
Ambient blade coating of mixed cation, mixed halide perovskites without dripping: in situ investigation and highly efficient solar cells†
Ming-Chun
Tang‡
 a,
Yuanyuan
Fan‡
b,
Dounya
Barrit
a,
Xiaoming
Chang
b,
Hoang X.
Dang
c,
Ruipeng
Li
d,
Kai
Wang
a,
Detlef-M.
Smilgies
e,
Shengzhong (Frank)
Liu
b,
Stefaan
De Wolf
a,
Thomas D.
Anthopoulos
*a,
Kui
Zhao
*b and
Aram
Amassian
*ac
a,
Yuanyuan
Fan‡
b,
Dounya
Barrit
a,
Xiaoming
Chang
b,
Hoang X.
Dang
c,
Ruipeng
Li
d,
Kai
Wang
a,
Detlef-M.
Smilgies
e,
Shengzhong (Frank)
Liu
b,
Stefaan
De Wolf
a,
Thomas D.
Anthopoulos
*a,
Kui
Zhao
*b and
Aram
Amassian
*ac
aKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Physical Science and Engineering Division (PSE), Thuwal, 23955-6900, Saudi Arabia. E-mail: thomas.anthopoulos@kaust.edu.sa; aamassi@ncsu.edu
bKey Laboratory of Applied Surface and Colloid Chemistry, National Ministry of Education, Shaanxi Key Laboratory for Advanced Energy Devices, Shaanxi Engineering Lab for Advanced Energy Technology, School of Materials Science and Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: zhaok@snnu.edu.cn
cDepartment of Materials Science and Engineering, North Carolina State University, Raleigh, NC 27695, USA
dNSLS-II, Brookhaven National Laboratory (BNL), Upton, NY 11973, USA
eCornell High Energy Synchrotron Source (CHESS), Cornell University, Ithaca, NY 14850, USA
First published on 4th December 2019
Abstract
Perovskite photovoltaics have made extraordinary strides in efficiency and stability thanks to process and formulation developments like anti-solvent dripping and mixed-cation mixed-halide compositions. Solar cell fabrication through low-cost scalable methods, such as blade coating, cannot accommodate anti-solvent dripping and needs to be performed in an ambient atmosphere. Consequently, their efficiency has lagged behind that of spin-cast devices, fabricated in an inert atmosphere and with carefully timed anti-solvent dripping to control nucleation and growth. In this study, we demonstrate formamidinium (FA)-dominated mixed-halide mixed-cation perovskite solar cells fabricated by blade coating in ambient air (T = 23 °C and RH ≈ 50%) without the benefits of anti-solvent dripping or a moisture-free environment. We investigated the solidification process during blade coating of single-cation (FAPbI3) and increasingly complex mixed-cation mixed-halide (FA0.8MA0.15Cs0.05PbI2.55Br0.45, MA is methylammonium) perovskites in situ using time-resolved grazing incidence wide-angle X-ray scattering (GIWAXS). We found that the perovskite precursor composition and the blade coating temperature profoundly influence the crystallization mechanism and whether halide segregation occurs or not. The inclusion of Br− suppresses the non-perovskite 2H phase, promoting instead PbI2 together with the intermediate 6H phase and 3C phase of FAPbI2.55Br0.45. Addition of Cs+ suppresses these intermediates and promotes the direct crystallization of the perovskite 3C phase FA0.8MA0.15Cs0.05PbI2.55Br0.45 when coating at elevated temperature, unlike when anti-solvent dripping is used at room temperature. Through control of ink formulation and coating conditions, we demonstrate blade coated perovskite solar cells with a champion power conversion efficiency (PCE) of 18.20% as compared with FAPbI3 perovskites, which yield a PCE of 12.35% under similar conditions without the benefit of anti-solvent dripping. This study provides valuable insight into the crystallization pathway of mixed-cation mixed-halide formulations without anti-solvent dripping under high-temperature processing conditions that enable the translation of perovskites toward upscalable ambient manufacturing under high throughput conditions.
Introduction
Hybrid organic–inorganic metal-halide perovskites have impressed the semiconductor community thanks to their remarkable optoelectronic properties, including low exciton binding energy, a sharp absorption onset, and a small Urbach energy despite being prepared via low-cost solution-processing approaches.1–4 Hybrid organic–inorganic metal-halide perovskites feature the characteristic AMX3 structure, where A is an organic or inorganic monovalent cation, most often methylammonium (MA+), formamidinium (FA+) or cesium (Cs+), M is a metal cation, such as lead (Pb2+) or tin (Sn2+), and X is a monovalent anion such as chloride (Cl−), bromide (Br−), iodide (I−), and a mixture.5,6 Hybrid perovskite thin films are most commonly processed through a one-step spin-coating method, whereby a precursor ink is cast onto a substrate and spun up, an anti-solvent is dripped at an opportune time and the resulting film is subsequently annealed to acquire the desirable perovskite phase. To date, anti-solvent dripping has been the primary approach applied to one-step spin-coating planar heterojunction small-scale perovskite solar cells with certified power conversion efficiency (PCE) now surpassing 25%.7 Recently, hybrid perovskite compositions have been predominantly based on FA+ as the major cation since the α-FAPbI3 perovskite has a bandgap of 1.48 eV, which is closer to the ideal bandgap of Shockley–Queisser single-junction solar cells (1.34 eV) than that of classical MAPbI3 (1.57 eV).8 However, the photo-active α-FAPbI3 perovskite is thermodynamically unstable in ambient air and tends to transform into a non-perovskite yellow δ-FAPbI3 phase. The incorporation of MA+, Cs+, and Rb+ has been demonstrated to alleviate the issue of phase instability and results in more stable and efficient multi-component perovskite solar cells.9,10 Moreover, mixed-cation (MA+, FA+, and Cs+) and mixed-halide (I− and Br−) based perovskite solar cells are more reproducible and thermally more stable with higher solar cell performance and have a wider antisolvent processing window.11,12Despite the current success of high-efficiency small-scale devices via spin coating combined with anti-solvent dripping, scalable printing methods are needed to address upscaling and manufacturability challenges including material and solvent waste, limitation to small-area fabrication, and overall lack of reproducible device-to-device performance.13–19 Scalable solution-processing techniques adapted to polycrystalline hybrid perovskite thin films to date include blade coating, slot-die coating, dip coating, spray-coating, and inkjet printing.16,19–25 To date, the blade coating process remains one of the most popular and facile implementations for fabricating high-quality hybrid perovskite thin films and effort has been made to improve the device efficiency and stability.13,15,26,27 For instance, Huang's group reported a high PCE of ≈19% for MA-rich mixed cation and halide (MA0.6FA0.38Cs0.02PbI2.975Br0.025) perovskite photovoltaics inside a N2 glove box.27 The device performance is highly related to the perovskite morphology and crystallization, which, in turn, influence optoelectronic properties such as defect and trap state densities.28–30 Despite the apparent simplicity of the blade coating process, the mechanisms underlying the film formation from precursor inks to solid-state thin films as well as the process–structure relationships are poorly understood. This is partly due to reliance upon ex situ characterization methods, which do not capture the phase transformation behaviour or the formation of intermediate phases. The ability to investigate film formation from the blade coating process via in situ techniques is likely to be an essential step toward enabling transition to scalable manufacturing.
For this purpose, in situ time-resolved grazing incidence wide-angle X-ray scattering (GIWAXS) has recently emerged as a powerful technique to investigate and track the microstructure evolution of perovskite precursor inks during solution-casting to form solid-state perovskite thin films and to investigate the crystallization distribution and orientation in real time. Gong et al. examined a classical MAPbI3 perovskite solution using in situ GIWAXS methods and identified the existence of sol–gel disordered nanoparticles, solvated crystalline intermediates, and the polycrystalline perovskite phase during mini-slot-die printing.31 Our group revealed, through in situ investigations of film formation during spin-coating and blade coating, that significant differences exist in the phase transformation pathway of MAPbI3 perovskite in these two processes.19,32,33 We further showed that the processing temperature mediates the structure, halide composition, and polymorph of the solvated intermediate phase, while high-temperature processing (>100 °C) circumvented the crystalline solvated phase entirely, thus changing the phase transformation behavior.19,33,34 FA-rich mixed cation and mixed halide perovskites (FA1−x−yMAxCsyPbI3−zBrz) have recently demonstrated superior device performance, stability and ease of processing as compared to classical MAPbI3 and FAPbI3, but they also benefit from the crucial anti-solvent dripping step which mediates their phase transformation and can kinetically suppress halide segregation.9,35 The incompatibility of dripping with blade coating and meniscus-guided coating techniques in general requires alternative means of control over the phase transformation of the perovskite phase.36 A comprehensive understanding of the phase transition including microstructural and phase evolutions is missing for conditions representative of such scalable manufacturing processes for these complex perovskite compounds. During blade coating, the elevated stage temperature has been found to affect the quality of hybrid perovskite thin films critically in the case of MAPbI3, which otherwise tends to crystallize through solvated intermediate phases that cause pinhole formation.19,32 However, the phase transition is drastically different in FA-rich perovskites, which form non-solvated intermediate hexagonal phases which promote halide segregation in mixed halide perovskites. We, therefore, seek to provide a rational pathway toward the translation of mixed halide mixed cation perovskites toward scalable manufacturing under ambient conditions.
