
The emerging applications of click chemistry reactions in the modification of industrial polymers
Mehmet
Arslan
 *a,
Gokhan
Acik
ab and
Mehmet Atilla
Tasdelen
*a
*a,
Gokhan
Acik
ab and
Mehmet Atilla
Tasdelen
*a
aDepartment of Polymer Engineering, Faculty of Engineering, Yalova University, 77100 Yalova, Turkey. E-mail: mehmet.arslan@yalova.edu.tr; tasdelen@yalova.edu.tr
bDepartment of Chemistry, Faculty of Sciences and Letters, Piri Reis University, 34940 Istanbul, Turkey
First published on 6th June 2019
Abstract
Polymer modification has been practiced since the very early technological development of human kind by transforming natural resources into value-added commodities. Ever since the middle of the last century in which petroleum-based polymers indisputably became widespread in diverse technological applications, the advances in polymer modification increasingly yielded more scientific attention to impute different and mostly desirable properties to industrial polymers. By cutting across the traditional boundaries of laboratory scale synthesis of reactive polymers, the functionalization and modification of large scale-produced commodity polymers might provide unique opportunities in the fabrication of advanced materials. In this sense, efficient chemical methodologies always have great relevance in the covalent manipulation of polymer substrates to impart desired properties. In the current manuscript, we aim to highlight the emerging applications of highly versatile click chemistry-based strategies in the modification of major industrial polymers by analysing the synthetic approaches and the resulting material properties.
 Mehmet Arslan | Mehmet Arslan obtained his undergraduate degree from Bilkent University, Department of Chemistry, Turkey. He completed his PhD in 2015 at Bogazici University, Turkey. He currently works at Yalova University, Department of Polymer Engineering, Turkey as an academician. His research primarily focuses on the fabrication of reactive polymeric materials for key biomedical applications such as drug delivery and biomolecular immobilization. He holds interest in developing new condensation and multi-component polymerization methods by adapting versatile synthetic organic transformations in polymer chemistry. |
 Gokhan Acik | Gokhan Acik graduated from Trakya University, earning a B.Sc. degree in the Department of Chemistry in 2009. He received his M.Sc. degree from the Chemistry Program of Istanbul Technical University in 2011 and Ph.D degree from the Department of Polymer Engineering of Yalova University in 2019. His research primarily focuses on polymer synthesis and their characterization, photopolymerization and electrospinning techniques. He is a co-author of more than 15 original research papers. |
 Mehmet Atilla Tasdelen | Dr Mehmet Atilla Tasdelen completed his B.Sc. (2000) degree at the Chemistry Department of Ege University, and M.Sc (2002) and Ph.D (2008) degrees at the Polymer Science and Technology Programme of Istanbul Technical University under the supervision of Prof. Yusuf Yagci. He joined Yalova University, Department of Polymer Engineering in 2010 as an assistant professor and he was promoted to full professor in 2018. His research is intensively focused on the design, synthesis and characterization of novel architecturally hybrid, complex, functional and well defined macromolecular structures via various polymerization techniques. He is a co-author of more than 100 original research papers and several book chapters. |
Introduction
Polymer modification refers to the chemical manipulation of polymers to induce functional diversity.1,2 The chemical tailoring of a polymer molecular structure is usually intended to impute enhanced properties to the processed material such as reactivity, thermal stability, biological resistance and response, compatibility, physical impact response, flexibility, rigidity and so on. The polymer modification is an interdisciplinary phenomenon that combines the chemistry of materials with materials science, engineering, physics, biochemistry and medicine to optimise the existing properties for the on-demand end-goal applications.Industrial polymers can be defined as chemical compounds that are produced on a large scale from natural or purely synthetic building blocks. In this respect, a wide variety of materials including commodity plastics, elastomers, thermosets, fibers, adhesives, and surface coatings constitute the major intermediate products that are employed in a tremendous variety of applications. From the very early introduction of the plant or animal based resins in material fabrication to highly-advanced applications of commodity polymers, the chemical manipulation tool in all along with synthesis, processing and modification steps results in attaining maximum effectiveness of final material properties. Considering the structural diversity of commodity polymers, a maximum effectiveness of the final material properties could be achieved with a good understanding of the structure–property relationship. This often requires a good correlation between synthesis, modification and processing steps in material fabrication.
The majority of industrial polymers are produced in large scale synthesis in chemical plants by using petroleum-derived monomers. In intended end-user applications, the final polymer properties are generally determined by largely physical processing of raw materials such as formulating, polymer blending, hot extruding or molding.3 To impart more value-added properties there is not much room to functionalize or modify the materials during the manufacturing process. Since the specialty polymers are specifically designed and produced to meet the demands, this inevitably brings sophisticated production design and high cost. In order to utilize the low-cost and abundant industrial commodity polymers in specific applications, the importance of post-polymerization modification emerges as a virtual tool to manipulate the chemical structure of the existing polymers. Most of the industrial polymers contain reactive functional groups on their polymer structure or such functionalities can be generated using different physical and chemical techniques. As such, this abundant and low-cost feed-stock offers great utility in the fabrication of materials with specific properties.4
The history of polymer modification has been unarguably shaped by the chemical knowledge since the early developments in polymer science. Starting from the 1940s, every decade has witnessed tremendous progress in developing breakthrough chemical reaction knowledge and better understanding of the structure–property relationship of materials.2 In the last few decades, various intriguing living/controlled polymerization techniques and ‘click’ chemistry methodologies have opened new avenues in both synthesis and functionalization of polymers. Judicious implementation of highly efficient chemical reactions in polymer modification leads to versatile material fabrication for the specific needs of applications.5
In this study, we aim to review the applications of some highly efficient and versatile chemical transformations, together denoted as click chemistry reactions, in design, synthesis, and applications of chemically-modified industrial or commodity polymers. Considering the high-volume production and low-cost amenities, the click tailoring of industrial polymers has gained increasing attention recently. By emphasizing the current utilization of click reactions in the on-demand modification of industrial polymers rather than custom-made lab-scale special synthesis (Fig. 1), common synthetic approaches, and their drawbacks and limitations have been summarized.
 | ||
| Fig. 1 Rationalizing modified polymer synthesis by uncovering click chemistry tailoring of industrial/commodity polymers. | ||
The applications of click chemistry methodologies in the modification of industrial polymers
Click chemistry methodologies comprise a series of chemical reactions that share some important aspects such as high reaction rates and conversions, mild reaction conditions and experimental simplicity as well as molecular selectivity, orthogonality and stereospecificity.6 In polymer chemistry applications of click reactions, these prominent features are essential benefits that led to triumph in macromolecular synthesis since large polymer chains are usually principal building blocks. In particular, in building complex polymer architectures7 and polymer-based nanofabrication,8 highly efficient click reactions have been proven as indispensable chemical tools. Each click transformation requires and thus allows different reaction conditions (such as metal-catalyzed or catalyst-free reaction mechanisms, thermal or UV-activations, reversible reaction coordinates, etc.) during the synthesis which could help in deciding optimum design and implementation.Among the most widely applied click chemistry methodologies in polymer science are cycloaddition reactions of alkyne and azide containing compounds. These reactions could proceed under transition metal-catalyzed conditions (copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC)) or can be driven by the intrinsic ring strain of cyclic alkynes (strain-promoted alkyne–azide cycloaddition (SPAAC)). Due to prominent advantages such as high selectivity, rapid and quantitative transformations and orthogonality, the CuAAC click reactions have gained immense attraction in polymer science for fabrication for various polymer constructs5,9–11 or functionalization of polymeric materials.12 The Diels–Alder (DA) and hetero Diels–Alder (HDA) reactions carry important aspects of click methodology due to which atom economic cycloaddition reactions between diene and dienophile counterparts are employed in carbon–carbon bond formation reactions. The high efficiency, catalyst-free reaction conditions and thermo-reversibility are prominent advantages of these reactions due to which polymer science has extensively exploited Diels–Alder reactions in the fabrication and functionalization of various macromolecular platforms.13–15 Thiol-X reactions that might infer the reactions of thiols with alkenes, alkynes, bromo compounds, epoxides or some fluoro compounds have gained acceptance in polymer chemistry procuring efficient, reliable and robust procedures.16–20 Diverse applications of these reactions include fabrication and functionalization of polymer architectures,21–24 polymer-coated nanoparticles,25,26 and crosslinked materials.27–30 The click methodologies that are applied in polymer science are not limited to these common reactions. A set of other reactions including hydrazone and oxime-based carbonyl chemistries31 and sulfur(VI) fluoride exchange reactions32,33 have also found employment for the synthesis and modification of macromolecular architectures. Apparently, the click chemistry concept is an ongoing chemical phenomenon that perpetually postulates new chemical reactions due to which polymer science shows promise for the utilization of these reactions.
On the other hand, the industrial or commodity polymers span a wide variety of materials including plastics, elastomers, fibers, resins, thermosets and surface coatings. The world production of the polymer industry almost reached 350 million tons in 2017 and both production and processing efforts have been gradually increasing annually (according to the data presented by the Association of Plastics Manufacturers in Europe, PlasticsEurope). Enormous production of these polymeric materials leads to diverse applications from daily-life commodities to high technology devices, and biomedical and nanomaterial applications. In this context, highly efficient click reactions have gained increasing attention for appropriate modifications of these polymers to impart new properties for the aimed use (Table 1). In the following parts, we aim to summarize the click chemistry-based modification and functionalization efforts of important industrial polymers by analysing their application-wise advantages and limitations.
| Polymers | Relevant click methodologies | Modification routes |
|---|---|---|
| Polyethylene | Thiol–ene | Modification of commercially available vinyl-terminated PE |
| Polypropylene | CuAAC | Installation of azide functionalities onto halogenated PP |
| Poly(vinyl chloride) | CuAAC | Direct installation of azide functionalities |
| Unsaturated polyolefins | Thiol–ene, triazolinedione click | Direct addition to backbone double bonds |
| Polyacrylonitrile | Azide–nitrile cycloaddition | Direct modification of side chain nitrile groups with azides |
| Poly(meth)acrylates | CuAAC, thiol–ene | Installation of clickable groups via transesterification |
| Poly(acrylic acid) | CuAAC | Installation of alkyne and azide groups via esterification |
| Poly(vinyl alcohol) | CuAAC, thiol–ene | Installation of clickable groups via transesterification |
| Poly(ethylene terephthalate) | CuAAC | Installation of clickable groups via esterification on chain-end carboxylic acids |
| Polysulfones | CuAAC | Installation of azide groups onto the chloromethylated backbone |
Polyethylene
As a durable polymer, PE is conveniently applied in various biomedical applications such as hip/knee replacement implants and cell/tissue supports. However, PE generally displays low wetting properties in aqueous media which entails appropriate modification of the polymer surface to improve its wettability and bioadhesion. The main difficulty arising here is the chemical structure of PE that possesses linear hydrocarbon chains with a lack of apparent reactive functionality. Although the surface treatment of PE with different oxidative techniques such as plasma or ion beams can induce some reactive species in the treated parts which undergo consecutive chemical reactions, this destructive method is not practically applicable in all cases.35 Therefore, a limited range of polyethylene products that bear reactive functionalities on polymer chains (such as ethylene/vinyl acetate copolymers) are industrially produced. Recently, a versatile strategy based on the synthesis of end-functional PE by using catalytic coordination polymerization has been developed.36 In laboratory synthesis, a range of PEs carrying clickable azido,37–39 alkyne40 and alkene41–43 end-groups or side chains were reported. In a study, Li and co-workers demonstrated the installation of trimethoxysilane (TMS) groups at vinyl-terminated PE via radical thiol–ene reaction (Fig. 2).44 It was demonstrated that the quantitative thiol–ene end-group modification could be achieved under simple experimental conditions. This polymer (TMS-PE) is subsequently used in the fabrication of nanocomposites with TMS-modified multiwall carbon nanotubes (TMS-MWNTs). Although there are specific studies that demonstrate the laboratory synthesis of PEs with either end-chain or side-chain clickable groups, there exists still a lack of batch and pristine clickable PEs with commercial availability.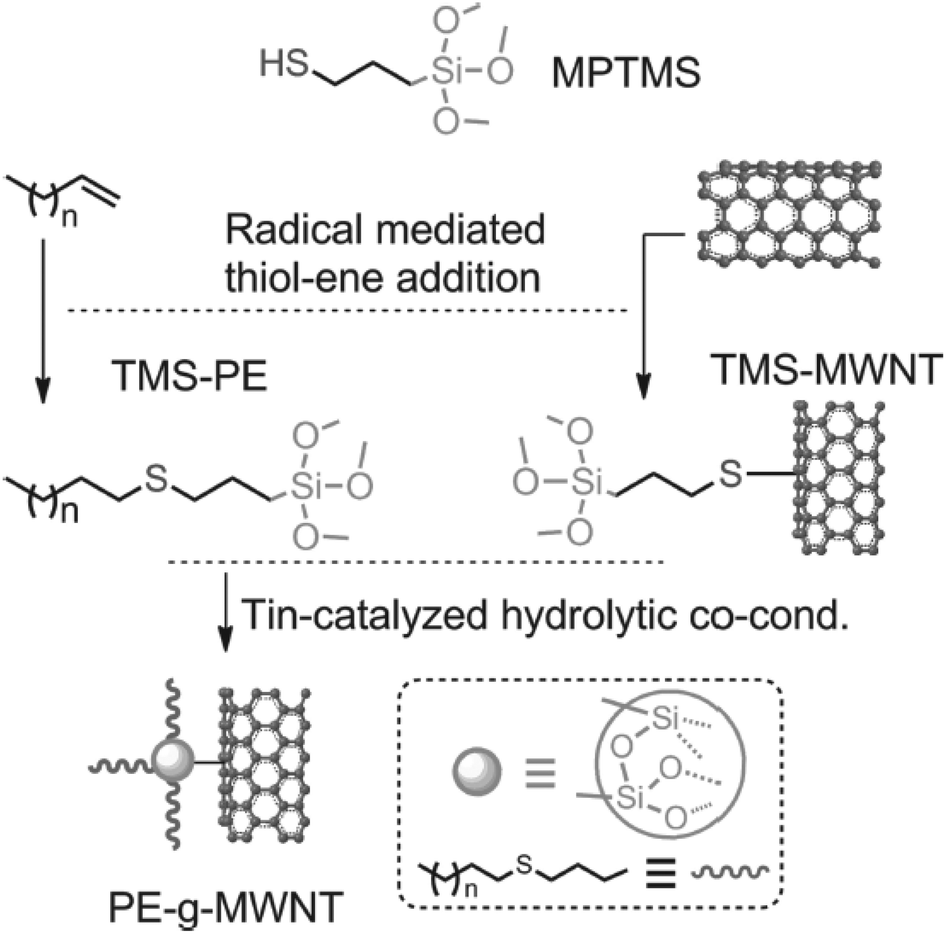 | ||
| Fig. 2 Thiol–ene end-group modification of vinyl-terminated PE and utilization of the functional polymer in MWNT-based nanocomposite fabrication. Reproduced from ref. 44 with permission from WILEY-VCH Verlag GmbH & Co., copyright [2016]. | ||
Polypropylene
Polypropylene (PP) is a thermoplastic polymer which offers a highly crystalline molecular structure. It is mainly produced by the polymerization of propylene gas using Ziegler–Natta catalysts and its worldwide production estimates reached up to 68 million metric tons as of 2015.34 The main commodity applications of PP include fibers and fabrics for home furnishings, bottles and containers, toys, and automobile parts. Although polypropylene exhibits similar properties to polyethylene, it shows higher melting temperatures due to increased polymer crystallinity and has a chemical structure that is more oxidation-sensitive. In terms of the important biotechnology, nanocomposite and membrane applications, it is often essential to properly modify the PP surface to induce more enhanced polarity, etched surface morphology, antifouling properties and hydrophilic character.45The gamma irradiation, UV-irradiation, microwave and plasma treatments are among the high-energy surface modification techniques that induce reactive groups on the PP surface. In terms of click surface modification of PP, bromination is usually accompanied by these methods to insert bromo functionalities onto the surface. These bromo groups can be subsequently transformed into various clickable groups. Chlorinated polypropylenes with various chlorination degrees are also commercially available.
In one of the examples of early click-modification, Yu and co-workers demonstrated a multi-step approach for grafting polyacrylamide (PAAm) chains onto the PP macroporous membrane.46 The strategy involved UV-induced bromination, azidation and click coupling of alkyne functional PAAm on the polymer surface (Fig. 3). The bromination of the PP surface was conveniently managed by a gas phase free-radical photochemical pathway. In the mechanism, bromine radicals were first generated by homolytic bond cleavage upon UV exposure. The bromine radicals then abstract hydrogen from the polymer backbone resulting in a radical center which subsequently reacts with the bromine molecule.47 The azide modification of the bromo groups was conducted with about 40% efficiency under SN2-type nucleophilic exchange conditions. On the other hand, the Cu(I)-catalyzed click reaction of alkyne functional PAAm onto azide functional PP surface has revealed a much lower coupling efficiency due to the low interaction of reactive sites. The authors suggested that the low coupling efficiency was due to the low interaction of reactive sites. In the following studies, the group extended the strategy to click couple poly(2-acrylamido-2-methyl propane sulfonic acid),48 methoxy poly(ethylene glycol)49 and zwitterionic polymers50 onto the PP membrane surface. It was reported that the introduction of hydrophilic polymers onto the membrane surface provided a reduced water contact angle, improved permeation performance and enhanced antifouling characteristics. Zhou and co-workers utilized a similar approach to modify the PP membrane surface with poly(N-vinyl-2-pyrrolidone) via one-pot reversible-addition fragmentation chain-transfer (RAFT) polymerization and CuAAC click chemistry.51
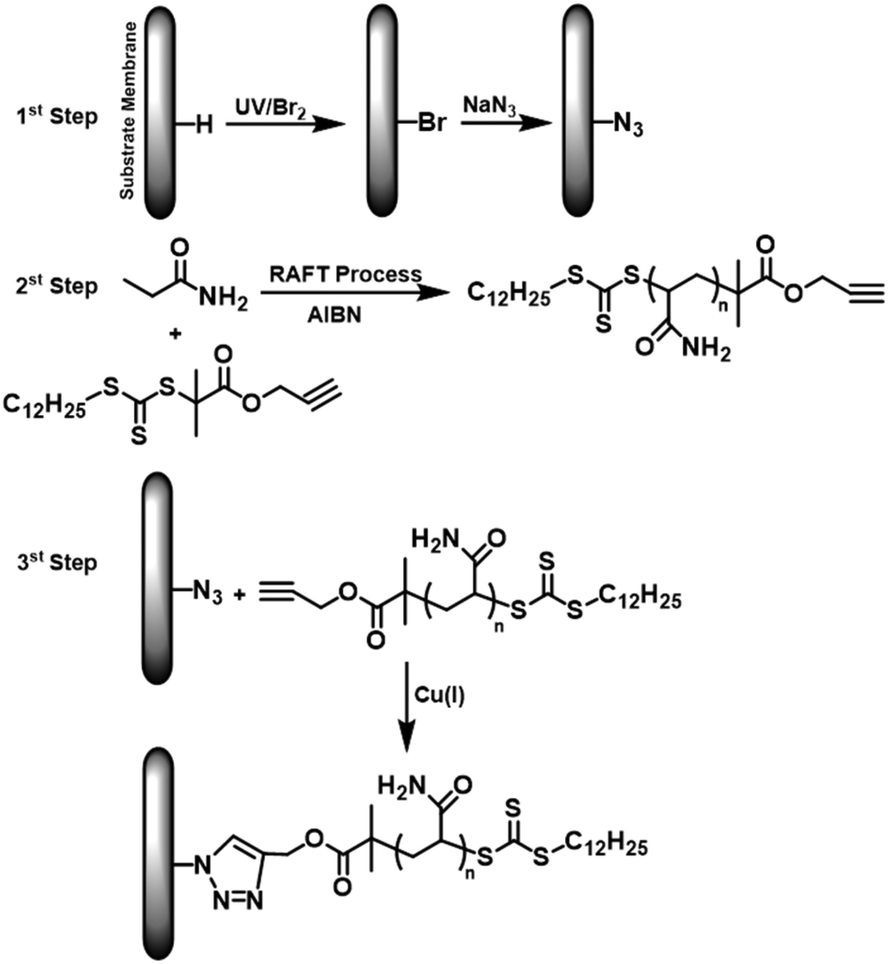 | ||
| Fig. 3 Modification of the PP membrane surface by installation of CuAAC-clickable azide groups and subsequent alkyne–PAAm conjugation. Reproduced from ref. 46 with permission from Elsevier, copyright [2012]. | ||
Besides UV-irradiation, Boyer and co-workers used gamma-irradiation to generate radicals on the PP film surface.52 Surface-initiated atom transfer radical polymerization (ATRP) in the presence of tethered bromine and iodine groups was utilized to obtain polystyrene (PSt) and poly(methyl acrylate) (PMA) brushes. This approach was extended to the introduction of azide groups onto the PMA brushes which were subsequently CuAAC-click conjugated with propargyl alcohol. Functionalization of the bromo end-groups of PP-grafted poly(methyl acrylate) was also studied via thio-bromo click reaction using 2-aminoethanethiol as the model compound. The successful click modifications of the polymer-grafted PP surfaces were established by X-ray photoelectron spectrometry (XPS), Fourier transform infrared (FTIR) spectroscopy and contact angle measurements.
Plasma treatment is a highly efficient method to introduce functional groups onto the unreactive polyolefins. By this technique, the surface energy of the targeted zone is lowered, which results in better wetting capability and enhanced adhesion properties. In plasma treatment, peroxide/hydroperoxide groups can be created at the material surface in which these reactive groups can be utilized in further functionalization or modification. Wang et al. employed this approach to modify the PP surface with reactive peroxide/hydroperoxide groups.53 A combined UV irradiation in the presence of the 3-(trimethylsilyl) propargyl methacrylate monomer led to polymer grafted microporous polypropylene membranes with protected alkyne groups (Fig. 4). After deprotection, the alkyne units were used for the glycosylation of the surface via UV-induced thiol–yne click reaction. According to XPS analysis, 48% thiol–yne efficiency was obtained in the immobilization of lectin-binding glycosyl groups.
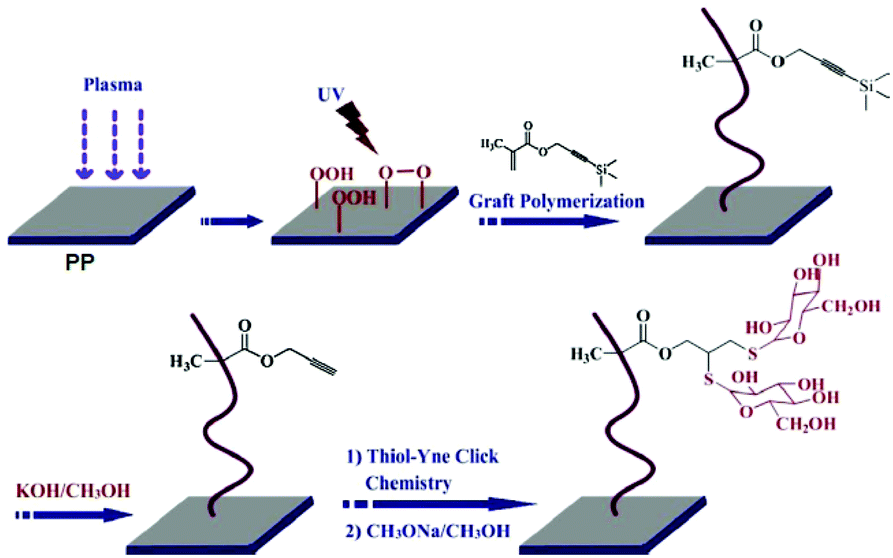 | ||
| Fig. 4 Construction of the glycosylated PP membrane surface by thiol–yne click chemistry. Reproduced from ref. 53 with permission from Elsevier, copyright [2013]. | ||
In another study, microwave plasma treatment of PE and PP in the presence of maleic anhydride was conducted to immobilize reactive anhydride units onto the polymer surface.54 These anhydride groups were further modified to obtain clickable propargyl units amenable to CuAAC functionalization. As an alternative to high energy surface treatment methods, direct C–H activation of commercial PP using stable hypervalent iodide sources has proven useful for azide group installation. Bielawski and co-workers reported up to 3 mol% azide functionalization of PP by the azidoiodinane mediated radical pathway.55 However, the radical mechanism ineluctably results in chain cleavage of the polymer.
On the other hand, commercially available chlorinated polypropylene can be directly used to install various functionalities including clickable groups along the polymer structure. This may provide synthetic utility in the chemical modification of PP by reducing the surface treatment efforts. The CuAAC click modification of chlorinated PP was demonstrated by Tasdelen and co-workers by azidation of chloro groups first and subsequent conjugation of alkyne end-functional poly(ethylene glycol) (PEG) or poly(ε-caprolactone) (PCL) polymers.56 The strategy was recently extended to the synthesis of polypropylene-graft-poly(L-lactide) copolymers57 and installation of quaternary ammonium58 and fluorinated compounds59 onto chlorinated PP.
Poly(vinyl chloride)
Poly(vinyl chloride) (PVC) is an important commodity polymer and its global production reached 38 million tons as of 2015.34 It is industrially produced by free-radical polymerization of vinyl chloride that can be done via bulk, solution, suspension or emulsion polymerization. The important characteristics of PVC are low flammability and high structural rigidity that provide flexural strength in material fabrication. The major applications involve manufacturing of pipes, conduits, wire cable insulations, siding, and window and door frames. In combination with suitable plasticizers PVC finds more consumer applications such as production of floor tiles, hoses, curtains and imitation leather. In advanced studies, PVC finds use in the fabrication of membranes for diverse applications.As a chlorine containing polymer, PVC has chemical derivatization routes similar to those of halogenated PE and PP. In the click chemistry applications of PVC modification, chloro groups have been conveniently transformed into azide functionalities via nucleophilic substitution. The foregoing azide groups are CuAAC clickable functionalities that can be exploited in further derivatization.
Yagci and co-workers demonstrated the azidation approach for the synthesis of thermally-curable PVC, where side-chain benzoxazine functionalities were installed along the PVC backbone (Fig. 5).60 Alkyne functional benzoxazine was efficiently clicked onto azide-containing PVC and the obtained polymer was readily transformed to thermosets by simple heating without a curing agent.
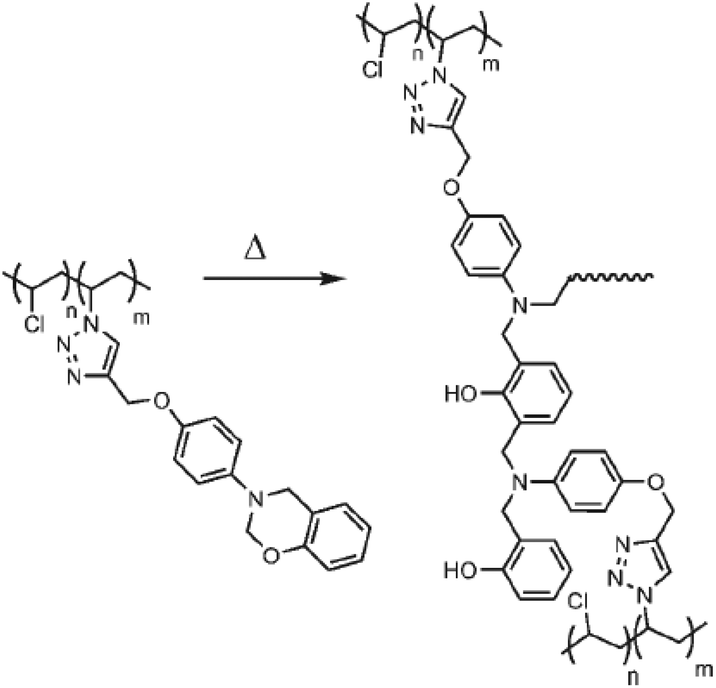 | ||
| Fig. 5 CuAAC click modification of PVC with benzoxazine units and thermal activation for crosslinking process. Reproduced from ref. 60 with permission from WILEY-VCH Verlag GmbH & Co., copyright [2008]. | ||
Although PVC has some superior mechanical and physical properties and high chemical and abrasion resistance, its structural weakness is low UV resistance. Upon exposure to UV or sunlight, PVC is susceptible to photochemical decomposition due to structural anomalies and defects originating from the manufacturing process. In particular, in outdoor applications, UV-absorbent chemical compounds are usually formulated with PVC by physical mixing for inhibiting photo-degradation. In order to prevent the leakage of such UV-absorbers from the polymer mixture, a feasible strategy might be covalent attachment of these compounds onto the polymeric skeleton. In a study, a CuAAC click installation of UV-absorber benzophenone groups onto azidated-PVC was reported by Huang et al. (Fig. 6).61 It was shown that benzophenone-immobilized PVC is compatible with an unmodified polymer and stabilizes PVC against photo-degradation for up to 200 h UV-irradiation.
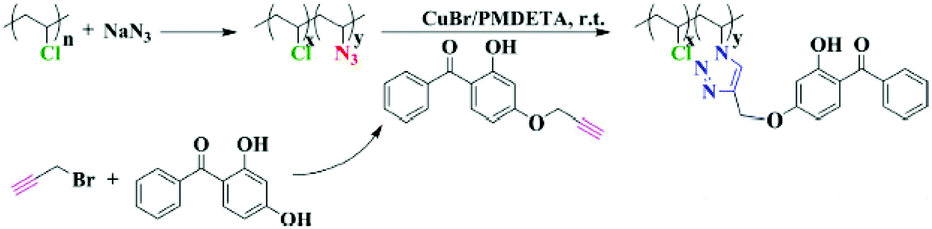 | ||
| Fig. 6 CuAAC click modification of PVC to install UV-absorber groups. Reproduced from ref. 61 with permission from Springer Nature Publishing AG, copyright [2016]. | ||
Another important concern in the commodity applications of PVC originates from the highly rigid polymeric structure. In order to obtain soft and elastic materials, PVC is usually formulated with certain small organic compounds known as plasticizers. Phthalates, phosphates, adipates, maleates and other compounds are used as additives in PVC processing to increase processability and flexibility. Similar to UV-absorbers, a common problem is the migration of plasticizers to the PVC surface causing progressive loss of properties. Also, serious health hazards might be encountered due to the leakage of toxic organic compounds. To decrease the glass transition temperature of PVC and avoid leakage, a useful strategy is covalent linking of plasticizers to PVC chains. In this sense, cardanol (phenolic compounds with long fatty tails)62,63 hyperbranched polyglycerol,64 and triethyl citrate65 based plasticizers were covalently bound to azide functional PVC via CuAAC click reaction.
In a recent study, Finn and co-workers systematically studied surface functionalization of flexible PVC tubing via CuAAC and azide–nitrile cycloaddition click reactions.66 By employing a series of phase transfer catalysts, the optimum conditions were analyzed for chemical modification of catheter tubing to install azide and cyano groups (Fig. 7). Subsequently, click modification and alkylation of functional surfaces were investigated by using various alkynes and an alkyl halide. Notably, PVC tubing was efficiently functionalized via CuAAC click reaction, demonstrating the efficiency of the surface modification methodology without changing the properties of the bulk preformed material.
 | ||
| Fig. 7 Modification of the flexible PVC tubing surface by combination of nucleophilic substitution and click chemistry reactions. Reproduced from ref. 66 with permission from the American Chemical Society, copyright [2018]. | ||
Tasdelen and co-workers extended the CuAAC click modification of PVC to a photochemical version of CuAAC click reaction.67 Azidated-PVC was efficiently grafted with alkyne functional polymers implemented under UV-initiated conditions. The authors suggested that this alternative reaction mechanism might be more beneficial for thermally sensitive materials rather than conventional thermal processes. A copper-free analogue of azide–alkyne cycoladdition for attachment of dialkyl acetylenedicarboxylate-based plasticizers onto azidated-PVC was also reported.68
CuAAC click modification of azidated-PVC has been exemplified in attachment of ferrocene units for electrochemical ion sensors,69 K+-selective crown ether ionophores for potentiometric and optical sensors,70 PEG units for biocompatible ion-selective electrodes,71 fluorescein dyes as membrane visual probes,72 copolymers with antibacterial groups,73,74 PEG-stearate for thermal energy storage materials,75 thioxanthone groups for graft polymers76 and poly(N-isopropylacrylamide) for electrospun membranes.77 The triazole groups resulting from the CuAAC click reaction of azidated-PVC with various alkynes were utilized in metal ion extraction applications.78
An attractive one-step strategy for PVC surface functionalization by combination of sulfur(VI)-fluoride exchange (SuFEx) click chemistry and benzophenone photochemistry reactions was recently reported.79 By taking advantage of UV-mediated tethering of benzophenone groups onto PVC polymer chains, a series of functional groups were successfully installed on the polymer surface (Fig. 8). This strategy features one-pot coupling of the sulfonyl fluoride group present on the benzophenone derivative with various silyl ether containing functional molecules.
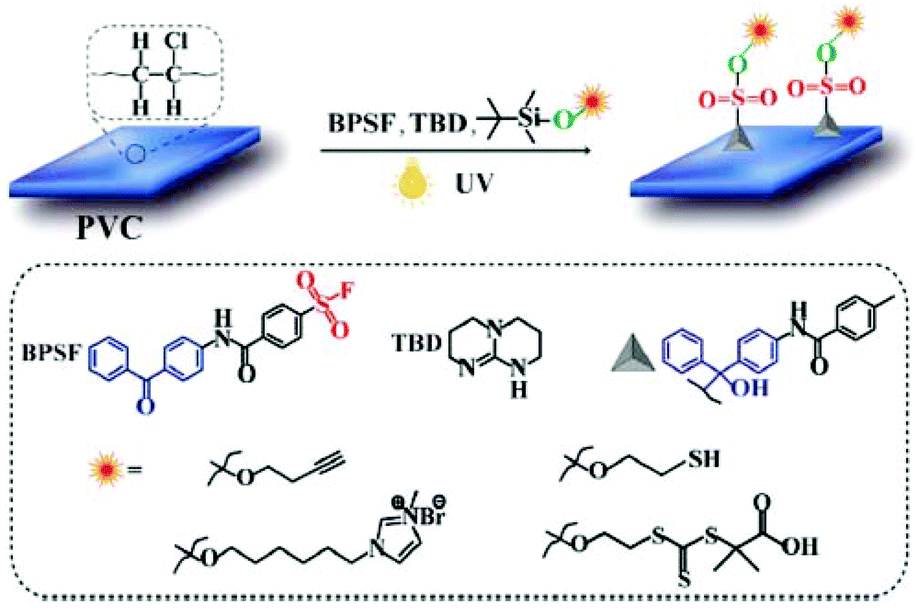 | ||
| Fig. 8 One-pot modification of the PVC surface by combination of SuFEx click reaction and benzophenone photochemistry. Reproduced from ref. 79 with permission from The Royal Society of Chemistry, copyright [2019]. | ||
Fluoropolymers
Fluorinated polymers or fluoropolymers contain multiple carbon–fluorine bonds in their chain structure. These polymers are characterized by their high resistance to solvents, high melting temperatures, very low coefficient of friction and outstanding resistance to oxygen, ozone and heat. Certain fluorinated polymers having elastomeric properties are distinguished as fluoroelastomers. The principal applications of fluoropolymers include temperature-resistant rings, seals and gaskets. They are frequently laminated as a protective layer onto various surfaces, especially in outdoor applications. Due to their exceptional inertness, resistance and stability, fluorinated polymers are employed in biomedical research, electrochemical energy storage and conversion, and manufacture of microchips and integrated circuits.Functionalization of fluoropolymers is desirable in biomedical applications to impart biocompatibility and cellular responses. The conventional approaches for fluoropolymer surface modification include plasma or ion irradiation. In conjunction with click-mediated tailoring, this method was employed for gamma radiation-induced poly(vinylidene fluoride) (PVDF) surface modification by grafting a (2,3,4,5,6)-pentafluorostyrene polymer.80 Subsequently, thiol/para-fluoro click reaction allowed the attachment of a thiol bearing molecule onto the crosslinked PVDF. According to the surface analysis, increased wettability adjustment was maintained as determined by water contact angle measurements. In an elegant study, alkyne surface functionalization of fluoropolymer Dyneon THV (a polymer of tetrafluoroethylene, hexafluoropropylene and vinylidene fluoride) was achieved by the use of scanning electrochemical microscopy (SECM).81 The local reduction of the THV fluoropolymer using a SECM tip led to the formation of surface alkyne functions which were subsequently modified by an azide-containing dye via CuAAC click coupling. In another study, surface argon plasma treatment of Dyneon THV to introduce bromo groups was demonstrated.82 This modification was followed by azidation of the surface to CuAAC click grafting of alkyne functional molecules.
An operationally benign procedure for functionalization of fluoropolymers by combining fluorous interactions and CuAAC click reaction was demonstrated by Cai and co-workers.83 Functional molecules including the alkynyl group and long perfluorinated hydrocarbon tails were tethered onto fluoropolymer surfaces via stable non-covalent fluorous interactions. These surfaces were then successfully modified with the azide group containing antimicrobial peptides and PEG molecules via CuAAC reaction chemistry.
In an elegant study, crosslinking of DuPont's perfluoro elastomer Kalrez® towards robust film formation was achieved by nitrile–azide cycloaddition click reaction.84 The side chain nitrile groups present on the fluoroelastomers were efficiently reacted with a bis-azido fluorinated curing agent through tetrazole formation. It was shown that improved thermal degradation properties were achieved resulting from cured films compared to those from uncured fluoroelastomers.
Reactive alkene units can be generated on PVDF polymer chains by the dehydrofluorination process using suitable bases. These alkene groups can be considered as clickable functionalities to offer radical-initiated thiol additions. On the other hand, due to the electron-deficient nature of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF– double bond, the addition reaction proceed in nucleophilic thiol–ene pathway (Fig. 9).85
CF– double bond, the addition reaction proceed in nucleophilic thiol–ene pathway (Fig. 9).85
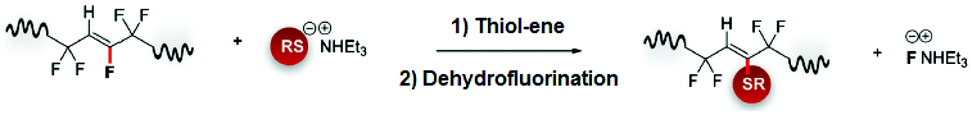 | ||
| Fig. 9 Nucleophilic thiol–ene addition/dehydrofluorination steps for the double bond containing PVDF polymer modification. Reproduced from ref. 85 with permission from WILEY-VCH Verlag GmbH & Co., copyright [2018]. | ||
Polystyrene
Polystyrene is a rigid and relatively brittle thermoplastic polymer produced by free-radical polymerization of the styrene monomer. It is a key material in various applications including insulation, packaging materials and food containers which this makes a substantial production effort of the polymer reaching 25 million tons as of year 2015.34 Polystyrene is usually blended with elastomers such as polybutadiene to improve the impact strength and reduce brittleness. Due to its important characteristics such as high melting temperatures, processability, dimensional stability, chemical resistance, low dielectric constant and low gas permeability, the polymer also gained interest in scientific research. However, polystyrene still has several drawbacks associated with its chemical structure that require additional modifications in practical uses. High rigidity and the absence of polar groups are important concerns towards improving the mechanical properties, adhesion and compatibility with polar substrates. The progress in the chemical modification of polystyrene has been reviewed elsewhere.86 The chemical tailoring of polystyrene usually targets the aromatic benzene ring, especially for Friedel–Crafts type alkylation, acylation and halogenation reactions. Convenient modification routes are the benzylic bromination and chlorination in which the resulting haloalkyl groups can be employed in polymer grafting.87 Chloromethylation is also applied for functional diversification at benzene rings.88Although there are enormous click chemistry applications of styrenic polymers in scientific research that are mainly based on the laboratory scale polymerization of styrene with functional monomers,89 interestingly, there are no reports in the literature that show the click chemistry functionalization of commercial polystyrene. The advantages associated with the low-cost and abundant commercial polystyrene on the other hand can enable practical utilization towards efficient click manipulation. It is perceptible that chloromethylation or benzylic halogenation of commercial polystyrene would allow further functionalization by the installation of clickable groups onto the polymer structure.
Unsaturated polyolefin elastomers
Elastomers are important subgroups of industrial polymers that are characterized by their viscoelastic behavior and weak intermolecular forces resulting in low Young's modulus and high failure strain. Although the term elastomer is used interchangeably with rubber, the latter is usually referred to as vulcanized material which is produced from unsaturated elastomeric polymers by crosslinking. Unsaturated polyolefin elastomers involve natural polyisoprene, polybutadiene, polychloroprene, styrene–butadiene copolymers, butadiene–acrylonitrile copolymers and many others.Since unsaturated polyolefin elastomers carry reactive double bonds in their polymer structure, they have been subject to vast industrial and research studies for both chemical modification and crosslinking. Although the reactive alkene units in their polymer structure are primarily employed in vulcanization processes, these reactive handles are also amenable to covalent functionalization.90
In click chemistry-based methods, the reactive alkene units on unsaturated elastomers are bare substrates for thiol–ene modification. Since unactivated double bonds (i.e. alkenes without electron-withdrawing groups directly attached to the double bond) undergo radical mediated thiol–ene reactions with thiols, this click modification approach has been utilized in this context. It is important to note that performing the photoaddition of thiyl radicals onto the double bonds of unsaturated polyolefin elastomers often gives desired and side products concurrently, due to intramolecular cyclizations (Fig. 10). The extent of the formation of side products is strictly related to the experimental conditions. For example, the thiol–ene functionalization efficiency of 1,2-polybutadiene (PB) was reported to reach 80% or higher when employing high concentrations of reactants and low temperatures.91
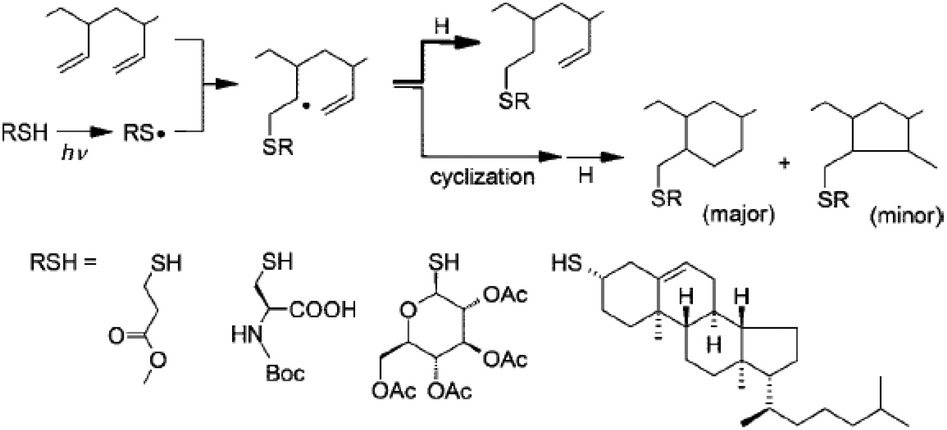 | ||
| Fig. 10 Radical thiol–ene functionalization of an unsaturated 1,2-polybutadiene elastomer depicting the main reaction product and cyclization-derived ring segments. Reproduced from ref. 91 with permission from the American Chemical Society, copyright [2008]. | ||
Besides thiol–ene modification of unsaturated elastomers, backbone residing alkene groups have also been manipulated by other click chemistry methodologies such as the inverse electron-demand Diels–Alder reaction and nitrile oxide/alkene cycloadditions. In a recent study, Du Prez, De Clerck and co-workers employed triazolinedione click reaction to produce elastomeric styrene–butadiene–styrene (SBS) copolymer membranes by electrospinning (Fig. 11).92 The thermo-mechanical properties of electrospun fibers were modified by simple covalent modification or crosslinking of alkene groups with triazolinediones containing molecules. Triazolinedione modification or crosslinking of unsaturated rubbers offers simultaneous click modification and creation of “sacrificial” H-bonds by urazole groups that could be harnessed in shape memory and energy dissipation, significantly improving the mechanical properties.93
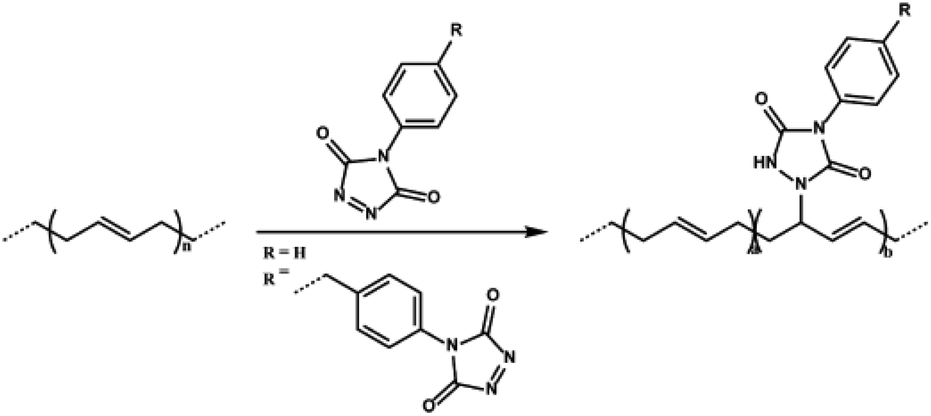 | ||
| Fig. 11 Triazolinedione click modification of the SBS elastomer. Reproduced from ref. 92 with permission from the American Chemical Society, copyright [2015]. | ||
Installation of other clickable groups onto unsaturated polyolefin elastomers via sequential modifications is also possible. The CuAAC click modification of unsaturated elastomers by bromo or epoxide group attachment that allows further azidation has been demonstrated.94
A direct thiol–ene grafting of DA clickable furan groups has attracted attention towards the thermally-reversible curing efforts of such unsaturated elastomers. Recently, Shi and co-workers reported thiol–ene and Diels–Alder sequential click modification of polybutadiene towards the fabrication of thermally recyclable polymers.95 Attachment of furan groups via radical thiol–ene reaction and further DA crosslinking by a bismaleimide crosslinker resulted in a polybutadiene elastomer network with tunable mechanical properties (Fig. 12).
 | ||
| Fig. 12 Sequential click functionalization of polybutadiene towards thermally-reversible crosslinking. Reproduced from ref. 95 with permission from the American Chemical Society, copyright [2015]. | ||
To provide an overview of the compositions and click chemistry-mediated modification applications of unsaturated polyolefin elastomers, a collection of examples are summarized in Table 2.
| UPE | Click reaction | Applications | Ref. |
|---|---|---|---|
| Polybutadiene (PB) | Thiol–ene | UV, visible light or sunlight-aided modification with thiols; hydrophilic modification of nanoporous crosslinked polymers; composite formation; crosslinking of electrospun fibers | 91 and 96–101 |
| Polybutadiene (PB) | Thiol–ene/Diels–Alder | Reversibly crosslinked materials | 95 and 102 |
| Polybutadiene (PB) | Triazolinedione click | Crosslinked networks with flexible and rigid chains | 103 |
| Polybutadiene (PB) | CuAAC | Surface modification of nanoporous polymers; modification with self-curable benzoxazine groups | 94 and 104 |
| Polybutadiene (PB) | Tetrazine click | Antioxidant foams | 105 |
| Natural rubber (NR) | Thiol–ene | Antioxidant grafting | 106 and 107 |
| Natural rubber (NR) | Nitrile oxide cycloaddition | Catalyst-free crosslinking and decrosslinking | 108 |
| Polyisoprene (IR) | Thiol–ene | Crosslinked nanolayer deposition | 109 |
| Nitrile–butadiene rubber (NBR) | Thiol–ene | Nanocomposite formation; grafting on the AFM tip to study interfacial interaction between nanofillers and elastomers | 110 and 111 |
| Styrene–butadiene rubber (SBR) | Thiol–ene | Modification with ionic liquids; silica nanoparticle modification; blending with silicone rubber; alcohol and carboxylic acid functionalization; crosslinking | 112–118 |
| Styrene–butadiene rubber (SBR) | Triazolinedione click | Crosslinking with adaptive recovery | 93 |
| Styrene–butadiene rubber (SBR) | CuAAC | Installation of H-bonding groups for supramolecular network formation | 119 and 120 |
| Styrene–butadiene-styrene rubber (SBS) | Thiol–ene | Modification with carboxylic acids and other polar groups, alkyl thiols or benzoxazines to tailor mechanical and morphological properties | 121–125 |
| Styrene–butadiene-styrene rubber (SBS) | Triazolinedione click | Modification or crosslinking of electrospun fibers | 92 |
| Styrene–butadiene-styrene rubber (SBS) | CuAAC | Installation of quaternary ammonium groups for anion exchange membranes | 126 |
| Styrene–isoprene–styrene rubber (SIS) | Thiol–ene | PEG grafting; crosslinking | 127 and 128 |
| Ethylene–propylene rubber | Diels–Alder | Reversibly crosslinked materials | 129 |
Polyacrylonitrile
Polyacrylonitrile (PAN) is an important acrylic polymer produced by free-radical polymerization of acrylonitrile through suspension methods. Most of the industrially produced PAN (3 million tons global production output) is employed in the fabrication of acrylic fibers that are soft, flexible and wool-like; hence the most common use of PAN is as a wool replacement synthetic fiber. Due to its low cost, excellent chemical resistance, thermal stability and low gas permeability, PAN has been used for ultrafiltration membranes, electrospun nanofibers, energy storage devices and other advanced applications.As is seen in many industrial polymers that have hydrophobic polymer structures, appropriate modification of PAN to induce utility is often required, especially for separation membrane applications. A common drawback of hydrophobic polymers in membrane fabrication is that membrane fouling originates from the adsorption or deposition of hydrophobic compounds on material surfaces or pores. This often results in flux decline in the feed stream, which causes poor membrane performance and loss in mechanical properties during the long time use. To avoid or limit this problem decorating the membrane surface with antifouling substrates and hydrophilic compounds is usually carried out.
In the click chemistry applications of industrial PAN modification, azide–nitrile cycloaddition has been employed as the sole click approach. The present nitrile dangling groups of the polymer structure can undergo cycloaddition reactions with azide anions of sodium azide. This reaction can be implemented in very good yields and short reaction times giving 5-substituted tetrazoles. Alkylation of heterocycles with various methods has been demonstrated for side chain grafting of PAN with tetrazole groups,130 hydrophilic polymers (Fig. 13),131 cationic metal species132,133 and hydrophobic alkyl tails.134 Interestingly, no studies that employ direct functionalization of nitrile groups with azide group containing substances have been reported yet.
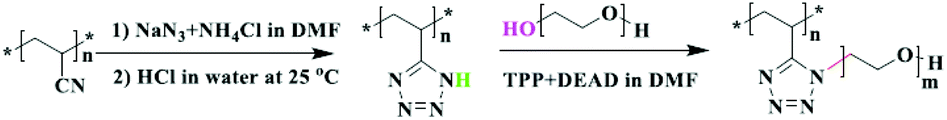 | ||
| Fig. 13 Azide–nitrile click functionalization of PAN through tetrazole formation and subsequent N-alkylation. Reproduced from ref. 131 with permission from Springer Nature Publishing AG, copyright [2018]. | ||
Polyacrylates and polymethacrylates
Polyacrylates and polymethacrylates are produced at an industrial scale by free-radical polymerization of acrylic/methacrylic monomers through bulk or solution processed methods. The important members of this family include poly(methyl methacrylate) and poly(methyl acrylate) as well as poly(2-hydroxyethyl methacrylate) and poly(cyanoacrylate)s.Although polyacrylates and polymethacrylates are indispensable materials that have found vast applications in polymer research, introducing functionalities to these polymers is often challenging. In laboratory synthesis, functional (meth)acrylates can be prepared by employing functional monomers along with polymerization of non-functional monomers.135 However, the utilization of industrial (meth)acrylate polymers requires efficient post-polymerization modification methods to install reactive groups. In this sense, transesterification reactions of side chain ester groups might be feasible for introducing functionalities by either direct grafting of target substrates or attachment of reactive groups, including clickable functions. This approach was recently reported by demonstrating the efficiency of catalytic transesterification in functionalization of poly(methyl acrylate) (Fig. 14).136 Several alcohols containing clickable alkene, alkyne and azide groups were successfully grafted onto the side chains of the polymer by employing efficient zinc catalysts.
 | ||
| Fig. 14 Transesterification-based synthetic strategy to functional polyacrylates. Reproduced from ref. 136 with permission from WILEY-VCH Verlag GmbH & Co., copyright [2017]. | ||
Poly(acrylic acid)
Poly(acrylic acid) (PAA) is a major industrial polymer produced on a large scale from acrylic acid by free-radical polymerization methods. PAA and its derivatives have found numerous applications in the pharmaceutical, cosmetic, adhesive, detergent and paint sectors, especially as thickening, dispersing, suspending and emulsifying agents. The weak anionic polyelectrolyte structure with the ability to absorb and retain water makes PAA suitable for the fabrication of polymeric gels and ion-exchange materials.The presence of carboxylic acid groups at the side chains of PAA is purposive in the direct functionalization of polymers via esterification and amidation. Besides, several nucleophilic substitution reactions can be implemented by the carboxylate function. In this context, the modification of PAA with CuAAC clickable groups has been demonstrated by grafting azide or alkyne group containing amino compounds via amidation. Layer-by-layer construction of PAA films on various surfaces such as silicon wafer and gold electrodes was studied by CuAAC crosslinking of PAA chains to covalently stabilize thin films (Fig. 15).137,138 The gold electrode surface with a deposited PAA layer was employed as a nanoreactor to embed palladium nanoparticles.139
 | ||
| Fig. 15 Layer-by-layer construction of PAA films on the substrate surface by CuAAC click crosslinking of alkyne or azide modified polymers. Reproduced from ref. 138 with permission from the American Chemical Society, copyright [2011]. | ||
Poly(vinyl alcohol)
Poly(vinyl alcohol) (PVA) is a synthetic water-soluble polymer prepared by the hydrolysis of polyvinylacetate. As an important commodity polymer, PVA has diverse applications in the adhesive, coating, paper and textile industries, as well as biomedical and pharmaceutical applications. To date, advanced applications cover membrane fuel cells, tissue engineering scaffolds, drug delivery formulations, separation technologies and catalysis. Functionalization of PVA congruents with the desired applications via conventional esterification, carbamation, and etherification reactions can be carried out by using side chain alcohol groups.140The click chemistry-mediated modification of PVA has been utilized for mainly grafting clickable functional groups onto polymer chains via carbamate formation, esterification or azidation routes. In an early example, Hilborn and co-workers demonstrated carbonyldiimidazole pre-activation of PVA alcohol groups which were then reacted with various amines, terminated with clickable alkyne, azide and furan functional groups.141 Azide- and alkyne-modified PVA polymers were subsequently crosslinked by the CuAAC click reaction to give hydrogels. A similar carbamate formation strategy for azide installation was reported for CuAAC attachment of an alkyne-functional radiographic component onto PVA.142 Pre-activation of hydroxyl groups by mesyl or tosyl chloride to introduce azide groups at the side chains was also reported.143–145
The sequential modifications of PVA to introduce maleimide,146 thiol,147,148 and hydrazide149 groups for Diels–Alder, thiol–ene, thiol–yne and hydrazone formation-based click tailoring have been reported.
Poly(ethylene terephthalate)
Poly(ethylene terephthalate) (PET) is a semicrystalline polymer possessing large benzene rings in its structure giving notable stiffness and strength to the polymer chains. It is among the top produced industrial polymers in which the global output reached 33 million tons as of 2015.34 The stiff polymer structure and orderly arrangement of chains by stretching make PET fibers highly resistant to deformation. The outstanding fiber properties of PET make it applicable in various textile and fabric products. Molten PET can be shaped by most of the common thermoplastic processing techniques into bottles and food containers. PET has also been employed in the production of important biomedical apparatus such as surgical drapes, vascular grafts and ligament prostheses.PET fibers need to be modified for creating feature-added materials for special applications and for overcoming some structural drawbacks such as low moisture regain of hydrophobic polymers and for avoiding static electricity, pilling and dyeing problems. The allocated modification approaches of PET employ physical, chemical and plasma surface treatment methods.150
Due to the absence of functionalizable groups besides aromatic benzene rings, the click chemistry-mediated applications of PET modification target surface –COOH groups. Installation of CuAAC-clickable alkyne groups via amidation could be accomplished on fabric or membrane form PET specimens (Fig. 16).151–153 However, more efficient and benign click modification approaches especially targeting the aromatic rings still need more comprehensive research.
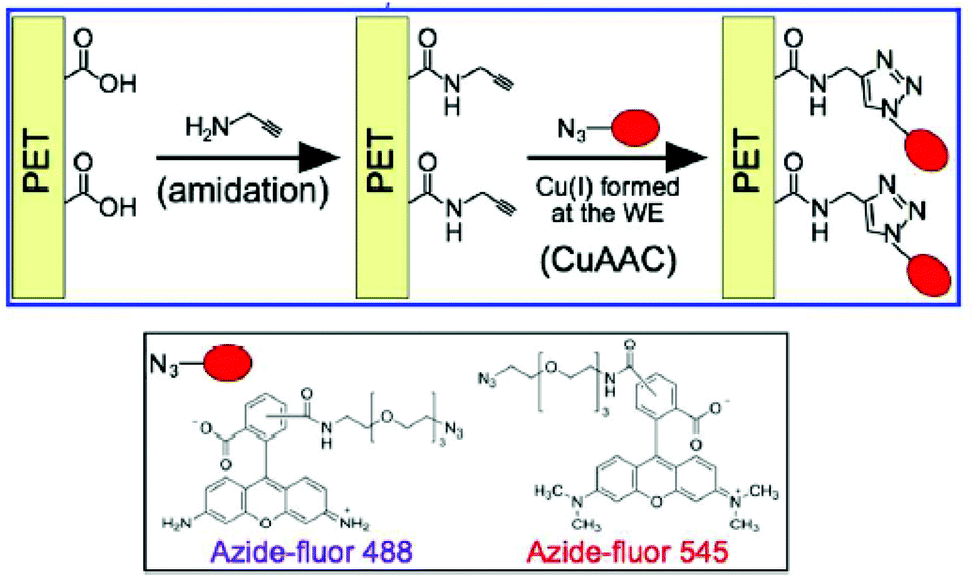 | ||
| Fig. 16 Installation of alkyne groups on PET surface carbonyls and CuAAC click attachment of fluorescent dye molecules. Reproduced from ref. 153 with permission from the American Chemical Society, copyright [2017]. | ||
Polysulfones
Polysulfones (PSU) are among the engineering thermoplastics that have favorable high temperature properties and excellent chemical resistance to hydrolysis, acids and bases. The outstanding properties make PSUs applicable in medical devices, food processing, membrane technologies, feeding systems, and the automotive and electronic industries.The hydrophobic nature and especially high stiffness of aromatic PSUs, as well as the demand to introduce functionality often necessitates appropriate covalent modification. The common modification routes include sulfonation, halogenation, Friedel–Crafts alkylation/acylation reactions etc. A straightforward and commonly employed route is halomethylation of aromatic rings that makes functional alkyl halogen groups amenable to further derivatization.
In the context of click chemistry modification of PSUs, a much sought strategy relies on the installation of CuAAC clickable azide groups to the chloromethylated polymer structure. Subsequent derivatizations of azidated polymers by click coupling with alkyne functional substrates have been reported to tune the material properties especially in membrane applications. These include the grafting of polar groups,154,155 peptides,156 pyrene,157 zwitterionic complexes,158,159 hydrophilic/antifouling polymers160,161 and hydrophobic organic molecules.162,163 A thiol–ene approach for the modification of chloromethylated PSU with vinyl groups and subsequent radical thiol addition was recently reported.164
Conclusions
The click chemistry-mediated modification approaches of industrial polymers have gained increasing interest, especially in the last decade. This interest originates from the fine tuning of common, low-cost and abundant commodity polymers with highly efficient click reactions towards the fabrication of novel polymeric materials for various applications. In comparison with laboratory scale efforts to synthesize functional polymers, efficient covalent manipulations might provide great utility in modification, diversification and property enhancement of commodity polymers that are already produced at an industrial scale.The ideal polymer modification by click chemistry approaches however has some prerequisites. The first important concern is the efficient installation of clickable functional groups on polymer structures. Certain industrial polymers i.e. olefinic rubbers bearing thiol-reactive alkene groups on the chain structure can be directly employed as substrates in thiol–ene based modification reactions. Commercially available chlorinated polymers on the other hand provide facile installation of various clickable functionalities amenable to click chemistry tailoring.
However, this is not the case for most of the commodity polymers and attaching clickable groups by tedious multi-step procedures is a must. Polymers devoid of apparent reactive groups on polymer structures such as polyethylene and polypropylene require harsh and destructive surface treatments to create functionalities for subsequent exploitation. Radical mediated surface modification techniques and plasma methods often cause chain cleavage that may alter the polymer properties. In this sense, further research studies for the optimum installation of clickable groups onto various commodity polymers might be fundamental.
A common drawback of CuAAC click modification is the requirement to use catalytic copper(I) salts and suitable ligands. Although the metal catalysts are employed in substantially low amounts (typically 1 to 5% mol ratio of the respective alkyne and azide groups),165 the removal of the catalytic species is essential to reduce any deteriorating effects on the physical and mechanical properties as well as the oxidative stability of the polymers.166 In addition to solvent precipitation and washing efforts, proper treatment of modified polymers, surfaces and solid scaffolds with copper chelating agents might result in efficient removal of the residual copper species. Employment of heterogeneous167 and resin-supported copper(I) species168 could also maintain more convenient and reusable catalytic systems.
The undesired environmental effects such as heat, light, oxidation agents and various organic/inorganic chemicals can cause polymer degradation and change in the properties over time. These factors could be more important in the case of modified polymers since the modification processes can cause structurally weak points on the polymer chains. Covalent attachment of organic molecules to the main chain polymers might lead to susceptible chemical bonds being attacked by atmospheric oxygen, ozone and water. Although chemical degradation or aging of polymers is often inevitable, these deteriorating effects can be minimized or delayed by careful control of the reaction conditions, functionalization degree and purification. Stabilization of the modified polymers against photo-oxidation and ultraviolet light by using suitable additives might result in a better lifecycle. Research studies emphasizing the proper structural analysis of commodity polymers under various click modification conditions would be a worthwhile contribution to the field.
A subtle approach for efficient production of polymer nanocomposites has relied on the utilization of click chemistry reactions in the fabrication process.8 Composite materials exhibit some superior properties of the constituent matrix and fillers along with showing some additional properties. Advantages of industrial polymers can be inherited in (nano)composites as matrix materials along with efficient click chemistry mediated fabrication efforts.
Considering the high structural, mechanical and functional diversity of industrial polymers, this abundant and low-cost feed-stock could offer great utility in the fabrication of materials with specific properties. Success in overcoming the limitations and a good understanding of the interactions between polymer structures and ever-growing synthetic methods might open new avenues in the modification and property enhancement of polymeric materials.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed., 2009, 48(1), 48–58 CrossRef CAS.
- K. A. Günay, P. Theato and H. A. Klok, in Functional Polymers by Post-Polymerization Modification: Concepts, Guidelines, and Applications, Wiley-VCH, Weinheim, 2013 Search PubMed.
- L. F. Ramos-De Valle, in Handbook of Polymer Synthesis, Characterization, and Processing, Wiley-VCH, Weinheim, 2013 Search PubMed.
- J. B. Williamson, S. E. Lewis, R. R. Johnson, I. M. Manning and F. Leibfarth, Angew. Chem., Int. Ed., 2018 DOI:10.1002/ange.201810970.
- C. Barner-Kowollik and A. J. Inglis, Macromol. Chem. Phys., 2009, 210, 987–992 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40(11), 2004–2021 CrossRef CAS.
- M. Arslan and M. A. Tasdelen, Chem. Africa, 2018 DOI:10.1007/s42250-018-0030-8.
- M. Arslan and M. Tasdelen, Polymers, 2017, 9, 499 CrossRef PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- M. Arslan, O. Gok, R. Sanyal and A. Sanyal, Macromol. Chem. Phys., 2014, 215, 2237–2247 CrossRef CAS.
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, Polym. Chem., 2018, 9, 2853–2867 RSC.
- G. K. Such, A. P. R. Johnston, K. Liang and F. Caruso, Prog. Polym. Sci., 2012, 37, 985–1003 CrossRef CAS.
- G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4103–4120 CAS.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- M. Arslan, Macromol. Chem. Phys., 2018, 219, 1800087 CrossRef.
- Thiol-X Chemistries in Polymer and Materials Science, ed. A. Lowe and C. Bowman, The Royal Society of Chemistry, 2013 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- Y. Zhenglong, C. Qiuyun, Z. Dan and B. Yilong, Prog. Chem., 2012, 24, 395–404 Search PubMed.
- G. Delaittre and L. Barner, Polym. Chem., 2018, 9, 2679–2684 RSC.
- I. I. Yilmaz, M. Arslan and A. Sanyal, Macromol. Rapid Commun., 2012, 33, 856–862 CrossRef CAS PubMed.
- M. Arslan, T. N. Gevrek and A. Sanyal, Maleimide containing thiol-reactive polymers: Synthesis and functionalization, in Functional Polymers, Apple Academic Press, New York, 2017 Search PubMed.
- M. Arslan, T. N. Gevrek and A. Sanyal, Design and synthesis of maleimide group containing polymeric materials via the Diels-Alder/Retro Diels-Alder strategy, in Functional Polymers by Post-Polymerization Modification, ed. P. Theato and H. Klok, Wiley-VCH Verlag GmbH & Co., 2013 Search PubMed.
- O. C. J. Andrén and M. Malkoch, Polym. Chem., 2017, 8, 4996–5001 RSC.
- M. Arslan, T. N. Gevrek, J. Lyskawa, S. Szunerits, R. Boukherroub, R. Sanyal, P. Woisel and A. Sanyal, Macromolecules, 2014, 47, 5124–5134 CrossRef CAS.
- Y. Oz, M. Arslan, T. N. Gevrek, R. Sanyal and A. Sanyal, ACS Appl. Mater. Interfaces, 2016, 8, 19813–19826 CrossRef CAS PubMed.
- M. Arslan, T. N. Gevrek, A. Sanyal and R. Sanyal, RSC Adv., 2014, 4, 57834–57841 RSC.
- M. Arslan, D. Aydin, A. Degirmenci, A. Sanyal and R. Sanyal, ACS Omega, 2017, 2, 6658–6667 CrossRef CAS.
- M. Arslan, T. N. Gevrek, R. Sanyal and A. Sanyal, Eur. Polym. J., 2015, 62, 426–434 CrossRef CAS.
- M. Arslan, T. Yirmibesoglu and M. Celebi, J. Turk. Chem. Soc., Sect. A, 2018, 5, 1327–1336 CrossRef.
- J. Collins, Z. Xiao, M. Müllner and L. A. Connal, Polym. Chem., 2016, 7, 3812–3826 RSC.
- J. C. Brendel, L. Martin, J. Zhang and S. Perrier, Polym. Chem., 2017, 8, 7475–7485 RSC.
- T. Abdul Fattah, A. Saeed and F. Albericio, J. Fluor. Chem., 2018, 213, 87–112 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- M. Lehocký, H. Drnovská, B. Lapčíková, A. M. Barros-Timmons, T. Trindade, M. Zembala and L. Lapčík, Colloids Surf., A, 2003, 222, 125–131 CrossRef.
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793–800 RSC.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311–9313 CrossRef CAS PubMed.
- M. M. Unterlass, E. Espinosa, F. Boisson, F. D'Agosto, C. Boisson, K. Ariga, I. Khalakhan, R. Charvet and J. P. Hill, Chem. Commun., 2011, 47, 7057–7059 RSC.
- M. Ciftci, D. Wang, M. Buchmeiser and Y. Yagci, Macromol. Chem. Phys., 2017, 218, 1700279 CrossRef.
- M. Hong, J. Y. Liu, B. X. Li and Y. S. Li, Macromolecules, 2011, 44, 5659–5665 CrossRef CAS.
- H. Li, A. Guo, Q. Li, W. Wang, J. Li and Y. Zhang, Macromol. Chem. Phys., 2017, 218, 1600449 CrossRef.
- S. Norsic, C. Thomas, F. D'Agosto and C. Boisson, Angew. Chem., Int. Ed., 2015, 54, 4631–4635 CrossRef CAS PubMed.
- Z. Xiao, C. W. Bennett and L. A. Connal, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1957–1960 CrossRef CAS.
- H. Li, A. Guo, Q. Li, W. Wang, J. Li and Y. Zhang, Macromol. Chem. Phys., 2017, 218, 1600449 CrossRef.
- Q. Yang, Z. K. Xu, Z. W. Dai, J. L. Wang and M. Ulbricht, Chem. Mater., 2005, 17, 3050–3058 CrossRef CAS.
- X. M. Wu, L. L. Wang, Y. Wang, J. S. Gu and H. Y. Yu, J. Membr. Sci., 2012, 421–422, 60–68 CrossRef CAS.
- S. Balamurugan, A. B. Mandale, S. Badrinarayanan and S. P. Vernekar, Polymer, 2001, 42, 2501–2512 CrossRef CAS.
- Y. Wang, L. L. Wang, X. C. He, Z. J. Zhang, H. Y. Yu and J. S. Gu, J. Colloid Interface Sci., 2014, 435, 43–50 CrossRef CAS PubMed.
- L. L. Wang, J. J. Wu, Z. B. Zhang, J. Zhou, X. C. He, H. Y. Yu and J. S. Gu, Sep. Purif. Technol., 2016, 164, 81–88 CrossRef CAS.
- J. J. Wu, J. Zhou, J. Q. Rong, Y. Lu, H. Dong, H. Y. Yu and J. S. Gu, Chin. J. Polym. Sci., 2018, 36, 528–535 CrossRef CAS.
- J. Zhou and B. Hu, J. Appl. Polym. Sci., 2015, 132, 42649 Search PubMed.
- O. Foster, A. H. Soeriyadi, M. R. Whittaker, T. P. Davis and C. Boyer, Polym. Chem., 2012, 3, 2102–2111 RSC.
- C. Wang, Y. Fan, M. X. Hu, W. Xu, J. Wu, P. F. Ren and Z. K. Xu, Colloids Surf., B, 2013, 110, 105–112 CrossRef CAS PubMed.
- H. A. Pearson and M. W. Urban, J. Mater. Chem. B, 2014, 2, 2084–2087 RSC.
- D. Liu and C. W. Bielawski, Polym. Int., 2017, 66, 70–76 CrossRef CAS.
- G. Acik, E. Sey and M. A. Tasdelen, eXPRESS Polym. Lett., 2018, 12, 418–428 CrossRef CAS.
- G. Acik, C. Altinkok and M. A. Tasdelen, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2595–2601 CrossRef CAS.
- G. Acik, C. Altinkok, H. Olmez and M. A. Tasdelen, Prog. Org. Coat., 2018, 125, 73–78 CrossRef CAS.
- G. Acik, C. E. Cansoy and M. A. Tasdelen, J. Appl. Polym. Sci., 2019, 136, 47072 CrossRef.
- B. Kiskan, G. Demiray and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3512–3518 CrossRef CAS.
- Z. Huang, A. Ding, H. Guo, G. Lu and X. Huang, Sci. Rep., 2016, 6, 25508 CrossRef PubMed.
- P. Yang, J. Yan, H. Sun, H. Fan, Y. Chen, F. Wang and B. Shi, RSC Adv., 2015, 5, 16980–16985 RSC.
- P. Jia, M. Zhang, L. Hu, R. Wang, C. Sun and Y. Zhou, Polymers, 2017, 9, 621 CrossRef PubMed.
- K. W. Lee, J. W. Chung and S. Y. Kwak, Macromol. Rapid Commun., 2016, 37, 2045–2051 CrossRef CAS PubMed.
- P. Jia, L. Hu, G. Feng, C. Bo, M. Zhang and Y. Zhou, Mater. Chem. Phys., 2017, 190, 25–30 CrossRef CAS.
- J. M. Beveridge, H. M. Chenot, A. Crich, A. Jacob and M. G. Finn, Langmuir, 2018, 34, 10407–10412 CrossRef CAS PubMed.
- G. Demirci and M. A. Tasdelen, Eur. Polym. J., 2015, 66, 282–289 CrossRef CAS.
- A. Earla and R. Braslau, Macromol. Rapid Commun., 2014, 35, 666–671 CrossRef CAS PubMed.
- M. Pawlak, E. Grygolowicz-Pawlak and E. Bakker, Anal. Chem., 2010, 82, 6887–6894 CrossRef CAS PubMed.
- Y. Liu, Y. Xue, H. Tang, M. Wang and Y. Qin, Sens. Actuators, B, 2012, 171–172, 556–562 CrossRef CAS.
- M. Pawlak, E. Grygolowicz-Pawlak, G. A. Crespo, G. Mistlberger and E. Bakker, Electroanalysis, 2013, 25, 1840–1846 CrossRef CAS.
- M. Pawlak, G. Mistlberger and E. Bakker, J. Mater. Chem., 2012, 22, 12796–12801 RSC.
- J. Lafarge, N. Kébir, D. Schapman and F. Burel, React. Funct. Polym., 2013, 73, 1464–1472 CrossRef CAS.
- J. Lafarge, N. Kébir, D. Schapman, V. Gadenne and F. Burel, Cellulose, 2013, 20, 2779–2790 CrossRef CAS.
- B. Oktay, ChemistrySelect, 2018, 3, 11737–11743 CrossRef CAS.
- H. Akat and M. Ozkan, eXPRESS Polym. Lett., 2011, 5, 318–326 CrossRef CAS.
- J. W. Guo, Z. Y. Lin, C. J. Chang, C. H. Lu and J. K. Chen, Appl. Surf. Sci., 2018, 439, 313–322 CrossRef CAS.
- A. Ouerghui, H. Elamari, M. Dardouri, S. Ncib, F. Meganem and C. Girard, React. Funct. Polym., 2016, 100, 191–197 CrossRef CAS.
- W. Liu, Y. Dong, S. Zhang, Z. Wu and H. Chen, Chem. Commun., 2019, 55, 858–861 RSC.
- L. Dumas, E. Fleury and D. Portinha, Polymer, 2014, 55, 2628–2634 CrossRef CAS.
- C. Slim, E. Ratajovà, S. Griveau, F. Kanoufi, D. Ferraro, C. Perréard, F. D'Orlyé, A. Varenne and F. Bedioui, Electrochem. Commun., 2015, 60, 5–8 CrossRef CAS.
- Y. Ladner, F. D'Orlyé, C. Perréard, B. Da Silva, C. Guyon, M. Tatoulian, S. Griveau, F. Bedioui and A. Varenne, Plasma Processes Polym., 2015, 60, 5–8 CrossRef.
- C. M. Santos, A. Kumar, S. S. Kolar, R. Contreras-Caceres, A. McDermott and C. Cai, ACS Appl. Mater. Interfaces, 2013, 5, 12789–12793 CrossRef CAS PubMed.
- G. Tillet, G. Lopez, M. H. Hung and B. Améduri, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1171–1173 CrossRef CAS.
- S. Tan, D. Li, Y. Zhang, Z. Niu and Z. Zhang, Macromol. Chem. Phys., 2018, 219, 1700632 CrossRef.
- M. Jaymand, Polym. Chem., 2014, 5, 2663–2690 RSC.
- A. V. Vivek and R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3818–3832 CrossRef CAS.
- G. D. Jones, Ind. Eng. Chem., 1952, 44, 2686–2693 CrossRef CAS.
- W. Guo, L. Xiong, C. M. Reese, D. V. Amato, B. J. Thompson, P. K. Logana and D. L. Patton, Polym. Chem., 2017, 8, 6778–6785 RSC.
- J. C. Brosse, I. Campistron, D. Derouet, A. El Hamdaoui, S. Houdayer, D. Reyx and S. Ritoit-Gillier, J. Appl. Polym. Sci., 2000, 78, 1461–1477 CrossRef CAS.
- N. Ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
- S. Van Der Heijden, K. De Bruycker, R. Simal, F. Du Prez and K. De Clerck, Macromolecules, 2015, 48, 6474–6481 CrossRef CAS.
- J. Huang, L. Zhang, Z. Tang, S. Wu, N. Ning, H. Sun and B. Guo, Macromol. Rapid Commun., 2017, 38, 1600678 CrossRef.
- F. Guo, K. Jankova, L. Schulte, M. E. Vigild and S. Ndoni, Langmuir, 2010, 26, 2008–2013 CrossRef CAS.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- B. Korthals, M. C. Morant-Miñana, M. Schmid and S. Mecking, Macromolecules, 2010, 43, 8071–8078 CrossRef CAS.
- A. Berthold, K. Sagar and S. Ndoni, Macromol. Rapid Commun., 2011, 32, 1259–1263 CrossRef CAS PubMed.
- H. Rosilo, E. Kontturi, J. Seitsonen, E. Kolehmainen and O. Ikkala, Biomacromolecules, 2013, 14, 1547–1554 CrossRef CAS.
- M. W. Thielke, E. P. Bruckner, D. L. Wong and P. Theato, Polymer, 2014, 55, 5596–5599 CrossRef CAS.
- W. Wu, X. Zeng, H. Li, X. Lai and Z. Yan, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51, 229–239 CrossRef CAS.
- J. Xu and C. Boyer, Macromolecules, 2015, 48, 520–529 CrossRef CAS.
- E. Trovatti, T. M. Lacerda, A. J. F. Carvalho and A. Gandini, Adv. Mater., 2015, 27, 2242–2245 CrossRef CAS PubMed.
- A. Muller, Polym. Int., 1996, 41, 251–257 CrossRef.
- M. Kukut, B. Kiskan and Y. Yagci, Des. Monomers Polym., 2009, 12, 167–176 CrossRef CAS.
- R. E. Bagge, T. C. Mauldin, D. J. Boday, B. M. Kobilka and D. A. Loy, Chem. Mater., 2017, 29, 7953–7960 CrossRef CAS.
- N. Y. Ning, Z. P. Zheng, L. Q. Zhang and M. Tian, eXPRESS Polym. Lett., 2015, 9, 490–495 CrossRef CAS.
- W. Wu, X. Zeng, H. Li, X. Lai, F. Li and J. Guo, J. Macromol. Sci., Part B: Phys., 2014, 53, 1244–1257 CrossRef CAS.
- H. Sogawa, T. Tsutsuba, S. Monjiyama, C.-G. Wang and T. Takata, Polym. Chem., 2018, 9, 4382–4385 RSC.
- R. C. Fraser, A. Carletto, M. Wilson and J. P. S. Badyal, ACS Appl. Mater. Interfaces, 2016, 8, 21832–21838 CrossRef CAS PubMed.
- C. Pan and P. Liu, Ind. Eng. Chem. Res., 2018, 57, 4949–4954 CrossRef CAS.
- X. Li, Y. Feng, G. Chu, N. Ning, M. Tian and L. Zhang, Compos. Commun., 2018, 7, 36–41 CrossRef.
- Y. Lei, Z. Tang, L. Zhu, B. Guo and D. Jia, J. Appl. Polym. Sci., 2012, 123, 1252–1260 CrossRef CAS.
- Y. Lei, Z. Tang, L. Zhu, B. Guo and D. Jia, Polymer, 2011, 52, 1337–1344 CrossRef CAS.
- Y. Ye, C. Zhang, M. Tian, Z. Du and J. Mi, J. Phys. Chem. C, 2015, 119, 20957–20966 CrossRef CAS.
- Z. Sun, Q. Huang, Y. Wang, L. Zhang and Y. Wu, Ind. Eng. Chem. Res., 2017, 56, 1471–1477 CrossRef CAS.
- X. Zhang, Z. Tang and B. Guo, Polymer, 2018, 144, 57–64 CrossRef CAS.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 24224–24231 CrossRef CAS PubMed.
- Y. Liu, Z. Tang, Y. Chen, C. Zhang and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 2992–3001 CrossRef CAS PubMed.
- E. Wittenberg and V. Abetz, Polymer, 2017, 121, 304–311 CrossRef CAS.
- E. Wittenberg, A. Meyer, S. Eggers and V. Abetz, Soft Matter, 2018, 14, 2701–2711 RSC.
- M. Tian, H. Yan, H. Sun, L. Zhang and N. Ning, RSC Adv., 2016, 6, 96190–96195 RSC.
- J. Bai, Z. Shi, J. Yin and M. Tian, Macromolecules, 2014, 47, 2964–2973 CrossRef CAS.
- P. Mandal, S. Ponnupandian, S. Choudhury and N. K. Singha, Rubber Chem. Technol., 2017, 90, 550–561 CrossRef CAS.
- K. Lee, J. Lee, S. Choi, K. Char and J. W. Choi, ACS Energy Lett., 2019, 4, 94–101 CrossRef CAS.
- C. Ellingford, R. Zhang, A. M. Wemyss, C. Bowen, T. McNally, Ł. Figiel and C. Wan, ACS Appl. Mater. Interfaces, 2018, 10, 38438–38448 CrossRef CAS.
- L. Liu, D. Li, Y. Xing and N. Li, J. Membr. Sci., 2018, 564, 428–435 CrossRef CAS.
- E. Keleş, B. Hazer and F. B. Cömert, Mater. Sci. Eng., C, 2013, 33, 1061–1066 CrossRef PubMed.
- Q. Z. Zhong, L. W. Zhang, Y. Ou, B. H. Wu, L. S. Wan and Z. K. Xu, Mater. Chem. Front., 2017, 1, 1073–1078 RSC.
- L. M. Polgar, M. Van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- W. Zhao, L. Liu, L. Wang and N. Li, RSC Adv., 2016, 6, 72133–72140 RSC.
- B. Liu, Y. Liu, Y. Wang, H. Man, W. Wang, H. Chen and L. Bai, J. Polym. Res., 2017, 25, 12 CrossRef.
- W. Wang, Y. Wang, Y. Liu, L. Jiang, L. Bai, H. Chen and Z. Cheng, J. Appl. Polym. Sci., 2017, 134, 45490 CrossRef.
- W. xiang Wang, Y. Liu, Y. xue Wang, H. Chen and L. Jiu Bai, Chem. Pap., 2018, 72, 191–200 CrossRef.
- W. Liang, Y. Chenyang, Z. Bin, W. Xiaona, Y. Zijun, Z. Lixiang, Z. Hongwei and L. Nanwen, J. Membr. Sci., 2019, 569, 157–165 CrossRef CAS.
- A. Das and P. Theato, Macromolecules, 2015, 48, 8695–8707 CrossRef CAS.
- J. G. Kim, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2554–2560 CrossRef CAS.
- G. Rydzek, J. S. Thomann, N. Ben Ameur, L. Jierry, P. Mésini, A. Ponche, C. Contal, A. E. El Haitami, J. C. Voegel, B. Senger, P. Schaaf, B. Frisch and F. Boulmedais, Langmuir, 2016, 26, 2816–2824 CrossRef PubMed.
- C. R. Kinnane, G. K. Such and F. Caruso, Macromolecules, 2011, 44, 1194–1202 CrossRef CAS.
- M. Villalba, M. Bossi, M. Del Pozo and E. J. Calvo, Langmuir, 2016, 32, 6836–6842 CrossRef CAS PubMed.
- S. Moulay, Polym.-Plast. Technol. Eng., 2015, 54, 1289–1319 CrossRef CAS.
- D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS.
- N. R. B. Boase, S. T. Smith, K. S. Masters, K. Hosokawa, S. B. Crowe and J. V. Trapp, React. Funct. Polym., 2018, 132, 81–88 CrossRef CAS.
- B. N. Gacal, B. Koz, B. Gacal, B. Kiskan, M. Erdogan and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1317–1326 CrossRef CAS.
- W. Chen, K. Achazi, B. Schade and R. Haag, J. Controlled Release, 2015, 205, 15–24 CrossRef CAS PubMed.
- H. Awada and C. Daneault, Appl. Sci., 2015, 5, 840–850 CrossRef CAS.
- C. Gaina, V. Gaina and D. Ionita, Polym. Bull., 2016, 73, 2019–2038 CrossRef CAS.
- S. Baudis, D. Bomze, M. Markovic, P. Gruber, A. Ovsianikov and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2060–2070 CrossRef CAS.
- B. Oktay, E. Baştürk, M. V. Kahraman and N. K. Apohan, React. Funct. Polym., 2018, 127, 10–19 CrossRef CAS.
- M. H. Alves, C. J. Young, K. Bozzetto, L. A. Poole-Warren and P. J. Martens, Biomed. Mater., 2012, 7, 2 Search PubMed.
- M. Gao, L. Sun, Y. Guo, J. Shi and J. Zhang, Chem. Phys. Lett., 2017, 689, 179–184 CrossRef CAS.
- B. Yameen, M. Ali, M. Álvarez, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Polym. Chem., 2010, 1, 183–192 RSC.
- L. Li, N. Zhao and S. Liu, Polymer, 2012, 53, 67–78 CrossRef CAS.
- H. Coceancigh, K. H. Tran-Ba, N. Siepser, L. A. Baker and T. Ito, Langmuir, 2017, 33, 11998–12006 CrossRef CAS PubMed.
- C. Gaina, V. Gaina and D. Ionita, Polym. Int., 2011, 60, 296–303 CrossRef CAS.
- Y. Xie, R. Tayouo and S. P. Nunes, J. Appl. Polym. Sci., 2015, 132, 41549 Search PubMed.
- Y. He, H. Hoi, C. D. Montemagno and S. Abraham, J. Appl. Polym. Sci., 2018, 135, 46678 CrossRef.
- H. Toiserkani, G. Yilmaz, Y. Yagci and L. Torun, Macromol. Chem. Phys., 2010, 211, 2389–2395 CrossRef CAS.
- C. Xu, S. Gu, J. Huang, W. Xu, H. Xia, J. Du, H. Liu, H. Yang, Y. Zhou and Z. Bai, J. Appl. Polym. Sci., 2015, 132, 41327 Search PubMed.
- S. Gu, H. Xia, J. Du, L. Yang, Y. Cai, Y. Zhou and J. Huang, Fibers Polym., 2016, 17, 161–165 CrossRef CAS.
- G. Yilmaz, H. Toiserkani, D. O. Demirkol, S. Sakarya, S. Timur, Y. Yagci and L. Torun, J. Polym. Sci., Part A: Polym. Chem., 2010, 49, 110–117 CrossRef.
- K. Kim, B. K. Jung, T. Ko, T. H. Kim and J. C. Lee, J. Membr. Sci., 2018, 554, 232–243 CrossRef CAS.
- R. Sood, A. Donnadio, S. Giancola, A. Kreisz, D. J. Jones and S. Cavaliere, ACS Appl. Mater. Interfaces, 2016, 8, 16897–16906 CrossRef CAS PubMed.
- C. Dizman, C. Altinkok and M. A. Tasdelen, Des. Monomers Polym., 2017, 20, 293–299 CrossRef CAS PubMed.
- E. J. Park, W. H. Lee and C. Bae, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3237–3243 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- R. H. Lambeth, S. J. Pederson, M. Baranoski and A. M. Rawlett, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5752–5757 CrossRef CAS.
- B. H. Lipshutz and B. R. Taft, Angew. Chem., 2006, 118, 8415–8418 CrossRef.
- S. I. Presolski, S. K. Mamidyala, F. Manzenrieder and M. G. Finn, ACS Comb. Sci., 2012, 14, 527–530 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |