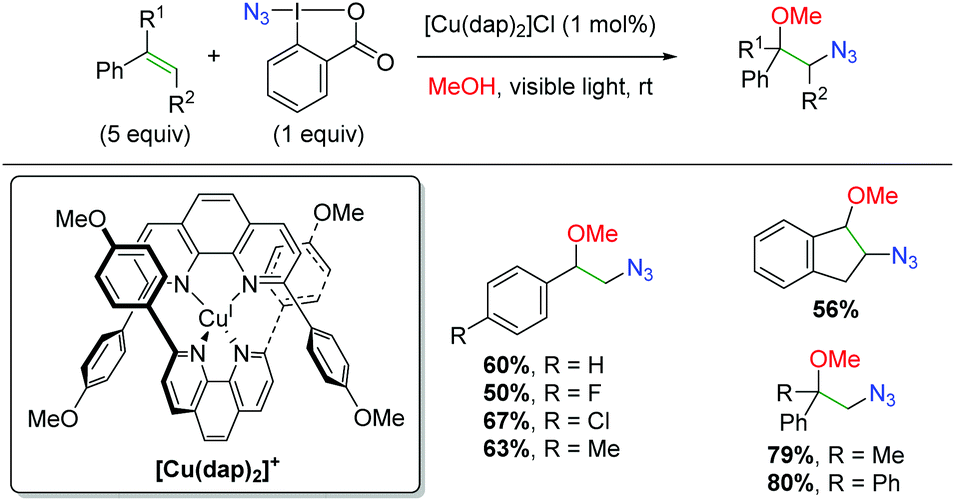
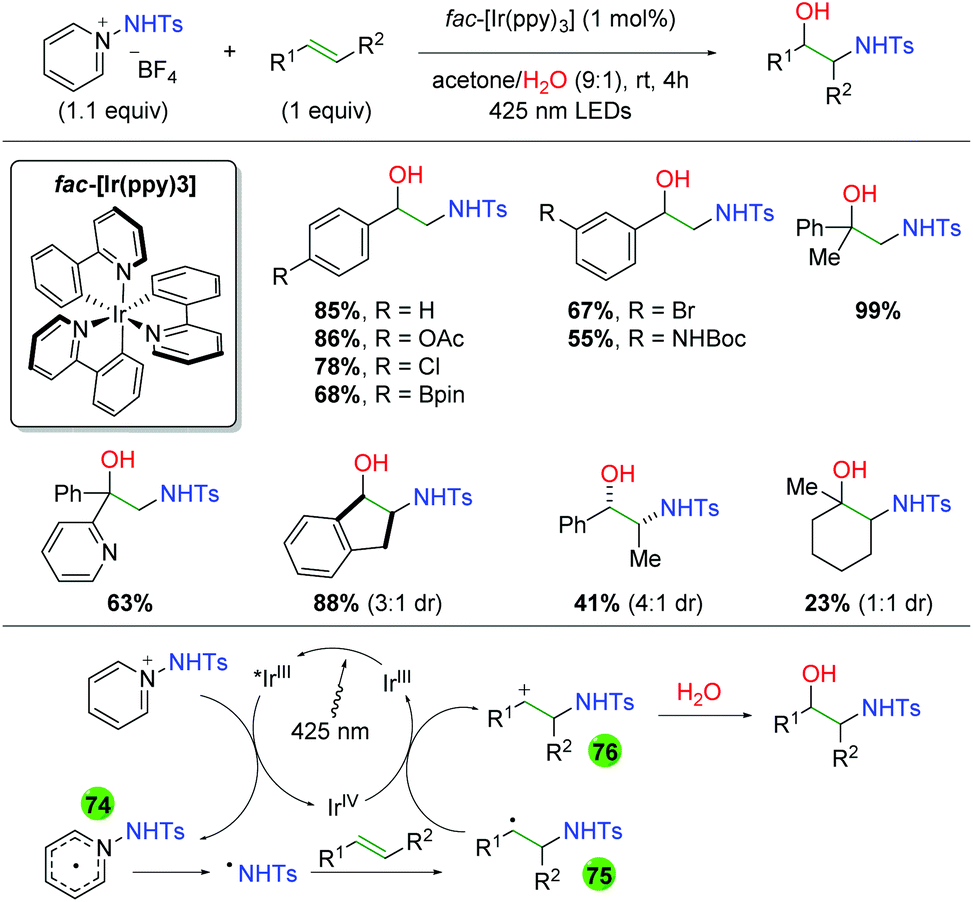
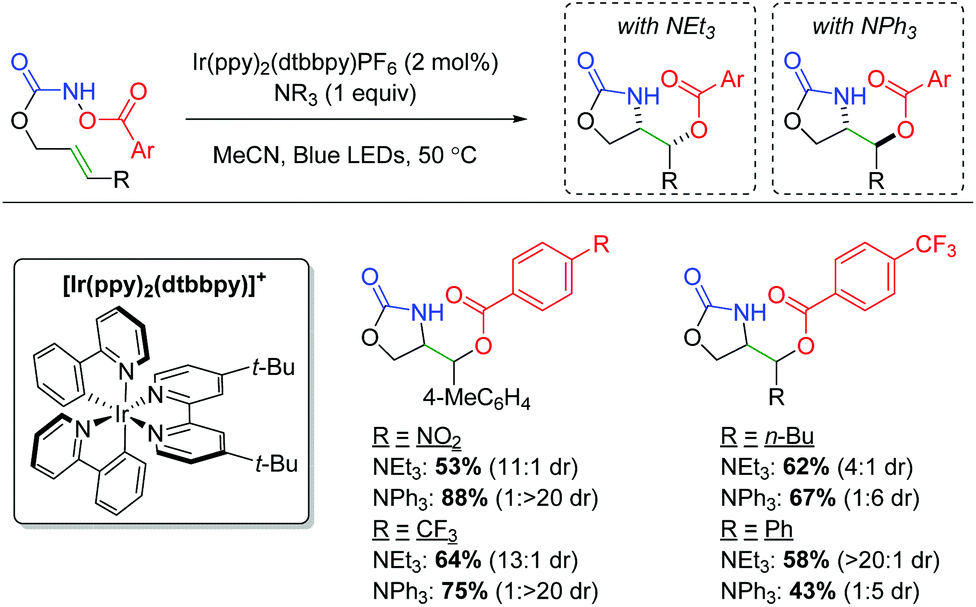
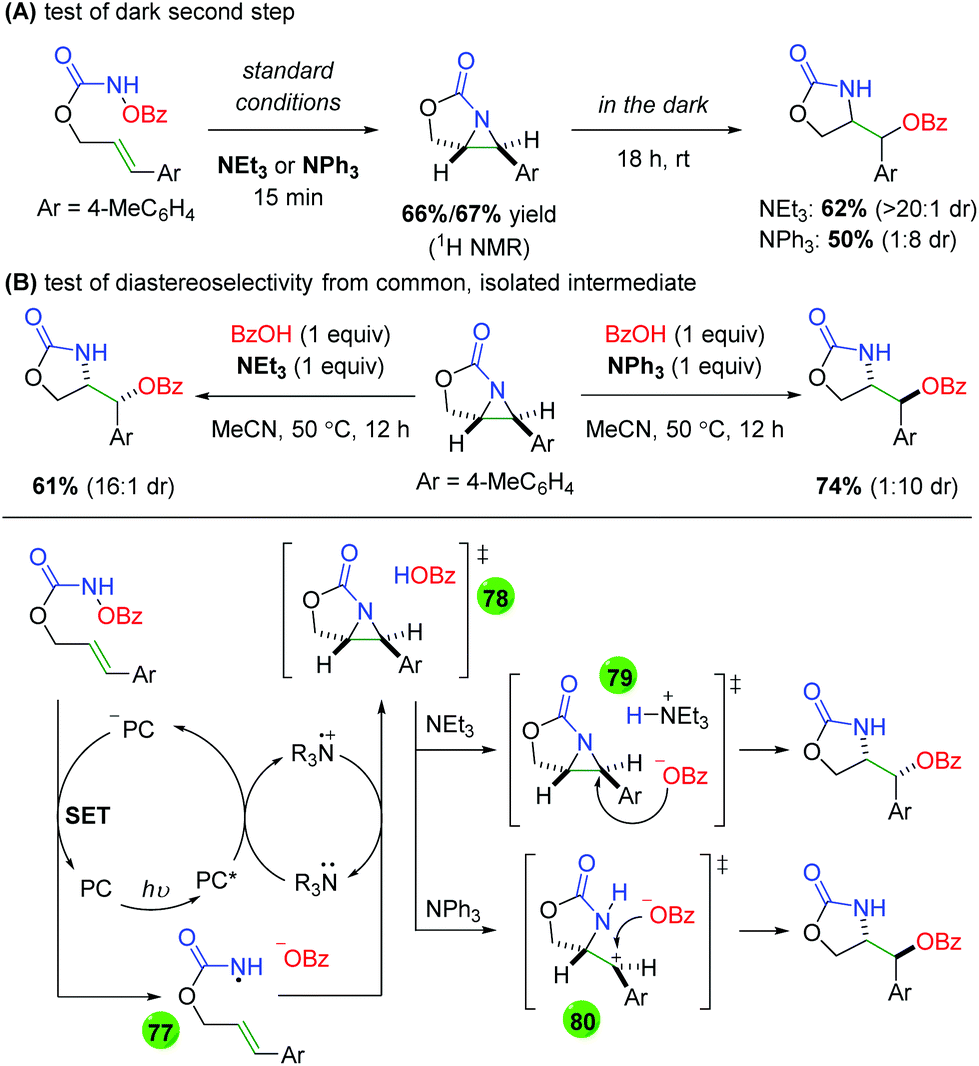
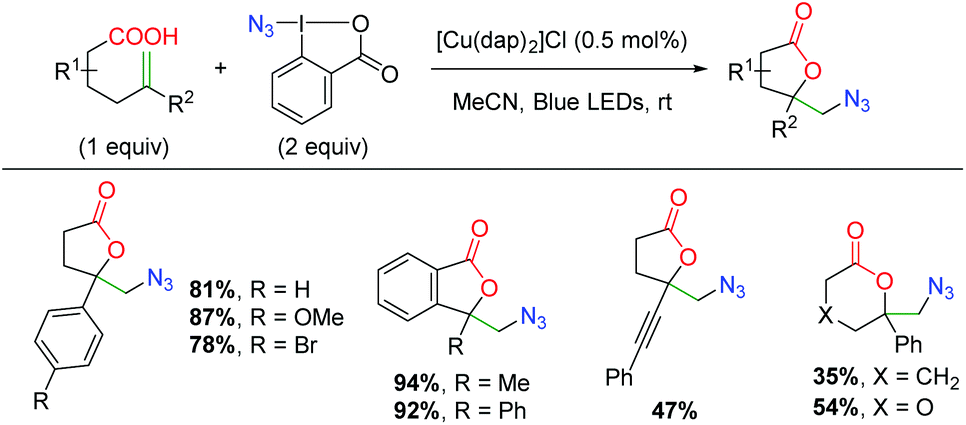
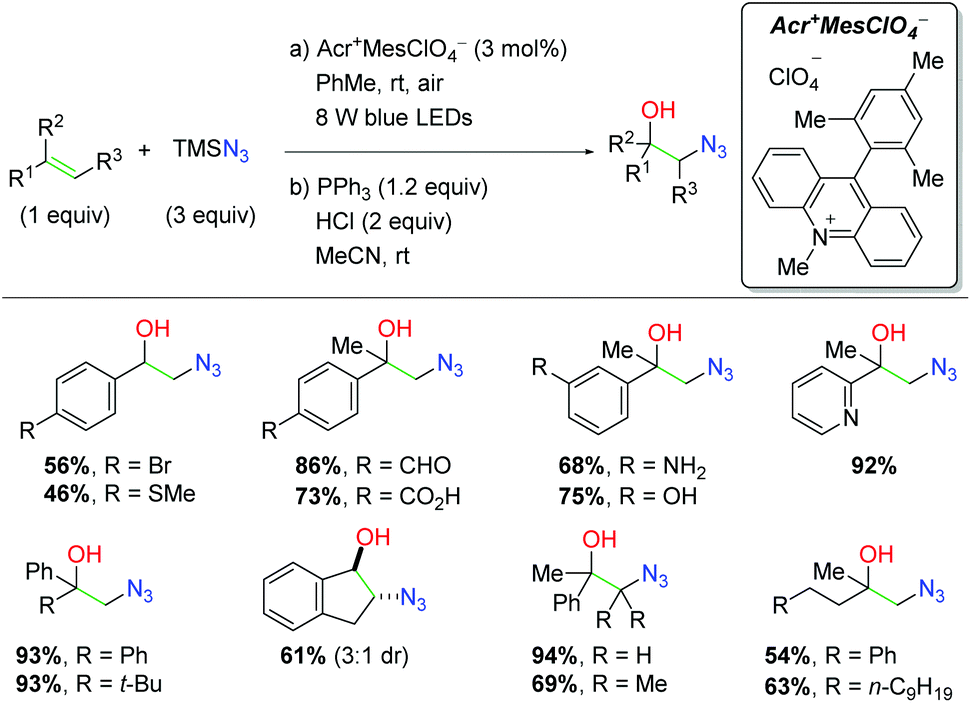
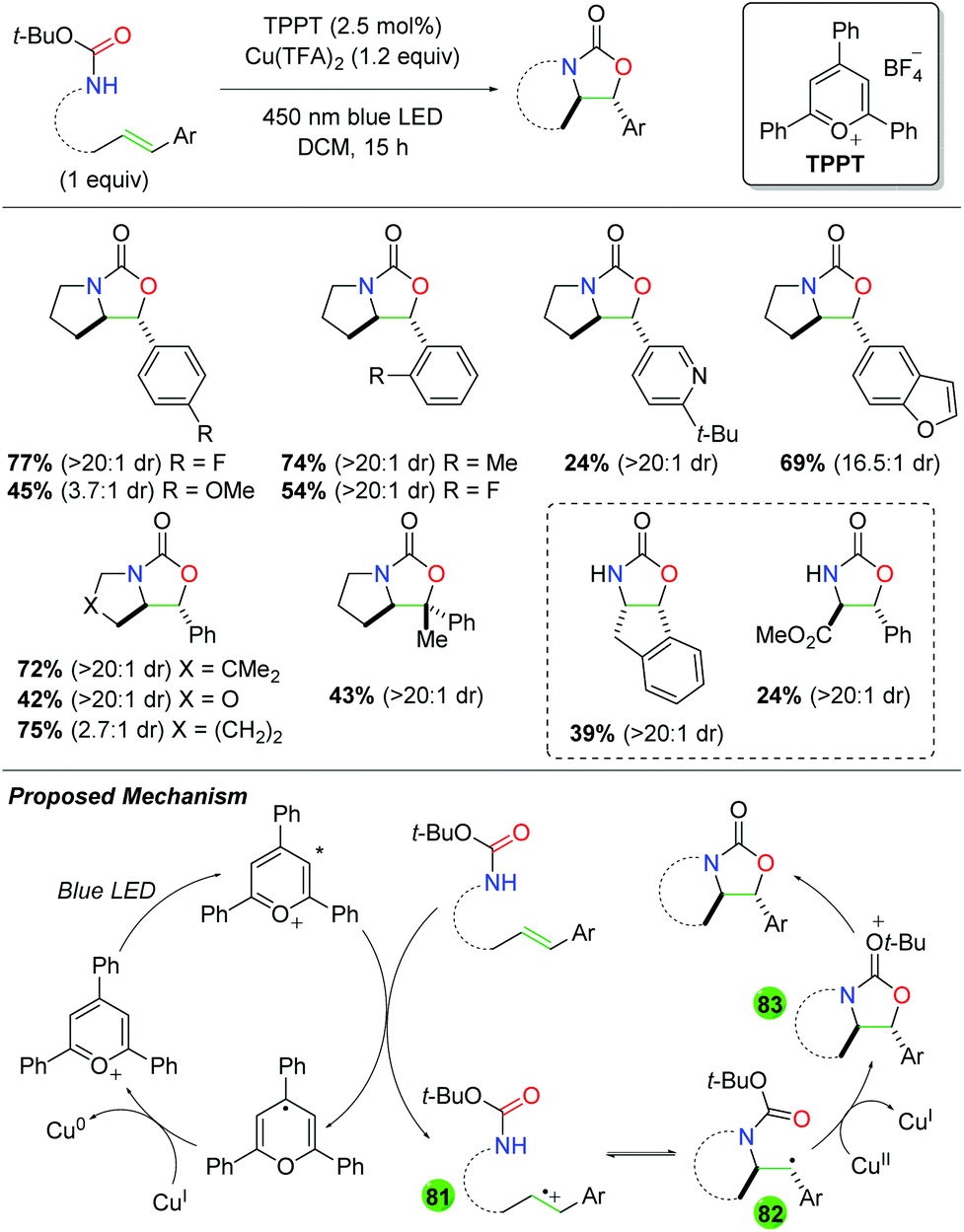
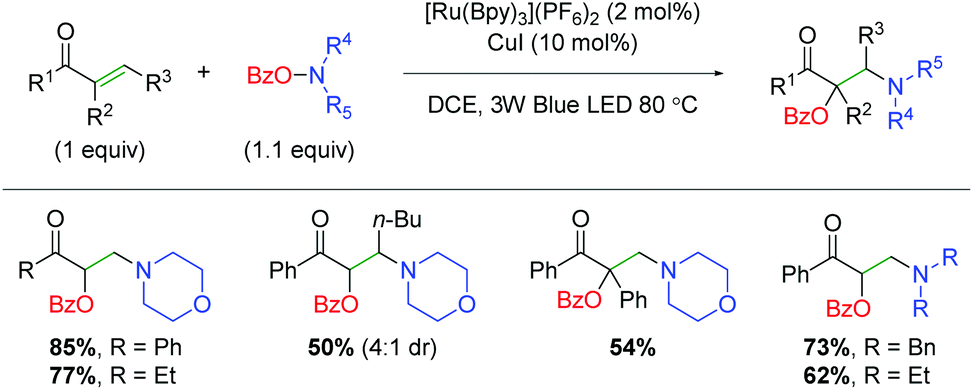
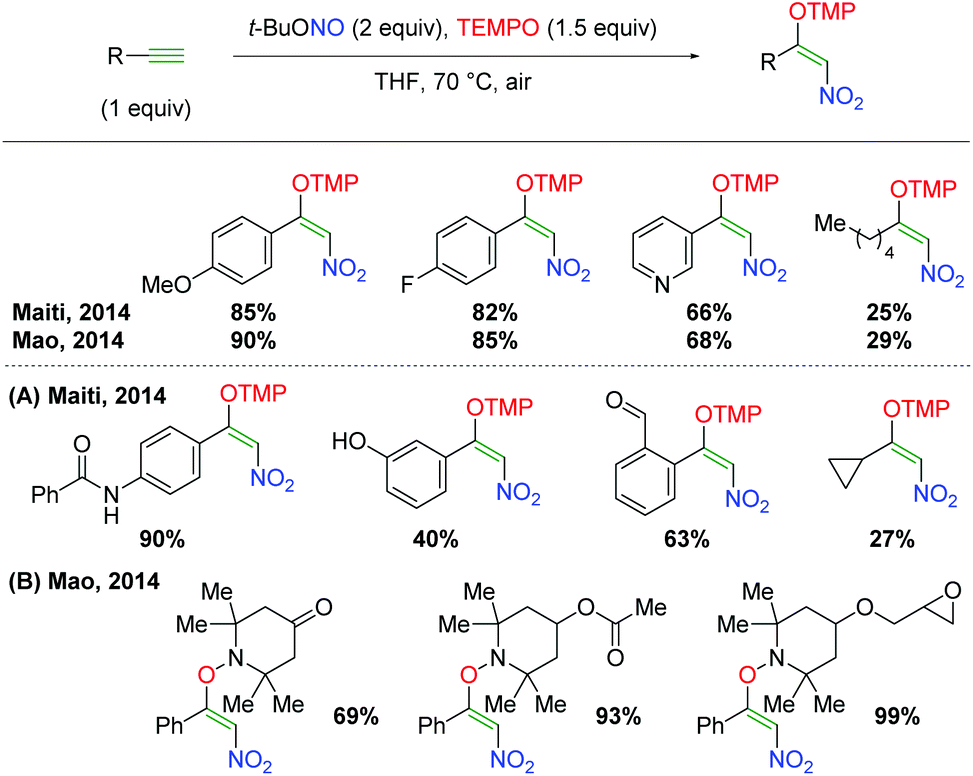
DOI:
10.1039/D0OB01938K
(Review Article)
Org. Biomol. Chem., 2021,
19, 46-81
Beyond osmium: progress in 1,2-amino oxygenation of alkenes, 1,3-dienes, alkynes, and allenes
Received
21st September 2020
, Accepted 30th October 2020
First published on 30th October 2020
Abstract
Olefin 1,2-difunctionalization has emerged as a popular strategy within modern synthetic chemistry for the synthesis of vicinal amino alcohols and derivatives. The advantage of this approach is the single-step simplicity for rapid diversification, feedstock nature of the olefin starting materials, and the possible modularity of the components. Although there is a vast number of possible iterations of 1,2-olefin difunctionalization, 1,2-amino oxygenation is of particular interest due to the prevalence of both oxygen and nitrogen within pharmaceuticals, natural products, agrochemicals, and synthetic ligands. The Sharpless amino hydroxylation provided seminal results in this field and displayed the value in achieving methods of this nature. However, a vast number of new and novel methods have emerged in recent decades. This review provides a comprehensive review of modern advances in accomplishing 1,2-amino oxygenation of alkenes, 1,3-dienes, alkynes, and allenes that move beyond osmium to a range of other transition metals and more modern strategies such as electrochemical, photochemical, and biochemical reactivity.
 Brett N. Hemric | Brett N. Hemric obtained his Ph.D. from Duke University in 2018 after working with Prof. Qiu Wang on copper-catalyzed amino oxygenation of alkenes and 1,3-dienes. In 2019, he began working as a postdoctoral research associate at the University of Illinois at Urbana-Champaign with Prof. Scott E. Denmark, investigating de novo pharmaceutical synthesis and selenium redox chemistry. He is an ardent advocate for intentional and effective teaching and mentorship, both in the classroom and laboratory. His research interests are centered on the use of umpolung reagents for the discovery of economical, straightforward, and robust reactivity. |
1. Introduction
Nitrogen-containing compounds are one of the most ubiquitous classes in modern organic chemistry due to their prevalence in pharmaceuticals, natural products, agrochemicals, catalysts, and ligands. Modern pharmaceuticals are particularly intertwined with nitrogen-containing compounds; of the top 150 drugs in 2016 by number of prescriptions, 81% contain at least one nitrogen atom and 46% contain at least one non-conjugated/aromatic nitrogen.1 Consequently, it is no surprise that drug discovery reaction demands mirror this trend, as C–N bond formation accounts for 37% of drug discovery reactions.2 This amount is impressive given the precedence of commonplace reactions such as Suzuki couplings, esterification, and hydrolysis. With such a high demand in pharmaceutical development for improvements on side effects and off-target activity, it is vital that synthetic chemists continue to push the boundaries, develop new reactions, and increase the accessible chemical space.
Among common nitrogen-containing compounds in modern pharmaceuticals and agrochemicals, the 1,2-amino alcohol moiety holds a privileged position. From mainstream drugs such as Advair, ProAir®, Xarelto®, Lopressor, and Prezista®, to untested, experimental drugs such as Ifenprodil and Sutezolid, the 1,2-amino alcohol derivatives are involved in a broad range of applications and treatments from asthma/COPD to anti-tuberculosis. Of particular note is the prevalence of the 2-amino-1-arylethanol scaffold (shaded) in the oxyamino subclass. Already boasting pharmaceuticals such as the aforementioned Advair and ProAir®, this subclass has given rise to a large number of bioactive molecules with a broad variety of pharmaceutical activities.
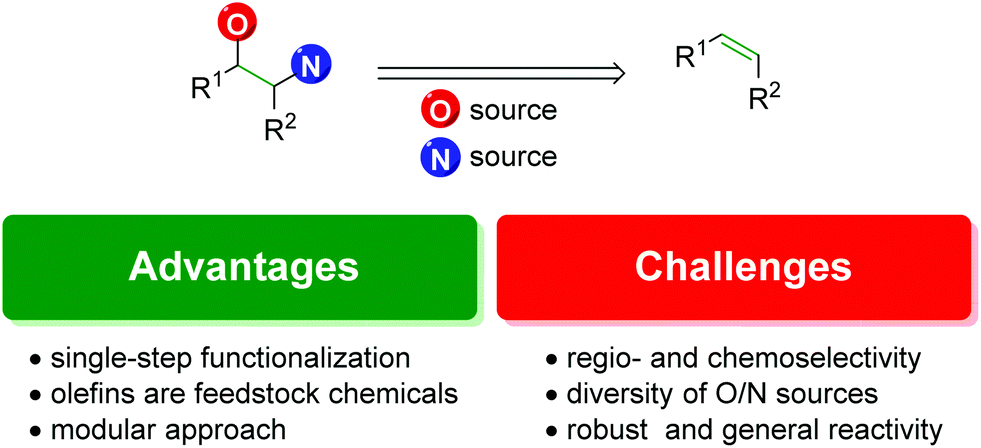
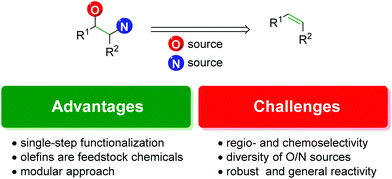
With the precedence and potential of the vicinal amino alcohol substitution pattern in pharmaceuticals, agrochemicals, and ligand development, straightforward and logical routes for the synthesis of such scaffolds is in high demand. One of the most forthright approaches to accomplish this is the 1,2-difunctionalization of alkenes (Fig. 1).3
 |
| | Fig. 1 Advantages and challenges in alkene amino oxygenation. | |
The advantages of the approach are highlighted by the ability to achieve the product in a single-step. Although there are many other viable approaches to the 1,2-oxyamino scaffold that rely on a sequence of steps to realize the product, olefin 1,2-difunctionalization provides a single-step approach that circumvents any potential incompatibility of the desired compound with secondary (or tertiary) reactions in a sequence.
Secondly, olefins are readily accessible feedstock chemicals. Many methods exist to access them, such as starting from carbonyl groups (Wittig reaction and derivatives), alcohols (elimination reactions), or other olefins (olefin metathesis). With such a broad number of options to produce alkenes, it is a desirable starting point for a reaction.
Finally, it offers a potentially modular approach with the ability to choose individual starting materials from each class (olefin, oxygen source, and nitrogen source) and achieve complex scaffolds bearing any combination of the components.
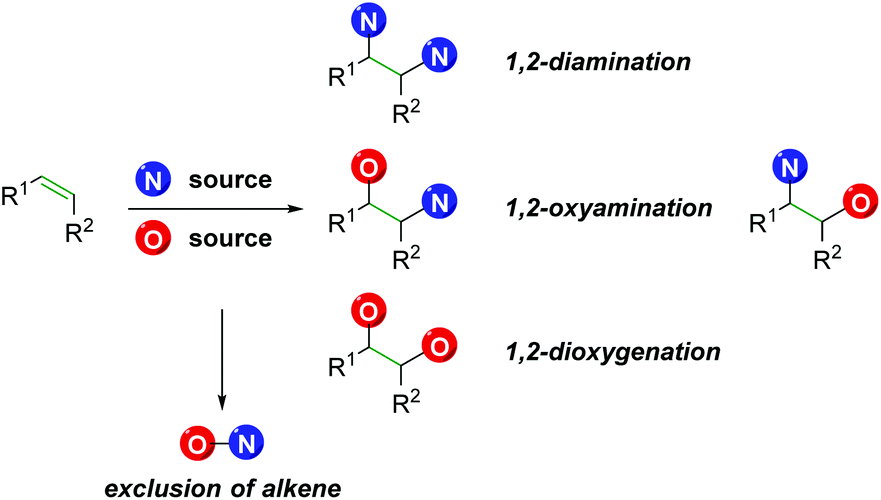
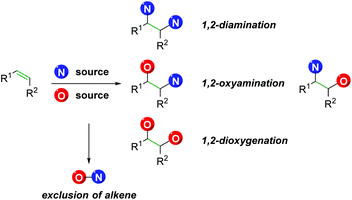
However, there exist a number of challenges to the 1,2-olefin difunctionalization approach. First, the regio- and chemo-selectivity of the transformation can be problematic, owing to the traditionally nucleophilic nature of both oxygen and nitrogen (Scheme 1). To accomplish the olefin difunctionalization, one must achieve cooperative reactivity between the oxygen source, the nitrogen source, and the olefin, while also maintaining selective addition to the olefin. Overcoming any potential diamination, dioxygenation, or nitrogen–oxygen reactivity (exclusion of the olefin) can be challenging. Controlling the regioselectivity of the addition without relying on contrived conditions can also be a challenge, as a clear trend must be established for the initial addition to the olefin.
 |
| | Scheme 1 Challenges of undesired reactivity in 1,2-alkene difunctionalization. | |
Secondly, a method must seek to achieve variety in both the oxygen and nitrogen sources utilized within the transformation. To achieve the cooperative reactivity mentioned above, the diversity of one of the heteroatom sources is often compromised for the sake of the reaction, leading to a rigid component that cannot be varied to the operator's liking.
Finally, it is a challenge to establish a transformation that shows robust, consistent, and straightforward reactivity. All new methods are only as good as their application and ability to be performed by any competent user anywhere in the world under imperfect conditions. When developing a method, chemists should seek to establish transformations that are of general use to the chemistry community as a whole, not simply those within the discovering lab or institution.
Note that because of the dearth of methods to accomplish oxyamination of unsaturated C–C bonds, this review will focus on the 1,2 (vicinal) difunctionalization of alkenes, 1,3-dienes, alkynes, and allenes. Accordingly, it will not deal with (1) more activated systems such as silyl enol ethers and indoles, (2) difunctionalizations producing substitutions other than vicinal, or (3) halogen-substituted olefins. Furthermore, because of the extensive attention that has already been devoted to the Sharpless amino hydroxylation and other osmium-catalyzed methods, those will not be covered in detail here, but rather the reader is directed to a number of reviews on the subject.4
2. 1,2-Amino oxygenation of alkenes and 1,3-dienes
2.1 Palladium catalysis
Initial work in the area of palladium-catalyzed alkene oxyamination was reported by Bäckvall and co-workers in 1975 using a procedure of stoichiometric 1,2-aminopalladation, followed by stoichiometric oxidation and reductive elimination (Scheme 2).5 This initial work provided good selectivity for the syn diastereomer with symmetric alkenes, but suffered from very poor regioselectivity with internal alkyl/inactivated alkenes, such as 1-butene. Furthermore, this procedure only engaged a narrow range of amine substrates and demonstrated only acetic acid as the oxygen species.
 |
| | Scheme 2 Palladium-promoted alkene oxyamination (Bäckvall, 1975–1982).5 | |
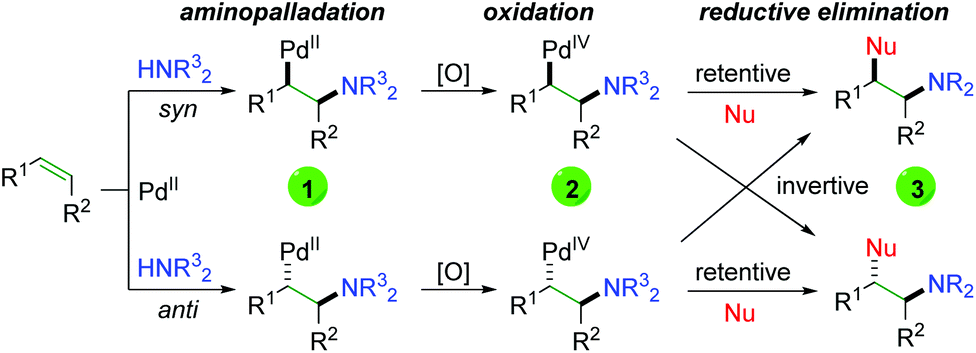
Although Bäckvall's original oxyamination condition requires stoichiometric amounts of palladium, the approach (Scheme 3) laid the groundwork for forthcoming Pd-catalyzed alkene oxyamination procedures. The general procedure begins with palladium(II) coordination to the alkene, followed by aminopalladation in either a syn or anti fashion to generate a 1,2-aminopalladium species (1), being careful to avoid β-hydride elimination. Oxidation of this palladium(II) species generates palladium(IV) (2), which undergoes reductive elimination in either a retentive or invertive manner to generate the product (3) and regenerate palladium(II). This system accounts for the two relative products generated (syn and anti), regardless of whether the reaction undergoes a cis or trans aminopalladation. With this in mind, it is often difficult to ascertain the exact pathway, simply from the type of product generated.
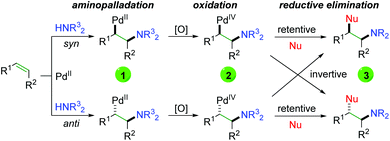 |
| | Scheme 3 General catalytic system for 1,2-alkene oxyamination. | |
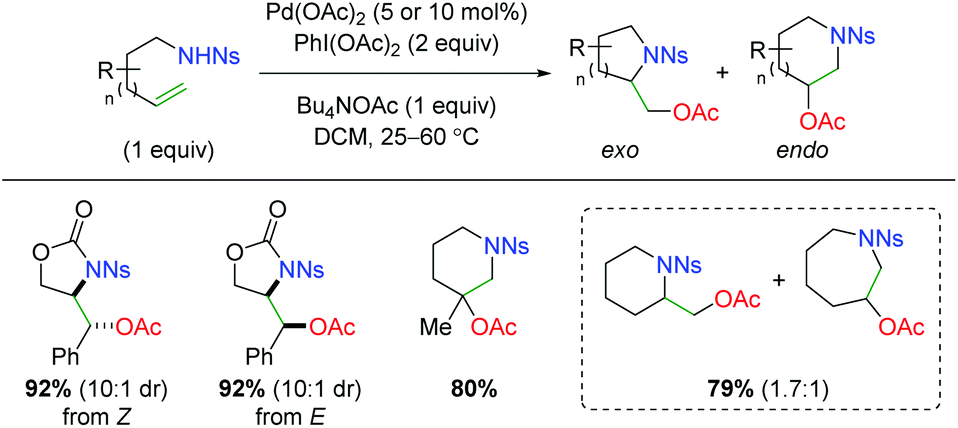
In 2005, Sorensen disclosed a catalytic alkene oxyamination reaction using palladium(II) acetate with sulfonamide-tethered alkenes (Scheme 4).6 From his testing of E and Z alkenes, he found a net anti addition of the nitrogen and oxygen substituents and proposed a trans aminopalladation with retentive reductive elimination to explain the diastereoselectivity. In regards to the substrate scope, this method showed poor 5-exo vs. 6-endo (and 6-exo vs. 7-endo) selectivity and only examined a fairly narrow scope of alkene substrate backbones.
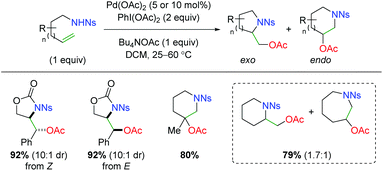 |
| | Scheme 4 Palladium-catalyzed alkene oxyamination (Sorensen, 2005).6 | |
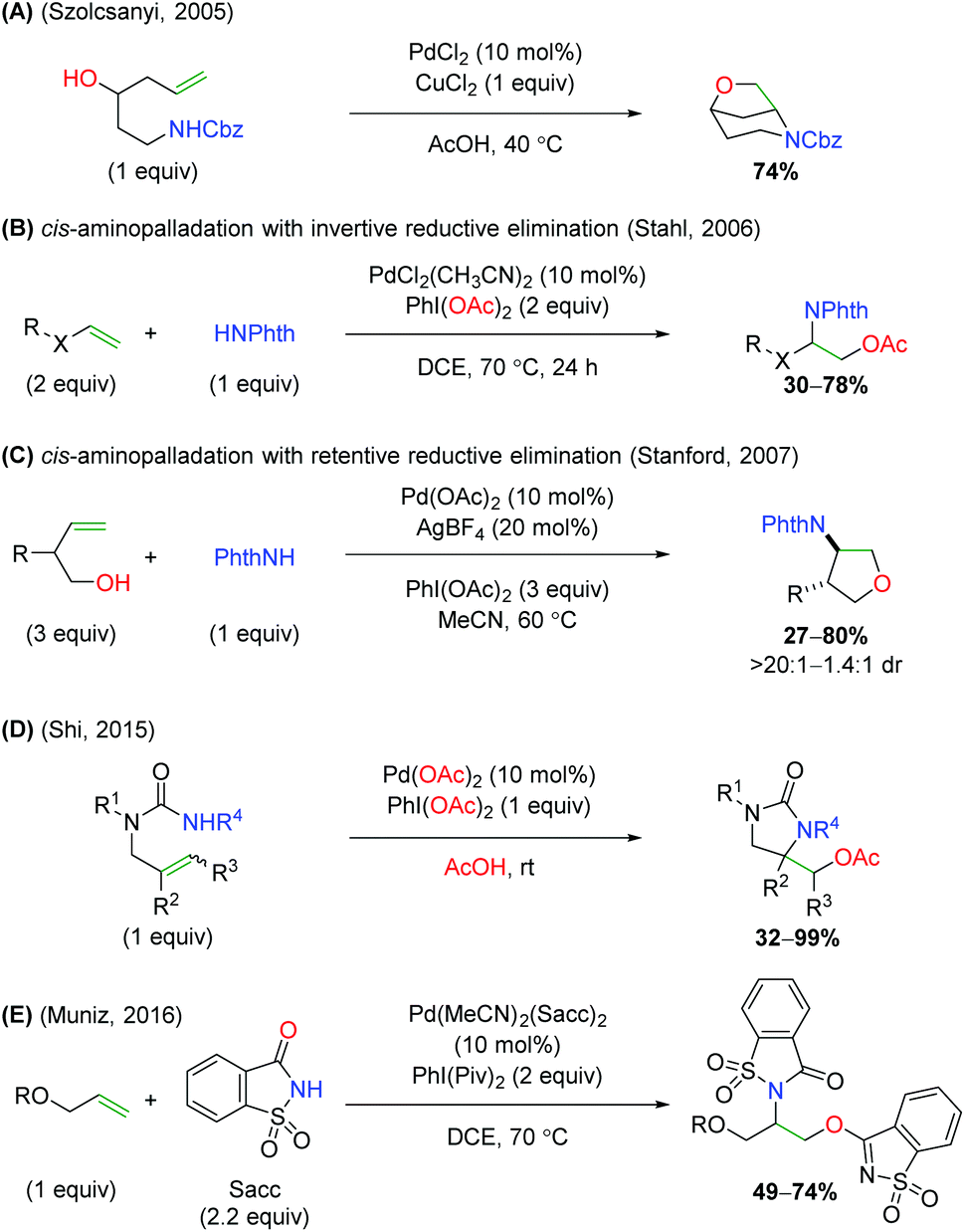
In the same year, the Szolcsányi group reported formation of a bicyclic piperidine product via intramolecular aminopalladation, followed by an intramolecular reductive elimination (Scheme 5A).7 However, they reported only one substrate (for the optimized conditions) and did not undertake any mechanistic studies. A year later, the Stahl lab produced a similar transformation using a three-component, intermolecular approach (Scheme 5B).8 While this approach was able to overcome the requirement of tethering the amine to the alkene, the alkene scope was limited to a narrow range of alkoxy terminal alkenes and acetate oxygen trapping. Mechanistic studies with a select internal alkene (Z-cinnamyl methyl ether) showed that the reaction was initiated through syn-aminopalladation, with an invertive reductive elimination to generate anti products.9 Soon thereafter, Sanford and co-workers demonstrated that alkene-tethered alcohols were viable oxygen trapping partners, expanding the scope from the previously use acetate group (Scheme 5C).10 The scope was strictly limited to substitutions at the allylic position and used phthalimide as the sole amine source to generate 5-endo products, with generally good diastereoselectivity. Similar to the Stahl paper, internal alkene mechanistic studies showed the initiation step being cis-aminopalladation, with retentive reductive elimination producing the anti-diastereomer products. As part of a study on palladium vs. copper catalyst-divergent methods, the Shi group disclosed an amino oxygenation using carbamates with acetate as the terminal trap (Scheme 5D).11 While the scope showed good tolerance of a variety of substitutions on the backbone and alkene, no mechanistic studies were conducted to examine the method of aminopalladation or reductive elimination. The Muñiz laboratory demonstrated an interesting intermolecular alkene oxyamination reaction while investigating an alkene diamination reaction using saccharin and sulfonamides (Scheme 5E).12 They found, in the presence of excess saccharin (Sacc) and using a specialized palladium(II) catalyst, that they were able to achieve aminopalladation with the nitrogen of saccharin, followed by reductive elimination of with the amide oxygen to generate a unique scaffold. Similar to the Stahl study, the scope was limited to a narrow range of alkoxy terminal alkenes.
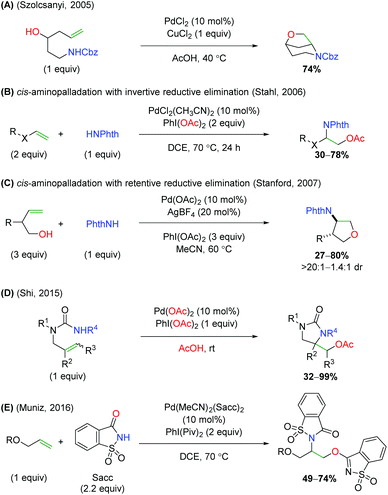 |
| | Scheme 5 Further palladium-catalyzed alkene oxyamination. | |
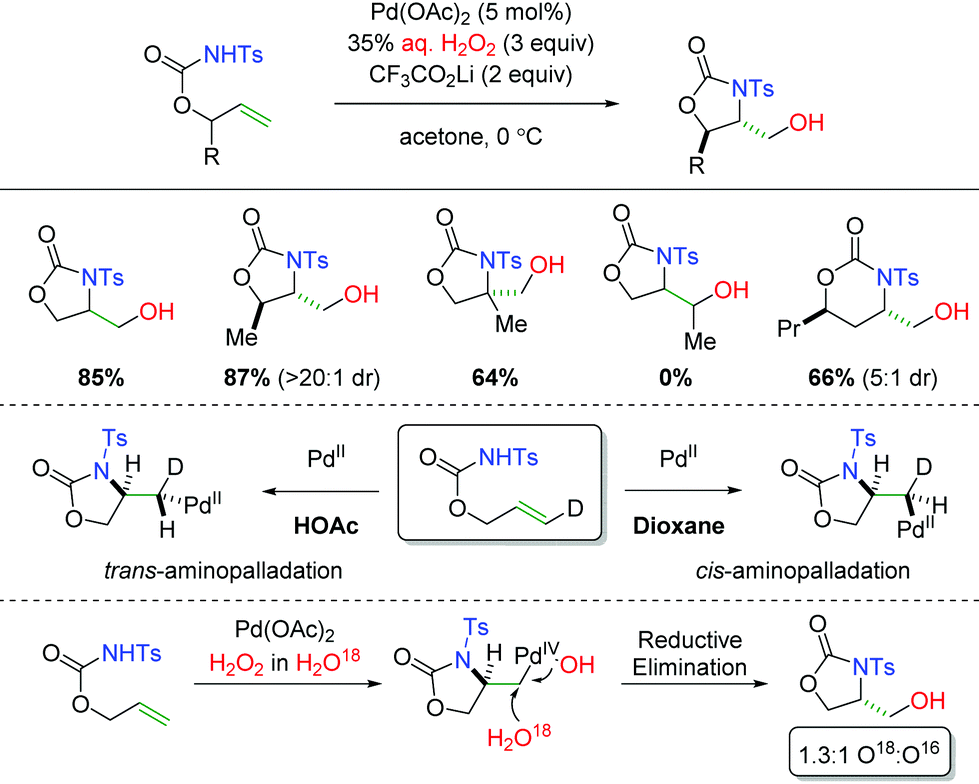
In 2014, the Liu group demonstrated using carbamates as the amine source with aqueous H2O2 as the oxygen trapping agent (Scheme 6).13 The use of H2O2 as the oxygen source and oxidant allowed for exclusion of a more harsh external oxidant. The substrate scope showed excellent diastereoselectivity in 5-exo products and was tolerant of substitution at the α-position of the alkene. However, the reaction was not tolerant of substitution at the β-position of the alkene and the diastereoselectivity was diminished in 6-exo products. Interestingly, they were able to demonstrate that changing the solvent from dioxane to acetic acid resulted in a switch from cis-aminopalladation to trans-aminopalladation. Furthermore, they also showed through isotopic labeling of H2O18 in the H2O2 solution that the hydroxyl group generated in the product was a mixture of both water and hydrogen peroxide. The hydrogen peroxide would provide a retentive reductive elimination, while the water gives an invertive reductive elimination.
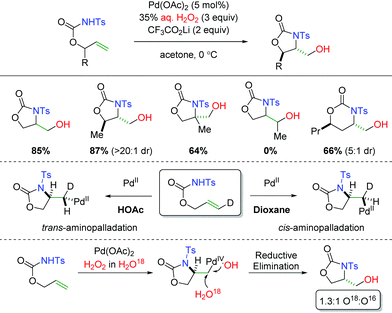 |
| | Scheme 6 Palladium-catalyzed alkene amino hydroxylation (Liu, 2014).13 | |
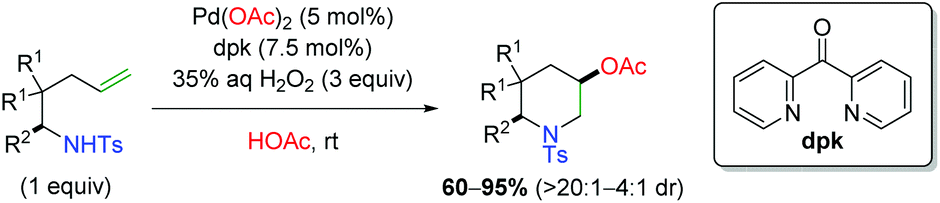
Shortly thereafter, Liu and co-workers released a follow-up paper detailing further use of H2O2 as a green oxidant (Scheme 7), this time with formation of piperidine scaffolds similar to the original Sorensen study (Scheme 4).14 The scope largely improves upon the Sorensen work with generally excellent diastereoselectivity, though the scope is deficient of internal alkenes. Despite this shortcoming, their analysis of deuterium-labeled substrates argued for an anti-aminopalladation, with retentive reductive elimination.
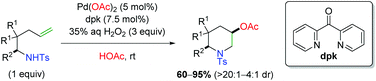 |
| | Scheme 7 Formation of piperidine scaffolds with H2O2 as an oxidant (Liu, 2015).14 | |
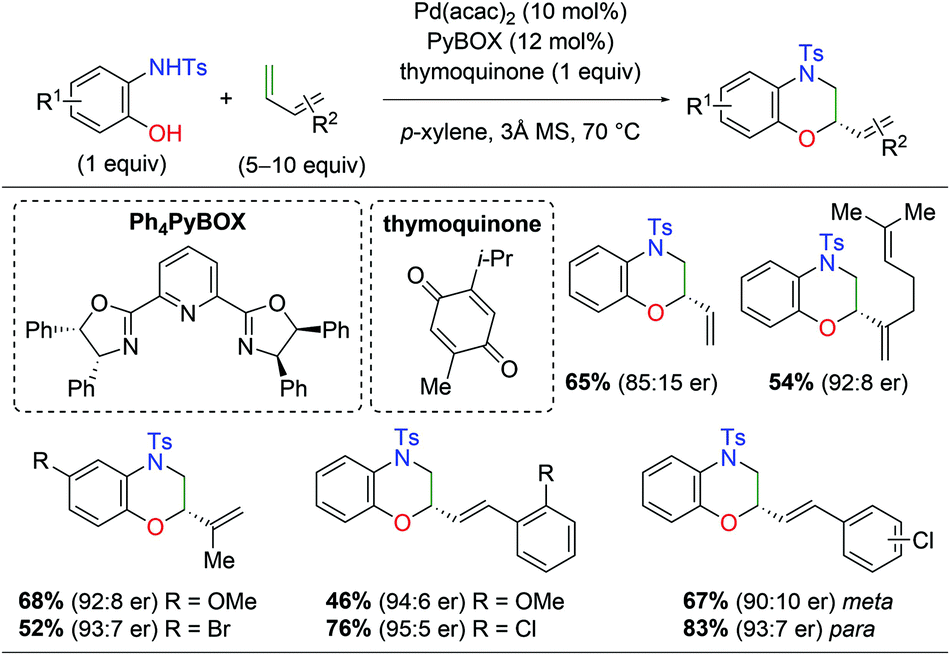
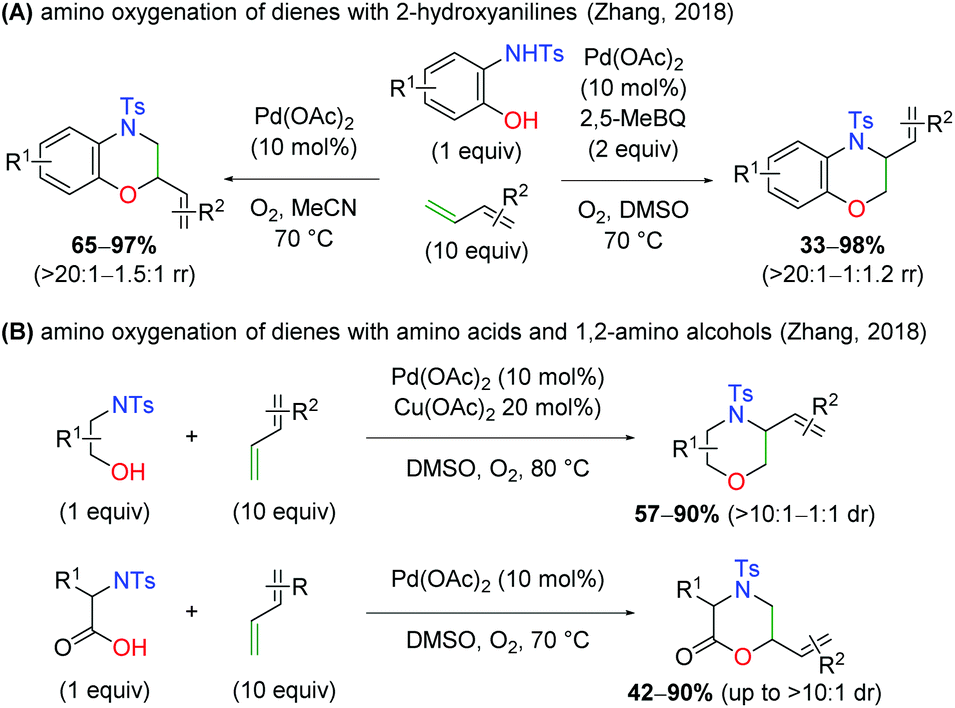
With regard to palladium-catalyzed amino oxygenation of 1,3-dienes, a 2018 paper detailed an enantioselective amino oxygenation protocol using 2-hydroxyanilines to create morpholine scaffolds with olefin selectivity for the 1,3-diene and regioselectivity for the N,O-addition (Scheme 8).15 The authors found that this method worked on a variety of anilines containing electron-donating and withdrawing substituents. Furthermore, the reaction tolerated a range of mono-substituted 1,3-dienes, including alkene-bearing myrcene, giving good enantioselectivity and complete regioselectivity in almost every case. However, or di/tri-substituted dienes or internal systems were noted.
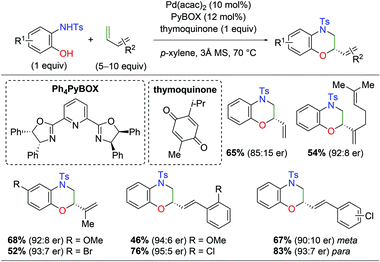 |
| | Scheme 8 Regioselective Pd-catalyzed asymmetric amino oxygenation of 1,3-dienes (Gong, 2018).15 | |
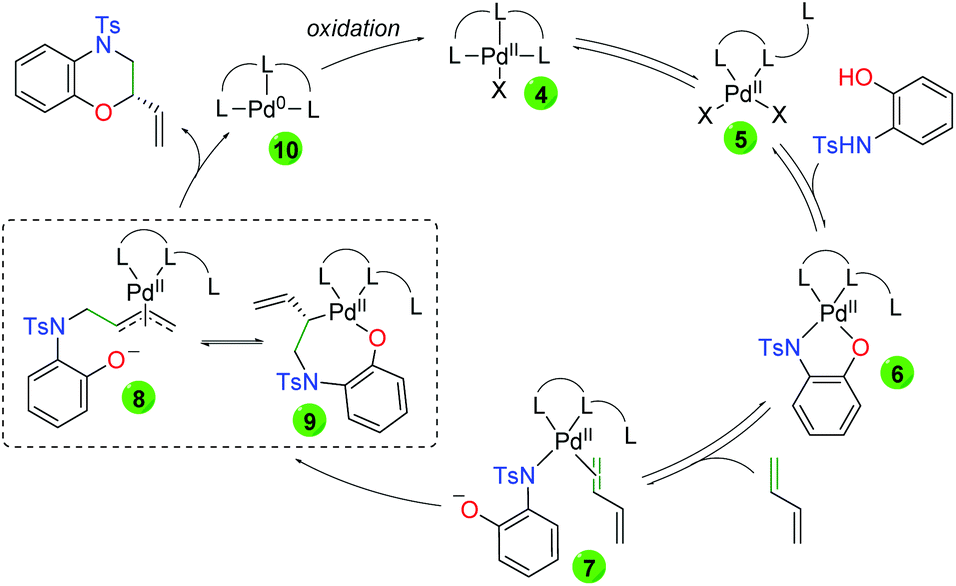
Mechanistically, the authors propose initial ligand tri-coordination of Pd(II) in equilibrium with ligand di-coordination (Scheme 9). Addition of the 2-hydroxyaniline leads to a tetra coordinate Pd(II) complex (6), a complex observed by HRMS. This complex is in equilibrium with the loss of the anionic oxygen and diene coordination (7). Regioselective migratory insertion of the aniline leads to two possible equilibrium intermediates, anionic π-allyl 8 and palladacycle 9. In either case, the final product can be generated. From 8, attack of the anionic oxygen on the anionic π-allyl system would generate the product enantioselectively due to the environment of the chiral ligand. From 9, retentive reductive elimination would generate the final product. In both cases, the generated Pd(0) is oxidized back to Pd(II) by the thyoquinone.
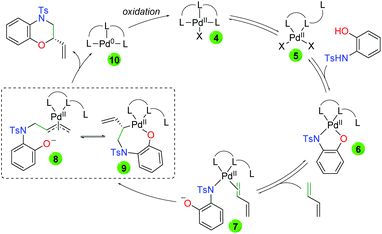 |
| | Scheme 9 Proposed mechanism for palladium-catalyzed asymmetric amino oxygenation of 1,3-dienes. | |
Months later, the Zhang laboratory disclosed a similar concept (Scheme 10A).16 While this method was not enantioselective, it did provide a divergent protocol that allowed for the formation of either constitutional isomer from the N,O-addition to the 1,3-diene. Using only O2 as an oxidant in a sealed tube, the protocol gives the branched product at the oxygen addition site, while in the presence of O2 and 2,5-dimethylbenzoquinone as oxidants, the branched product is observed at the site of nitrogen addition. The 2-hydroxy aniline scope is very similar to the Gong case above, but the 1,3-diene scope features di-substituted dienes and internal systems, though with poor yield in some cases. Later that year, the same group released protocols for use of aliphatic 1,2-amino alcohols and amino acids in an analogous manner to the 2-hydroxy anilines previously (Scheme 10B).17 When using 1,2-amino alcohols, the branched product is observed at the site of nitrogen addition, while amino acids supply the product branched at the site of oxygen addition. While some diastereoselectivity is observed, this is entirely a substrate-dependent phenomenon.
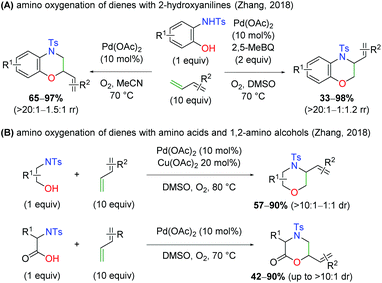 |
| | Scheme 10 Pd-Catalyzed amino oxygenation of 1,3-dienes using various 1,2-amino alcohols (Zhang, 2018).16,17 | |
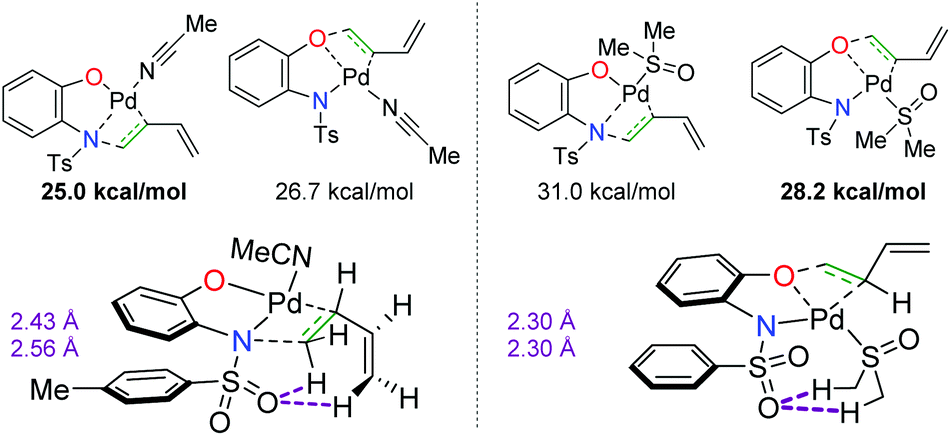
In a complementary publication to the divergent 2-hydroxy aniline paper, the authors conducted DFT studies to determine the origin of the divergence (Scheme 11).18 They found that the benzoquinone additive has no effect on the overall regioselectivity of the reaction, but the coordination of the solvent to the Pd complex dictates the migratory insertion step of the oxygen or nitrogen nucleophile onto the diene. It was found that interactions between the sulfonamide oxygen on the substrate were crucial. When acetonitrile was used as the solvent, the sulfonamide oxygen displayed stabilizing interactions with the C–H bonds on the opposite end of the diene. In the case of DMSO, the sulfonamide oxygen interacted with C–H bonds on the methyl groups of DMSO, thus switching the regioselectivity of the diene addition to opposite the sulfonamide.
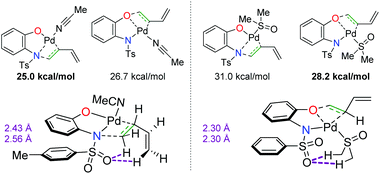 |
| | Scheme 11 DFT studies elucidating the origin of the regioselectivity of the diene amino oxygenation.18 | |
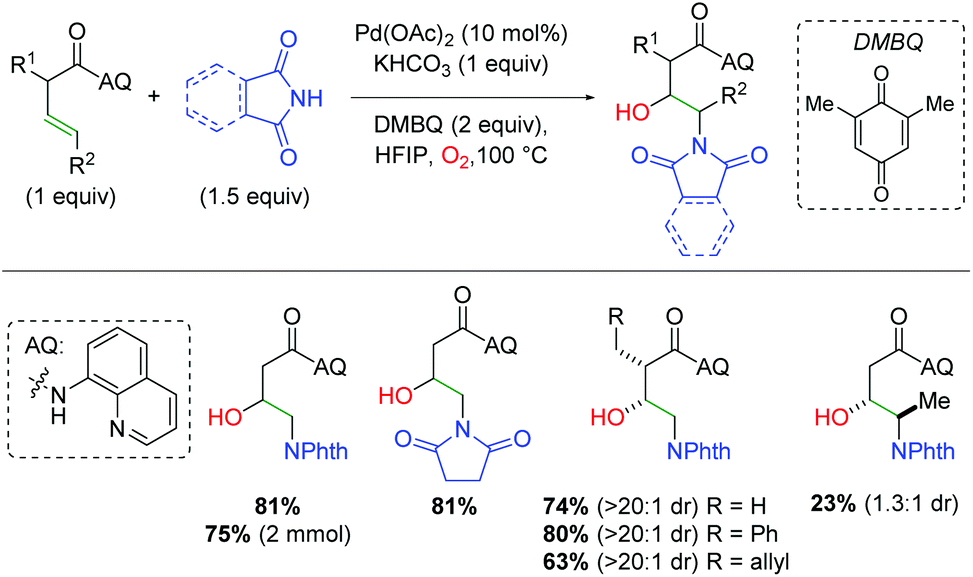
In 2018, the Engle group utilized a bidentate quinoline amide directing group to accomplish palladium-catalyzed alkene difunctionalization. They applied this template to the amino oxygenation of alkenes using phthalimides and molecular oxygen in hexafluoroisopropyl alcohol (HFIP) (Scheme 12).19 The use of phthalimide or succinimide derivatives as nitrogen nucleophiles proved successful, though the authors noted that other nucleophiles that had been previously successful in their templated strategy are not successful in this method. They hypothesized that this arises from some manner of synergistic coordination of the already-installed phthalimide, enhancing the O2 addition step. Regarding the structural elements of the unsaturated amide, α-substitution showed excellent diastereoselectivity for the product, though the authors noted that very sterically encumbered α-substitution is not tolerated. While more sterically accessible internal alkenes are viable in the reaction in significantly reduced yield, less accessible internal alkenes are not viable in the reaction. Mechanistic studies verified molecular oxygen as the source of the hydroxy and eliminated a hydroamination/β-hydroxylation mechanism as a viable reaction pathway. The mechanism is assumed to involve directed aminopalladation, followed by some form of oxidative addition of O2, either as O2 or a peroxide form.
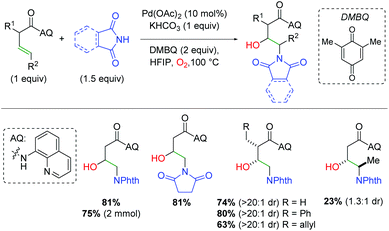 |
| | Scheme 12 Templated, palladium-catalyzed alkene amino oxygenation (Engle, 2018).19 | |
2.2 Rhodium catalysis
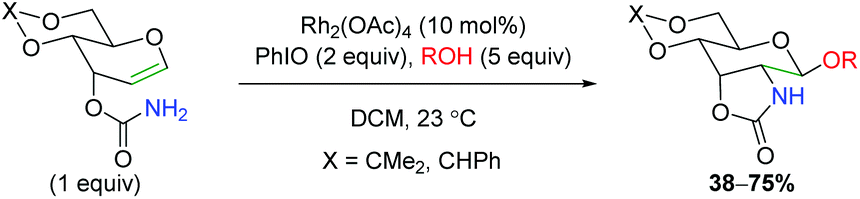
Despite the utility of using rhodium nitrene and carbene complexes, only a few examples exist of rhodium-catalyzed alkene oxyamination. The first example of such a transformation comes via an amidoglycosylation of a dihydropyran derivative using exogenous alcohol (Scheme 13).20 Unfortunately, the reaction scope shown is quite limited, as only two types of dihydropyran scaffolds were tested with four different alcohols. The reaction is proposed to proceed through a nitrene-mediated aziridine formation, but no mechanistic experiments were performed. This method was further expounded upon several years later with DFT studies to provide more mechanistic insights.21
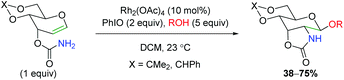 |
| | Scheme 13 Rhodium-catalyzed amidoglycosylation (Rojas, 2002).20 | |
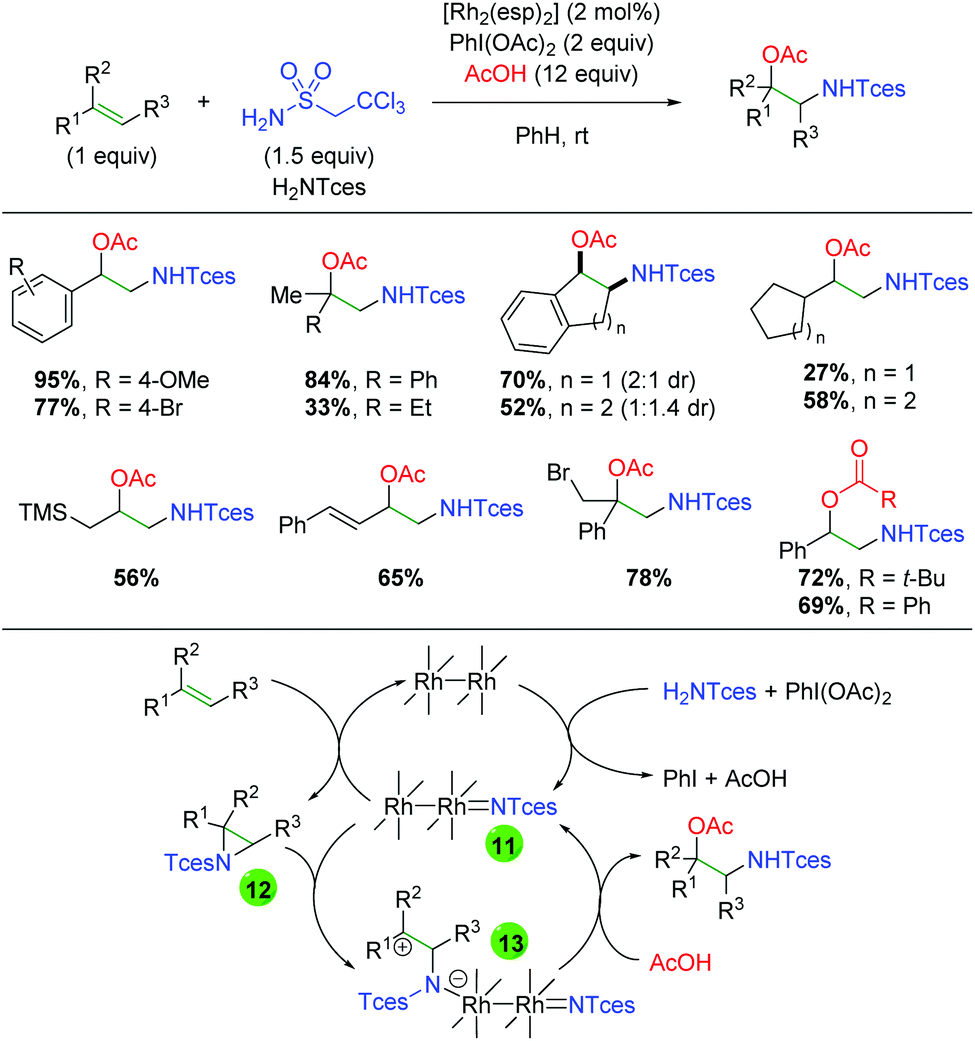
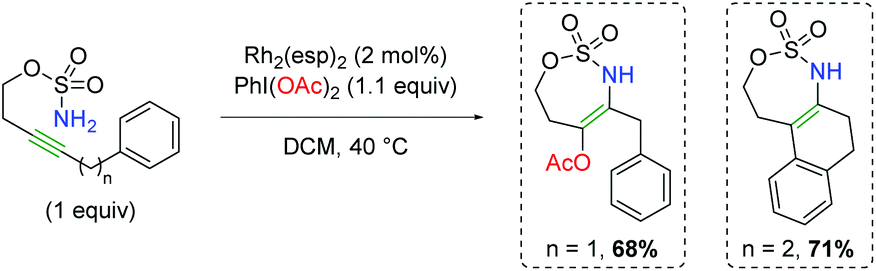
In 2014, Dauban and co-workers published a report detailing a rhodium-catalyzed intermolecular alkene amino oxygenation using sulfonamides and a large excess of carboxylic acids (Scheme 14).22 The reaction exhibits a fairly broad substrate scope, as both internal and external alkenes are shown to participate. Furthermore, the scope is not strictly limited to aryl alkenes, with even alkyl alkenes being viable partners. The reaction is also effective with 1,3-dienes, as well as highly reactive functional groups like trimethylsilane and primary alkyl bromides. The authors determined that the product can be smoothly generated from the corresponding aziridine, but only in the presence of all reaction components. Accordingly, they proposed that the Rh2esp2 complex forms a nitrene (11) from a combination of the H2NTces and PhI(OAc)2. This nitrene then combines with the alkene to form an aziridine (12). They then proposed combination with a second equivalent of the nitrene species (11) might act as a Lewis acid to promote opening of the aziridine (13), which is trapped by the acetic acid to generate the product and re-generate the nitrene (11). Recently, a greener procedure was disclosed of this exact transformation, exchanging PBS buffer for the benzene solvent used in the original method.23
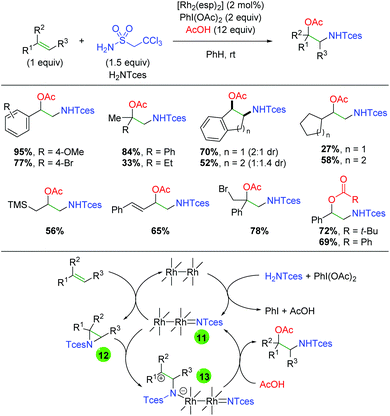 |
| | Scheme 14 Rhodium-catalyzed alkene amino oxygenation (Dauban, 2014).22 | |
A related work was done on carbamate-tethered 1,3-dienes using rhodium diacetate (Scheme 15).24 By tethering the diene and carbamate, the formation of a putative aziridine is limited to only one of the two olefin sites. However, this does not limit the nucleophilic attack, as an SN2 or SN2′ mechanism could be operable. Accordingly, the authors were able to create a protocol that skewed the products toward either the 1,2-addition or the 1,4-addition by changing the composition of the rhodium catalyst and the iodine(III) reagent. When using the conditions to favor 1,2-addition, fair to good yields are achieved in complete diastereoselectivity for several different olefin substitutions. Unfortunately, the ratios for 1,2 vs. 1,4-product left a considerable amount to be desired from the standpoint of synthetic utility.
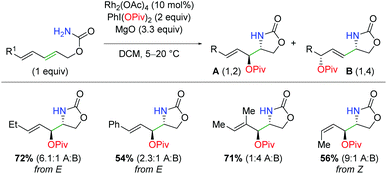 |
| | Scheme 15 Rhodium-catalyzed amino oxygenation of carbamate-tethered dienes (Castillón, 2014).24 | |
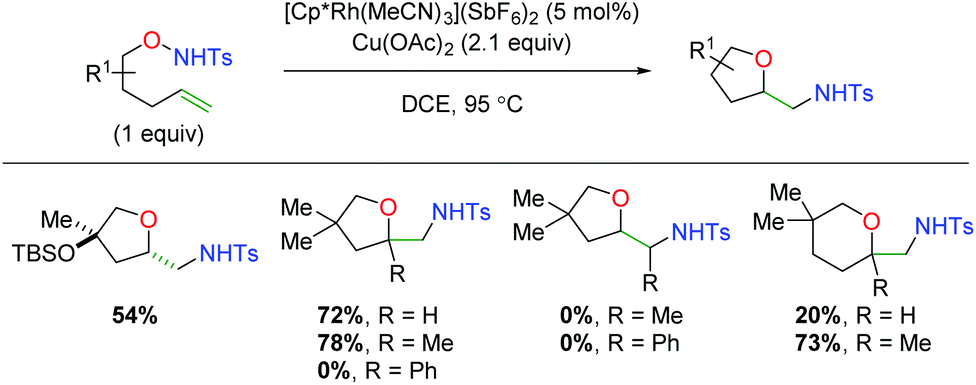
In 2018, another rhodium-catalyzed method was published using alkene-tethered N-alkoxysulfonamides (Scheme 16).25 This paper offers the unique aspect of using a rhodium(III) complex, whereas the previous examples rely on rhodium(II). This reaction engages a range of substitutions on the 5-exo product backbone, but substitutions on the alkene (α-methyl notwithstanding), produce no desired product. However, in the case of 6-exo products, substitution at the α-position of the alkene produces a substantial increase in product yield. Despite some mechanistic work performed by the authors and subsequent computational studies,26 the exact mechanism for this transformation remains inconclusive, though a [3 + 2] cyclization of the alkene with an O–N–Ru adduct is suggested as the most operable pathway.
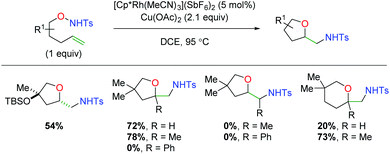 |
| | Scheme 16 Rhodium(III)-catalyzed alkene amino oxygenation using alkene-tethered N-alkoxysulfonamides (Cossy, 2018).25 | |
2.3 Copper catalysis
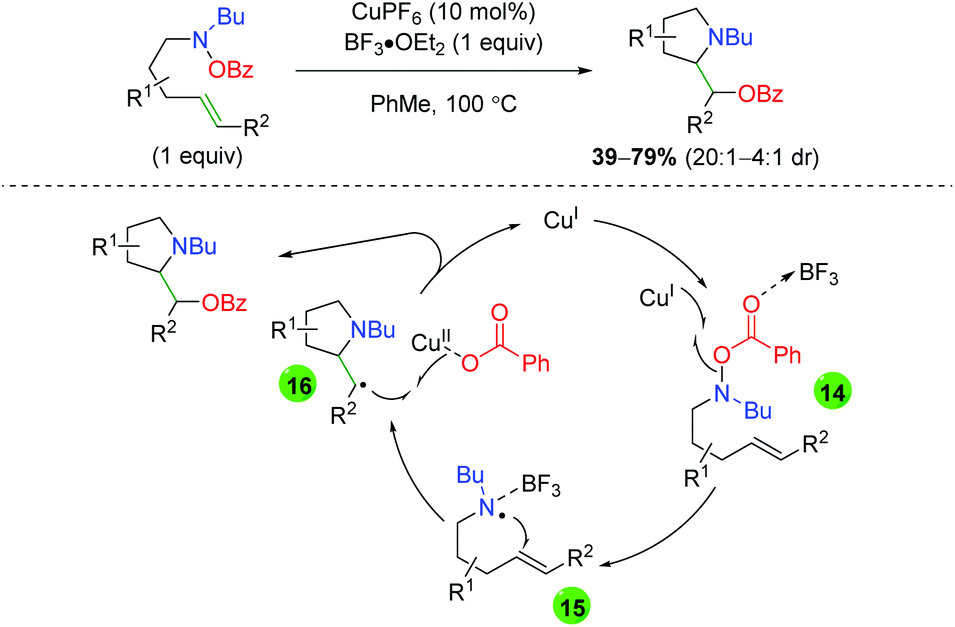
An initial report for copper catalyzed alkene oxyamination was published by the Göttlich laboratory in 2002 using O-benzoylhydroxylamine-tethered alkenes and Lewis acid additive BF3·OEt2 (Scheme 17).27 After trying a number of potential nitrogen-centered radical precursors, they settled on O-benzoylhydroxylamines. Although they showed a very limited scope of substitutions on a 5-exo product-generating backbone, they did note the lack of 6-endo product formation within their system. Furthermore, they proposed a mechanism for this transformation, starting with formation of a nitrogen-centered radical through single-electron transfer with the copper(I) catalyst (14). This nitrogen radical then undergoes radical addition (15) to generate a 5-exo cyclization product with β-position radical. This radical can then be terminated through a single-electron addition from the copper(II) benzoate species to generate the product and re-form the active copper(I) catalyst (16).
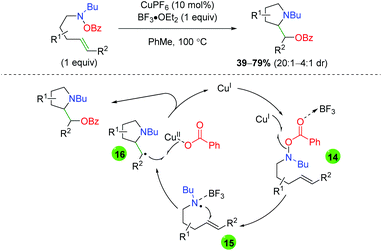 |
| | Scheme 17 Copper-catalyzed alkene oxyamination using O-benzoylhydroxylamine-tethered alkenes (Göttlich, 2002).27 | |
In 2016, the Wang group parlayed this concept of copper catalysis on O-benzoylhydroxylamines, applying it to an alkene amino lactonization method with unsaturated carboxylic acids (Scheme 18).28 This method requires slightly lower temperatures than the previous work, but is capable of undergoing 5-, 6-, and even 7-exo cyclizations on a variety of backbone scaffolds in the absence of BF3 Lewis acids. Notably, substitution at the α-position of the alkene is vital to achieving good yields, as mono-substituted alkenes produce appreciably lower yields than their 1,1-disubstituted analogs. Internal alkenes are also viable in the reaction, even without the α-substitution, while tri-substituted alkenes produce among the highest yields. Interestingly, stilbene scaffolds favor the 6-endo product over the expected 5-exo product. One clear deficiency of this method is the variety of amino groups that can be incorporated. Although piperidine analogs produced good yields, alternate ring sizes and acyclic amines showed a dramatic reduction in yield. In the same year, Wang and co-workers disclosed an intermolecular variant incorporating carboxylic acids and styrenyl alkenes (Scheme 18).29 This method exhibits a number of deficits, namely the poor reactivity of non-styrenyl alkenes and poor yields for non-piperidine amino sources. This method also requires an increase in catalyst loading compared to the lactonization method and works best with relatively acidic carboxylic acids. However, this protocol was successful in incorporating internal alkenes in good yields, much like the lactonization method.
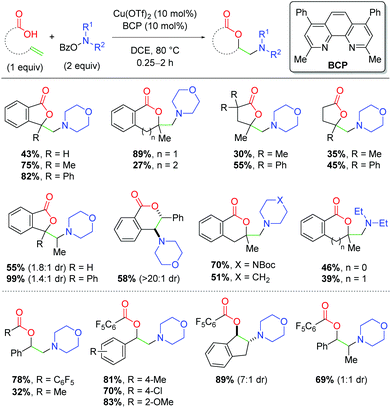 |
| | Scheme 18 Copper-catalyzed amino lactonization using O-benzoylhydroxylamines (Wang, 2016).28,29 | |
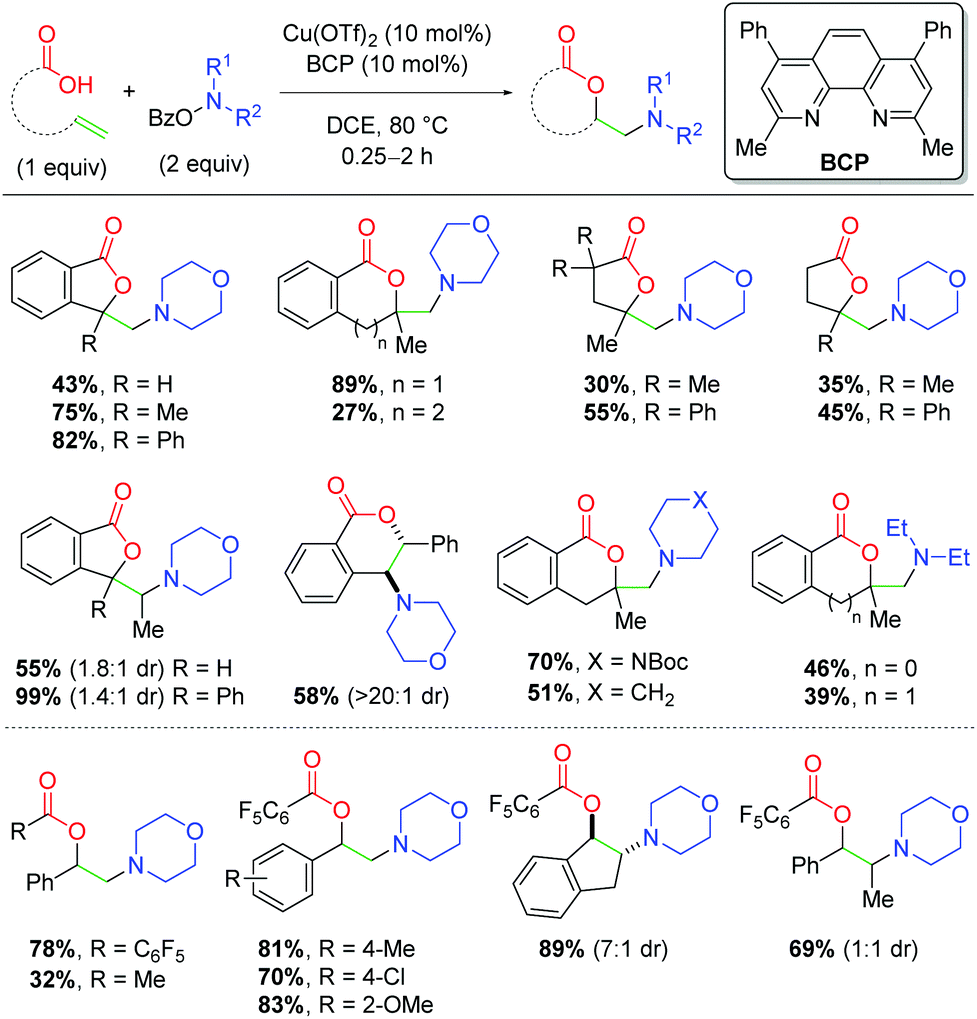
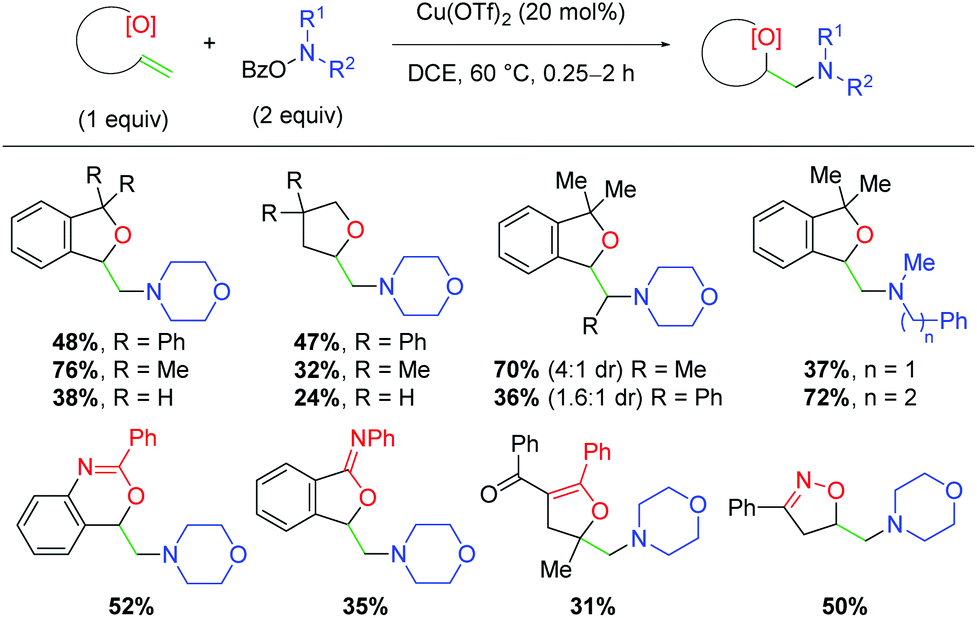
Several years later, the authors disclosed yet another alkene amino oxygenation protocol using a variety of different alkene-tethered oxygen nucleophiles (Scheme 19).30 This work was optimized on the nucleophilic trapping with benzylic alcohols to yield 5-exo products, finding that tertiary alcohols produce higher yields than the primary analogs. The authors also observe good reactivity with internal alkenes, consistent with previous efforts. Promisingly, this protocol offers better yields for a number of amino sources, a deficiency in the authors’ previous methods. Furthermore, a number of other oxygen sources are compatible as nucleophiles, such as secondary amides, 1,3-diones, and oximes. Although the yields of these unoptimized substrates are modest, they offered a clear picture of the possibilities within this method for incorporating a variety of oxygen nucleophiles and potentially other heteroatom nucleophiles.
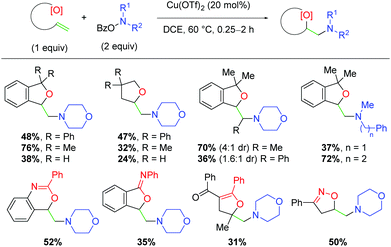 |
| | Scheme 19 Copper-catalyzed amino oxygenation using O-benzoylhydroxylamines and various oxygen nucleophiles (Wang, 2019).30 | |
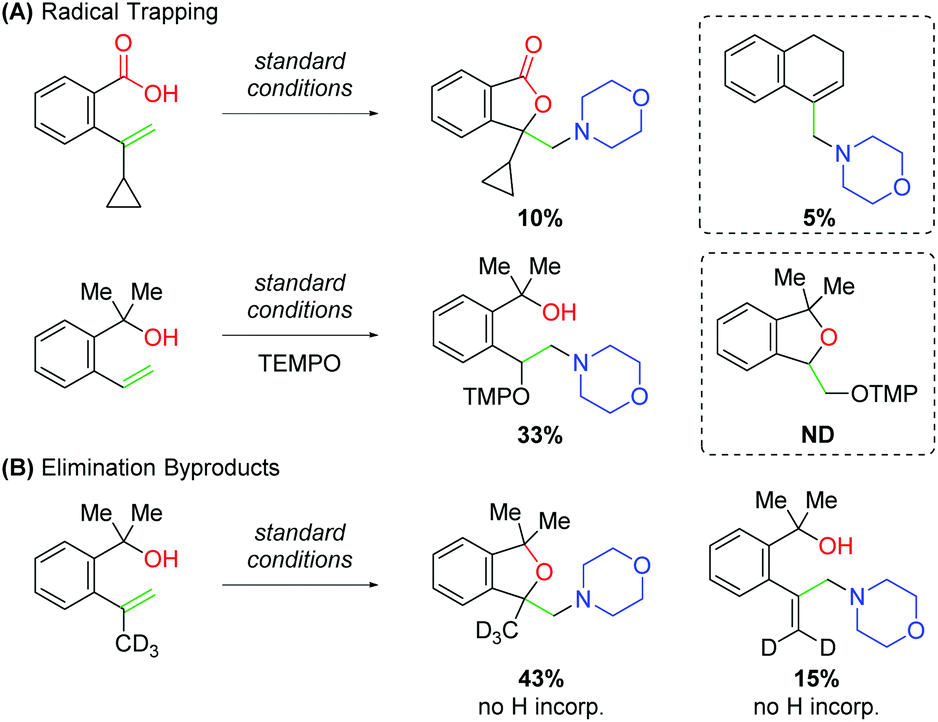
While studying this amino oxygenation reaction, Wang and co-workers noted several mechanistic points that shaped their understanding of the mechanism (Scheme 20). First, they tested unsaturated carboxylic acid substrates bearing cyclopropyl groups, both at the α- and β-positions on the alkene and observed ring-opened products only in the case of the 1,1-disubstituted substrate. Furthermore, when TEMPO was introduced to the reaction with unsaturated alcohol substrates, trapping was only observed for the uncyclized product. Both these results speak to the presence of a carbon-centered radical following an amino-initiation event. When studying the unsaturated alcohols, the authors noted that 1,1-disubstituted alkenes produce allylic amines. Deuterium labeling experiments confirmed that this occurs through amino addition to the alkene followed by elimination.
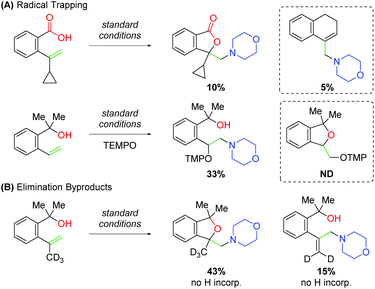 |
| | Scheme 20 Mechanistic insights and proposed mechanism for the amino oxygenation of alkenes. | |
Accordingly, the authors proposed a mechanism for this transformation (Scheme 21). First, disproportionation of Cu(II) provides Cu(I), which undergoes oxidative addition to the O-benzoylhydroxylamine to generate either a Cu(III) amino intermediate or Cu(II) with an amino radical (17). This intermediate adds to the alkene substrate to generate a Cu(III)/carbon-centered radical (18/19) that can be trapped by the oxygen nucleophile to generate the amino oxygenation product.
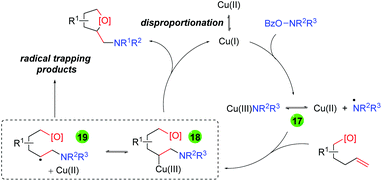 |
| | Scheme 21 Proposed mechanism for the copper-catalyzed alkene amino oxygenation using O-benzoylhydroxylamines. | |
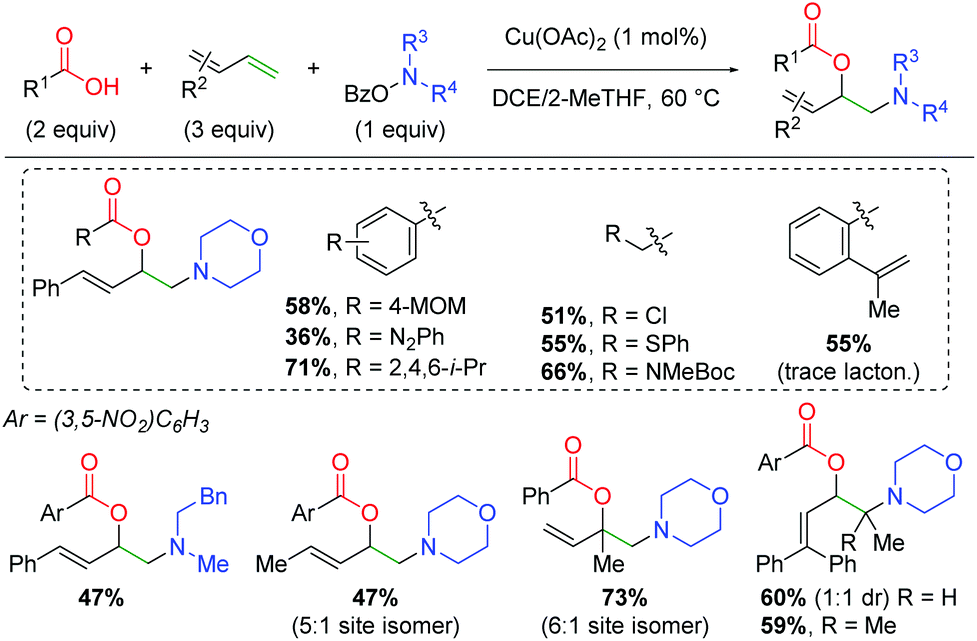
Furthermore, the Wang laboratory reported details on the application of their amino-initiated method to the 1,2-amino oxygenation of 1,3-dienes (Scheme 22).31 This method follows a similar reaction manifold as their previous copper-catalyzed methods, but proved viable with only 1 mol% catalyst loading. An extensive scope of carboxylic acids with a variety of functional groups was shown, including diazo, chloro, sulfide, and amino acid. Furthermore, when the alkene-bearing substrate from the authors’ previous lactonization method was tested, it participates in the 1,3-diene reaction and only undergoes the expected amino lactonization reaction in amounts only observable by gas chromatography. As with most of the authors’ amino-initiation methods, the amino group scope is quite poor outside of piperidine derivatives and select aliphatic substrates. For the scope of 1,3-dienes, a variety of terminal and aliphatic 1,3-dienes are reactive, giving fair yields with reasonable 1,2-addition selectivity for reaction at one of the olefins over the other option. Finally, terminal 1,3-dienes are successful, giving fair yield even with terminally tetrasubstituted 1,3-dienes.
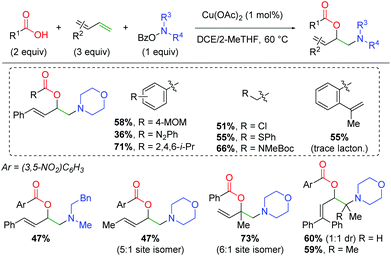 |
| | Scheme 22 Intermolecular copper-catalyzed amino oxygenation of dienes using O-benzoylhydroxylamines (Wang 2019).31 | |
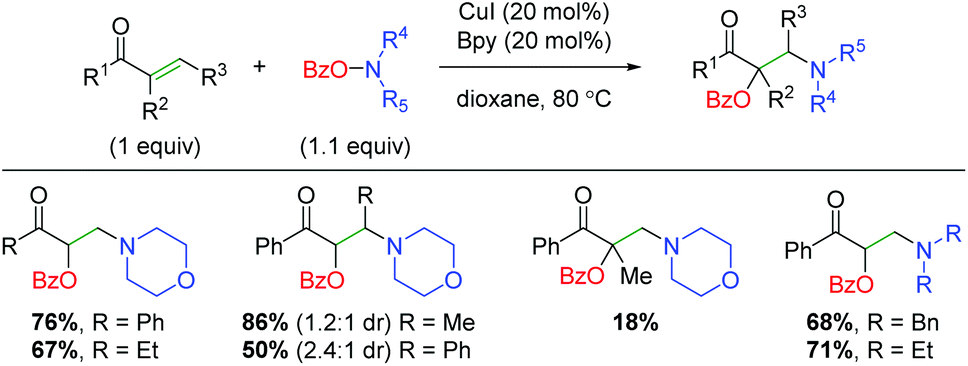
Soon after Wang and co-workers disclosed the amino lactonization report, Feng, Loh, and co-workers demonstrated a similar strategy for addition of the benzoyl and amine groups across activated (α,β-unsaturated) alkenes in an intermolecular fashion (Scheme 23).32 This approach shows a reasonable substrate scope of both aryl and alkyl ketones with a good tolerance for different amines. It also demonstrates internal alkenes as tolerated, though the yields are severely diminished when using α-substituted alkenes. This method does showcase better yields for a wider range of amino groups than several of the Wang group's methods. Mechanistically, the authors propose a similar process to the Wang laboratory's papers, with oxidative addition of the O-benzoylhydroxylamine onto copper and initial addition of the amino group to the alkene.
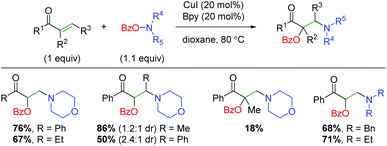 |
| | Scheme 23 Copper-catalyzed oxyamination of activated alkenes using O-benzoylhydroxylamines (Feng, Loh, 2016).32 | |
In 2007, the Yoon group disclosed a novel strategy for alkene oxyamination using copper catalysts in concert with sulfonamide oxaziridines (“Davis’ oxaziridine”, Scheme 24).33 This clever approach leverages the labile nature of the N–O bond in the oxaziridine to initiate addition to the alkene by the more electronegative oxygen atom, followed by trapping with the sulfonamide nitrogen to generate a range of oxazolidine products. The scope of the transformation for alkenes is fairly broad, inasmuch as styrenyl alkenes were used (a limited number of alkyl alkenes were also presented). In a second publication, a scope of 1,3-dienes was also presented. This 1,3-diene scope is rather broad, showing both aryl and alkyl dienes with good to excellent alkene site selectivity. Note that the intrinsic nature of the oxaziridine addition makes 1,4-addition quite unlikely and thus simplifies the possible outcomes.
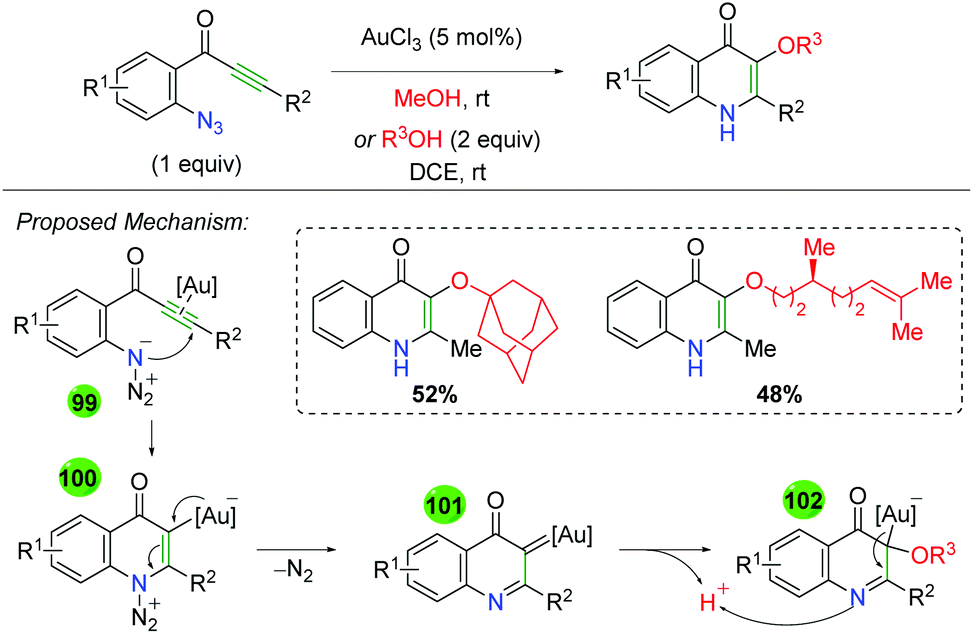
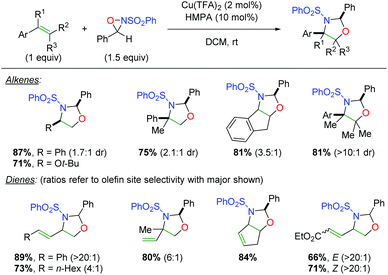 |
| | Scheme 24 Copper-catalyzed alkene oxyamination using oxaziridines (Yoon, 2007).33 | |
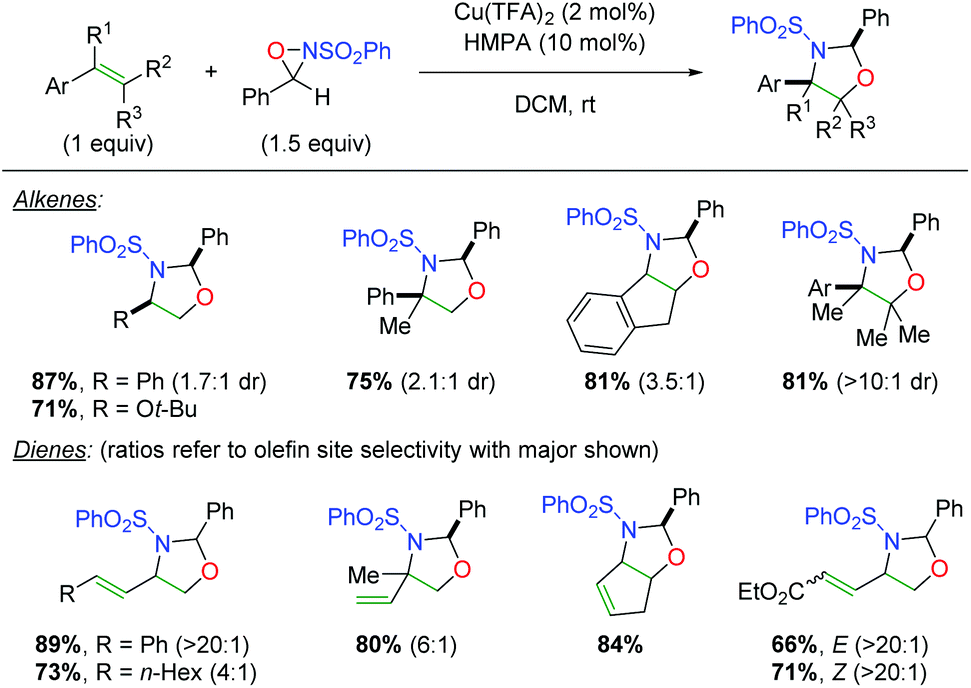
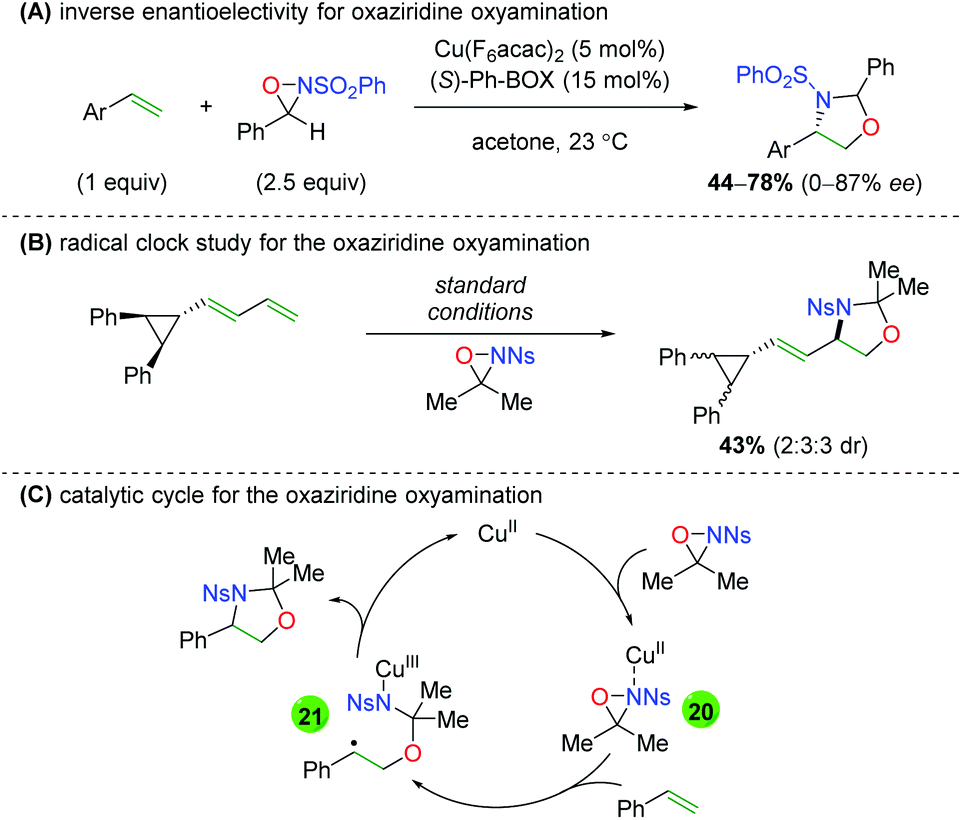
Soon thereafter, Yoon and co-workers reported an enantioselective version of this transformation (Scheme 25A).34 The reaction is shown only with styrenyl alkenes and exhibits fair yields, but poor to good enantioselectivities (note that better enantioselectivities are found for the syn diastereomer than the anti diastereomer). Furthermore, the authors also examined the potential mechanism for the reaction by testing a cyclopropyl radical clock (Scheme 25B).35 Starting with a diastereomerically pure diene, they observed a scrambling of diastereomers, indicating a ring-opening process. They noted that radical cyclopropyl ring openings are stereorandom, while cationic ring openings are stereospecific, implying a radical intermediate. Accordingly, they proposed a mechanism starting with copper(II) (they noted that copper(I) produced no reaction), which coordinates to the sulfonamide of the oxaziridine (20). Homolytic cleavage of the N–O bond and oxygen addition to the alkene generates carbon radical intermediate 21, which closes via homolytic cleavage of the N–Cu bond to generate the product and regenerate copper(II).
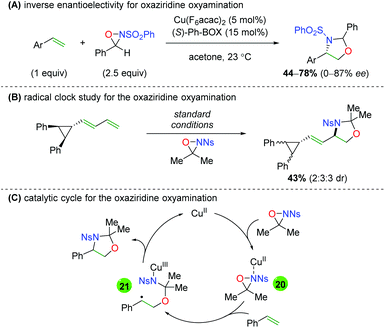 |
| | Scheme 25 Enantioselective alkene oxyamination and mechanistic proposal (Yoon).34,35 | |
The year after the Yoon laboratory's initial report, the Chemler group published an enantioselective copper-catalyzed alkene oxyamination method using sulfonamide-tethered alkenes with terminal TEMPO trapping (Scheme 26).36 This method accommodates a range of substitutions on the alkene backbone and α-position of the alkene, though substrates with β-substitution on the alkene were not shown.
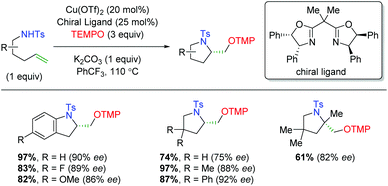 |
| | Scheme 26 Copper-catalyzed alkene oxyamination using sulfonamide-tethered alkenes and TEMPO (Chemler, 2008).36 | |
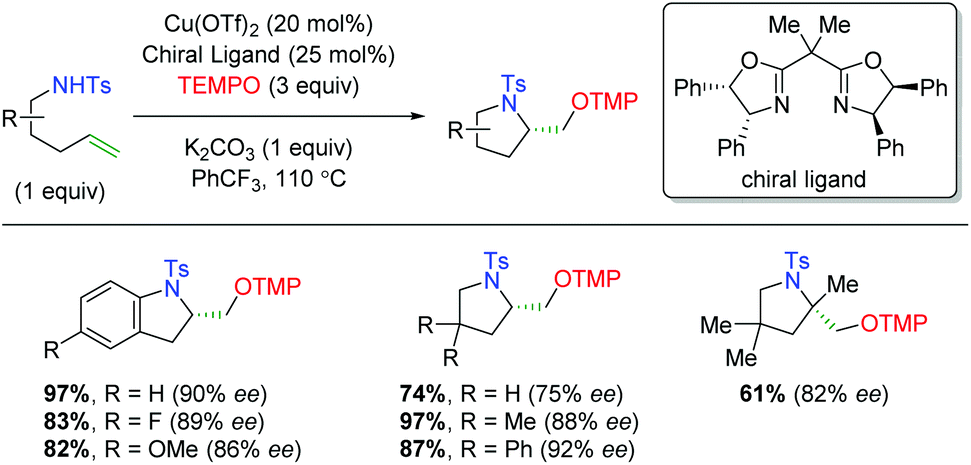
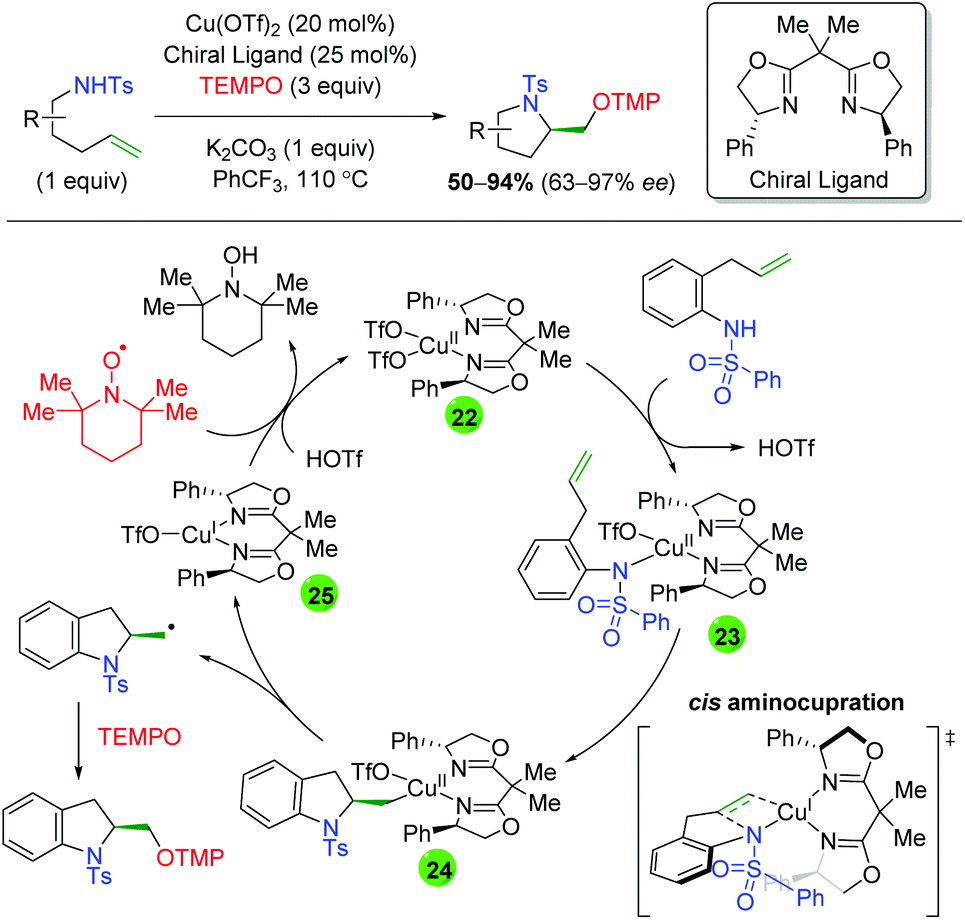
In 2013, Chemler and co-workers released a follow-up paper showing a complementary method to their earlier work producing the opposite enantiomer and providing a more definite proposal for the mechanism of their transformation (Scheme 27).37 Their proposed mechanism begins with coordination of the copper(II) species to the chiral ligand (22), then with coordination to the sulfonamide (23). They then propose the rate-limiting step (base on KIE experiments) as a syn-aminocupration controlled by the chiral ligand (24). This copper(II) species then undergoes homolytic cleavage to generate a terminal carbon radical and copper(I) species (25). They propose that both species are acted upon by TEMPO, with the carbon radical being quenched to generate the product, while the copper(I) species is oxidized to copper(II) to regenerate the active catalyst.
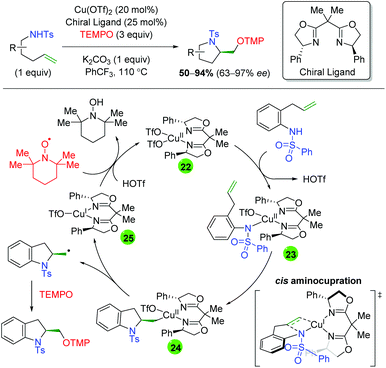 |
| | Scheme 27 Copper-catalyzed oxyamination showing opposite enantioselectivity and proposed mechanism (Chemler, 2013).37 | |
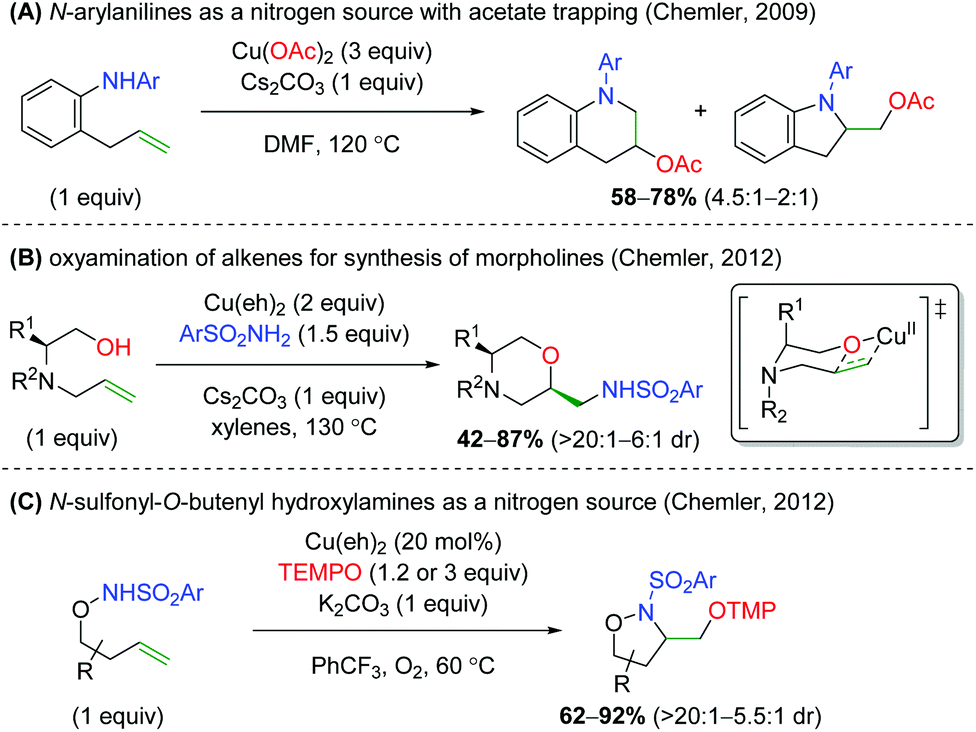
The Chemler laboratory has also reported three additional alkene oxyamination methods (Scheme 28). The first method uses N-aryl anilines as a nitrogen source in place of the previous sulfonamide and terminates with a reductive elimination from the acetate ligands on copper (Scheme 28A).38 Unfortunately, this reaction is more of a proof-of-concept and shows only three substrates while using three equivalents of copper(II) acetate. The second method uses excess amounts of copper like the first method, but uses reduced amino acid scaffolds and sulfonamides to generate morpholine skeletons (Scheme 28B).39 The authors proposed that this reaction proceeds through a similar mechanism as the previous work, but with an oxycupration event in place of the aminocupration reported previously. The third method is analogous to their group's original work, using N-sulfonyl-O-butenyl hydroxylamines as nitrogen nucleophiles with the TEMPO trapping profile seen in their original alkene oxyamination method (Scheme 28C).40
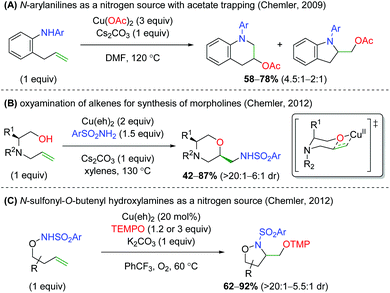 |
| | Scheme 28 Additional copper-catalyzed cyclizations from Chemler and Co-workers.38–40 | |
In 2010, Blakey and co-workers published a oxyamination strategy (Scheme 29)41 using similar sulfonamide-tethered alkenes to the Chemler group's work (Scheme 26), but using an oxidation strategy similar to that seen in the palladium(II/IV) chemistry (see section 2.1). While the regioselectivity of the reaction (5-exo vs. 6-endo) is not always high and displays a substrate dependence, the proposed reaction mechanism is more interesting. The reaction starts with copper(I), which then coordinates to the sulfonamide and is oxidized to copper(III) by PhI(OAc)2 (26). A free acetate group then attacks the alkene at the position that leads to the highest stability for the resulting positive charge to generate a Cu(III) species (27). This complex readily undergoes reductive elimination to yield the product and regenerate copper(I).
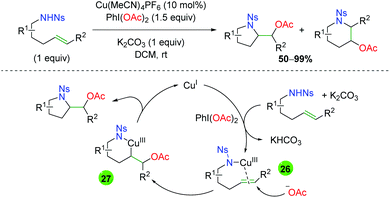 |
| | Scheme 29 Alkene oxyamination with sulfonamide-tethered alkenes using a copper(I/III) oxidative pathway (Blakey, 2010).41 | |
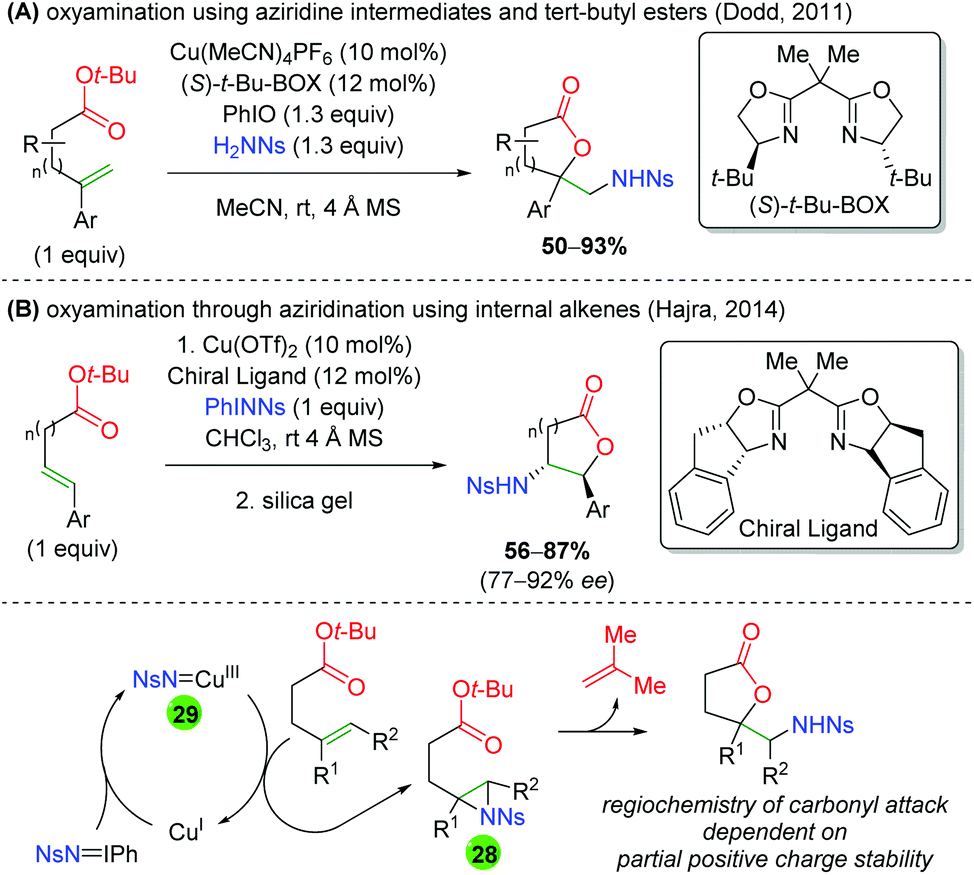
In 2011, the Dodd laboratory combined copper-catalyzed aziridination with ester trapping to effect an alkene oxyamination in 5-exo or 6-exo fashion (Scheme 30A).42 Despite using a chiral ligand ((S)-t-Bu-BOX), the reaction is not enantioselective. The authors stated that this ligand is necessary, though no additional BOX ligands were shown, and the authors note that no further attempts were made to improve the enantioselectivity. Their scope is mostly limited to different aryl groups on the alkene and showed almost exclusively unsubstituted ester backbones. Three years later, the Hajra group copied this procedure and slightly amended it to internal alkenes, observing the expected reversal of the regioselectivity to favor 5-endo and 6-endo products (Scheme 30B).43 Their scope is also limited to changing the aryl group on the alkene, as all substrates were unsubstituted, with a single exception. However, their reaction displays excellent diastereoselectivity and good enantioselectivity. Regarding the mechanism of these transformations, it can be imagined that copper(I) reacts with the nitrene (either generated in situ or preformed) to form a copper nitrene species (28). This copper nitrene can then affect aziridination of the alkene to 29 and regenerate copper(I). Finally, the regioselectivity of the ester trap can be easily explained by stability of the partial positive charge formed upon a nucleophilic attack on the aziridine. In both cases the carbonyl attacks at the carbon stabilized by the aryl group. Note that Hajra and co-workers reported silica gel (or excess copper) as being necessary to form the final lactone product for electron-deficient substrates.
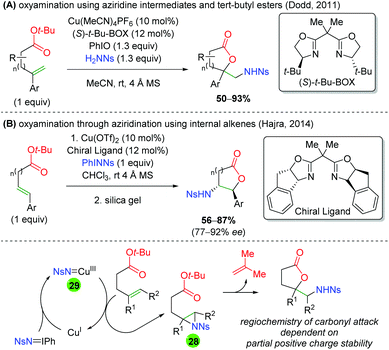 |
| | Scheme 30 Alkene oxyamination with aziridine formation and tert-butyl esters.42,43 | |
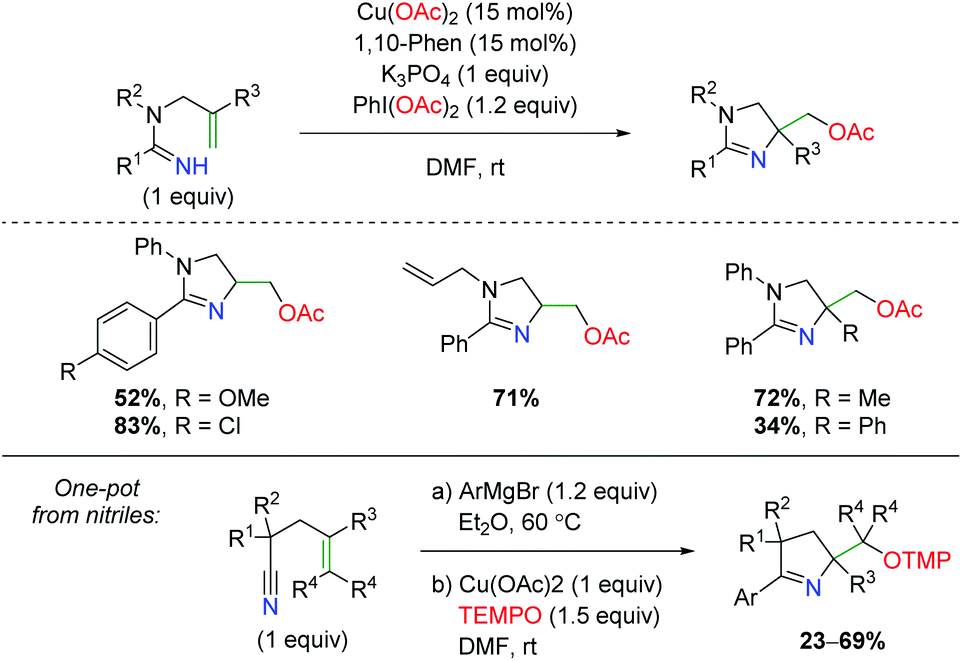
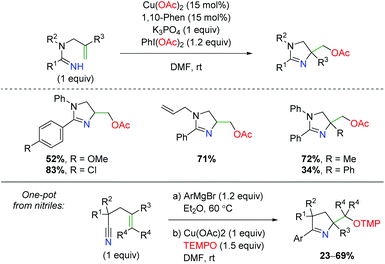
Another unique approach to alkene amino oxygenation is the use of alkene-tethered amidine as a nitrogen source with acetate trapping through terminal reductive elimination (Scheme 31).44 The substrate scope is confined exclusively to aryl groups at the R1 carbonyl position, but does demonstrate some variation on the amide nitrogen in the backbone and at the α-position on the alkene. However, no internal alkenes are shown. Furthermore, the Chiba group also demonstrated that this reaction is amenable to a one-pot approach starting with nitriles.45 Attack of the nitrile with an aryl Grignard, followed by addition of copper and TEMPO, furnished a variety of dihydropyrroles with range of substitutions on the backbone and alkene.
 |
| | Scheme 31 Copper-catalyzed dihydroimidazole formation (Chiba, 2012).44,45 | |
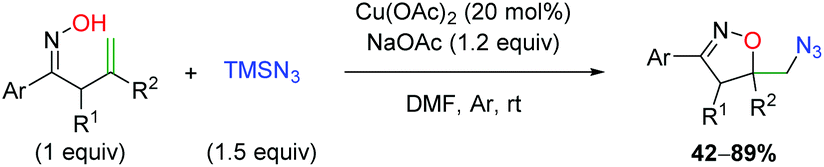
In 2014, Wang and co-workers disclosed a copper-catalyzed oxyamination using oximes as an oxygen cyclization group, followed by trapping with TMS azide (Scheme 32).46 This method shows moderate substrate tolerance, as substitutions at the α-position of the alkene produce a slight decrease in yield over the unsubstituted case. However, substitutions at the β-position of the alkene show a β-hydride elimination product following oxygen cyclization. The mechanism of this transformation remains unconfirmed, but mechanism studies point toward the possibility of a base-catalyzed cyclization, followed by a copper-catalyzed azide trap.
 |
| | Scheme 32 Copper-catalyzed oxyamination using oximes (Wang, 2014).46 | |
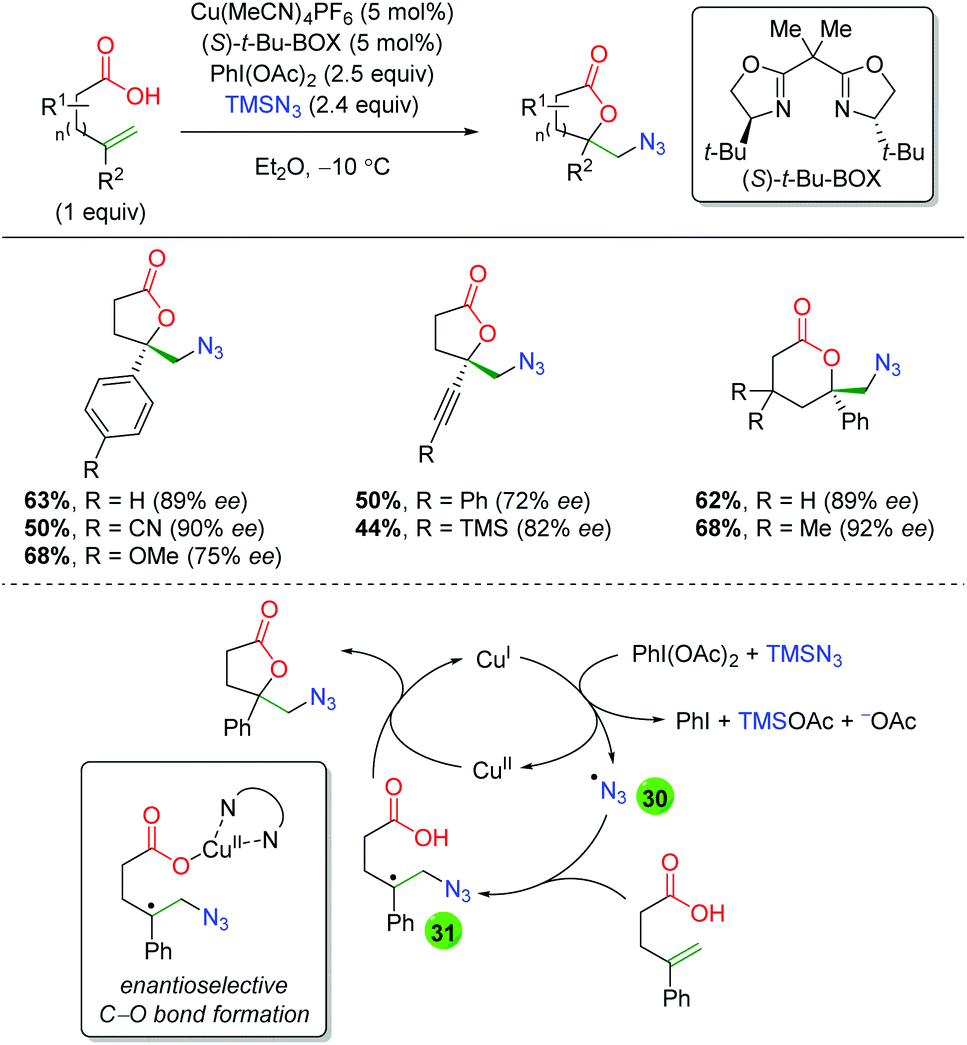
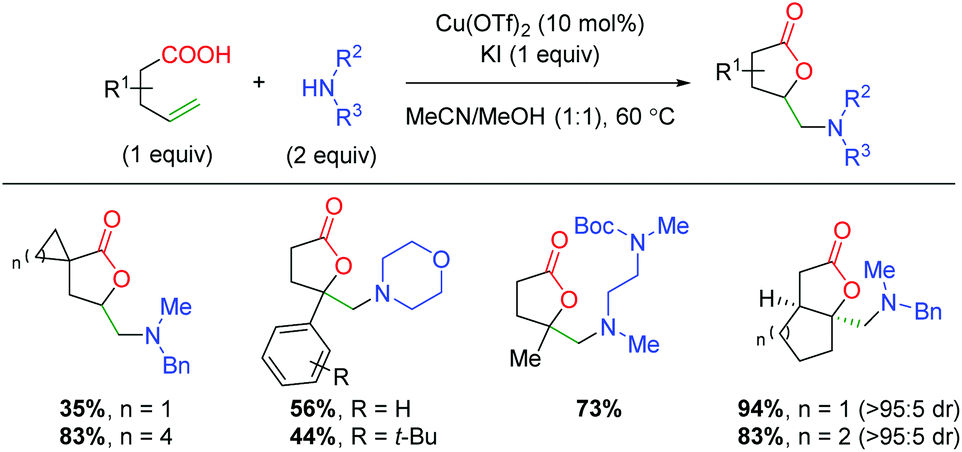
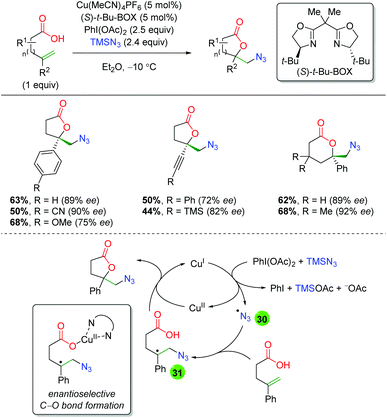
In 2015, the Buchwald laboratory used TMS azide as an initiator, rather than the terminal trap just mentioned, to extend their previous lactonization efforts47 to other groups, such as azides (Scheme 33).48 The substrate scope of this transformation is almost exclusively confined to 5-exo and 6-exo substrates lacking backbone substitution and containing either aryl or alkyne groups at the α-position of the alkene. The yields are fair to very good, with enantioselectivities ranging from 72 to 92%. As mentioned above, the proposed mechanism begins with generation of an azide radical (30) from the combination of copper(I), PhI(OAc)2, and TMS azide. The azide radical adds to the alkene, generating a carbon radical stabilized by the α-aryl group (31). The lactone ring is then closed through an enantioselective C–O bond forming event directed by the chiral ligand bound to copper(II).
 |
| | Scheme 33 Copper-catalyzed enantioselective azido-lactonization (Buchwald, 2015).48 | |
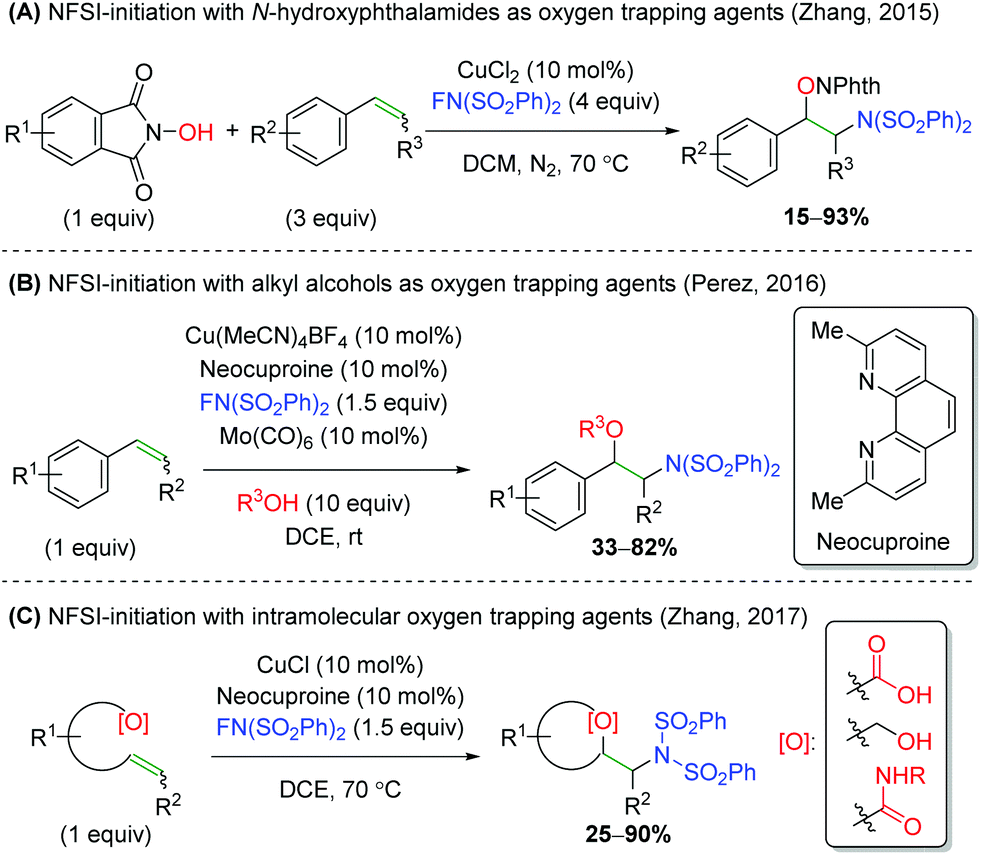
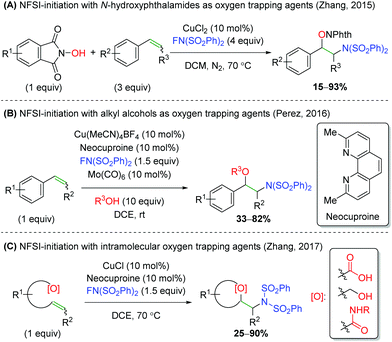
Another strategy employed by several groups has been using NFSI (N-fluorobenzenesulfonamide) as a nitrogen-initiation agent, followed by trapping with various oxygen nucleophiles (Scheme 34). First, the Zhang laboratory demonstrated the use of N-hydroxyphthalimides as oxygen trapping agents (Scheme 34A).49 This method shows good tolerance of substitutions on styrene, but lower yields for substitution at the β-position on the alkene. Furthermore, Pérez and co-workers disclosed a similar strategy, using a large excess of alcohols to effect a similar transformation, but with improved tolerance for substitution at the β-position of the alkene (Scheme 34B).50 Interestingly, the authors note that the reaction does not proceed without catalytic Mo(CO)6. This oxidant was not needed in the first NFSI example above, but that is potentially offset by the excess NFSI used. Finally, a second Zhang laboratory released an NFSI-initiated reaction involving an intramolecular trap using carboxylic acids, alcohols, and amides (Scheme 34C).51 The yields for this reaction are fairly good, but the alcohols and amides showed almost exclusively yields below 50%. Though a reaction mechanism was proposed for each of the three cases presented, there has been little mechanistic investigation performed and each group proposed slightly differing mechanisms.
 |
| | Scheme 34 Copper-catalyzed, NFSI-initiated alkene amino oxygenation.49–51 | |
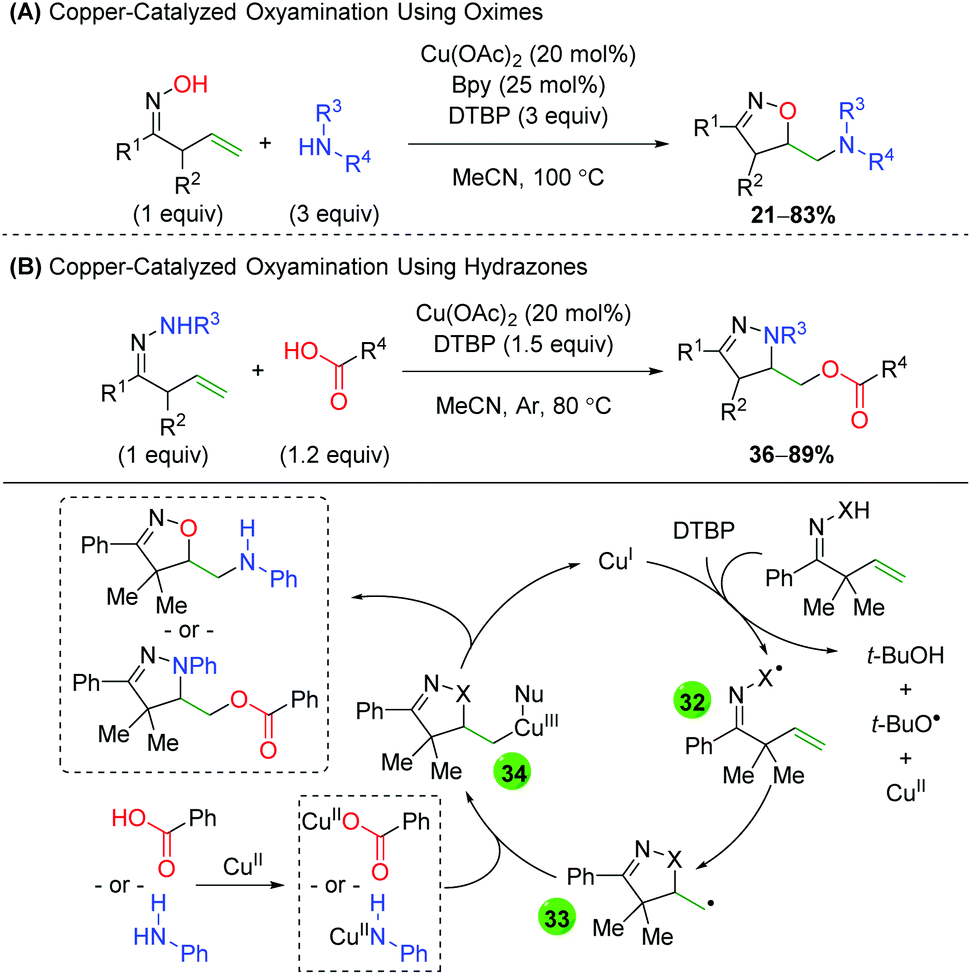
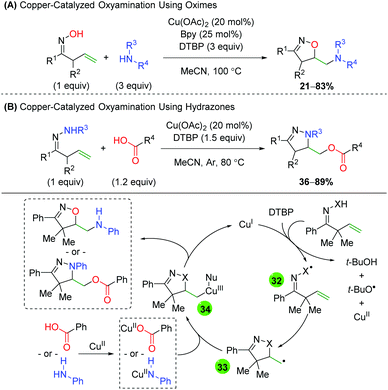
The Han group, which has extensively studied alkene difunctionalization of unsaturated oximes and hydrazones under transition metal-free conditions, produced protocols for copper-catalyzed versions to expand the scope of nucleophilic trapping partners (Scheme 35).52 Both protocols feature a similar alkene scope, though the hydrazone method (B) does feature one 6-exo example. The oxime method (A) showcases a very broad scope of both alkyl and aryl amines, with the formation of both secondary and tertiary amines. The hydrazone method (B) also shows a broad scope for the trapping reagent, as a wide range of alkyl and aryl carboxylic acids participated in the reaction. For the mechanism, the authors propose a single electron transfer (SET) from copper(I), with DTBP abstracting a proton to generate either a nitrogen or oxygen-centered radical (32). The 5-exo ring closing results in a terminal carbon radical (33), which then coordinates with the copper(II) species bearing the respective nucleophile (34). Reductive elimination then generates the product and regenerates copper(I). This same strategy was recently employed using molecular oxygen as the terminal trapping agent to achieve 1,2-amino alcohols.53
 |
| | Scheme 35 Copper-catalyzed radical alkene oxyamination using oximes and hydrazones with amines and carboxylic acids (Han, 2016/2018).52 | |
In 2018, Li and co-workers disclosed an aerobic copper-catalyzed one-pot iodo-lactonization/amination (Scheme 36).54 The scope of the transformation is quite broad, with a large number of aliphatic 5-exo products shown. The reaction appears to tolerate substitution at both the α- and β-positions on the alkene and tolerates a large number of amines (though only one primary amine is shown). The authors note that an oxygen atmosphere is important to the reaction and propose the formation of an iodolactone (which was isolated), before simple SN2 attack displacement of the iodine. While this may be considered a formal oxyamination, it is nonetheless relevant to the discussion of this topic.
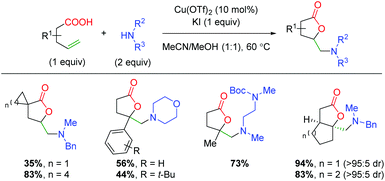 |
| | Scheme 36 Copper-catalyzed iodo-lactonization with amination (Li, 2018).54 | |
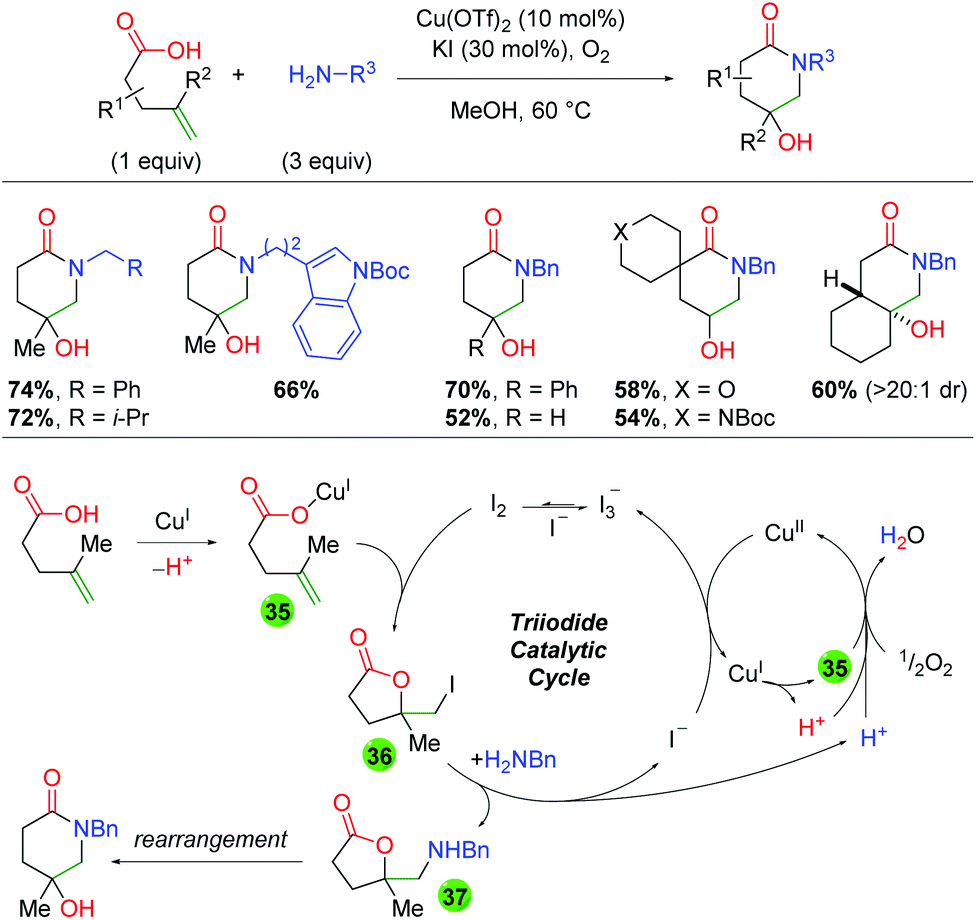
Two years later, the same laboratory published a method combining copper and iodine catalysis in an oxidative atmosphere to create piperidone scaffolds through formal 5-exo oxyamination and rearrangement (Scheme 37).55 The method incorporates a range of primary amine sources and shows good tolerance for an array of substitutions on the backbone and alkene. The authors conducted UV-Vis studies to elucidate the presence of an I3− species that they believe to be an iodine reservoir within the catalytic cycle. Furthermore, they utilized deuterium labeling studies to determine that the oxy iodination of the alkene is a anti addition, which they note points towards an iodine-initiated pathway. They also note that coordination of the carboxylic acid substrate (or another carboxylic acid) is vital to reaction turnover. In regards to the mechanism, the authors propose initial coordination of the acid substrate to Cu(I) (35). This reacts with I2 to give a net 5-exo anti addition across the alkene (36). The iodine is then displaced by the primary amine (37), where it can rearrange to the piperidone product. The displaced iodine is oxidized by Cu(II) to generate I3−, which is the iodine reservoir. The Cu(I) coordinates with a carboxylic acid and is oxidized to Cu(II) by molecular oxygen. The authors note that this oxidation by O2 also serves as a “proton sponge” to enhance the nucleophilicity of species, such as the amine, within the reaction.
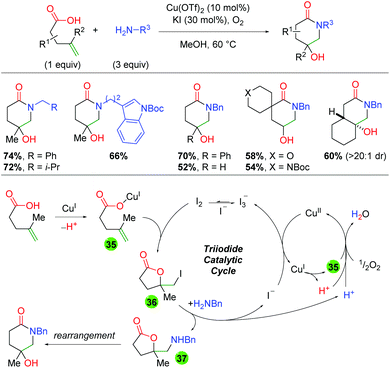 |
| | Scheme 37 Copper and iodine-catalyzed 4-hydroxy piperidone formation (Li, 2020).55 | |
2.4 Platinum catalysis
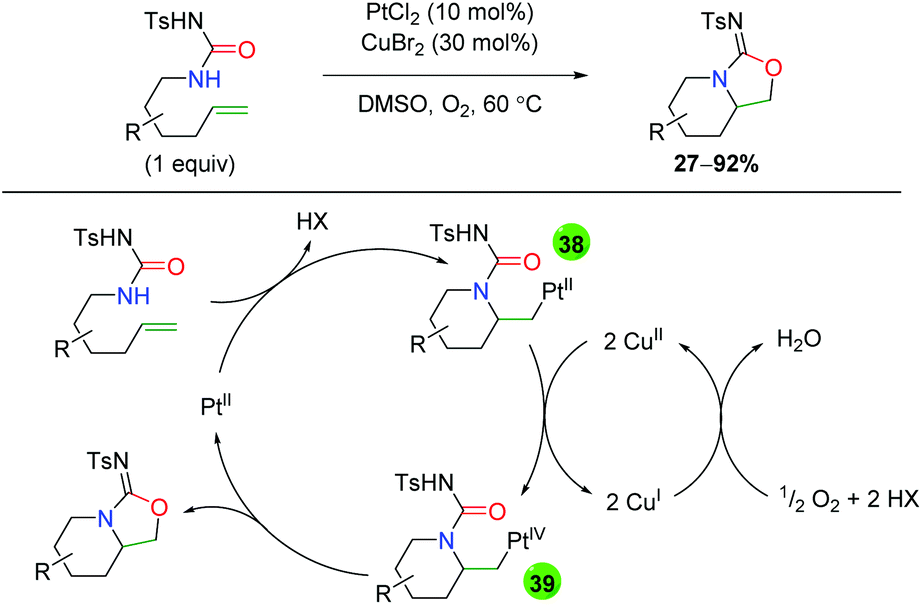
The sole example of platinum-catalyzed alkene oxyamination comes from the Muñiz group using urea-tethered alkenes (Scheme 38).56 The substrate scope is very rigid, showing minimal substitution along the formed piperidine ring and a single example of substitution at the α-position on the alkene. Given that copper was also added in catalytic amounts, the mechanism is of interest. The authors propose a platinum(II)-assisted 6-exo ring formation to generate platinum(I) intermediate 38, which is oxidized to platinum(IV) intermediate 39 by copper(II). Intermediate 39 then undergoes reductive elimination to form the product and platinum(II), which the copper is oxidized back to copper(II) by atmospheric oxygen. Notably, the authors tested the reaction with platinum(IV) to determine if perhaps this was the active species in the reaction, but found a slower reaction rate when using platinum(IV) as the active catalyst. Interestingly, no other oxidants were mentioned outside of copper(II).
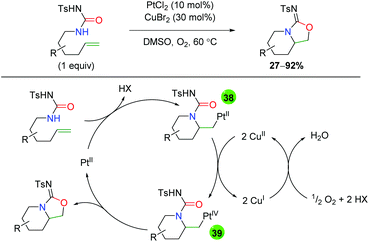 |
| | Scheme 38 Platinum-catalyzed amino oxygenation using urea (Muniz, 2009).56 | |
2.5 Iron catalysis
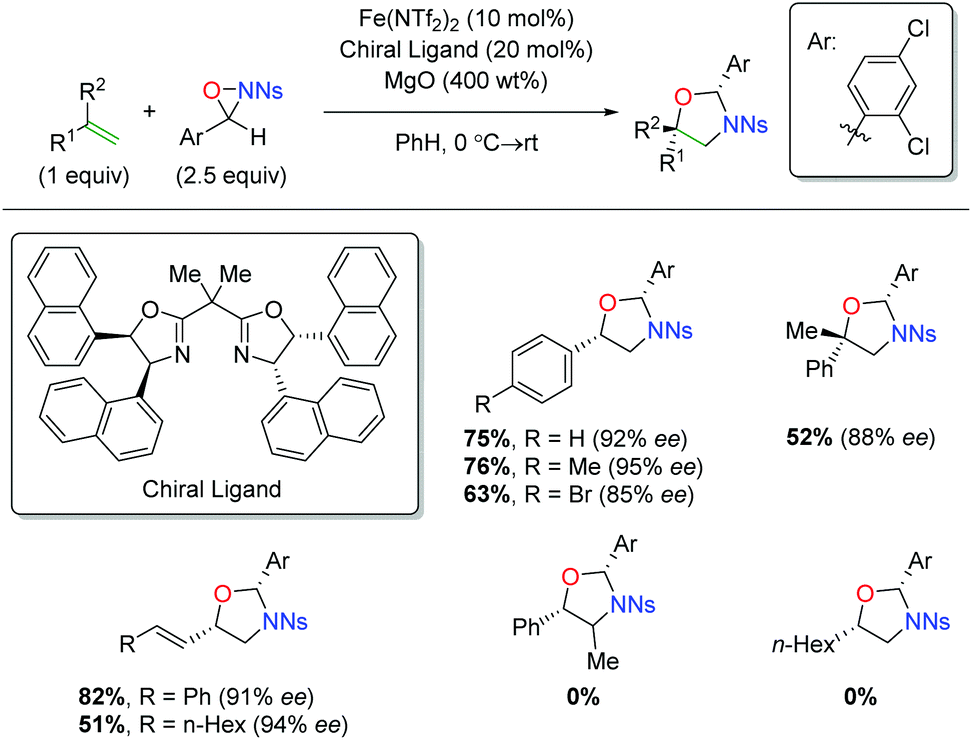
Shortly after releasing their copper-catalyzed alkene oxyamination procedure using oxaziridines, the Yoon group released a procedure using iron catalysts to achieve regioselectivity that was complementary to their copper case (Scheme 39).57 Their initial investigation did not result in enantioselective product formation,58 but their follow-up protocol used a chiral BOX ligand to affect enantioselectivity. The substrate scope of this transformation is limited to terminal aryl alkenes, though it is tolerant of electronic perturbation within that framework. The reaction is tolerant of substitution at the α-position of the alkene and also tolerates both aryl and alkyl 1,3-dienes. Notably, the mechanism for this switch in regiochemistry remains unclear at this time.
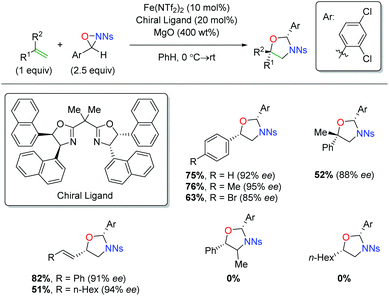 |
| | Scheme 39 Iron-catalyzed alkene oxyamination using oxaziridines (Yoon, 2012).57 | |
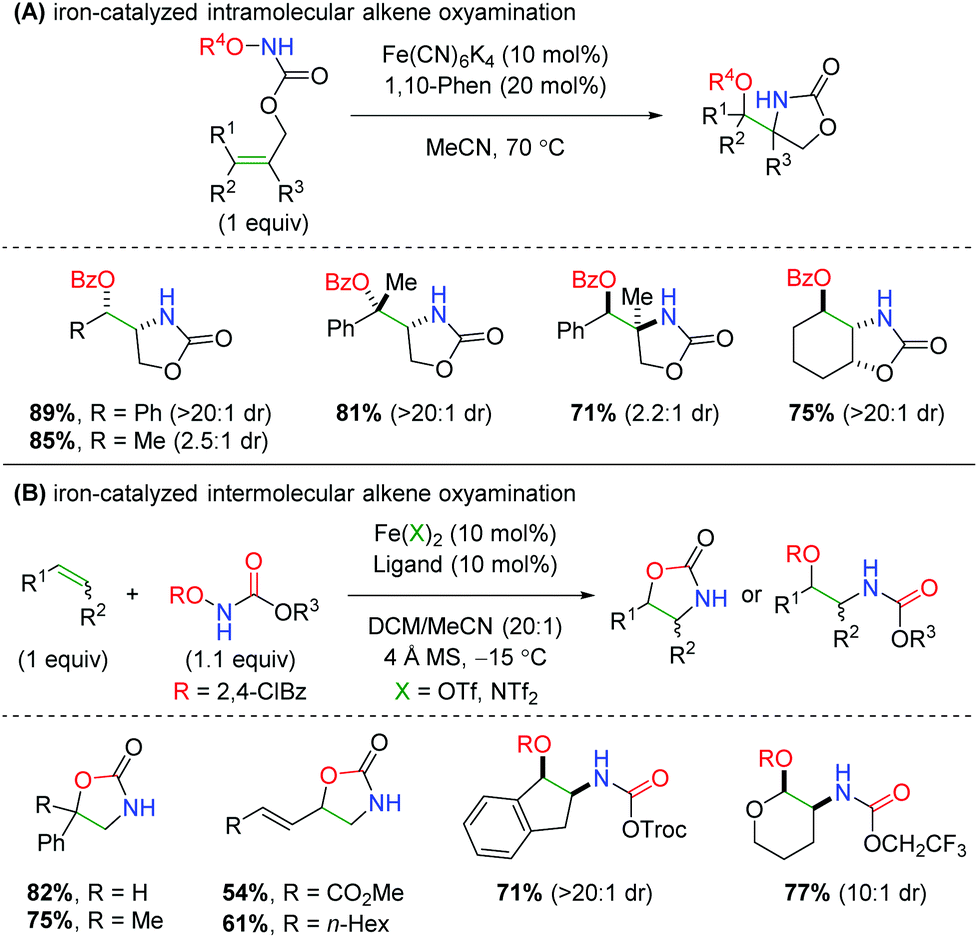
In 2013, Xu and co-workers disclosed an iron-catalyzed oxyamination procedure utilizing alkene-tethered N-benzoylcarbamates to produce oxazolidinones (Scheme 40A).59 The authors found that using iron(II) catalyst Fe(CN)6K4 is unique in its ability to produce both a high yielding reaction and high diastereoselectivity. This intramolecular procedure exhibits a good substrate scope within the confines of a tethered scaffold, as most yields are over 70% with excellent diastereoselectivity. The authors also show examples of enantioselectivity for several substrates, but in a proof-of-concept fashion using a chiral BOX ligand. The next year, the same laboratory published an intermolecular version of this chemistry using a variety of N-benzoylcarbamate derivatives, several different ligands, and two different iron catalysts (Scheme 40B).60 The scope of olefins that participate in this procedure is generally excellent, displaying both high yield and diastereoselectivity for a variety of alkenes and 1,3-dienes. As in their previous work, the authors also demonstrate two proof-of-concept examples for an enantioselective reaction using chiral PyBOX ligands. However, one of the curiosities of this procedure is the seemingly unpredictable nature of the products formed, as some olefins produce the expected oxazolidinone, while others produce the ring-opened product. The authors propose that more radical stabilizing olefins give ring-opened products, while less stabilizing olefins give oxazolidinone products. However, it is unclear how this is corroborated by their substrate scope. Furthermore, the number of unique combinations of iron catalyst, ligand, and carbamate used within the substrate scope could be problematic in instances when a substrate that was not previously investigated is desired.
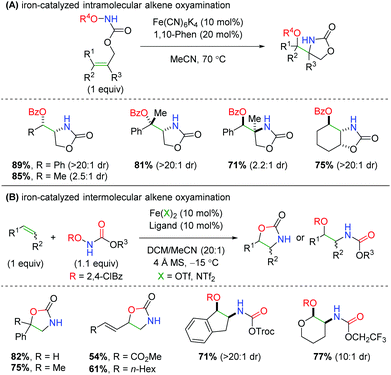 |
| | Scheme 40 Iron-catalyzed alkene oxyamination using N-benzoylcarbamates (Xu, 2013/2014).59,60 | |
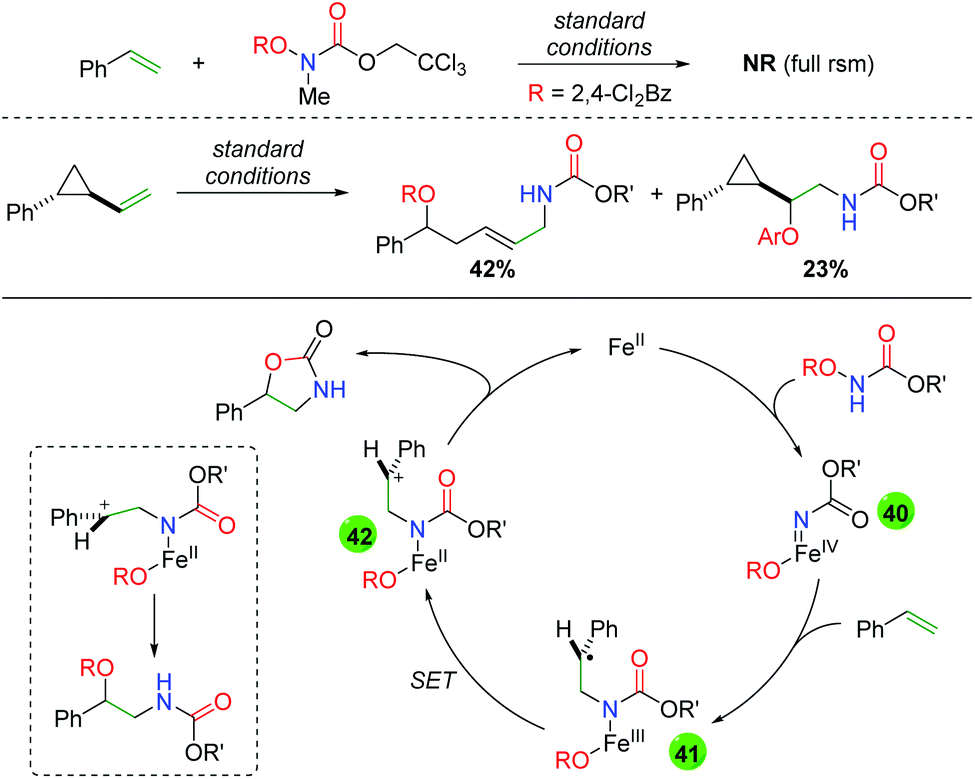
In order to further probe a potential mechanism, the Xu group examined several aspects of their transformation (Scheme 41). First, they substituted a methyl group on the nitrogen in their N-benzoylcarbamate and observed full starting material recovery, leading them to propose the secondary amide as necessary in the reaction. Secondly, they performed a cyclopropyl radical clock test and found a mixture of ring-opened and unopened products. With these experiments in mind, they proposed that the iron(II) catalyst interacts with the N-benzoylcarbamate to form an iron-nitrene species (40), which then adds to the alkene to form intermediate 41. From this carbon radical intermediate, they propose a single electron transfer event by the iron center to generate carbocation intermediate 42, which can be attacked either by the carbonyl of the carbamate (to form the oxazolidinone) or by the pendant benzoyl group to form the uncyclized oxyamino product.
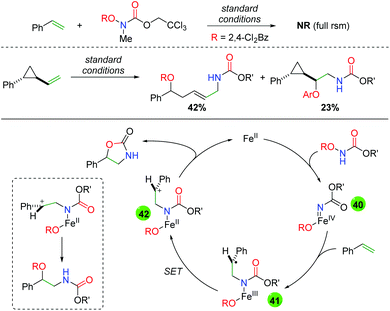 |
| | Scheme 41 Mechanism probes and proposed mechanism for the iron-catalyzed alkene oxyamination. | |
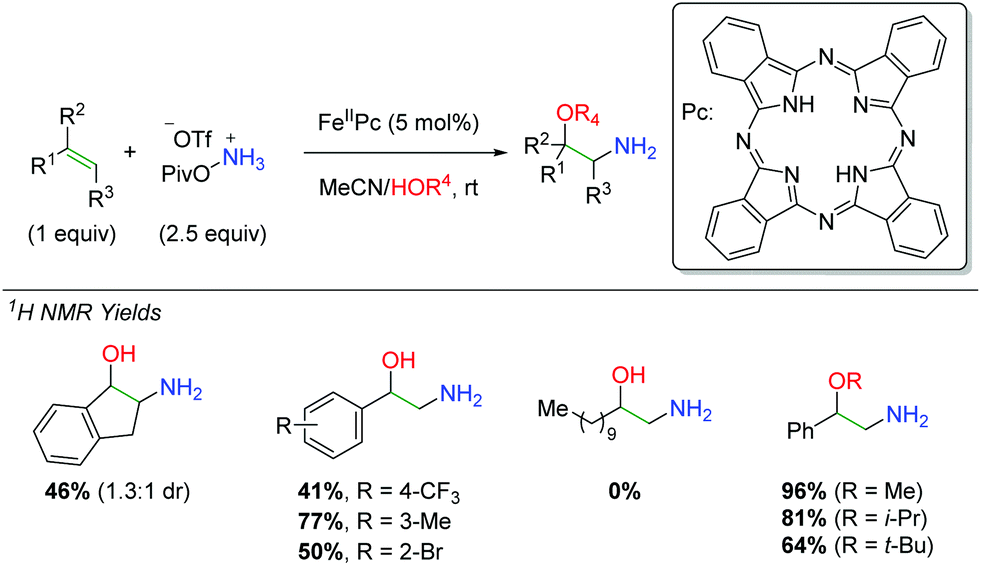
In 2016, Morandi and co-workers examined the use of iron phthalocyanine catalysts to affect an amino oxygenation of alkenes resulting in free alcohol and amine (Scheme 42).61 For the nitrogen source, they chose a PivONH3OTf salt and used alcohol or water as a co-solvent for the oxygen trapping agent. The procedure works fairly well, though most of the products had to be N-Boc protected before isolation, somewhat eliminating a key selling point of this approach. The reaction is tolerant of some β- and α-substitutions on the alkene as well as a number of different substituted styrenes. However, the reaction fails to work effectively in the absence of a styrenyl alkene system.
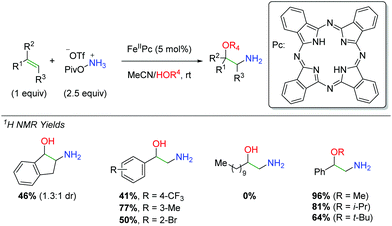 |
| | Scheme 42 Iron-catalyzed synthesis of 2-amino-1-phenylethanols from alkenes (Morandi, 2016).61 | |
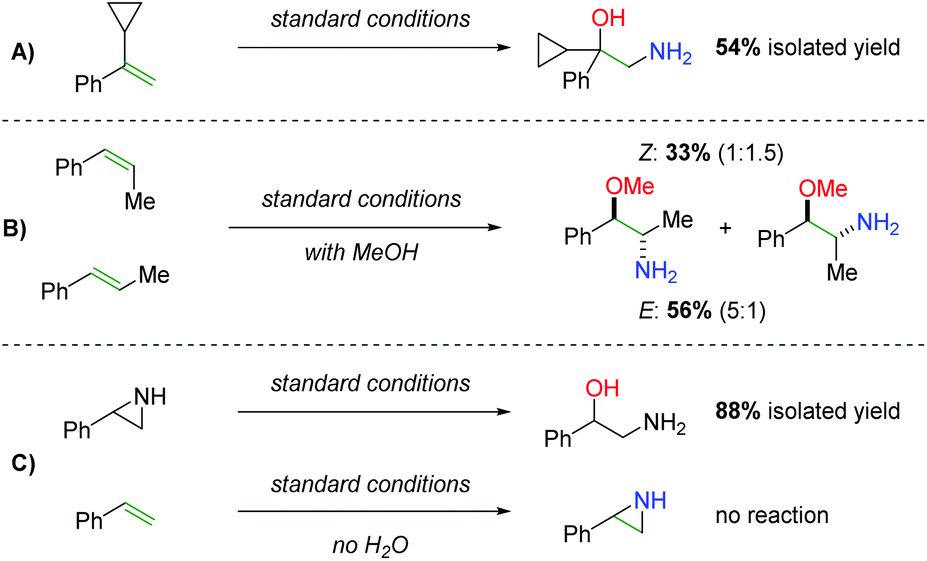
Regarding the mechanism, the authors did not observe ring opening products with a radical clock substrate (Scheme 43A) and they were able to observe marginal diastereospecificity originating from anti addition to each alkene geometry (Scheme 43B). Furthermore, they found that when a hypothetical aziridine intermediate was exposed to the reaction conditions, the expected product could be obtained in good yield (Scheme 43C). However, when they reacted the alkene with the standard conditions in the absence of water, they were unable to observe either the desired product or an aziridine product, suggesting that water (or an alcohol) plays a role in the initial nitrogen addition step.
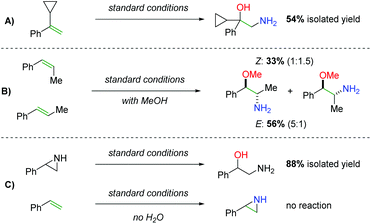 |
| | Scheme 43 Mechanistic probes in the synthesis of 2-amino-1-phenylethanols. | |
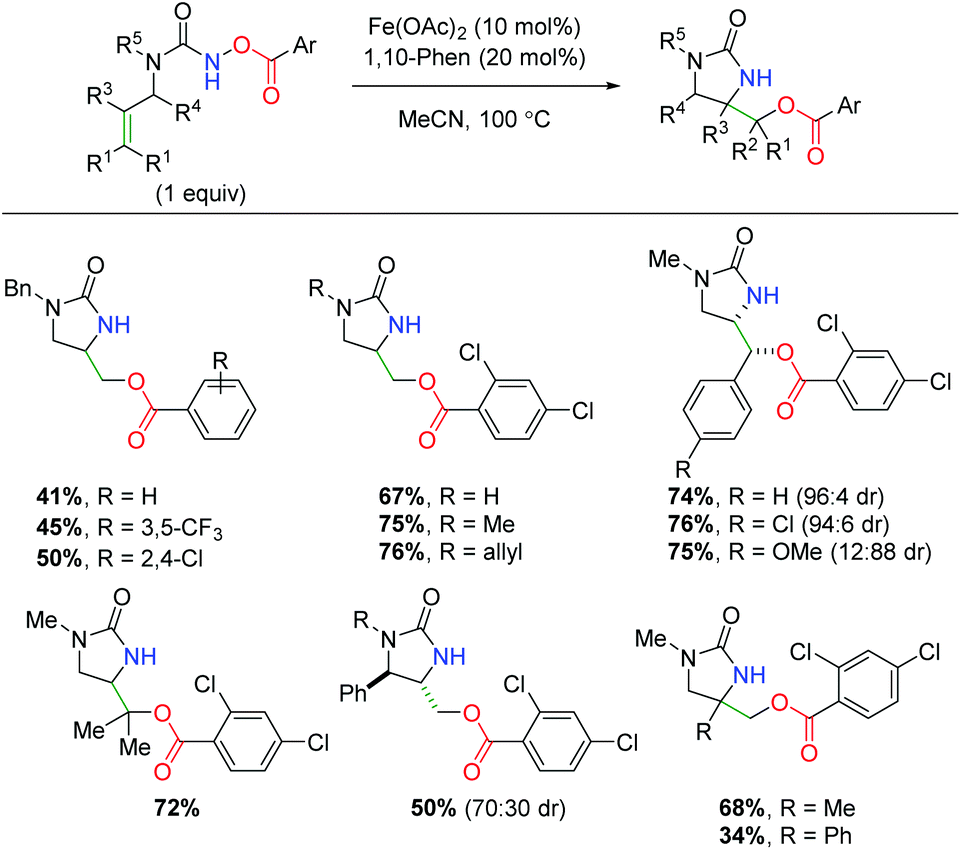
In 2018, another iron-catalyzed procedure was reported using alkene-tethered N-benzoylureas to affect an alkene amino oxygenation by splitting the labile N–O bond (Scheme 44).62 The reaction shows a fairly impressive scope within the intrinsic confines of 5-exo cyclized products (6-exo was not shown). The benzoyl group appears to be limited to benzoic acid derivatives, while the urea nitrogen within the backbone can tolerate several substitutions. Regarding substitution on the alkene, substituents were shown at all positions, giving good yields with good to great diastereoselectivity. The authors demonstrated that the high temperature was necessary to overcome stalling at the aziridine, proposing the aziridine as a putative mechanistic intermediate by demonstrating its competence within the reaction with exogenous benzoic acid. Like Xu and co-workers, the authors also proposed an iron-nitrene as a possible intermediate in the mechanism, but were unable to observe a carbon radical via TEMPO radical trapping.
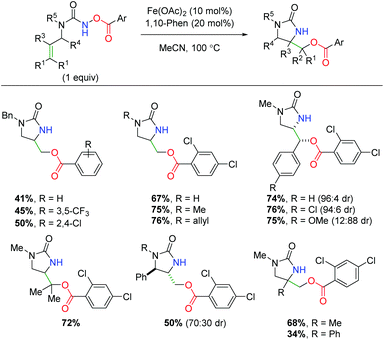 |
| | Scheme 44 Iron-catalyzed alkene oxyamination with N-benzoylureas (Prestat, 2018).62 | |
2.6 Gold catalysis
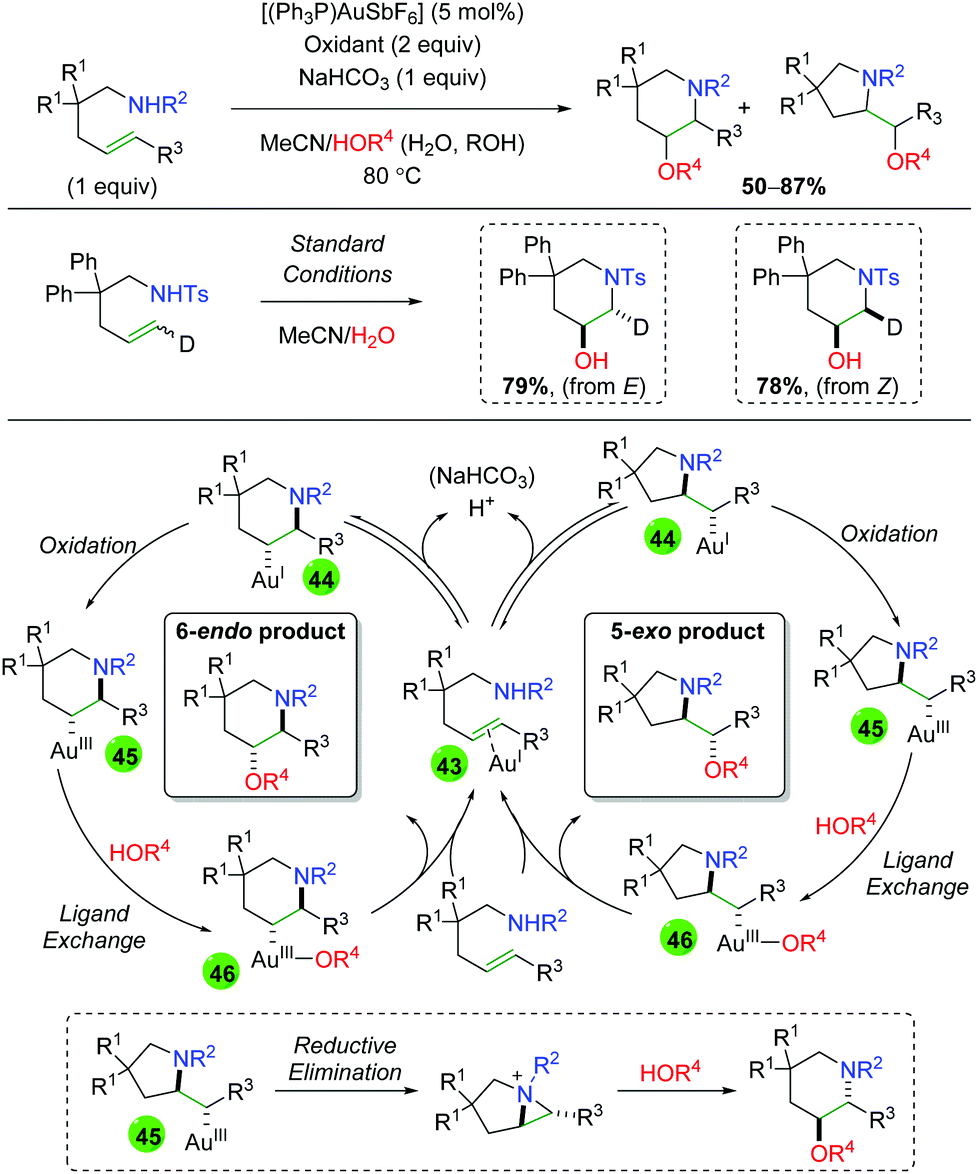
Despite the use of gold catalysts in previous alkene functionalization reactions, there exists only a single example of a gold-catalyzed reaction in the alkene 1,2-oxyamination literature. The Nevado laboratory published a 1,2-amino oxygenation using sulfonamide-tethered alkenes, with water or alcohol as the co-solvent and oxygen nucleophile (Scheme 45).63 Their method displays fair to good yields for several different backbone types as well as alkene substitutions, but often displays a mixture of both 6-endo and 5-exo products. When the authors examined the diastereoselectivity of the reaction using both E and Z deuterium-labeled substrates, they found that each constitutional isomer leads to a separate (and exclusive) diastereomer. From this result and others, they proposed a mechanism for the generation of both 6-endo and 5-exo products. Starting with π-acid coordination of the gold(I) to the alkene (43), a reversible trans-aminoauration generates either 6-endo or 5-exo intermediate 44. Oxidation of the gold center from gold(I) to gold(III) generates intermediate 45. Following ligand exchange with water or alcohol to generate intermediate 46, reductive elimination furnishes the final product and re-generates gold(I). The authors also noted the possibility that 5-exo intermediate 45 could undergo an aziridinium-forming reductive elimination from the pyrrolidine nitrogen. This aziridinium could then be attacked intermolecularly by the oxygen nucleophile to generate the 6-endo product.
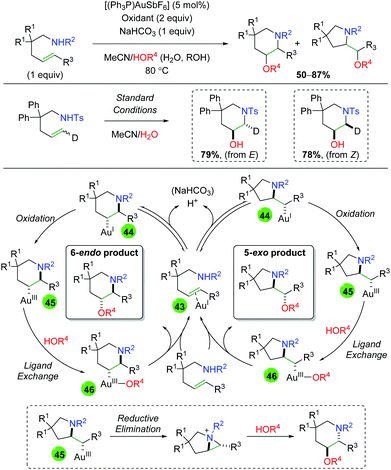 |
| | Scheme 45 Gold-catalyzed alkene amino oxygenation (Nevado, 2011).63 | |
2.7 Manganese catalysis
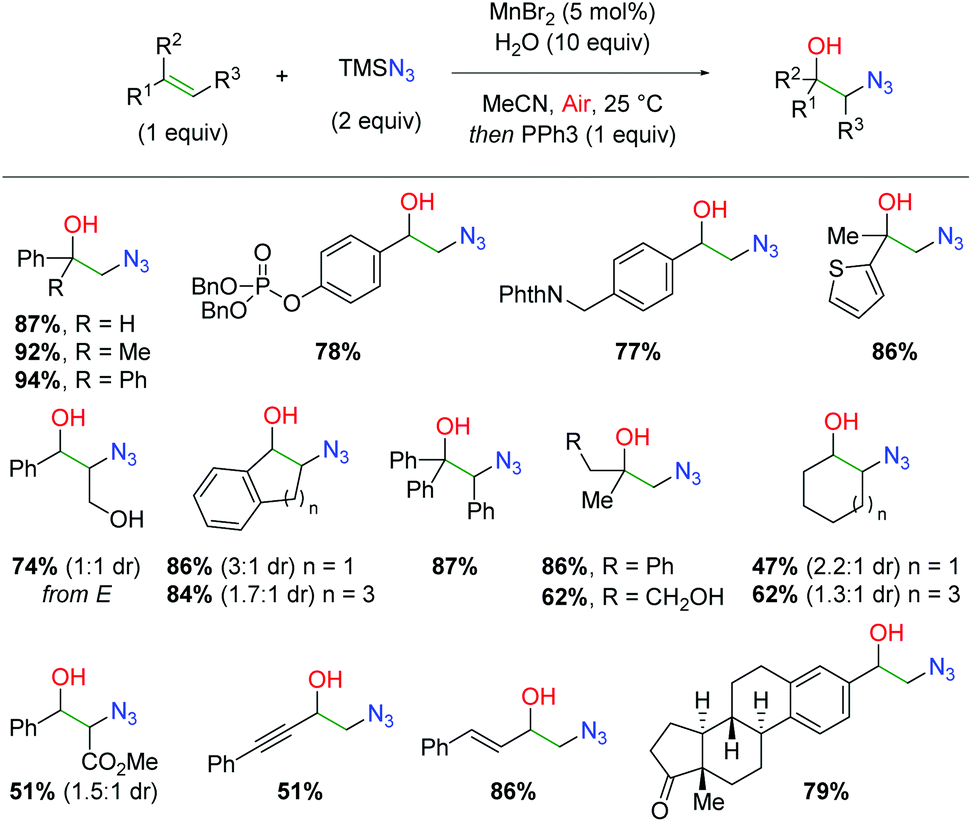
While manganese has seen ubiquitous use as a stoichiometric oxidant, its use as a catalyst has been much more limited. In 2015, the Jiao group released a method for formation of 1,2-azido alcohols from alkenes using a manganese catalyst under aerobic conditions (Scheme 46).64 This strategy follows a similar approach as an alkene scission strategy developed by the same group using TEMPO oxidation for formation of nitrile-tethered ketones from alkenes.65 In this case, the authors hoped to achieve the same azido and molecular oxygen addition to the alkene, but avoid further scission of the C–C bond. This approach was successful and resulted in an extensive substrate scope. Terminal 1,1-disubstituted alkenes (including known radical retardant diphenylethylene) are tolerated along with functional groups such as phosphates, phthalimides, and thiophenes. Internal alkenes are also tolerated, including cinnamyl alcohol and even triphenyl substituted alkenes. Aliphatic alkenes of both terminal and internal varieties work with only a slight drop in yield. Finally, α,β-unsaturated alkenes, enynes, and dienes are viable partners and more complex scaffolds, such as steroids, give appreciable yields under these conditions.
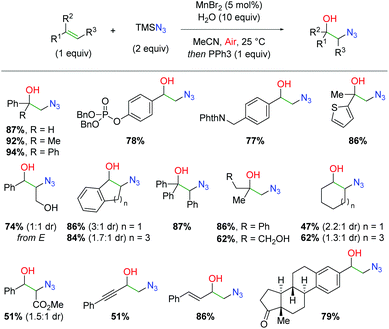 |
| | Scheme 46 Manganese-catalyzed alkene azido oxygenation (Jiao, 2015).64 | |
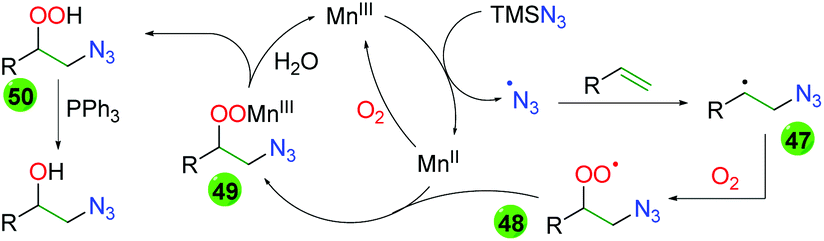
Extensive DFT calculations and some labeling experiments delivered a plausible mechanism for this manganese-catalyzed transformation (Scheme 47). Initial oxidation of Mn(II) to Mn(III) by molecular oxygen starts the catalytic cycle. Mn(III) oxidizes TMS azide to the azido radical, which adds to the terminal end of the alkene (47). The resulting carbon-centered radical then joins with molecular oxygen to create a peroxy radical (48), which coordinates and oxidizes the Mn(II) catalyst (49). Release of the Mn(III) catalyst through protonation of the peroxy species creates peroxy alcohol 50, which can be reduced by triphenylphosphine.
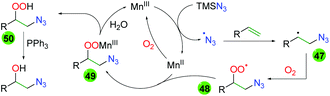 |
| | Scheme 47 Mechanism of the manganese-catalyzed azido oxygenation of alkenes. | |
2.8 Iridium catalysis
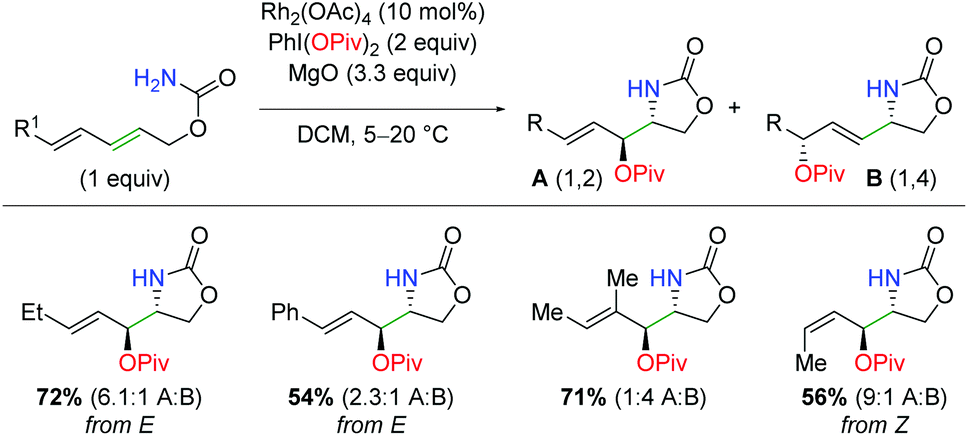
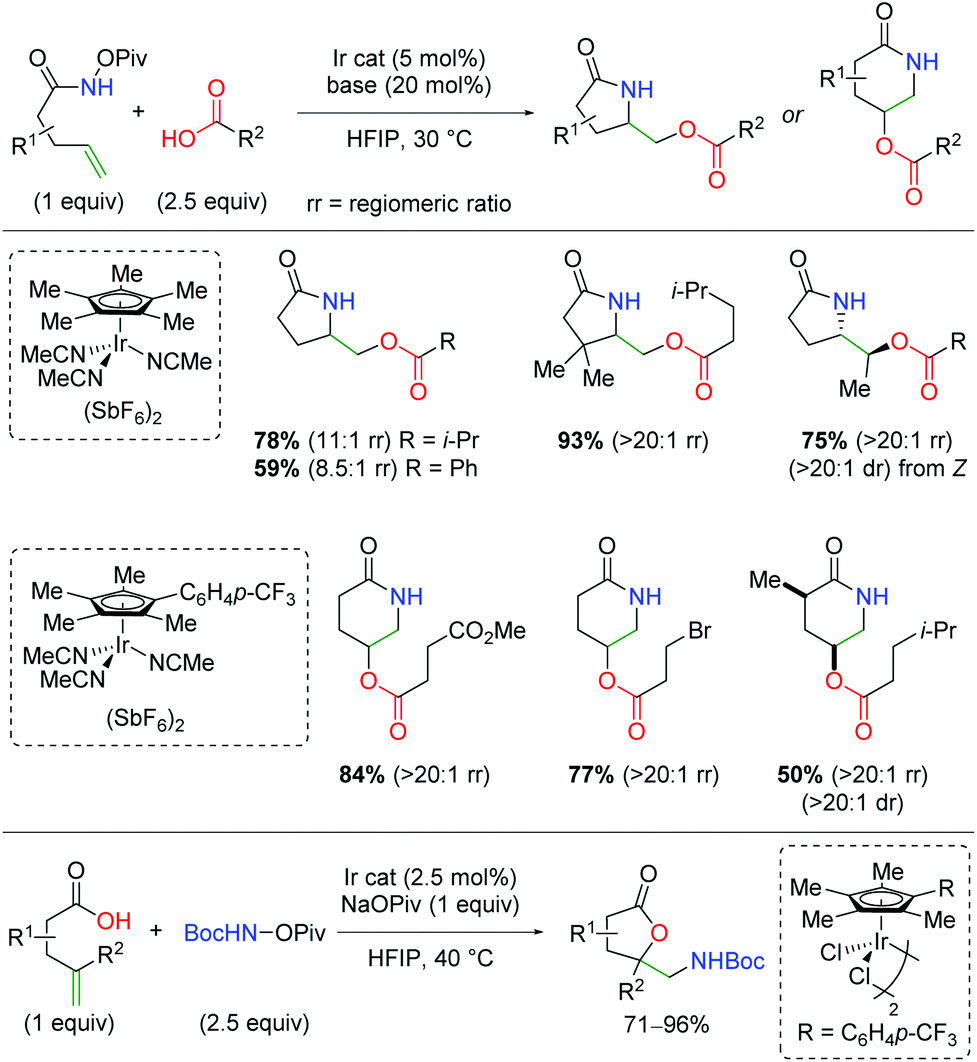
In 2019, the first example of iridium-catalyzed alkene oxyamination was reported by Rovis and co-workers (Scheme 48).66 This method utilized unsaturated N-pivoyl amides with external carboxylic acids to provide a regiodivergent reaction for access to either the 5-exo or 6-endo product, dictated by the choice of ligand. The authors investigated a number of iridium ligands, finding that the standard Cp* gives the 5-exo product preferentially, while the p-CF3 phenyl analog gives the 6-endo product in excellent regioselectivity. Outside of the excellent diastereoselectivity of internal alkenes, substitutions on the unsaturated amides was not fully investigated. A variety of carboxylic acids are incorporated, including those bearing primary alkyl bromides and esters. Furthermore, unsaturated carboxylic acids combined with N-Boc-O-Piv hydroxylamine give the 5-exo lactone products in good to excellent yields.
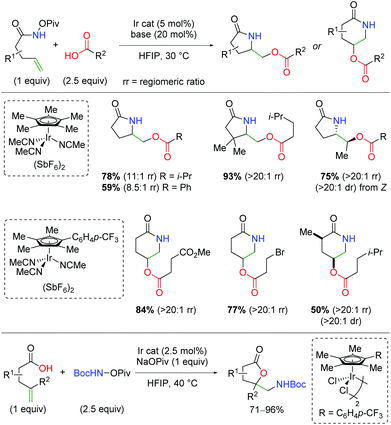 |
| | Scheme 48 Ir-Catalyzed oxyamination of alkenes with regioselective control by ligand choice (Rovis, 2019).66 | |
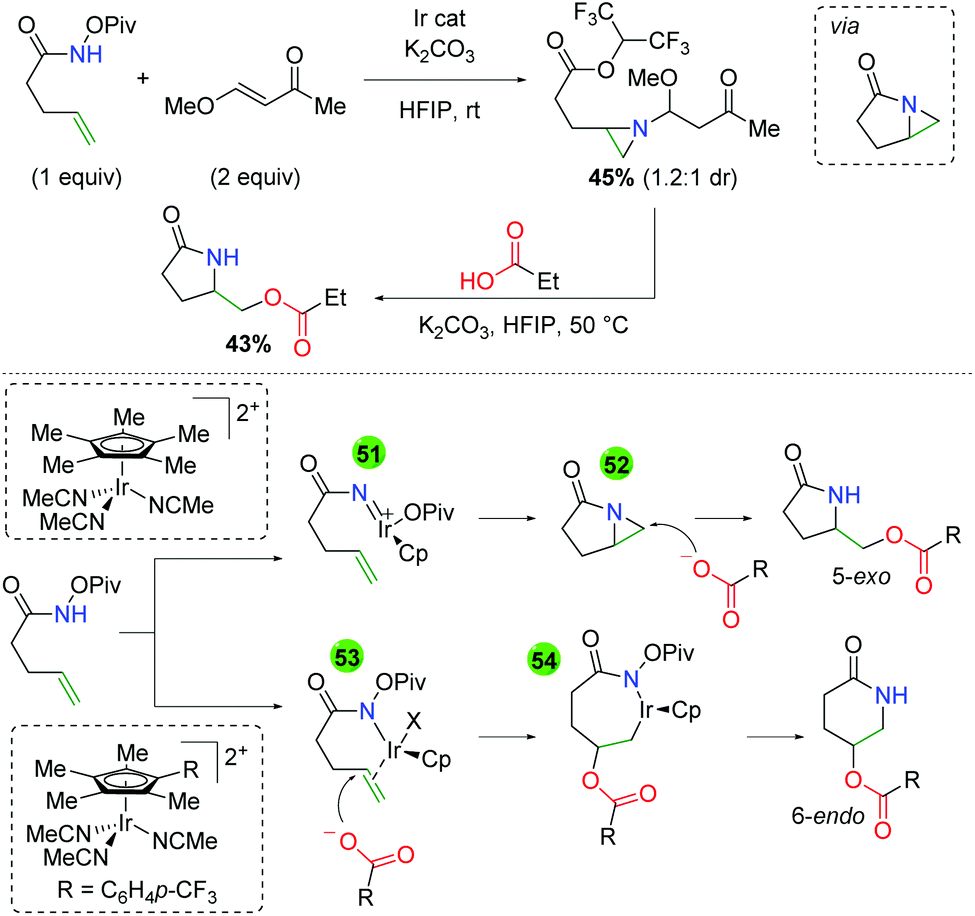
The authors also noted several observations within their studies that sheds light on the potential mechanism of the reaction (Scheme 49). When the unsaturated amide is reacted with a vinylogous ester, an aziridine compound containing a HFIP ester is isolated. It is presumed that this compound arises from the opening of an azabicyclo[3.1.0] intermediate with HFIP and aziridine addition to the α,β unsaturated ketone. When this compound is exposed to the reaction conditions in the absence of the iridium catalyst, the desired product is achieved, suggesting that this azabicyclo[3.1.0] intermediate is a catalytically competent intermediate. The authors proposed two mechanisms for the presence of the 5-exo and 6-endo products. When the standard Cp* ligand was used, it is sufficiently electron donating and leads to the formation of a nitrene (51), which creates the azabicyclo[3.1.0] (52) and is opened by the acid nucleophile for the 5-exo product. When the electron withdrawn Cp ligand is used, the iridium serves as a π-acid, activating the alkene for carboxylic acid attack (53). The resulting iridicycle (54) can then collapse to the 6-endo product.
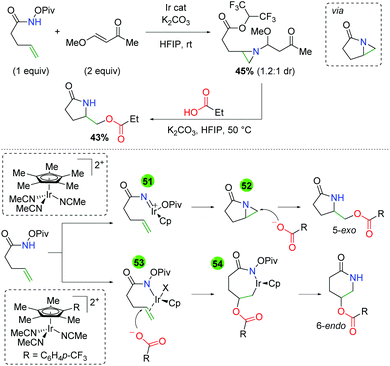 |
| | Scheme 49 Mechanistic insights into the regioselective control of the Ir-catalyzed alkene oxyamination. | |
2.9 Oxidation using iodine, azodicarboxylate, TEMPO, peroxide, or manganese (transition metal free)
2.9.1 Iodine methods.
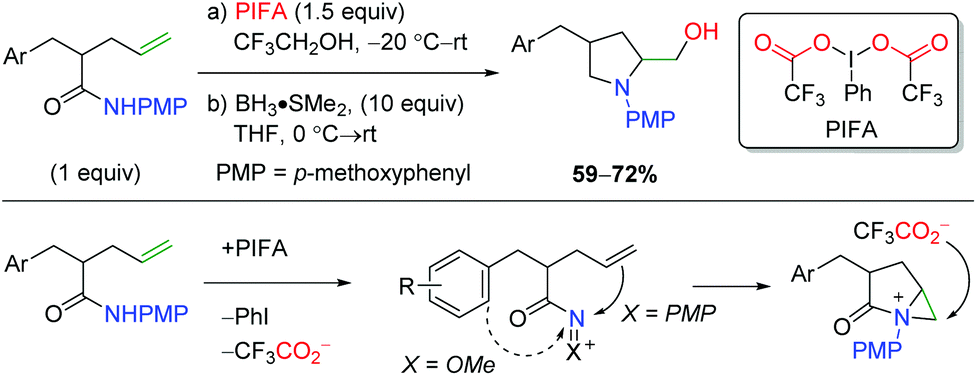
An initial example of transition-metal free alkene oxyamination was reported by the SanMartin laboratory in 2003 and 2004 using PMP-protected amides to affect amidohydroxylation (Scheme 50).67 Although this is truly an amidohydroxylation, the resulting amide must be reduced to the amine with BH3 due to product instability. These methods do not display an extensive substrate scope, but the authors did propose a mechanistic hypothesis. First, the PIFA hypervalent iodide reagent would oxidize the substrate to create a cationic intermediate stabilized by the amide substitution. They found that the PMP group is vital in promoting attack by the alkene, as N-methoxy leads to either ortho or ipso aryl nucleophilic addition. Aziridinium formation from the alkene addition leads to nucleophilic attack by the exogenous trifluoroacetate to generate the trifluoroacetate product, which is cleaved by a basic workup to generate the product.
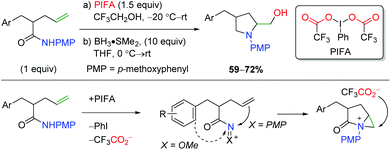 |
| | Scheme 50 Hypervalent iodine-mediated alkene amidohydroxylation (SanMartin, 2003/2004).67 | |
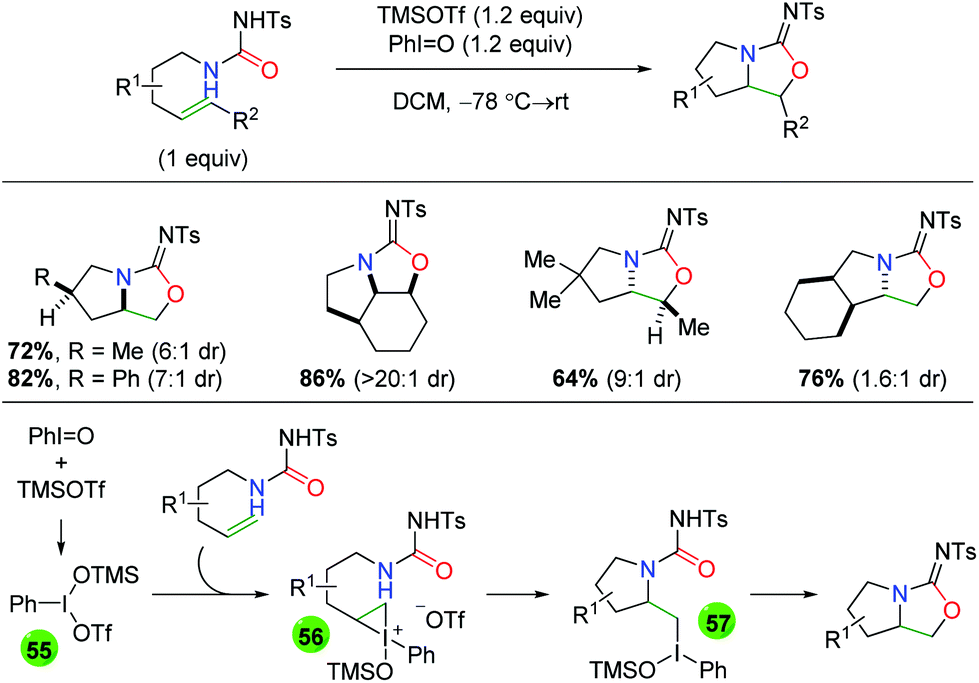
In 2008, Michael and co-workers produced a protocol using urea-tethered alkenes with an iodide(III) oxidant to effect both the nitrogen and oxygen functionalities of the urea (Scheme 51).68 The authors note that the choice of N-tosyl is important for the formation of the amino oxygenation product over the diamination product or a 7-exo byproduct. The reaction features a good scope of substitutions along the substrate backbone, as well as substitutions at the α and β-positions on the alkene to assemble some interesting multi-cyclic heteroatom ring systems. The diastereoselectivity is generally good, with one substrate-controlled example of excellent diastereoselectivity. Mechanistically, the reaction is hypothesized to be initiated by combination of PhIO and TMSOTf to generate the reactive iodide(III) reagent (55), which combines with the alkene to create the iodonium ion intermediate 56. Following intramolecular nucleophilic attack of the iodonium ion by the amide nitrogen to generate tertiary iodide(III) intermediate 57, a second intramolecular attack is conducted by the urea carbonyl to generate the product. It is worth noting that an identical strategy was utilized several years later by the Wirth group using chiral hypervalent iodine reagents to generate an enantioselective method.69 However, while it was touted as “highly stereoselective”, this approach only delivers enantiomeric excesses greater than 79% for one substrate and has generally low yields.
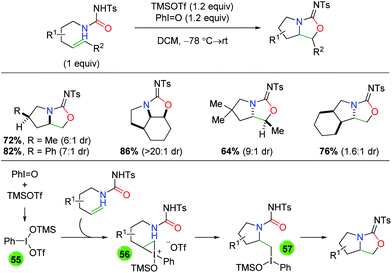 |
| | Scheme 51 Urea-tethered amino oxygenation with hypervalent iodide (Michael, 2008).68 | |
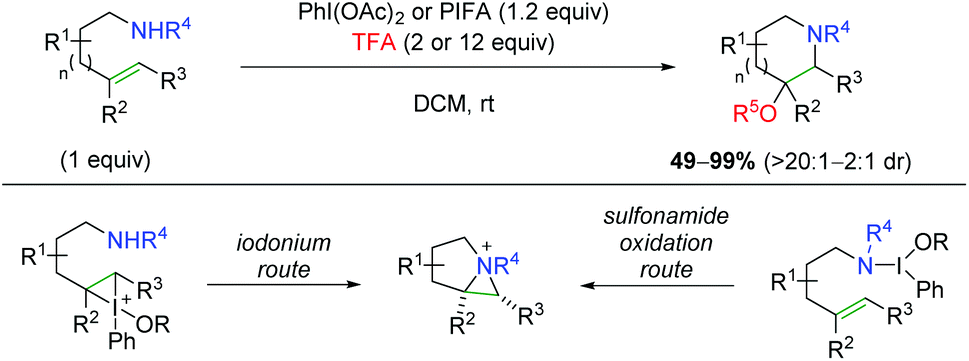
Shortly after their initial publication, the Michael group released another report detailing a similar strategy, but with an external oxygen nucleophile for the aminotrifluoroacetoxylation and hydroxylation of alkenes (Scheme 52).70 This strategy is very effective for anti addition of a variety of substrates and alkene substitutions generally in a 6-endo fashion, but even showcasing a 7-endo substrate. For the mechanism of the 6-endo formation, the authors hypothesized that an aziridinum ion intermediate was likely formed, either through attack of an iodonium ion intermediate (similar to their previous case), or through a more direct oxidation of the sulfonamide nitrogen.
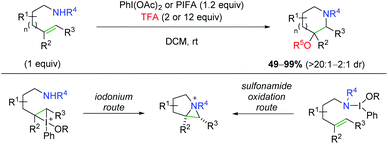 |
| | Scheme 52 Aminotrifluoroacetoxylation and hydroxylation of alkenes (Michael, 2010).70 | |
In a paper submitted the same day as the Michael group's above, the Wardrop laboratory disclosed a similar procedure for the oxyamidation of alkenes using alkene-tethered Weinreb amides (Scheme 53).71 The authors also propose that this transformation occurs through an aziridinium ion intermediate, similar to the conclusion of the Michael group. The authors found a series of both exo and endo products, but there appears to be an explanation based on the proposed mechanism. In the case of 1,2-disubsituted (internal) alkenes, the 5-exo product is preferred in a trans configuration, while 1,1-disubstituted alkenes prefer the 6-endo product. In the case of trisubstituted alkenes with both α and β-substitution, a mixture of the 5-exo and 6-endo products is obtained. The method is also transferrable for the formation of bicyclic substrates, which generally follows the same product projection trends as the previous substrates.
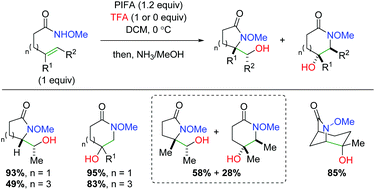 |
| | Scheme 53 Lactam formation with alkene-tethered amides (Wardrop, 2010).71 | |
2.9.2 Diazodicarboxylate methods.
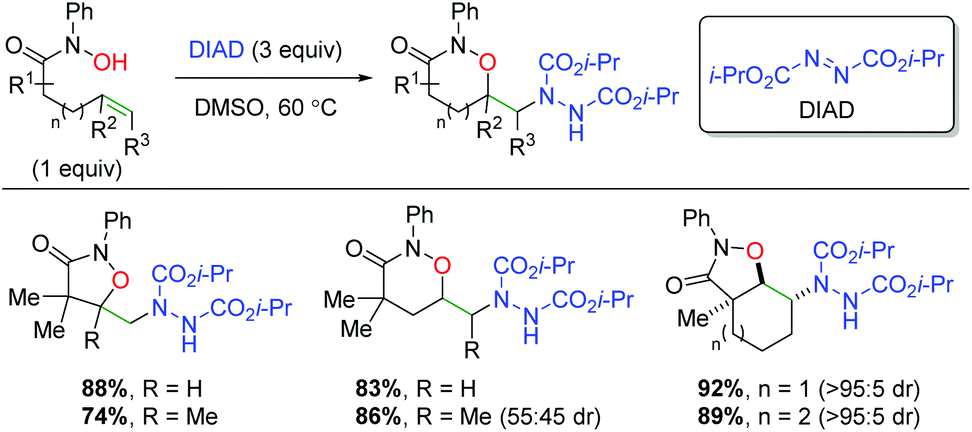
Building on their previous work with alkene dioxygenation,72 the Alexanian laboratory published an alkene oxyamination protocol using hydroxamic acids and diisopropyl azodicarboxylate (DIAD) to achieve a variety of 1,2-oxazinan-3-one skeletons (Scheme 54).73 The reaction exhibits good yields for the alkyl backbone substrates shown, both terminal and internal. For the internal substrates, very little diastereoselectivity is shown in substrates containing internal, acyclic alkenes. However, switching to internal cyclic alkenes (e.g. cyclohexene) exhibited excellent diastereoselectivity in a trans configuration.
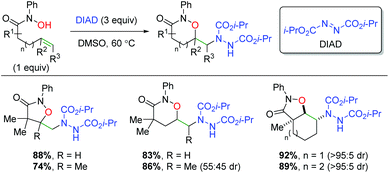 |
| | Scheme 54 Alkene oxyamination using hydroxamic acids and DIAD (Alexanian, 2011).73 | |
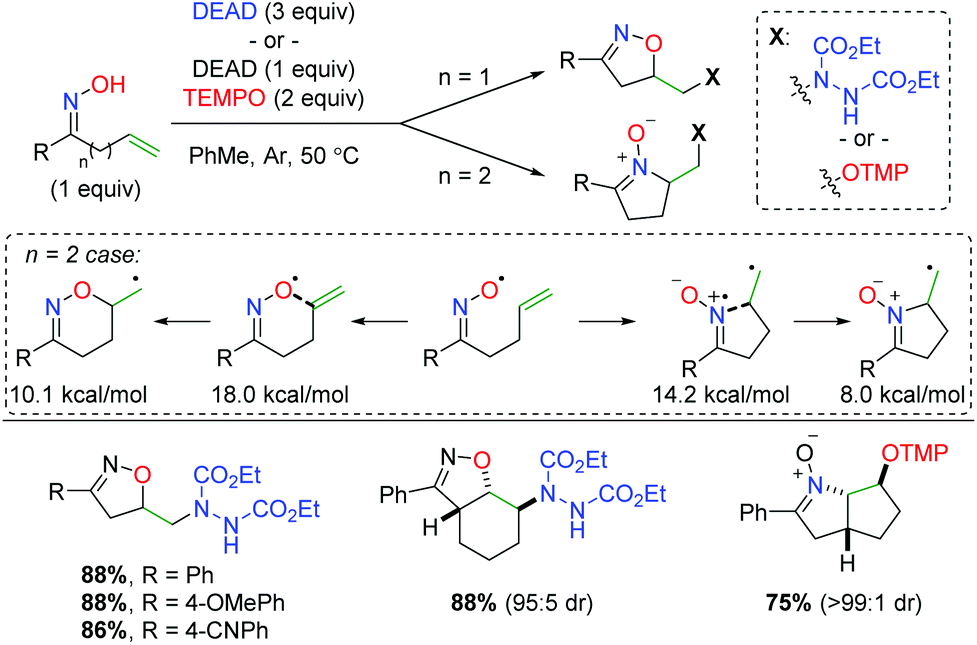
The Han group (previously mentioned for their copper-catalyzed strategies) initially found that an oxime radical could be easily generated in the presence of diethyl azodicarboxylate (DEAD) and cyclize on a pendant alkene (Scheme 55).74 They also identified both DEAD and TEMPO as good radical traps for the terminal carbon radical generated after the oxime addition. Importantly, they discovered that the tether length between the oxime and alkene created a dichotomy of products. In their initial test, they found a 5-exo product with the oxime oxygen initiating the cyclization. However, when an extra carbon is added to the tether, the imine nitrogen in the oxime initiates another 5-exo addition. When they evaluated the energy profile of this reaction, they found that the activation energy of the 6-exo cyclization from the oxygen is 3.8 kcal mol−1 higher than the 5-exo cyclization from the nitrogen, while the 6-endo product is 2.1 kcal mol−1 higher than the 5-exo product. Regarding the substrate scope, the limited number of oxyamination examples showed good yields with generally excellent diastereoselectivity for cyclic internal alkenes.
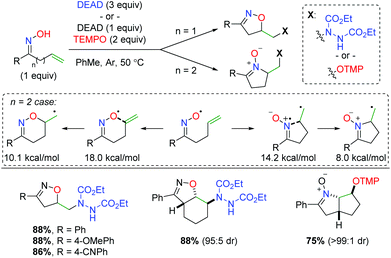 |
| | Scheme 55 Divergent alkene oxyamination strategy using unsaturated oximes with TEMPO or DEAD (Han, 2012).74 | |
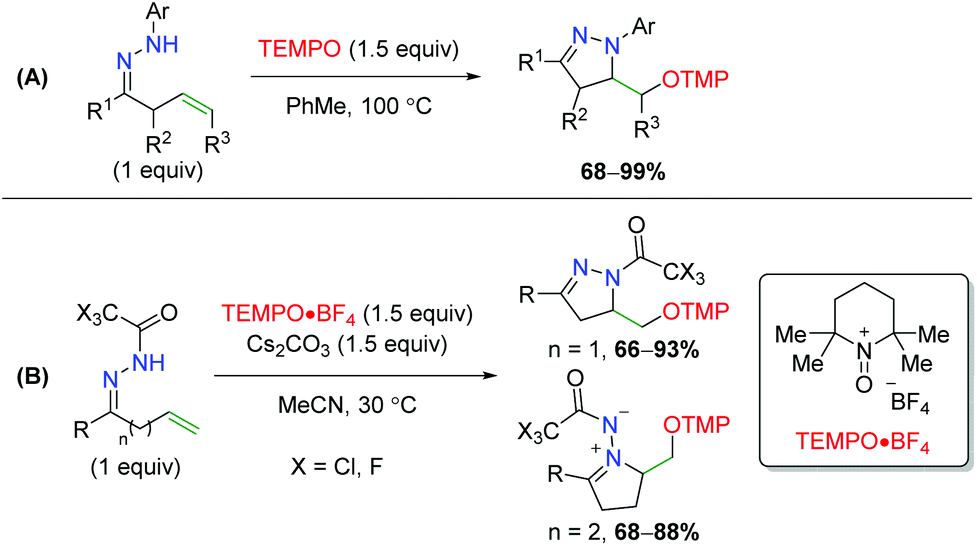
2.9.3 TEMPO methods.
Soon thereafter, Han an co-workers transferred their strategy from oximes to hydrazones; first with N-aryl hydrazones, then with N-trihaloacetyl hydrazones (Scheme 56).75 First, they noted that N-aryl hydrazones are effective in achieving the 5-exo product seen previously in good yields. Secondly, they determined that using an N-trifluoroacetyl or trichloroacetyl group allows them to achieve both the 5-exo and 6-exo products.
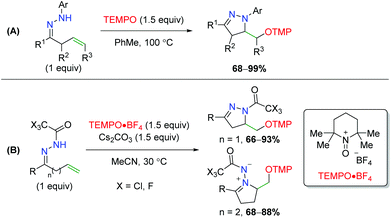 |
| | Scheme 56 Divergent alkene oxyamination using unsaturated hydrazones (Han, 2013/2014).75 | |
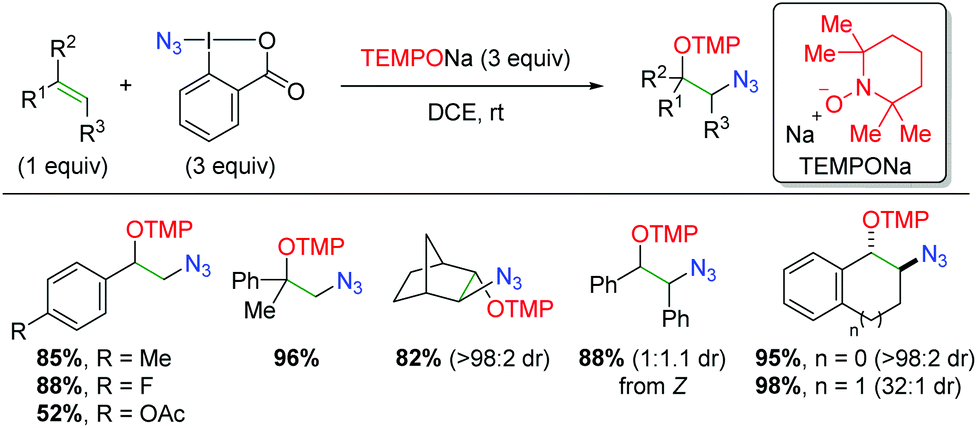
In 2013, the Studer laboratory published an azido oxygenation version of their previous alkene trifluoromethyl oxygenation,76 using the Zhdankin reagent (iodine(III) azide reagent) and TEMPONa (Scheme 57).77 The authors propose that TEMPONa reduces the azide reagent to generate both an azido radical and TEMPO. The azido radical then simply adds to the alkene, with TEMPO terminating the reaction. Regarding the substrate scope of the reaction, the reactivity is centered on styrene derivatives, though the reaction is tolerant of some alkyl olefins. The reaction is also compatible with substitution at the α or β-positions of the alkene. The yields are generally quite good, but the diastereoselectivity varies from good to excellent based on the substrate.
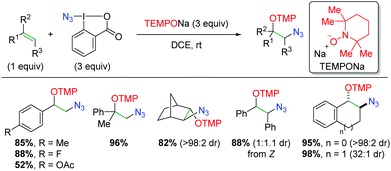 |
| | Scheme 57 Azido oxygenation of alkenes using Zhdankin reagent and TEMPONa (Studer, 2013).77 | |
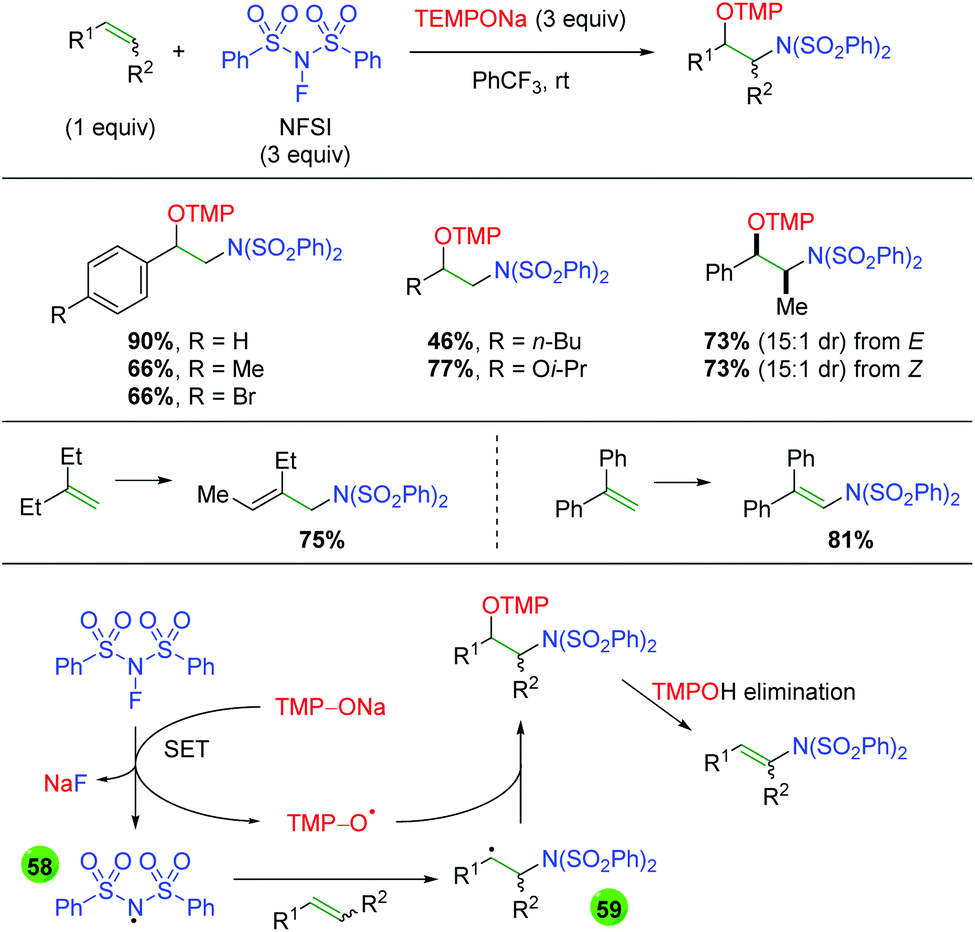
The Studer group built on this concept of TEMPONa reduction of nitrogen reagents in a second paper detailing the reduction of NFSI by TEMPONa to create an NFSI nitrogen radical for addition to the alkene, with terminal trapping by TEMPO (Scheme 58).78 The reaction works well with terminal alkenes of both aromatic and aliphatic nature and the reaction displays good yield and diastereoselectivity for internal alkenes of both E and Z geometry. Interestingly, when the authors tested 1,1-disubstituted alkenes, they found that the TMP adduct underwent elimination to give allylic amines or enamines in good yields. As mentioned previously, this transformation is proposed to occur first through SET reduction of NFSI by TEMPONa to generate NaF, TEMPO, and a nitrogen-centered radical (58). The nitrogen-centered radical then adds to the alkene to give carbon-centered radical intermediate 59, which re-joins with TEMPO to give the desired product.
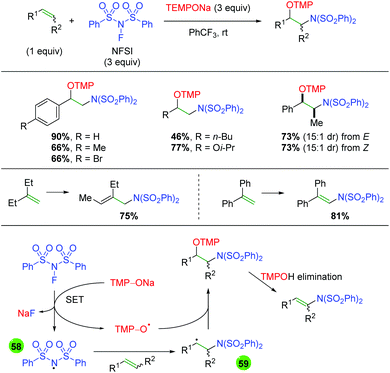 |
| | Scheme 58 Alkene amino oxygenation using NFSI and TEMPONa (Studer, 2015).78 | |
2.9.4 Peroxide methods.
A unique strategy was published by Zhu and co-workers using TBHP (tert-butyl hydrogen peroxide) to initiate an amine addition to the alkene with trapping by water (Scheme 59).79 This procedure was first shown with alkene dioxygenation, where the proposed mechanism involved generation of a carboxylate radical. In this case, it is likely an amino radical generated on the benzotriazole derivative, which adds to the alkene. The scope of this transformation is strictly limited to styrene derivatives, but does accommodate both α and β-substituted alkenes. The authors also demonstrated the compatibility of several different types of amines, though the scope is narrowly centered around benzotriazole-like scaffolds.
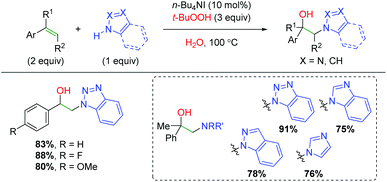 |
| | Scheme 59 Amino oxygenation of alkenes using TBHP and benzotriazoles (Zhu, 2013).79 | |
Another unique metal-free approach comes from a collaboration between the Tomkinson laboratory and GlaxoSmithKline using malanoyl peroxides as an oxygen source (Scheme 60).80 This platform was initially shown for dioxygenation of alkenes,81 but the authors extended it to sulfonamide-tethered alkenes to achieve an anti, 5-endo addition. The substrate scope of this transformation appears to be fairly limited by the 5-endo cyclization, as only styrenyl internal alkenes without backbone elements were used. However, the diastereoselectivity is fairly good and complementary diastereoselectivity was observed when comparing E and Z alkenes. Mechanistically, this transformation is proposed to begin through attack of the peroxide by the alkene (60) to generate a cationic intermediate (61), which is attacked by the carboxylate carbonyl. This transient dioxonium ion intermediate (62) is then attacked of the sulfonamide, generating the anti addition configuration of the product (63). The pendant ester is then cleaved by the secondary NaOH hydrolysis step to furnish the final product.
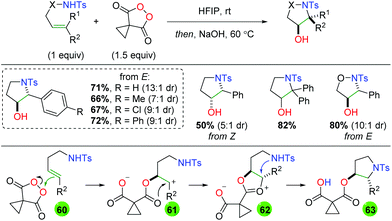 |
| | Scheme 60 Alkene oxyamination using malanoyl peroxides (Tomkinson, 2018).80 | |
Excitingly, the Tomkinson group was successful in achieving this transformation as a three-component reaction with N-Boc-N-tosyl carbamates (Scheme 61).82 The authors first demonstrated this transformation on functionalized stilbenes with good to excellent yields, then investigated styrenes with equally positive results. Interestingly, when terminal styrenes were used, the regioselectivity of the transformation was poor. The authors reasoned (using a similar mechanistic understanding as their previous work) that although the nucleophilic addition of the carbamate to the dioxonium ion will be favored electronically at the stabilized benzylic position, it could still exhibit some addition at the more sterically accessible terminal position. When β-functionalized styrenes are utilized, the regioselectivity is exclusive with the nitrogen at the benzylic carbon, since the increased steric bulk around the β-position discourages nucleophilic attack. Furthermore, the authors disclosed that oxazolidinone cyclization products could be achieved when O-methyl carbamate sulfonamides were used, instead of O-tert-butyl (Boc), and the products treated with trifluoroacetic acid.
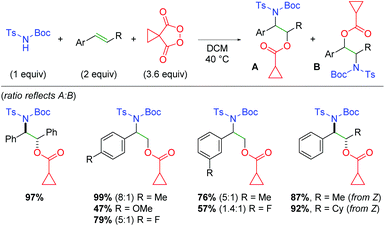 |
| | Scheme 61 Intermolecular alkene oxyamination using malanoyl peroxides (Tomkinson, 2020).82 | |
2.9.5 Miscellaneous oxidative methods.
In 2015, Xia and co-workers disclosed an oxidative method for oxy azidation of alkenes involving a proposed oxidation of N-hydroxyphthalimide to promote addition to various styrenyl alkenes with TMS azide trapping (Scheme 62).83 Initially, the authors proposed using catalytic copper salts, but found that the reaction is successful with just (diacetoxy)iodobenzene as an oxidant. The substrate scope exhibits tolerance of a number of functionalities such as halides esters, ethers, nitro, and imidazole functionalities, though the yields are better with electron-withdrawing substituents on the styrene aryl ring. Alkyl alkenes do not facilitate good yields of the desired product, as norbornene and allyl phenyl ether were successful in 30% yield. Although some radical-trapping mechanistic studies were conducted, the mechanism of this transformation is merely putative at this time.
 |
| | Scheme 62 Oxy azidation of alkenes using N-hydroxyphthalimides and oxidants. (Xia, 2015).83 | |
In 2019, Niggemann and co-workers published a protocol using stoichiometric manganese with a TEMPO radical trap for the cyclization of aniline-tethered alkenes, forming pyrrolidine scaffolds (Scheme 63).84 The yields for this transformation are good for substitution of the aniline aryl ring, as well as the aryl group on the alkene. Only a few variations on the pyrrolidine backbone are shown, with mixed results, though a piperazine product is shown in good yield. Mechanistically, this transformation was proposed as initiated by oxidation of TEMPO by manganese acetate. The oxidized TEMPO+ in turn oxidizes the aniline to an aminium radical cation, which adds to the alkene. The resulting radical is trapped with TEMPO in a diastereoselective fashion. The authors rationalize the complete diastereoselectivity as resulting from the aryl bulk of the aniline, though it is not clear how the possible free rotation of a radical intermediate and potential π,π stacking from the two aryl groups might factor into this.
 |
| | Scheme 63 Manganese oxidation of aniline-tethered alkenes with TEMPO trapping (Niggemann, 2019).84 | |
2.10 Aziridine/epoxide formation with oxygen/nitrogen nucleophilic ring opening
In 2005, the Johnston laboratory disclosed a formal anti amino oxygenation of alkenes that relied on in situ formation of a Brønsted acid-promoted aziridine (Scheme 64).85 They first optimized the aziridination for Michael acceptors, then applied the method to a system with a tethered oxygen nucleophile to accomplish the anti addition. Overall, the scope is limited by the acetamide/carbamate scaffold, but this paper focused on the novelty of the Brønsted acid-mediated aziridination.
 |
| | Scheme 64 Amino oxygenation through aziridine formation (Johnston, 2005).85 | |
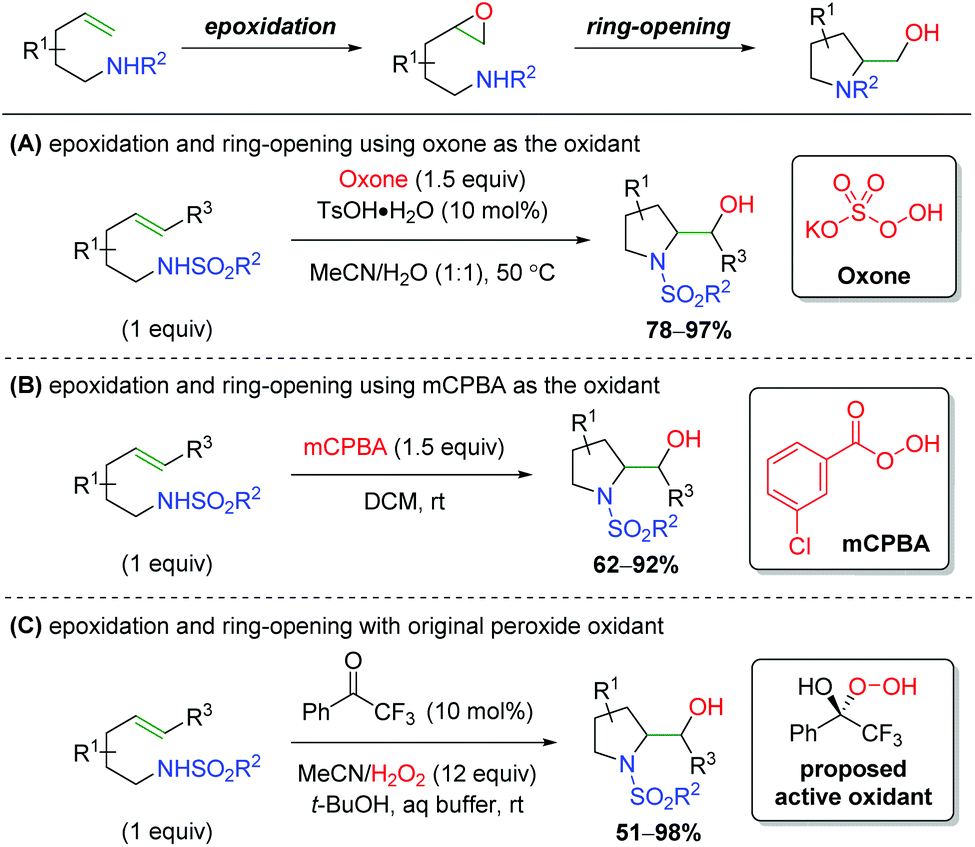
The inverse approach, facilitating an alkene epoxidation followed by nucleophilic attack from a tethered sulfonamide in a 5-exo fashion, has been achieved by a number of research groups (Scheme 65). While there was one report in 1990 hypothesizing to have achieved this on two substrates,86 the first clear example comes from Togo and co-workers, who utilized oxone as the oxidant, with tosylic acid acting as a Brønsted acid to promote the epoxidation (Scheme 65A).87 Much like the aziridination of the Johnston laboratory, the authors found that a Brønsted acid, specifically tosylic acid, is vital in promoting the epoxidation. Several years later, the Li group reported using mCPBA (meta-chloroperoxybenzoic acid) to facilitate the epoxidation, attacking with a similar sulfonamide tether as in the first example (Scheme 65B).88 The conditions/components for the reaction are quite simple and there is a fairly expansive 5-exo scope with good yields. Additionally, the authors synthesized the epoxide intermediate and showed that simply stirring this substrate in DCM at room temperature does not produce the desired reaction, suggesting a promoter is necessary to initiate attack of the epoxide, though no mechanism was proposed. Finally, the Kokotos laboratory presented a unique take on this strategy, using a ketone organocatalyst with excess H2O2 to effect the epoxidation and subsequent ring opening (Scheme 65C).89 They propose the active oxidant as a hemiketal from peroxide addition to the ketone, based on their previous work with this system.90 This strategy produces a good range of products, mostly centered around indolines, with fair to good yield, but lacks an expansive investigation of functional group tolerance.
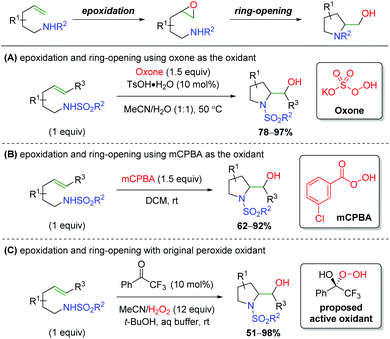 |
| | Scheme 65
In situ epoxidation/nitrogen ring opening sequence.87–89 | |
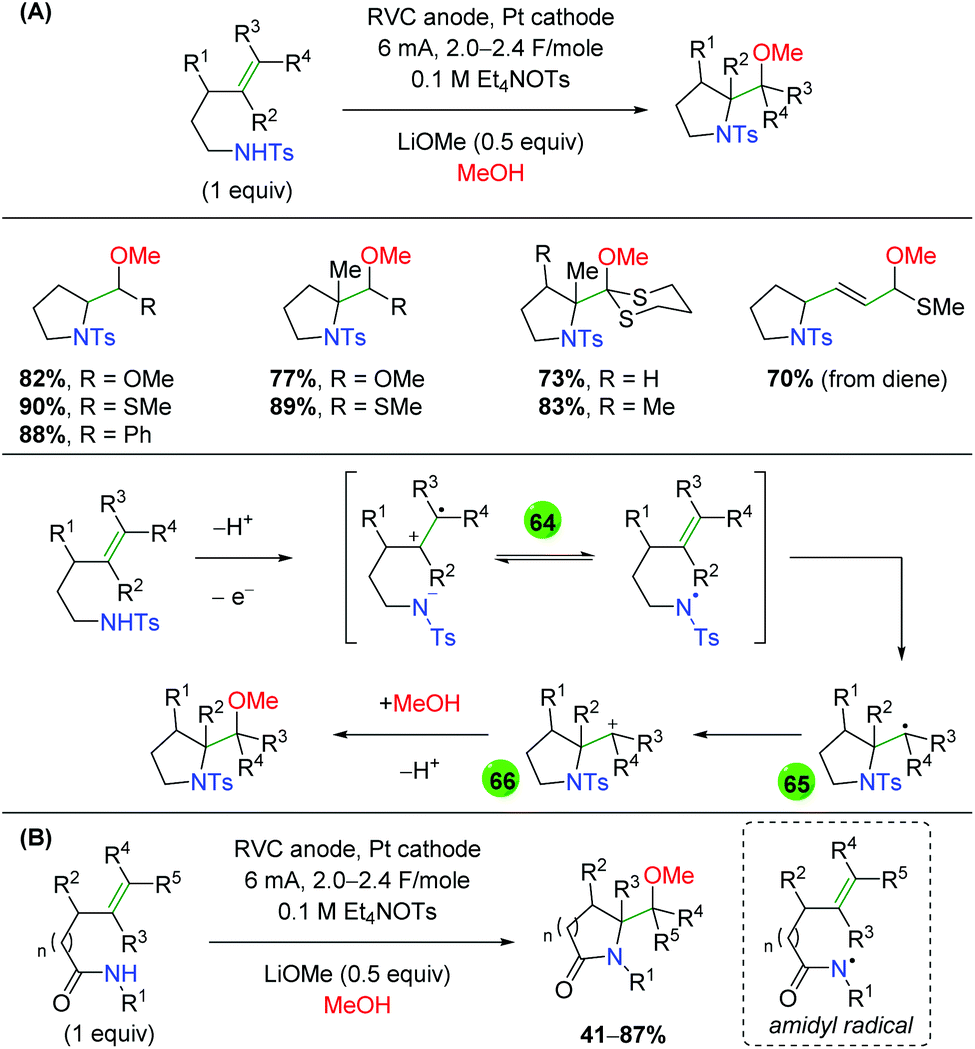
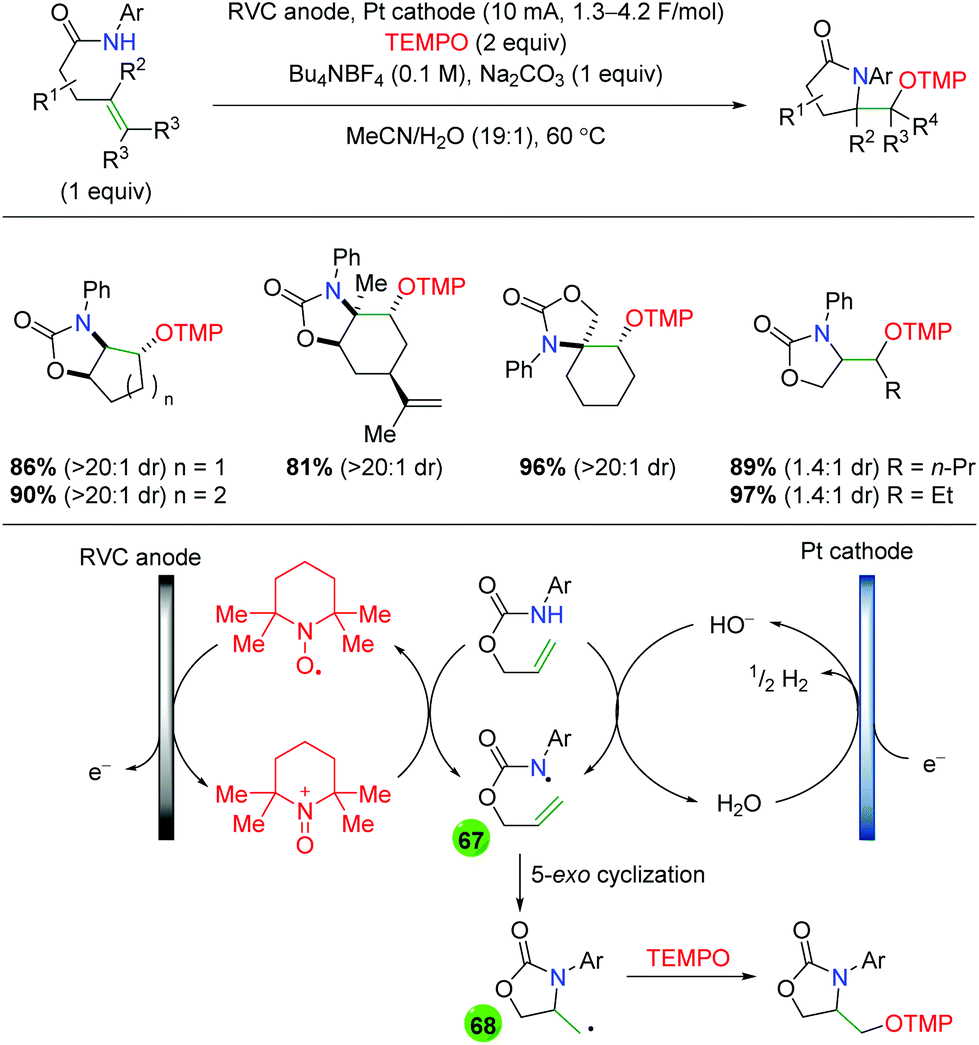
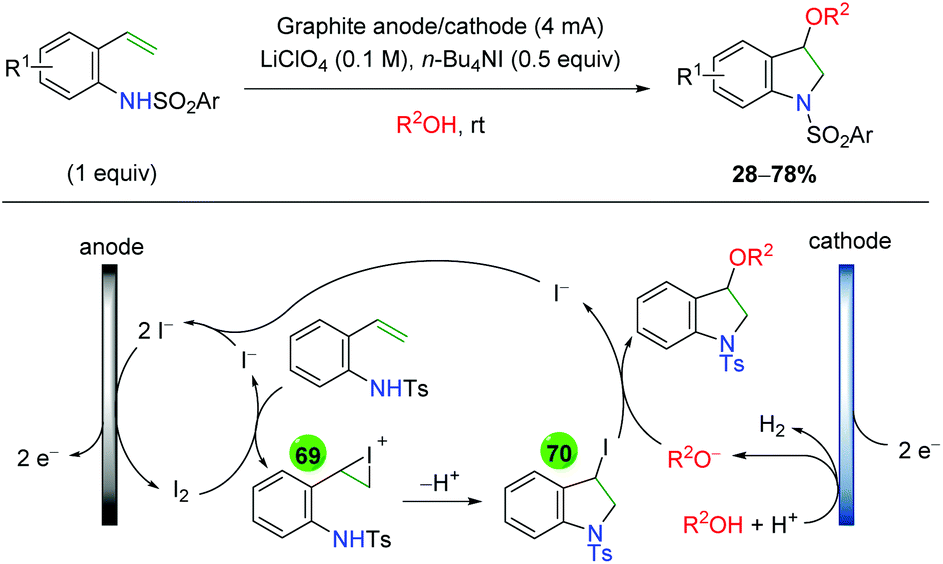
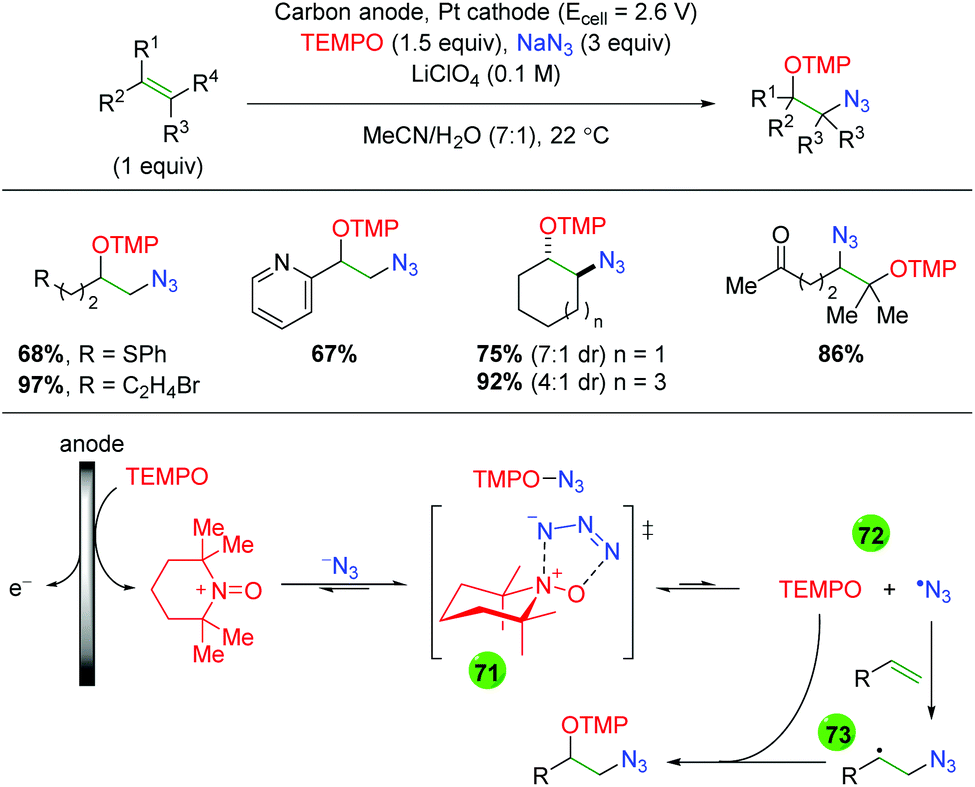
2.11 Electrochemical methods
In 2008, the Moeller laboratory published the first electrochemical oxidation strategy for amino oxygenation of alkenes using sulfonamide-tethered alkenes with a methanol solvent trap (Scheme 66A).91 The reaction builds upon analogous carbo-oxidation reactions from their group,92 with the carbon nucleophile being replaced by a sulfonamide in this instance. The first publication featured initial exploration, which was expanded upon in a follow-up paper two years later. Ultimately, the scope for this transformation is very narrow, as internal alkenes are required to be electron-rich, bearing either methoxy or mercaptan derivatives, for the initial electrochemical oxidation to function properly. Only a few substitutions on the backbone are demonstrated and a single diene substrate was evaluated with both papers showing a similar, narrow substrate compatibility. Mechanistically, the reaction is proposed to begin with an oxidation of the electron-rich alkene to a radical cation, which could be in equilibrium with a sulfonamide radical (64). This species then cyclizes to generate a 5-exo product with a resulting carbon-centered radical (65). Further oxidation of the carbon-centered radical to a cation (66), sets up the final intermolecular methanol trap with subsequent deprotonation to generate the final product. Several years later, the Moeller laboratory released another example of this strategy using amide-tethered alkenes (Scheme 66B).93 The scope of this transformation is very similar to the previous reports, but the authors provided additional calculations to conclude that the reaction is being initiated by formation of an amidyl radical, which adds to the alkene to generate a carbon-centered radical, which is trapped by methanol after being oxidized to the cation.
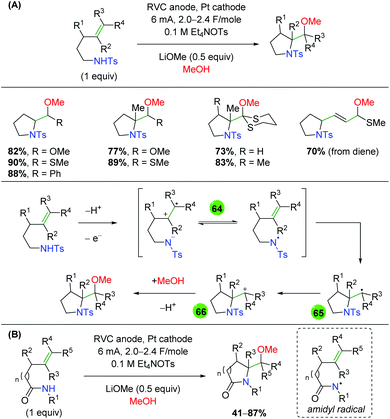 |
| | Scheme 66 Electrochemical amino oxygenation of alkenes (Moeller, 2008–2014).91,93 | |
Shortly after the Moeller laboratory's work using amides, Xu and co-workers disclosed an electrochemical amino oxygenation using amide-tethered unactivated alkenes (Scheme 67).94 By not requiring an electron-rich alkene (a deficiency of the Moeller laboratory method), the authors were able to expand the range of alkene sources tolerated in the reaction. The substrate scope focused on internal, cyclic alkenes, which are formed in good yields with excellent diastereoselectivity. They also showed that the reaction is tolerant of pendant alkene groups. While the reaction shows good diastereoselectivity with internal, cyclic alkenes, this selectivity is lost when the cyclic nature of the alkenes is exchanged with acyclic E alkenes. Mechanistically, the authors measured the oxidation potentials of the substrate and TEMPO with cyclic voltammetry, demonstrating that TEMPO is more readily oxidized to its oxoammonium cation form than the amide substrate is oxidized to an amidyl radical. Accordingly, they propose that TEMPO is oxidized to the oxoammonium cation at the RVC anode, followed by oxidation of the amide substrate to the amidyl radical (67), regenerating TEMPO. Furthermore, control experiments found that the reaction is ineffective with just Na2CO3 as a base, leading the authors to propose hydroxide (formed from water reduction at the cathode) as necessary for deprotonation of the amide. The amidyl radical adds to the alkene in a 5-exo fashion and the resulting terminal carbon radical (68, verified by cyclopropyl radical clock experiments) is trapped by TEMPO to generate the final product.
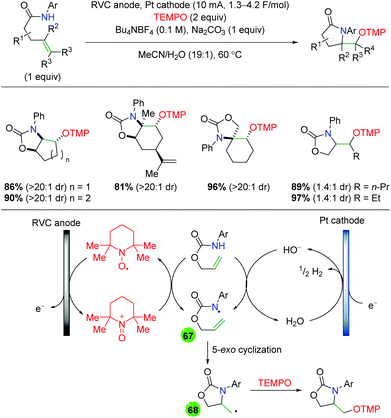 |
| | Scheme 67 Electrochemical amino oxygenation of unactivated alkenes (Xu, 2014).94 | |
Another example of electrochemical alkene amino oxygenation is shown by Sun and co-workers in an amino iodination followed by oxygen nucleophilic attack for a formal amino oxygenation (Scheme 68).95 The scope of this reaction is quite narrow, with all substrates containing the aromatic backbone with either methanol or ethanol being the only alcohols incorporated. This reaction is proposed to start with oxidation of the catalytic iodine source at the anode, creating I2, which generates an iodonium ion intermediate on the alkene (69). This intermediate is attacked by the pendant sulfonamide to generate, upon deprotonation, an amino iodine substrate (70). Reduction of the alcohol at the cathode leads to an alkoxide, which easily displaces the iodine to generate the final product.
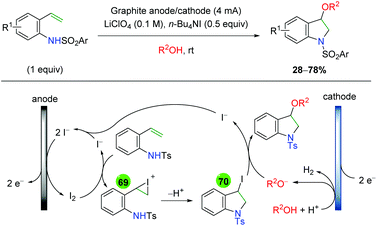 |
| | Scheme 68 Formal electrochemical alkene amino oxygenation through alkene amino iodination with nucleophilic oxygen displacement (Sun, 2016).95 | |
In 2018, the Lin group conducted an extensive study detailing an azido oxygenation of alkenes through a TEMPO-azide charge transfer complex (Scheme 69).96 With their previous experience developing electrochemical alkene diazidation reactions,97 the authors used TEMPO as the secondary trap to affect an azido oxygenation. The scope of the reaction is exceptionally broad and tolerates of a number of functional groups, such as primary alkyl bromides and thioethers, that are incompatible, challenging, or ignored by most methods, specifically oxidative ones. Internal alkenes engage the reaction with fair diastereoselectivity and substrates with a higher degree of substitution at the β-position showcase the azide residing at the less hindered position. However, the mechanism of this transformation was painstakingly probed. The authors propose aniodic oxidation of TEMPO to the oxoammonium form, followed by azide addition to form a TEMPO-azide complex (71). They proposed this intermediate through UV-Vis and cyclic voltammetry experiments, which showed a signal for the combined complex unique from each individual component, and this hypothesis agrees with DFT modeling studies. The authors then proposed an equilibrium between the TEMPO-azide complex and TEMPO with an azide radical (72), proposing that the azide radical would add to the alkene. They backed up this claim with kinetics studies showing an inverse fist order rate for TEMPO, zero order for the sodium azide, and first order for the alkene. This pointed toward an equilibrium between the TEMPO-azide complex and their free radical forms, with the TEMPO-azide complex being favored, as free TEMPO would inhibit the azido radical addition to alkenes. Finally, they proposed a terminal TEMPO trap of the carbon-centered radical (73) to generate the product.
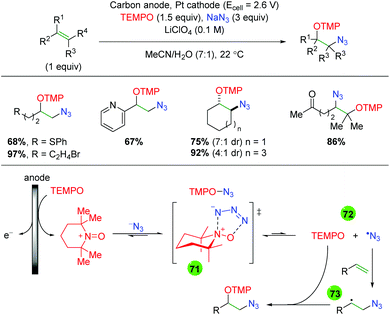 |
| | Scheme 69 Electrochemical alkene azido oxygenation through a TEMPO-azide charge transfer complex (Lin, 2018).96 | |
2.12 Photocatalysis
Due to the fairly recent rise of photochemistry within reaction methodology, only a number of examples for photochemical alkene oxyamination exist. The first example of this came in 2015 from the Greaney laboratory pairing a photoredox approach with Zhdankin reagent and solvent trapping by methanol (Scheme 70).98 For this procedure, the authors used a copper(I) catalyst with two 2,9-methoxyphenanthroline ligands, [Cu(dap)2], as their photoredox catalyst. The reaction is limited to styrenyl alkenes, but shows tolerance of both 1,1- and 1,2-disubstituted alkenes. The authors did not perform an in-depth analysis of the mechanism but, based on previous work from other laboratories,99 they proposed that the reaction would initiate via single electron reduction of Zhdankin reagent to generate a azido radical, which would add to the alkene. The resulting carbon-centered radical could be trapped by the alcohol solvent similar to their previous work with diaryl iodonium salts.100 Of note, the authors found that in the absence of the light source, they achieved diazidation in good to excellent yields.
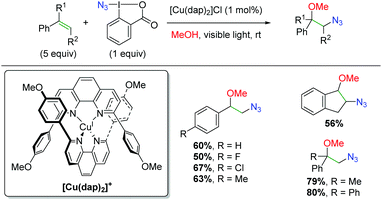 |
| | Scheme 70 Photochemical alkene azido oxygenation using Zhdankin reagent (Greaney, 2015).98 | |
Only months later, Akita and co-workers published an amino hydroxylation of alkenes using an iridium photocatalyst and N-sulfonamide pyridine salts (Scheme 71).101 This process shows good reactivity with styrenyl alkenes and tolerates a range of functional groups, including esters, amides, boronates, and pyridine. Furthermore, the reaction works on 1,1- and 1,2-disubstituted alkenes, though the reaction displays only minimal diastereoselectivity and gives appreciably less product in acyclic, internal alkenes. While the reaction does work on aliphatic alkenes, the yields are much lower, leaving a clear area for improvement. Regarding the mechanism, the authors envision the excited Ir(III) photocatalyst reducing the N-sulfonamide pyridine salt (74), which can release a sulfone radical. This radical then undergoes normal electronically-dictated addition to the alkene to generate a carbon-centered radical (75). Oxidization of this species by the Ir(IV) photocatalyst regenerates the Ir(III) catalyst, while the resulting cation (76) is trapped by water to generate the final product.
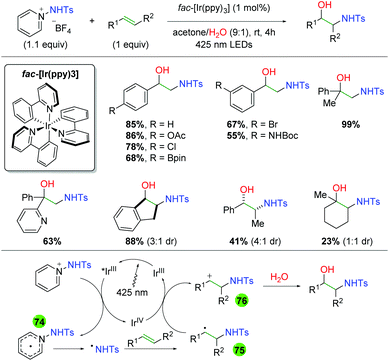 |
| | Scheme 71 Amino oxygenation of alkenes using iridium photocatalyst (Akita, 2015).101 | |
A year later, Lu and co-workers released another iridium photocatalyzed method using alkene-tethered, N-benzoyl carbamates to effect an amine-ester addition across the alkene (Scheme 72).102 Interestingly, the authors found that the diastereoselectivity can be controlled by the choice of amine base. When triethylamine is used, a net anti addition across the alkene is observed, but when triphenylamine is used, a net syn addition is observed. Regarding the substrate scope, the authors focused on the anti/syn addition, varying the benzoyl group, as well as the substituent on the terminal end of the alkene. In both cases, they did not show any true electron-donating groups, other than alkyl chains.
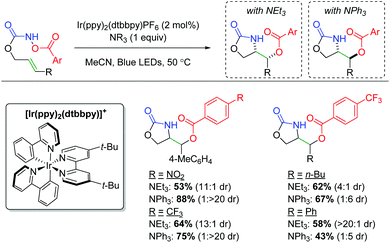 |
| | Scheme 72 Amino oxygenation of alkenes using iridium photocatalyst (Lu, 2016).102 | |
With this unique switch in syn/anti addition, the authors were interested in the reaction mechanism and the origins of the diastereoselectivity, so they undertook several mechanistic experiments (Scheme 73). First, they ran the reaction for 15 min and observed the same aziridine isomer, regardless of the amine base, in nearly identical yield by 1H NMR (Scheme 73A). They continued with the reaction in the dark for 18 hours and now observed the expected, syn/anti diastereomers for the triphenyl/triethyl amine bases, implying that the origins of selectivity occur after the aziridine formation (Scheme 73A). To eliminate any iridium catalysis input from the diastereo-determining step, they tested the reaction of the isolated aziridine with benzoic acid as an additive in the absence of the iridium catalyst (Scheme 73B). The triethyl and triphenyl amine bases gave anti and syn products, respectively, confirming that the second part of the mechanism was polar and diastereo-determining in nature. From this information, they proposed a mechanism beginning with single electron transfer from the reduced, low valent photocatalyst to generate an amido nitrogen radical (77). The low valent photocatalyst is then regenerated through excitation by the light source and subsequent reduction by an electron donor (amine). The amido radical then undergoes aziridination with oxidation from the amine electron acceptor and deprotonation by the benzoate anion (78). As shown from the mechanistic testing, this is the same result for both amine bases. For the origin of diastereoselectivity, the authors proposed that triethylamine can deprotonate the benzoate to initiate a SN2 pathway (79), yielding the anti product, while the less basic aniline nitrogen of triphenylamine would not be protonated, leading to benzoate protonation of the aziridine. This could result in equilibrium generation of a carbocation and potential SN1 mechanism (80) to generate the syn product due to steric repulsion of the alkene aryl group and the amide tether.
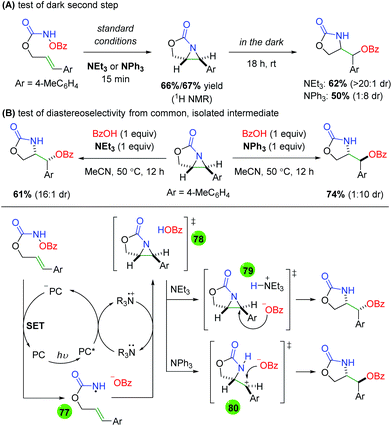 |
| | Scheme 73 Mechanistic testing for iridium alkene amino oxygenation reaction. | |
Waser and co-workers employed a similar strategy to the Greaney laboratory (Zhdankin reagent and Cu(dap)2), exchanging the alcohol solvent used by Greaney for an intramolecular carboxylic acid to effect an azido lactonization (Scheme 74),103 analogous to the copper-catalyzed work of the Buchwald group (Scheme 33). The scope of this transformation is quite similar to the Buchwald study, as each substrate has a phenyl ring at the α-position of the alkene, the examples with an alkyne notwithstanding. However, the authors did show examples containing a phenyl backbone, one substrate class not explored by Buchwald. Mechanistically, no investigations were performed, though the authors did speculate that there could be multiple processes at play.
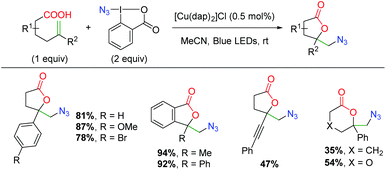 |
| | Scheme 74 Azido lactonization of alkenes using copper photocatalyst (Waser, 2017).103 | |
Months later, the Lu group released a second photo-catalyzed alkene amino oxygenation, this time using a three-component azido hydroxylation using TMS azide and air (Scheme 75).104 In contrast with their previous iridium photocatalyst method, this protocol utilized the organo photocatalyst Acr+MesClO4−, which was the only tested photocatalyst to exhibit the desired reactivity. The substrate scope of the reaction is quite broad, as halides, thioethers, aldehydes, even carboxylic acids are tolerated. Pyridyl styrenes are successful, as well as 1,1-disubstituted alkenes such as known radical polymerization retardant 1,1-diphenylethylene. Internal alkenes, such as indene, are also viable substrates and even tetrasubstituted alkenes work with only slightly diminished yields from their 1,1-disubstituted analog. Alkyl alkenes were also tolerated, giving fair yields. Mechanistically, the reaction is known to occur through a radical process, with the excited photocatalyst oxidizing the TMS azide and alkene to generate azide addition and a carbon-centered radical, which is trapped by molecular oxygen.
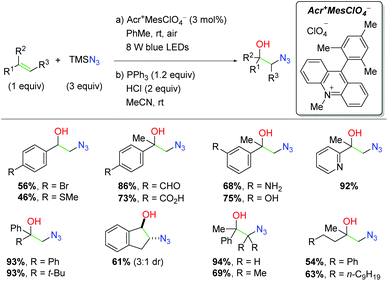 |
| | Scheme 75 Azido hydroxylation of alkenes using copper photocatalyst (Lu, 2017)).104 | |
In 2018, Yoon and co-workers merged their interests in copper-catalyzed alkene amino oxygenation and photocatalysis to create an alkene amino oxygenation strategy using a pyrylium photocatalyst and unsaturated carbamates (Scheme 76).105 The use of simple carbamates as the nitrogen source is notable, as previous photochemical methods have relied upon pre-oxidized nitrogen sources. Irradiating the commercially available 2,4,6-triphenylpyrylium tetrafluoroborate (TPPT) photocatalyst produces a variety of oxazolidinone scaffolds with average to good yields for internal E-styrenyl alkenes with a number of backbone substitutions. Impressively, several intermolecular examples were also noted, starting from simple O-tert-butyl carbamate. Mechanistic inquiries determined that the copper salt is necessary as a two-electron oxidant for net turnover the photocatalyst. Accordingly, the mechanism is believed to begin with oxidation of the alkene by the excited photocatalyst to yield a radical cation (81), which can be attacked by the nitrogen of the carbamate in a reversible process. Oxidation of the resulting carbon-centered radical (82) by Cu(II) yields a benzylic cation that can be trapped by the carbamate carbonyl (83). Loss of a tert-butyl cation yields the final amino oxygenation product. It is also proposed that the Cu(I) resulting from oxidation of the benzylic radical could undergo oxidation of photocatalyst for turnover, though the authors noted that this is not completely proven over the possibility that Cu(I) disproportionates to Cu(II) and Cu(0). A follow-up study from the same authors noted that the reaction can be rendered catalytic in Cu(II) by employing stoichiometric Ag(I) salts.106 They note that the reaction is incompatible without the Cu(II) salts and conclude that Ag(I) must be unsuitable as the terminal oxidant.
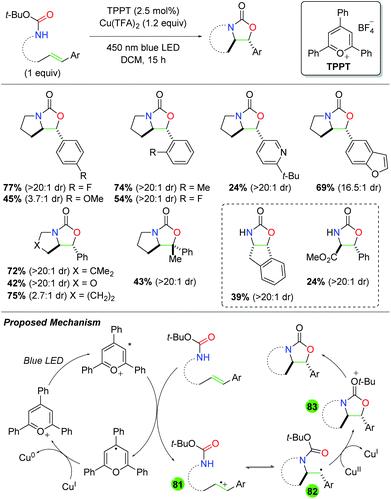 |
| | Scheme 76 Photocatalyzed amino oxygenation of carbamate-tethered alkenes (Yoon, 2018).105 | |
In 2020, the Zhang laboratory reported a photochemical method that provides amino oxygenation of enones (Scheme 77).107 This method is very similar to the previous Loh method (Scheme 23)32 but uses a lower temperature due to the photochemical catalyst. The yields are very similar to the Loh method, though the authors did report better success with α-substituted alkenes than the exclusively copper-catalyzed method.
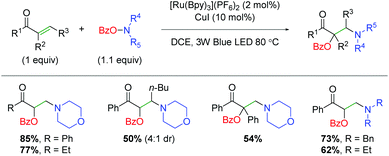 |
| | Scheme 77 Photocatalyzed amino oxygenation of enones with O-benzoylhydroxylamines (Zhang, 2020).107 | |
2.13 Biocatalysis
In 2016, the Li group developed the first biocatalytic method for amino oxygenation of styrenes using E. coli expressing different enzymatic “modules” (Scheme 78).108 The authors developed a series of enzymes that would recognize simple molecular skeleton inputs and convert them to the input needed by the next module. By adding different selections of these modules to E. coli, they were able to create an “assembly line” that produces different intermediates for the next enzyme and results in a mix-and-match program to create unique E. coli strains for delivering different outputs. In addition to 1,2-amino alcohols, they were also able to create outputs of α-hydroxy acids and α-amino acids. For the styrene oxyamination, a module containing a styrene monooxygenase and epoxide hydrolase converted the styrene into an asymmetric 1,2-diol. This diol was then recognized by another module featuring an alcohol dehydrogenase and transaminase that converted the 1,2-diol to a 1,2-amino alcohol. The reaction proceeds in generally good conversion (65–86%), though chloro and bromo-containing styrenes produce poor conversion (18–36%). In all cases, the 1,2-amino alcohols are produced in excellent enantioselectivity.
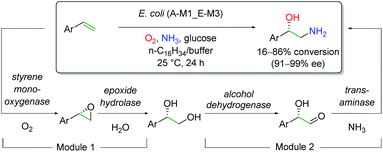 |
| | Scheme 78 Asymmetric oxyamination of styrenes using a two-stage enzymatic cascade (Li, 2016).108 | |
Several years later, Arnold and co-workers disclosed another biocatalyzed method for creating 1,2-amino alcohols from styrenes, this time using a hemoprotein evolved from cytochrome c (Scheme 79).109 Notably, this method is complementary to the Li method detailed above, as they generate opposite enantiomers for the styrenyl 1,2-amino alcohols. Since the authors planned to start from a porphyrin ring in cytochrome c, this protocol adopted the same nitrogen source (O-pivaloylhydroxylamine salts) as the 1,2-amino alcohol protocol by Morandi and co-workers 3 years prior, which used iron phthalocyanines (Scheme 42).61 Starting from the wild type, cytochrome c was subjected to a series of mutations to increase its promiscuity and improve its aptness for the desired reaction. The cytochrome c mutant TQL was ultimately the most successful variant found and was used for the substrate scope. Overall, the reaction generates a range of yields for amino alcohols, though para-substituted styrenes are most applicable for the reaction. TQL is also successful in tolerating 1,1-disubstituted alkenes as well as internal alkenes, though the 1,1-disubstituted alkene results in poor conversion. Mechanistically, the authors suggest formation of an iron nitrenoid intermediate that can form a stereospecific aziridine in reaction with the styrene. This aziridine can then be opened by water present in the aqueous buffer to yield the product. However, they acknowledge that other mechanisms cannot be ruled out at this time.
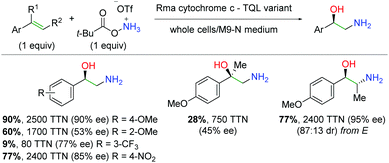 |
| | Scheme 79 Directed evolution of a cytochrome c variant for 1,2-amino oxygenation of styrenes (Arnold, 2019).109 | |
2.14 Halogen/chalcogen redox catalysis
In 2019, the Li group discovered a method for using a catalytic iodide source with stoichiometric oxidant to accomplish amino oxygenation through formation of oxazolines from intermolecular, unprotected ureas (Scheme 80).110 Mechanistically, the authors believe that this transformation occurs through oxidation of the iodide source to I+, which forms an iodonium ion on the alkene. The nitrogen from the urea then opens the iodonium ring and the carbonyl oxygen of the urea displaces the resulting alkyl iodide to regenerate the iodide catalyst. The reaction proceeds with excellent regio- and diastereoselectivity and works on an exceptionally wide scope of both urea and alkene substrates, tolerating internal, terminal, and 1,1-disubstituted alkenes. Interestingly, when styrenyl alkenes are used, selectivity of the oxygen for the terminal alkene position is observed, but this trend is reversed when alkyl alkenes were used. The authors justified this as a difference between the steric and electronic bias of the iodonium ion system for initial nucleophilic attack by the urea. In the case of a styrenyl alkene, the iodonium ion is susceptible to attack at the stabilized benzylic position, while the iodonium ion of an alkyl alkene is less stabilized and nucleophilic attack is dictated by the less sterically encumbered site.
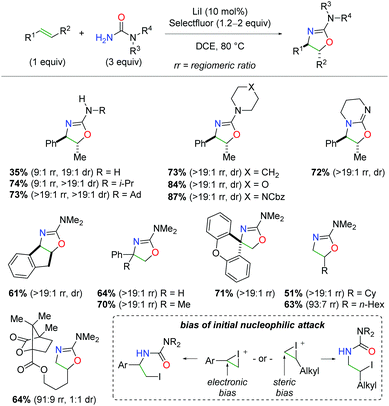 |
| | Scheme 80 Oxazoline formation through oxidative iodine catalysis (Li, 2019).110 | |
In 2019, while studying enantio- and diastereoselective diamination of alkenes using Lewis base catalysts and urea derivatives, Denmark and co-workers found that their selenium redox method could be adapted for oxyamination through use of a intermolecular carbamate (Scheme 81).111 This process was illustrated on a unsymmetrical stilbene using a chiral diselenide as the Lewis base catalyst and source of enantioinduction. Mechanistically, this reaction can be envisioned as following a similar route as the diamination. First, oxidation of the diselenide reveals the active selenium(II) catalyst (84). The catalyst then adds to the alkene, forming an enantioenriched seleniranium ion (85). Nucleophilic opening of the seleniranium ion by the carbamate furnishes intermediate 86, which can be oxidized by the N-fluoropyridine to selenium(IV) (87). This oxidation allows the carbamate oxygen to nucleophilically displace the selenium(IV) moiety to form the oxazolidinone product and regenerate the selenium(II) catalyst. While this entry was but a single example of this strategy, development of this result into a general method promises to reveal a powerful strategy for 1,2-amino oxygenation, as it is the only enantioselective example in alkene amino oxygenation that does not require transition metals.
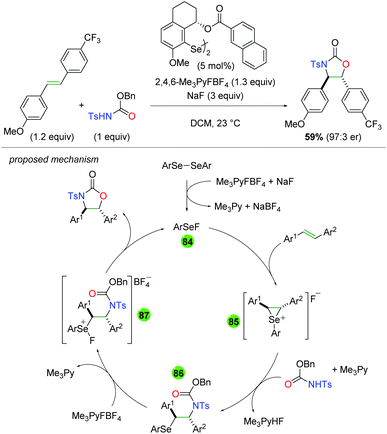 |
| | Scheme 81 Alkene oxyamination using Lewis base catalysts and intermolecular carbamates (Denmark, 2019).111 | |
A year later, the Michael laboratory also discovered an alkene amino oxygenation reaction while working on a Lewis base catalyzed diamination method. From their standard unsaturated ester scaffold, the authors found that when certain esters were tethered to the alkene, a rearrangement would occur to yield a 1,2-oxyamino compound (Scheme 82).112 While a substantial substrate scope was not explored, varying the chain length provides little change in the yield. Mechanistically, the authors proposed that the ester opens a putative seleniranium ion (88), followed by nucleophilic addition of the sulfonamide to supplant the ester (89). As with their diamination, nucleophilic attack of another sulfonamide displaces the selenium Lewis base (89) and generates the product. Furthermore, the authors found that using protected carbonates instead of esters allows for the generation of cyclic carbonates in good yields for the narrow range of substrates tested.
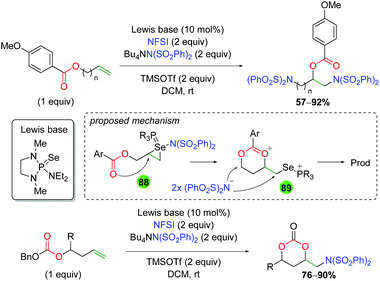 |
| | Scheme 82 Lewis base catalyzed amino oxygenation of alkenes through ester rearrangement (Michael, 2020).112 | |
3. 1,2-Amino oxygenation of alkynes
Although there are a number of methods for 1,2-addition of oxygen and nitrogen across alkynes that are driven thermodynamically by resulting aromaticity, such as oxazoles, pyrroles, and furans, this review will only address alkyne 1,2-amino oxygenation methods that are not aromaticity-inducing.
3.1 Rhodium catalysis
Within these parameters, the first example of a 1,2 alkyne difunctionalization was reported by the Blakey laboratory in 2009 (Scheme 83).113 While the authors were investigating the trapping of a rhodium nitrene/alkyne intermediate with aryl rings in a Friedel–Crafts arylation-type process, they noted that when the distance between the aryl group and alkyne is lowered, the acetate on the iodobenzene diacetate reagent acts as a nucleophile and supplies a 1,2-oxyamino product. While this was not investigated any further within this report, it nevertheless marks the first example of alkyne 1,2-amino oxygenation.
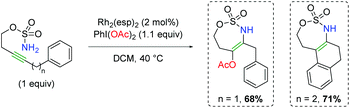 |
| | Scheme 83 1,2-Amino oxygenation of sulfonamide-tethered alkynes with rhodium catalysis (Blakey, 2009).113 | |
Several procedures have been developed to utilize copper(I)-catalyzed azide–alkyne cycloaddition (click chemistry) in sequence with rhodium catalysis to accomplish a formal alkyne 1,2-amino oxygenation (Scheme 84).114 While these studies focused on step-wise procedures starting from the 1,2,3-triazole, one-pot procedures were demonstrated for at least one substrate in both cases.
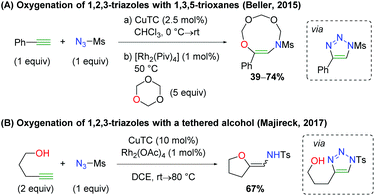 |
| | Scheme 84 Oxygenation of 1,2,3-triazole intermediates via rhodium catalysis.114 | |
3.2 Gold catalysis
Later in 2009, the Toste group reported an alkyne oxyamination method using gold catalysis to activate propargyl esters for attack by azomethine imines (Scheme 85).115 While the structural requirements of the substrates do not lend themselves to particularly significant variation, some substitutions were shown on the azomethine, imine, and propargyl ester with good to great yields in all cases (imine notwithstanding). The diastereoselectivity is good but not great in most cases, although some excellent diastereoselectivities are shown. Mechanistically, the reaction begins through activation of the propargyl group by the gold catalyst as a π-acid (90). Migration of the benzoate group creates a cationic π-allyl intermediate (91) that is in equilibrium with a gold carbene (92). This carbene species can be attacked by the anionic nitrogen of the azomethine (93). Elimination of the gold catalyst allows for nucleophilic attack of the electrophilic carbon on the imine and cyclization to the final product. The origin of diastereoselectivity can be explained at the final addition step into the imine (93). When the gold allyl nucleophile as above the imine, it has a steric conflict with the substituent on the azomethine ring. This strain is mitigated when the gold allyl nucleophile is below the imine.
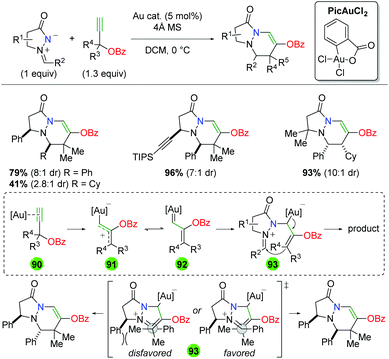 |
| | Scheme 85 1,2-Amino oxygenation of propargyl esters through benzoate migration (Toste, 2009).115 | |
An interesting amino oxygenation of alkynes was reported in 2014 by Gong and co-workers using alkyne-tethered boronic acids with diazo dicarboxylates (Scheme 86).116 The reaction that the authors were attempting to create involved gold activation of the alkyne and subsequent nucleophilic cyclization by the boronic acid (94). The authors then envisioned that a nucleophilic attack on the boron by the diazo compound would collapse the masked enol to attack the diazine (95) and yield a hydrazine product (96). However, the initial gold activation and boronic acid attack likely resulted in a gold intermediate that was not protonated off. This species likely attacked the diazene (97) to yield an atropisomeric nitrogen product with the oxaborinine ring (98). The substrate scope of this transformation contains the expected aryl substitutions and variation of the alkyne substituents, but the key feature is the good to great atropisomer enantioselectivity imparted by the chiral BINAP ligand on the gold catalyst.
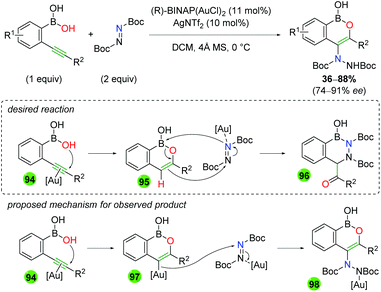 |
| | Scheme 86 1,2-Amino oxygenation of boronic acid-tethered alkynes with gold catalysis (Gong, 2014).116 | |
Several years later, the Patil laboratory was working on the synthesis of organic fluorophores and utilized a gold-catalyzed method for amino oxygenation of alkynes using tethered 2-alkyoxypyridine derivatives (Scheme 87).117 The procedure is proposed as an Au(I)/(III) pathway with the Selectfluor serving as an oxidant for catalyst turnover. The Au(III) activates the alkyne for nucleophilic attack by the pyridine and loss of the tert-butyl group on the ether exposes the oxygen nucleophile. The scope of this transformation is quite broad for the types of aryl groups that can be incorporated, though the authors note that some electron-rich aryl groups can undergo fluorine incorporation from the Selectfluor oxidant. Mechanistically, there are several proposals by the authors, though only a small number of control experiments were conducted. Several years later, the same group released a second method to avoid the aryl fluorination seen in the previous method.118 However, this method is not catalytic and requires stoichiometric copper salts.
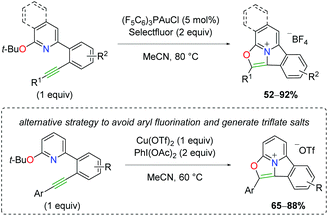 |
| | Scheme 87 Gold-catalyzed 1,2-amino oxygenation of alkyne-tethered 2-hydroxy pyridines (Patil, 2017/2019).117,118 | |
In 2020, a method was developed for alkyne amino oxygenation using gold catalysts with azide-tethered alkynes and intermolecular alcohol trapping (Scheme 88).119 The authors propose that the alkyne is activated by the gold(III) catalyst for nucleophilic attack by the internal nitrogen on the azide (99). Formation of a gold carbene extrudes molecular nitrogen to generate a cyclic imine (101). The gold carbene can then be attacked by the exogenous alcohol. Release of the gold catalyst generates the cyclic α,β-unsaturated ketone product after protonation of the resulting aniline nitrogen anion (102). While the substrate scope for the aryl ring and alkyne substitution display much of the anticipated variation with good yields with methanol as the solvent, it is notable that this procedure also incorporates various primary and tertiary alcohols with DCE as a solvent, showcasing the lack of influence that steric repulsion plays on the alcohol nucleophilic trapping step.
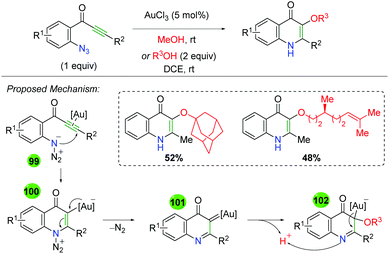 |
| | Scheme 88 Gold-catalyzed 1,2-amino oxygenation of azide-tethered alkynes (Xu, 2020).119 | |
3.3 Nitro radical-initiated methods
Starting in 2014, a series of papers exploited the use of nitro radicals for addition to terminal alkynes with terminal TEMPO radical trapping to generate geometrically-selective amino oxygenation methods. Both the Maiti120 and Mao121 groups almost simultaneously disclosed their findings and demonstrated nearly identical substrate compatibility for different types of alkynes, including aryl, pyridyl, and alkyl groups (Scheme 89). Although this is due to the identical nature of their conditions, it also speaks to the reproducibility and transferability of this method. However, a few unique and notable results from each method are also highlighted. The Maiti group demonstrated compatibility of amides, alcohols, and aldehydes on the alkyne, while the Mao group demonstrated many different types of TEMPO reagents, including ketones, esters, and epoxides. Mechanistically, this reaction is initiated by generation of a nitro radical, which adds to the terminal end of the alkyne. The resulting vinyl radical is then trapped by TEMPO. The Mao group argues that the selectivity for the E constitutional isomer is due to the vinyl radical p orbital, rather than the p orbitals in the alkene, being conjugated with the aryl ring, thereby stabilizing it on a lifetime that allows for TEMPO trapping.
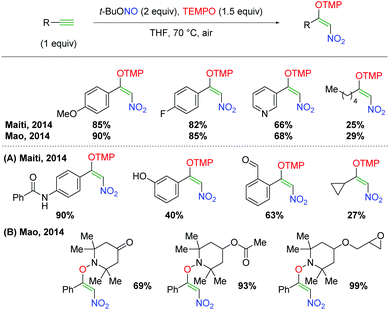 |
| | Scheme 89 Nitro oxygenation of terminal alkynes via nitro radicals.120,121 | |
A year later, a series of similar products were achieved using a similar procedure, starting from arylpropiolic acids and using silver(I) salts (Scheme 90).122 The premise of this reaction is similar to the nitro radical work above, but starts with decarboxylation of the alkyne with the silver salts. This intermediate is then reacted as in the previous work and the vinyl silver species is turned over via terminal protonation of the C–Au bond.
 |
| | Scheme 90 Nitro oxygenation of arylpropiolic acids via nitro radicals (He, 2015).122 | |
3.4 Miscellaneous methods
In 2010, another serendipitous discovery of an alkyne amino oxygenation was disclosed by Narasaka and co-workers when studying allylic amination with N-benzoate sulfonamides. The authors then tested alkynes and found that the tether length between the alkyne and sulfonamide dictates the product of the reaction (Scheme 91).123 A longer, 4 carbon tether between the alkyne and sulfonamide results in the expected pyrrolidine product. However, when the tether is shortened by a carbon, an amino oxygenation product is achieved through transfer of the benzoate group from the sulfonamide. As this product was not the object of their research, it was not pursued further.
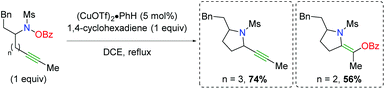 |
| | Scheme 91 1,2-Amino oxygenation of propargyl esters through benzoate migration (Narasaka, 2010).123 | |
Over the next few years several different methods were disclosed using iodine(III) reagents to achieve alkyne amino oxygenation. First, the Liu group used ynones and primary amines with iodobenzene diacetate to achieve E constitutional isomer products (Scheme 92A).124 While a putative mechanistic rationale was proposed, exact details were not explicitly established. Furthermore, when the Muñiz laboratory was developing a procedure for formation of ynamides from terminal alkynes using a mixed NTs2/OAc iodobenzene oxidant, they found that having a tethered amide provides nucleophilic oxygenation (Scheme 92B).125 The authors proposed that the presence of acetic acid could induce a 6-exo-dig cyclization from the ynamide intermediate. Finally, Van der Eycken and co-workers reacted alkyne-tethered N-Boc guanidines using iodobenzene diacetate to induce cyclization of the Boc group onto the alkyne (Scheme 92C).126 The authors found that the identity of the aryl group on the alkyne is vital, as using more electron-rich groups induces an additional cyclization product.
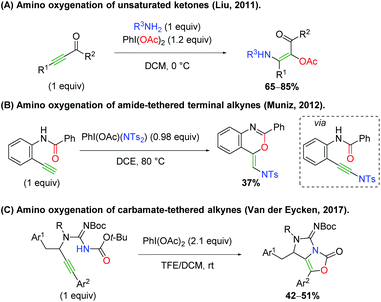 |
| | Scheme 92 1,2-Amino oxygenation of alkynes using iodine(III) reagents.124–126 | |
In 2019, a ruthenium-catalyzed method was developed by the Chikhalia laboratory as part of a study pursuing cyclooxygenase (COX) inhibitors (Scheme 93).127 To forge these morpholine analogs, the authors utilized cyclization of 2-hydroxyanilines onto internal alkynes. Regarding substrate variability, the aromatic backbone is compulsory for the phenol/aniline reactivity, but a fair array of substitution within those confines is shown to be tolerated. Furthermore, alkynes other than diphenylacetylenes are shown, such as dialkyl and diester. Heteroatoms are also tolerated, in addition to secondary anilines. Mechanistically, this reaction was proposed to start from a Ru(II) catalyst and undergo two rounds of ligand exchange with the 2-hydroxyaniline (104). The acetylene is then added via migratory insertion, either from the oxygen or nitrogen ligand coordination (105). Reductive elimination from either intermediate delivers the amino oxygenation product and a Ru(0) catalyst (106), which is oxidized back to Ru(II) by a mixture of Cu(OAc)2 and O2.
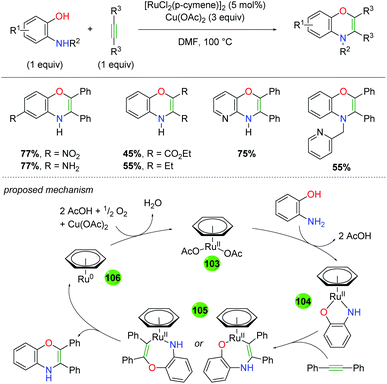 |
| | Scheme 93 Ruthenium-catalyzed 1,2-amino oxygenation of alkynes using 2-hydroxyanilines (Chikhalia, 2019).127 | |
4. 1,2-Amino oxygenation of allenes
While allenes have not been extensively studied in the context of 1,2-difunctionalization reactions due to competing 1,3 reactivity and the stability of the resulting products, there exist a number of interesting examples. The first reported example was reported in 2007 by the Shi group using iodobenzene diacetate to react phthalhydrazide and vinylidenecyclopropanes (Scheme 94).128 The authors were also studying the same amino oxygenation reaction on methylidenecyclopropanes to create tetrahydropyridizinium products. While only a very narrow range of allene substitutions were investigated, the yields are very good for the vinylidenecyclopropanes tested. It is also important to note that methylidene cyclopropanes are often considered a separate class of alkene.
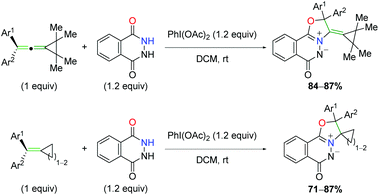 |
| | Scheme 94 Vinylidenecyclopropane and methylidene 1,2-amino oxygenation using phthalhydrazides (Shi, 2007).128 | |
In 2011, Blakey and co-workers published a three-component amino oxygenation of allenes using rhodium catalysis with nitrones and sulfonamide-tethered allenes (Scheme 95).129 A range of α-substituted allenes and benzaldehyde-derived nitrones were tested with average yields of the bicyclo [4.3.1] 1,2,4-oxadiazinane system with the terminal vinyl group from the allene remaining intact. Notably, when terminally di-substituted allenes are introduced to the reaction, the bicyclic system is not observed. Instead, a fused 7 + 6 ring system is observed in average to good yield. These results can be rationalized through the proposed mechanism. Initial rhodium nitrene (107) addition to the allene yields a cationic π-allyl species (108), which rearranges to an iminocyclopropane species (109). The imine nitrogen then adds reversibly into the nitrone to yield 110, which is in equilibrium with cationic π-allyl species 111. The product is generated by the anionic oxygen trapping of this species, depending on the ability of the allene substituents to stabilize the cation. When the internal substituents are stabilizing, the anionic oxygen attacks at this position and the bicyclic system is achieved. When the terminal substituent is stabilizing, the fused ring system is achieved. Unfortunately, no substrates with competing stabilizing effects were tested to determine the extent of this stabilization on the product distribution.
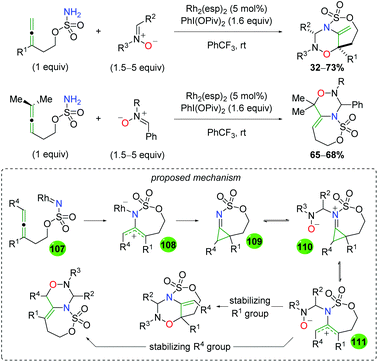 |
| | Scheme 95 Rhodium-catalyzed three-component amino oxygenation of allenes using nitrones (Blakey, 2011).129 | |
Later in 2011, Schomaker and co-workers were working on trifunctionalization of allenes using carbamate-tethered allenes to create methylene aziridines (Scheme 96A).130 These intermediates, achieved through rhodium catalysis, can be trapped by a number of nucleophiles including carboxylic acids and alcohols (with catalytic scandium). A year later, this step-wise process was rendered one-pot using excess equivalents of the nucleophilic partner (Scheme 96B).131 As mentioned above, this method was developed as part of a stereoselective trifunctitonalization of allenes, allowing for the setting of three contiguous stereocenters in a single step. As this difunctionalization was not the main pursuit, only a small substrate scope was demonstrated. However, the high yields of the carboxylic acids in the trapping step of the stepwise process offer promise that more carboxylic acids and alcohols can be seamlessly incorporated into the one-pot process than just the acetic acid and methanol shown. For their trifunctionalization work, the authors developed a road map that provided access to any stereoisomer. This platform was applied toward efforts in the total synthesis of Jogyamycin as the initial steps in setting the stereochemistry for two of the three stereogenic centers (Scheme 96C).132
 |
| | Scheme 96 Rhodium-catalyzed amino oxygenation of allenes via methylene aziridines (Schomaker).130–132 | |
Several years later, the Ma laboratory disclosed a radical nitro oxygenation of allenes (Scheme 97).133 The method used silver nitrite as a radical source and TEMPO as a terminal radical trapping reagent. Regarding the substrate scope, the chemical space of allenes is limited to terminal, mono-substituted allenes, though the authors demonstrated a range of aromatic, heteroaromatic, and alkyl-substituted allenes with fair to good yield and excellent E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratios. The origin of this constitutional isomer selectivity is found in the proposed mechanism: a nitro radical (generated from heating of silver nitrite) adds to the allene, generating two possible radical π-allyl species. Due to the steric repulsion of the nitro group, the intermediate with the allene substituent anti would be favored. From this radical intermediate, the TEMPO radical scavenger would add at the terminal position of the radical π-allyl due to the steric repulsion of the allene substituent to generate the E product. Trapping of the opposite syn radical π-allyl species would give rise to the minor Z isomer.
:Z ratios. The origin of this constitutional isomer selectivity is found in the proposed mechanism: a nitro radical (generated from heating of silver nitrite) adds to the allene, generating two possible radical π-allyl species. Due to the steric repulsion of the nitro group, the intermediate with the allene substituent anti would be favored. From this radical intermediate, the TEMPO radical scavenger would add at the terminal position of the radical π-allyl due to the steric repulsion of the allene substituent to generate the E product. Trapping of the opposite syn radical π-allyl species would give rise to the minor Z isomer.
 |
| | Scheme 97 Radical nitro oxygenation of allenes via Ag(NO2) and TEMPO (Ma, 2014).133 | |
5. Conclusions
In conclusion, this review has detailed 1,2-amino oxygenation reactions of alkenes, 1,3-dienes, alkynes, and allenes. 1,2-Amino oxygenation of olefins presents a rapid method of assembling molecular complexity in a straightforward and efficient manner. Although the initial Sharpless asymmetric amino hydroxylation reaction has proven to be a powerful and timeless synthetic tool for simultaneous forging of C–O and C–N bonds, recent years have brought a vast array of exciting methods for accomplishing this valuable transformation in novel and orthogonal ways. Nevertheless, there are still opportunities for the supplementing of these existing methods and construction of unique approaches. Specifically, there is still a great need for reaction platforms that exhibit high levels of enantioselectivity for a broad range of substrate types. Within the focus of alkyne and allene functionalization, there is still an exceptional range of chemical space that has yet to be explored and is limited only by our current understanding and interest.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author would like to acknowledge the insights, guidance, and encouragement provided by Prof. Qiu Wang, Prof. Scott E. Denmark, Prof. Jennifer L. Roizen, Prof. Steven W. Baldwin, and Prof. Ross A. Widenhoefer.
References
-
(a) N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87(12), 1348–1349 CrossRef CAS;
(b) Njardarson Group. Top Pharmaceuticals Poster. njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster (accessed 2018).
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54(10), 3451–3479 CrossRef CAS.
-
(a) T. Courant and G. Masson, J. Org. Chem., 2016, 81(16), 6945–6952 CrossRef CAS;
(b) S. Ortgies and A. Breder, ACS Catal., 2017, 7(9), 5828–5840 CrossRef CAS;
(c) J. B. Parry, N. Fu and S. Lin, Synlett, 2018, 29(03), 257–265 CrossRef CAS;
(d) G. S. Sauer and S. Lin, ACS Catal., 2018, 8(6), 5175–5187 CrossRef CAS;
(e) H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37(3), 292–301 CAS;
(f) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49(1), 32–48 RSC;
(g) A. Matviitsuk, J. L. Panger and S. E. Denmark, Angew. Chem., Int. Ed., 2020, 59(45), 19796–19819 CrossRef CAS;
(h) G. J. P. Perry, T. Jia and D. J. Procter, ACS Catal., 2020, 10(2), 1485–1499 CrossRef CAS;
(i) X. Qi and T. Diao, ACS Catal., 2020, 10(15), 8542–8556 CrossRef CAS;
(j) A. Whyte, A. Torelli, B. Mirabi, A. Zhang and M. Lautens, ACS Catal., 2020, 10(19), 11578–11622 CrossRef CAS.
-
(a) P. O'Brien, Angew. Chem., Int. Ed., 1999, 38(3), 326–329 CrossRef;
(b) D. Nilov and O. Reiser, Adv. Synth. Catal., 2002, 344(10), 1169–1173 CrossRef CAS;
(c) T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem. – Eur. J., 2011, 17(1), 58–76 CrossRef CAS.
-
(a) J.-E. Bäckvall, Tetrahedron Lett., 1975, 16(26), 2225–2228 CrossRef;
(b) J. E. Backvall and E. E. Bjoerkman, J. Org. Chem., 1980, 45(14), 2893–2898 CrossRef;
(c) J. E. Backvall and S. E. Bystroem, J. Org. Chem., 1982, 47(6), 1126–1128 CrossRef.
- E. J. Alexanian, C. Lee and E. J. Sorensen, J. Am. Chem. Soc., 2005, 127(21), 7690–7691 CrossRef CAS.
- P. Szolcsányi and T. Gracza, Chem. Commun., 2005,(31), 3948–3950 RSC.
- G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2006, 128(22), 7179–7181 CrossRef CAS.
- C. Martínez, Y. Wu, A. B. Weinstein, S. S. Stahl, G. Liu and K. Muñiz, J. Org. Chem., 2013, 78(12), 6309–6315 CrossRef.
- L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46(30), 5737–5740 CrossRef CAS.
- W.-H. Rao, X.-S. Yin and B.-F. Shi, Org. Lett., 2015, 17(15), 3758–3761 CrossRef CAS.
- C. Martínez, E. G. Pérez, Á. Iglesias, E. C. Escudero-Adán and K. Muñiz, Org. Lett., 2016, 18(12), 2998–3001 CrossRef.
- H. Zhu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136(5), 1766–1769 CrossRef CAS.
- H. Zhu, P. Chen and G. Liu, Org. Lett., 2015, 17(6), 1485–1488 CrossRef CAS.
- H.-C. Shen, Y.-F. Wu, Y. Zhang, L.-F. Fan, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2018, 57(9), 2372–2376 CrossRef CAS.
- K. Wen, Z. Wu, B. Huang, Z. Ling, I. D. Gridnev and W. Zhang, Org. Lett., 2018, 20(6), 1608–1612 CrossRef CAS.
- K. Wen, Z. Wu, B. Chen, J. Chen and W. Zhang, Org. Biomol. Chem., 2018, 16(31), 5618–5625 RSC.
- Y. Li, Z. Wu, Z. Ling, H. Chen and W. Zhang, Org. Chem. Front., 2019, 6(4), 486–492 RSC.
- T. Zeng, Z. Liu, M. A. Schmidt, M. D. Eastgate and K. M. Engle, Org. Lett., 2018, 20(13), 3853–3857 CrossRef CAS.
- E. Levites-Agababa, E. Menhaji, L. N. Perlson and C. M. Rojas, Org. Lett., 2002, 4(5), 863–865 CrossRef CAS.
- R. Lorpitthaya, Z.-Z. Xie, J.-L. Kuo and X.-W. Liu, Chem. – Eur. J., 2008, 14(5), 1561–1570 CrossRef CAS.
- G. Dequirez, J. Ciesielski, P. Retailleau and P. Dauban, Chem. – Eur. J., 2014, 20(29), 8929–8933 CAS.
- Y. Shi, Y. Wang, X. Lu, Y. Zhang, Y. Wu and F. Zhong, Green Chem., 2019, 21(4), 780–784 RSC.
- J. Guasch, Y. Díaz, M. I. Matheu and S. Castillón, Chem. Commun., 2014, 50(55), 7344–7347 RSC.
- J. Escudero, V. Bellosta and J. Cossy, Angew. Chem., Int. Ed., 2018, 57(2), 574–578 CrossRef CAS.
- S. Bi, Y. Wang, Y.-Y. Jiang and Y. Liu, J. Organomet. Chem., 2019, 880, 253–260 CrossRef CAS.
- M. Noack and R. Göttlich, Chem. Commun., 2002,(5), 536–537 RSC.
- B. N. Hemric, K. Shen and Q. Wang, J. Am. Chem. Soc., 2016, 138(18), 5813–5816 CrossRef CAS.
- B. N. Hemric and Q. Wang, Beilstein J. Org. Chem., 2016, 12, 22–28 CrossRef CAS.
- B. N. Hemric, A. W. Chen and Q. Wang, J. Org. Chem., 2019, 84(3), 1468–1488 CrossRef CAS.
- B. N. Hemric, A. W. Chen and Q. Wang, ACS Catal., 2019, 9(11), 10070–10076 CrossRef CAS.
- S. Ren, S. Song, L. Ye, C. Feng and T.-P. Loh, Chem. Commun., 2016, 52(68), 10373–10376 RSC.
-
(a) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129(7), 1866–1867 CrossRef CAS;
(b) D. J. Michaelis, M. A. Ischay and T. P. Yoon, J. Am. Chem. Soc., 2008, 130(20), 6610–6615 CrossRef CAS.
- D. J. Michaelis, K. S. Williamson and T. P. Yoon, Tetrahedron, 2009, 65(26), 5118–5124 CrossRef CAS.
- T. Benkovics, J. Du, I.
A. Guzei and T. P. Yoon, J. Org. Chem., 2009, 74(15), 5545–5552 CrossRef CAS.
- P. H. Fuller, J. W. Kim and S. R. Chemler, J. Am. Chem. Soc., 2008, 130(52), 17638–17639 CrossRef CAS.
- M. C. Paderes, J. B. Keister and S. R. Chemler, J. Org. Chem., 2013, 78(2), 506–515 CrossRef CAS.
- E. S. Sherman and S. R. Chemler, Adv. Synth. Catal., 2009, 351(3), 467 CrossRef CAS.
- F. C. Sequeira and S. R. Chemler, Org. Lett., 2012, 14(17), 4482–4485 CrossRef CAS.
- S. D. Karyakarte, T. P. Smith and S. R. Chemler, J. Org. Chem., 2012, 77(17), 7755–7760 CrossRef CAS.
- D. E. Mancheno, A. R. Thornton, A. H. Stoll, A. Kong and S. B. Blakey, Org. Lett., 2010, 12(18), 4110–4113 CrossRef CAS.
- D. Karila, L. Leman and R. H. Dodd, Org. Lett., 2011, 13(21), 5830–5833 CrossRef CAS.
- S. Hajra, S. M. S. Akhtar and S. M. Aziz, Chem. Commun., 2014, 50(52), 6913–6916 RSC.
- S. Sanjaya and S. Chiba, Org. Lett., 2012, 14(20), 5342–5345 CrossRef CAS.
- S. Sanjaya, S. H. Chua and S. Chiba, Synlett, 2012, 23(11), 1657–1661 CrossRef CAS.
- L. Zhu, H. Yu, Z. Xu, X. Jiang, L. Lin and R. Wang, Org. Lett., 2014, 16(6), 1562–1565 CrossRef CAS.
-
(a) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134(30), 12462–12465 CrossRef CAS;
(b) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52(48), 12655–12658 CrossRef CAS.
- R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137(25), 8069–8077 CrossRef CAS.
- Y. Li, X. Zhou, G. Zheng and Q. Zhang, Beilstein J. Org. Chem., 2015, 11, 2721–2726 CrossRef CAS.
- C. Herrera-Leyton, M. Madrid-Rojas, J. J. López, Á. Cañete, P. Hermosilla-Ibáñez and E. G. Pérez, ChemCatChem, 2016, 8(12), 2015–2018 CrossRef CAS.
- J. Xie, Y.-W. Wang, L.-W. Qi and B. Zhang, Org. Lett., 2017, 19(5), 1148–1151 CrossRef CAS.
-
(a) R.-H. Liu, D. Wei, B. Han and W. Yu, ACS Catal., 2016, 6(10), 6525–6530 CrossRef CAS;
(b) R.-H. Liu, Z.-Q. Wang, B.-Y. Wei, J.-W. Zhang, B. Zhou and B. Han, Org. Lett., 2018, 20(14), 4183–4186 CrossRef CAS.
- S. Chen, W. Chen, X. Chen, G. Chen, L. Ackermann and X. Tian, Org. Lett., 2019, 21(19), 7787–7790 CrossRef CAS.
- F. Wu, S. Stewart, J. P. Ariyarathna and W. Li, ACS Catal., 2018, 8(3), 1921–1925 CrossRef CAS.
- F. Wu, J. P. Ariyarathna, N.-E. Alom, N. Kaur and W. Li, Org. Lett., 2020, 22(3), 884–890 CrossRef CAS.
- K. Muniz, A. Iglesias and Y. Fang, Chem. Commun., 2009,(37), 5591–5593 RSC.
- K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134(30), 12370–12373 CrossRef CAS.
- K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2010, 132(13), 4570–4571 CrossRef CAS.
- G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135(9), 3343–3346 CrossRef CAS.
- D. F. Lu, C. L. Zhu, Z. X. Jia and H. Xu, J. Am. Chem. Soc., 2014, 136(38), 13186–13189 CrossRef CAS.
- L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55(6), 2248–2251 CrossRef CAS.
- A.-D. Manick, S. Aubert, B. Yalcouye, T. Prangé, F. Berhal and G. Prestat, Chem. – Eur. J., 2018, 24(44), 11485–11492 CrossRef CAS.
- T. de Haro and C. Nevado, Angew. Chem., Int. Ed., 2011, 50(4), 906–910 CrossRef CAS.
- X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137(18), 6059–6066 CrossRef CAS.
- T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135(32), 11692–11695 CrossRef CAS.
- H. Lei, J. H. Conway, C. C. Cook and T. Rovis, J. Am. Chem. Soc., 2019, 141(30), 11864–11869 CrossRef CAS.
-
(a) S. Serna, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartín, Tetrahedron Lett., 2003, 44(17), 3483–3486 CrossRef CAS;
(b) S. Serna, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartin, Tetrahedron, 2004, 60(31), 6533–6539 CrossRef CAS.
- B. M. Cochran and F. E. Michael, Org. Lett., 2008, 10(21), 5039–5042 CrossRef CAS.
- U. Farid and T. Wirth, Angew. Chem., Int. Ed., 2012, 51(14), 3462–3465 CrossRef CAS.
- H. M. Lovick and F. E. Michael, J. Am. Chem. Soc., 2010, 132(4), 1249–1251 CrossRef CAS.
- D. J. Wardrop, E. G. Bowen, R. E. Forslund, A. D. Sussman and S. L. Weerasekera, J. Am. Chem. Soc., 2010, 132(4), 1188–1189 CrossRef CAS.
- V. A. Schmidt and E. J. Alexanian, Angew. Chem., Int. Ed., 2010, 49(26), 4491–4494 CrossRef CAS.
- V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133(30), 11402–11405 CrossRef CAS.
- B. Han, X.-L. Yang, R. Fang, W. Yu, C. Wang, X.-Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51(35), 8816–8820 CrossRef CAS.
-
(a) X.-Y. Duan, X.-L. Yang, R. Fang, X.-X. Peng, W. Yu and B. Han, J. Org. Chem., 2013, 78(21), 10692–10704 CrossRef CAS;
(b) X. Y. Duan, N. N. Zhou, R. Fang, X. L. Yang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2014, 53(12), 3158–3162 CrossRef CAS.
- Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51(33), 8221–8224 CrossRef CAS.
- B. Zhang and A. Studer, Org. Lett., 2013, 15(17), 4548–4551 CrossRef CAS.
- Y. Li, M. Hartmann, C. G. Daniliuc and A. Studer, Chem. Commun., 2015, 51(26), 5706–5709 RSC.
- Q. Xue, J. Xie, P. Xu, K. Hu, Y. Cheng and C. Zhu, ACS Catal., 2013, 3(6), 1365–1368 CrossRef CAS.
- C. Alamillo-Ferrer, J. M. Curle, S. C. Davidson, S. C. C. Lucas, S. J. Atkinson, M. Campbell, A. R. Kennedy and N. C. O. Tomkinson, J. Org. Chem., 2018, 83(12), 6728–6740 CrossRef CAS.
-
(a) J. C. Griffith, K. M. Jones, S. Picon, M. J. Rawling, B. M. Kariuki, M. Campbell and N. C. O. Tomkinson, J. Am. Chem. Soc., 2010, 132(41), 14409–14411 CrossRef CAS;
(b) C. Alamillo-Ferrer, M. Karabourniotis-Sotti, A. R. Kennedy, M. Campbell and N. C. O. Tomkinson, Org. Lett., 2016, 18(13), 3102–3105 CrossRef CAS.
- J. M. Curle, M. C. Perieteanu, P. G. Humphreys, A. R. Kennedy and N. C. O. Tomkinson, Org. Lett., 2020, 22(4), 1659–1664 CrossRef CAS.
- X.-F. Xia, Z. Gu, W. Liu, H. Wang, Y. Xia, H. Gao, X. Liu and Y.-M. Liang, J. Org. Chem., 2015, 80(1), 290–295 CrossRef CAS.
- S. Gao and M. Niggemann, Adv. Synth. Catal., 2019, 361(7), 1549–1553 CrossRef CAS.
- J. M. Mahoney, C. R. Smith and J. N. Johnston, J. Am. Chem. Soc., 2005, 127(5), 1354–1355 CrossRef CAS.
- K. Raner, B. Skelton, A. Ward and A. White, Aust. J. Chem., 1990, 43(3), 609–616 CrossRef CAS.
- K. Moriyama, Y. Izumisawa and H. Togo, J. Org. Chem., 2012, 77(21), 9846–9851 CrossRef CAS.
- G.-Q. Liu, L. Li, L. Duan and Y.-M. Li, RSC Adv., 2015, 5(75), 61137–61143 RSC.
- A. Theodorou and C. G. Kokotos, Adv. Synth. Catal., 2017, 359(9), 1577–1581 CrossRef CAS.
- E. Voutyritsa, A. Theodorou, M. G. Kokotou and C. G. Kokotos, Green Chem., 2017, 19(5), 1291–1298 RSC.
-
(a) H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2008, 130(41), 13542–13543 CrossRef CAS;
(b) H. C. Xu and K. D. Moeller, J. Am. Chem. Soc., 2010, 132(8), 2839–2844 CrossRef CAS.
- F. Tang and K. D. Moeller, J. Am. Chem. Soc., 2007, 129(41), 12414–12415 CrossRef CAS.
- H. C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79(1), 379–391 CrossRef CAS.
- F. Xu, L. Zhu, S. Zhu, X. Yan and H. C. Xu, Chem. – Eur. J., 2014, 20(40), 12740–12744 CrossRef CAS.
- S. Liang, C.-C. Zheng, X.-G. Luo, F.-Z. Ren, H.-Y. Tian, B.-G. Sun and R. D. Little, Green Chem., 2016, 18, 2222 RSC.
- J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauvire, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140(39), 12511–12520 CrossRef CAS.
- N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357(6351), 575–579 CrossRef CAS.
- G. Fumagalli, P. T. G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54(39), 11481–11484 CrossRef CAS.
-
(a) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18(24), 7336–7340 CrossRef CAS;
(b) A. Baralle, L. Fensterbank, J.-P. Goddard and C. Ollivier, Chem. – Eur. J., 2013, 19(33), 10809–10813 CrossRef CAS;
(c) X.-J. Tang and W. R. Dolbier, Angew. Chem., Int. Ed., 2015, 54(14), 4246–4249 CrossRef CAS;
(d) D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54(24), 6999–7002 CrossRef CAS.
- G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2013, 15(17), 4398–4401 CrossRef CAS.
- K. Miyazawa, T. Koike and M. Akita, Chem. – Eur. J., 2015, 21(33), 11677–11680 CrossRef CAS.
- X. Ren, Q. Guo, J. Chen, H. Xie, Q. Xu and Z. Lu, Chem. – Eur. J., 2016, 22(52), 18695–18699 CrossRef CAS.
- S. Alazet, F. Le Vaillant, S. Nicolai, T. Courant and J. Waser, Chem. – Eur. J., 2017, 23(40), 9501–9504 CrossRef CAS.
- B. Yang and Z. Lu, ACS Catal., 2017, 7(12), 8362–8365 CrossRef CAS.
- N. L. Reed, M. I. Herman, V. P. Miltchev and T. P. Yoon, Org. Lett., 2018, 20(22), 7345–7350 CrossRef CAS.
- N. L. Reed, M. I. Herman, V. P. Miltchev and T. P. Yoon, Beilstein J. Org. Chem., 2019, 15, 351–356 CrossRef CAS.
- S. Li, Q. Chen, W. Li, G. Gu and J. Zhang, Chin. J. Chem., 2020, 38, 1116–1122 CrossRef CAS.
- S. Wu, Y. Zhou, T. Wang, H.-P. Too, D. I. C. Wang and Z. Li, Nat. Commun., 2016, 7(1), 11917 CrossRef CAS.
- I. Cho, C. K. Prier, Z.-J. Jia, R. K. Zhang, T. Görbe and F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58(10), 3138–3142 CrossRef CAS.
- F. Wu, N.-E. Alom, J. P. Ariyarathna, J. Naß and W. Li, Angew. Chem., Int. Ed., 2019, 58(34), 11676–11680 CrossRef CAS.
- Z. Tao, B. B. Gilbert and S. E. Denmark, J. Am. Chem. Soc., 2019, 141(48), 19161–19170 CrossRef CAS.
- J. R. Tabor, D. C. Obenschain and F. E. Michael, Chem. Sci., 2020, 11(6), 1677–1682 RSC.
- A. R. Thornton, V. I. Martin and S. B. Blakey, J. Am. Chem. Soc., 2009, 131(7), 2434–2435 CrossRef CAS.
-
(a) J. Pospech, R. Ferraccioli, H. Neumann and M. Beller, Chem. – Asian J., 2015, 10(12), 2624–2630 CrossRef CAS;
(b) J. M. Bennett, J. D. Shapiro, K. N. Choinski, Y. Mei, S. M. Aulita, E. W. Reinheimer and M. M. Majireck, Tetrahedron Lett., 2017, 58(12), 1117–1122 CrossRef CAS.
- N. D. Shapiro, Y. Shi and F. D. Toste, J. Am. Chem. Soc., 2009, 131(33), 11654–11655 CrossRef CAS.
- R. Guo, K.-N. Li, B. Liu, H.-J. Zhu, Y.-M. Fan and L.-Z. Gong, Chem. Commun., 2014, 50(41), 5451–5454 RSC.
- A. C. Shaikh, D. S. Ranade, P. R. Rajamohanan, P. P. Kulkarni and N. T. Patil, Angew. Chem., Int. Ed., 2017, 56(3), 757–761 CrossRef CAS.
- A. C. Shaikh, M. E. Varma, R. D. Mule, S. Banerjee, P. P. Kulkarni and N. T. Patil, J. Org. Chem., 2019, 84(4), 1766–1777 CrossRef CAS.
- J. Huang, H. Su, M. Bao, L. Qiu, Y. Zhang and X. Xu, Org. Biomol. Chem., 2020, 18(20), 3888–3892 RSC.
- U. Dutta, S. Maity, R. Kancherla and D. Maiti, Org. Lett., 2014, 16(24), 6302–6305 CrossRef CAS.
- H. Yan, G. Rong, D. Liu, Y. Zheng, J. Chen and J. Mao, Org. Lett., 2014, 16(24), 6306–6309 CrossRef CAS.
- H. Yan, J. Mao, G. Rong, D. Liu, Y. Zheng and Y. He, Green Chem., 2015, 17(5), 2723–2726 RSC.
- W.-M. Liu, Z.-H. Liu, W.-W. Cheong, L.-Y. T. Priscilla, Y. Li and K. Narasaka, Bull. Korean Chem. Soc., 2010, 31(3), 563–569 CrossRef CAS.
- W. Liu, C. Chen and Q. Zhang, Org. Biomol. Chem., 2011, 9(19), 6484–6486 RSC.
- J. A. Souto, P. Becker, Á. Iglesias and K. Muñiz, J. Am. Chem. Soc., 2012, 134(37), 15505–15511 CrossRef CAS.
- G. Tian, P. Fedoseev and E. V. Van der Eycken, Chem. – Eur. J., 2017, 23(22), 5224–5227 CrossRef CAS.
- M. M. Shaikh, A. P. Patel, S. P. Patel and K. H. Chikhalia, New J. Chem., 2019, 43(26), 10305–10317 RSC.
- L.-P. Liu, J.-M. Lu and M. Shi, Org. Lett., 2007, 9(7), 1303–1306 CrossRef CAS.
- A. H. Stoll and S. B. Blakey, Chem. Sci., 2011, 2(1), 112–116 RSC.
- L. A. Boralsky, D. Marston, R. D. Grigg, J. C. Hershberger and J. M. Schomaker, Org. Lett., 2011, 13(8), 1924–1927 CrossRef CAS.
- C. S. Adams, L. A. Boralsky, I. A. Guzei and J. M. Schomaker, J. Am. Chem. Soc., 2012, 134(26), 10807–10810 CrossRef CAS.
- N. C. Gerstner, C. S. Adams, R. D. Grigg, M. Tretbar, J. W. Rigoli and J. M. Schomaker, Org. Lett., 2016, 18(2), 284–287 CrossRef CAS.
- C. Xue, C. Fu and S. Ma, Chem. Commun., 2014, 50(97), 15333–15336 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab
ab