Herein, we compare and contrast four representative perovskite systems (FAPbI3, FAPbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI3, and FA0.8MA0.15Cs0.05PbI2.55Br0.45) in terms of their dynamical microstructure evolutions and conversion processes during blade coating by performing in situ GIWAXS measurements. With this investigation, we unveil the relationship between the solidification pathway and the effect of the mixed cations and halides at different temperatures without resorting to anti-solvent dripping. We adopt the widely used nomenclature for metal oxide perovskites proposed by Gratia et al. to specify the crystal structure of phases associated with the formation of perovskite thin films.37 For example, intermediate phases such as hexagonal 2H, 4H, and 6H are often referred to as the δ phase, while the cubic 3C perovskite phase is often referred to as the α phase. The acronyms 2H, 4H, 6H, and 3C refer to the well-established Ramsdell notation widely used for describing oxide perovskite polytypes.38 In the case of FAPbI3, blade coating at 120 °C reveals the formation of a yellow non-perovskite 2H (100) phase formed within 10 s. By incorporating a small amount of Br− into FAPbI3, the 2H (100) phase is suppressed at the expense of the desired perovskite 3C (100) phase, but it forms weakly and is accompanied by PbI2 formation, pointing to halide segregation. Incorporation of MA+ and Cs+ into FAPbI3 ink (resulting in FA0.8MA0.15Cs0.05PbI3 composition) promotes the formation of the non-perovskite 2H (100) phase and perovskite 3C (100) phase from the disordered sol–gel precursor. By addition of Cs+ and MA+ into the system to obtain FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite ink – one of the most popular perovskite compositions yielding efficient solar cells nowadays – the undesired intermediate phases (2H, 6H, and PbI2) are suppressed when processing at elevated temperature and give way to the formation of the desired perovskite 3C phase exclusively without the need for anti-solvent dripping. Using these insights, we achieved maximum/average PCEs of 18.2%/16.4% for FA0.8MA0.15Cs0.05PbI2.55Br0.45 based on planar heterojunction n–i–p devices made with the blade coated hybrid perovskite films in ambient air with 50 R.H.%. In comparison we obtained maximum/average PCEs of 16.3%/14.1% for FA0.8MA0.15Cs0.05PbI3, 15.8%/13.9% for FA0.85MA0.15PbI2.55Br0.45, 13.9%/11.6% for FAPbI2.55Br0.45, and 12.4%/9.0% for FAPbI3.
Results and discussion
Crystallization behavior of FA1−x−yMAxCsyPbI3−zBrz inks via blade coating
Blade coating experiments were conducted using a custom-built setup described elsewhere,35,39 and were computer-controlled remotely while performing both in situ and ex situ GIWAXS measurements at the D1 beamline of the Cornell High Energy Synchrotron Source (CHESS).19,33,35,36,39–46 The integration time for individual GIWAXS snapshots was 0.2 s, allowing a frame rate of 5 Hz, sufficient to capture the phase transformation behaviour in blade coating experiments performed in this study.We began our investigations by comparing the blade coating of FA-dominated triiodide and mixed halide systems, including FAPbI3 and FA0.8MA0.15Cs0.05PbI3, as well as FAPbI2.55Br0.45 and FA0.8MA0.15Cs0.05PbI2.55Br0.45. All formulations were prepared with the same molarity (1.2 M) in a mixture of N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) solvents with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. In Fig. 1a–d, we present the time evolutions of 2D X-ray scattering intensity with respect to the scattering vector (q) for the perovskite ink formulations mentioned above. Representative 2D GIWAXS snapshots collected at 5, 20, and 60 s during blade coating at 120 °C of all perovskite ink formulations are also shown in Fig. S1.† The length of the scattering vector q is determined by using the following equation:
:1 volume ratio. In Fig. 1a–d, we present the time evolutions of 2D X-ray scattering intensity with respect to the scattering vector (q) for the perovskite ink formulations mentioned above. Representative 2D GIWAXS snapshots collected at 5, 20, and 60 s during blade coating at 120 °C of all perovskite ink formulations are also shown in Fig. S1.† The length of the scattering vector q is determined by using the following equation: 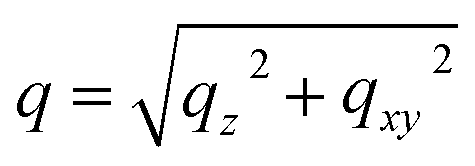 , where:
, where: 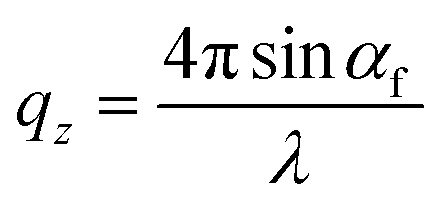 ,
, 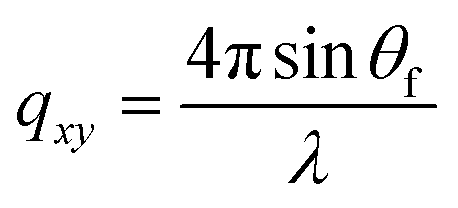 , and λ is the wavelength.43,47,48 The emergent waves with momentum can be described by the in-plane exit angle αf and the out-of-plane angle θf (with respect to the scattering plane).49 Blade coating experiments were performed on compact TiO2-coated glass substrates for 60 s at a coating speed of 25 mm s−1. The sample holder was pre-heated to 120 °C and the as-cast films were subjected to an additional annealing step at 100 °C for 10 minutes, after which an additional ex situ GIWAXS measurement was performed.
, and λ is the wavelength.43,47,48 The emergent waves with momentum can be described by the in-plane exit angle αf and the out-of-plane angle θf (with respect to the scattering plane).49 Blade coating experiments were performed on compact TiO2-coated glass substrates for 60 s at a coating speed of 25 mm s−1. The sample holder was pre-heated to 120 °C and the as-cast films were subjected to an additional annealing step at 100 °C for 10 minutes, after which an additional ex situ GIWAXS measurement was performed.
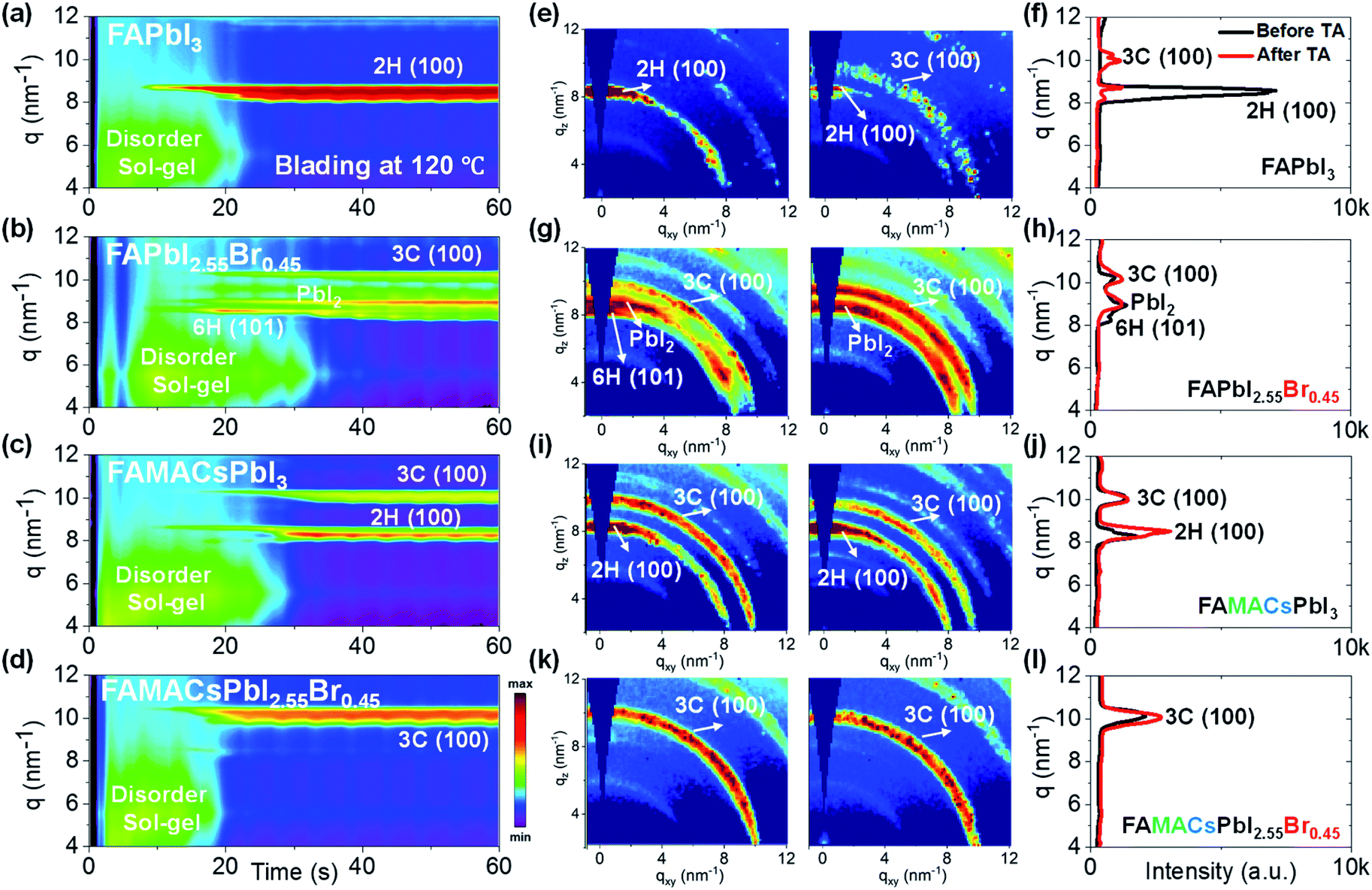 | ||
| Fig. 1 In situ 2D GIWAXS scattering evolution vs. time for various perovskite precursor formulations showing ink-to-solid transformation during blade coating for (a) FAPbI3, (b) FAPbI2.55Br0.45, (c) FA0.8MA0.15Cs0.05PbI3, and (d) FA0.8MA0.15Cs0.05PbI2.55Br0.45. Representative 2D GIWAXS snapshots taken at the final stage of blade coating as well as post-annealing (10 minutes at 100 °C) together with a plot of q vs. intensity are shown for (e and f) FAPbI3, (g and h) FAPbI2.55Br0.45, (i and j) FA0.8MA0.15Cs0.05PbI3, and (k and l) FA0.8MA0.15Cs0.05PbI2.55Br0.45. Scattering features associated with the disordered sol–gel phase and yellow non-perovskite 2H (100) phase (q = 8.5 nm−1) are identified along with the (100) diffraction of PbI2 (q = 9.0 nm−1) and perovskite 3C (110) phase diffraction (q = 10.2 nm−1). | ||
The time evolution of the scattering features of FAPbI3 (Fig. 1a) reveals a solvated, colloidal sol precursor during the first 20 s, as indicated by the formation of a scattering halo at low q values (4 to 8 nm−1).50 The hexagonal (non-perovskite) 2H (100) phase9,12,37,38 (q ≈ 8.5 nm−1) forms around 10 s, coexisting with the colloidal sol state for ∼10 s. The 2H (100) phase scattering intensifies significantly with the disappearance of the sol scattering signal for t > 20 s and is subsequently the only scattering feature observed in the as-cast film, indicating that the ink has solidified and converted to the 2H (100) phase. The 2H phase formed in the as-cast film tends to be highly textured with the c-axis normal to the substrate plane (Fig. 1e). Upon annealing, the FAPbI3 film converts partially to the perovskite 3C (100) phase and exhibits weak diffraction intensity (Fig. 1f). This is not surprising as the annealing temperature required for full conversion has been reported to be above 150 °C.51,52
Addition of a small amount of Br− into the formulation (nominally FAPbI2.55Br0.45) complicates the crystallization outcome considerably (Fig. 1b). The as-cast film yields three phases: the 6H (101) phase9,12,37,38 forms most prominently at first, along with PbI2 (q = 9.0 nm−1) and the perovskite 3C (100) phase9,12,37,38 (q = 10.2 nm−1). However, the 6H (101) phase seems unstable as its intensity weakens significantly after ∼30 s (Fig. 1g). Upon thermal annealing, the 6H (101) phase disappears completely, leaving behind a partially converted film composed of a mixture of crystalline PbI2 and 3C (100) perovskite phases (Fig. 1h). Addition, instead, of small fractions of MA+ and Cs+ into the triiodide formulation (nominally FA0.8MA0.15Cs0.05PbI3) (Fig. 1c) suppresses PbI2 formation and promotes the formation of the perovskite 3C (100) phase in addition to the 2H (100) phase (Fig. 1i). Thermal annealing does not appreciably alter the as-cast film's state and appears to promote 2H (100) phase formation (Fig. 1j).
In Fig. 1d, we reveal that simultaneously incorporating small amounts of MA+ and Cs+ into the previous FAPbI2.55Br0.45 ink (resulting in the FA0.8MA0.15Cs0.05PbI2.55Br0.45 nominal composition) tends to form the desired perovskite 3C (100) phase directly from the disordered sol precursor without forming intermediate non-perovskite phases, such as 2H and 6H phases and PbI2. It is important to recognize that the phase transformation of the solution by blade coating at elevated temperature is very different from that of spin coating and appears to emulate the effects of anti-solvent dripping without kinetic quenching.9,53 In contrast to the previous three Cs-free perovskite formulations, the Cs-containing formulation (FA0.81MA0.14Cs0.05PbI2.55Br0.45) starts to show the perovskite phase (3C (100) phase, q = 10.2 nm−1) at around 20 s during blade coating. The as-cast FA0.81MA0.14Cs0.05PbI2.55Br0.45 thin film exhibits a strong feature of the 3C (100) perovskite phase, which further increases its diffraction intensity after thermal annealing (Fig. 1k and l). Importantly, the non-perovskite 2H phase has a negligible signal, indicating that the 3C phase dominates the microstructure. Thus, we found that the main factors that could have an enormous impact on the microstructural evolution relate very much to the original composition of the perovskite ink before heat treatment, and that the presence of Cs+ and Br− promotes the formation of the perovskite 3C phase exclusively without other intermediate phases.
We summarize the in situ GIWAXS findings in Table 1. FAPbI3 formulation forms the non-perovskite yellow 2H (100) phase from a disordered sol precursor. Addition of a small amount of Br− (FAPbI2.55Br0.45) suppresses the 2H phase and generates the desired 3C perovskite phase together with undesirable PbI2 crystals, indicative of halide segregation in the as-cast film. Further addition of Cs+ (FA0.8MA0.15Cs0.05PbI2.55Br0.45) further suppresses the intermediate non-perovskite 2H phase and promotes single perovskite 3C phase formation.
| Nominal formula | FAPbI3 | FAPbI2.55Br0.45 | FAMACsPbI3 | FAMACsPbI2.55Br0.45 |
|---|---|---|---|---|
| Phases present | 2H | 6H, PbI2, 3C | 2H, 3C | 3C |
| Perovskite phase formed? | No | Yes | Yes | Yes |
| Non-perovskite phase(s) present? | Yes | Yes | Yes | No |
Impact of coating temperature on phase formation
The processing temperature was previously reported to play an essential role in the phase transformation behaviour of perovskite films.19,32–34 Blade coating at elevated temperature has yielded higher quality films and better photovoltaic performance than films prepared at a lower temperature. To understand the impact of the blading temperature on the phase conversion of mixed-halide mixed-cation systems, we have compared the phase transformation behaviours of FA0.8MA0.15Cs0.05PbI2.55Br0.45 formulations blade coated at low (60 °C) and high (120 °C) temperatures. Fig. 2a and b show 2D maps (with integration of each time slice) of the scattering intensity q (ordinate; 4 < q < 12 nm−1) tracked over time (abscissa; 0 < t < 360 s for blading at 60 °C and 0 < t < 60 s for blade coating at 120 °C, respectively). Representative 2D GIWAXS snapshots collected during blade coating at 60 and 120 °C are summarized in Fig. S2.†Fig. 2a shows the time evolution of phase transformation at 60 °C. Prior to ≈210 s, we observe the wet sol colloidal precursor, as indicated by the scattering halo at low q values (4 to 7 nm−1).19,32 Subsequently, two diffraction features appear close to the 3C perovskite: a Br-poor 3C perovskite α1 phase at q = 9.9 nm−1 and a Br-rich 3C perovskite α2 phase (at q = 10.3 nm−1). They are accompanied by the formation of PbI2 crystals (q = 9.0 nm−1) and the non-perovskite 4H phase (100)9,12,37,38 (q ≈ 8.36 nm−1). These results prove that the phase transformation behaviour promotes halide segregation in the perovskite phase, resulting simultaneously in Br-poor and Br-rich 3C structures as PbI2 crystals and the non-perovskite 4H phase form, respectively. This leads to perovskite 3C phases exhibiting different energetics and bandgaps, which are expected to act as recombination sites with a negative impact on the performance of solar cells. On the other hand, blade coating at 120 °C (Fig. 2b) forms the desired perovskite 3C (100) phase directly and exclusively, providing no apparent cause for halide segregation. The 3C phase forms directly from the disordered sol at around 20 s. The fact that non-perovskite phases are not observed suggests that thermal energy favours the nucleation and growth of the entropically stabilized mixed halide mixed cation 3C phase and that rapid drying of the solution due to hot casting does not quench the system into alternative phases.9,53 A schematic representation of the solidification pathway of FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite thin films at low and elevated temperatures is shown in Fig. 2c. At low temperature, slow drying leads to the formation of halide-segregated perovskite 3C phases (Br-poor and Br-rich), along with non-perovskite PbI2 crystals and the 4H (100) phase. The film achieves poor surface coverage as per scanning electron microscopy (SEM) images (Fig. S3†). At high temperature, the perovskite crystals nucleate and grow directly from the colloidal sol, forming compact films as confirmed in the SEM micrographs shown in Fig. 3a. Representative 2D GIWAXS snapshots at the final stage of blade coating (at 360 s for 60 °C and at 60 s for 120 °C) for the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite thin films are plotted in Fig. 2d. We further plot in Fig. 2e the intensity versus q (at 60 s) and the time-evolution of intensity at q = 10.2 nm−1, confirming that the perovskite 3C phase forms a single intense phase and does so very rapidly at 120 °C, whereas low-temperature processing leads to the formation of competing phases with distinct halide composition which cause halide segregation when converted to the perovskite phase.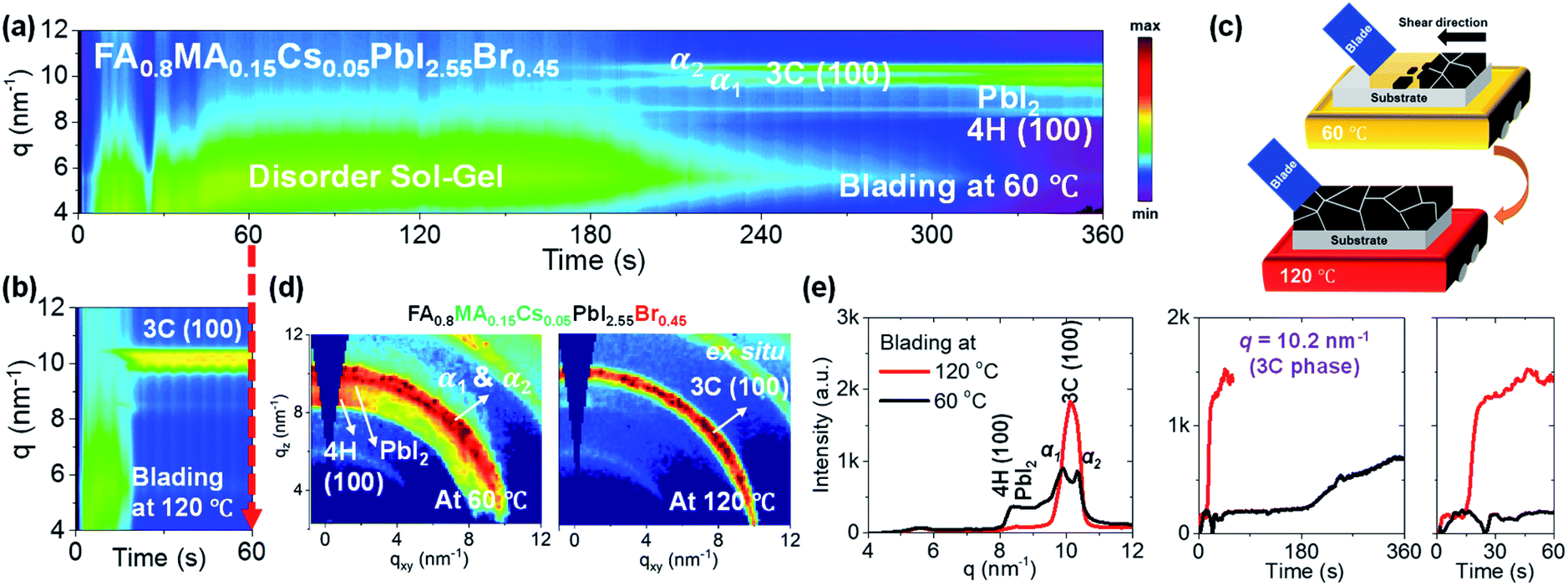 | ||
| Fig. 2 In situ 2D GIWAXS intensity map of the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite precursor inks showing ink-to-solid transformation during blade coating (a) at 60 °C and (b) 120 °C. (c) Schematic representation of the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite thin film formation mechanism at low 60 °C and high 120 °C blade coating temperatures. (d) Representative 2D GIWAXS snapshots taken at the final stage of blade coating perovskite films for FA0.8MA0.15Cs0.05PbI2.55Br0.45 at 60 °C and 120 °C, respectively. (e) Scattering vector q versus intensity distribution of the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite film at the final stage of blade coating and time evolution of the diffraction intensity related to the perovskite 3C (100) phase (q = 10.2 nm−1) for FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite ink at 60 °C and 120 °C. | ||
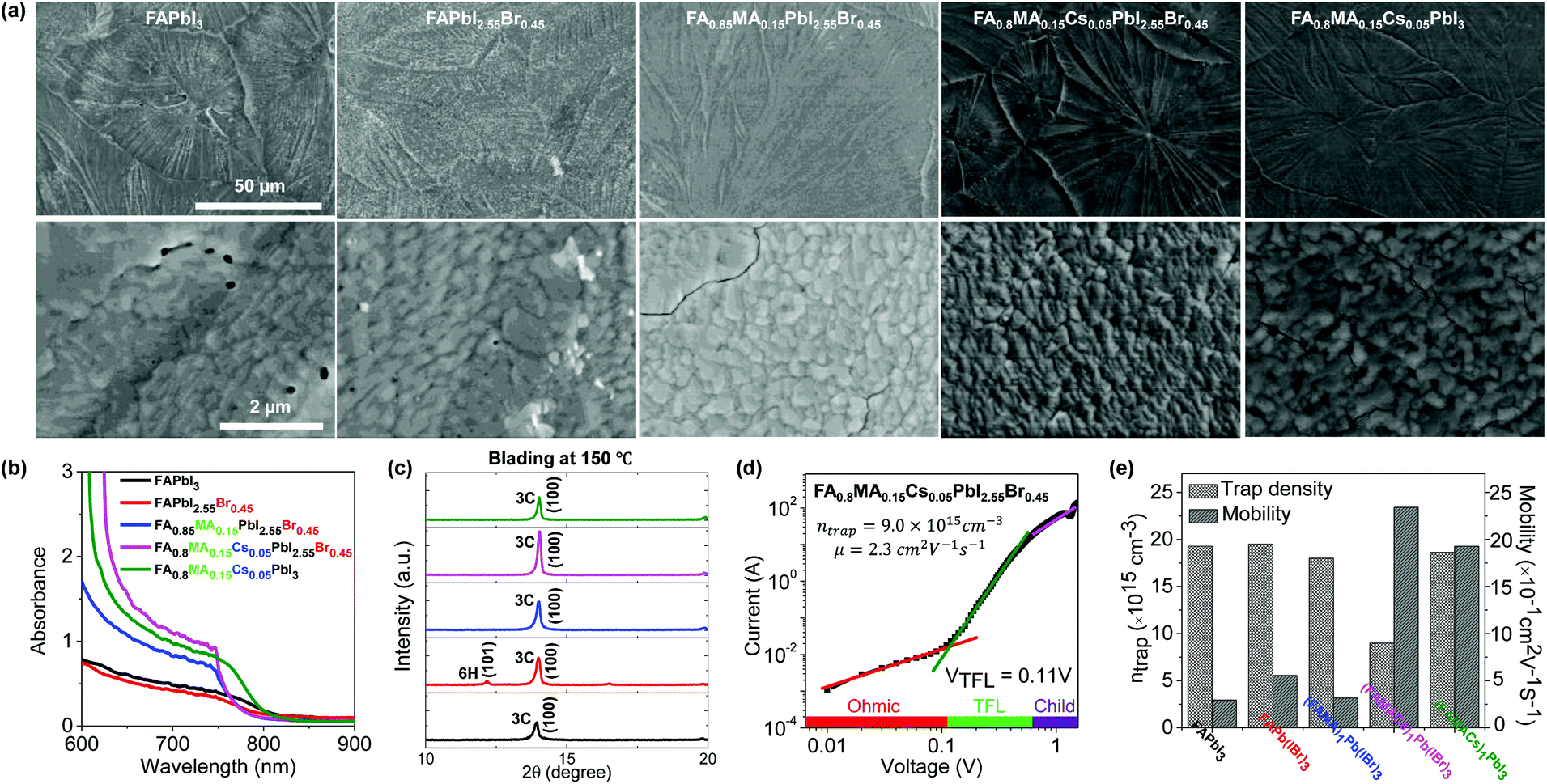 | ||
| Fig. 3 Comparison of FAPbI3, FAPbI2.55Br0.45, FA0.85MA0.15PbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 films via blade coating. (a) Top-view SEM images of perovskite films prepared using the blade coating method at 150 °C (top row shows low magnification while the bottom row shows higher magnification images). (b and c) Absorption spectra and XRD patterns of blade coated hybrid perovskite as-cast thin films with different precursor inks. (d) Dark I–V measurement of the electron-only device displaying VTFL kink point behavior for the representative FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite thin film. (e) Trap density and electron mobility. | ||
Previous UV-Vis absorbance studies of lead iodide and lead-bromide solutions in DMF and DMSO have revealed significant differences in the formation of lead-solvent complexes known as iodiplumbates and bromoplumbates. Lead bromide tends to complex more weakly with solvents and forms metal-rich clusters, whereas lead iodide complexes strongly with DMSO.54–56 It is reasonable to assume that a slow drying sol precursor containing a mixture of lead iodide and lead bromide in the presence of DMSO and DMF should promote halide segregation by preferential loss of solvent from the bromoplumbate sol while iodoplumbate sol remains highly solvated, thus promoting the formation of Br-rich and I-rich regions of the as-cast sol and leading to the formation of PbI2 and other phases. Hot casting enhances the drying kinetics while also providing thermal energy in favor of the phase transformation of the thermodynamically favored mixed halide, mixed cation perovskite phase.
Morphology and optoelectronic properties
We have investigated the resultant thin film morphology of all five formulations after coating and thermal annealing with the help of SEM. At low magnification, the films appear as continuous and compact films with large domains across the substrate (Fig. 3a). However, inspection at higher magnification reveals several pinholes in FAPbI3 and FAPbI2.55Br0.45 films and extended cracks between domains in FA0.85MA0.15PbI2.55Br0.45 and FA0.8MA0.15Cs0.05PbI3 films. Under these blade coating conditions, it appears that only FA0.8MA0.15Cs0.05PbI2.55Br0.45 films exhibit high structural quality, free of pinholes or cracks. The origin of these pinholes and cracks is not entirely understood but is not entirely surprising given the volume change that intermediate phases must undergo to transform into the 3C phase. Furthermore, the thermal expansion coefficient of hybrid perovskites tends to be an order of magnitude higher than that of typical inorganic substrates,57 causing cracking-induced stress relaxation during cooling, not unlike other semiconductors exhibiting large mismatch of thermal expansion coefficients.58We investigate the structure and properties of the above films (Fig. 3b, c, and S6†), as well as additional films prepared at an elevated coating temperature (150 °C, to overcome the challenges of the change processing translation from low to high humidity ambient conditions). This was found to promote the formation of higher quality films than at 120 °C, which we attribute tentatively and partly to humid ambient conditions in our lab (T = 23 °C and RH ≈ 50%), as well as to the higher processing temperature which promotes the formation of the thermodynamically favored phase and at the same time rapidly dries the solution, thus kinetically inhibiting precursor segregation. Optical photographs of all five representative blade coated perovskite thin films at 120 and 150 °C are summarized in Fig. S4a.† The spectral absorbance of FAPbI3 and FAPbI2.55Br0.45 films does not exhibit a clear absorption onset compared to the other perovskite systems. We attribute this to incomplete perovskite phase conversion after blade coating at 150 °C and possible decomposition in humid ambient air, as evidenced by the appearance of 2H, 6H and PbI2 phases in ex situ XRD analysis (Fig. 3c and S6†). By contrast, the FA0.8MA0.15Cs0.05PbI3 solid-state thin film exhibits a characteristic perovskite absorbance onset at around 800 nm, while the addition of a small quantity of Br− for the FA0.85MA0.15PbI2.55Br0.45 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 films resulted in a blue shift of the bandgap onset (absorbance onset is around 770 nm). The variation of cations was found to have little impact on the bandgap of the studied perovskites, but the addition of Cs+ into the perovskite structure increases the absorbance of FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite thin films at λ < 750 nm, owing to their propensity to form the 3C phase. The FA0.8MA0.15Cs0.05PbI2.55Br0.45 film shows the most intense 3C phase reflection (14.0°) among all perovskite cases studied, whereas FAPbI3 and FAPbI2.55Br0.45 films show evidence of non-perovskite intermediate 2H and PbI2 phases when blade coating at 120 °C. However, FA0.85MA0.15PbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI2.55Br0.45, and FA0.8MA0.15Cs0.05PbI3 perovskite solutions formed the desired perovskite 3C phase exclusively when blade coated at 150 °C, improving upon the outcome of deposition at 120 °C.
We have investigated how the composition of the perovskite film influences its trap state density and charge mobility using space charge limited current (SCLC) measurements. We collected the dark current–voltage characteristics for electron-only devices with a glass/fluorine-doped tin oxide (FTO)/compact titanium dioxide (c-TiO2)/blade coated perovskite/[6,6]-phenyl-C61-butyric acid methyl ester (PCBM)/silver (Ag) architecture for various perovskite thin films (see Fig. S5† for the other perovskites).50Fig. 3d illustrates the dark current–voltage characteristics of the representative mixed-cation mixed-halide perovskite FA0.8MA0.15Cs0.05PbI2.55Br0.45, indicating a linear ohmic response at a low bias (<0.11 V), a trap-filling regime (0.12 to 0.61 V) and a trap-free SCLC regime (>0.62 V). The trap state density was determined by the trap-filled limit voltage using the following equation: 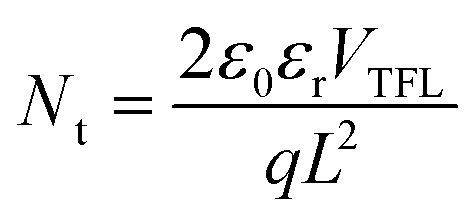 , where ε0 is the vacuum permittivity, εr is the relative dielectric constant, VTFL is the onset voltage of the trap-filled limit region, q is the elemental charge, and L is the thickness of the film.59 We found that the trap-state densities are similar for FAPbI3 and FAPbI2.55Br0.45 perovskite films, which are on the order of 2 × 1016 cm−3. Trap densities decrease slightly to 1.8 × 1016, 1.9 × 1016 and 0.9 × 1016 cm−3 for FA0.85MA0.15PbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI3, and FA0.8MA0.15Cs0.05PbI2.55Br0.45, respectively (Fig. 3e). These results indicate that the introduction of Cs+ and Br− simultaneously remediates defects in the perovskite thin film. This is consistent with in situ observations that a single perovskite 3C phase forms during blade coating, resulting in ion uniformity throughout the film, with the benefit of reduced charge recombination and possibly increased FF in solar cell devices.
, where ε0 is the vacuum permittivity, εr is the relative dielectric constant, VTFL is the onset voltage of the trap-filled limit region, q is the elemental charge, and L is the thickness of the film.59 We found that the trap-state densities are similar for FAPbI3 and FAPbI2.55Br0.45 perovskite films, which are on the order of 2 × 1016 cm−3. Trap densities decrease slightly to 1.8 × 1016, 1.9 × 1016 and 0.9 × 1016 cm−3 for FA0.85MA0.15PbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI3, and FA0.8MA0.15Cs0.05PbI2.55Br0.45, respectively (Fig. 3e). These results indicate that the introduction of Cs+ and Br− simultaneously remediates defects in the perovskite thin film. This is consistent with in situ observations that a single perovskite 3C phase forms during blade coating, resulting in ion uniformity throughout the film, with the benefit of reduced charge recombination and possibly increased FF in solar cell devices.
The intrinsic electron mobility was estimated using the SCLC method (Fig. 3e).60 The electron mobility is found to be 0.3 cm2 V−1 s−1 for the FAPbI3 film. Introduction of Br−, MA+, and Cs+ together enhances the electron mobility significantly. The electron mobility increases to 0.6 cm2 V−1 s−1 with the addition of Br− and further increases for FA0.8MA0.15Cs0.05PbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 films, to 1.9 and 2.3 cm2 V−1 s−1, respectively. These results further show that the microstructural benefits of Cs+ and Br− addition also impact the carrier mobility of the film. Meanwhile, the FA0.8MA0.15Cs0.05PbI2.55Br0.45 ink eliminates all intermediate non-perovskite phases and promotes the perovskite 3C phase formation at elevated temperature, emulating the effects of anti-solvent dripping at room temperature.9,53 SCLC measurements confirm that simultaneous introduction of Cs+ and Br− in the perovskite structure remediates trap states and enhances the carrier mobility in the perovskite thin film.
Photovoltaic device performance and stability
We fabricated planar n–i–p type perovskite solar cells in ambient air (T = 23 °C and RH ≈ 50%) using blade coated perovskite layers in the following configuration (Fig. 4c): glass/FTO/c-TiO2/blade coated perovskite/2,2′,7,7′-tetrakis-(N,N-di-pmethoxyphenylamine)-9,9′-spirobifluorene (sprio-OMeTAD)/gold (Au)/Ag. All solar cells were tested under standard AM 1.5G (air mass 1.5 global 1 sun) illumination in ambient air (T = 23 °C, RH ≈ 50%) with a scan rate of 0.1 V s−1, and the photovoltaic figures of merit are collected from 30–40 devices for each set of conditions (Fig. 4a), with the statistical distribution of PCE shown in Fig. 4d and summarized in Table S1.† The FAPbI3 device exhibits a PCE of 9.03 ± 2.44%, with a short-circuit current (JSC) of 20.86 ± 0.82 mA cm−2, a fill factor (FF) of 60.63 ± 1.87%, and an open-circuit voltage (VOC) of 0.97 ± 0.01 V. We observed statistically meaningful PCE improvements with a small amount of Br− halide addition for the FAPbI2.55Br0.45 devices to 11.64 ± 0.89%. Addition of a small amount of MA+ for the FA0.85MA0.15PbI2.55Br0.45 devices further improves the PCE to 13.89 ± 0.84%. Incorporation of Cs+ to yield the FA0.8MA0.15Cs0.05PbI2.55Br0.45 film leads to an enhancement of PCE to 16.40 ± 1.22%, which is the highest value among the five perovskite devices. Interestingly, without Br− (i.e., FA0.8MA0.15Cs0.05PbI3), PCE decreases to 14.10 ± 0.83%, which is a solid proof of the critical importance of Cs+ and Br−. This may be attributed to the introduction of Br− which remediates defects in the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite layer, resulting in higher electron mobility and lower trap density (Fig. 3e). Moreover, this is consistent with time-resolved investigations that a single perovskite 3C phase forms in the FA0.8MA0.15Cs0.05PbI2.55Br0.45 case (Fig. 1), with the benefit of higher performance in solar cells. Representative reverse and forward J–V scans of FAPbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 solar cells are shown in Fig. S7.† Due to the synergistic addition of Cs+ and Br−, it is clear that not only the PCE of devices increases but the hysteresis of J–V curves massively reduces in FA0.8MA0.15Cs0.05PbI2.55Br0.45 solar cells. The poor performance of the FAPbI3 device results from bad film quality as shown in Fig. 3a. The significant improvement in efficiency is mainly attributed to the enhancement in FF and JSC, partially due to improved film quality and optoelectronic properties (Fig. 3). These results highlight the need to have the coexistence of small amounts of Cs+ and Br−. We achieved a champion cell with a PCE of 18.20%, VOC = 1.09 V, JSC = 22.50 mA cm−2 and FF = 74.23% for the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite solar cell (Fig. 4b), which is one of the highest values reported to date for the FA-rich perovskite devices fabricated at a high speed under realistic conditions. The other representative J–V curves of the remaining perovskite devices under AM 1.5G illumination are shown in Fig. S4b.† We further demonstrated promising light-soaking stability of the cell, with a stabilized PCE of ≈17.70% under 8 minute continuous illumination (Fig. 4c). This stable illumination stability is due to suppressed phase conversion from the perovskite 3C phase to non-perovskite phases mainly assisted by the addition of Cs+ and Br− ions.61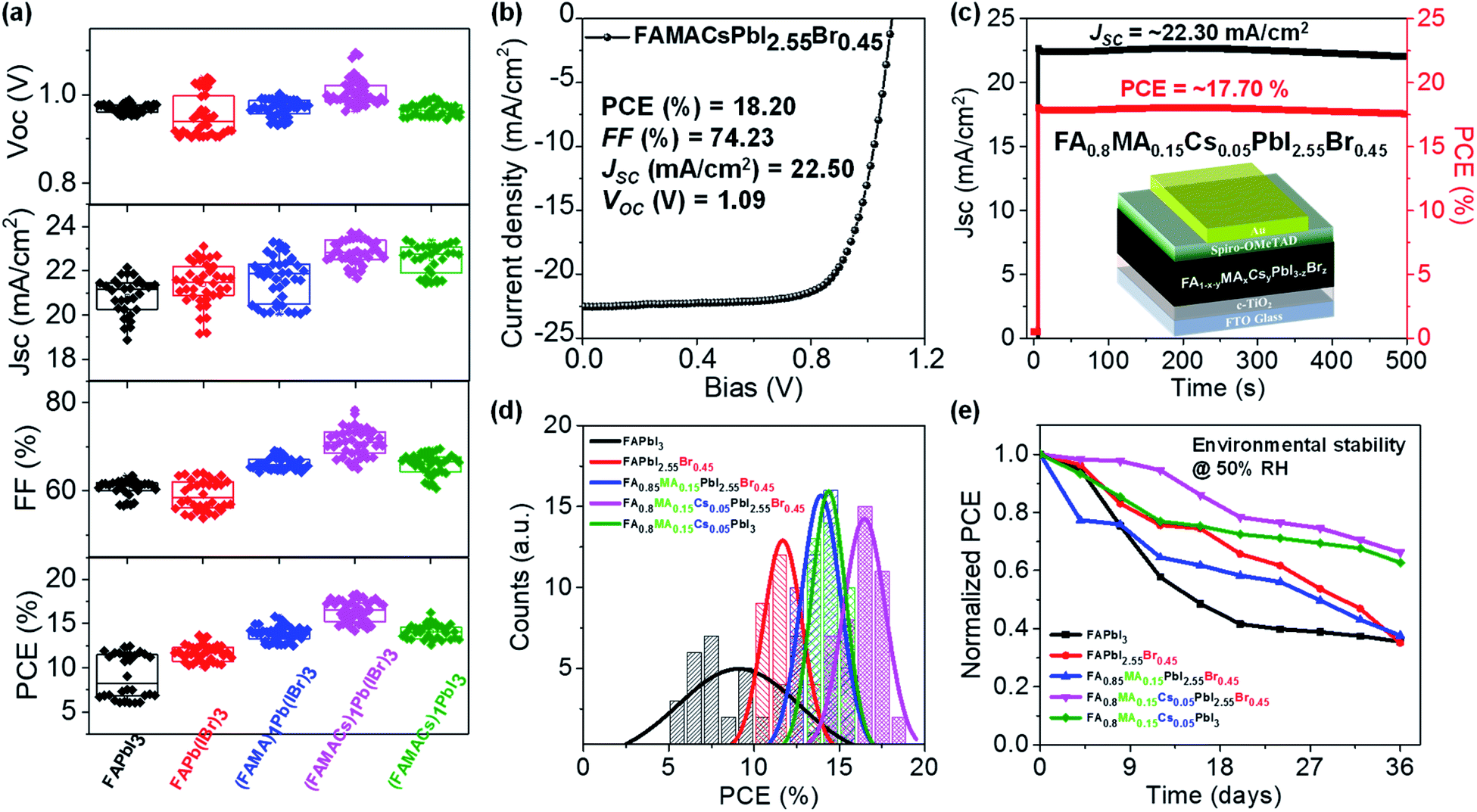 | ||
| Fig. 4 Solar cell architecture and characterization. (a) Statistics of 30–40 devices for each FA1−x−yMAxCsyPbI3−zBrz perovskite solar cell. (b) J–V curve for a peak efficiency of 18.20% obtained in the FA0.8MA0.15Cs0.05PbI2.55Br0.45 perovskite solar cell under AM 1.5G illumination. (c) The stabilized power output of JSC and PCE is monitored over time at the maximum power point with the solar cell architecture. (d) Comparison of photovoltaic PCE distribution employing blade coated perovskite solar cells prepared at 150 °C (approximately 30–40 devices for each composition were measured). (e) Recorded long-term environmental stability of the corresponding FAPbI3, FAPbI2.55Br0.45, FA0.85MA0.15PbI2.55Br0.45, FA0.8MA0.15Cs0.05PbI2.55Br0.45, and FA0.8MA0.15Cs0.05PbI3 perovskite solar cells exposed to an ambient environment with 50% relative humidity without encapsulation. | ||
The environmental stability of the corresponding perovskite solar cells was further evaluated. Solar cell devices without encapsulation were exposed to an ambient environment with ≈50% relative humidity in the dark at room temperature. The normalized PCEs versus time were recorded periodically and are reported in Fig. 4e. The FA0.8MA0.15Cs0.05PbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 devices exhibit significantly improved environmental stability in contrast to the other ones. For example, the PCEs retain 36%, 35%, 38%, 66% and 63% of their initial value for the FAPbI3, FAPbI2.55Br0.45, FA0.85MA0.15PbI2.55Br0.45 FA0.8MA0.15Cs0.05PbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 solar cells after 36 day ambient exposure under 50% RH conditions without encapsulation. The far superior ambient stability of FA0.8MA0.15Cs0.05PbI3 and FA0.8MA0.15Cs0.05PbI2.55Br0.45 devices is partially attributed to fewer phase impurities present in the as-prepared films61 and to better phase stability since the addition of Cs+ and Br− together with high-temperature processing helps to form an entropically stabilized phase.9,53
Conclusions
In summary, we systemically investigated blade coating of FA-based mixed-halide mixed-cation perovskite films to assess how they form without anti-solvent dripping but in the presence of elevated processing temperatures. We focused our attention on the roles played by Cs+ and Br− additions in the crystallization pathway in blade coating, associated halide segregation, microstructure, morphology, carrier mobility, trap state formation and solar cell performance. In situ time-resolved GIWAXS measurements performed during blade coating of FA-based perovskite formulations at different temperatures reveal a microstructural evolution that is extremely sensitive to the cation and halide formulation as well as to the coating temperature. FAPbI3 forms the non-perovskite 2H phase from the disordered colloidal sol. Addition of a small amount of Br− to form FAPbI2.55Br0.45 strongly suppresses the 6H phase and generates the desired perovskite 3C phase, but concurrently with the PbI2 phase. Meanwhile, further addition of Cs+ to form FA0.8MA0.15Cs0.05PbI2.55Br0.45 at elevated temperature eliminates all intermediate non-perovskite phases and promotes the perovskite phase formation. SCLC measurements confirm that simultaneous introduction of Cs+ and Br− in the perovskite structure remediates trap states and enhances the carrier mobility in the perovskite thin film. We go on to show that the blade coated solar cell performance is closely linked to the initial formulation of perovskite inks and the performance results obtained are supported by in situ GIWAXS observation of the microstructure evolution and whether a single perovskite or multiple non-perovskite phases form or not. FA0.8MA0.15Cs0.05PbI2.55Br0.45 solar cells blade coated in ambient air yielded a peak efficiency of 18.20%. To the best of our knowledge, our study is the first one to use in situ GIWAXS to shed light on the role of cation (FA+, MA+, and Cs+) and halide (I−, Br−) mixing as well as the role of coating temperature in the crystallization pathway of mixed-halide mixed-cation perovskites during blade coating. This insight provides an in-depth understanding of perovskite ink drying and phase transformation without anti-solvent dripping, which is critical and very meaningful for perovskite ink design in the future to achieve improved reproducibility in low-cost and large-scale manufacturing of hybrid perovskite photovoltaics.Author contributions
M.-C. T. and A. A. designed and performed most of the time-resolved investigations. Y. F. and X. C. conducted device fabrication and measurements of optoelectronic properties and SEM analysis. A. A., K. Z., and T. D. A. supervised the work. M.-C. T., D. B., R. L., and D.-M. S. assisted with in situ GIWAXS measurements. H. D., K. W., S. L., S. D. W., and all the authors contributed to the writing and revisions of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the King Abdullah University of Science and Technology (KAUST), the North Carolina State University (NCSU), the National Key Research and Development Program of China (2017YFA0204800 and 2016YFA0202403), the National Natural Science Foundation of China (61604092, 61674098), the National University Research Fund (Grant No. GK261001009, GK201603055), the 111 Project (B14041), and the National 1000 Talents Plan Program (1110010341). GIWAXS measurements were performed at the D-line at the Cornell High Energy Synchrotron Source (CHESS) at Cornell University. CHESS is supported by the NSF via NSF Award DMR-1332208.References
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2015, 9, 106–112 CrossRef CAS.
- A. Miyata, A. Mitioglu, P. Plochocka, O. Portugall, J. T.-W. Wang, S. D. Stranks, H. J. Snaith and R. J. Nicholas, Nat. Phys., 2015, 11, 582–587 Search PubMed.
- F. Hao, C. C. Stoumpos, R. P. Chang and M. G. Kanatzidis, J. Am. Chem. Soc., 2014, 136, 8094–8099 CrossRef CAS PubMed.
- T. Liu, K. Chen, Q. Hu, R. Zhu and Q. Gong, Adv. Energy Mater., 2016, 6, 1600457 CrossRef.
- V. Gonzalez-Pedro, E. J. Juarez-Perez, W. S. Arsyad, E. M. Barea, F. Fabregat-Santiago, I. Mora-Sero and J. Bisquert, Nano Lett., 2014, 14, 888–893 CrossRef CAS PubMed.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 RSC.
- N. R. E. Laboratory, Best Research-Cell Efficiencies Chart, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies-190416.pdf, accessed 05 May, 2019 Search PubMed.
- M. T. Weller, O. J. Weber, J. M. Frost and A. Walsh, J. Phys. Chem. Lett., 2015, 6, 3209–3212 CrossRef CAS.
- H. X. Dang, K. Wang, M. Ghasemi, M.-C. Tang, M. De Bastiani, E. Aydin, E. Dauzon, D. Barrit, J. Peng, D.-M. Smilgies, S. De Wolf and A. Amassian, Joule, 2019, 3, 1746–1764 CrossRef CAS.
- N. Pellet, P. Gao, G. Gregori, T. Y. Yang, M. K. Nazeeruddin, J. Maier and M. Gratzel, Angew. Chem., Int. Ed., 2014, 53, 3151–3157 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- K. Wang, M. C. Tang, H. X. Dang, R. Munir, D. Barrit, M. De Bastiani, E. Aydin, D. M. Smilgies, S. De Wolf and A. Amassian, Adv. Mater., 2019, 31, e1808357 CrossRef PubMed.
- Y. Deng, Q. Dong, C. Bi, Y. Yuan and J. Huang, Adv. Energy Mater., 2016, 6, 1600372 CrossRef.
- J. H. Kim, S. T. Williams, N. Cho, C.-C. Chueh and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1401229 CrossRef.
- Z. Yang, C.-C. Chueh, F. Zuo, J. H. Kim, P.-W. Liang and A. K. Y. Jen, Adv. Energy Mater., 2015, 5, 1500328 CrossRef.
- Y. Deng, E. Peng, Y. Shao, Z. Xiao, Q. Dong and J. Huang, Energy Environ. Sci., 2015, 8, 1544–1550 RSC.
- Y. Deng, Q. Wang, Y. Yuan and J. Huang, Mater. Horiz., 2015, 2, 578–583 RSC.
- K. A. Bush, A. F. Palmstrom, Z. J. Yu, M. Boccard, R. Cheacharoen, J. P. Mailoa, D. P. McMeekin, R. L. Z. Hoye, C. D. Bailie, T. Leijtens, I. M. Peters, M. C. Minichetti, N. Rolston, R. Prasanna, S. Sofia, D. Harwood, W. Ma, F. Moghadam, H. J. Snaith, T. Buonassisi, Z. C. Holman, S. F. Bent and M. D. McGehee, Nat. Energy, 2017, 2, 17009 CrossRef CAS.
- Y. Zhong, R. Munir, J. Li, M.-C. Tang, M. R. Niazi, D.-M. Smilgies, K. Zhao and A. Amassian, ACS Energy Lett., 2018, 3, 1078–1085 CrossRef CAS.
- S.-H. Turren-Cruz, M. Saliba, M. T. Mayer, H. Juárez-Santiesteban, X. Mathew, L. Nienhaus, W. Tress, M. P. Erodici, M.-J. Sher, M. G. Bawendi, M. Grätzel, A. Abate, A. Hagfeldt and J.-P. Correa-Baena, Energy Environ. Sci., 2018, 11, 78–86 RSC.
- M. Adnan and J. K. Lee, Sci. Rep., 2018, 8, 2168 CrossRef PubMed.
- P. Fan, D. Gu, G. X. Liang, J. T. Luo, J. L. Chen, Z. H. Zheng and D. P. Zhang, Sci. Rep., 2016, 6, 29910 CrossRef CAS PubMed.
- Z. Li, T. R. Klein, D. H. Kim, M. Yang, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Rev. Mater., 2018, 3, 18017 CrossRef CAS.
- M. Park, W. Cho, G. Lee, S. C. Hong, M. C. Kim, J. Yoon, N. Ahn and M. Choi, Small, 2019, 15, e1804005 CrossRef PubMed.
- A. J. Huckaba, Y. Lee, R. Xia, S. Paek, V. C. Bassetto, E. Oveisi, A. Lesch, S. Kinge, P. J. Dyson, H. Girault and M. K. Nazeeruddin, Energy Technol., 2019, 7, 317–324 CrossRef CAS.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Energy, 2017, 2, 17038 CrossRef CAS.
- S. Tang, Y. Deng, X. Zheng, Y. Bai, Y. Fang, Q. Dong, H. Wei and J. Huang, Adv. Energy Mater., 2017, 7, 1700302 CrossRef.
- J. Sun, J. Wu, X. Tong, F. Lin, Y. Wang and Z. M. Wang, Adv. Sci., 2018, 5, 1700780 CrossRef PubMed.
- J. M. Ball and A. Petrozza, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- Y. Hu, E. M. Hutter, P. Rieder, I. Grill, J. Hanisch, M. F. Aygüler, A. G. Hufnagel, M. Handloser, T. Bein, A. Hartschuh, K. Tvingstedt, V. Dyakonov, A. Baumann, T. J. Savenije, M. L. Petrus and P. Docampo, Adv. Energy Mater., 2018, 8, 1703057 CrossRef.
- Q. Hu, L. Zhao, J. Wu, K. Gao, D. Luo, Y. Jiang, Z. Zhang, C. Zhu, E. Schaible, A. Hexemer, C. Wang, Y. Liu, W. Zhang, M. Gratzel, F. Liu, T. P. Russell, R. Zhu and Q. Gong, Nat. Commun., 2017, 8, 15688 CrossRef CAS PubMed.
- R. Munir, A. D. Sheikh, M. Abdelsamie, H. Hu, L. Yu, K. Zhao, T. Kim, O. E. Tall, R. Li, D. M. Smilgies and A. Amassian, Adv. Mater., 2017, 29, 1604113 CrossRef PubMed.
- J. Li, R. Munir, Y. Fan, T. Niu, Y. Liu, Y. Zhong, Z. Yang, Y. Tian, B. Liu, J. Sun, D.-M. Smilgies, S. Thoroddsen, A. Amassian, K. Zhao and S. Liu, Joule, 2018, 2, 1313–1330 CrossRef CAS.
- X. Zhang, R. Munir, Z. Xu, Y. Liu, H. Tsai, W. Nie, J. Li, T. Niu, D.-M. Smilgies, M. G. Kanatzidis, A. D. Mohite, K. Zhao, A. Amassian and S. Liu, Adv. Mater., 2018, 30, 1707166 CrossRef PubMed.
- G. Giri, R. Li, D. M. Smilgies, E. Q. Li, Y. Diao, K. M. Lenn, M. Chiu, D. W. Lin, R. Allen, J. Reinspach, S. C. Mannsfeld, S. T. Thoroddsen, P. Clancy, Z. Bao and A. Amassian, Nat. Commun., 2014, 5, 3573 CrossRef CAS PubMed.
- Y. Fan, J. Fang, X. Chang, M.-C. Tang, D. Barrit, Z. Xu, Z. Jiang, J. Wen, H. Zhao, T. Niu, D.-M. Smilgies, S. Jin, Z. Liu, E. Q. Li, A. Amassian, S. Liu and K. Zhao, Joule, 2019, 3, 2485–2502 CrossRef CAS.
- P. Gratia, I. Zimmermann, P. Schouwink, J. H. Yum, J. N. Audinot, K. Sivula, T. Wirtz and M. K. Nazeeruddin, ACS Energy Lett., 2017, 2, 2686–2693 CrossRef CAS.
- L. S. Ramsdell, Am. Mineral., 1947, 32, 64–82 CAS.
- D. M. Smilgies, R. P. Li, G. Giri, K. W. Chou, Y. Diao, Z. A. Bao and A. Amassian, Phys. Status Solidi RRL, 2013, 7, 177–179 CrossRef CAS.
- Y. Zhang, P. Wang, M.-C. Tang, D. Barrit, W. Ke, J. Liu, T. Luo, Y. Liu, T. Niu, D.-M. Smilgies, Z. Yang, Z. Liu, S. Jin, M. G. Kanatzidis, A. Amassian, S. F. Liu and K. Zhao, J. Am. Chem. Soc., 2019, 141, 2684–2694 CrossRef CAS PubMed.
- D. Barrit, P. Cheng, M.-C. Tang, K. Wang, H. Dang, D.-M. Smilgies, S. F. Liu, T. D. Anthopoulos, K. Zhao and A. Amassian, Adv. Funct. Mater., 2019, 1807544 CrossRef CAS.
- T. Niu, J. Lu, M.-C. Tang, D. Barrit, D.-M. Smilgies, Z. Yang, J. Li, Y. Fan, T. Luo, I. McCulloch, A. Amassian, S. Liu and K. Zhao, Energy Environ. Sci., 2018, 11, 3358–3366 RSC.
- Y. Zhong, R. Munir, A. H. Balawi, A. D. Sheikh, L. Yu, M.-C. Tang, H. Hu, F. Laquai and A. Amassian, ACS Energy Lett., 2016, 1, 1049–1056 CrossRef CAS.
- M.-C. Tang, D. Barrit, R. Munir, R. Li, J. M. Barbé, D.-M. Smilgies, S. Del Gobbo, T. D. Anthopoulos and A. Amassian, Sol. RRL, 2019, 3, 1800305 CrossRef.
- T. Niu, J. Lu, X. Jia, Z. Xu, M. C. Tang, D. Barrit, N. Yuan, J. Ding, X. Zhang, Y. Fan, T. Luo, Y. Zhang, D. M. Smilgies, Z. Liu, A. Amassian, S. Jin, K. Zhao and S. Liu, Nano Lett., 2019, 19, 7181–7190 CrossRef CAS PubMed.
- Y. Fan, J. Fang, X. Chang, M.-C. Tang, D. Barrit, Z. Xu, Z. Jiang, J. Wen, H. Zhao, T. Niu, D.-M. Smilgies, S. Jin, Z. Liu, E. Q. Li, A. Amassian, S. Liu and K. Zhao, Joule, 2019, 3, 2485–2502 CrossRef CAS.
- L. A. Perez, K. W. Chou, J. A. Love, T. S. van der Poll, D. M. Smilgies, T. Q. Nguyen, E. J. Kramer, A. Amassian and G. C. Bazan, Adv. Mater., 2013, 25, 6380–6384 CrossRef CAS PubMed.
- J. L. Baker, L. H. Jimison, S. Mannsfeld, S. Volkman, S. Yin, V. Subramanian, A. Salleo, A. P. Alivisatos and M. F. Toney, Langmuir, 2010, 26, 9146–9151 CrossRef CAS PubMed.
- J. Schlipf and P. Müller-Buschbaum, Adv. Energy Mater., 2017, 7, 1700131 CrossRef.
- T. Niu, J. Lu, R. Munir, J. Li, D. Barrit, X. Zhang, H. Hu, Z. Yang, A. Amassian, K. Zhao and S. F. Liu, Adv. Mater., 2018, 30, e1706576 CrossRef PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- J. A. Steele, H. Yuan, C. Y. X. Tan, M. Keshavarz, C. Steuwe, M. B. J. Roeffaers and J. Hofkens, ACS Nano, 2017, 11, 8072–8083 CrossRef CAS PubMed.
- K. Wang, M. C. Tang, H. X. Dang, R. Munir, D. Barrit, M. De Bastiani, E. Aydin, D. M. Smilgies, S. De Wolf and A. Amassian, Adv. Mater., 2019, e1808357 CrossRef PubMed.
- A. Sharenko, C. Mackeen, L. Jewell, F. Bridges and M. F. Toney, Chem. Mater., 2017, 29, 1315–1320 CrossRef CAS.
- L.-M. Wu, X.-T. Wu and L. Chen, Coord. Chem. Rev., 2009, 253, 2787–2804 CrossRef CAS.
- J. C. Hamill, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2018, 3, 92–97 CrossRef CAS.
- C. Ge, M. Hu, P. Wu, Q. Tan, Z. Chen, Y. Wang, J. Shi and J. Feng, J. Phys. Chem. C, 2018, 122, 15973–15978 CrossRef CAS.
- Y. Mei, P. J. Diemer, M. R. Niazi, R. K. Hallani, K. Jarolimek, C. S. Day, C. Risko, J. E. Anthony, A. Amassian and O. D. Jurchescu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6739–E6748 CrossRef CAS PubMed.
- R. H. Bube, J. Appl. Phys., 1962, 33, 1733–1737 CrossRef CAS.
- Q. Han, S. H. Bae, P. Sun, Y. T. Hsieh, Y. M. Yang, Y. S. Rim, H. Zhao, Q. Chen, W. Shi, G. Li and Y. Yang, Adv. Mater., 2016, 28, 2253–2258 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta12890e |
| ‡ M.-C. Tang and Y. Fan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |