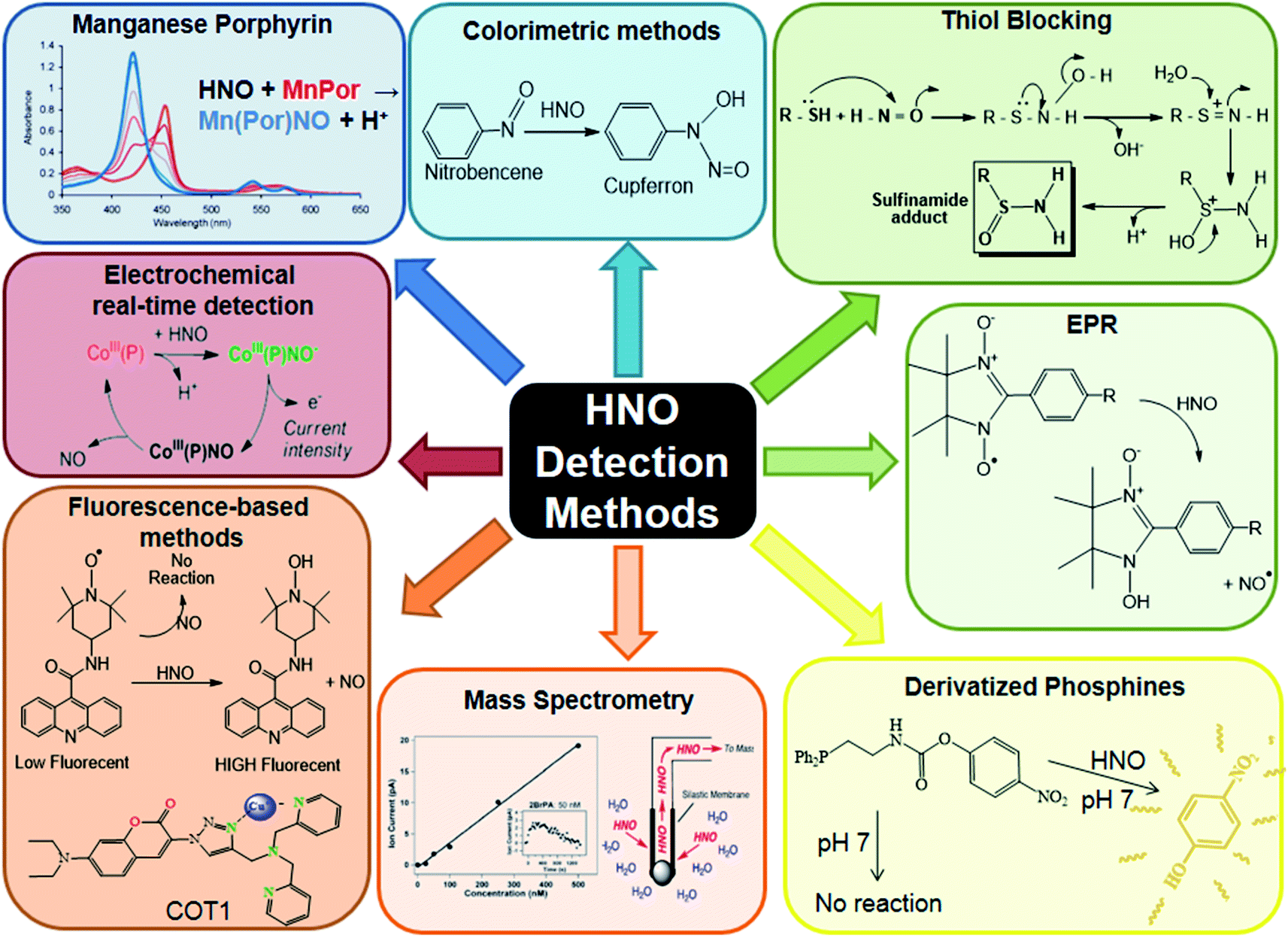 | ||
| Fig. 4 HNO detection methods. Reproduced from ref. 211. | ||
Perspectives and outlook
Less than ten years ago it was thought that NO˙ was impossible to reduce to HNO by biological agents. This has been assessed not only by state-of-the-art theoretical calculations, but also by showing experimentally that NO˙ can be reduced to HNO by mild reducing agents found in biological media, such as: aromatic and pseudo-aromatic alcohols, including Vitamins C and E, and medicaments such as aspirin, piroxicam and paracetamol.32 Thiols, H2S and HS− also produce HNO, at a faster rate, and aliphatic amines react slow at room temperature and ambient pressure. Therefore, not only HNO can be produced by these agents, but would allow the persistence of HNO in reducing biological environments once produced by nitric oxide reduction or other reactions, and its transportation in free form. Azanone could even be transported, for example, by reduced hemes present in the body.111 Moreover, HNO endogenous formation is not only a possibility, but a reality: it has been observed to form spontaneously by human platelet aggregation.87,88,206All these findings were made thanks to the use of HNO trapping with metalloporphyrins (an indirect method),155 and the use of an electrochemical HNO sensor developed in 2013,187 which allows the direct HNO detection in real time, allowing to obtain kinetic data.
The mechanistic issues related to NO˙ reduction and HNO formation are still open: recent findings suggest the possibility of two successive additions of NO to the RXH substrate, or even the direct reaction with the dimer (NO)2.207 On one hand, known reactions of NO˙ such as its oxidation with O2,208 and its reaction with SO32−,209 are thought to proceed through this type of mechanism. On the other hand, it has been found that the formation of (NO)2 is favored in the presence of aromatic rings or negatively charged molecules such as CH3S−.5 Moreover, the reaction of S2− with (NO)2 is exothermic by as much as 84 kcal mol−1, as shown by theoretical calculations.207 Although S2− is not present at measurable concentration in aqueous solutions, even at high pH values, the reaction could be carried out in organic solvents or by reaction of solid Na2S with NO. This is not a biologically relevant issue, but a chemical one: the product would be “Thio-Angeli's salt” ON![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NSO2−, which can be expected to be a new HNO donor, such as the well-known Angeli's salt (ONNO22−).
NSO2−, which can be expected to be a new HNO donor, such as the well-known Angeli's salt (ONNO22−).
Regarding medical uses, HNO dimerizes in solution, but in gas phase at low concentrations that reaction is expected to be slow. Therefore, it could be used for respiratory treatments (nowadays NO˙ is used for COVID-19 infections and other pulmonary diseases) or heart treatment: Bristol–Myers–Squibb is developing HNO donors to avoid cardiac arrest.210 Moreover, the chemistry of HNO(g) is not well known. Since the N–H bond is weak, it could produce NO˙ in the gas phase at low pressures. Therefore, both HNO and NO˙ would be administered with just one solid HNO donor, by reaction with a gas such as NH3, ethylamine, or HCl, which could be easily removed by a trapping agent.179
In summary, azanone arrived to the scene to be the non-radical and reducing offspring of nitric oxide, with properties which are different from his father's, but quite interesting and potentially useful, no doubt at all.
Author contributions
CMG, AM and PV: investigation and writing – original draft; SS, JP and FD: writing-review & editing; FD: conceptualization and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The acknowledgements come at the end of an article after the conclusions and before the notes and references. The research reported in this publication was supported by the Ministerio de Ciencia y Tecnología e Innovación Productiva (PICT-2015-3854 and PICT-2017-1930) and the Universidad de Buenos Aires (UBACYT) project # 20020170100595BA. All authors are CONICET members. C. G., A. M., and P. V. are doctoral students, while S. S., J. P. and F. D. are faculty researchers.Notes and references
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci., 1987, 84, 9265–9269 CrossRef CAS PubMed.
- S. Moncada, R. M. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109–142 CAS.
- V. Shafirovich and S. V. Lymar, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7340 CrossRef CAS PubMed.
- D. Srivastava, C. H. Turner, E. E. Santiso and K. E. Gubbins, J. Phys. Chem. B, 2018, 122, 3604–3614 CrossRef CAS PubMed.
- Y.-L. Zhao, M. D. Bartberger, K. Goto, K. Shimada, T. Kawashima and K. N. Houk, J. Am. Chem. Soc., 2005, 127, 7964–7965 CrossRef CAS PubMed.
- J. Ivanic, M. W. Schmidt and B. Luke, J. Chem. Phys., 2012, 137, 214316 CrossRef PubMed.
- S. V. Lymar, V. Shafirovich and G. A. Poskrebyshev, Inorg. Chem., 2005, 44, 5212–5221 CrossRef CAS PubMed.
- J. R. B. Gomes, M. D. M. C. Ribeiro da Silva and M. A. V. Ribeiro da Silva, J. Phys. Chem. A, 2004, 108, 2119–2130 CrossRef CAS.
- B. Lopez, M. Shinyashiki, T. Han and J. Fukuto, Free Radicals Biol. Med., 2007, 42, 482–491 CrossRef CAS PubMed.
- D. A. Bazylinski and T. C. Hollocher, J. Am. Chem. Soc., 1985, 107, 7982–7986 CrossRef CAS.
- J. Rosenthal and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 5536–5537 CrossRef CAS PubMed.
- J. M. Fukuto, A. J. Hobbs and L. J. Ignarro, Biochem. Biophys. Res. Commun., 1993, 196, 707–713 CrossRef CAS PubMed.
- R. Smulik-Izydorczyk, A. Mesjasz, A. Gerbich, J. Adamus, R. Michalski and A. Sikora, Nitric Oxide, 2017, 69, 61–68 CrossRef CAS PubMed.
- M. I. Jackson, T. H. Han, L. Serbulea, A. Dutton, E. Ford, K. M. Miranda, K. N. Houk, D. A. Wink and J. M. Fukuto, Free Radicals Biol. Med., 2009, 47, 1130–1139 CrossRef CAS PubMed.
- M. Venâncio, F. Doctorovich and W. Rocha, J. Phys. Chem. B, 2017, 121, 6618–6625 CrossRef PubMed.
- N. Paolocci, M. I. Jackson, B. E. Lopez, K. Miranda, C. G. Tocchetti, D. A. Wink, A. J. Hobbs and J. M. Fukuto, Pharmacol. Ther., 2007, 113, 442–458 CrossRef CAS PubMed.
- F. L. M. Ricciardolo, P. J. Sterk, B. Gaston and G. Folkerts, Physiol. Rev., 2004, 84, 731–765 CrossRef CAS PubMed.
- E. G. DeMaster, F. N. Shirota and H. T. Nagasawa, Alcohol, 1985, 2, 117–121 CrossRef CAS PubMed.
- J. Fukuto, P. Gulati and H. T. Nagasawa, Biochem. Pharmacol., 1994, 47, 922–924 CrossRef CAS PubMed.
- J. C. Irvine, R. H. Ritchie, J. L. Favaloro, K. L. Andrews, R. E. Widdop and B. K. Kemp-Harper, Trends Pharmacol. Sci., 2008, 29, 601–608 CrossRef CAS PubMed.
- P. Ramakrishnan Geethakumari, M. J. Schiewer, K. E. Knudsen and Wm. K. Kelly, Curr. Treat. Option On., 2017, 18, 37 CrossRef PubMed.
- J. Galizia, M. P. Acosta, E. Urdániz, M. A. Martí and M. Piuri, Tuberculosis, 2018, 109, 35–40 CrossRef CAS PubMed.
- P. Pagliaro, D. Mancardi, R. Rastaldo, C. Penna, D. Gattullo, K. M. Miranda, M. Feelisch, D. A. Wink, D. A. Kass and N. Paolocci, Free Radicals Biol. Med., 2003, 34, 33–43 CrossRef CAS PubMed.
- B. K. Kemp-Harper, J. D. Horowitz and R. H. Ritchie, Drugs, 2016, 76, 1337–1348 CrossRef CAS PubMed.
- J. C. Hartman, C. L. del Rio, J. E. Reardon, K. Zhang and H. N. Sabbah, JACC, 2018, 3, 625–638 Search PubMed.
- S. R. Roof, Y. Ueyama, R. Mazhari, R. L. Hamlin, J. C. Hartman, M. T. Ziolo, J. E. Reardon and C. L. del Rio, Front. Physiol., 2017, 8, 894 CrossRef PubMed.
- C. X. Qin, J. Anthonisz, C. H. Leo, N. Kahlberg, A. Velagic, M. Li, E. Jap, O. L. Woodman, L. J. Parry, J. D. Horowitz, B. K. Kemp-Harper and R. H. Ritchie, Antioxid. Redox Signaling, 2020, 32, 60–77 CrossRef CAS PubMed.
- D. J. Carlsson, R. Brousseau, C. Zhang and D. M. Wiles, Polyolefin oxidation: Quantification of alcohol and hydroperoxide products by nitric oxide reactions, 1987, vol. 17 Search PubMed.
- M. Graetzel, S. Taniguchi and A. Henglein, Ber. Bunsen-Ges., 1970, 74, 1003–1010 CAS.
- W. Pryor, D. Church and C. Govindan, J. Org., 1982, 156–159 CrossRef CAS.
- S. A. Suarez, N. I. Neuman, M. Muñoz, L. Álvarez, D. E. Bikiel, C. D. Brondino, I. Ivanović-Burmazović, J. Lj. Miljkovic, M. R. Filipovic, M. A. Martí and F. Doctorovich, J. Am. Chem. Soc., 2015, 137, 4720–4727 CrossRef CAS PubMed.
- M. Hamer, S. A. Suarez, N. I. Neuman, L. Alvarez, M. Muñoz, M. A. Marti and F. Doctorovich, Inorg. Chem., 2015, 54, 9342–9350 CrossRef CAS PubMed.
- F. Doctorovich, D. Bikiel, J. Pellegrino, S. A. Suárez, A. Larsen and M. A. Martí, Coord. Chem. Rev., 2011, 255, 2764–2784 CrossRef CAS.
- Q. C. Yong, S. W. Lee, C. S. Foo, K. L. Neo, X. Chen and J.-S. Bian, Am. J. Physiol.: Heart Circ. Physiol., 2008, 295, H1330–H1340 CrossRef CAS PubMed.
- J. W. Elrod, J. W. Calvert, J. Morrison, J. E. Doeller, D. W. Kraus, L. Tao, X. Jiao, R. Scalia, L. Kiss, C. Szabo, H. Kimura, C.-W. Chow and D. J. Lefer, Proc. Natl. Acad. Sci., 2007, 104, 15560–15565 CrossRef CAS PubMed.
- I. Ivanovic-Burmazovic and M. R. Filipovic, Inorg. Chem., 2019, 58, 4039–4051 CrossRef CAS PubMed.
- M. M. Cortese-Krott, G. G. C. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci., 2015, 112(34), E4651–E4660 CrossRef CAS PubMed.
- J. P. Marcolongo, M. F. Venâncio, W. R. Rocha, F. Doctorovich and J. A. Olabe, Inorg. Chem., 2019, 58, 14981–14997 CrossRef CAS PubMed.
- S. A. Suarez, P. Vargas and F. A. Doctorovich, J. Inorg. Biochem., 2021, 216, 111333 CrossRef CAS PubMed.
- S. A. Suarez, M. Muñoz, L. Alvarez, M. F. Venâncio, W. R. Rocha, D. E. Bikiel, M. A. Marti and F. Doctorovich, J. Am. Chem. Soc., 2017, 139, 14483–14487 CrossRef CAS PubMed.
- M. Hamer, S. Suarez, M. Muñoz, L. Álvarez, M. Marti and F. Doctorovich, Pure Appl. Chem., 2020, 92(12), 2005–2014 CrossRef CAS.
- C. T. Aravindakumar, M. De Ley and J. Ceulemans, J. Chem. Soc., Perkin Trans. 2, 2002, 663–669 RSC.
- G. Carrone, J. Pellegrino and F. Doctorovich, Chem. Commun., 2017, 53, 5314–5317 RSC.
- Y. Zhou, R. B. Cink, R. S. Dassanayake, A. J. Seed, N. E. Brasch and P. Sampson, Angew. Chem., 2016, 128, 13423–13426 CrossRef.
- Y. Zhou, R. B. Cink, Z. A. Fejedelem, M. Cather Simpson, A. J. Seed, P. Sampson and N. E. Brasch, Eur. J. Org. Chem., 2018, 2018, 1745–1755 CrossRef CAS.
- Y. Zhou, R. B. Cink, A. J. Seed, M. C. Simpson, P. Sampson and N. E. Brasch, Org. Lett., 2019, 21, 1054–1057 CrossRef CAS PubMed.
- M. Kawaguchi, T. Tani, R. Hombu, N. Ieda and H. Nakagawa, Chem. Commun., 2018, 54, 10371–10374 RSC.
- C.-K. Chiang, K.-T. Chu, C.-C. Lin, S.-R. Xie, Y.-C. Liu, S. Demeshko, G.-H. Lee, F. Meyer, M.-L. Tsai, M.-H. Chiang and C.-M. Lee, J. Am. Chem. Soc., 2020, 142, 8649–8661 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- G. Carrone, A. Mazzeo, E. Marceca, J. Pellegrino, J. Zarenkiewicz, J. P. Toscano and F. Doctorovich, J. Inorg. Biochem., 2021, 111535 CrossRef CAS.
- M. Bodenstein, Angew. Chem., 1927, 40, 174–177 CrossRef CAS.
- P. Harteck, Ber. Dtsch. Chem. Ges., 1933, 66, 423–426 CrossRef.
- A. v. Nagel, Zeitschrift für Elektrochemie und angewandte physikalische Chemie, 1930, 36, 754–757 Search PubMed.
- M. a. A. Clyne and B. A. Thrush, Trans. Faraday Soc., 1961, 57, 69–78 RSC.
- M. a. A. Clyne and B. A. Thrush, Discuss. Faraday Soc., 1962, 33, 139–148 RSC.
- K. Sirsalmath, S. A. Suárez, D. E. Bikiel and F. Doctorovich, J. Inorg. Biochem., 2013, 118, 134–139 CrossRef CAS PubMed.
- M. G. Bryukov, A. A. Kachanov, R. Timonnen, J. Seetula, J. Vandoren and O. M. Sarkisov, Chem. Phys. Lett., 1993, 208, 392–398 CrossRef CAS.
- R. Smulik, D. Dębski, J. Zielonka, B. Michałowski, J. Adamus, A. Marcinek, B. Kalyanaraman and A. Sikora, J. Biol. Chem., 2014, 289, 35570–35581 CrossRef CAS PubMed.
- M. Hamer, M. A. Morales Vásquez and F. Doctorovich, in The Chemistry and Biology of Nitroxyl (HNO), ed. F. Doctorovich, P. J. Farmer and M. A. Marti, Elsevier, Boston, 2017, pp. 1–9 Search PubMed.
- P. Wang and T.-S. Chung, J. Membr. Sci., 2015, 474, 39–56 CrossRef CAS.
- Z. Ding, L. Liu, Z. Li, R. Ma and Z. Yang, J. Membr. Sci., 2006, 286, 93–103 CrossRef CAS.
- Z. Xie, T. Duong, M. Hoang, C. Nguyen and B. Bolto, Water Res., 2009, 43, 1693–1699 CrossRef CAS PubMed.
- M. S. EL-Bourawi, M. Khayet, R. Ma, Z. Ding, Z. Li and X. Zhang, J. Membr. Sci., 2007, 301, 200–209 CrossRef CAS.
- R. A. Krasuski, J. J. Warner, A. Wang, J. K. Harrison, V. F. Tapson and T. M. Bashore, J. Am. Coll. Cardiol., 2000, 36, 2204–2211 CrossRef CAS PubMed.
- E. Öztürk, S. Haydin, İ. C. Tanıdır, İ. Özyılmaz, Y. Ergül, E. Erek, A. Güzeltaş, E. Ödemiş and İ. Bakır, Turk Kardiyol Dern Ars, 2016, 44, 196–202 Search PubMed.
- United States Patent, 6358536-Nitric oxide donor compositions, methods, apparatus, and kits for preventing or alleviating vasoconstriction or vasospasm in a mammal, 2002 Search PubMed.
- J. Á. Monsalve-Naharro, E. Domingo-Chiva, S. García Castillo, P. Cuesta-Montero and J. M. Jiménez-Vizuete, Farm. Hosp., 2017, 41, 292–312 Search PubMed.
- H. Gerlach, D. Pappert, K. Lewandowski, R. Rossaint and K. J. Falke, Intensive Care Med., 1993, 19, 443–449 CrossRef CAS PubMed.
- L. Chen, P. Liu, H. Gao, B. Sun, D. Chao, F. Wang, Y. Zhu, G. Hedenstierna and C. G. Wang, Clin. Infect. Dis., 2004, 39, 1531–1535 CrossRef CAS PubMed.
- A. P. Hunt, A. E. Batka, M. Hosseinzadeh, J. D. Gregory, H. K. Haque, H. Ren, M. E. Meyerhoff and N. Lehnert, ACS Catal., 2019, 9, 7746–7758 CrossRef CAS PubMed.
- D. J. Stuehr, Annu. Rev. Pharmacol. Toxicol., 1997, 37, 339–359 CrossRef CAS PubMed.
- A. Machha and A. N. Schechter, Nutr. Rev., 2012, 70, 367–372 CrossRef PubMed.
- Y. Ishimura, Y. T. Gao, S. P. Panda, L. J. Roman, B. S. S. Masters and S. T. Weintraub, Biochem. Biophys. Res. Commun., 2005, 338, 543–549 CrossRef CAS PubMed.
- H. H. H. W. Schmidt, H. Hofmann, U. Schindler, Z. S. Shutenko, D. D. Cunningham and M. Feelisch, Proc. Natl. Acad. Sci., 1996, 93, 14492–14497 CrossRef CAS PubMed.
- D. R. Arnelle and J. S. Stamler, Arch. Biochem. Biophys., 1995, 318, 279–285 CrossRef CAS PubMed.
- L. V. Ivanova, D. Cibich, G. Deye, M. R. Talipov and Q. K. Timerghazin, ChemBioChem, 2017, 18, 726–738 CrossRef CAS PubMed.
- M. Kirsch, A.-M. Büscher, S. Aker, R. Schulz and H. de Groot, Org. Biomol. Chem., 2009, 7, 1954–1962 RSC.
- D. a. Wink, J. a Cook, S. Y. Kim, Y. Vodovotz, R. Pacelli, M. C. Krishna, a Russo, J. B. Mitchell, D. Jourd’heuil, a M. Miles and M. B. Grisham, J. Biol. Chem., 1997, 272, 11147–11151 CrossRef CAS PubMed.
- D. Bykov, M. Plog and F. Neese, JBIC, J. Biol. Inorg. Chem., 2014, 19, 97–112 CrossRef CAS PubMed.
- O. Einsle, A. Messerschmidt, R. Huber, P. M. H. Kroneck and F. Neese, J. Am. Chem. Soc., 2002, 124, 11737–11745 CrossRef CAS PubMed.
- N. Lehnert, H. T. Dong, J. B. Harland, A. P. Hunt and C. J. White, Nat. Rev. Chem., 2018, 2, 278–289 CrossRef CAS.
- M. Eberhardt, M. Dux, B. Namer, J. Miljkovic, N. Cordasic, C. Will, T. I. Kichko, J. de la Roche, M. Fischer, S. A. Suárez, D. Bikiel, K. Dorsch, A. Leffler, A. Babes, A. Lampert, J. K. Lennerz, J. Jacobi, M. A. Martí, F. Doctorovich, E. D. Högestätt, P. M. Zygmunt, I. Ivanovic-Burmazovic, K. Messlinger, P. Reeh and M. R. Filipovic, Nat. Commun., 2014, 5, 1–17 Search PubMed.
- K. M. Miranda, R. W. Nims, D. D. Thomas, M. G. Espey, D. Citrin, M. D. Bartberger, N. Paolocci, J. M. Fukuto, M. Feelisch and D. A. Wink, J. Inorg. Biochem., 2003, 93, 52–60 CrossRef CAS PubMed.
- M. G. Espey, K. M. Miranda, D. D. Thomas and D. A. Wink, Free Radicals Biol. Med., 2002, 33, 827–834 CrossRef CAS PubMed.
- J. A. Reisz, E. Bechtold and S. B. King, Dalton Trans., 2010, 39, 5203–5212 RSC.
- L. Álvarez, S. A. Suárez, P. J. González, C. D. Brondino, F. Doctorovich and M. A. Martí, Inorg. Chem., 2020 DOI:10.1021/acs.inorgchem.9b02750.
- M. C. Maccaferro, S. B. Suárez Freire, S. Suárez, M. A. Martí, P. C. Ivani, M. A. Schattner and R. G. Pozner, presented in part at the XIII Congreso Argentino de Hemostasia y Trombosis y IV Curso Educacional de la ISTH, Buenos Aires, September, 2018 Search PubMed.
- Y. Doctorovich, S. Suárez, F. Doctorovich, M. Schattner and R. Pozner, XIV Congreso, Argentino de Hemostasia y Trombosis, Buenos Aires, 2021 Search PubMed.
- Z. Miao, J. A. Reisz, S. M. Mitroka, J. Pan, M. Xian and S. B. King, Bioorg. Med. Chem. Lett., 2015, 25, 16–19 CrossRef CAS PubMed.
- A. L. Speelman and N. Lehnert, Acc. Chem. Res., 2014, 47, 1106–1116 CrossRef CAS PubMed.
- E. Fujita, C. K. Chang and J. Fajer, J. Am. Chem. Soc., 1985, 107, 7665–7669 CrossRef CAS.
- M. A. Rhine, A. V. Rodrigues, R. J. B. Urbauer, J. L. Urbauer, T. L. Stemmler and T. C. Harrop, J. Am. Chem. Soc., 2014, 136, 12560–12563 CrossRef CAS PubMed.
- S.-H. Cheng and Y. Su, Inorg. Chem., 1994, 33, 5847–5854 CrossRef CAS.
- D. Lancon and K. M. Kadish, J. Am. Chem. Soc., 1983, 105, 5610–5617 CrossRef CAS.
- L. W. Olson, D. Schaeper, D. Lancon and K. M. Kadish, J. Am. Chem. Soc., 1982, 104, 2042–2044 CrossRef CAS.
- I. K. Choi, Y. Liu, D. W. Feng, K. J. Paeng and M. D. Ryan, Inorg. Chem., 1991, 30, 1832–1839 CrossRef CAS.
- Z. Wei and M. D. Ryan, Inorg. Chem., 2010, 49, 6948–6954 CrossRef CAS PubMed.
- N. Kundakarla, S. Lindeman, Md. H. Rahman and M. D. Ryan, Inorg. Chem., 2016, 55, 2070–2075 CrossRef CAS PubMed.
- Y. Liu and M. D. Ryan, J. Electroanal. Chem., 1994, 368, 209–219 CrossRef CAS.
- Md. H. Rahman and M. D. Ryan, Inorg. Chem., 2017, 56, 3302–3309 CrossRef CAS PubMed.
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, J. Am. Chem. Soc., 2016, 138, 104–107 CrossRef CAS PubMed.
- J. Lee and G. B. Richter-Addo, J. Inorg. Biochem., 2004, 98, 1247–1250 CrossRef CAS PubMed.
- J. Pellegrino, S. E. Bari, D. E. Bikiel and F. Doctorovich, J. Am. Chem. Soc., 2010, 132, 989–995 CrossRef CAS PubMed.
- B. Hu and J. Li, Angew. Chem., Int. Ed., 2015, 54, 10579–10582 CrossRef CAS PubMed.
- R. G. Serres, C. A. Grapperhaus, E. Bothe, E. Bill, T. Weyhermüller, F. Neese and K. Wieghardt, J. Am. Chem. Soc., 2004, 126, 5138–5153 CrossRef CAS PubMed.
- L. E. Goodrich, S. Roy, E. E. Alp, J. Zhao, M. Y. Hu and N. Lehnert, Inorg. Chem., 2013, 52, 7766–7780 CrossRef CAS PubMed.
- J. Pellegrino, R. Hübner, F. Doctorovich and W. Kaim, Chem.–Eur. J., 2011, 17, 7868–7874 CrossRef CAS PubMed.
- R. Lin and P. J. Farmer, J. Am. Chem. Soc., 2000, 122, 2393–2394 CrossRef CAS.
- C. E. Immoos, F. Sulc, P. J. Farmer, K. Czarnecki, D. F. Bocian, A. Levina, J. B. Aitken, R. S. Armstrong and P. A. Lay, J. Am. Chem. Soc., 2005, 127, 814–815 CrossRef CAS PubMed.
- M. R. Kumar, D. Pervitsky, L. Chen, T. Poulos, S. Kundu, M. S. Hargrove, E. J. Rivera, A. Diaz, J. L. Colón and P. J. Farmer, Biochemistry, 2009, 48, 5018–5025 CrossRef CAS PubMed.
- A. Mazzeo, J. Pellegrino and F. Doctorovich, J. Am. Chem. Soc., 2019, 141, 18521–18530 CrossRef CAS PubMed.
- C. E. McKenna, W. G. Gutheil and W. Song, Biochim. Biophys. Acta, 1991, 1075, 109–117 CrossRef CAS.
- H. Seki, M. Hoshino and S. Kounose, J. Chem. Soc., Faraday Trans., 1996, 92, 2579–2583 RSC.
- M. H. Barley and T. J. Meyer, J. Am. Chem. Soc., 1986, 108, 5876–5885 CrossRef CAS PubMed.
- N. O. Codesido, T. Weyhermüller, J. A. Olabe and L. D. Slep, Inorg. Chem., 2014, 53, 981–997 CrossRef CAS PubMed.
- Y. Fang, Y. G. Gorbunova, P. Chen, X. Jiang, M. Manowong, A. A. Sinelshchikova, Y. Yu. Enakieva, A. G. Martynov, A. Yu. Tsivadze, A. Bessmertnykh-Lemeune, C. Stern, R. Guilard and K. M. Kadish, Inorg. Chem., 2015, 54, 3501–3512 CrossRef CAS PubMed.
- B. H. Solis, A. G. Maher, D. K. Dogutan, D. G. Nocera and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 485–492 CrossRef CAS PubMed.
- L. Yang, Y. Ling and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 13814–13817 CrossRef CAS PubMed.
- R. L. Khade, Y. Yang, Y. Shi and Y. Zhang, Angew. Chem., Int. Ed., 2016, 55, 15058–15061 CrossRef CAS PubMed.
- A. C. Montenegro, V. T. Amorebieta, L. D. Slep, D. F. Martín, F. Roncaroli, D. H. Murgida, S. E. Bari and J. A. Olabe, Angew. Chem., Int. Ed., 2009, 48, 4213–4216 CrossRef CAS PubMed.
- M. H. Rahman, Y. Liu and M. D. Ryan, Inorg. Chem., 2019, 58, 13788–13795 CrossRef CAS PubMed.
- S. Amanullah and A. Dey, Chem. Sci., 2020, 11, 5909–5921 RSC.
- J.-H. Lee, H. Fan, M. Pink and K. G. Caulton, New J. Chem., 2007, 31, 838–840 RSC.
- C. Gaviglio, Y. Ben-David, L. J. Shimon, F. Doctorovich and D. Milstein, Organometallics, 2009, 28, 1917–1926 CrossRef CAS.
- J. Pellegrino, C. Gaviglio, D. Milstein and F. Doctorovich, Organometallics, 2013, 32, 6555–6564 CrossRef CAS.
- Unpublished results.
- A. Y. Verat, M. Pink, H. Fan, B. C. Fullmer, J. Telser and K. G. Caulton, Eur. J. Inorg. Chem., 2008, 2008, 4704–4709 CrossRef.
- B. C. Fullmer, M. Pink, H. Fan, X. Yang, M.-H. Baik and K. G. Caulton, Inorg. Chem., 2008, 47, 3888–3892 CrossRef CAS PubMed.
- E. Fogler, I. Efremenko, M. Gargir, G. Leitus, Y. Diskin-Posner, Y. Ben-David, J. M. L. Martin and D. Milstein, Inorg. Chem., 2015, 54, 2253–2263 CrossRef CAS PubMed.
- A. M. Tondreau and J. M. Boncella, Polyhedron, 2016, 116, 96–104 CrossRef CAS.
- D. Himmelbauer, M. Mastalir, B. Stöger, L. F. Veiros, M. Pignitter, V. Somoza and K. Kirchner, Inorg. Chem., 2018, 57, 7925–7931 CrossRef CAS PubMed.
- D. Himmelbauer, B. Stöger, L. F. Veiros, M. Pignitter and K. Kirchner, Organometallics, 2019, 38, 4669–4678 CrossRef CAS.
- J. Pecak, W. Eder, B. Stöger, S. Realista, P. N. Martinho, M. J. Calhorda, W. Linert and K. Kirchner, Organometallics, 2020, 39, 2594–2601 CrossRef CAS PubMed.
- A. Choualeb, A. J. Lough and D. G. Gusev, Organometallics, 2007, 26, 3509–3515 CrossRef CAS.
- E. Fogler, M. A. Iron, J. Zhang, Y. Ben-David, Y. Diskin−Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Inorg. Chem., 2013, 52, 11469–11479 CrossRef CAS PubMed.
- J. Pecak, B. Stöger, M. Mastalir, L. F. Veiros, L. P. Ferreira, M. Pignitter, W. Linert and K. Kirchner, Inorg. Chem., 2019, 58, 4641–4646 CrossRef CAS PubMed.
- C. M. Gallego, C. Gaviglio, Y. Ben-David, D. Milstein, F. Doctorovich and J. Pellegrino, Dalton Trans., 2020, 49, 7093–7108 RSC.
- A. L. Speelman, B. Zhang, C. Krebs and N. Lehnert, Angew. Chem., Int. Ed., 2016, 55, 6685–6688 CrossRef CAS PubMed.
- D. R. Lang, J. A. Davis, L. G. F. Lopes, A. A. Ferro, L. C. G. Vasconcellos, D. W. Franco, E. Tfouni, A. Wieraszko and M. J. Clarke, Inorg. Chem., 2000, 39, 2294–2300 CrossRef CAS PubMed.
- L. M. Baltusis, K. D. Karlin, H. N. Rabinowitz, J. C. Dewan and S. J. Lippard, Inorg. Chem., 1980, 19, 2627–2632 CrossRef CAS.
- M. R. Walter, S. P. Dzul, A. V. Rodrigues, T. L. Stemmler, J. Telser, J. Conradie, A. Ghosh and T. C. Harrop, J. Am. Chem. Soc., 2016, 138, 12459–12471 CrossRef CAS PubMed.
- A. K. Patra, K. S. Dube, B. C. Sanders, G. C. Papaefthymiou, J. Conradie, A. Ghosh and T. C. Harrop, Chem. Sci., 2012, 3, 364–369 RSC.
- D. Sellmann, T. Gottschalk-Gaudig, D. Häußinger, F. W. Heinemann and B. A. Hess, Chem.–Eur. J., 2001, 7, 2099–2103 CrossRef CAS.
- N. Levin, N. O. Codesido, J. P. Marcolongo, P. Alborés, T. Weyhermüller, J. A. Olabe and L. D. Slep, Inorg. Chem., 2018, 57, 12270–12281 CrossRef CAS PubMed.
- J. Perdoménico, M. M. Ruiz, N. O. Codesido, A. G. D. Candia, J. P. Marcolongo and L. D. Slep, Dalton Trans., 2021, 50, 1641–1650 RSC.
- K. M. Miranda, Coord. Chem. Rev., 2005, 249, 433–455 CrossRef CAS.
- H. T. Nagasawa, E. G. DeMaster, B. Redfern, F. N. Shirota and D. J. W. Goon, J. Med. Chem., 1990, 33, 3120–3122 CrossRef CAS PubMed.
- J. M. Fukuto, M. D. Bartberger, A. S. Dutton, N. Paolocci, D. A. Wink and K. N. Houk, Chem. Res. Toxicol., 2005, 18, 790–801 Search PubMed.
- J. Y. Cho, A. Dutton, T. Miller, K. N. Houk and J. M. Fukuto, Arch. Biochem. Biophys., 2003, 417, 65–76 CrossRef CAS PubMed.
- J. Yoo and J. M. Fukuto, Biochem. Pharmacol., 1995, 50, 1995–2000 CrossRef CAS PubMed.
- M. P. Doyle, S. N. Mahapatro, R. D. Broene and J. K. Guy, Oxidation and reduction of hemoproteins by trioxodinitrate(II), The role of nitrosyl hydride and nitrite, accessed 21 July 2020, DOI:10.1021/ja00210a047.
- E. H. Lan, B. C. Dave, J. M. Fukuto, B. Dunn, J. I. Zink and J. S. Valentine, J. Mater. Chem., 1999, 9, 45–53 RSC.
- F. Sulc, C. E. Immoos, D. Pervitsky and P. J. Farmer, J. Am. Chem. Soc., 2004, 126, 1096–1101 CrossRef CAS PubMed.
- I. Spasojević, I. Batinić-Haberle and I. Fridovich, Nitric Oxide, 2000, 4, 526–533 CrossRef PubMed.
- S. E. Bari, M. A. Martí, V. T. Amorebieta, D. A. Estrin and F. Doctorovich, J. Am. Chem. Soc., 2003, 125, 15272–15273 CrossRef CAS PubMed.
- M. A. Martí, S. E. Bari, D. A. Estrin and F. Doctorovich, J. Am. Chem. Soc., 2005, 127, 4680–4684 CrossRef PubMed.
- S. A. Suárez, M. A. Martí, P. M. De Biase, D. A. Estrin, S. E. Bari and F. Doctorovich, Polyhedron, 2007, 26, 4673–4679 CrossRef.
- L. Álvarez, S. A. Suarez, D. E. Bikiel, J. S. Reboucas, I. Batinić-Haberle, M. A. Martí and F. Doctorovich, Inorg. Chem., 2014, 53, 7351–7360 CrossRef PubMed.
- I. Boron, S. A. Suárez, F. Doctorovich, M. A. Martí and S. E. Bari, J. Inorg. Biochem., 2011, 105, 1044–1049 CrossRef CAS PubMed.
- K. P. Dobmeier, D. A. Riccio and M. H. Schoenfisch, Anal. Chem., 2008, 80, 1247–1254 CrossRef CAS PubMed.
- R. Smulik-Izydorczyk, K. Dębowska, J. Pięta, R. Michalski, A. Marcinek and A. Sikora, Free Radicals Biol. Med., 2018, 128, 69–83 CrossRef CAS PubMed.
- P. S.-Y. Wong, J. Hyun, J. M. Fukuto, F. N. Shirota, E. G. DeMaster, D. W. Shoeman and H. T. Nagasawa, Biochemistry, 1998, 37, 5362–5371 CrossRef CAS PubMed.
- D. W. Shoeman, F. N. Shirota, E. G. DeMaster and H. T. Nagasawa, Alcohol, 2000, 20, 55–59 CrossRef CAS PubMed.
- K. M. Miranda, N. Paolocci, T. Katori, D. D. Thomas, E. Ford, M. D. Bartberger, M. G. Espey, D. A. Kass, M. Feelisch, J. M. Fukuto and D. A. Wink, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9196 CrossRef CAS PubMed.
- B. Shen and A. M. English, Biochemistry, 2005, 44, 14030–14044 CrossRef CAS PubMed.
- S. Donzelli, M. G. Espey, D. D. Thomas, D. Mancardi, C. G. Tocchetti, L. A. Ridnour, N. Paolocci, S. B. King, K. M. Miranda, G. Lazzarino, J. M. Fukuto and D. A. Wink, Free Radicals Biol. Med., 2006, 40, 1056–1066 CrossRef CAS PubMed.
- G. Keceli, C. D. Moore, J. W. Labonte and J. P. Toscano, Biochemistry, 2013, 52, 7387–7396 CrossRef CAS PubMed.
- S. Donzelli, M. G. Espey, W. Flores-Santana, C. H. Switzer, G. C. Yeh, J. Huang, D. J. Stuehr, S. B. King, K. M. Miranda and D. A. Wink, Free Radicals Biol. Med., 2008, 45, 578–584 CrossRef CAS PubMed.
- J. A. Reisz, C. N. Zink and S. B. King, J. Am. Chem. Soc., 2011, 133, 11675–11685 CrossRef CAS PubMed.
- U. Samuni, Y. Samuni and S. Goldstein, J. Am. Chem. Soc., 2010, 132, 8428–8432 CrossRef CAS PubMed.
- Y. Samuni, U. Samuni and S. Goldstein, J. Inorg. Biochem., 2013, 118, 155–161 CrossRef CAS PubMed.
- A. A. Bobko, A. Ivanov and V. V. Khramtsov, Free Radical Res., 2013, 47, 74–81 CrossRef CAS PubMed.
- Y. Xia, A. J. Cardounel, A. F. Vanin and J. L. Zweier, Free Radicals Biol. Med., 2000, 29, 793–797 CrossRef CAS PubMed.
- A. M. Komarov, D. A. Wink, M. Feelisch and H. H. H. W. Schmidt, Free Radicals Biol. Med., 2000, 28, 739–742 CrossRef CAS PubMed.
- G. Hoch and B. Kok, Arch. Biochem. Biophys., 1963, 101, 160–170 CrossRef CAS PubMed.
- C. Tu, E. R. Swenson and D. N. Silverman, Free Radicals Biol. Med., 2007, 43, 1453–1457 CrossRef CAS PubMed.
- M. R. Cline, C. Tu, D. N. Silverman and J. P. Toscano, Free Radicals Biol. Med., 2011, 50, 1274–1279 CrossRef CAS PubMed.
- T. A. Chavez and J. P. Toscano, in The Chemistry and Biology of Nitroxyl (HNO), ed. F. Doctorovich, P. J. Farmer and M. A. Martí, Elsevier, 2017, pp. 255–267 Search PubMed.
- G. Carrone, A. Mazzeo, J. Pellegrino, S. Suárez, E. Marceca, J. Zarenkiewicz, and J. P. Toscano, submitted.
- W. R. Scheidt and J. L. Hoard, J. Am. Chem. Soc., 1973, 95, 8281–8288 CrossRef CAS PubMed.
- S. Kelly, D. Lancon and K. M. Kadish, Inorg. Chem., 1984, 23, 1451–1458 CrossRef CAS.
- P. J. Farmer and F. Sulc, J. Inorg. Biochem., 2005, 99, 166–184 CrossRef CAS PubMed.
- F. Roncaroli and R. van Eldik, J. Am. Chem. Soc., 2006, 128, 8042–8053 CrossRef CAS PubMed.
- K. Shimazu, M. Takechi, H. Fujii, M. Suzuki, H. Saiki, T. Yoshimura and K. Uosaki, Thin Solid Films, 1996, 273, 250–253 CrossRef CAS.
- T. Akiyama, H. Imahori and Y. Sakata, Chem. Lett., 1994, 23, 1447–1450 CrossRef.
- S. A. Suárez, M. H. Fonticelli, A. A. Rubert, E. de la Llave, D. Scherlis, R. C. Salvarezza, M. A. Martí and F. Doctorovich, Inorg. Chem., 2010, 49, 6955–6966 CrossRef PubMed.
- S. A. Suárez, D. E. Bikiel, D. E. Wetzler, M. A. Martí and F. Doctorovich, Anal. Chem., 2013, 85, 10262–10269 CrossRef PubMed.
- B. F. Doctorovich, S. A. Suárez and M. F. Martí, HNO Biosensor, 2018 Search PubMed.
- M. Eberhardt, M. Dux, B. Namer, J. Miljkovic, N. Cordasic, C. Will, T. I. Kichko, J. de la Roche, M. Fischer, S. A. Suárez, D. Bikiel, K. Dorsch, A. Leffler, A. Babes, A. Lampert, J. K. Lennerz, J. Jacobi, M. A. Martí, F. Doctorovich, E. D. Högestätt, P. M. Zygmunt, I. Ivanovic-Burmazovic, K. Messlinger, P. Reeh and M. R. Filipovic, Nat. Commun., 2014, 5, 4381 CrossRef CAS PubMed.
- Y. Zhou, K. Liu, J.-Y. Li, Y. Fang, T.-C. Zhao and C. Yao, Org. Lett., 2011, 13, 1290–1293 CrossRef CAS PubMed.
- Y. Zhou, Y.-W. Yao, J.-Y. Li, C. Yao and B.-P. Lin, Sens. Actuators, B, 2012, 174, 414–420 CrossRef CAS.
- U.-P. Apfel, D. Buccella, J. J. Wilson and S. J. Lippard, Inorg. Chem., 2013, 52, 3285–3294 CrossRef CAS PubMed.
- H. Lv, R. Ma, X. Zhang, M. Li, Y. Wang, S. Wang and G. Xing, Tetrahedron, 2016, 72, 5495–5501 CrossRef CAS.
- A. T. Wrobel, T. C. Johnstone, A. Deliz Liang, S. J. Lippard and P. Rivera-Fuentes, J. Am. Chem. Soc., 2014, 136, 4697–4705 CrossRef CAS PubMed.
- P. Rivera-Fuentes and S. J. Lippard, Acc. Chem. Res., 2015, 48, 2927–2934 CrossRef CAS PubMed.
- D. Maiti, A. S. M. Islam, A. Dutta, M. Sasmal, C. Prodhan and M. Ali, Dalton Trans., 2019, 48, 2760–2771 RSC.
- A. S. M. Islam, M. Sasmal, D. Maiti, A. Dutta, S. Ganguly, A. Katarkar, S. Gangopadhyay and M. Ali, ACS Appl. Bio Mater., 2019, 2, 1944–1955 CrossRef CAS.
- M. R. Cline and J. P. Toscano, J. Phys. Org. Chem., 2011, 24, 993–998 CrossRef CAS.
- J. A. Reisz, C. N. Zink and S. B. King, J. Am. Chem. Soc., 2011, 133, 11675–11685 CrossRef CAS PubMed.
- Z. Miao and S. B. King, in The Chemistry and Biology of Nitroxyl (HNO), ed. F. Doctorovich, P. J. Farmer and M. A. Martí, Elsevier, 2017, pp. 225–238 Search PubMed.
- K. Kawai, N. Ieda, K. Aizawa, T. Suzuki, N. Miyata and H. Nakagawa, J. Am. Chem. Soc., 2013, 135, 12690–12696 CrossRef CAS PubMed.
- Z. Chai, D. Liu, X. Li, Y. Zhao, W. Shi, X. Li and H. Ma, Chem. Commun., 2021, 57, 5063–5066 RSC.
- H. Wang, C. Liu, Z. He, P. Li, W. Zhang, W. Zhang and B. Tang, Anal. Chem., 2021, 93, 6551–6558 CrossRef CAS PubMed.
- N. W. Pino, J. Davis, Z. Yu and J. Chan, J. Am. Chem. Soc., 2017, 139, 18476–18479 CrossRef CAS PubMed.
- J. Alday, A. Mazzeo and S. Suarez, Inorg. Chim. Acta, 2020, 510, 119696 CrossRef CAS.
- P. C. Ivani, Tesis de Grado, Universidad de Buenos Aires. Facultad de Ciencias Exactas y Naturales, 2017 Search PubMed.
- N. I. Neuman, M. F. Venâncio, W. R. Rocha, D. E. Bikiel, S. A. Suárez and F. A. Doctorovich, submitted.
- D. S. Bohle, Curr. Opin. Chem. Biol., 1998, 2, 194–200 CrossRef CAS PubMed.
- D. Littlejohn, K. Y. Hu and S. G. Chang, Inorg. Chem., 1986, 25, 3131–3135 CrossRef CAS.
- United States Patent, 10517847-Nitroxyl donors with improved therapeutic index, 2019 Search PubMed.
- F. Doctorovich, D. E. Bikiel, J. Pellegrino, S. A. Suárez and M. A. Martí, Acc. Chem. Res., 2014, 47(10), 2907–2919 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |