
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid production of block copolymer nano-objects via continuous-flow ultrafast RAFT dispersion polymerisation†
Sam
Parkinson
 ,
Stephen T.
Knox
,
Richard A.
Bourne
and
Nicholas J.
Warren
*
,
Stephen T.
Knox
,
Richard A.
Bourne
and
Nicholas J.
Warren
*
School of Chemical and Process Engineering, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, West Yorkshire, UK. E-mail: n.warren@leeds.ac.uk
First published on 29th April 2020
Abstract
Ultrafast RAFT polymerisation is exploited under dispersion polymerisation conditions for the synthesis of poly(dimethylacrylamide)-b-poly(diacetoneacrylamide) (PDMAmx-b-PDAAmy) diblock copolymer nanoparticles. This process is conducted within continuous-flow reactors, which are well suited to fast reactions and can easily dissipate exotherms making the process potentially scalable. Transient kinetic profiles obtained in-line via low-field flow nuclear magnetic resonance spectroscopy (flow-NMR) confirmed the rapid rate of polymerisation whilst still maintaining pseudo first order kinetics. Gel permeation chromatography (GPC) reported molar mass dispersities, Đ < 1.3 for a series of PDMAmx-b-PDAAmy diblock copolymers (x = 46, or 113; y = 50, 75, 100, 150 and 200) confirming control over molecular weight was maintained. Particle characterisation by dynamic light scattering (DLS) and transmission electron microscopy (TEM) indicated successful preparation of spheres and a majority worm phase at 90 °C but the formation of vesicular morphologies was only possible at 70 °C. To maintain the rapid rate of reaction at this lower temperature, initiator concentration was increased which was also required to overcome the gradual ingress of oxygen into the PFA tubing which was quenching the reaction at low radical concentrations. Ill-defined morphologies observed at PDAAm DPs close to the worm-vesicle boundary, combined with a peak in molar mass dispersity suggested poor mixing prevented an efficient morphological transition for these samples. However, by targeting higher PDAAm DPs, the additional monomer present during the transition plasticises the chains to facilitate formation of vesicles at PDAAm DPs of ≥300.
Introduction
Heterogeneous RAFT polymerisation technologies have been widely reported over the last 15 years since they allow rational production of block copolymer spheres, worms and vesicles via polymerisation-induced self-assembly (PISA).1–5 PISA is also highly versatile, since many different monomers can be polymerised in a wide range of solvents, directly resulting in block copolymer nanoparticles with tuneable size, morphology and functionality.6–9 These materials have extremely attractive properties which have resulted in a broad range of applications including as cell storage or growth media,10–12 viscosity modifiers,13 friction reducing agents,14,15 and as nano-reactors.16 This rapid growth in applicability means cost effective scale-up of PISA synthesised polymers is desirable. Otherwise manufacture on the scales required for incorporation in commercial products will be difficult, which is an issue often encountered for controlled radical polymerisations.17Decreasing reaction time (increased rate of reaction) while maintaining polymerisation control would be highly desirable for achieving the cost-effective preparation of block copolymers. An elegant approach of accelerating RAFT polymerisation was reported by Gody et al.18 who demonstrated it was possible to significantly increase the rate of RAFT solution polymerisation by combining fast propagating (high kp) monomers with an initiator that provides high radical flux. This can be achieved through rapid thermal decomposition of an initiator (in this case VA-044) at reaction temperatures well above the 10 h half-life. This approach is referred to as ultrafast RAFT and allows for the synthesis of soluble multi-block copolymers within minutes without compromising control over molecular weight or molar mass dispersity (Đ). Its potential has been recently demonstrated by conducting on extremely small scale in microvolume vials (2 μL)19 and with a commercially available parallel batch reactor system.20 To our knowledge, this ultrafast methodology has yet to be reported for a heterogeneous polymerisation. An important complication introduced when using ultrafast RAFT, is that the accelerated reaction rates can produce large exotherms. Not only can such temperature variations affect polymer quality,17,21 they can also be hazardous on scale-up.22 Within the relatively low volume reactions already reported,18,20,23 this was considered an advantage since it aided in deoxygenation by causing the solvent to boil.18
The efficient heat transfer within flow reactors has been shown to enable scalable production for highly exothermic processes without compromising product quality or safety.24,25 This approach also brings improved temporal control over the reaction, which results in significantly higher reproducibility26–28 and is considered a key technology in the drive for scalable, sustainable and precise polymer synthesis.29–35 There have been many reports on the use of flow reactors for conducting RAFT polymerisation: early work which focused on continuous-flow solution RAFT polymerisation demonstrated its potential for the scalable polymerisation of a wide variety of monomers achieving narrow Đ.36,37 Of late, flow platforms have been combined with new generation RAFT technologies such as photo-induced RAFT38 and oxygen tolerant PET-RAFT,39 while so-called reactor telescoping has enabled the preparation of multi block copolymers by sequential polymerisation of different monomers.23,40,41
Conducting heterogeneous RAFT polymerisation in flow reactors has also become more widely reported. For example, the RAFT emulsion polymerisation42 and RAFT dispersion polymerisation43 of methyl methacrylate (MMA) has been used to produce well-defined spherical particles. Although possible, it is widely reported that the production of higher-order morphologies (e.g. worms and vesicles) via RAFT aqueous emulsion polymerisation is difficult44 and to our knowledge there are no reports of successful synthesis of these in flow reactors. However, continuous-flow RAFT dispersion polymerisation can be used to generate higher order morphologies using both visible light mediated45–47 and thermally initiated RAFT polymerisation.48 Although reaction times as low as 45 minutes have been reported,47 they are typically of the order of hours, thus making scale-up using flow less desirable. Combining RAFT-PISA with ultrafast polymerisation offers the opportunity for increased productivity, cost-effectiveness and therefore accessibility of these advanced materials for practical applications.
Aqueous PISA of poly(dimethyl acrylamide)-b-poly(diacetone acrylamide) (PDMAm-b-PDAAm) is one of the most widely reported and predictable systems, allowing easy access to the full range of morphologies.48–56 Conveniently, this is an all-acrylamide system, meaning the monomers have high kp values and thus are ideally suited to ultrafast RAFT polymerisation.18 Herein, we evaluate whether ultrafast RAFT polymerisation of diacetone acrylamide (DAAm) in the presence of a PDMAmx macromolecular chain transfer agent can be conducted in a flow reactor. Conveniently, the ability to monitor the reaction conversion on short timescales using online Flow-NMR enables detailed kinetic monitoring despite short reaction times.49 Following this, we explore the production of a series of PDMAmx-b-PDAAmy diblock copolymer nano-objects.
Experimental
Materials
Diacetone acrylamide (DAAm, 99%) was purchased from Alfa Aesar (UK). 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044, 99%) was purchased from Wako Chemicals (USA). 3-((((1-Carboxyethyl)thio)carbonothioyl)thio) propanoic acid (CTTP, 90%) was purchased from Boron Molecular (USA). Dimethyl acrylamide (DMAm, 99%), 4,4′-azobis(4-cyanovaleric acid) (ACVA, 99%), deuterated methanol (CD3OD, 99.8%) and deuterium oxide (D2O, 99.9%) were purchased from Sigma Aldrich (UK).Continuous-flow reactor configuration
All experiments were performed on an automated flow platform (Fig. 1) consisting of a single Jasco PU-980 HPLC pump, a 5 mL PFA (2.5 m, 1.6 mm ID) reactor coiled around an aluminium block which was heated by 2 Elmatic Max K cartridge heaters controlled by a Eurotherm 3210 process controller, and a back pressure regulator placed downstream from the reactor coil. For the transient kinetic studies, a low-field benchtop NMR instrument was placed downstream from the reactor coil to directly measure monomer conversion.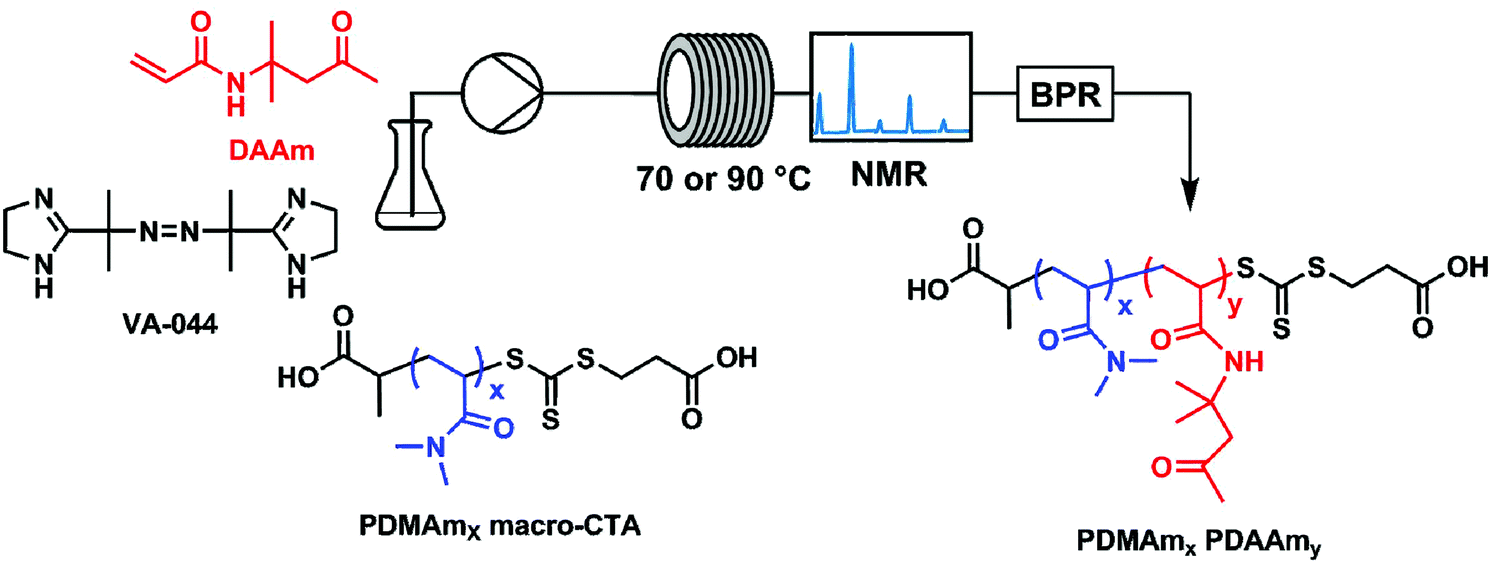 | ||
| Fig. 1 Continuous-flow reactor configuration and chemical structures for the synthesis of polydimethyl acrylamide (PDMAm)-b-polydiacetone acrylamide (PDAAm) nano-objects. | ||
Synthesis of PDMAmx macro-CTA
A typical synthesis of a PDMAmx macro-CTA with a target DP = 100 macro-CTA is as follows: dimethyl acrylamide (20 g, 0.2 mol, 100 eq.), CTTP (0.51 g, 2 mmol 1 eq.), ACVA (0.05 g, 40 μmol 0.1 eq.) were added to a round bottom flask and dissolved in water (48 ml) to give a 30% w/w reaction solution. A stirrer bar was added and then the flask was sealed and sparged with nitrogen for 20 minutes. The sealed flask was then immersed in an oil bath at 70 °C and left for 50 minutes after which it was removed from the oil bath and quenched by exposure to oxygen. 1H NMR analysis indicated 93% monomer conversion and GPC indicated the number average molecular weight, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 and molar mass dispersity, Đ = 1.11. End group analysis of the polymer indicated a final DP of 113. No further purification was performed and the macro-CTA solution was used as is for further chain extension experiments.
400 and molar mass dispersity, Đ = 1.11. End group analysis of the polymer indicated a final DP of 113. No further purification was performed and the macro-CTA solution was used as is for further chain extension experiments.
High resolution transient flow kinetic studies
A typical protocol for the transient kinetic profiling experiment was as follows: for PDMAm113-b-PDAAm50 synthesis, DAAm (3 g, 18 mmol, 50 eq.), PDMAm113 macro-CTA (3.6 g, 350 μmol 1 eq.), VA-044 (2.3 mg, 7 μmol, 0.02 eq.) were added to a round bottom flask and dissolved in water (26 ml) to give a 20% w/w reaction solution. To this, 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (0.6 g) was added to act as an internal standard. The flask was sealed and sparged with nitrogen for 20 minutes. A Jasco PU-980 HPLC pump inlet tube was then inserted into the sealed flask and the solution was pumped through a 5 ml, tubular PFA reactor at a flow rate 10 ml min−1 for 90 seconds, the flow rate was then reduced to 0.25 ml min−1 which equated to a mean residence time of 20 minutes. The outlet of the reactor was connected to a section of PFA tubing which passed directly through the NMR spectrometer.Continuous-flow synthesis of PDMAmx-b-PDAAmy copolymers
For a target composition of PDMAm113-b-PDAAm100, diacetone acrylamide (3 g, 18 mmol, 100 eq.), PDMAm113 macro-CTA (1.8 g, 180 μmol, 1 eq.), VA-044 (2.3 mg, 3.5 μmol, 0.02 eq.) were added to a round bottom flask and dissolved in water (26 ml) to give a 20% w/w reaction solution. The flask was sealed and sparged with nitrogen for 30 minutes. The HPLC pump inlet tube was then inserted into the sealed flask and the solution was pumped through a PFA tubular reactor at 90 °C with a retention time of 20 minutes. The polymer was collected in multiple vials at the reactor outlet.Batch synthesis of PDMAm46-b-PDAAm500 copolymers
DAAm (3 g, 18 mmol, 500 eq.), PDMAm113 macro-CTA (0.36 g, 35 μmol 1 eq.), VA-044 (0.2 mg, 0.7 μmol, 0.02 eq.) were added to a vial and dissolved in water (13 mL) to give a 20% w/w reaction solution. A stirrer bar was added to vial and the vial was the sealed. The sealed vial was sparged for 15 minutes and then placed in an oil bath with stirring at either 70 °C or 90 °C for 20 minutes.Dynamic light scattering measurements were conducted at 25 °C using a Malvern Instruments Zetasizer Nano series instrument. Light scattering was detected at 173° and hydrodynamic diameters were determined using the Stokes–Einstein equation, which assumes spherical, non-interacting particles.
Gel permeation chromatography measurements were conducted an Agilent 1260 Infinity system fitted with two 5 μm Mixed-C columns plus a guard column, a refractive index detector and an UV/Vis detector operating at 309 nm. DMF eluent contained 1.0% w/v lithium bromide (LiBr) at a flow rate of 1.0 mL min−1 at a temperature of 60 °C. A series of ten near-monodisperse poly(methyl methacrylate) standards (Mp ranging from 800 to 2200000 g mol−1) were employed as calibration standards in conjunction with RI detector for determining molecular weights.
Transmission electron microscopy (TEM) was conducted at 200 kV using a Tecnai F20 FEGTEM. TEM samples were prepared on 300 mesh continuous film copper grids at 0.1% w/w and stained with a 1% w/w uranyl acetate solution.
1H NMR spectra
Transient kinetic profiles were obtained using a Magritek Spinsolve Ultra 60 MHz benchtop NMR spectrometer. Upon exiting the flow reactor coil, the reaction mixture entered 1/8′′ PFA tubing which flowed directly through the spectrometer. A presaturation method was used to suppress solvent signals at 4.79 ppm (1 s saturation pulse of −65 dB, 7 μs excitation pulse, acquisition time of 6.4 s, repetition time of 10 s & number of scans was 2). All chemical shifts are reported in ppm (δ).For all other experiments polymer conversions were acquired using a Bruker 500 MHz. Samples were dissolved in D2O or CD3OD. All chemical shifts are reported in ppm (δ). The average number of scans accumulated per spectrum was 32.
Results and discussion
Prior attempts by our group, at performing “traditional” heterogeneous RAFT polymerisations in stainless-steel reactors have been successful, mainly producing spherical particles. However, despite successful synthesis of mixed phases of worms and vesicles, obtaining pure phases of these higher order morphologies was problematic.48 This was attributed to reactor fouling caused by an affinity of the polymer particles with the reactors stainless-steel tube walls. Here, the stainless-steel tubing was replaced with highly hydrophobic perflouroalkoxy (PFA) tubing (5 ml, 2.5 m, 1.6 mm ID). Despite its oxygen permeability,57 the material has already been used for successful synthesis of high order polymer nano-objects via PISA.45,46To obtain kinetic profiles of the continuous-flow ultrafast RAFT dispersion polymerisation experiments, transient kinetic studies were conducted for the chain extension of PDMAm113 macro-CTA with 50, 100 and 200 equivalents of DAAm (Fig. 2). Spectra were obtained by conducting flow-NMR at the reactor outlet.49 Conversion was then calculated (eqn (S1)†) using the integrals from the vinyl protons between 5.6 and 7.0 ppm, and the peak from the 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt internal standard at 0 ppm.
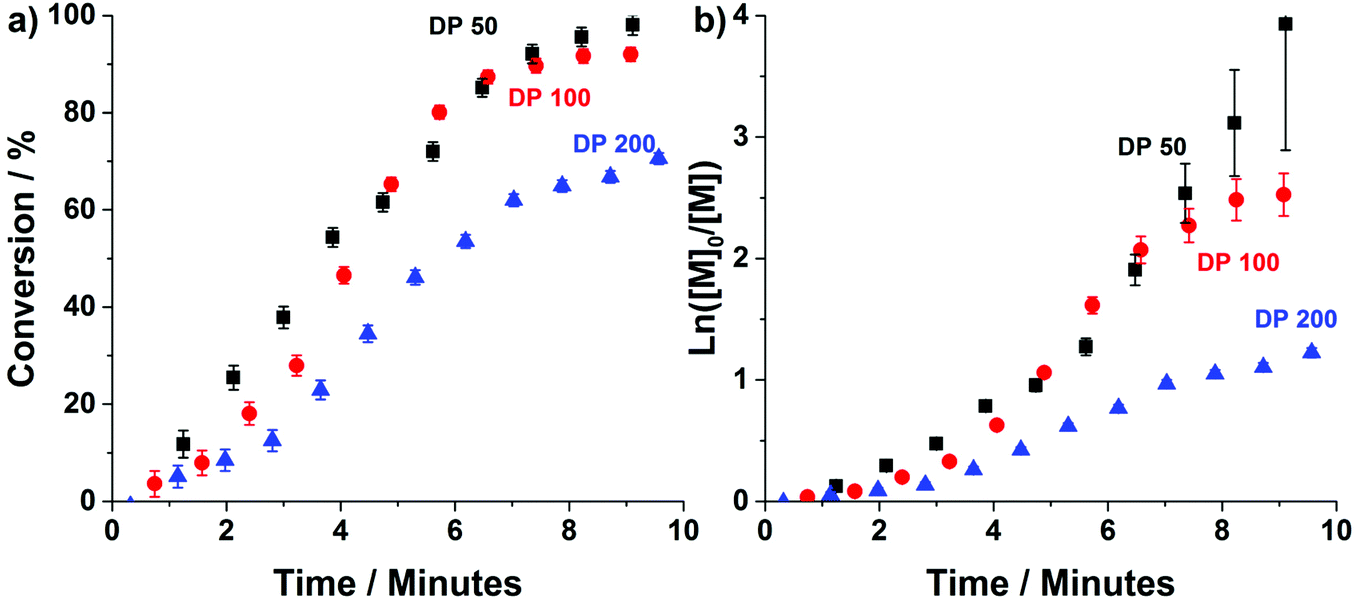 | ||
| Fig. 2 (a) Conversion vs. time and (b) semi-logarithmic rate plots for the continuous-flow RAFT aqueous dispersion polymerisation of diacetoneacrylamide (DAAm) using a PDMAm113 macro-CTA targeting PDAAm DPs of 50, 100 and 200. All reactions were conducted at 90 °C with total solids concentration of 20% w/w and [PDMAm113]:[VA-044] = 1:0.02. | ||
Conversions >90% are achieved within 8 minutes for target PDAAm DPs of 50 and 100, confirming that the high radical flux in this system results in the fast rate of reaction observed for ultrafast RAFT solution polymerisation.18 Despite the rapid rate of reaction, temporal resolution afforded by the flow-NMR makes it possible to discern the characteristic rate acceleration associated with the onset of micellisation during the PISA process.3 This occurs after approximately 3, 4 and 5 minutes for DPs 50, 100 and 200 respectively. In each example, this corresponds to a PDAAm DP of approximately 35, which is consistent with that observed for the same copolymer formulation prepared in flow using ACVA instead of VA-044.48 For both the slow (pre-nucleation) and fast (post-nucleation) regimes, the rate of reaction decreases with increasing DP due to reduced initiator concentration. For PDAAm DPs of 100 and 200, a third regime of rate deceleration is apparent towards the end of the reaction, at 8 minutes and 7 minutes respectively which relates to difference in cumulative radical generation for each of the target DPs (Fig. S1†). While a lower concentration of this magnitude would not usually alter the course of polymerisation (and indeed at short times shows a limited impact), the oxygen permeability of PFA tubing means that there is a critical radical flux below which the reaction is affected which is particularly relevant once a significant portion of the initiator has been consumed. (See also the later experiment relating to polymerisation at a lower temperature.)
From the kinetic data we can calculate a productivity of approximately 180 g per day (eqn (S2)†) using the flow methodology. This corresponds to a space–time yield (STY) of 36 g mL−1 day−1. Where 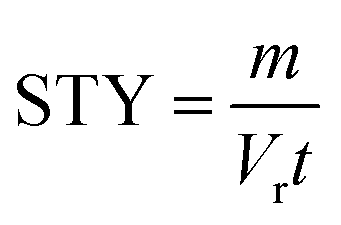 (m corresponds to the mass of product, Vr is the volume of reaction mixture and t is the reaction time). Commercially available flow reactors are typically available up to 40 mL in volume, which would allow production of >1 kg per day using this methodology assuming continuous operation for 24 hours.
(m corresponds to the mass of product, Vr is the volume of reaction mixture and t is the reaction time). Commercially available flow reactors are typically available up to 40 mL in volume, which would allow production of >1 kg per day using this methodology assuming continuous operation for 24 hours.
Conducting this synthesis beyond a moderate laboratory scale (e.g. in a 100 mL flask) would be unwise due to potential exotherms, so working above a STY 1 g mL−1 day−1 would be difficult without significant parallelisation of batch reactions.
To determine whether the traditional methods of exploring PISA formulations58 can be applied to this method, a series of PDMAm113-b-PDAAmy diblock copolymers were synthesised where y ranged from 50 to 200. To ensure high monomer conversion was attained in all these reactions, the residence time was set to 20 minutes, after which the product was collected at the reactor outlet and characterised by 1H NMR, GPC, DLS and TEM (Table S1†). For a target DP of 200, a cloudy liquid was obtained whereas for DPs of 150, 100, 75 and 50 clear gels were extruded from the reactor outlet. The properties of these dispersions may be explained by size and concentration dependant colloidal interactions.59
1H NMR studies (Fig. S2†) indicated high conversion (>90%) was achieved for target DPs of 50 and 100, however only 83% conversion was achieved for PDMAm113-b-PDAAm200. This can be explained by the decreased radical flux which reduces the system's tolerance to oxygen permeating through the PFA tubing.
Monomodal GPC chromatograms show a systematic shift to higher Mn with narrow dispersity and negligible macro-CTA contamination for all polymers (Fig. 3) further demonstrating the good RAFT control achieved despite fast reaction.
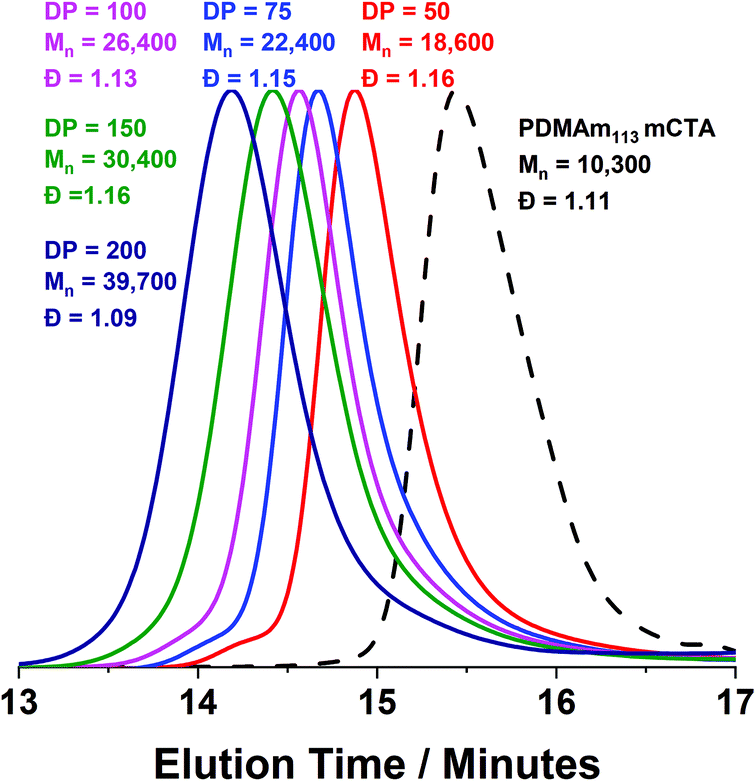 | ||
| Fig. 3 DMF GPC chromatograms for a series of PDMAm113-b-PDAAmy block copolymers synthesised in a flow reactor where y = 50 to 200. Polymerisation was conducted at 90 °C, 20% w/w solids and [PDMAm113]:[VA-044] = 50:1. GPC data was calibrated against a series of ten near-monodisperse poly(methyl methacrylate) standards. | ||
DLS and TEM studies enabled comparison of this system with that reported by Byard et al.55 who reported that PDMAmy DPs of above 65 produce only spherical particles due to kinetic trapping and steric stabilisation preventing sphere-sphere fusion which would be required to form higher-order morphologies.55 Analysing particles produced here by DLS (Fig. 4a) indicated that as PDAAm DP increases, there was a monotonic increase in Dh (31, 38, 40, 45 and 52 nm respectively) alongside low PDIs (<0.05), indicating a near-monodisperse population of particles. TEM images (Fig. 4b–d) confirmed that the monomodal peak in the DLS distribution corresponded to spherical particles with a particle size increase in accordance with increasing PDAAm DP.
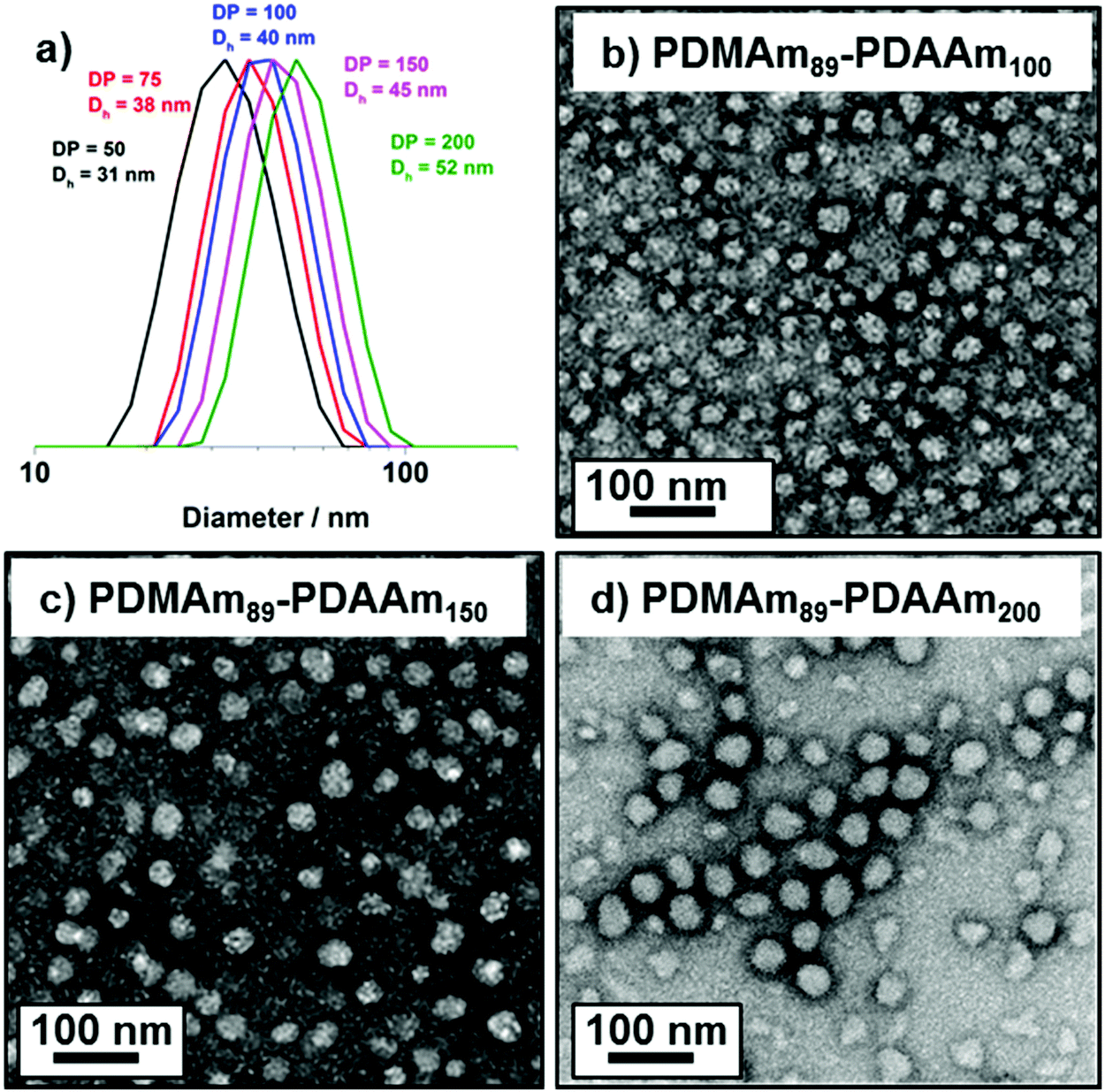 | ||
| Fig. 4 (a) DLS traces for PDMAm113-b-PDAAmy polymer nano-objects and representative TEM images for pure sphere morphologies block copolymers where y = (b) 100, (c) 150, (d) 200. All images were obtained from 0.1% w/w aqueous dispersions of diblock copolymer at pH 3. | ||
According to the reported phase diagram for PDMAmx-b-PDAAmy the synthesis of higher order morphologies requires a PDMAm macro-CTA with a mean DP below 65.55 A shorter stabilising block is necessary because if the stabilisng block becomes too long there is strong steric stabilisation upon micellar formation.2 This stabilisation prevents the fusion of spherical particles, which is the initial step in the formation of anisotropic worms.3
A series of polymers were therefore synthesised using a PDMAm46 macro-CTA. These attained high conversions (>90%) within 20 minutes up to a PDAAm DP of 100. Beyond this, the conversion decreased to 79% for a target PDAAm DP of 200, analogous to earlier observations with the PDMAm113 macro-CTA. GPC analysis indicated a systematic shift to higher Mn was observed in line with target DP in combination with low dispersities and good blocking efficiency (Fig. 5). DLS analysis of these polymers (Fig. 6a) provides limited information as such analysis is only appropriate for spherical morphologies. However, as PDAAm DP increases, there was an increase in Dh and PDI which likely indicates the presence of different (non-spherical) morphologies.60 Furthermore, DLS analysis of target PDAAm DPs of 150 and 200 polymers was not possible without passing the sample through a 1 μm syringe filter prior to analysis to remove precipitated polymer (Fig. S3†). For a final morphological classification, TEM images indicated a pure sphere phase at a PDAAm DP of 50, a majority worm phase at DP 75 and a mixed worm-vesicle phase at DP 100 (Fig. 6b–d). This is all in reasonable agreement with the reported phase diagram for this polymer system produced with ACVA in batch reactors.55 Yet for DP 150 and 200, which are predicted to be pure vesicle phases, a mixed phased consisting of worms, vesicles and lamellae was observed (Fig. S4†). This inability to synthesise a pure vesicle phase along with the formation of aggregates, leads to the conclusion that control over the polymer self-assembly is being compromised in the flow reactor despite achieving good control over the polymerisation.
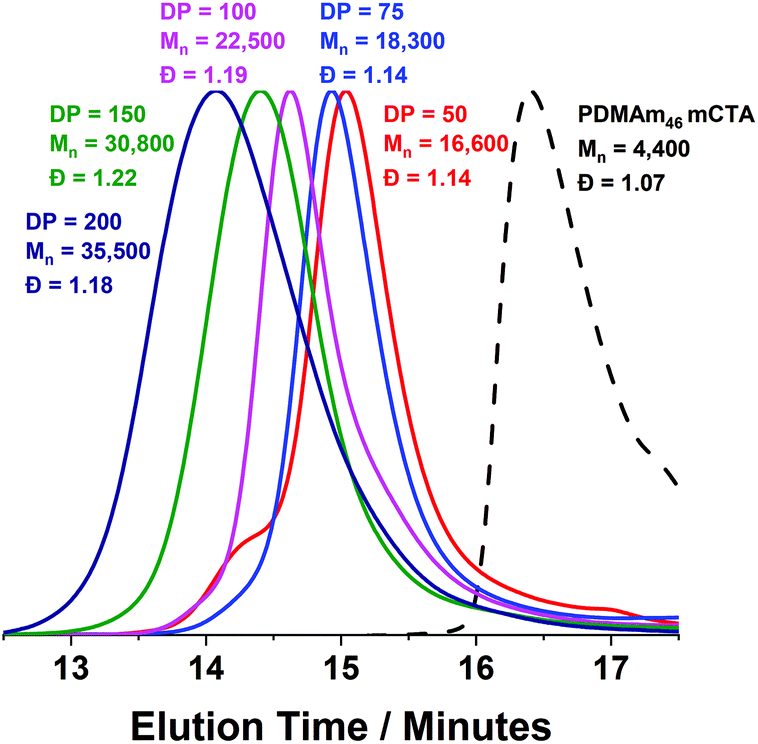 | ||
| Fig. 5 DMF GPC chromatograms for a series of PDMAm46-b-PDAAmy polymer nano-objects synthesised in a tubular reactor where y = 50 to 200. Reactions were conducted at 90 °C using RAFT aqueous dispersion polymerisation at 20% w/w solids and [PDMAm113]:[VA-044] = 50:1. GPC data was calibrated against a series of ten near-monodisperse poly(methyl methacrylate) standards. | ||
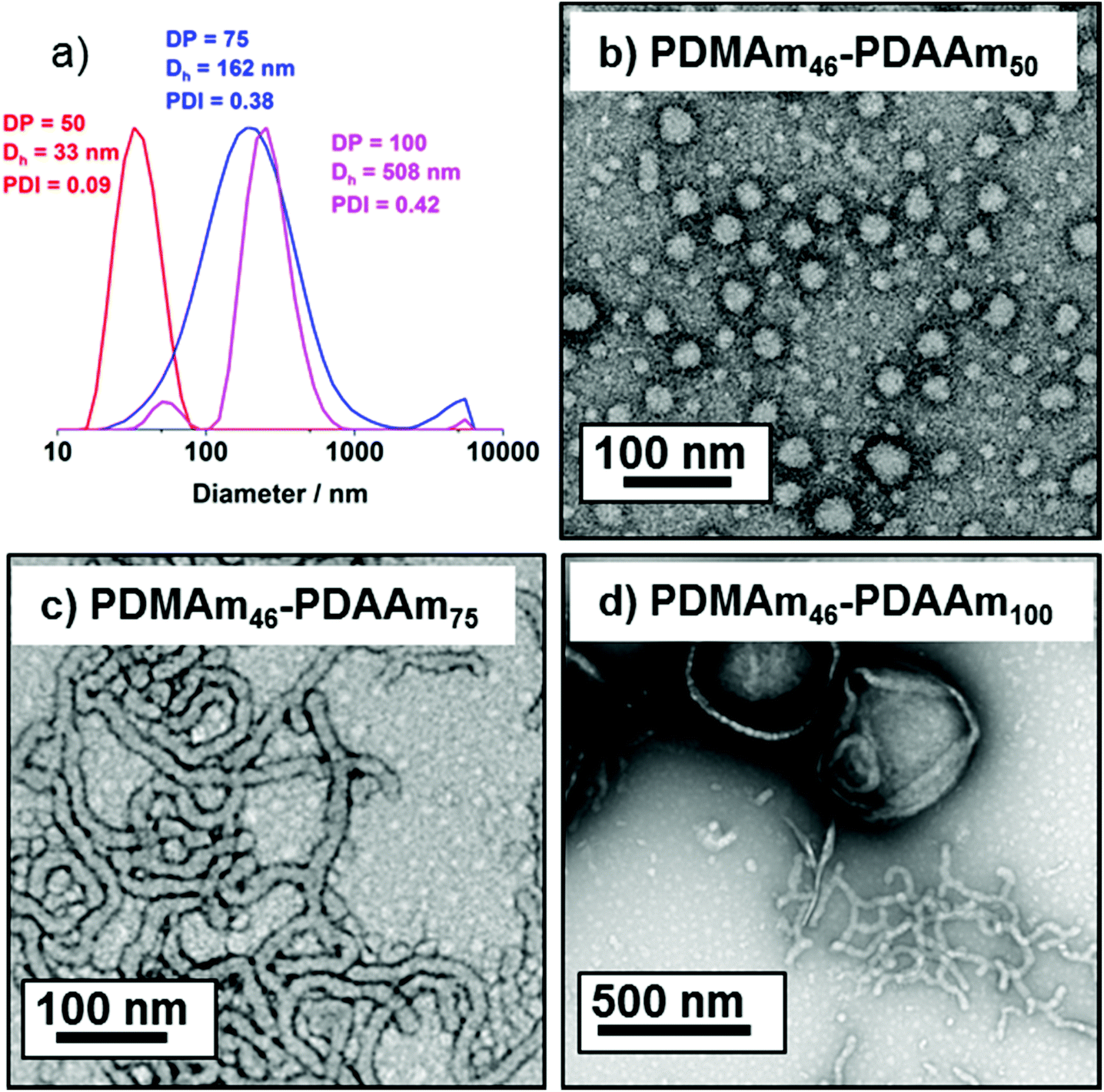 | ||
| Fig. 6 (a) DLS traces for PDMAm46-b-PDAAmy polymer nano-objects and representative transmission electron microscopy images for pure sphere morphologies block copolymers where y = (b) 50, (c) 75, (d) 100. All images were obtained from 0.1% w/w aqueous dispersions of diblock copolymer at pH 3. | ||
To determine the cause of this loss of control, a series of control experiments were performed: first, to determine whether conducting the polymerisation in a flow reactor was the cause, the synthesis was performed in batch at 90 °C. During this synthesis, phase separation occurred, where aggregates of polymer in precipitated from solution (Fig. S5a†). As the volume of material in any given area of the flow reactor is very low, the appearance of precipitate differs from batch – though phase separation was observed along the tubing in flow and the polymer aggregates formed were smaller. Temperature responsive behaviour has been reported previously for PDMAm-b-PDAAm block copolymer systems, generally involving transitions between worm and lamella phases from 20–70 °C.53 Therefore, the reaction temperature was reduced to 70 °C to attempt to eliminate any unwanted effect of the temperature.53 The resulting polymer solution was a homogenous white liquid, which would be expected from a vesicular dispersion (Fig. S5b†). The synthesis was then repeated in flow at this lower temperature with the hope of being able to form a pure vesicle phase. However, the sample collected at the reactor outlet was completely clear while NMR and GPC indicated that no polymerisation had occurred (Fig. S6†). This is attributed to the permeation of oxygen through the walls of the PFA reactor which was enough to quench the low concentration of radicals.36,57 To overcome this, the concentration of initiator was raised to give a similar radical flux to the reactions performed at 90 °C (see calculated initiator concentrations in Fig. S7†).
Using a VA-044 concentration of 0.25 mM, a series of polymers were synthesised at 70 °C, with DPs targeting a pure vesicle morphology.55 High conversions were obtained for all polymers in 20 minutes (>90%) until high target DPs (600 & 1000).55 GPC chromatograms show a shift to higher Mn (Fig. 7) with good blocking efficiency for all polymers apparent due to the absence of a peak due to residual macro-CTA. Molar mass dispersity initially increases up to DP 200 but then begins to decrease from 1.36 (at DP 200) to 1.17 (at DP 600). At a target DP of 1000, a much higher molar mass dispersity indicates a loss of RAFT control. DLS analyses of the polymers once again shows strange behaviour for DP 150 and 200, yet as DP increases, we observed ≥400 nm particles forming with PDI typical for similarly reported vesicle systems (Table S1 and Fig. S8†).55 Again, the target DP 1000 sample produced larger particles according to DLS, which is again comparable to previous observations for traditional (slower) PISA systems.61,62
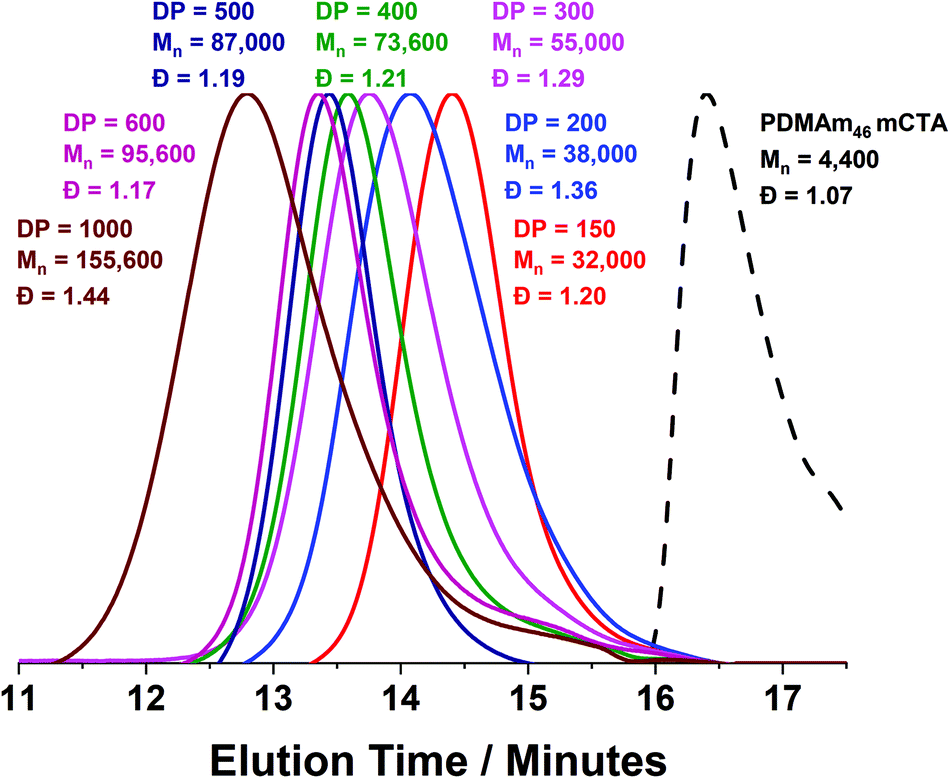 | ||
| Fig. 7 DMF GPC chromatograms for a series of PDMAm46-b-PDAAmy polymer nano-objects synthesised in a tubular flow reactor where y = 150 to 1000. Polymerisation was conducted at 70 °C using RAFT aqueous dispersion polymerisation at 20% w/w solids and [VA-044] = 0.25 mM. GPC data was calibrated against a series of ten near-monodisperse poly(methyl methacrylate) standards. | ||
TEM images (Fig. 8) confirmed the presence of a pure vesicle phase for all samples apart from PDMAm46-b-PDAAm200 which contained a mixture of large ill-defined aggregates, lamellar and vesicular structures.
 | ||
| Fig. 8 Representative transmission electron microscopy images of PDMAm46-b-PDAAmy block copolymers, synthesised in the flow reactor, where y = (a) 200, (b) 300, (c) 400, (d) 500, (e) 600 and (f) 1000. All images were obtained using 0.1% w/w of diblock copolymer at pH 3. | ||
The mechanisms through which block copolymers self-assemble offers some explanation for the observed aggregate formation around DP 200. It is well reported that evolution from spherical morphologies to worm/vesicle morphologies is driven by a reduction in the surface curvature,63 which occurs during PISA as the hydrophobic chain grows.64 However, as the hydrophobic chain increases in length it becomes more dehydrated and the mobility of the chain is reduced58 which can inhibit morphological evolution.65 This can be mitigated by adding a co-solvent;58 in this case, it is likely that residual monomer is facilitating morphological change. Poor hydrophobic chain solvation is likely a factor in the aggregate formation occurring for the target DP 200 system. When the target DP of the hydrophobic block is increased, there is more monomer present during the worm-vesicle phase transition thus facilitating formation of vesicle morphologies. Hence, it can be concluded that poor hydrophobic solvation is contributing to aggregate formation inside the reactor coil. However, aggregate formation for this block copolymer composition was not reported in batch, so there must be a second contributing factor.
Trends in PDMAm46-b-PDAAmy molar mass dispersity evolution (Fig. 9) offer additional insight into phenomena occurring which contribute to polymer aggregation. As PDAAm block length increases, the expected linear increase in molecular weight (Mn) is observed, indicating good RAFT polymerisation control. However a clear local maximum in dispersity is observed at a PDAAm DP of 200, which is uncharacteristic of the RAFT process. It has previously been reported that the rate of mixing has a considerable effect on the dispersity of polymers synthesised in flow reactors.66 It can be assumed that the PFA reactor described here operates in the laminar regime (since the Reynolds number, Re ≪ 2000) and mixing within these systems occurs solely through diffusion.67 Given the lack of mechanical agitation, the associated increase in the viscosity during the sphere-to-worm transition limits the diffusive mixing which in turn increases the dispersity of the polymers.68 This viscosity change also affects the morphological transitions; continuous agitation in batch provides additional kinetic energy which helps the system overcome the barrier for worm-vesicle transition. The lack of agitation in the flow reactor results in the polymeric nanoparticles getting trapped during the worm-to-vesicle transition. Precipitation then occurs due to the colloidal instability of the ill-defined particles. To confirm the importance of mixing, two control syntheses of PDMAm46-b-PDAAm200 were conducted in batch, with and without mechanical stirring to mimic physical and diffusive mixing respectively (Fig. S9†). Whilst both reactions yielded comparable polymers, upon dilution for DLS analysis, aggregates were visually observed for the diffusively mixed sample. Furthermore, a significant disparity in Dh was observed: the diffusively mixed polymerisation (550 nm) was nearly double that of the continuously mixed polymerisation (350 nm). While the effect in batch was not as drastic as in the flow reactor, the observations elude to the fact that during the flow synthesis both inadequate mixing in the reactor and poor hydrophobic core mobility contribute to precipitate formation for block copolymer compositions which should theoretically produce nano-objects near the worm-vesicle phase boundary.
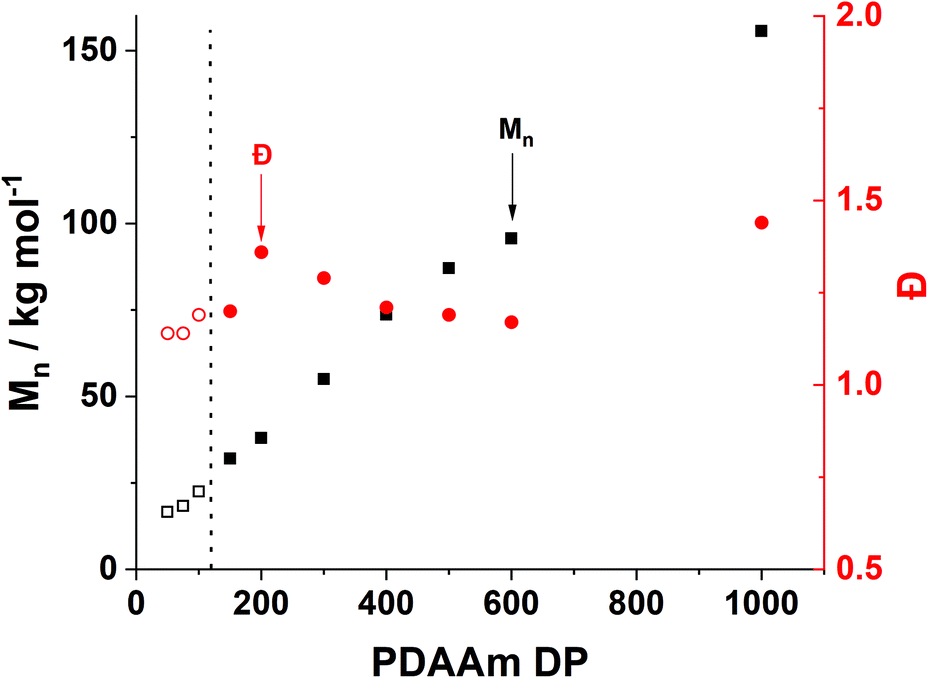 | ||
| Fig. 9 Evolution of molar mass and dispersity versus PDAAm DP (y) for a series of PDMAm46-b-PDAAmy polymers synthesised in the flow reactor with respect to increasing PDAAm DP. The residence time was set to 20 minutes and reactions were performed at either 70 °C (filled) or 90 °C (hollow). Reaction solution were made at 20% w/w total final solids and adjusted to pH 3. | ||
Conclusions
In summary, we have shown for the first time that it is possible to conduct ultrafast RAFT dispersion polymerisation to produce PDMAmx-b-PDAAmy nano-objects by PISA. Furthermore, by utilising continuous-flow reactors, the process is potentially scalable, since exotherms associated with such reactions (which would be hazardous in large batch reactors) can be dissipated. The rate of polymerisation was significantly increased by using the low temperature initiator, VA-044 at 90 °C as judged by Flow-NMR. This technique also revealed that high conversions were achieved within 8 minutes. Conversion and semi-logarithmic profiles obtained were characteristic of RAFT dispersion polymerisation, with a rate acceleration observed at a PDAAm DP of 35. However, the oxygen permeability caused a subtle decrease in rate at high conversions for PDAAm DPs of 100 and 200. Nevertheless, a series of well-defined PDMAmx-b-PDAAmy block copolymers were then synthesised with high conversion using a fixed residence time of 20 minutes. Depending on their composition, these polymers underwent PISA to form spherical, worm and mixed phase nano-objects, as determined by DLS and TEM. However, it was not possible to obtain a pure vesicle phase under these reaction conditions. Instead, it was necessary to lower the reaction temperature to 70 °C and increase the concentration of VA-044 initiator which maintains sufficient radical flux to overcome the ingress of oxygen through the PFA tubing. Under these conditions, a series of diblock copolymer vesicle dispersions could be obtained in under 20 minutes with no detrimental effect on molar mass dispersity. Some loss of control over particle formation and aggregate formation was still observed for polymer compositions around the reported worm/vesicle phase boundary. Through observation of molecular weight trends, it was determined that this aggregate formation to a complex amalgam of both the PISA mechanism and mixing within the flow reactor. We anticipate that the procedures described will be highly relevant for those wishing to efficiently produce block copolymers and block copolymer nano-objects over multiple scales.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for providing a studentship for SP through the Doctoral Training Account and support for NJW and STK through an EPSRC New Investigator Award (EP/S000380/1) and the University of Leeds for a University Academic Fellowship for NJW. RAB was supported by the Royal Academy of Engineering under the Research Chairs and Senior Research Fellowships scheme (RCSRF1920\9\38).References
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef CAS.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- H. Zhou, C. Liu, C. Gao, Y. Qu, K. Shi and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1517–1525 CrossRef CAS.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed.
- K. A. Simon, N. J. Warren, B. Mosadegh, M. R. Mohammady, G. M. Whitesides and S. P. Armes, Biomacromolecules, 2015, 16, 3952–3958 CrossRef CAS PubMed.
- M. Sponchioni, C. T. O'Brien, C. Borchers, E. Wang, M. N. Rivolta, N. J. W. Penfold, I. Canton and S. P. Armes, Chem. Sci., 2020, 11, 232–240 RSC.
- M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Angew. Chem., Int. Ed., 2017, 56, 1746–1750 CrossRef CAS PubMed.
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS PubMed.
- P. J. Docherty, M. J. Derry and S. P. Armes, Polym. Chem., 2019, 10, 603–611 RSC.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 RSC.
- T. Junkers, Macromol. Chem. Phys., 2017, 218, 1600421 CrossRef.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- J. Tanaka, P. Gurnani, A. B. Cook, S. Häkkinen, J. Zhang, J. Yang, A. Kerr, D. M. Haddleton, S. Perrier and P. Wilson, Polym. Chem., 2019, 10, 1186–1191 RSC.
- P. Gurnani, T. Floyd, J. Tanaka, C. Stubbs, D. Lester, C. Sanchez-Cano and S. Perrier, Polym. Chem., 2020, 11, 1230–1236 RSC.
- N. Micic, A. Young, J. Rosselgong and C. Hornung, Processes, 2014, 2, 58–70 CrossRef CAS.
- M. Fujita, Y.-I. Izato, Y. Iizuka and A. Miyake, Process Saf. Environ. Prot., 2019, 129, 339–347 CrossRef CAS.
- A. Kuroki, I. Martinez-Botella, C. H. Hornung, L. Martin, E. G. L. Williams, K. E. S. Locock, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 3249–3254 RSC.
- P. Barthe, C. Guermeur, O. Lobet, M. Moreno, P. Woehl, D. M. Roberge, N. Bieler and B. Zimmermann, Chem. Eng. Technol., 2008, 31, 1146–1154 CrossRef CAS.
- A. A. Kulkarni, Beilstein J. Org. Chem., 2014, 10, 405–424 CrossRef PubMed.
- C. Zhang, J. Zhang and G. Luo, J. Flow Chem., 2016, 6, 309–314 CrossRef CAS.
- I. Dobrosavljevic, E. Schaer, J. M. Commenge and L. Falk, Chem. Eng. Process., 2016, 105, 46–63 CrossRef CAS.
- A. Laybourn, A. M. López-Fernández, I. Thomas-Hillman, J. Katrib, W. Lewis, C. Dodds, A. P. Harvey and S. W. Kingman, Chem. Eng. J., 2019, 356, 170–177 CrossRef CAS.
- R. P. Andrews, Nature, 1986, 319, 429–430 CrossRef CAS.
- I. R. Baxendale, J. Chem. Technol. Biotechnol., 2013, 88, 519–552 CrossRef CAS.
- D. Cambie, C. Bottecchia, N. J. Straathof, V. Hessel and T. Noel, Chem. Rev., 2016, 116, 10276–10341 CrossRef CAS PubMed.
- G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708–5723 CrossRef CAS PubMed.
- S. V. Ley, Chem. Rec., 2012, 12, 378–390 CrossRef CAS PubMed.
- S. V. Ley and I. R. Baxendale, CHIMIA Int. J. Chem., 2008, 62, 162–168 CrossRef CAS.
- R. M. Myers, D. E. Fitzpatrick, R. M. Turner and S. V. Ley, Chem. – Eur. J., 2014, 20, 12348–12366 CrossRef CAS PubMed.
- C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo and S. H. Thang, Org. Process Res. Dev., 2011, 15, 593–601 CrossRef CAS.
- C. H. Hornung, X. Nguyen, G. Dumsday and S. Saubern, Macromol. React. Eng., 2012, 6, 458–466 CrossRef CAS.
- B. Wenn and T. Junkers, Macromolecules, 2016, 49, 6888–6895 CrossRef CAS.
- N. Corrigan, A. Almasri, W. Taillades, J. T. Xu and C. Boyer, Macromolecules, 2017, 50, 8438–8448 CrossRef CAS.
- E. Baeten, J. J. Haven and T. Junkers, Polym. Chem., 2017, 8, 3815–3824 RSC.
- M. Rubens, P. Latsrisaeng and T. Junkers, Polym. Chem., 2017, 8, 6496–6505 RSC.
- Z. Li, W. Chen, Z. Zhang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2015, 6, 1937–1943 RSC.
- J. Y. Peng, C. Tian, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2017, 8, 1495–1506 RSC.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- N. Zaquen, W. A. A. W. Azizi, J. Yeow, R. P. Kuchel, T. Junkers, P. B. Zetterlund and C. Boyer, Polym. Chem., 2019, 10, 2406–2414 RSC.
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165–5172 CrossRef CAS.
- N. Zaquen, H. Zu, A. M. N. B. P. H. A. Kadir, T. Junkers, P. B. Zetterlund and C. Boyer, ACS Appl. Polym. Mater., 2019, 1, 1251–1256 CrossRef CAS.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 RSC.
- S. T. Knox, S. Parkinson, R. Stone and N. J. Warren, Polym. Chem., 2019, 10, 4774–4778 RSC.
- H. Shi, T. Qiu, H. D. Ou-Yang, H. Xu, Q. Lu, Y. Zheng, K. Liu, L. He, L. Guo and X. Li, J. Colloid Interface Sci., 2019, 545, 220–230 CrossRef CAS PubMed.
- G. Mellot, P. Beaunier, J.-M. Guigner, L. Bouteiller, J. Rieger and F. Stoffelbach, Macromol. Rapid Commun., 2019, 40, 1800315 CrossRef PubMed.
- P. Biais, P. Beaunier, F. Stoffelbach and J. Rieger, Polym. Chem., 2018, 9, 4483–4491 RSC.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- S. J. Byard, A. Blanazs, J. F. Miller and S. P. Armes, Langmuir, 2019, 35, 14348–14357 CrossRef CAS PubMed.
- F. W. Giacobbe, J. Appl. Polym. Sci., 1990, 39, 1121–1132 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- S. Kaga, N. P. Truong, L. Esser, D. Senyschyn, A. Sanyal, R. Sanyal, J. F. Quinn, T. P. Davis, L. M. Kaminskas and M. R. Whittaker, Biomacromolecules, 2017, 18, 3963–3970 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- C. Tanford, J. Phys. Chem., 1972, 76, 3020–3024 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- J. van Stam, S. Creutz, F. C. De Schryver and R. Jérôme, Macromolecules, 2000, 33, 6388–6395 CrossRef CAS.
- J. Morsbach, A. H. E. Müller, E. Berger-Nicoletti and H. Frey, Macromolecules, 2016, 49, 5043–5050 CrossRef CAS.
- V. L. Streeter and E. B. Wylie, Fluid mechanics, McGraw-Hill, New York, 1985 Search PubMed.
- T. G. Hiss and E. L. Cussler, AIChE J., 1973, 19, 698–703 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00276c |
| This journal is © The Royal Society of Chemistry 2020 |