
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Current applications of kinetic resolution in the asymmetric synthesis of substituted pyrrolidines
Sian S.
Berry
and
Simon
Jones
 *
*
Department of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield S3 7HF, UK. E-mail: simon.jones@sheffield.ac.uk
First published on 17th November 2021
Abstract
Chiral substituted pyrrolidines are key elements in various biologically active molecules and are therefore valuable synthetic targets. One traditional method towards enantiomerically pure compounds is the application of kinetic resolution. In this review, current KR methodology used in the synthesis of substituted pyrrolidines is surveyed, including enzymatic methods, cycloadditions and reduction of ketones.
 Sian Berry | Sian Berry was born in London, UK in 1995. Sian received her undergraduate degree from the University of Sheffield, completing her final year project in 2018 working on the asymmetric reduction of α-imino esters under the supervision of Professor Simon Jones. She began her Ph.D. studies in the same year in Prof Jones’ laboratory on the asymmetric synthesis of amino acids via trichlorosilane mediated reductions. |
 Simon Jones | Professor Jones obtained his BSc Chemistry from Southampton University and Ph.D. from University of Wales, Cardiff. He worked in postdoctoral positions at Arizona State University and at the Dyson Perrins Laboratory, University of Oxford, before starting his first academic position at the University of Newcastle upon Tyne. He moved to the University of Sheffield in 2003, where he is now Professor of Chemistry. His research interests lie in asymmetric organic synthesis and in construction of biological probes. |
1. Introduction
Pyrrolidines are present in many pharmaceuticals and natural products (Fig. 1).1–4 The combination of a structural scaffold that has defined shape, together with functional groups that allow for further diversification leading to arrays of products, makes them a popular target for synthesis. Current synthetic routes to construct these systems are numerous and varied, including cycloaddition and cyclization reactions, metal catalysed C–H activations, and reductions of enamines, alkenes, and various carbonyl compounds, to name but a few.5–10 A prominent strategy that exists amongst these methods is the use of kinetic resolution (KR) derived protocols. The use of these in synthetic chemistry have been long documented and can offer significant advantages in strategies where asymmetric synthetic techniques cannot be employed because of poor substrate compatibility or lack of appropriate methodology. In simplistic terms, KR involves the resolution of a racemic mixture employing a chemical transformation that differentiates between the two enantiomers of substrate based on differences in relative reaction rates of each enantiomer. Thus, in traditional KR, one enantiomer is transformed to a different product, leaving one enantiomer untouched. This immediately brings challenges of separating the transformed product from the starting material and also limits the maximum yield to 50% unless recycling protocols can be introduced. Innovations in this strategy that include dynamic kinetic resolution (DKR) and parallel kinetic resolution (PKR) variants that offer increased yield over traditional processes by circumventing the need for separation and/or loss of starting material (Scheme 1).11–14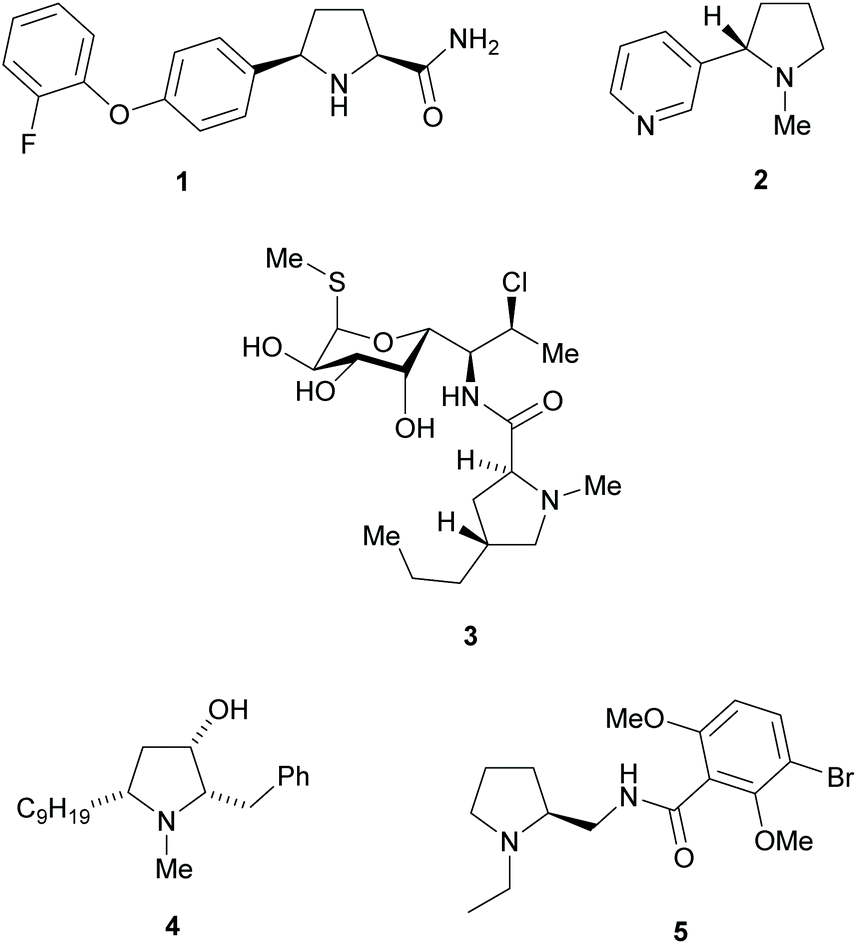 | ||
| Fig. 1 Pyrrolidine containing natural products and pharmaceuticals, Vixotrigine 1, Nicotine 2, Clindamycin 3, (+)-Preussin 4 and Remoxipride 5. | ||
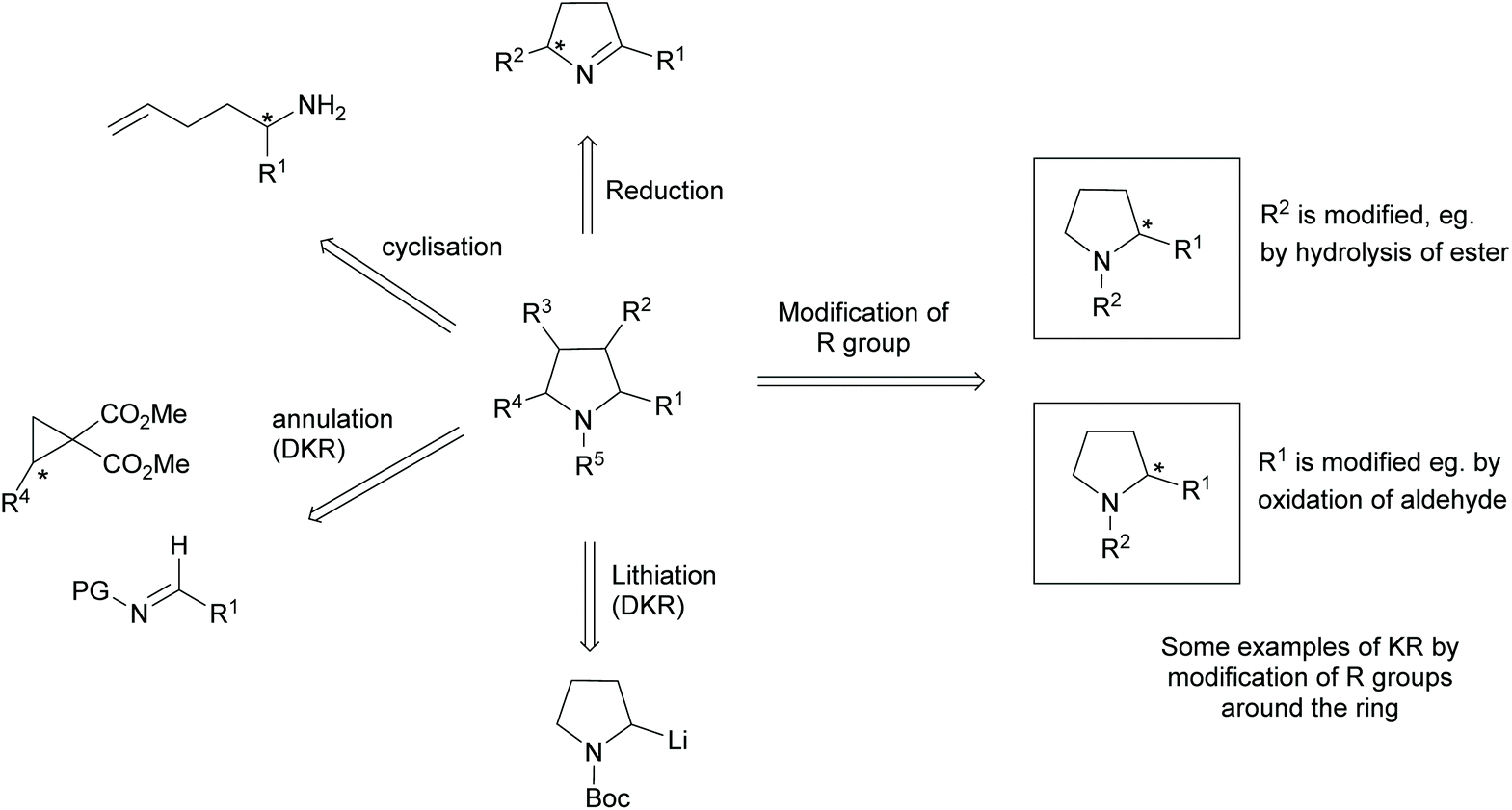 | ||
| Scheme 1 Examples of kinetic resolutions in pyrrolidine synthesis. | ||
In this review, the current KR methods involved in the asymmetric synthesis of pyrrolidines are reviewed categories by the substitution of their respective parent pyrrolidines framework.
2. Monosubstituted pyrrolidines
In 1994, Beak et al., reported on the asymmetric deprotonation of N-Boc-pyrrolidines via lithiation/substitution employing (−)-sparteine and sec-butyllithium (sBuLi), providing access to 2-substituted-N-tert-butylcarbonyl-pyrrolidines (N-Boc).15 It was noted that upon warming the enantioselectivity decreased suggesting racemization of the organolithium species at higher temperatures.16 From this, Coldham and co-workers developed a dynamic kinetic resolution (DKR) employing chiral diamine ligand L1 (Scheme 2).17,18 Transmetalation of stannane 6 with excess nBuLi, addition of chiral ligand L1 with nBuLi, followed by slow addition of trimethylsilyl chloride (TMSCl) at −20 °C formed 2-substituted-pyrrolidine 7 in a moderate 54% yield but with good enantioselectivity of 92% ee.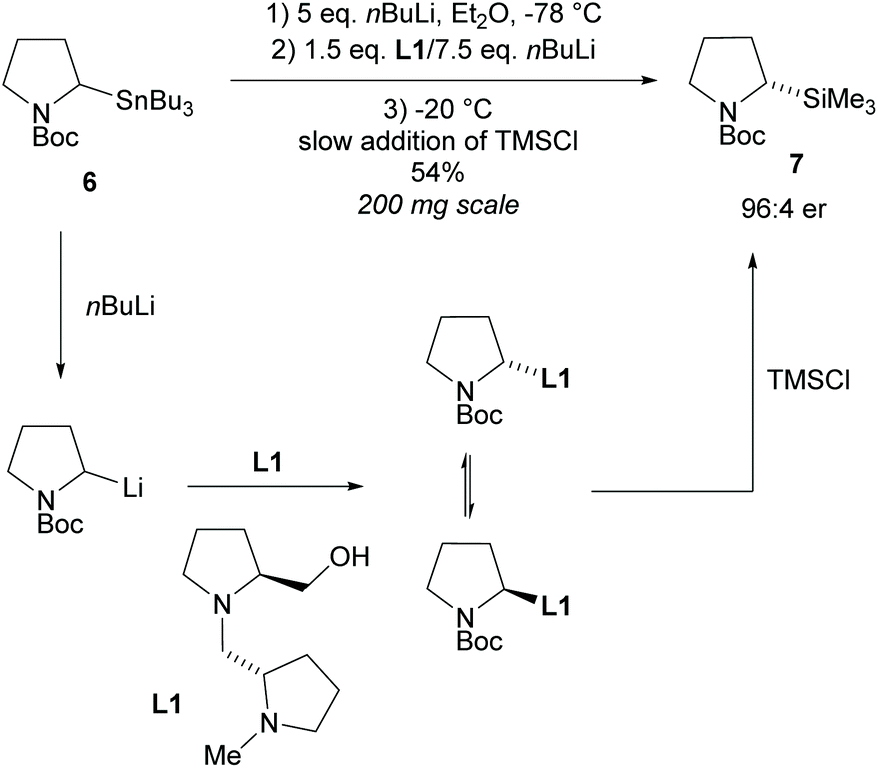 | ||
| Scheme 2 Dynamic kinetic resolution of N-Boc pyrrolidine 6 reported by Coldham et al. | ||
The opposite enantiomer of pyrrolidine 7 was obtained in a similar yield and enantioselectivity through the use of the diastereoisomer of diamine L1. Many methods involving lithiation of N-protected-pyrrolidines operate via dynamic thermodynamic resolution (DTR) with asymmetric deprotonation and so will not be discussed here.7,19–22
Biocatalytic methods for the kinetic resolution of racemic pyrrolidines provide easy access to enantiomerically enriched pyrrolidines under mild conditions; often performing to high degrees of selectivity at mild temperatures and pH under atmospheric pressure.23 Unfortunately, the high selectivity comes with a loss of substrate diversity, with many of the enzymes only performing well with one or two pyrrolidine substrates.
Hydrolases can be utilised in the hydrolysis of esters closely attached to the pyrrolidine ring (Scheme 3). Racemic proline derivatives were resolved by both Kazlauskas et al. and Sugai et al. employing Aspergillus niger lipase (ANL) and Candida antarctica lipase B (CAL-B) repectively.24,25 Good selectivity was obtained by Kazlauskas for the unprotected amino acid (S)-9 with the noted requirement of purified ANL due to lower selectivity when using the crude enzyme. Sugai obtained excellent enantioselectivity for the N-benzylacetyl amino acid (R)-11. Changing the protecting group to N-benzyloxycarbonyl (Cbz) afforded similarly good results, however, no reactivity was observed with an N-carbamoyl protected amino ester. In 2004, Larchevêque et al. resolved β-amino esters using B. cepacia lipase, obtaining the best results with N-Boc protected pyrrolidine 12.26 Alternative pyrrolidines, including a N-benzylacetyl protected β-amino ester, were attempted, however, very poor selectivity was observed. In 2005, Hu et al. resolved 2-methyl-pyrrolidine 14via hydrolysis of the oxalamic ester using a protease obtained from Aspergillus species.27 Excellent enantioselectivity was obtained for the remaining oxalamic ester. Two pyrrolidines containing aromatic substituents at the 2-position were also resolved, obtaining good enantioselectivities. In 2015, Petri et al. used phosphatidylserine (PS) from B. cepacia to resolve 3-substituted pyrrolidine (±)-16 obtaining 3-hydroxypyrrolidine (R)-17 in excellent enantioselectivity.28
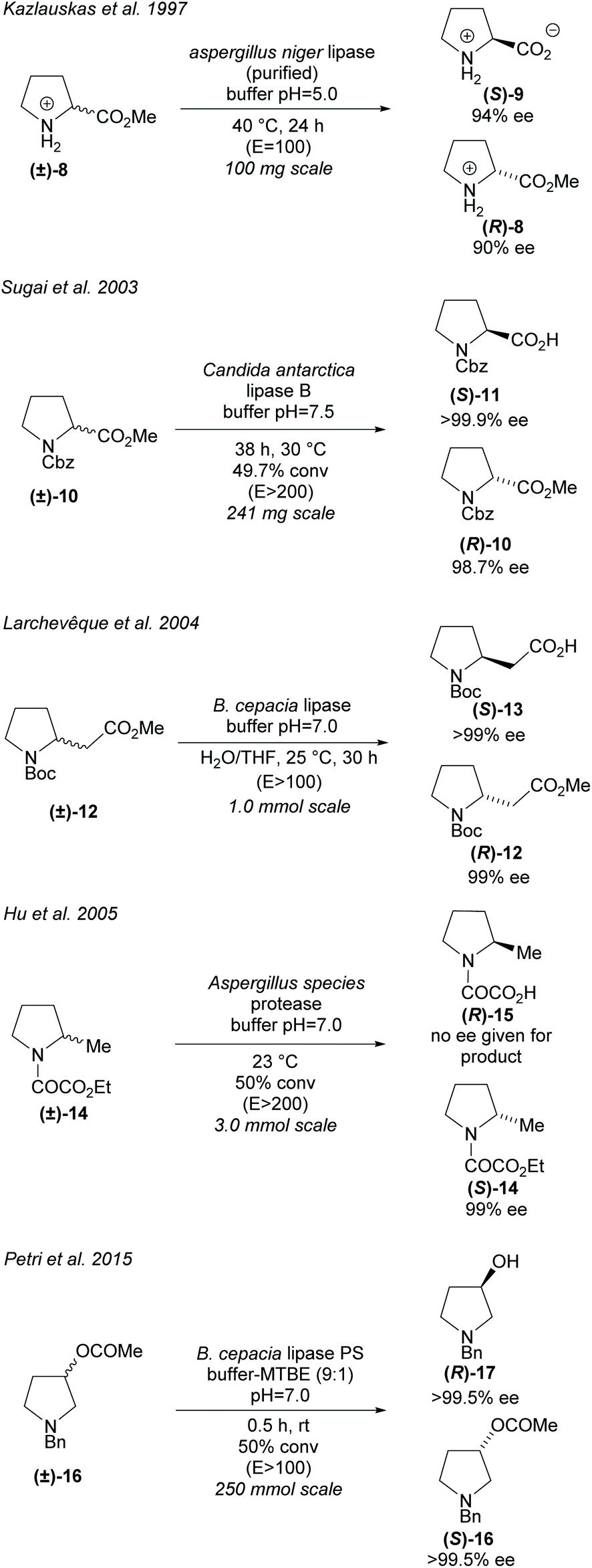 | ||
| Scheme 3 Hydrolysis of various monosubstituted pyrrolidines. | ||
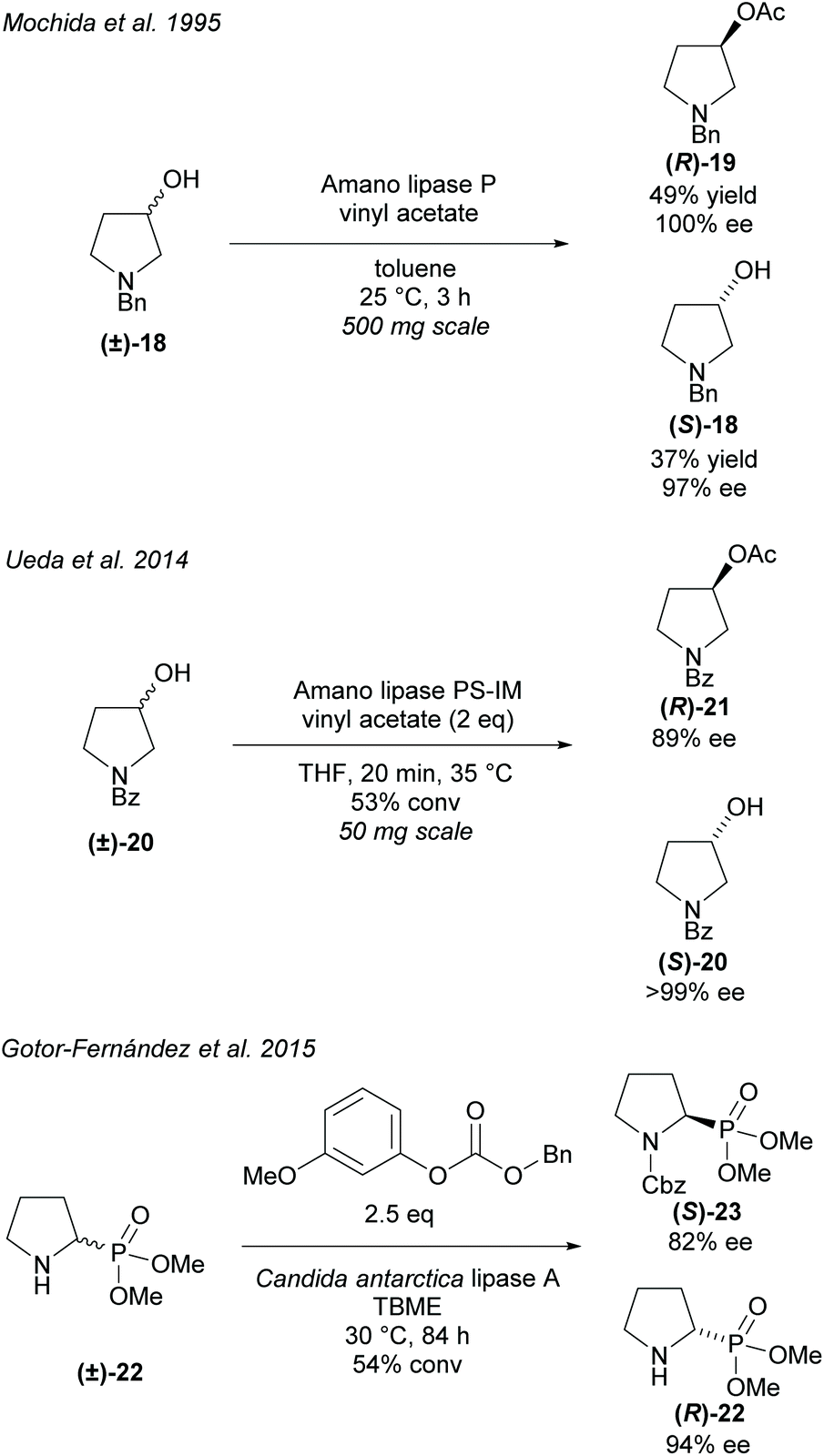
Alternatively, hydrolyses can be used in organic solvents to promote asymmetric acetylation of racemic alcohols (Scheme 4). Use of organic solvents allows for easier extraction of products when compared to hydrolysis methods conducted in buffers. Resolution of 3-hydroxy-pyrrolidines were conducted by Mochida et al. and Ueda et al. employing Amano lipases P and PS-IM (immobilised), respectfully.29,30 Mochida's acetylation of racemic 3-hydroxy-pyrrolidine (±)-18 achieved excellent enantioselectivity for both the acetylated product (R)-19 and the remaining starting material (S)-18. Similarly, excellent enantioselectivities were obtained by Ueda for the returned starting material (S)-20, however, lower enantioselectivity was obtained for the acetylated pyrrolidine (R)-21.
 | ||
| Scheme 4 Acetylation of various monosubstituted pyrrolidines. | ||
Resolution of racemic cyclic aminophosphonate (±)-22 was conducted by Gotor-Fernández et al. employing Candida antarctica lipase type A (CAL-A) obtaining good enantioselectivities for both the benzyl carbamate product (S)-23 and the returned starting material (R)-22.31
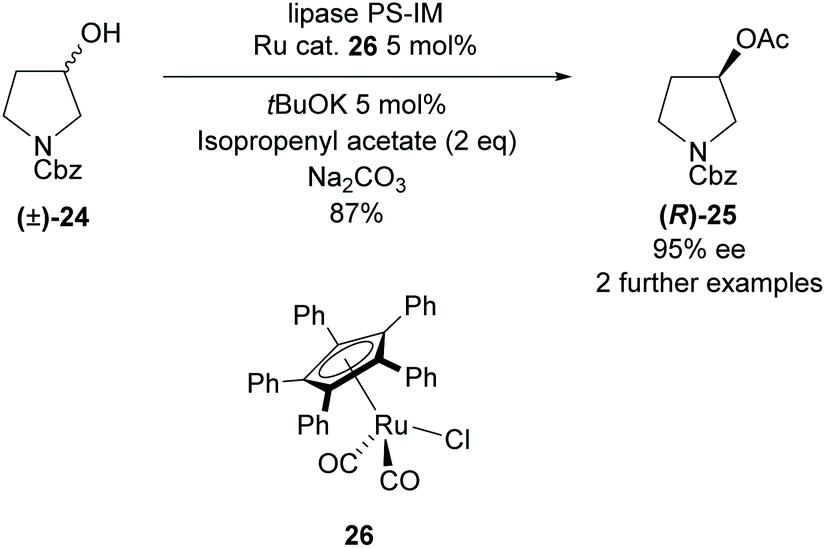
All methods involving enzymatic resolution of racemic pyrrolidines discussed so far have a theoretical maximum yield of 50% for the enantiopure product and returned starting material. In 2011, Bäckvall et al. employed lipase PS-IM in the acetylation of 3-hydroxy-pyrrolidine (±)-24 with a ruthenium catalyst to racemise the starting material (Scheme 5).32 The racemisation catalysis allows dynamic kinetic resolution allowing for significantly higher yields than traditionally available with enzymatic methodologies. They obtained a good yield of 87% with excellent enantioselectivity of 95% for N-Cbz protected pyrrolidine (R)-25.
 | ||
| Scheme 5 Acetylation of racemic alcohol 24 involving ruthenium catalysed racemisation. | ||
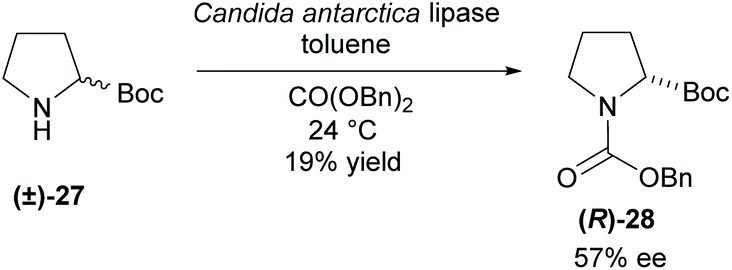
Protection of amines via enzymatic transesterification has been significantly less investigated. One common problem associated with this method is the higher nucleophilicity of the amine compared to the commonly acetylated alcohols. In 1999, Wong et al. attempted to overcome this problem by employing less reactive dibenzyl carbonate as the protecting group source using the lipase CAL to catalyse the reaction (Scheme 6).33 Unfortunately, the N-protected pyrrolidine was obtained in a poor 19% yield with only moderate enantioselectivity. This was the only example provided.
 | ||
| Scheme 6 Enzyme catalysed amine protection by Wong et al. | ||
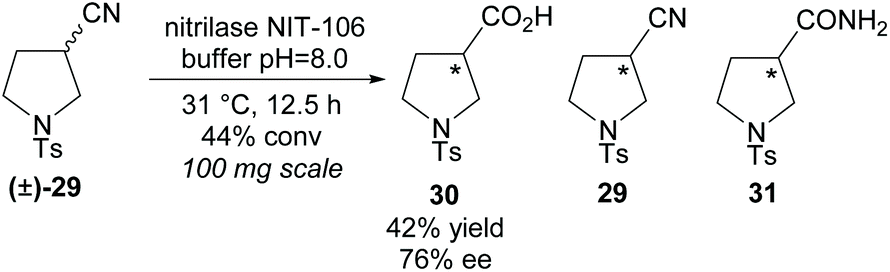
In 2007, Klempier et al. employed nitrilase NIT-106 in the synthesis of β-amino acid 30 (Scheme 7).34 Nitrilase catalysed hydrolysis of racemic 3-nitrile-pyrroline (±)-29 afforded the β-amino acid 30 in a good 42% yield at 44% conversion with a good enantioselectivity of 76% ee. Higher conversions at longer reaction times afforded the amide by-product 31. The stereochemistry of the remaining starting material nitrile or either of the products was not reported.
 | ||
| Scheme 7 Enzyme catalysed hydrolysis of nitrile pyrrolidine 29 reported by Klempier et al. | ||
An alternative functional group transformation is to utilize ω-transaminases to resolve racemic amines by converting one enantiomer of the amine to a ketone. In 2008, Bornscheuer et al. employed a ω-transaminase from Alcaligenes denitrificans (Ade-TA) to resolve 3-amino-N-Boc-pyrrolidine (±)-32 (Scheme 8).35 Excellent enantioselectivity was obtained for the remaining starting material with a moderate 39% yield at 50% conversion. Use of the carbamate protecting group (Boc) improved the efficiency and selectivity of the reaction when compared to the unprotected and amide (benzyl) protected substrates.
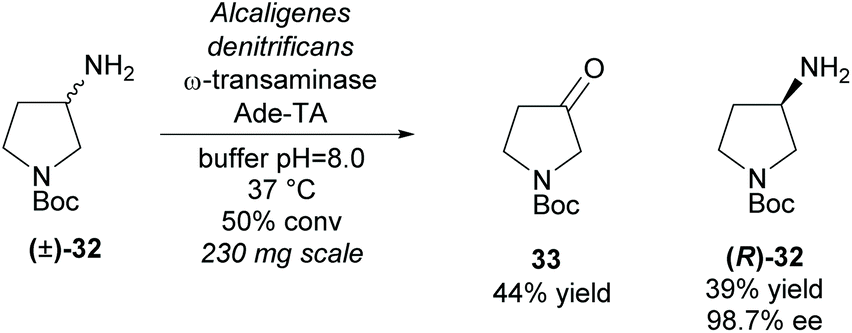 | ||
| Scheme 8 Conversion of amine to ketone employing ω-transaminase Ade-TA reported by Bornscheuer et al. | ||
Modification of functional groups attached to pyrrolidine rings can be achieved using redox methods. In 2008, Onomura and co-workers investigated the electrochemical oxidation of aminoalcohols and aminoaldehydes (Scheme 9).36 Racemic 2-substituted-pyrrolines were resolved employing a copper catalyst with (R,R)-Ph-BOX as the chiral ligand. Oxidation of the aminoaldehyde (±)-34 achieved a moderate 43% yield in good enantioselectivity however, the recovered starting aldehyde was obtained in a poor 27% ee. The oxidation of aminoalcohol (±)-36 was less successful, obtaining lower enantioselectivities and significantly lower yields for the ester product. Very poor enantioselectivity was obtained for the recovered amino alcohol (S)-36.
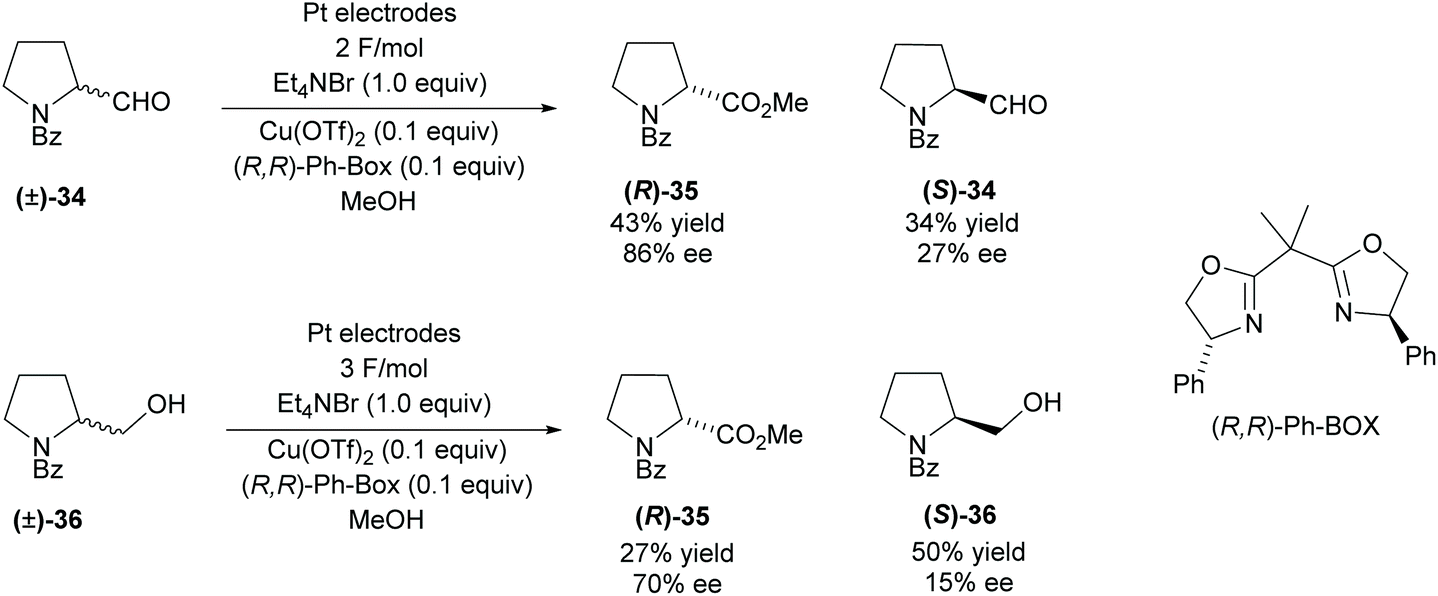 | ||
| Scheme 9 Electrochemical oxidation reported by Onomura et al. | ||
Monosubstituted pyrrolidine Dolaproine (Dap) is a key unit of Dolastatin 10, a marine natural product belonging to a family of antineoplastic peptides (Scheme 10).37 Genet and co-workers developed a ruthenium catalysed hydrogenation via dynamic kinetic resolution of β-keto-esters derived from (S)-Boc-proline as a route to access Dolaproine and its isomers.38–40 Initial work produced Boc-(2S,3R)-iso-Dap in good diastereoselectivity and excellent yield for the DKR of pyrrolidine salt 40 (Scheme 10).38 Ru[(R)-MeO-BIPHEP]2 was employed to access Boc-(2R,3S)-iso-Dap however, a mixture of diastereoisomers (2R,3S)-42, (2S,3R)-42 and (2S,3S)-42 was obtained with 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24:5 dr.39 Recrystallisation effectively removed one diastereoisomer, and subsequent functionalisation allowed for separation of the remaining diastereoisomers via chromatography on silica gel. Harsher reaction conditions by an increase in pressure from 10 bar to 100 bar and longer reaction times were employed to afford (2S,3S)-42 in 87% de from Boc-protected pyrrolidine 41. Changing the chiral ligand to (S)-SYNPHOS allowed for access to (2R,3R)-42, the isomer required for the synthesis of Dolastatin 10.40 A mixture of four diastereoisomers was obtained with moderate 59:33:5:4 dr. Separation by chromatography on silica gel afforded an inseparable mixture of (2R,3R)-42 and (2S,3R)-42 in 2:1 dr.
:24:5 dr.39 Recrystallisation effectively removed one diastereoisomer, and subsequent functionalisation allowed for separation of the remaining diastereoisomers via chromatography on silica gel. Harsher reaction conditions by an increase in pressure from 10 bar to 100 bar and longer reaction times were employed to afford (2S,3S)-42 in 87% de from Boc-protected pyrrolidine 41. Changing the chiral ligand to (S)-SYNPHOS allowed for access to (2R,3R)-42, the isomer required for the synthesis of Dolastatin 10.40 A mixture of four diastereoisomers was obtained with moderate 59:33:5:4 dr. Separation by chromatography on silica gel afforded an inseparable mixture of (2R,3R)-42 and (2S,3R)-42 in 2:1 dr.
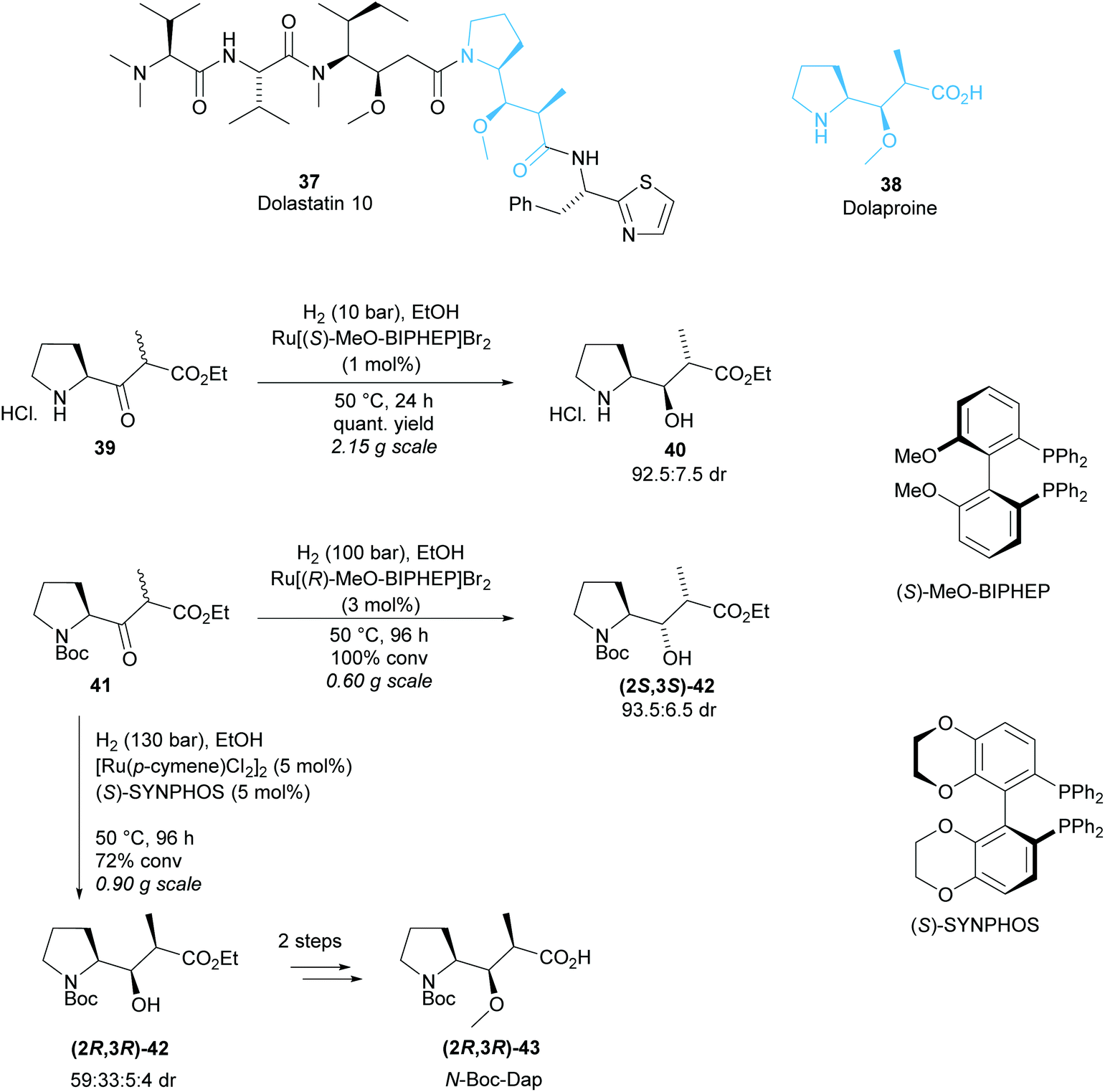 | ||
| Scheme 10 Synthesis of Dolaproine reported by Genet et al. | ||
Methylation of the mixture and separation by chromatography on silica gel afforded pure O-methylated-(2R,3R)-43 which was used in the total synthesis of Dolastatin 10.
In 2011, Ohkuma et al. employed a similar method, synthesising 2- and 3-monosubstituted pyrrolidines from racemic pyrrolidines containing a ketone (Scheme 11).41
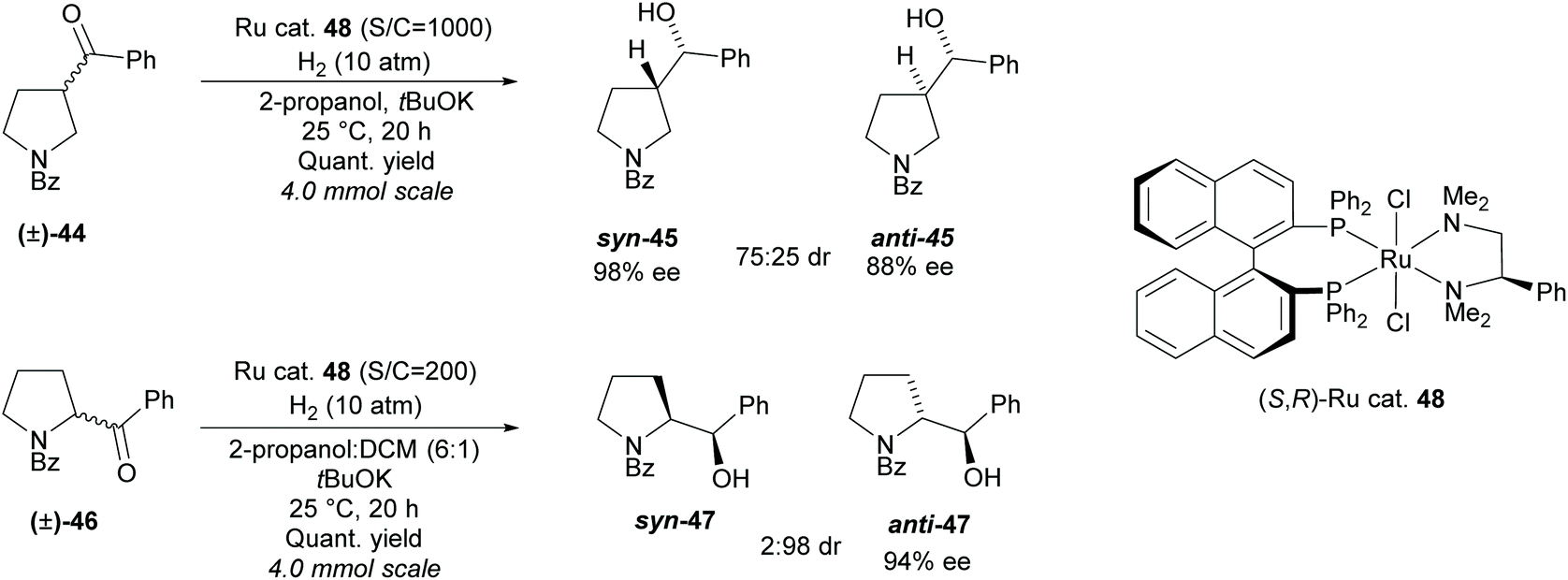 | ||
| Scheme 11 Ruthenium catalysed hydrogenation involving DKR reported by Ohkuma et al. | ||
A ruthenium catalyst 48 was employed in the hydrogenation, obtaining syn-3-substituted pyrrolidine 45 in moderate diastereoselectivity and excellent enantioselectivity and anti-2-substituted pyrrolidine 47 in excellent diastereoselectivity and good enantioselectivity.
3. Disubstituted pyrrolidines
Reduction of racemic disubstituted cyclic imines provides access to disubstituted pyrrolidines. In 1994, Buchwald and co-workers developed a resolution of disubstituted cyclic imines via titanocene catalysed asymmetric reduction (Scheme 12).42,43 2,5-Disubstituted pyrrolidines were afforded as single diastereoisomers in excellent enantioselectivity and moderate yields with the recovered imines also obtained in excellent enantioselectivities. 2,3-Disubstituted pyrrolidine 45 was obtained in good diastereoselectivity with excellent enantioselectivity observed for both cis- and trans-isomers. Good enantioselectivity for the remaining cyclic imine was obtained from the crude reaction mixture at 50% conversion, however, due to racemisation on deactivated silica gel during purification, the remaining cyclic imine was not isolated. The 2,4-disubstituted pyrrolidine 46 was obtained in lower diastereoselectivity but with similarly excellent enantioselectivity. The enantioselectivities for the recovered imine for both the 2,3- and 2,4-pyrrolidines was significantly lower than observed with the 2,5-pyrrolidines.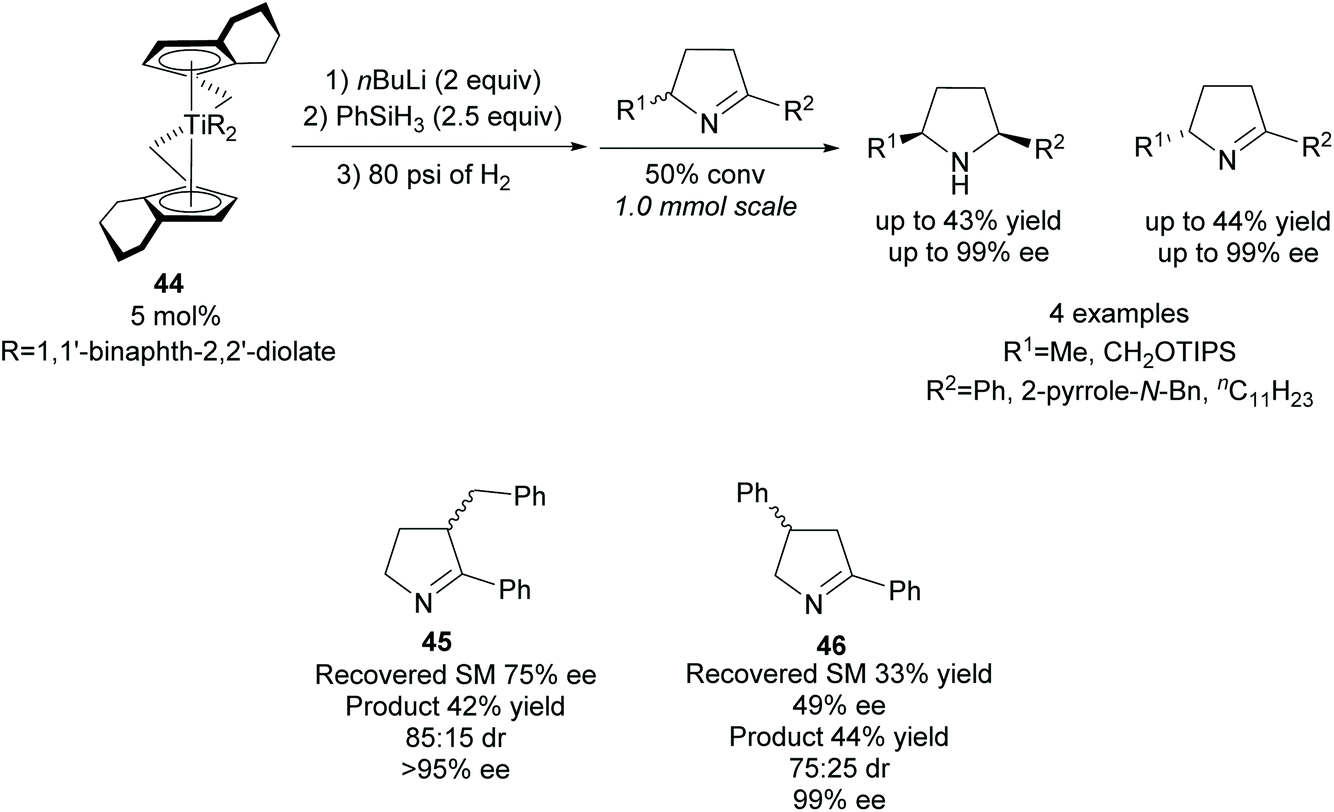 | ||
| Scheme 12 Reduction of disubstituted cyclic imines reported by Buchwald et al. | ||
Monosubstituted nitrile pyrrolidine 49 is a key intermediate in the synthesis of PF-00951966, a fluoroquinolone based broad-spectrum antibiotic discovered at Pfizer. In 2012, Lall and co-workers reported the synthesis of nitrile pyrrolidine 49via a modified Noyori asymmetric hydrogenation of racemic disubstituted β-keto-γ-lactam (±)-47 (Scheme 13).44 Excellent diastereoselectivity and enantioselectivity was obtained after recrystallisation affording (S,S)-lactam 48 in a good yield. Further functional group manipulations including reduction of the lactam afforded nitrile pyrrolidine 49 in 11 steps. This method of dynamic kinetic resolution using ruthenium catalysts was developed by Noyori et al. and first expanded to the reduction of β-keto-γ-lactams by Hattori et al. during the synthesis of 2-[N-imidoylpyrrolidinyl]carbapenems.45,46
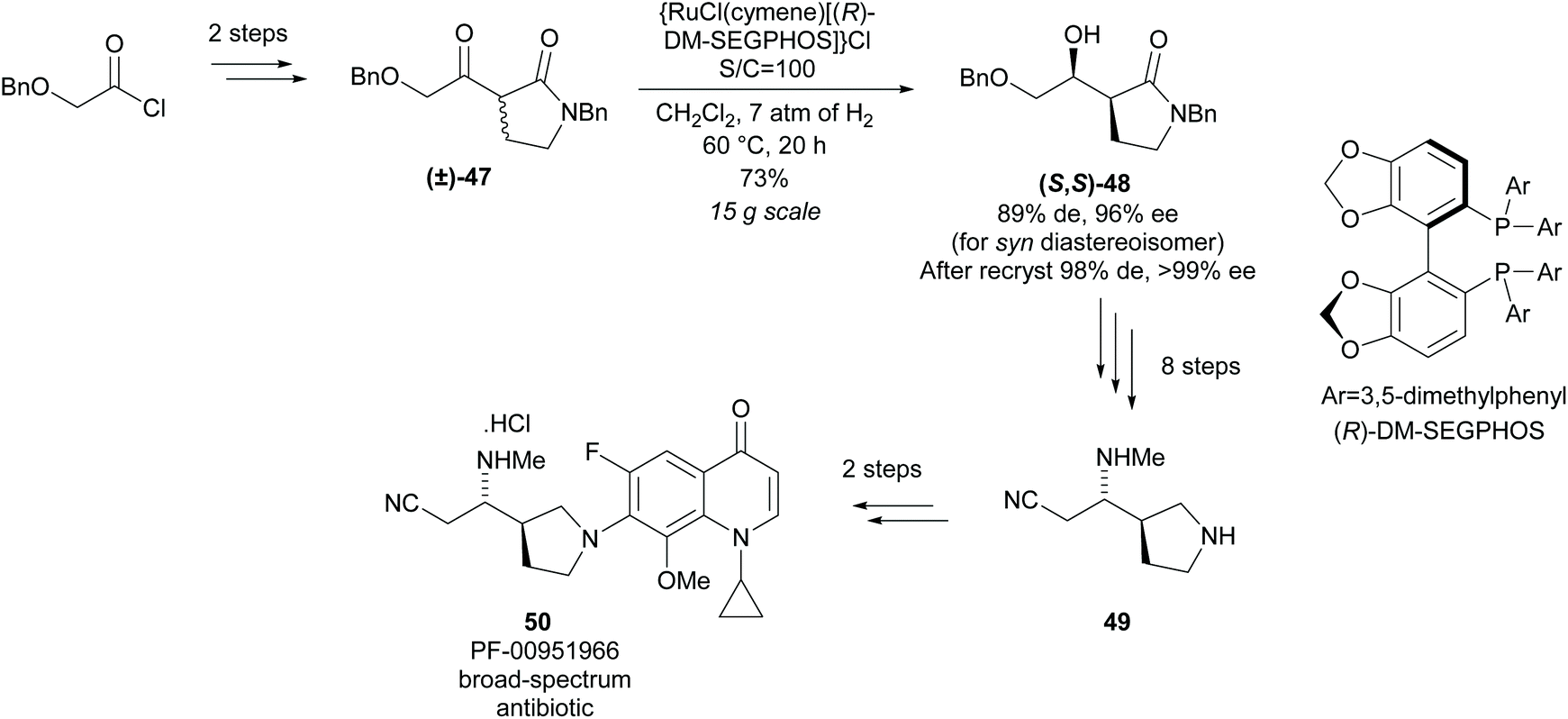 | ||
| Scheme 13 Synthesis of nitrile pyrrolidine 49 reported by Lall et al. | ||
In 2013, Magnus et al. at Eli Lilly employed a similar method of hydrogenation of racemic β-keto-γ-lactam (±)-51 in the synthesis of a pyrrolidine-based serotonin norepinephrine reuptake inhibitor 53 (Scheme 14).47 Good yields and selectivities were obtained for the β-hydroxy-γ-lactam 52. Further modifications obtained the pyrrolidine-based inhibitor in 7 steps.
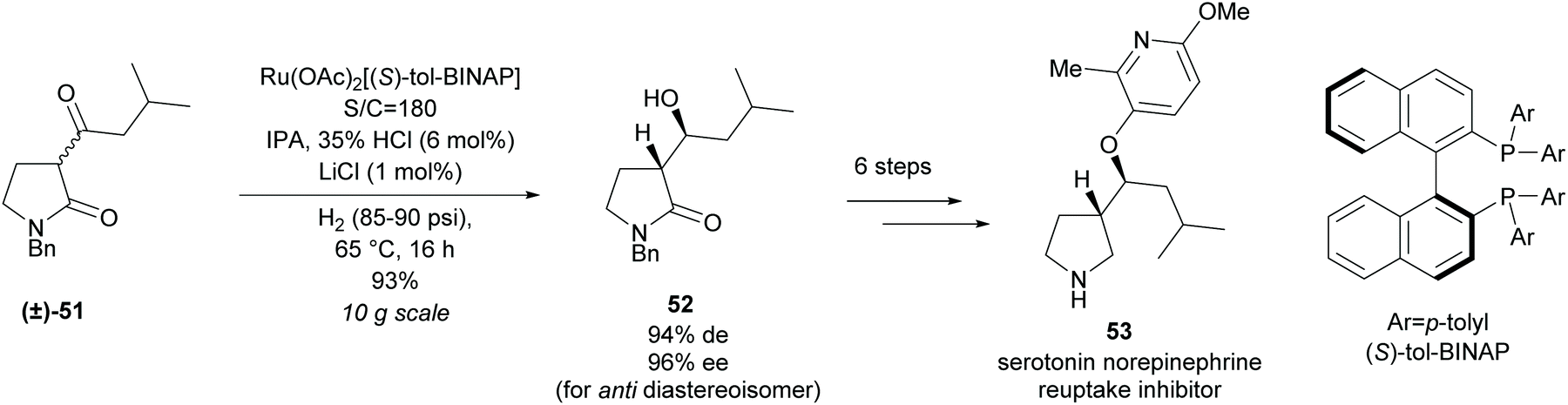 | ||
| Scheme 14 Synthesis of serotonin norepinephrine reuptake inhibitor 53 reported by Magnus et al. | ||
More recently, Xie et al. employed an iridium catalyst in the asymmetric hydrogenation of racemic β-keto-γ-lactams (Scheme 15).48 They obtained excellent yields and selectivities across the 12 examples. From β-hydroxy-γ-lactam 54, diamine 55, a key intermediate in the synthesis of fluoroquinolone antibiotic Premafloxacin 56, was synthesised in 6 steps.
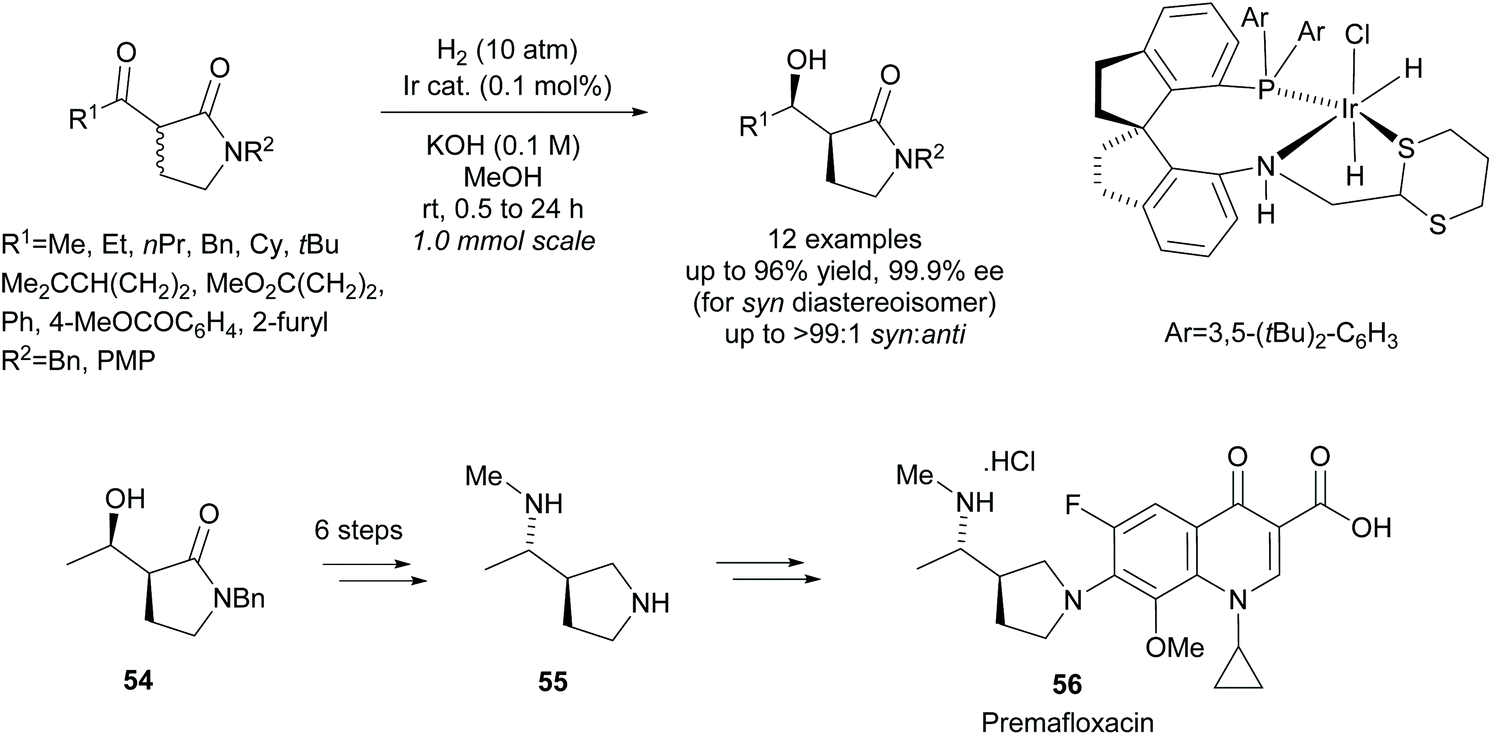 | ||
| Scheme 15 Synthesis of premafloxacin via iridium catalysed hydrogenation reported by Xie et al. | ||
Various methods of intramolecular cyclisation have been employed in the synthesis of disubstituted pyrrolidines. The resolution of racemic α-substituted-1-aminopent-4-enes via the asymmetric hydroamination of aminoalkenes was developed by Hultzsch et al. employing rare earth metal catalysts with various binaphtholate ligands (Scheme 16).49–54 Initial investigations employed binaphtholate catalyst 57 in the cyclisation affording a 2,5-disubstituted pyrrolidine in moderate diastereoselectivity and poor enantioselectivity.49 Building on this result, more sterically demanding binaphtholate ligands were developed incorporating 3,3′-bis(triarylsilyl) groups. In most cases, the highest enantioselectivities were observed using yttrium catalyst 59, with the exception of the α-phenyl-aminoalkene which obtained a higher selectivity for the recovered starting material employing a lutetium catalyst.51 In many cases, the enantioselectivity was only reported for the recovered aminoalkenes after the kinetic resolution. The same group also attempted the resolution of β- and γ-substituted-1-aminopent-4-enes, obtaining 2,4- and 2,3-disubstituted pyrrolidines respectfully.52 2,4-Disubstituted pyrrolidine 60 was obtained in poor diastereoselectivity with no enantioselectivities for either isomer reported. Poor enantioselectivity was observed for the recovered starting aminoalkene 60. Moderate enantioselectivity was reported for the recovered aminoalkene 62 however no diastereoselectivity or enantioselectivity was reported for the 2,3-disubstituted pyrrolidine 63.
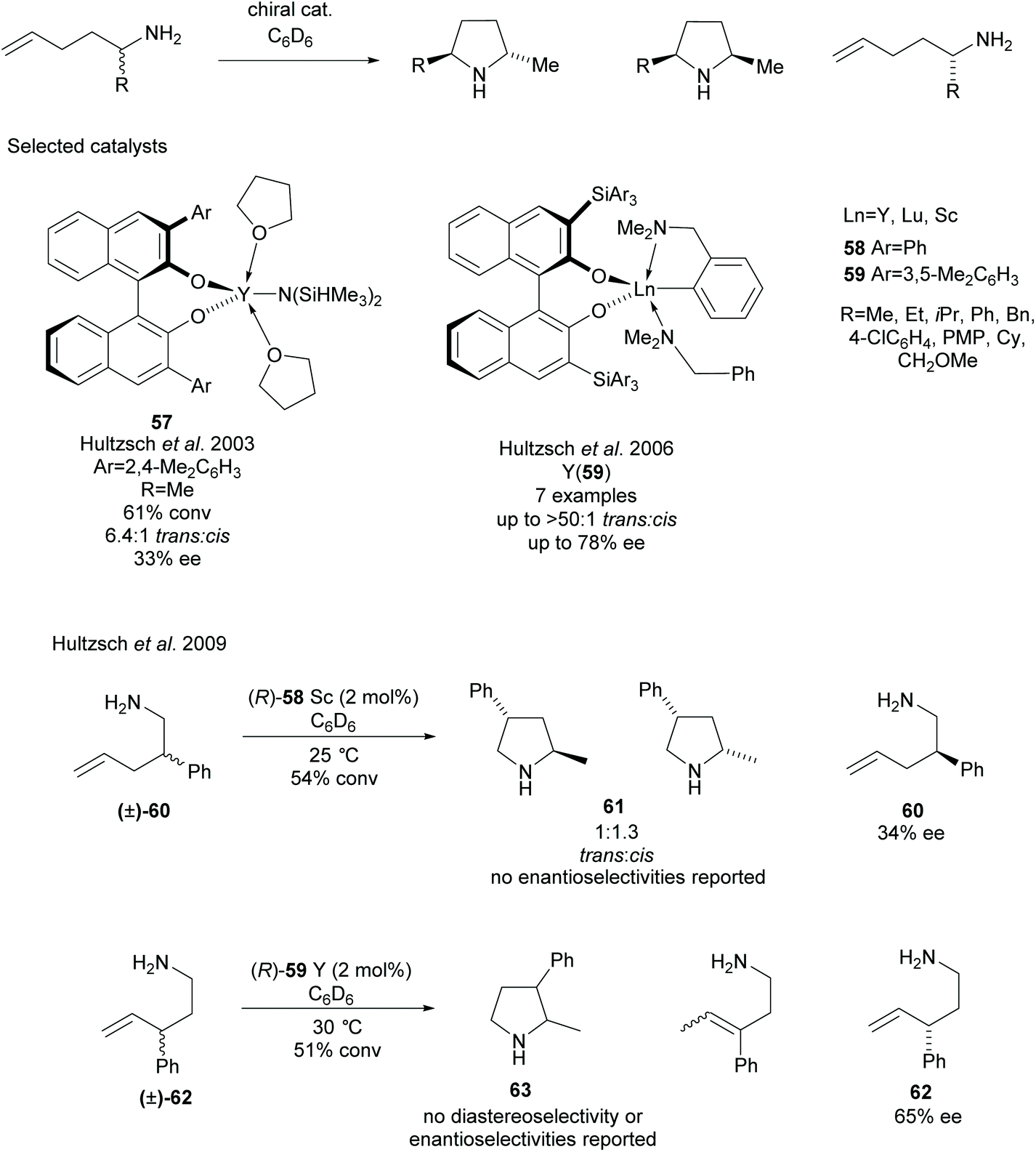 | ||
| Scheme 16 Asymmetric hydroamination of aminoalkenes reported by Hultzsch et al. | ||
In 2009, Gracza et al. reported the Pd catalysed carbonylative bicyclisation of N-protected 1-aminopent-4-ene-3-ols as a route to synthesize bicyclic pyrrolidines (Scheme 17).55
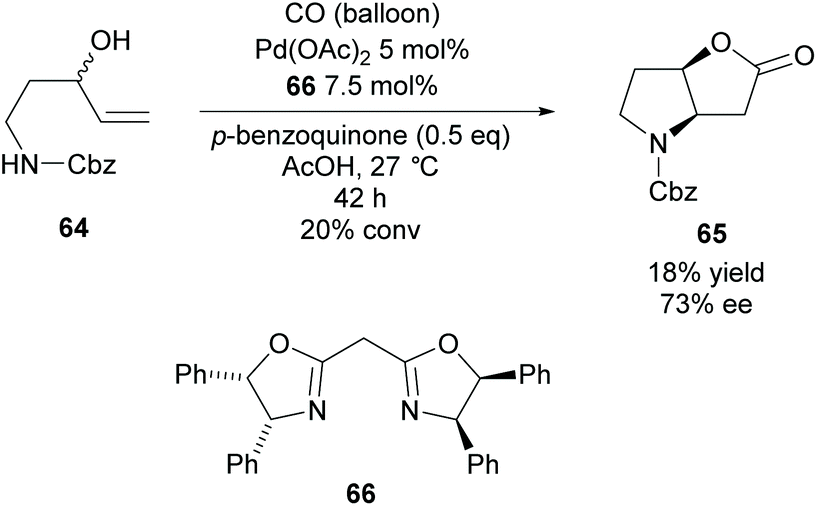 | ||
| Scheme 17 Carbonylative cyclisation reported by Gracza et al. | ||
They obtained the Cbz protected pyrrolidine 65 in a poor yield at a low conversion with good enantioselectivity. The yield could be improved with the use of p-toluenesulfonyl as the protecting group compared to carboxybenzyl however the enantioselectivity decreased to 60% ee.
(−)-Supinidine, a pyrrolizidine alkaloid, can be synthesised from disubstituted pyrrolidine 70. In 1991, Takahata and co-workers employed the Katsuki–Sharpless asymmetric epoxidation in the kinetic resolution of racemic N-protected 1-aminopent-4-ene-3-ol 67, a method the group developed to access chiral urethanes (Scheme 18).56–58 Disubstituted pyrrolidine 70 was obtained with good enantioselectivity and was subsequently used in the synthesis of (−)-supinidine in 12 further steps.
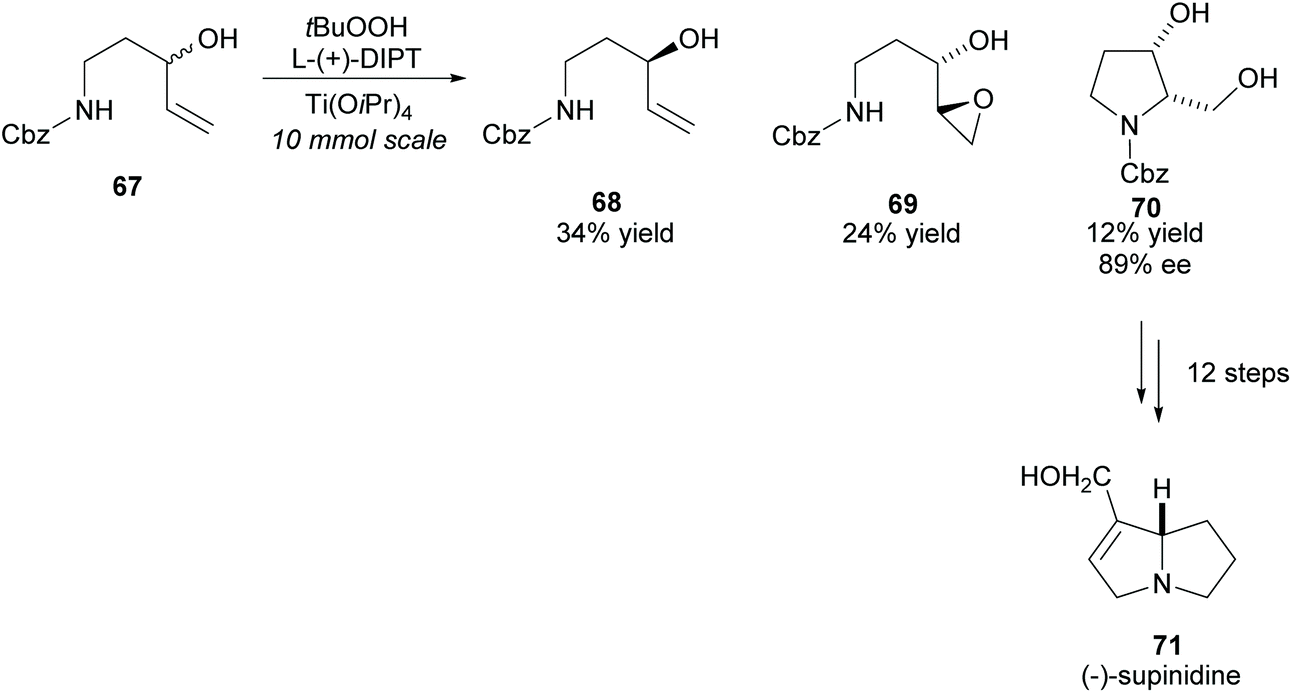 | ||
| Scheme 18 Synthesis of (−)-supinidine via Katsuki–Sharpless asymmetric epoxidation reported by Takahata et al. | ||
Cyclisation employing dynamic kinetic resolution within the aza-Cope rearrangement was developed by Overman et al. (Scheme 19).59N-Protected bicyclic pyrrolidine 73 was obtained in a good 89% yield over two steps in an excellent 99% ee. Hydrolysis of the formaldiminium ion after the aza-Cope rearrangement using standard methods failed to form the desired product and so dimedone was employed.60
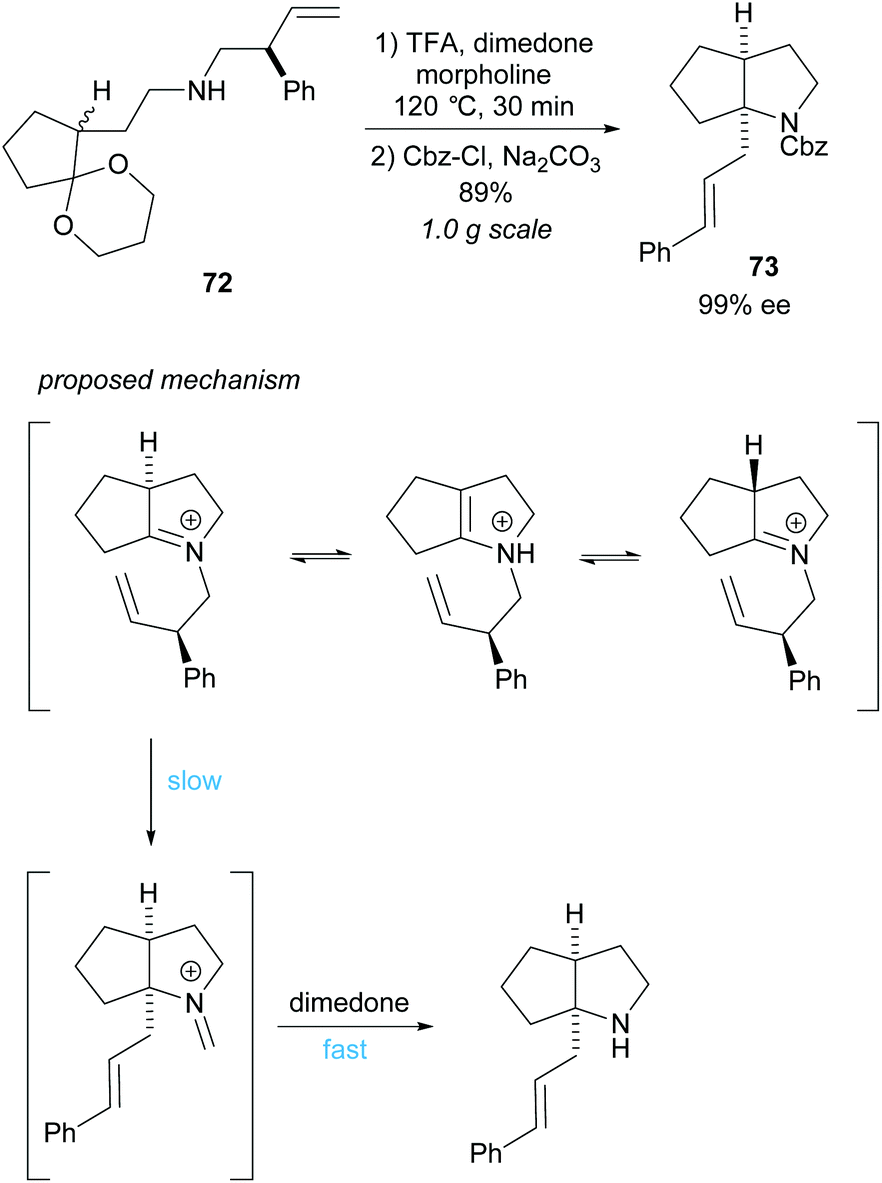 | ||
| Scheme 19 aza-Cope rearrangement reported by Overman et al. | ||
The dynamic kinetic resolution of racemic disubstituted pyrrolidine (±)-74 within an enantioselective Morita–Baylis–Hillman (MBH) cyclisation was investigated by Zakarian et al. (Scheme 20).61 When employing non-racemic pyrrolidine 74, significant racemisation was observed at temperatures above −30 °C during the MBH cyclisation. They suggested this occurred through acid-catalysed isomerisation of the cyclic iminium ion, however, the exact route of epimerisation is not currently known. After screening a series of chiral sufides, they obtainedbicyclic 75 in good enantioselectivity but poor yield. Further work is required to improve yields and make this a viable route.
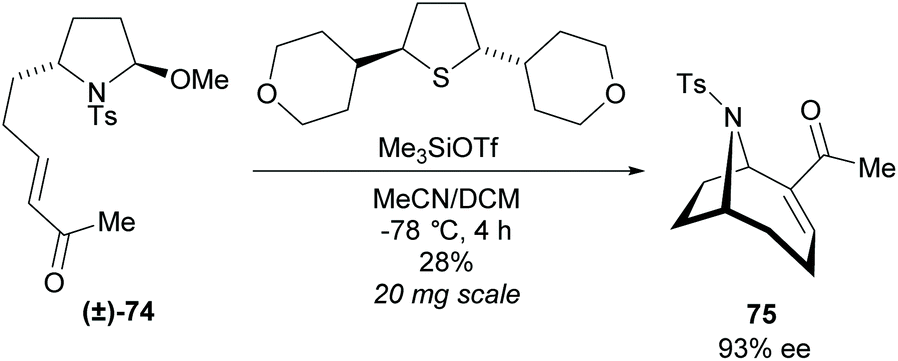 | ||
| Scheme 20 DKR of disubstituted pyrrolidine 74 reported by Zakarian et al. | ||
As with the monosubstituted pyrrolidines, enzymes are a popular technique to synthesise disubstituted pyrrolidines from racemic pyrrolidines in a highly selective way. The same issues of the limits of diversity while retaining selectivity are still prevalent. Akai et al. employed CAL-B in the esterification of racemic N-oxide (±)-76, which then underwent an intramolecular Diels–Alder reaction affording pyrrolidine 77 (Scheme 21).62,63 In two further steps they synthesised (−)-rosmarinecine, a component of several pyrrolizidine alkaloids. This method is the shortest reported total synthesis for (−)-rosmarinecine, achieving a 15% yield over 4 steps. Previous methods include synthesis from a furanose derivative, obtaining a higher yield of 18% but in 10 total steps, and intramolecular cycloaddition methods, obtaining the same 15% yield but in 8 steps.64–67N-Oxide 76 was shown to partially racemise and so the group attempted to develop a DKR. Higher yields could be obtained with a slight decrease in enantioselectivity, however the enantioselectivity was higher than that that would be obtained if no racemization were occurring.
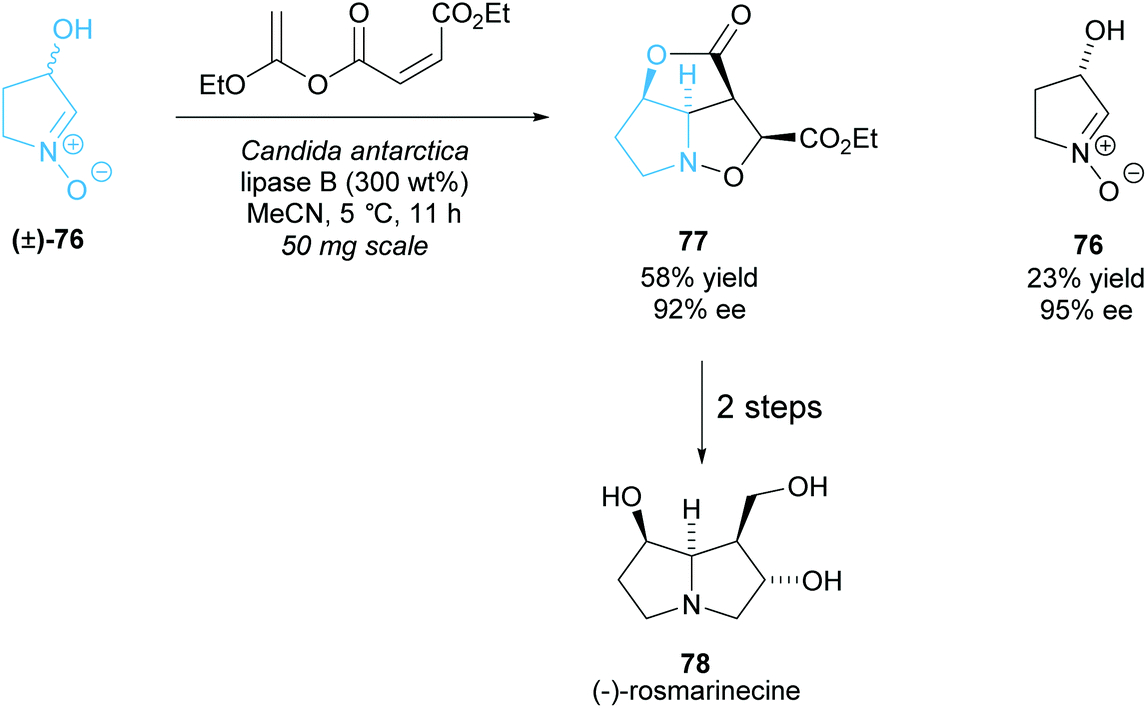 | ||
| Scheme 21 Synthesis of (−)-rosmarinecine via enzyme catalysed KR reported by Akai et al. | ||
Hydrolysis of esters closely attached to the pyrrolidine ring in disubstituted pyrrolidines is again a popular technique (Scheme 22). In 1996, Kawanami et al. employed PS to resolve racemic 2,5-diacetyl pyrrolidine trans-(±)-79 affording the monoacetyl pyrrolidine 80 in moderate enantioselectivity with the returned diacetyl 79 in excellent enantioselectivity.68 In 2007, Correia et al. hydrolysed racemic 3,4-disubstituted pyrrolidine trans-(±)-81 employing PS-AK obtaining excellent enantioselectivity for both the hydroxy product and remaining starting material.69 They used several different aromatic substituents obtaining excellent enantioselectivities for all examples. Faigl et al. has since used a similar method synthesizing the phenyl substituted analogue.70 CAL-B has been employed by Gotor et al. in the hydrolysis of 3,4-diacetylated pyrrolidine trans-(±)-83, brominated pyrrolidine trans-(±)-86, and amino pyrrolidine cis-(±)-88.71–73 Hydrolysis of racemic diacetate trans-(±)-83 afforded the dihydroxy-pyrrolidine 84 and the monoacetylated pyrrolidine 85 in excellent enantioselectivities, however, the returned diacetylated pyrrolidine 83 was obtained in significantly lower enantioselectivity.71 Improved enantioselectivity for diacetate 83 could be obtained with longer reaction times with a small decrease in enantioselectivity for the dihydroxy product. Bromohydrin 87 was obtained in excellent enantioselectivity after the hydrolysis of racemic methoxymethyl pyrrolidine trans-(±)-86.72 Similarly excellent enantioselectivity was obtained for cis-3-amino-4-hydroxy-pyrroldine 89 and its returned starting methoxymethyl pyrrolidine 88.73
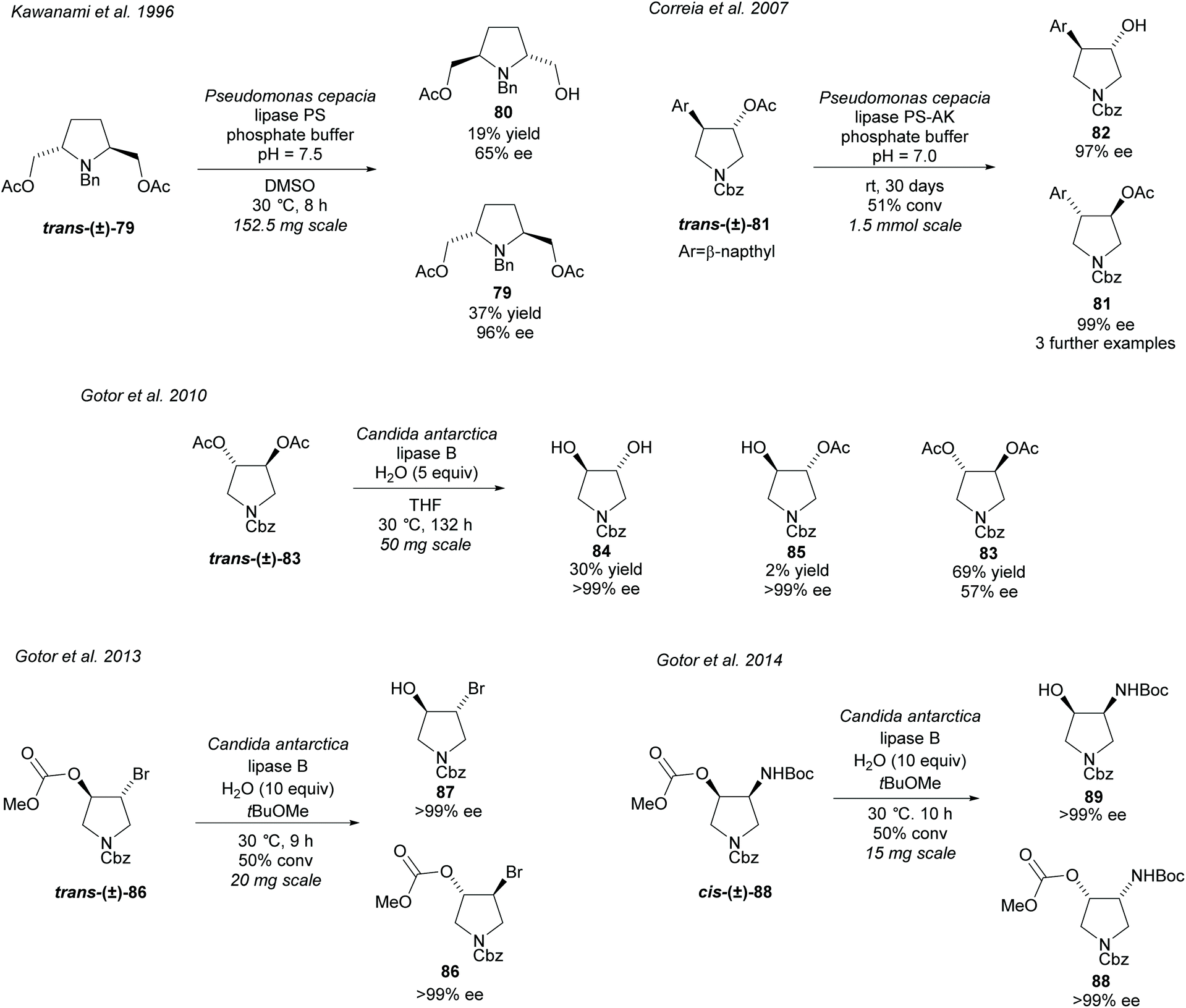 | ||
| Scheme 22 Hydrolysis of various disubstituted pyrrolidines. | ||
In 2005, Aggarwal and co-workers employed hydrolyses from Candida lipolytica and Rhizomucor meihei to resolve the cis- and trans-2,5-disubstituted pyrrolidine 90, respectively (Scheme 23).74 Excellent enantioselectivities were obtained for the returned esters with no reports of the enantioselectivities of the acid products.
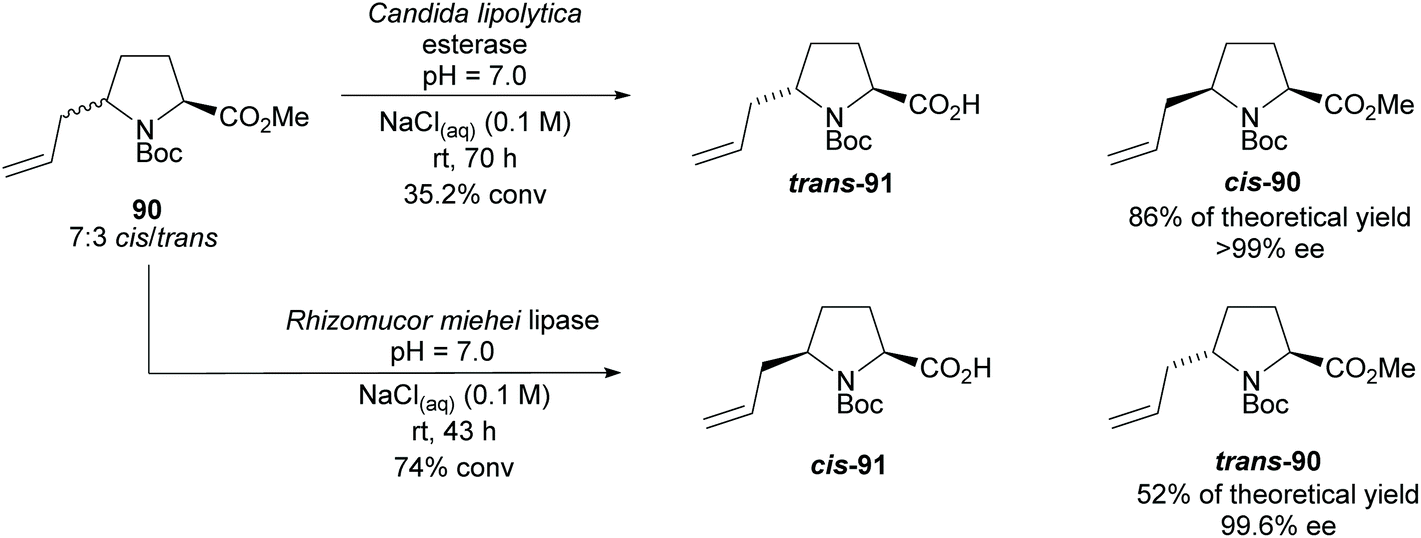 | ||
| Scheme 23 Resolution of disubstituted pyrrolidine 90via enzyme catalysed hydrolysis reported by Aggarwal et al. | ||
Transesterification of alcohols attached to a pyrrolidine ring promoted by lipases is another popular use of enzymes in the synthesis of disubstituted pyrrolidines (Scheme 24). In 1994, Sibi et al. acetylated racemic 2,5-disubstituted-dihydroxy pyrrolidine trans-(±)-92 employing PS lipase, affording the diacetate product 79 and the returned dihydroxy pyrrolidine 92 in excellent enantioselectivities.75 Poor enantioselectivity was obtained for the monoacetylated product 80. Kawanami et al. developed a method to obtain the monoacetylated pyrrolidine 93 in excellent enantioselectivity employing the same enzyme, with good a enantioselectivity also reported for the diacetylated product.76 Further work was conducted by the same group investigating the substituents of the aryl group of the N-protecting group, concluding that 3,5-dimethylbenzyl affords the best enantioselectivities.77 Kamal et al. employed lipases PS-C (lipase immobilized on ceramic particles) and PS-D (lipase immobilized on diatomaceous earth) in the resolution of trans- and cis-azido pyrrolidines 95, respectively.78,79 Good to excellent enantioselectivities were obtained for both the returned alcohols and the acetylated products. In 2006, Clinch et al. employed Novozyme® 435 (an immobilized form of CAL-B) in the resolution of 3,4-disubstituted pyrrolidine trans-(±)-97 obtaining good to excellent yields of 85% and 97% for the product ester 98 and remaining alcohol 97, respectively.80 These yields are presumed to be on the basis of a 50% maximum yield of either compound. This method was used by Whelligan et al. during the synthesis of new aza-nucleoside mimics, obtaining optical rotations in line with those reported by Clinch et al., however, they obtained lower yields of 83% and 75% for the remaining alcohol and product ester respectively.81 In 2013, Gotor et al. resolved trans-3,4-disubstituted pyrrolidine trans-(±)-99 using the lipase PLS-IM (immobilised) affording the acetylated product 100 and the returned alcohol 99 in good to excellent enantioselectivities.82 Acetylation of the unprotected primary amine was also attempted employing CAL-B although only moderate enantioselectivities were obtained for the returned free amine and monoacetylated product.
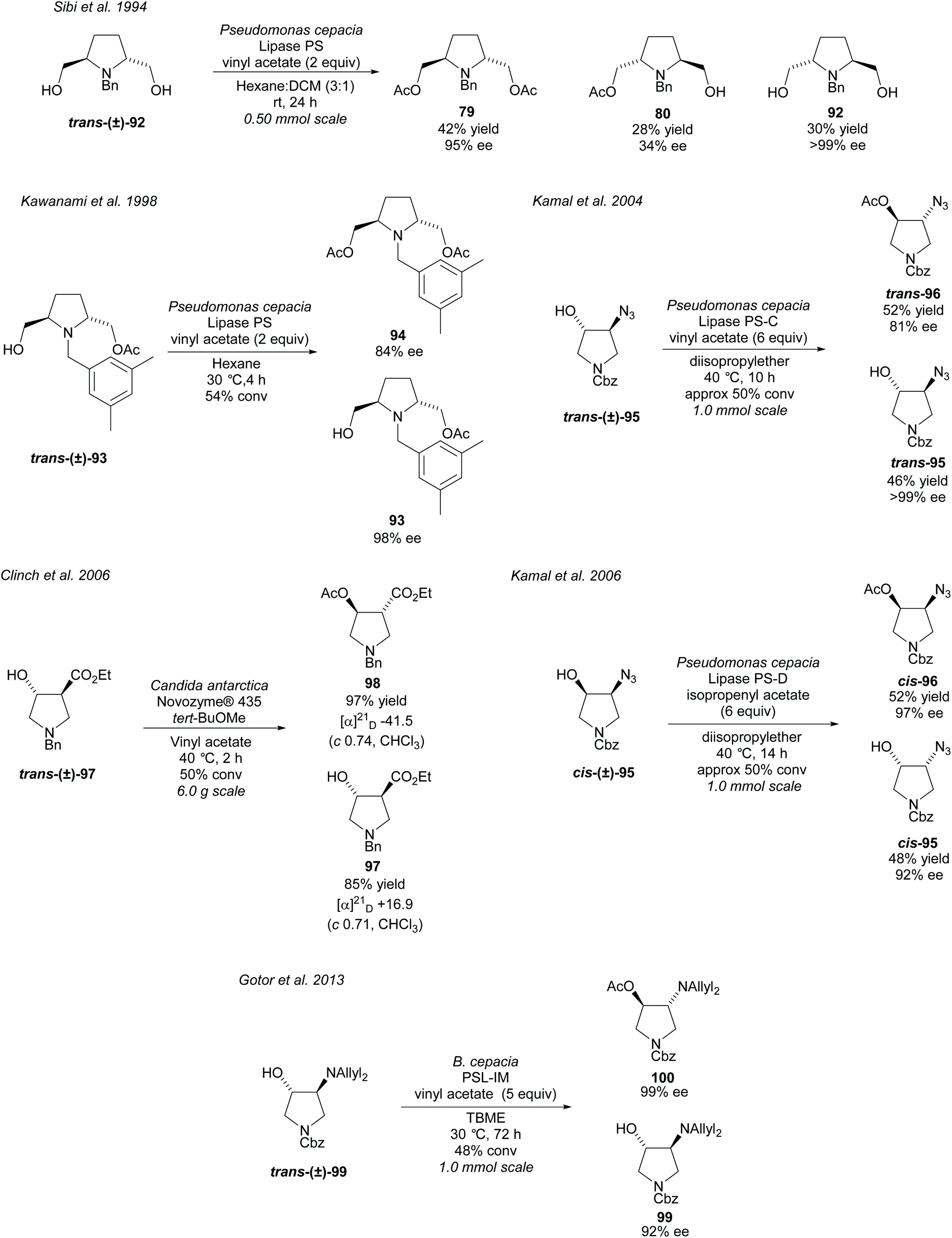 | ||
| Scheme 24 Enzyme mediated transesterification of various disubstituted pyrrolidines. | ||
Pyrrolidine based precursors to nitric oxide synthase inhibitors have been synthesised by Gotor and co-workers via enzyme catalysed esterification of racemic 3,4-disubstituted pyrrolidines (Scheme 25).83 The highest selectivities were observed during the resolution of trans-disubsituted pyrrolidine trans-(±)-101 employing CAL-A (IMMCALA-T2-150) obtaining the returned alcohol 102 in optically pure form. A pyridine regioisomer was also investigated, as both cis- and trans-isomers, affording lower enantioselectivities for the returned alcohols of both diastereoisomers. Hydrolysis of acetylated pyrrolidine 102 was also investigated however significantly lower enantioselectivities were obtained for both the returned acetylated pyridine and alcohol product. C–H Activation of racemic monosubstituted N-Boc pyrrolidines employing the rhodium catalysed decomposition of methyl aryldiazoacetates was reported by Davies et al. as a method to provide access to β-amino acid derivatives (Scheme 26).84,85 Excellent enantioselectivities and diastereoselectivities were obtained, exhibiting control over three stereogenic centres in the final disubstituted pyrrolidine. Limited substrate scope was investigated with variation only occurring at the 2-position in the racemic pyrrolidine with the exception of the kinetic resolution of pyrrolidine cis-(±)-105. Brigaud et al. employed phenyl magnesium bromide to selectively consume one diastereoisomer of a diastereomeric mixture of oxazolopyrrolidine rac-107 (Scheme 27).86 As the rate of addition to (S)-107 was faster than the addition to (R)-107, but less selective, (S)-107 could effectively be destroyed leaving (R)-oxazolopyrrolidine 107 to be recovered in excellent diastereoselectivity and good yield. (R,R)-Disubstituted pyrrolidine 108 was also obtained in excellent diastereoselectivity.
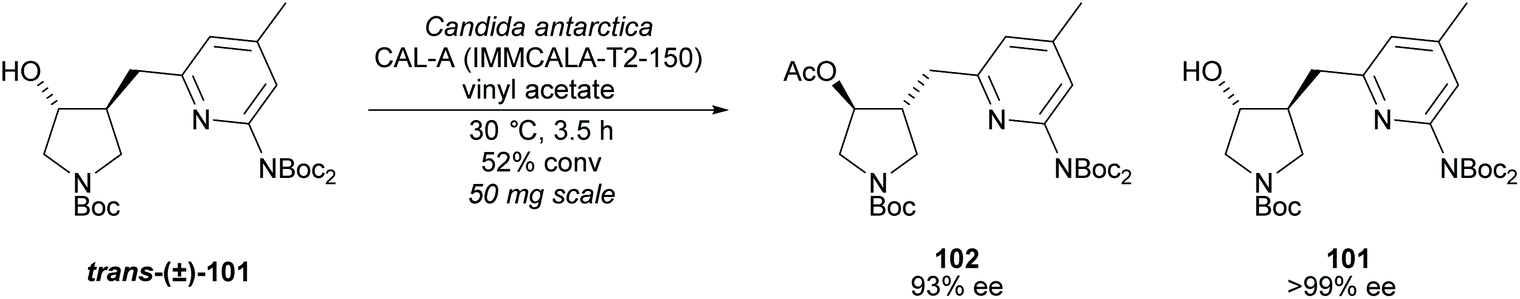 | ||
| Scheme 25 Nitric oxide synthase inhibitors synthesised by Gotor et al. via enzyme catalysed acetylation. | ||
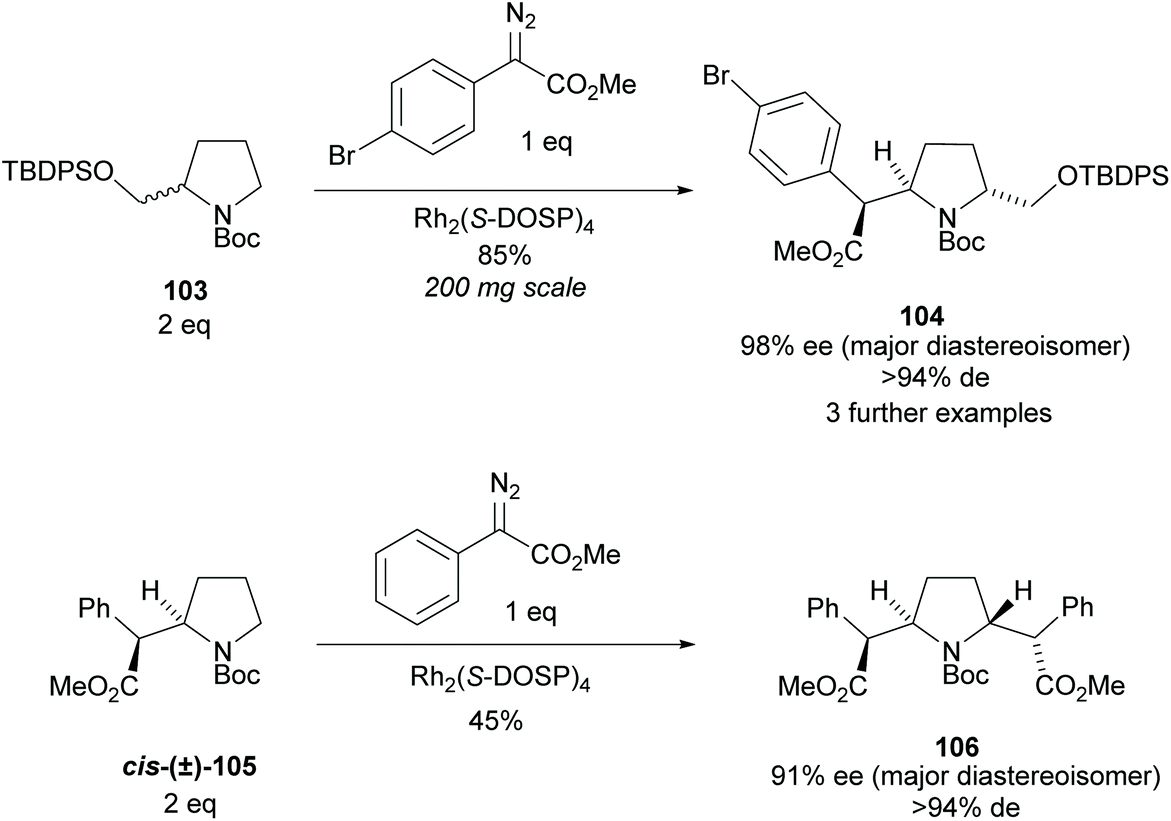 | ||
| Scheme 26 C–H activation of N-Boc pyrrolidines reported by Davies et al. | ||
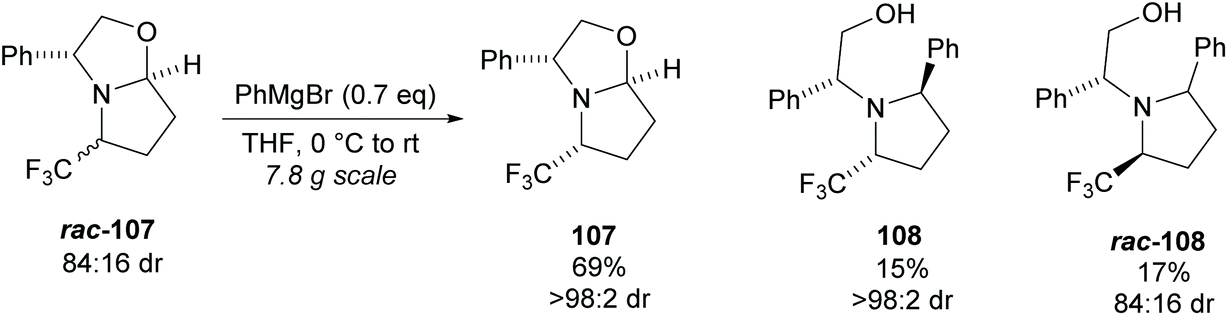 | ||
| Scheme 27 Phenyl magnesium bromide employed to consume one diastereoisomer reported by Brigaud et al. | ||
4. Trisubstituted pyrrolidines
Trisubstituted pyrrolidines have been accessed via an organocatalysed aza-Henry/aza-Michael cascade reaction by Huang et al. (Scheme 28).87 The reversable aza-Henry step provided a modest 2:1 ratio of trans:cis109 and in the subsequent aza-Michael cyclisation only the trans isomer reacted to form trisubstituted pyrrolidine 110. Using rac–trans–aza-Henry product 109 in an independent reaction provided pyrrolidine 110 in a moderate 50% ee and good 80% yield suggesting a moderately selective DKR. Good to excellent enantioselectivities and yield were observed across the 17 examples during the optimised cascade reaction employing modified cinchona alkaloid catalyst 111.
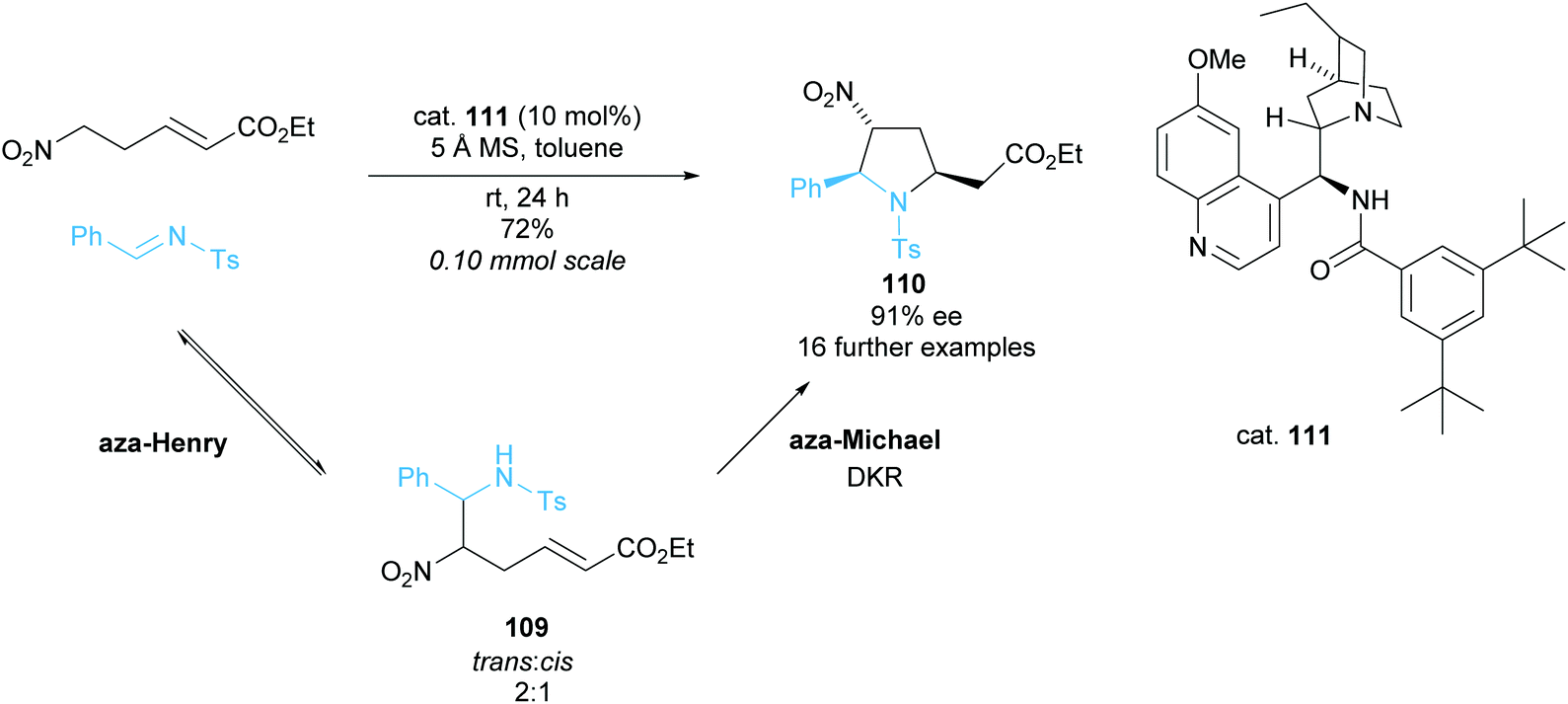 | ||
| Scheme 28 aza-Henry/aza-Michael cascade reaction reported by Huang et al. | ||
Imino-C-disaccharide precursors featuring a trisubstituted pyrrolidine subunit were synthesised by Cardona and co-workers (Scheme 29).88,89 A partial kinetic resolution of racemic five-membered ring nitrone (±)-cis-112 was achieved in a 1,3-diploar cycloaddition with isolevoglucosenone 114.88 Moderate enantioselectivity was obtained for the recovered nitrone. They adapted this method, employing both levoglucosenone 115 and its isomer 114, to achieve a parallel kinetic resolution (PKR) obtaining two distinct products in moderate to good yields.89 PKRs have the advantage of reacting both enantiomers at the same or similar rates to two different, separable products.14 Unlike simple KRs, the 1:1 ratio of the starting enantiomers is maintained throughout the reaction leading to higher enantioselectivities and higher yields.
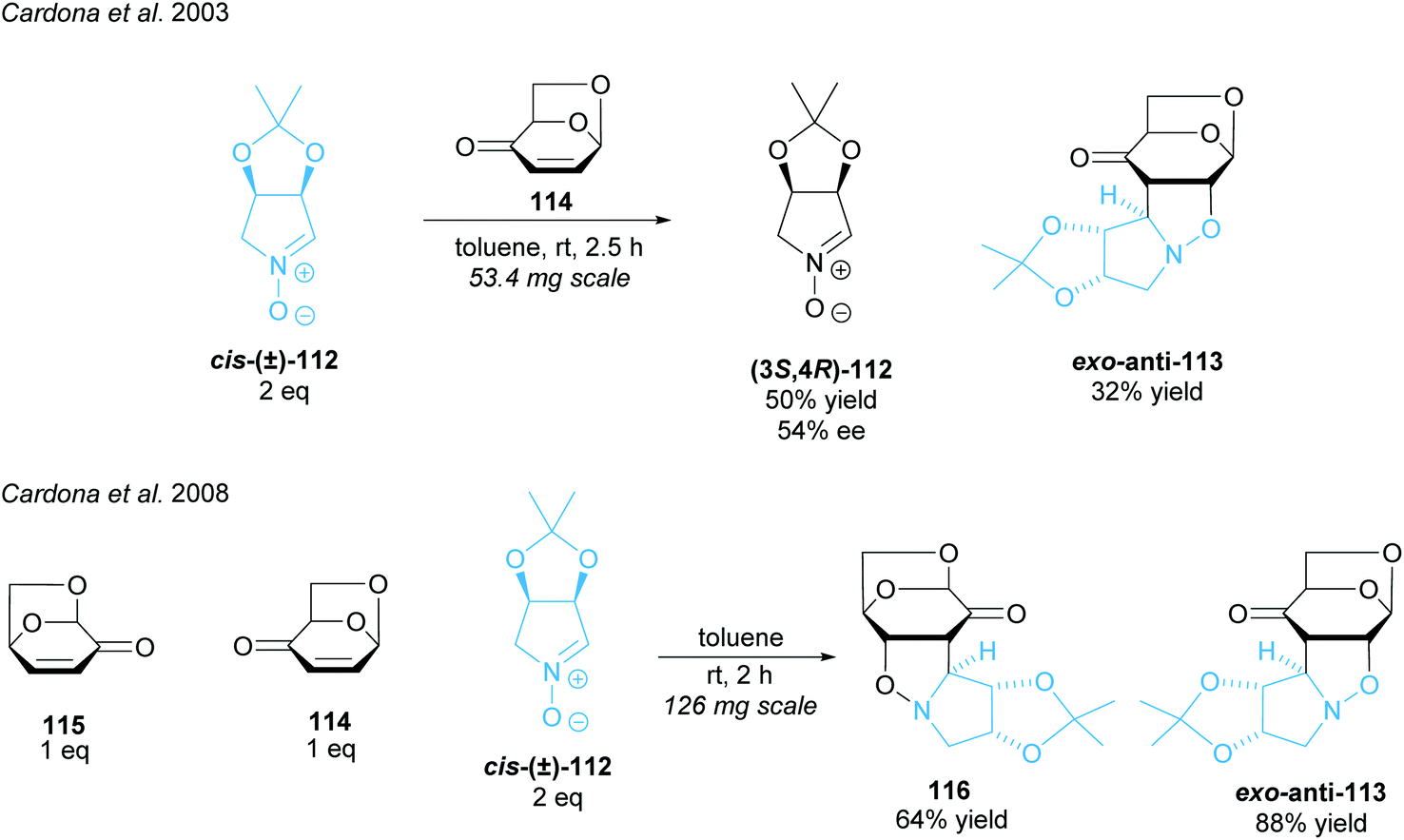 | ||
| Scheme 29 PKR method employing levoglucosenone and isolevoglucosenone reported by Cardona et al. | ||
The 1,3-dipolar cycloaddition of α,β-unsaturated δ- and γ-lactones to racemic nitrone cis-(±)-112 was investigated by Chmielewski et al. (Scheme 30).90 δ-Lactone 117 exhibited good yields with excellent diastereoselectivity, obtaining exo-cycloaddition product 118 exclusively, with (3R,4S)-nitrone 112 recovered in good enantioselectivity. δ-Lactones 119 and 120 achieved similarly good results, however, in the cycloaddition of γ-lactone 121 to racemic nitrone cis-(±)-112 both the exo and endo products were formed. In this case, the mismatched (3R,4S)-nitrone reacts, forming the endo product.
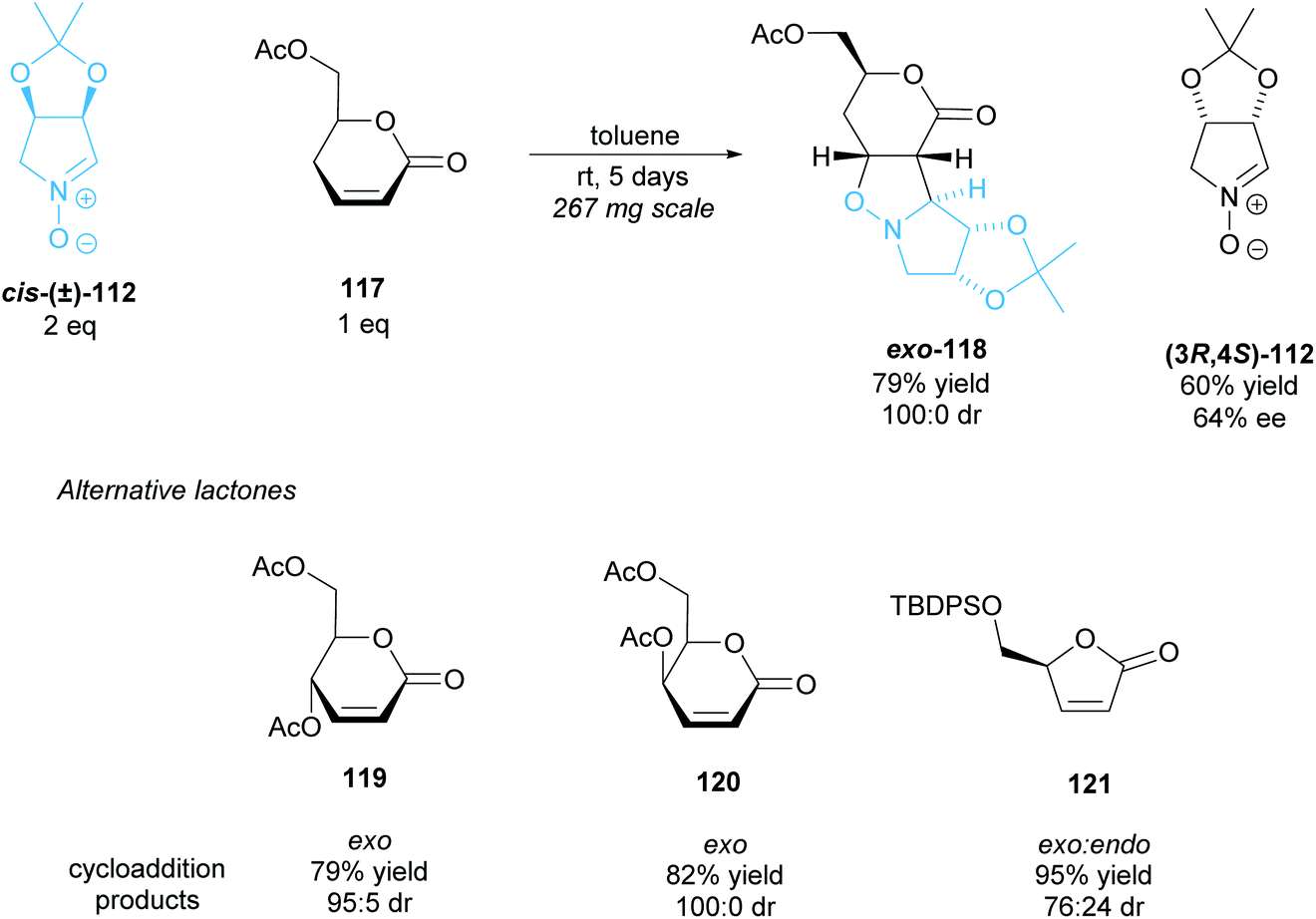 | ||
| Scheme 30 1,3-dipolar cycloaddition of racemic nitrone 112 to various lactones reported by Chmielewski et al. | ||
Use of enzymes in kinetic resolutions are a popular technique again for trisubstituted substrates. Most uses of enzymes in this class of pyrrolidines are aimed at synthesizing intermediates for nucleoside analogues, often for use as enzyme inhibitors. Mason et al. resolved lactone trans-(±)-122 using Novozyme® 435 obtaining the acid product 123 in a moderate yield with excellent enantioselectivity (Scheme 31).91 Similary excellent enantioselectivies were obtained for the recovered starting alcohol 122. From acid 123 in three further steps, they obtained disubstituted pyrrolidine 124 which is a key intermediate in the synthesis of a number of nucleoside analogues.
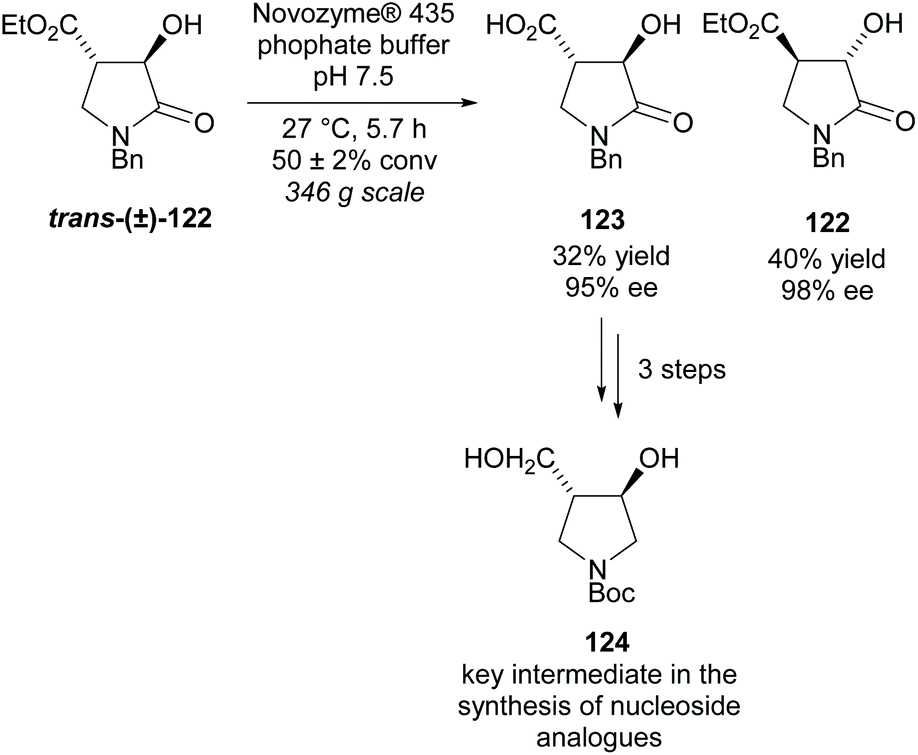 | ||
| Scheme 31 Enzyme catalysed hydrolysis reported by Mason et al. | ||
In 2008, Gotor et al. resolved bicyclic pyrrolidine (±)-125 employing CAL-A in an enzyme catalysed carbamate formation (Scheme 32).92 Excellent enantioselectivities were obtained for both the carbamate product 126 and the unreacted amino ester 125. In the same year, Mason et al. resolved fluoropyrrolidine (±)-127via CAL-B catalysed acetylation of an alcohol.93 Good enantioselectivity was obtained for the unreacted alcohol 127 with moderate yields for both the unreacted alcohol and acetylated product 128. The fluoropyrrolidine was subsequently used to synthesise a phosphorylase enzyme inhibitor.
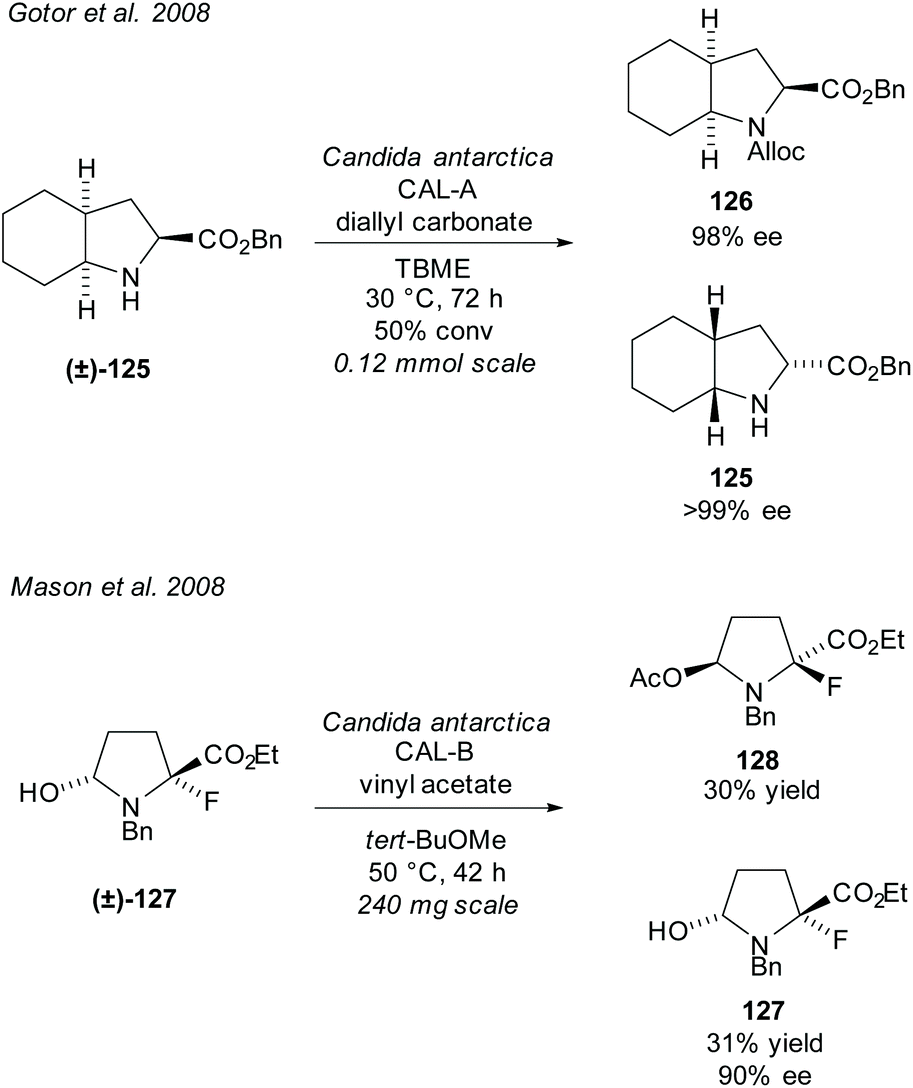 | ||
| Scheme 32 Alcohol and amine enzyme mediated protection. | ||
Takabe and co-workers resolved hydroxylactam rac-129via lipase PS-D catalysed acetylation (Scheme 33).94 Excellent yields and enantioselectivities were obtained for both the acetylated product 130 and unreacted alcohol 129. They developed a method to racemise acetate 130 producing racemic alcohol rac-129, creating a kinetic resolution/racemisation cycle. Through this, they could obtain alcohol 129 in 96% yield with >99% ee after 5 cycles. Alcohol 129 was transformed in 10 steps to polyhydroxylated indolizidine (−)-2-epi-lentiginosine 131, an inhibitor of glycosidases and was afforded in a 16% yield over 13 total steps.
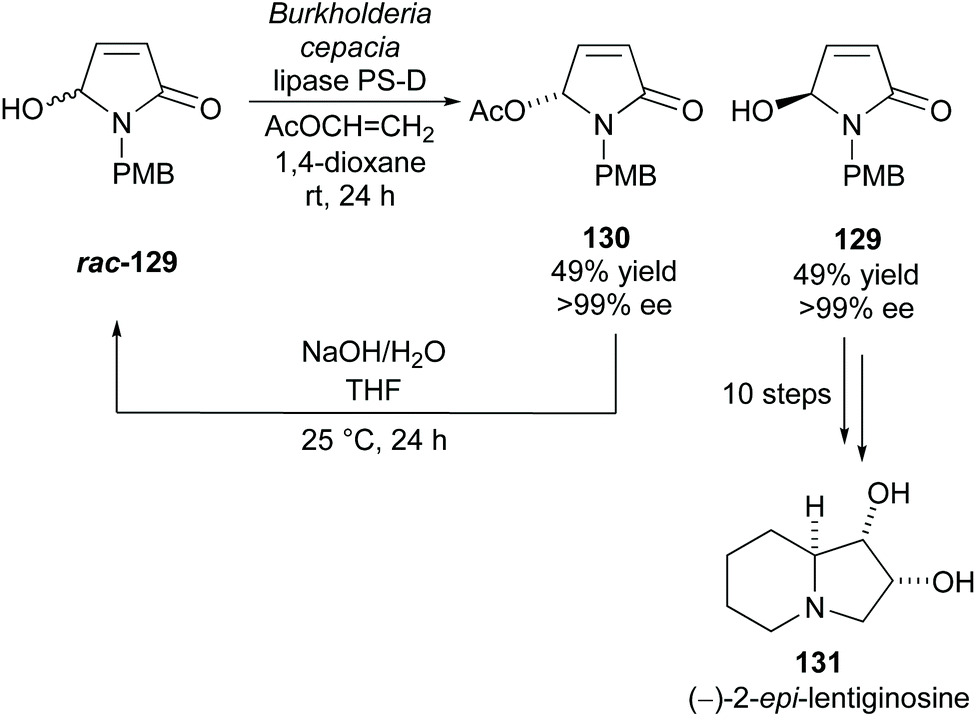 | ||
| Scheme 33 Synthesis of (−)-2-epi-lentiginosine reported by Takabe et al. | ||
In 2016, Juhl et al. developed a method for accessing 3,4-substituted fluoropyrrolidines by fluorination of pyrrolidines employing prolinol and imidazolidinone based organocatalysts to obtain the syn- and anti-fluoropyrrolidines, respectfully.95 They used this methodology in the resolution of racemic pyrrolidine rac-132, obtaining the anti-fluoropyrrolidine 133 in excellent enantioselectivity with a good yield (Scheme 34).
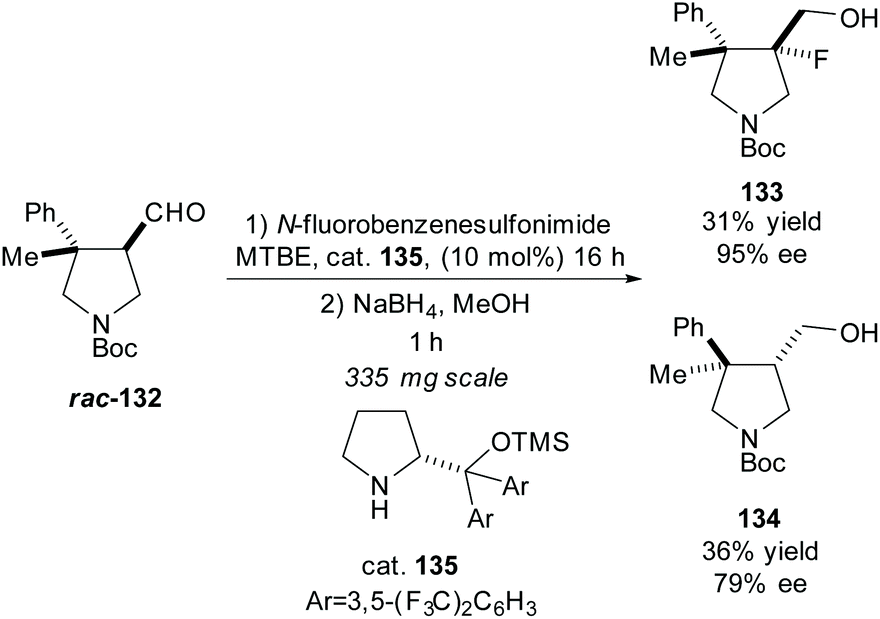 | ||
| Scheme 34 Fluorination of trisubstituted pyrrolidine rac-132 reported by Juhl et al. | ||
5. Multisubstituted pyrrolidines
Tetrasubstituted pyrrolidines have been accessed from racemic cyclopropanes via [3 + 2] annulation with aldimines. Initial work in this area achieved a highly diastereoselective reaction employing various ytterbium and scandium-based catalysts.96,97 In 2010, Johnson et al. developed a dynamic kinetic resolution variant of this reaction employing magnesium iodide and a chiral pyridine-based ligand (Scheme 35).98 2-Methoxybenzyl was selected as the protecting group, producing excellent enantioselectivities and yields. They obtained good diastereoselectivities with good yields and excellent enantioselectivities across the 11 examples.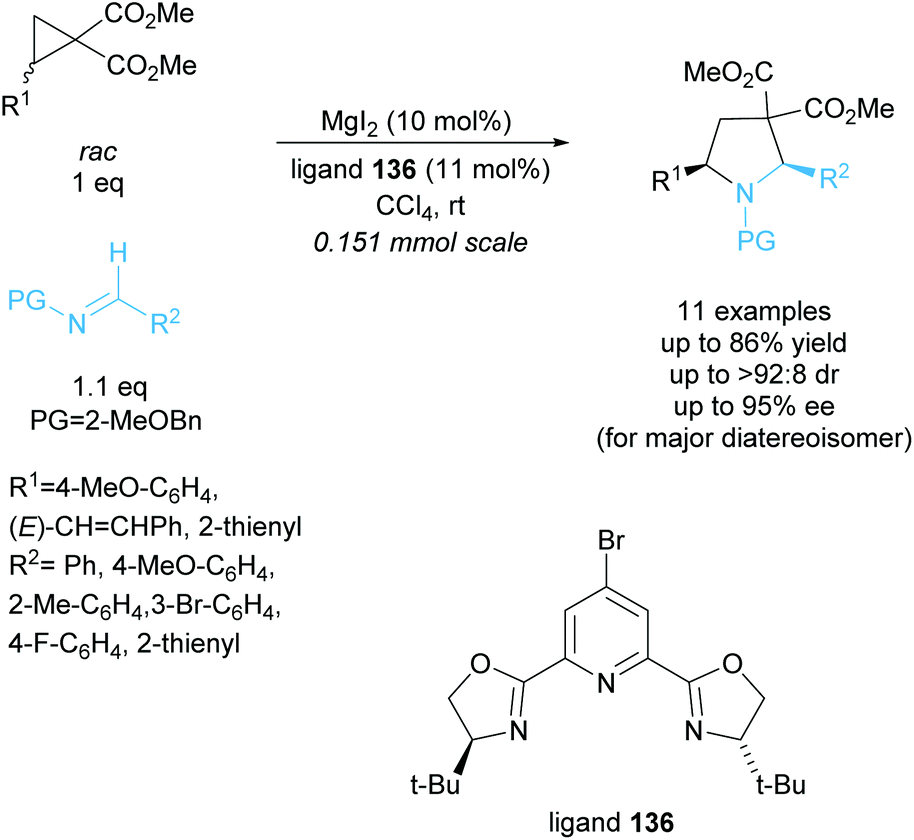 | ||
| Scheme 35 DKR [3 + 2] annulation of racemic cyclopropanes to aldimines reported by Johnson et al. | ||
In 2015, Chai et al. reported the [3 + 2] annulation of racemic aziridines with indoles employing a copper(I)/BINAP derived catalyst system (Scheme 36).99 The kinetic resolution obtained excellent yields, diastereoselectivities, and enantioselectivities across 25 examples. Under the optimised reaction conditions, 2-arylaziridines containing election donating groups proceeded more rapidly in the reaction than those containing electron withdrawing groups. 2-Alkylaziridines were reported to barely react and so are a limitation for this reaction system.
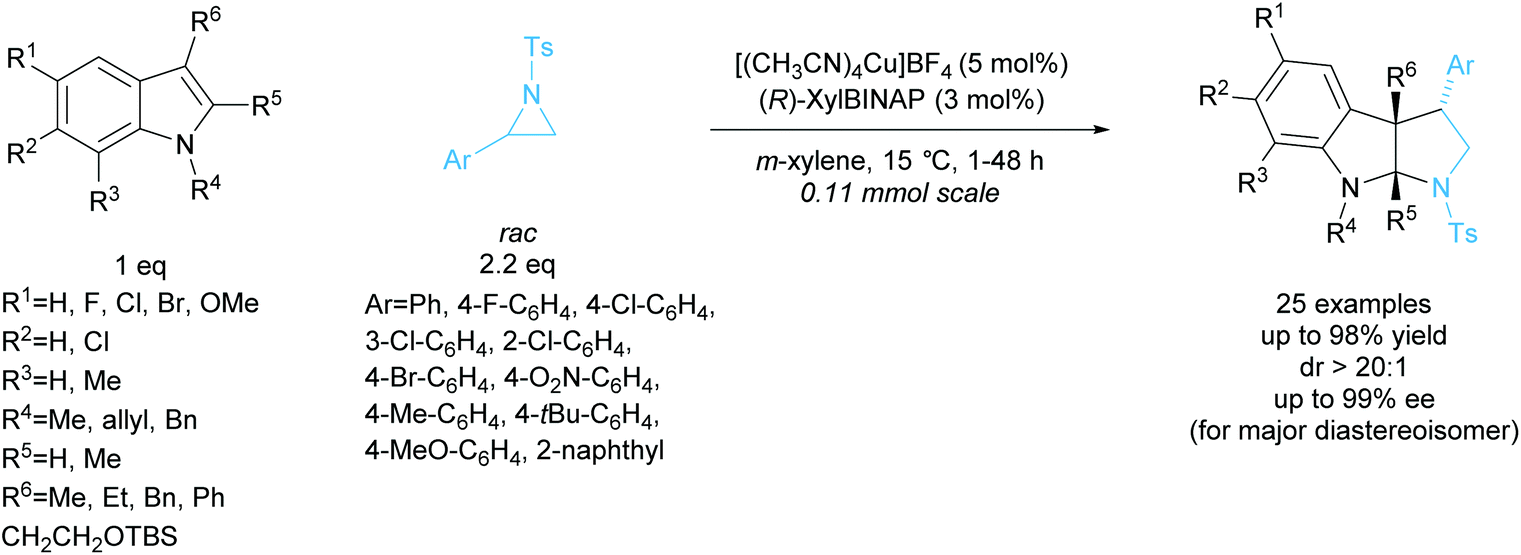 | ||
| Scheme 36 DKR [3 + 2] annulation of racemic 2-arylaziridines with indoles reported by Chai et al. | ||
Epoxy-pyrrolidines have been synthesised via epoxy-annulations of vinyl sulfonium salts with α-amido ketones by Aggarwal and co-workers (Scheme 37).100 They successfully achieved a kinetic resolution of racemic amido-ketone rac-137 obtaining epoxy-pyrrolidine 139 in a good yield with good diastereoselectivity and enantioselectivity. This was the only KR example reported. One popular technique for the synthesis of multisubstituted pyrrolidines is the 1,3-dipolar cycloaddition of azomethine ylides with various dipolarphiles.5
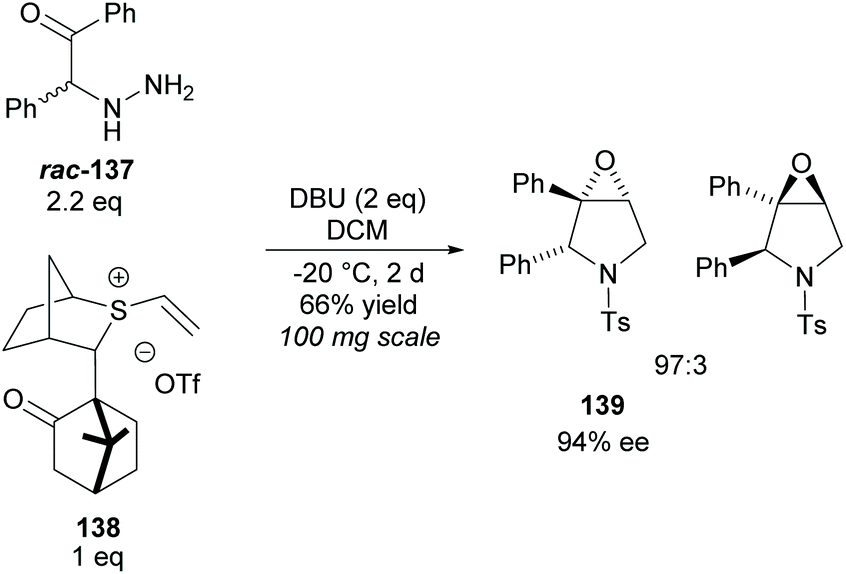 | ||
| Scheme 37 Synthesis of epoxy-pyrrolidines via KR of racemic amido-ketone rac-137 reported by Aggarwal et al. | ||
The kinetic resolution of racemic axially chiral allenes during 1,3-dipolar cycloadditions has been investigated by Gong et al. employing a bisphosphoric acid organocatalyst 140 (Scheme 38).101,102 They obtained excellent yields across 12 examples, affording the 3-methylenepyrrolidines in good enantioselectivities and the recovered starting allenes in excellent enantioselectivities.
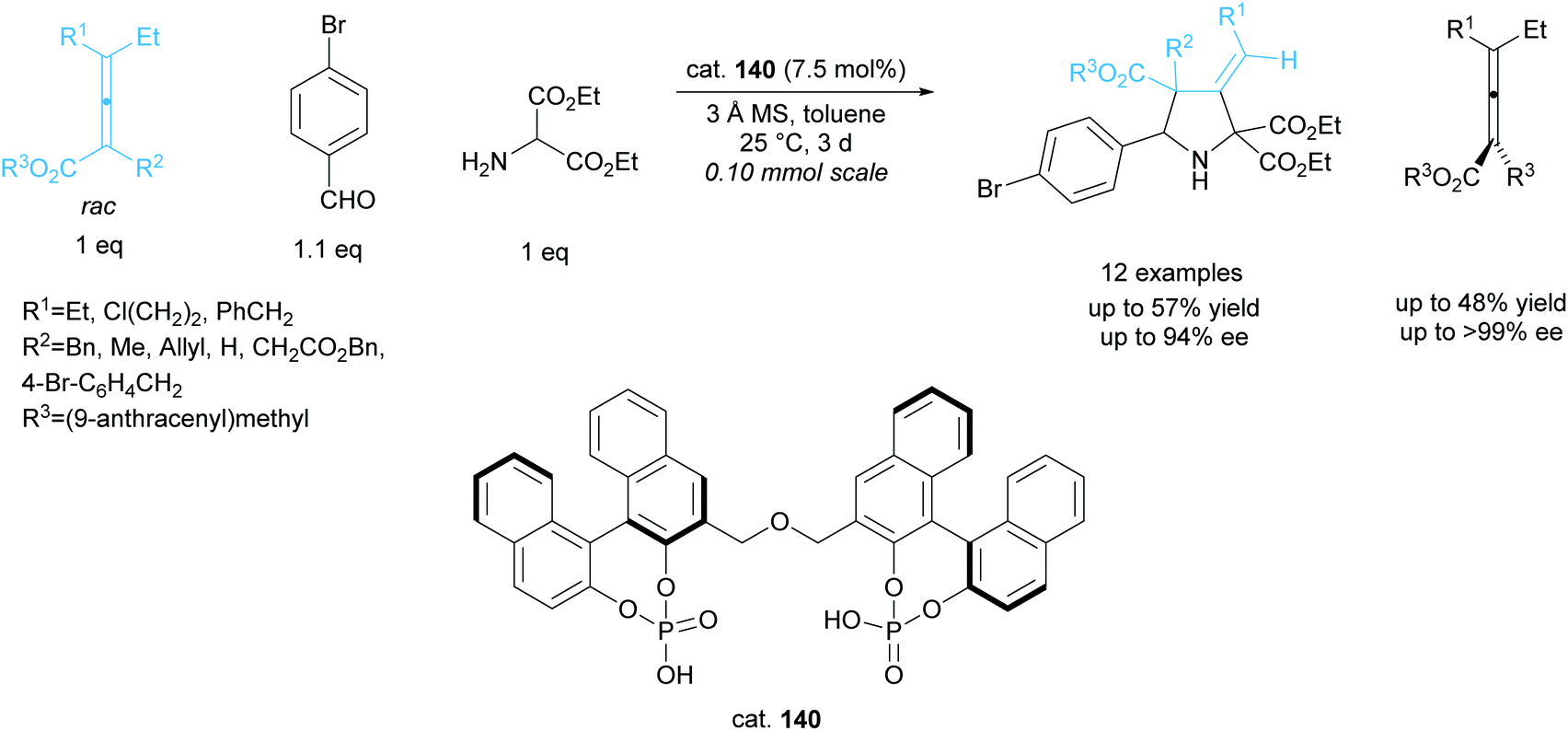 | ||
| Scheme 38 1,3-dipolar cycloaddition of azomethine ylides with racemic allenes reported by Gong et al. | ||
Waldmann et al. resolved racemic tropanes in a copper catalysed 1,3-dipolar cycloaddition reaction with various azomethine ylides aiming to synthesise polycyclic structures containing pyrrolidine and tropane fragments (Scheme 39).103 A series of azomethine ylides varying the aryl group provided good yields and excellent enantioselectivities for the polycyclic products. In the kinetic resolution of different tropanes under modified reaction conditions, excellent enantioselectivities of up to 99% ee were obtained for the recovered starting tropanes with similarly excellent enantioselectivities obtained for the polycyclic products. They went on to conduct one-pot sequential 1,3-dipolar cycloadditions using chiral ligand 141 in the first cycloaddition and rac-BINAP in the second cycloaddition. Each addition used a different azomethine ylide affording two distinct polycyclic products in excellent enantioselectivities of up to 99% ee and yields up to 49%.
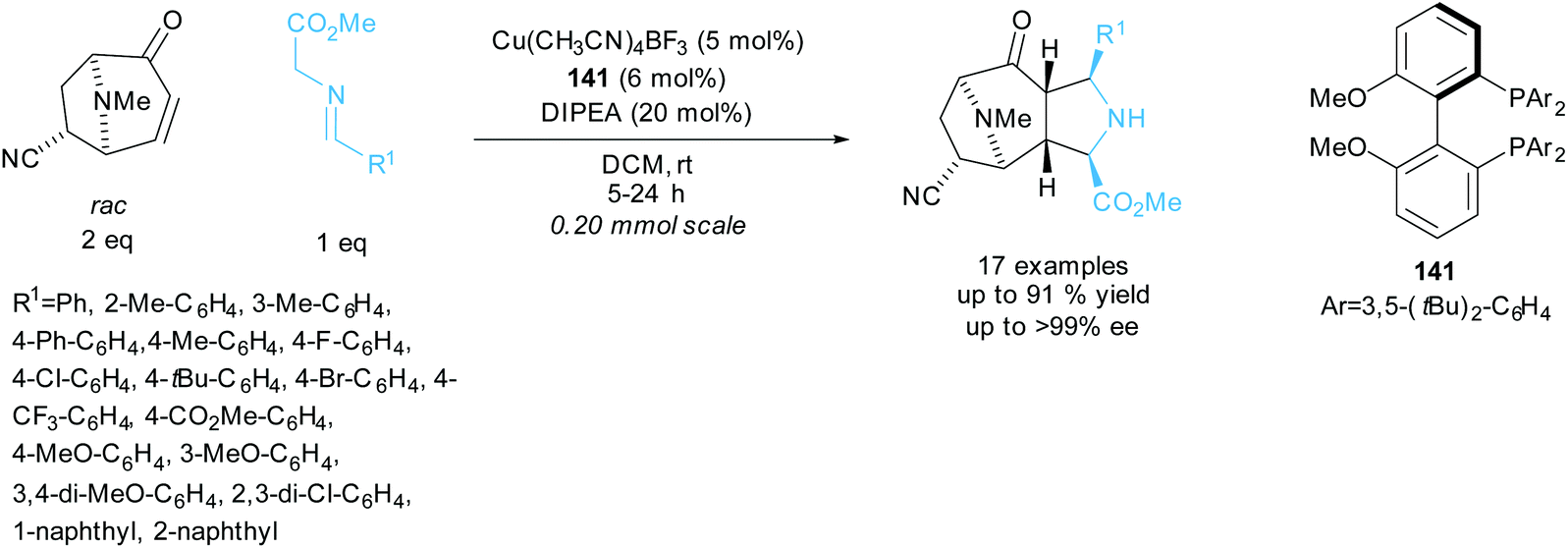 | ||
| Scheme 39 1,3-dipolar cycloaddition of azomethine ylides with racemic tropanes reported by Waldmann et al. | ||
In 2018, Wang and co-workers employed a similar technique to resolve racemic cyclopentenediones producing bicyclic pyrrolidines (Scheme 40).104 Silver catalysis was employed in the 1,3-cycloaddtion reaction between cyclopentenediones and azomethine ylides, achieving excellent enantioselectivities for both the recovered starting cyclopentenediones and the bicyclic pyrrolidine products. Good enantioselectivity was also obtained for the spiro-moiety containing cyclopentenedione 143.
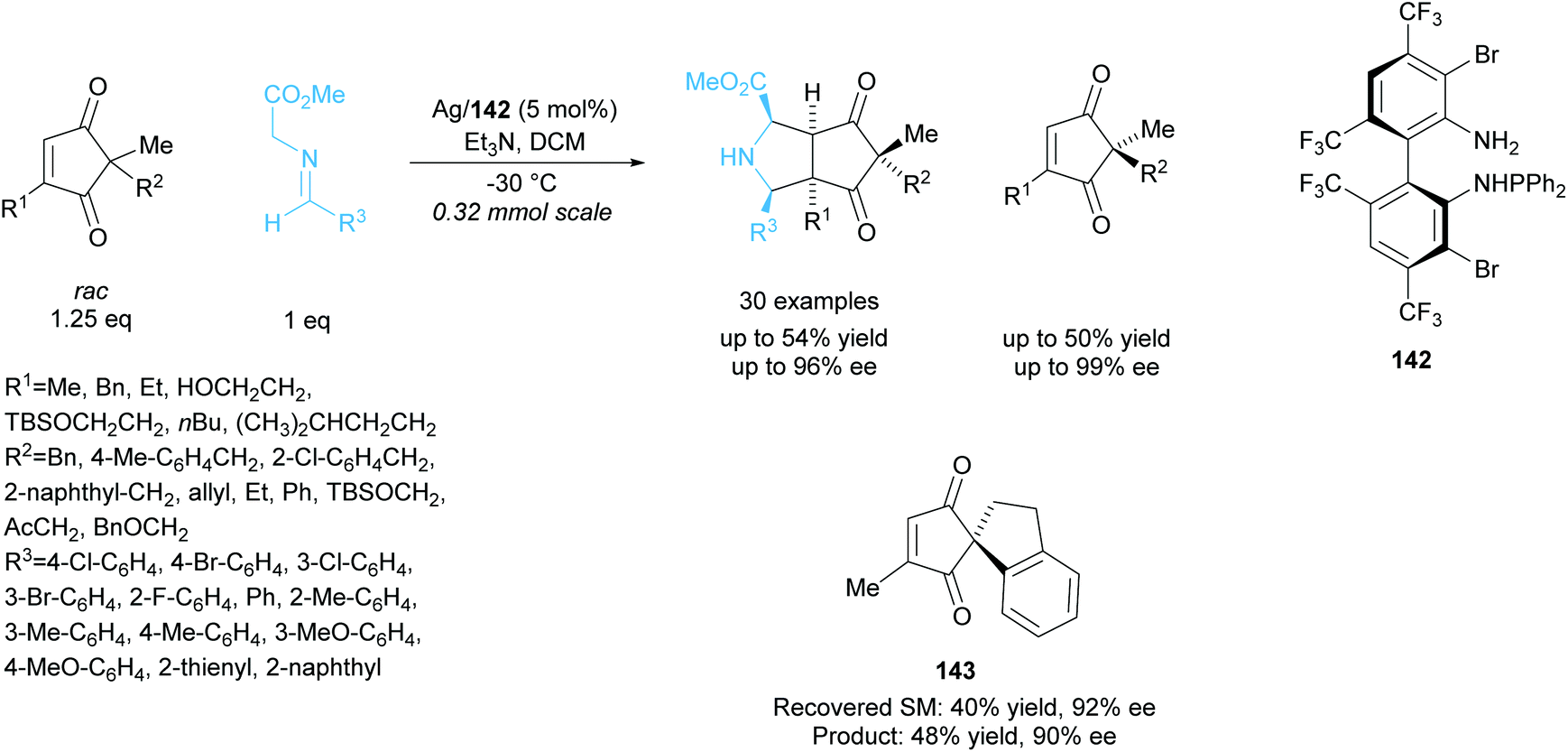 | ||
| Scheme 40 1,3-dipolar cycloaddition of azomethine ylides with racemic cyclopenenediones reported by Wang et al. | ||
Further work by the group employed biphenyl ligand 142 in the copper catalysed 1,3-dipolar cycloaddition between racemic alkylidene norcamphors and azomethine ylides (Scheme 41).105 Spirocyclic pyrrolidine products were obtained in good yields with moderate to excellent enantioselectivities. The kinetic resolution of a series of different alkylidene norcamphors achieved good to excellent enantioselectivities for both products and recovered starting materials. Fused norcamphors also exhibited excellent enantioselectivities.
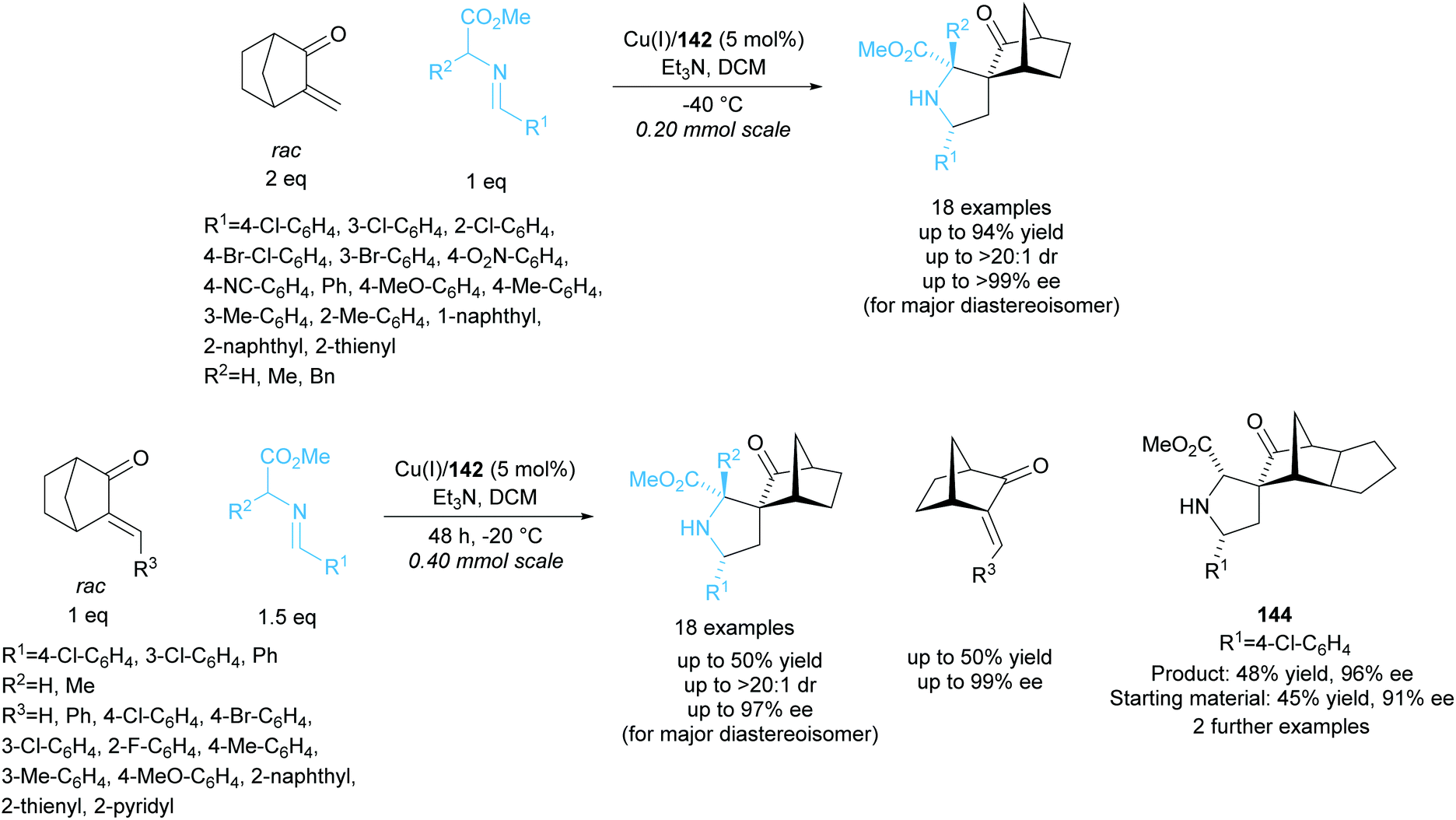 | ||
| Scheme 41 1,3-dipolar cycloaddition of azomethine ylides with racemic alkylidene norcamphors reported by Wang et al. | ||
The same group resolved a series of racemic exo-3-oxodicyclopentadienes in a copper catalysed 1,3-cycloaddition reaction with a glycine aldimino ester (Scheme 42).106 They obtained excellent enantioselectivities for both the recovered exo-3-oxodicyclopentadienes and polycyclic pyrrolidine products. One racemic endo-3-oxodicyclopentadiene was resolved under the same conditions and achieved excellent enantioselectivities of 95% ee for both recovered starting material and pyrrolidine product.
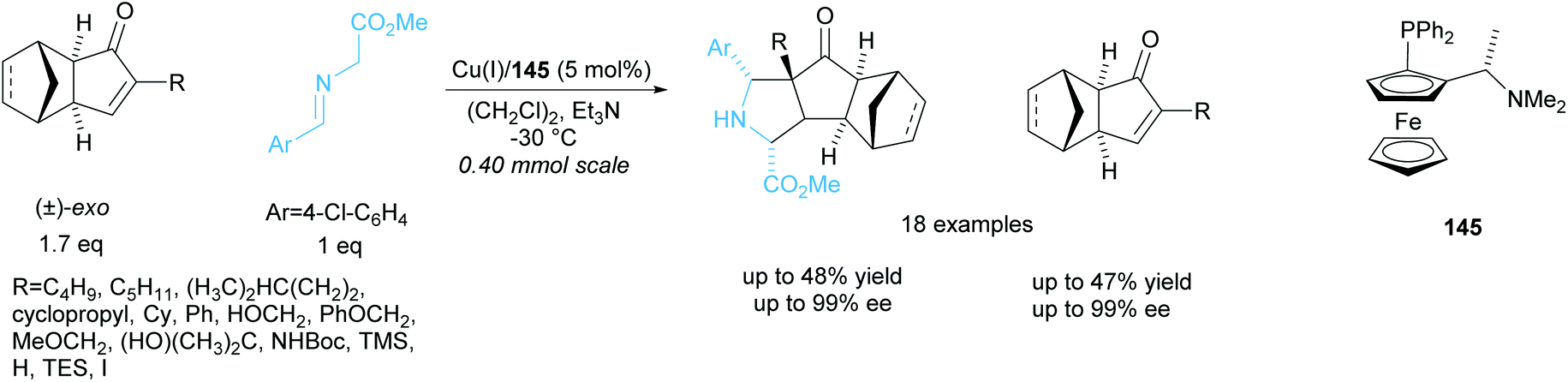 | ||
| Scheme 42 1,3-cycloaddition of racemic oxodicyclopentadienes with azomethine ylides reported by Wang et al. | ||
In 2018, Xu and co-workers developed a parallel kinetic resolution involving a copper catalysed 1,3-dipolar cycloaddition between racemic Morita–Baylis–Hillman (MBH) adducts rac-146 and glycine aldimino esters (Scheme 43).107 Pyrrolidines containing four stereogenic centres including one all-carbon quaternary were obtained in excellent diastereoselectivities and enantioselectivities. Glutamic acid derivatives were also produced during the reaction and were obtained in similarly excellent enantioselectivities. When MBH adducts containing aliphatic groups, ethyl or cyclohexyl, were used a significant decrease in enantioselectivity was observed, obtaining enantioselectivities as low as 42% ee for the glutamic acid product.
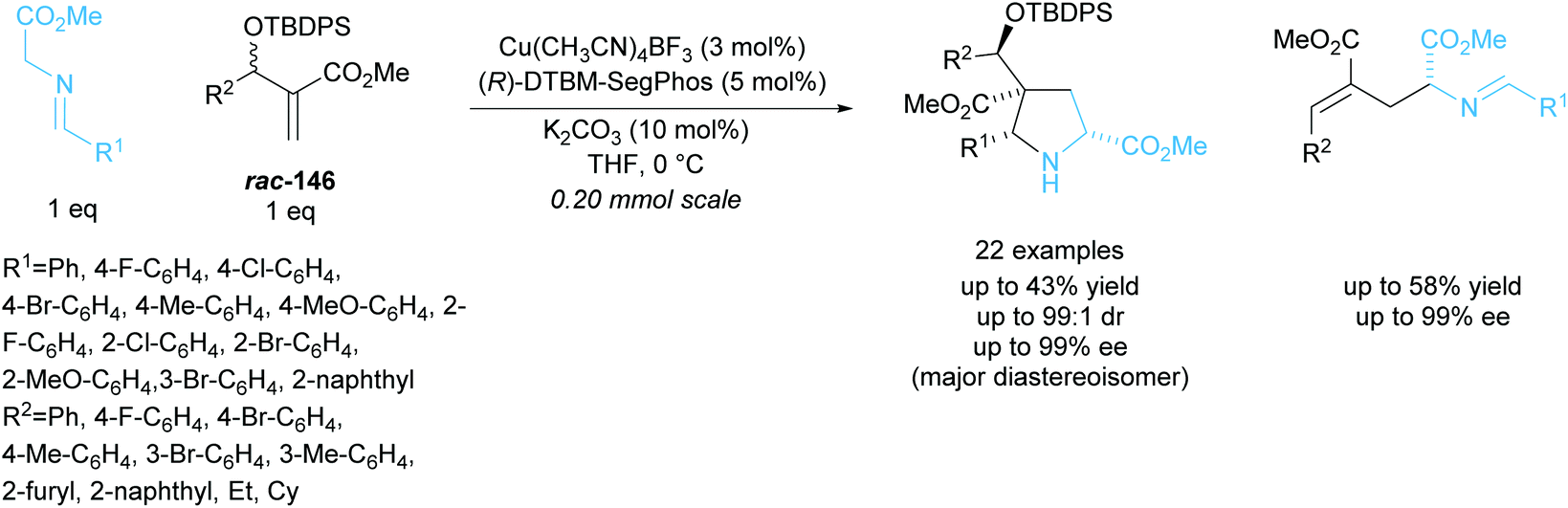 | ||
| Scheme 43 PKR 1,3-dipolar cycloaddition between MBH adducts and azomethine ylides reported by Xu et al. | ||
There are a very limited number of examples of enzymatic resolution of racemic multisubstituted pyrrolidines. Hideg et al. preformed acetylations of alcohols on the 3-position of three different pyrrolidines (Scheme 44).108 These all occurred with moderate enantioselectivities to good enantioselectivities obtained for each of the recovered alcohols and acetylated products.
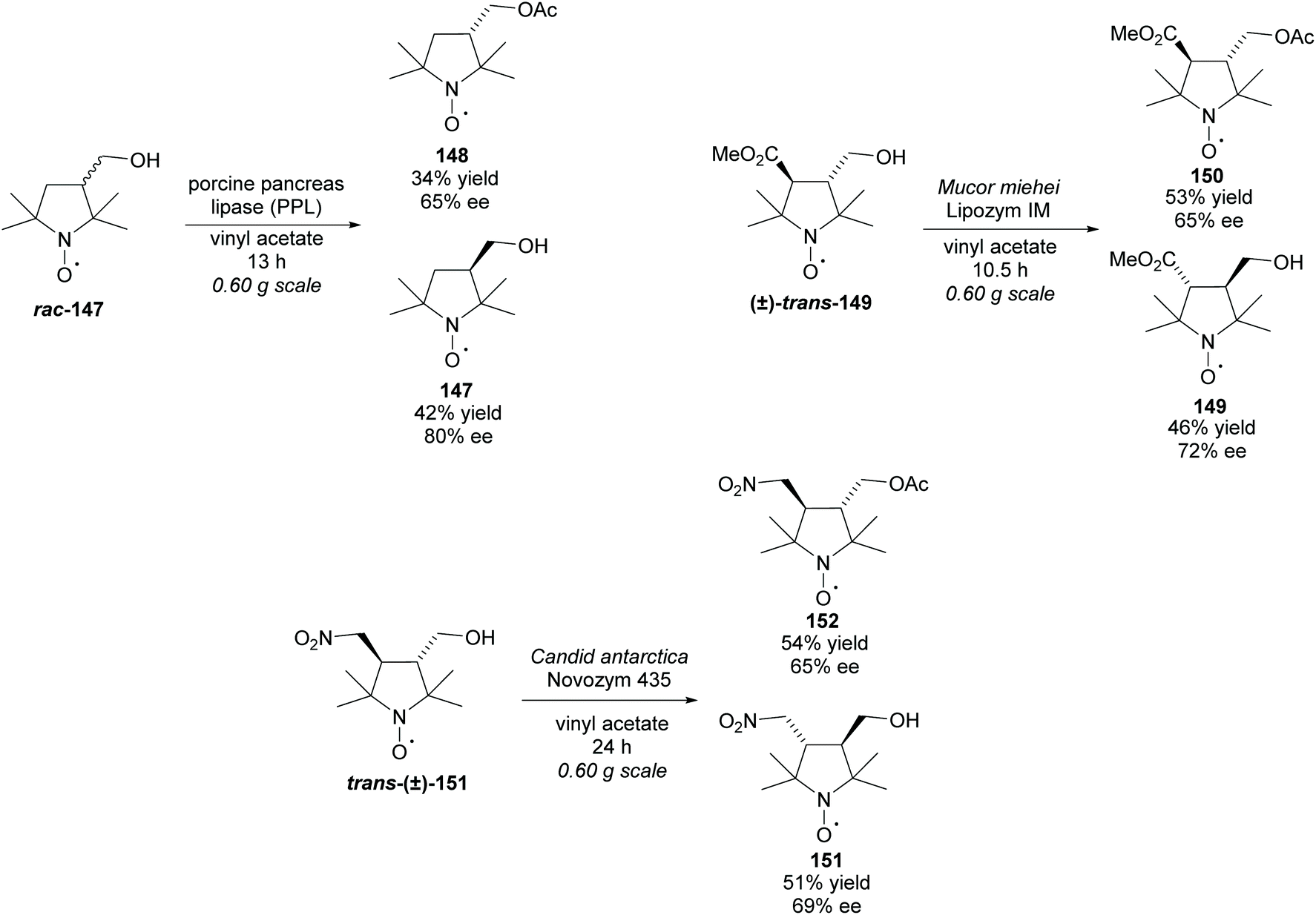 | ||
| Scheme 44 Enzymatic acetylation of various multisubstituted pyrrolidines reported by Hideg et al. | ||
6. Summary
Application of kinetic resolution remains an important technique to obtain enantiomerically pure substituted pyrrolidines. Enzymatic methodology is heavily relied upon for monosubstituted substrates and is regularly used for disubstituted substrates. The poor substrate diversity obtained through this technique is often outweighed by high selectivity. Whilst these biotransformations offer high selectivity, there is significant future scope in this area to develop better synthetic chemistry routes that are both selective and efficient in delivering the desired products with high diversity and complexity. In contrast, it is also important to note that although enzymes are widely used for some substrate classes, methods involving annulations and cycloadditions are favoured for heavily substituted substrates, with the added benefit of additional points of diversity. Therefore, there is scope from both the synthetic chemistry and biological chemistry community to identify and develop complementary techniques that can improve the efficiency of these transformations, and at the same time expand the diversity of products that can be obtained using this methodology.A final point to reflect on as illustrated in this review, is that traditional KR methods are far more commonly used than DKR and PKR methods despite the limit on overall yields, leaving significant scope for the development of new synthetic methodology in this area as well. Strategies and methodologies that embrace these methods offer significant advantages and would significantly enhance the atom efficiency in an area of continuing significant importance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC, RedBrick Molecular and University of Sheffield for support.Notes and references
- D. R. Witty, G. Alvaro, D. Derjean, G. M. P. Giblin, K. Gunn, C. Large, D. T. Macpherson, V. Morisset, D. Owen, J. Palmer, F. Rugiero, S. Tate, C. A. Hinckley and H. Naik, ACS Med. Chem. Lett., 2020, 11, 1678 CrossRef CAS PubMed.
- J. Auerbach, S. A. Weissman, T. J. Blacklock, M. R. Angeles and K. Hoogsteen, Tetrahedron Lett., 1993, 34, 931 CrossRef CAS.
- C. S. Pak and G. H. Lee, J. Org. Chem., 1991, 56, 1128 CrossRef CAS.
- R. D. Birkenmeyer and F. Kagan, J. Med. Chem., 1970, 13, 616 CrossRef CAS PubMed.
- L. Wei, X. Chang and C. J. Wang, Acc. Chem. Res., 2020, 53, 1084 CrossRef CAS PubMed.
- Z. Zhang, N. A. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS PubMed.
- E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel and B. U. W. Maes, Chem. – Eur. J., 2012, 18, 10092 CrossRef CAS PubMed.
- N. J. Race, I. R. Hazelden, A. Faulkner and J. F. Bower, Chem. Sci., 2017, 8, 5248 RSC.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
- M. Y. Han, J. Y. Jia and W. Wang, Tetrahedron Lett., 2014, 55, 784 CrossRef CAS.
- H. B. Kagan and J. C. Fiaud, in Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, John Wiley & Sons, Inc., New York, 1988, vol. 18, pp. 249–330 Search PubMed.
- J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS.
- E. Vedejs and X. Chen, J. Am. Chem. Soc., 1997, 119, 2584 CrossRef CAS.
- J. R. Dehli and V. Gotor, Chem. Soc. Rev., 2002, 31, 365 RSC.
- P. Beak, S. T. Kerrick, S. Wu and J. Chu, J. Am. Chem. Soc., 1994, 116, 3231 CrossRef CAS.
- P. Beak, D. R. Anderson, M. D. Curtis, J. M. Laumer, D. J. Pippel and G. A. Weisenburger, Acc. Chem. Res., 2000, 33, 715 CrossRef CAS PubMed.
- I. Coldham, J. J. Patel and G. Sanchez-Jimenez, Chem. Commun., 2005, 3083 RSC.
- I. Coldham, S. Dufour, T. F. N. Haxell, J. J. Patel and G. Sanchez-Jimenez, J. Am. Chem. Soc., 2006, 128, 10943 CrossRef CAS PubMed.
- T. K. Beng, T. I. Yousaf, I. Coldham and R. E. Gawley, J. Am. Chem. Soc., 2009, 131, 6908 CrossRef CAS PubMed.
- R. K. Dieter, C. M. Topping, K. R. Chandupatla and K. Lu, J. Am. Chem. Soc., 2001, 123, 5132 CrossRef CAS PubMed.
- R. K. Dieter, G. Oba, K. R. Chandupatla, C. M. Topping, K. Lu and R. T. Watson, J. Org. Chem., 2004, 69, 3076 CrossRef CAS PubMed.
- R. K. Dieter, N. Chen and R. T. Watson, Tetrahedron, 2005, 61, 3221 CrossRef CAS.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Rev., 2011, 111, 3998 CrossRef CAS PubMed.
- L. E. Janes and R. J. Kazlauskas, Tetrahedron: Asymmetry, 1997, 8, 3719 CrossRef CAS.
- M. Kurokawa, T. Shindo, M. Suzuki, N. Nakajima, K. Ishihara and T. Sugai, Tetrahedron: Asymmetry, 2003, 14, 1323 CrossRef CAS.
- C. Pousset, R. Callens, M. Haddad and M. Larchevêque, Tetrahedron: Asymmetry, 2004, 15, 3407 CrossRef CAS.
- S. Hu, D. Tat, C. A. Martinez, D. R. Yazbeck and J. Tao, Org. Lett., 2005, 7, 4329 CrossRef CAS PubMed.
- G. Tofani, A. Petri and O. Piccolo, Tetrahedron: Asymmetry, 2015, 26, 638 CrossRef CAS.
- A. Horiguchi and K.-I. Mochida, Biosci. Biotechnol. Biochem., 1995, 59, 1287 CrossRef CAS.
- S. Yamada, H. Shimada, R. Yamada, H. Shiratori-Takano, N. Sayo, T. Saito, H. Takano, T. Beppu and K. Ueda, Biotechnol. Lett., 2014, 36, 595 CrossRef CAS PubMed.
- A. Arizpe, M. Rodríguez-Mata, F. J. Sayago, M. J. Pueyo, V. Gotor, A. I. Jiménez, V. Gotor-Fernández and C. Cativiela, Tetrahedron: Asymmetry, 2015, 26, 1469 CrossRef CAS.
- R. Lihammar, R. Millet and J. E. Bäckvall, Adv. Synth. Catal., 2011, 353, 2321 CrossRef CAS.
- S. Takayama, S. T. Lee, S. C. Hung and C. H. Wong, Chem. Commun., 1999, 127 RSC.
- M. Winkler, D. Meischler and N. Klempier, Adv. Synth. Catal., 2007, 349, 1475 CrossRef CAS.
- M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2008, 350, 807 CrossRef.
- D. Minato, H. Arimoto, Y. Nagasue, Y. Demizu and O. Onomura, Tetrahedron, 2008, 64, 6675 CrossRef CAS.
- G. R. Pettit, Y. Kamano, C. L. Herald, A. A. Tuinman, F. E. Boettner, H. Kizu, J. M. Schmidt, L. Baczynskyi, K. B. Tomer and R. J. Bontems, J. Am. Chem. Soc., 1987, 109, 6883 CrossRef CAS.
- D. Lavergne, C. Mordant, V. Ratovelomanana-Vidal and J. P. Genet, Org. Lett., 2001, 3, 1909 CrossRef CAS PubMed.
- C. Mordant, S. Reymond, V. Ratovelomanana-Vidal and J. P. Genêt, Tetrahedron, 2004, 60, 9715 CrossRef CAS.
- C. Mordant, S. Reymond, H. Tone, D. Lavergne, R. Touati, B. B. Hassine, V. Ratovelomanana-Vidal and J. P. Genet, Tetrahedron, 2007, 63, 6115 CrossRef CAS.
- M. Akashi, N. Arai, T. Inoue and T. Ohkuma, Adv. Synth. Catal., 2011, 353, 1955 CrossRef CAS.
- A. Viso, N. E. Lee and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 9373 CrossRef CAS.
- J. Yun and S. L. Buchwald, Chirality, 2000, 12, 476 CrossRef CAS PubMed.
- M. S. Lall, G. Hoge, T. P. Tran, W. Kissel, S. T. Murphy, C. Taylor, K. Hutchings, B. Samas, E. L. Ellsworth, T. Curran and H. D. H. Showalter, J. Org. Chem., 2012, 77, 4732 CrossRef CAS PubMed.
- M. Kitamura, T. Ohkuma, M. Tokunaga and R. Noyori, Tetrahedron: Asymmetry, 1990, 1, 1 CrossRef CAS.
- K. Hattori, A. Yamada, S. Kuroda, T. Chiba, M. Murata and K. Sakane, Bioorg. Med. Chem. Lett., 2002, 12, 383 CrossRef CAS PubMed.
- N. A. Magnus, B. A. Astleford, D. L. T. Laird, T. D. Maloney, A. D. McFarland, J. R. Rizzo, J. C. Ruble, G. A. Stephenson and J. P. Wepsiec, J. Org. Chem., 2013, 78, 5768 CrossRef CAS PubMed.
- D. H. Bao, X. S. Gu, J. H. Xie and Q. L. Zhou, Org. Lett., 2017, 19, 118 CrossRef CAS PubMed.
- D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem. – Eur. J., 2003, 9, 4796 CrossRef CAS PubMed.
- D. V. Gribkov and K. C. Hultzsch, Chem. Commun., 2004, 730 RSC.
- D. V. Gribkov, K. C. Hultzsch and F. Hampel, J. Am. Chem. Soc., 2006, 128, 3748 CrossRef CAS PubMed.
- A. L. Reznichenko, F. Hampel and K. C. Hultzsch, Chem. – Eur. J., 2009, 15, 12819 CrossRef CAS PubMed.
- A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2013, 32, 1394 CrossRef CAS.
- H. N. Nguyen and K. C. Hultzsch, Eur. J. Org. Chem., 2019, 2019, 2592 CrossRef CAS PubMed.
- P. Koóš, I. Špánik and T. Gracza, Tetrahedron: Asymmetry, 2009, 20, 2720 CrossRef.
- H. Takahata, Y. Banba and T. Momose, Tetrahedron, 1991, 47, 7635 CrossRef CAS.
- H. Takahata, Y. Banba, M. Tajima and T. Momose, J. Org. Chem., 1991, 56, 240 CrossRef CAS.
- H. Takahata, Y. Banba and T. Momose, Tetrahedron: Asymmetry, 1990, 1, 763 CrossRef CAS.
- T. Ito, L. E. Overman and J. Wang, J. Am. Chem. Soc., 2010, 132, 3272 CrossRef CAS PubMed.
- Z. D. Aron and L. E. Overman, Org. Lett., 2005, 7, 913 CrossRef CAS PubMed.
- J. J. Lacharity, A. K. Mailyan, K. Y. Chen and A. Zakarian, Angew. Chem., Int. Ed, 2020, 59, 11364 Search PubMed.
- S. Akai, K. Tanimoto, Y. Kanao, S. Omura and Y. Kita, Chem. Commun., 2005, 2369 RSC.
- H. Nemoto, K. Tanimoto, Y. Kanao, S. Omura, Y. Kita and S. Akai, Tetrahedron, 2012, 68, 7295 CrossRef CAS.
- K. Tatsuta, H. Takahashi, Y. Amemiya and M. Kinoshita, J. Am. Chem. Soc., 1983, 105, 4096 CrossRef CAS.
- K. Tatsuta, S. Miyashita, K. Akimoto and M. Kinoshita, Bull. Chem. Soc. Jpn., 1982, 55, 3254 CrossRef CAS.
- S. E. Denmark, A. Thorarensen and D. S. Middleton, J. Am. Chem. Soc., 1996, 118, 8266 CrossRef CAS.
- S. E. Denmark, A. Thorarensen and D. S. Middleton, J. Org. Chem., 1995, 60, 3574 CrossRef CAS.
- Y. Kawanami, H. Moriya, Y. Goto, K. Tsukao and M. Hashimoto, Tetrahedron, 1996, 52, 565 CrossRef CAS.
- R. de L. Barreto, M. J. S. Carpes, C. C. Santana and C. R. D. Correia, Tetrahedron: Asymmetry, 2007, 18, 435 CrossRef CAS.
- F. Faigl, E. Kovács, D. Balogh, T. Holczbauer, M. Czugler and B. Simándi, Cent. Eur. J. Chem., 2014, 12, 25 CAS.
- J. A. Rodríguez-Rodríguez, R. Brieva and V. Gotor, Tetrahedron, 2010, 66, 6789 CrossRef.
- Á. Villar-Barro, V. Gotor and R. Brieva, Tetrahedron: Asymmetry, 2013, 24, 694 CrossRef.
- Á. Villar-Barro, V. Gotor and R. Brieva, Bioorg. Med. Chem., 2014, 22, 5563 CrossRef PubMed.
- V. K. Aggarwal, C. J. Astle, H. Iding, B. Wirz and M. Rogers-Evans, Tetrahedron Lett., 2005, 46, 945 CrossRef CAS.
- M. P. Sibi and J. L. Lu, Tetrahedron Lett., 1994, 35, 4915 CrossRef CAS.
- Y. Kawanami, N. Iizuna and K. Okano, Chem. Lett., 1998, 1231 CrossRef CAS.
- Y. Kawanami, N. Iizuna, K. Maekawa, K. Maekawa, N. Takahashi and T. Kawada, Tetrahedron, 2001, 57, 3349 CrossRef CAS.
- A. Kamal, A. A. Shaik, M. Sandbhor, M. S. Malik and H. Kaga, Tetrahedron Lett., 2004, 45, 8057 CrossRef CAS.
- A. Kamal, A. A. Shaik, M. Sandbhor, M. S. Malik and S. Azeeza, Tetrahedron: Asymmetry, 2006, 17, 2876 CrossRef CAS.
- K. Clinch, G. B. Evans, G. W. J. Fleet, R. H. Furneaux, S. W. Johnson, D. H. Lenz, S. P. H. Mee, P. R. Rands, V. L. Schramm, E. A. Taylor Ringia and P. C. Tyler, Org. Biomol. Chem., 2006, 4, 1131 RSC.
- E. Mas-Claret, B. Al-Yahyaei, S. Chu, R. M. Elliott, M. Imperato, A. Lopez, L. B. Meira, B. J. Howlin and D. K. Whelligan, Bioorg. Med. Chem., 2020, 28, 115507 CrossRef CAS PubMed.
- J. A. Rodríguez-Rodríguez, F. J. Quijada, R. Brieva, F. Rebolledo and V. Gotor, Tetrahedron, 2013, 69, 5407 CrossRef.
- Á. Villar-Barro, V. Gotor and R. Brieva, Green Chem., 2017, 19, 436 RSC.
- H. M. L. Davies and C. Venkataramani, Org. Lett., 2001, 3, 1773 CrossRef CAS PubMed.
- H. M. L. Davies, C. Venkataramani, T. Hansen and D. W. Hopper, J. Am. Chem. Soc., 2003, 125, 6462 CrossRef CAS PubMed.
- H. Lubin, J. Pytkowicz, G. Chaume, G. Sizun-Thomé and T. Brigaud, J. Org. Chem., 2015, 80, 2700 CrossRef CAS PubMed.
- T. Cheng, S. Meng and Y. Huang, Org. Lett., 2013, 15, 1958 CrossRef CAS PubMed.
- F. Cardona, A. Goti and A. Brandi, Org. Lett., 2003, 5, 1475 CrossRef CAS PubMed.
- F. Cardona, D. Lalli, C. Faggi, A. Goti and A. Brandi, J. Org. Chem., 2008, 73, 1999 CrossRef CAS PubMed.
- S. Stecko, K. Paśniczek, M. Jurczak, Z. Urbańczyk-Lipkowska and M. Chmielewski, Tetrahedron: Asymmetry, 2007, 18, 1085 CrossRef CAS.
- K. Clinch, G. B. Evans, R. H. Furneaux, D. H. Lenz, J. M. Mason, S. P. H. Mee, P. C. Tyler and S. J. Wilcox, Org. Biomol. Chem., 2007, 5, 2800 RSC.
- S. Alatorre-Santamaría, M. Rodriguez-Mata, V. Gotor-Fernández, M. C. de Mattos, F. J. Sayago, A. I. Jiménez, C. Cativiela and V. Gotor, Tetrahedron: Asymmetry, 2008, 19, 1714 CrossRef PubMed.
- J. M. Mason, A. S. Murkin, L. Li, V. L. Schramm, G. J. Gainsford and B. W. Skelton, J. Med. Chem., 2008, 51, 5880 CrossRef CAS PubMed.
- T. Muramatsu, S. Yamashita, Y. Nakamura, M. Suzuki, N. Mase, H. Yoda and K. Takabe, Tetrahedron Lett., 2007, 48, 8956 CrossRef CAS.
- K. Fjelbye, M. Marigo, R. P. Clausen and K. Juhl, Org. Lett., 2016, 18, 1170 CrossRef CAS PubMed.
- C. A. Carson and M. A. Kerr, J. Org. Chem., 2005, 70, 8242 CrossRef CAS PubMed.
- Y. B. Kang, Y. Tang and X. L. Sun, Org. Biomol. Chem., 2006, 4, 299 RSC.
- A. T. Parsons, A. G. Smith, A. J. Neel and J. S. Johnson, J. Am. Chem. Soc., 2010, 132, 9688 CrossRef CAS PubMed.
- Z. Chai, Y. M. Zhu, P. J. Yang, S. Wang, S. Wang, Z. Liu and G. Yang, J. Am. Chem. Soc., 2015, 137, 10088 CrossRef CAS PubMed.
- M. G. Unthank, B. Tavassoli and V. K. Aggarwal, Org. Lett., 2008, 10, 1501 CrossRef CAS PubMed.
- J. Yu, L. He, X. H. Chen, J. Song, W. J. Chen and L. Z. Gong, Org. Lett., 2009, 11, 4946 CrossRef CAS PubMed.
- J. Yu, W. J. Chen and L. Z. Gong, Org. Lett., 2010, 12, 4050 CrossRef CAS PubMed.
- H. Xu, C. Golz, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2016, 55, 7761 CrossRef CAS PubMed.
- H. C. Liu, L. Wei, R. Huang, H. Y. Tao, H. Cong and C. J. Wang, Org. Lett., 2018, 20, 3482 CrossRef CAS PubMed.
- C. Shen, Y. Yang, L. Wei, W. W. Dong, L. W. Chung and C. J. Wang, iScience, 2019, 11, 146 CrossRef CAS PubMed.
- X. Chang, X. S. Sun, C. Che, Y. Z. Hu, H. Y. Tao and C. J. Wang, Org. Lett., 2019, 21, 1191 CrossRef CAS PubMed.
- Y. Yuan, Z.-J. Zheng, L. Li, X.-F. Bai, Z. Xu, Y.-M. Cui, J. Cao, K.-F. Yang and L.-W. Xu, Adv. Synth. Catal., 2018, 360, 3002 CrossRef CAS.
- J. Bálint, V. Kiss, G. Egri, T. Kálai, Á. Demeter, M. Balog, E. Fogassy and K. Hideg, Tetrahedron: Asymmetry, 2004, 15, 671 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |