DOI:
10.1039/C8MT00237A
(Paper)
Metallomics, 2019,
11, 213-225
Cytoprotective effects of imidazole-based [S1] and [S2]-donor ligands against mercury toxicity: a bioinorganic approach†
Received
11th August 2018
, Accepted 12th November 2018
First published on 15th November 2018
Abstract
Here we report the coordination behaviour of an imidazole-based [S1]-donor ligand, 1,3-dimethyl-imidazole-2(3H)-thione (L1), and [S2]-donor ligand, 3,3′-methylenebis(1-methyl-imidazole-2(3H)-thione) (L2) or 4,4′-(3,3′-methylenebis-(2-thioxo-2,3-dihydro-imidazole-3,1-diyl))dibutanoic acid (L3), with HgX2 (X = Cl, Br or I) in solution and the solid state. NMR, UV-Vis spectroscopic, and single crystal X-ray studies demonstrated that L1 or L2 coordinated rapidly and reversibly to the mercury center of HgX2 through the thione moiety. Treatment of L2 with HgCl2 or HgBr2 afforded 16-membered metallacycle k1-(L2)2Hg2Cl4 or k1-(L2)2Hg2Br4 where two Cl or Br atoms are located inside the ring. In contrast, treatment of L2 with HgI2 afforded a chain-like structure of k1-[L2Hgl2]n, possibly due to the large size of the iodine atom. Interestingly, [S1] and [S2]-donor ligands (L1, L2, and L3) showed an excellent efficacy to protect liver cells against HgCl2 induced toxicity and the strength of their efficacy is in the order of L3 > L2 > L1. 30% decrease of ROS production was observed when liver cells were co-treated with HgCl2 and L1 in comparison to those cells treated with HgCl2 only. In contrast, 45% and 60% decrease of ROS production was observed in the case of cells co-treated with HgCl2 and thiones L2 and L3, respectively, indicating that [S2]-donor ligands L2 and L3 have better cytoprotective effects against oxidative stress induced by HgCl2 than [S1]-donor ligand L1. Water-soluble ligand L3 with N-(CH2)3CO2H substituents showed a better cytoprotective effect against HgCl2 toxicity than L2 in liver cells.
Significance to metallomics
Thiols, dithiols and other sulfhydryl-containing ligand interactions play a crucial role in the detoxification of mercury in cellular systems. Clinically used bidentate chelating agents DMPS and DMSA and the endogenous thiol glutathione are known to play significant roles in detoxification of Hg(II) and methylmercury. In this work, we have demonstrated the interaction of thione based [S1] and [S2]-donor ligands, inspired by bidentate chelating agents, with Hg(II) in solution and the solid state and also studied their efficacy to protect hepatocytes against Hg(II) toxicity. Our results showed that [S2]-donor ligands are, indeed, effective in reducing Hg(II) toxicity in liver cells.
|
1. Introduction
Heavy metals, especially mercury (Hg), cadmium (Cd), arsenic (As) and lead (Pb) are extremely toxic to the environment. Mercury and mercury related compounds including inorganic mercury like HgX2 (X = Cl, Br, and I) and organomercurial compound methylmercury (MeHg+), in particular, are highly toxic to humans and animals.1 Although all forms of mercury are toxic, the toxicity profile of mercury compounds depends mostly on the chemical or molecular form, the level of exposure, the duration of exposure, and the route of exposure.2 Mercury ions (Hg2+ or MeHg+) have a high affinity towards thiol or selenol groups of proteins including various essential antioxidant enzymes, thioredoxine reductase (TrxR), glutathione peroxidase (GPx), selenoprotein-P and glutathione reductase (GR).3–7 Most of the mercury, inorganic or organic form, within the various tissues and fluid compartments of mammals is bound to low molecular weight endogenous thiol molecules like L-cysteine and glutathione (GSH), which are present in large concentrations and facilitate the transport of mercury in various organs.8,9 The methylmercury–cysteine complex (MeHgCys) can easily cross the cellular membranes including placental and blood–brain barriers with the help of L-type large neutral amino acid transporter, LAT1.10 On the other hand, GSH is known to play a crucial role in removing inorganic mercury from hepatocytes into bile by forming a mercury–glutathione complex in the hepatocytes followed by the biliary secretion of this complex. Detailed investigation of interrelation between the biliary transport of GSH and of inorganic mercury by Clarkson et al. revealed that the increase in the rate of GSH secretion into bile after GSH administration is accompanied by an increase in the rate of mercury secretion into bile.11 As mercury toxicity is mostly associated with the inhibition of antioxidant enzymes and the reduced GSH concentration in the cellular system, it leads to the production of reactive oxygen species (ROS), causing oxidative damage of DNA, proteins and lipids, and ultimate cell death.12,13
Several bacteria are resistant to both inorganic and organomercury compounds due to the presence of the mercury resistance mer operon that codes for many Mer proteins including MerP, MerG, MerT, MerB, and MerA. The gene merB encodes an organomercurial lyase (MerB) that catalyses the protonolytic cleavage of carbon–mercury bonds. The cytosolic mercuric reductase MerA reduces inorganic mercury, Hg2+, to less toxic elemental Hg(0).14,15
Several synthetic molecules with vicinal thiols such as British antilewiste (BAL), sodium 2,3-dimercaptopropanesulfate (DMPS), and meso-2,3-dimercaptosuccinic acid (DMSA) have shown promising effects in removing mercury and other heavy metals like lead and arsenic and have been clinically used for mercury chelation (Fig. 1).16,17 This is probably due to the fact that Hg2+ might have strong preference for bis-thiolate coordination in addition to the chelate effect provided by these bidentate ligands, which is supported by the presence of the crystal structures of two cysteine coordinated mercury bound MerB, MerP, MerT, and the N-terminal domain of MerA.16,18 Nevertheless, the presence of more Hg2+–thiolate bonds (tri- or tetra-coordinated Hg–S bond) is also known in the literature.19,20 Notably, BAL has been discontinued due to the presence of side effects and low therapeutic index.21a Although DMPS and DMSA are not effective in chelating mercury in the brain, they have shown promising results in enhancing urinary excretion of mercury and thus, currently they are the best choice for treatments for mercury toxicity.21
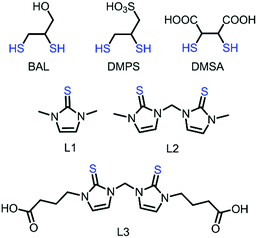 |
| | Fig. 1 Chemical structures of known Hg2+ chelating agents BAL, DMPS, and DMSA, and imidazole-based [S1] and [S2]-donor ligands L1, L2, and L3. | |
Recently, we have reported that imidazole-based [S2]-donor ligand 3,3′-methylenebis(1-methyl-1H-imidazole-2(3H)-thione) (L2) is more effective in cleaving the, otherwise inert, Hg–C bond of MeHg+ at high temperature but, less effective in cleaving the Hg–C bond of different MeHg+ species at room temperature (21 °C). We observed 50% Hg–C bond cleavage of MeHgI whereas only 15% Hg–C bond cleavage in the case of [MeHg]BF4, by L2 at 21 °C.22 Although imidazole based thiones with different sulfur donor groups, like [S2] and [S3]-donor, have recently been broadly studied to investigate their effect on Hg–C bond cleavage of organomercurials, RHg+, the coordination behaviour of [S2]-donor ligand L2 with inorganic mercury compounds HgX2 (X = Cl, Br, or I) in solution and the solid state has not been studied so far.23 Since the liver is the main mercury detoxification organ11d,24 we have employed HepG2 cells in our study to investigate the cytoprotective effects of imidazole-based thiones, 1,3-dimethyl-1H-imidazole-2(3H)-thione (L1), L2 and newly designed water soluble 4,4′-(3,3′-methylenebis(2-thioxo-2,3-dihydro-1H-imidazole-3,1-diyl))-dibutanoic acid (L3), against HgCl2-induced toxicity in cells. Herein, we report the coordination behaviour of [S1] and [S2]-donor ligands L1, L2, or L3 with mercury(II) halides in solution and the solid state. In addition, we have investigated the efficacy of these [S1] and [S2]-donor ligands to protect liver cells against HgCl2-induced toxicity.
2. Results and discussion
2.1 Nature of coordination of [S1] and [S2]-donor ligands
NMR experiment.
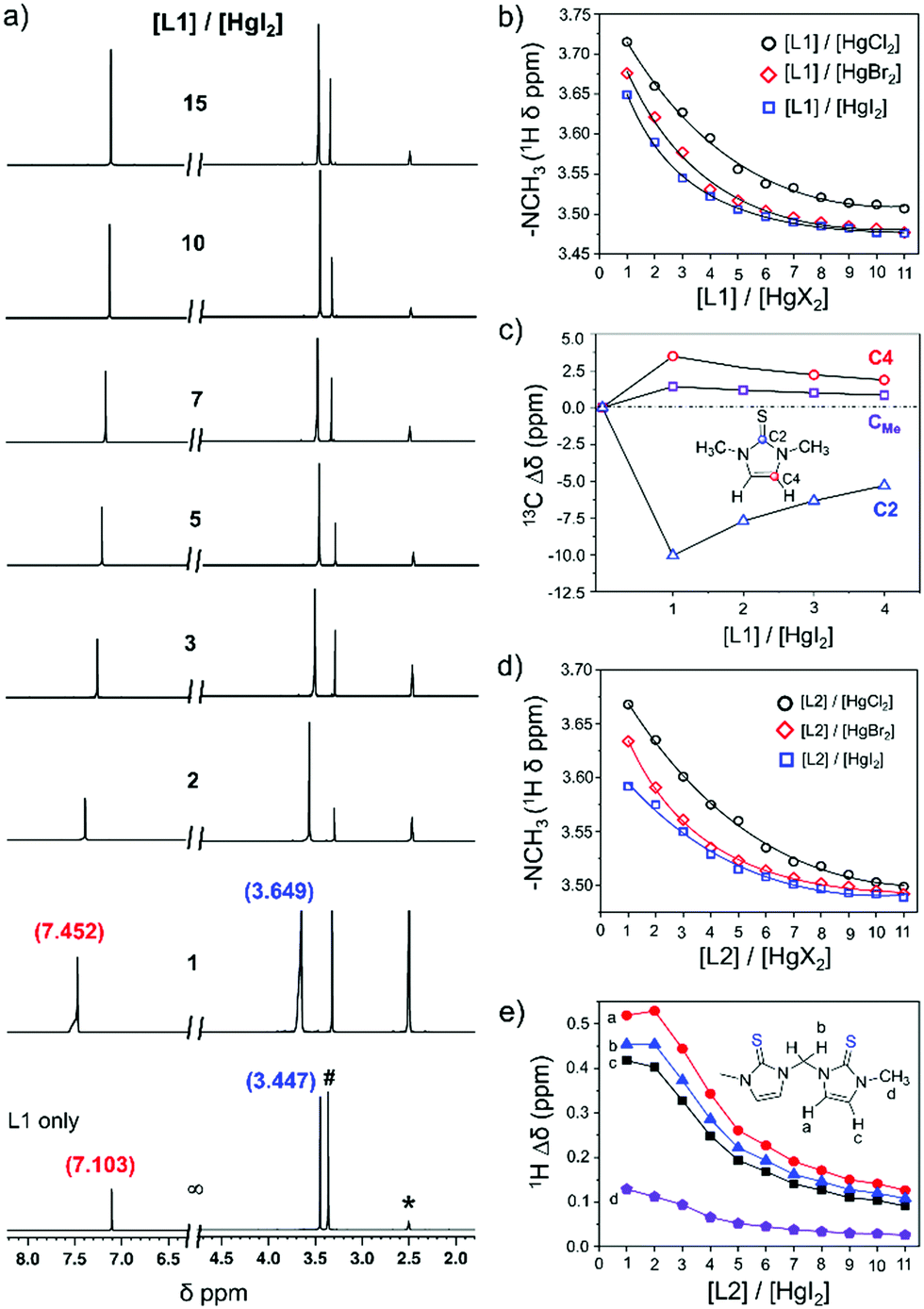
In order to understand the coordination behaviour of N,N-disubstituted imidazole-based [S1] and [S2] donor-ligands like L1 and L2 or L3 toward mercury(II) salts both in solution and the solid state we have employed HgX2 (X = Cl, Br, I) in our study and performed a series of experiments such as NMR, UV-Vis and single crystal X-ray studies as mentioned below. Here, it is pertinent to note that although the crystal structures of analogues of 1-methylimidazoline-2(3H)-thione, and 3,4,5,6-tetrahydropyrimidine-2-thione with mercury(II) halides have been reported in the literature,25 the coordination properties of [S1] and [S2]-donor ligands L1, L2, or L3 in solution and the solid state with HgX2 (X = Br or I), except HgCl2 for L1,25e and the effects of anion (X−; X = Cl, Br or l) on the coordination properties of these ligands toward HgX2 have not been studied. 1H NMR titration experiments between L1 or L2 and HgI2 at room temperature (21 °C) showed a significant downfield shift of proton resonance of both –NCH3 and olefinic protons of L1 or L2, indicating an interaction between the thione group of ligands to the mercury centre of HgI2, Fig. 2 and Fig. S1–S6 (ESI†). The resulting solution of L1 and HgI2, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, showed resonances for protons of –NCH3 and olefin at 3.649 and 7.452 ppm, respectively, which are 0.20 ppm and 0.35 ppm downfield chemical shifts in comparison to the resonances of those protons of free ligand L1 (3.447 ppm for –NCH3 and 7.103 ppm for olefinic proton), Fig. 2a. Interestingly, the gradual addition of excess L1 (1–11 equivalents) into the solution of HgI2 always resulted in a single set of resonances, with gradual shift of the resonances towards free ligand L1, suggesting that the interaction between L1 and Hg2+ is reversible in nature and rapid, faster than the NMR time scale at room temperature.23,26 The variation of proton resonances of –NCH3 proton and olefinic protons upon gradual addition of L1 (1–11 equivalents) into the solution of HgX2 (X = I, Br, or Cl) is summarized in Fig. 2b and Fig. S7 (ESI†). Interestingly, we found that the difference of chemical shift in the presence of 1 equivalent of HgX2 with respect to the free ligand (i.e. Δδ) increased with increasing electronegativity of the X atom, in the order of HgI2 < HgBr2 < HgCl2, as shown in Fig. 3 for Δδ of –NCH3 proton.27 Conversely, unlike proton resonance, in the case of 13C NMR we observed significantly large upfield shift (10.1 ppm) for C2 resonance of L1 upon treatment with HgI2 as illustrated in Fig. 2c and Fig. S8 (ESI†), indicating the decrease in double bond character of C
:1 molar ratio, showed resonances for protons of –NCH3 and olefin at 3.649 and 7.452 ppm, respectively, which are 0.20 ppm and 0.35 ppm downfield chemical shifts in comparison to the resonances of those protons of free ligand L1 (3.447 ppm for –NCH3 and 7.103 ppm for olefinic proton), Fig. 2a. Interestingly, the gradual addition of excess L1 (1–11 equivalents) into the solution of HgI2 always resulted in a single set of resonances, with gradual shift of the resonances towards free ligand L1, suggesting that the interaction between L1 and Hg2+ is reversible in nature and rapid, faster than the NMR time scale at room temperature.23,26 The variation of proton resonances of –NCH3 proton and olefinic protons upon gradual addition of L1 (1–11 equivalents) into the solution of HgX2 (X = I, Br, or Cl) is summarized in Fig. 2b and Fig. S7 (ESI†). Interestingly, we found that the difference of chemical shift in the presence of 1 equivalent of HgX2 with respect to the free ligand (i.e. Δδ) increased with increasing electronegativity of the X atom, in the order of HgI2 < HgBr2 < HgCl2, as shown in Fig. 3 for Δδ of –NCH3 proton.27 Conversely, unlike proton resonance, in the case of 13C NMR we observed significantly large upfield shift (10.1 ppm) for C2 resonance of L1 upon treatment with HgI2 as illustrated in Fig. 2c and Fig. S8 (ESI†), indicating the decrease in double bond character of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S of L1 upon coordination to the mercury center.28,29 In contrast, the C4 (3.5 ppm) and C5 (1.4 ppm) resonances shifted slightly downfield because of the increase in the double bond character of the CN groups in a 5-membered heterocycle ring, Fig. 2c and Table S1 (ESI†).
S of L1 upon coordination to the mercury center.28,29 In contrast, the C4 (3.5 ppm) and C5 (1.4 ppm) resonances shifted slightly downfield because of the increase in the double bond character of the CN groups in a 5-membered heterocycle ring, Fig. 2c and Table S1 (ESI†).
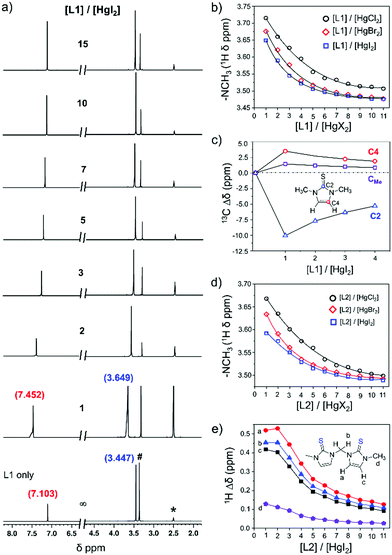 |
| | Fig. 2 (a) Stack spectra of 1H NMR titration of HgI2 (0.05 M) with various equivalents of L1. (b) Variation of the 1H NMR chemical shift of the methyl group (–NCH3) of L1 in the presence of different molar ratios of [L1]/[HgX2]. The concentration of HgX2 was 0.05 M and L1 was varied from 1 to 15 equiv. and ∞ (ligand only). (c) The difference of the 13C NMR chemical shift (Δδ) of the carbon resonances of L1 in the presence of HgI2 (0.05 M) against free Ligand L1. The [L1]/[HgI2] molar ratio was varied from 1 to 4, [L1]/[HgI2] = ∞ (free ligand only). (d) Variation of the 1H NMR chemical shift of the methyl group (–NCH3) of L2 in the presence of HgX2 (0.05 M) as a function of the molar ratio, as mentioned in (b). (e) The difference of 1H NMR chemical shift (Δδ) of –NCH3, –NCH2–N–, and olefinic protons of L2 in the presence of HgI2 (0.05 M) with respect to free Ligand L2. The [L2]/[HgI2] molar ratio was varied from 1 to 11, [L2]/[HgI2] = ∞ (free ligand only). All NMR experiments were performed in DMSO-d6 at 21 °C (* = DMSO-d6, # = water in DMSO-d6). 1H NMR for L1 only: 3.447 ppm for –NCH3 and 7.103 ppm for olefinic proton; 1H NMR for L2 only: 3.463 ppm for –NCH3, 7.118 and 7.395 ppm for olefinic protons, and 6.129 ppm bridged methylene (–NCH2N–) proton. | |
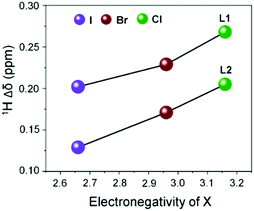 |
| | Fig. 3
1H NMR chemical shift of –NCH3vs. electronegativity of X (X = Cl, Br and I) from (1:1) complexes of L1 and L2 with HgX2. | |
Likewise, similar fluxional behaviour was also noticed in the case of [S2]-donor ligand L2 when it was treated with HgX2 (Fig. 2d). The titration profile between L2 and HgX2 showed significant amounts of downfield shift of resonances of both bridged methylene (–NCH2N–, Hb) and olefinic protons (Ha and Hc), and slight downfield shift of resonance of –NCH3 protons (Hd), Fig. 2e. In a 1:1 molar ratio ([L2]:[HgX2] = 1), the Δδ values for the bridged methylene protons (Hb) were 0.454, 0.454, and 0.447 ppm for HgI2, HgBr2, and HgCl2, respectively. Similarly, for olefinic proton Ha, Δδ values were 0.472, 0.494, and 0.519 ppm and the same values for the Hc proton were 0.418, 0.445, and 0.475 ppm for HgI2, HgBr2, and HgCl2, respectively. In contrast, the Δδ values for –NCH3 (Hd) were 0.129, 0.171, and 0.205 ppm for HgI2, HgBr2, and HgCl2, respectively, indicating a small chemical shift of –NCH3 proton resonance as compared to the bridged methylene and olefinic protons.
Significantly, on treatment of one equivalent of HgCl2, the C2 resonance of [S1]-donor ligand L1 was shifted significantly more in comparison to the C2 or C2′ (both CS moieties of L2 are equivalent) resonance of [S2]-donor ligand L2, Table 1 and Fig. 4a. 13.5 ppm upfield shift of the C2 resonance of L1 was observed against 7.33 ppm upfield shift of the C2 and C2′ resonances of L2, showing that the two CS groups of L2 are coordinating to the mercury centre of HgCl2 to an equal extent but to a lesser extent than the single CS group of L1. Furthermore, we have performed 199Hg NMR experiments to understand the nature of coordination between the sulphur atom of L1 or L2 to the mercury centre of HgCl2 as 199Hg NMR chemical shifts provide a sensitive probe for the complexation between them. Treatment of one equivalent of L2 into the solution of HgCl2 has shifted the 199Hg mercury NMR peak position drastically from −1501 ppm to −878 ppm (Δ199Hg = 632 ppm), showing the possibility of immediate formation of k2-fashioned di-sulfur coordinated 1:1 complex k2-L2HgCl2 which further converted into a thermodynamically more stable k1-fashioned di-sulfur coordinated 2:2 complex (k1-L2)2Hg2Cl4 (Table 1, Fig. 4b and Fig. S3, ESI,†Scheme 1), confirmed by single crystal X-ray experiment (vide infra). DFT calculations also suggest that k1-(L2)2Hg2Cl4 is almost 28.5 kcal mol−1 more stable than k2-L2HgCl2. In contrast, the treatment of one equivalent of L1 into the solution of HgCl2 shifted the 199Hg mercury NMR peak to 360 ppm only (from −1501 ppm to −1141 ppm), almost half of the Δ199Hg value for L2. However, addition of one more equivalent of L1 shifted the 199Hg mercury NMR position further to −902 ppm, indicating the formation of a di-sulfur coordinated 2:1 complex of (L1)2HgCl2 (Fig. S9, ESI†).28
Table 1 Variation of 13C NMR of the C2-carbon and 199Hg chemical shift values for titrationa of HgCl2 with L1 and L2 in DMSO-d6
| ([L]/[M]) |
L1 and HgCl2 complex (δ ppm) |
L2 and HgCl2 complex (δ ppm) |
|
13C of C2 |
199Hgb |
Δ199Hg |
13C of C2 |
199Hg |
Δ199Hg |
|
All NMR experiments were carried out at room temperature (21 °C) in DMSO-d6, [HgCl2] = 0.1 M and ligand concentrations were varied from 0.1 M to 0.4 M.
199Hg NMR of HgCl2 appeared at −1501 ppm. ∞ = ligand only.
|
| ∞ |
161.8 |
— |
|
162.73 |
— |
|
| 1 |
148.3 |
−1141 |
360 |
155.40 |
−878 |
623 |
| 2 |
151.6 |
−902 |
599 |
154.45 |
−900 |
601 |
| 3 |
153.8 |
−874 |
627 |
159.9 |
−869 |
632 |
| 4 |
156.1 |
−860 |
641 |
161.13 |
−867 |
634 |
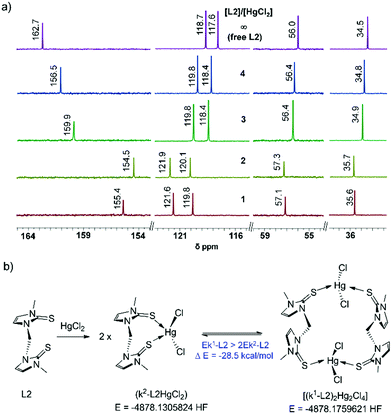 |
| | Fig. 4 (a) 13C NMR chemical shift variations of L2 (1 to 4 equiv.) in the presence of HgCl2 (1 equiv.). All 13C NMR experiments were performed in DMSO-d6 at 21 °C. (b) Scheme for an energetically stable complex of L2 with HgCl2. | |
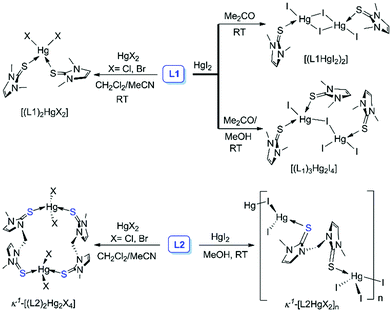 |
| | Scheme 1 Synthetic routes for the formation of (L1HgI2)2, (L1)3Hg2I4, (L1)2HgCl2, (L1)2HgBr2, k1-(L2)2Hg2Cl4, k1-(L2)2Hg2Br4 and k1-[L2HgI2]n. | |
UV-Vis experiment.
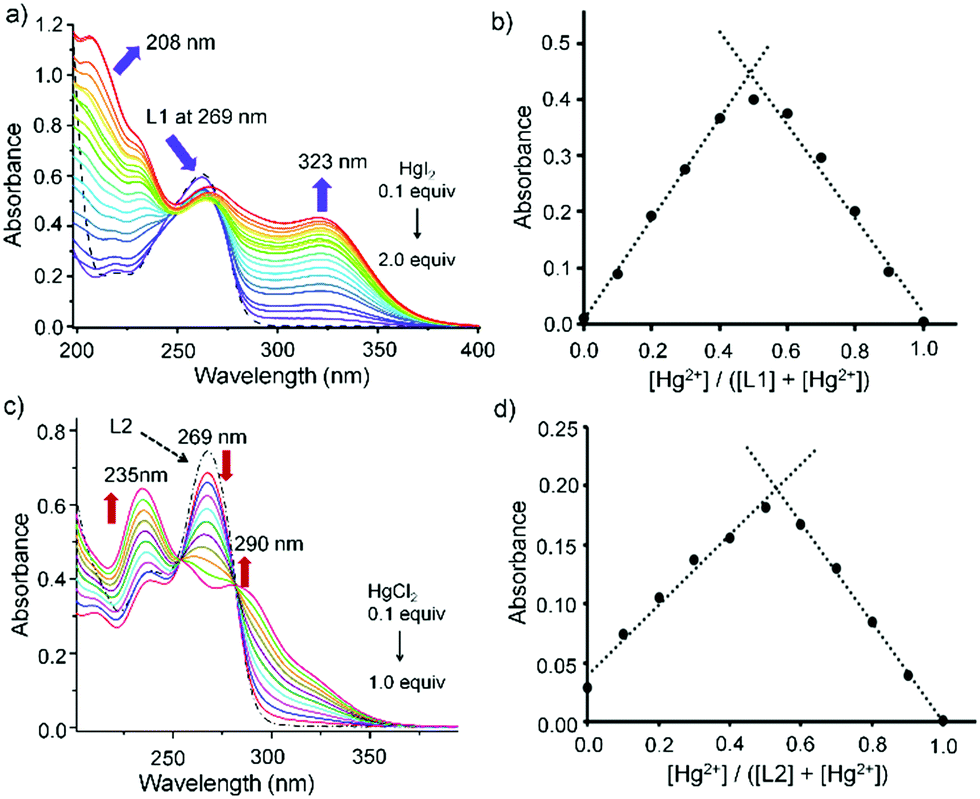
The nature of interaction between L1 or L2 with HgX2 was monitored by UV-Vis spectrophotometer by following the ligand to metal charge transfer transition bands, S → Hg, in the solution state. In solution both the thiones L1 and L2 showed an absorption band at 269 nm, which was attributed to n → π* electronic transition of CS and a shoulder absorption band at 235 nm corresponding to the π → π* electronic transition of the ligands (Fig. 5).30 Treatment of HgX2 into the solution of L1 or L2 (50 μM) showed a lower energy band that appeared in the range of 290 to 330 nm, which could be attributed to the ligand-to-metal charge-transfer (LMCT) band (Fig. S10, ESI†). For instance, upon gradual addition of HgI2 (0.1–2.0 equiv.) to the solution of L1 (50 μM), a new LMCT band at 323 nm gradually increased whereas the band at 269 nm of L1 decreased substantially (Fig. 5a and Fig. S10, ESI†). The Job's plot, obtained by varying the concentrations of L1 and HgI2, suggested the possible formation of 1:1 adduct L1HgI2 in solution with an inflection point at 0.5 with respect to 323 nm (Fig. 5b), which converted into a thermodynamically more stable product (L1HgI2)2, a dimer of L1HgI2, confirmed by single crystal X-ray study (vide infra). Likewise, gradual addition of HgCl2 (0.1–1.0 equiv.) into the solution of L2 showed the increase of an LMCT peak at 290 nm and the decrease of the peak at 269 nm of L2 (Fig. 5c and Fig. S12e, ESI†). Titration spectra of L2 and HgCl2 showed the generation of a new peak at 290 nm, which saturated with one equivalent of HgCl2 with the standard isobestic point, as shown in Fig. 5c. Formation of a 1:1 complex in solution was observed from the Job's plot of L2 and HgCl2, which showed an inflection point at 0.5 with respect to 290 nm (Fig. 5d), indicating the possible formation of a 1:1 complex of k2-L2HgCl2 in solution which later converted into a thermodynamically stable product of a 2:2 complex of k1-(L2)2Hg2Cl4 (vide infra). Interestingly, from the Job's plot, the initial formation of a 1:1 complex in solution was also noticed when L1 was treated with HgCl2, showing possible formation of a 1:1 adduct of L1HgCl2 (Fig. S11, ESI†), like L1HgI2, which slowly converted into a thermodynamically stable product of a 2:1 complex of (L1)2HgCl2 as confirmed by single crystal X-ray experiment.
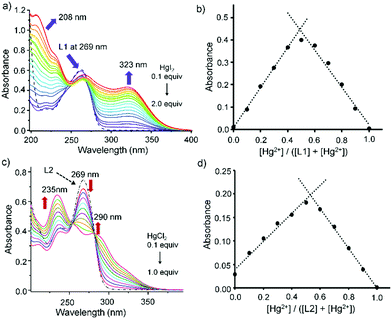 |
| | Fig. 5 (a) UV titration of L1 (5 × 10−5 M) with HgI2 up to 1 equiv. in acetonitrile. (b) Job's plot for the L1 (5 × 10−5 M) and HgI2 complex system. (c) UV titration of L2 (5 × 10−5 M) with HgCl2 (0.1–1 equiv.) in ACN. (d) Job's plot for the L2 (5 × 10−5 M) and HgCl2 complex system. | |
Single crystal X-ray study.
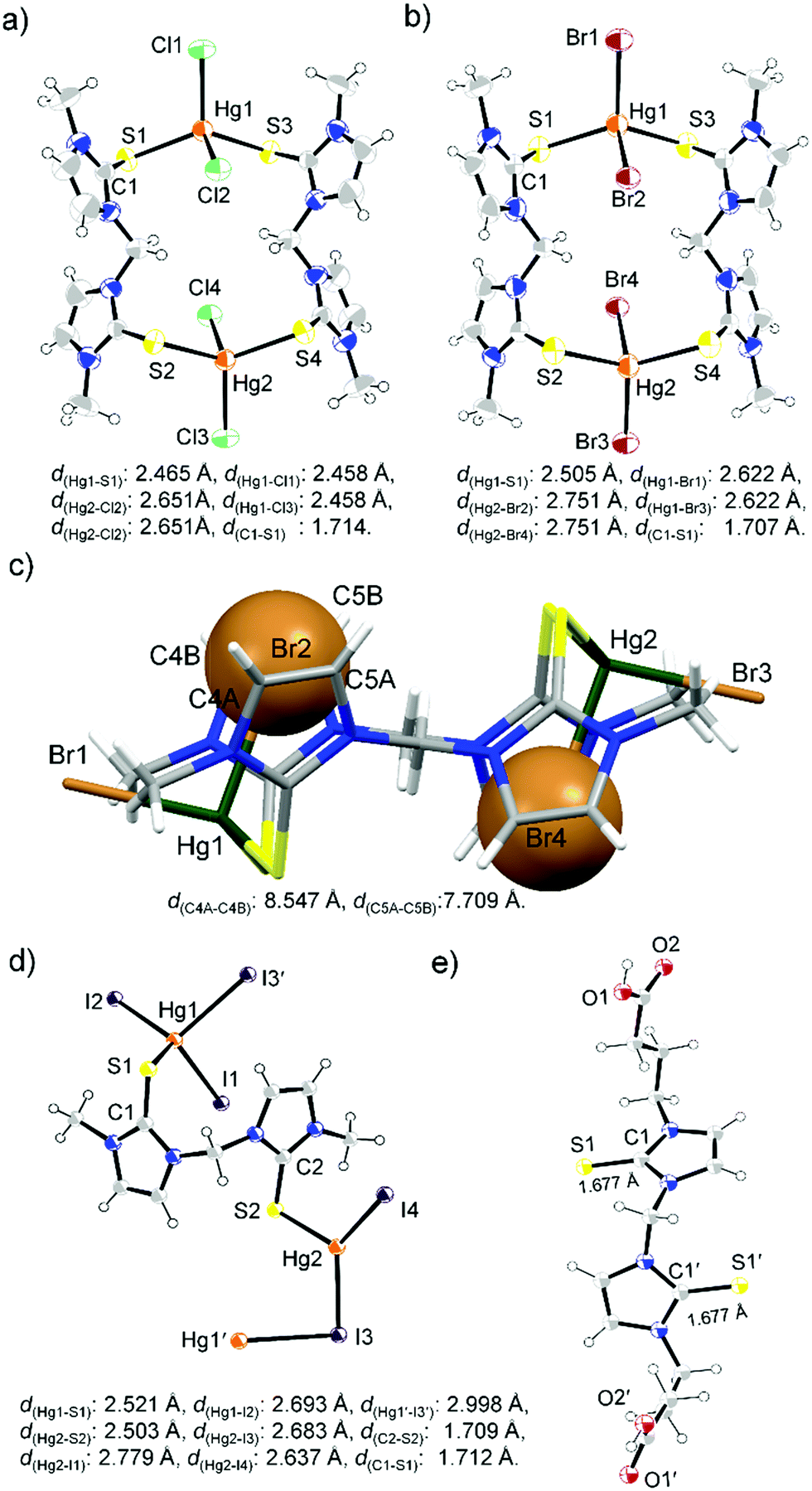
Considering the solution state coordination behaviour we intended to study the structural properties of the complexes between L1 and L2 with HgX2 in the solid state. Slow evaporation of the acetone solution of L1 and HgI2, 1:1 molar ratio, at room temperature afforded the formation of 1:1 complex [L1HgI2]2 (CCDC number: 1857557†), whereas excess L1 (4 equiv.) led to the formation of an unusual crystal of (L1)3Hg2I4, as illustrated in Scheme 1 and Fig. 6. The single crystal X-ray structure of [L1HgI2]2 showed the formation of a diamond shaped bridge between two asymmetric units of L1HgI2 through a weak covalent interaction between Hg and I atoms resulting in the formation of two different types of Hg–I bonds (d(Hg1–I1) = d(Hg1′–I1′): 2.694 Å; and for bridged Hg–I bonds d(Hg1–I2) = d(Hg1′–I2′): 2.844 Å and d(Hg1–I2′) = d(Hg1′–I2): 2.973 Å). The S atom of L1 strongly coordinated to the Hg centre with Hg–S and C–S bond distances of 2.471 Å and 1.705 Å, respectively, in [L1HgI2]2, indicating a slight elongation of the C–S bond upon coordination to the Hg centre in comparison to that in free L1 (d(CS): 1.689 Å). The formation of a binuclear complex might be due to the weak mercurophilic interaction between the two Hg centres (d(Hg1–Hg1′): 4.235 Å) of [L1HgI2]2.31 The crystal packing showed strong intermolecular hydrogen bonding (H-bonding) between the H atom of –NCH3 and the bridged iodine atom (d(H⋯l2): 3.137 Å), Fig. S13 (ESI†). The unusual crystal of (L1)3Hg2I4 formed due to a weak covalent interaction between the two unsymmetrical units, (L1)2HgI2 and L1HgI2, through the iodine atom of (L1)2HgI2 to the Hg atom of L1HgI2, leading to the distorted tetrahedral geometry at the Hg centres, with τ4 values of 0.81 (Hg1), and 0.87 (Hg2), Fig. 6 and Table 2. As a result, the bridged I atom formed longer Hg–I bonds (d(Hg1–I2): 3.222 Å; d(Hg2–I2): 2.811 Å) in comparison to the other Hg–I bonds in the complex (d(Hg1–I1): 2.689 Å; d(Hg2–I3): 2.745 Å; d(Hg2–I4): 2.765 Å). Three L1 units are coordinated unsymmetrically to the Hg centres with three different Hg–S bond lengths (d(Hg1–S1): 2.465 Å; d(Hg1–S2): 2.651 Å, d(Hg2–S3): 2.458 Å). The crystal packing of (L1)3Hg2I4 showed a chain like structure with intermolecular S⋯I interactions (Fig. S14, ESI†). On the other hand, mononuclear (L1)2HgX2, X = Cl25e or Br, types of crystals were obtained when L1 was treated with 0.5 or 1 equivalent of HgX2 in an ACN/DCM solvent mixture (1:1) at room temperature, as shown in Fig. 6c. (L1)2HgBr2 formed strong intermolecular H-bonding between the Br-atom of one molecular unit with the H-atom of –NCH3 and olefinic H atom of another molecular unit (d(Br1⋯Hb): 2.807 Å; d(Br⋯Ha): 2.904 Å) leading to the formation of a dimeric structure, as illustrated in Fig. 6d.
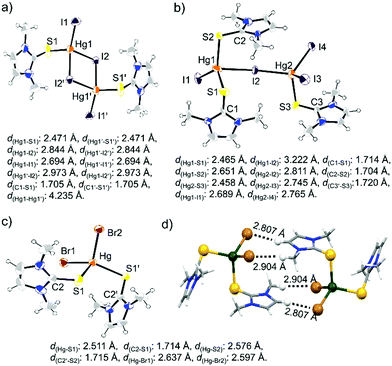 |
| | Fig. 6 Molecular structure of (a) [L1HgI2]2, (b) (L1)3Hg2I4, and (c) (L1)2HgBr2. (d) Image showing intermolecular H-bonding interaction between two units of (L1)2HgBr2. | |
Table 2 The Hg–S and Hg–X bond lengths and τ4 values in the complexes
| Compounds |
Bond length |
τ
4
|
| Hg–S (Å) |
Hg–X (Å) |
|
[L1HgI2]2
|
2.471 |
2.694, 2.844, 2.973 |
0.89 |
|
(L1)3Hg2I4
|
2.465, 2.651, 2.458 |
2.689, 3.222, 2.811, 2.745, 2.765 |
0.81, 0.87 |
|
(L1)2HgBr2
|
2.511, 2.576 |
2.637, 2.597 |
0.89 |
|
k
1-(L2)2Hg2Cl4
|
2.465 |
2.458, 2.651 |
0.90 |
|
k
1-(L2)2Hg2Br4
|
2.505 |
2.622, 2.751 |
0.88 |
|
k
1-[L2Hg2I2]n
|
2.503, 2.521 |
2.637, 2.683, 2.693, 2.779 |
0.91 |
Unlike L1, [S2]-donor ligand L2 afforded a chain or ring-like structure in the solid state on treatment with one equiv. of HgX2. X-ray structure analysis of the yellow colour diamond shaped single crystal of the complex between L2 and HgI2 confirmed the formation of a polymeric structure of k1-[L2HgI2]n, where two S atoms of L2 interact with the two geometrically different mercury centers, as shown in Fig. 7d. On the other hand, in the case of HgX2 where X = Cl or Br, we observed the formation of 16-membered metallacycle k1-(L2)2Hg2X4, with two mercury centres that are geometrically equivalent, in a tetrahedral geometry with τ4 values of 0.90, and 0.88 for k1-(L2)2Hg2Cl4 and k1-(L2)2Hg2Br4, respectively, Fig. 7a and b. In k1-(L2)2Hg2Cl4, the S atoms of two L2 ligands coordinated symmetrically (d(Hg–S1): 2.465 Å) with Hg centres of two HgCl2 units leading to the formation of a 16-membered ring-like structure, in which two Cl atoms are located inside of the metallocycle (sandwiched between the two 5-membered imidazole rings) with significantly elongated Hg–Cl bond lengths (d(Hg1–Cl2) = d(Hg1–Cl4) = 2.651 Å) and the other two Cl atoms are located outside of the ring with shorter Hg–Cl bond lengths (d(Hg1–Cl1) = d(Hg1–Cl3) = 2.458 Å) (Fig. S15, ESI†). Similar to k1-(L2)2Hg2Cl4, we also observed two different types of Hg–Br bonds in k1-(L2)2Hg2Br4, d(Hg1–Br1) = d(Hg1–Br3) = 2.622 Å; d(Hg1–Br2) = d(Hg1–Br4) = 2.751 Å. Br1 and Br3, oriented at the outside of the 16-membered metallacycle, formed a shorter Hg–Br bond, whereas Br2 and Br4 are sandwiched between two 5-membered imidazole rings of the metallacycle, as shown in Fig. 7c, and formed longer Hg–Br bonds. Crystal packing arrangement of k1-(L2)2Hg2Br4 showed that Br2 or Br4 are involved in strong H-bonding interaction with H-atoms of –NCH3 & olefin-H of another unit of the metallacycle, whereas the outer Br atom (Br1 or Br3) participated in halogen bonding (Br⋯S interaction) interaction with a S atom of another unit of the metallacycle, as shown in Fig. S16 (ESI†).
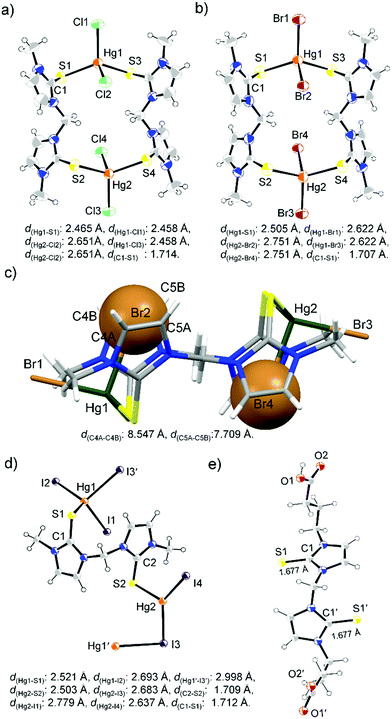 |
| | Fig. 7 ORTEP images of 16-membered metallacycles k1-(L2)2Hg2Cl4 (a) (solvent DMSO is omitted for clarity), and k1-(L2)2Hg2Br4 (b). (c) Mercury image of k1-(L2)2Hg2Br4 showing that two Br atoms are sandwiched between two imidazole rings. ORTEP images of the polymeric structure of k1-[L2HgI2]n (d) and L3 (e). | |
2.2 Cytoprotective effects of [S1] and [S2]-donor ligands against Hg(II) induced toxicity
Cytotoxicity.
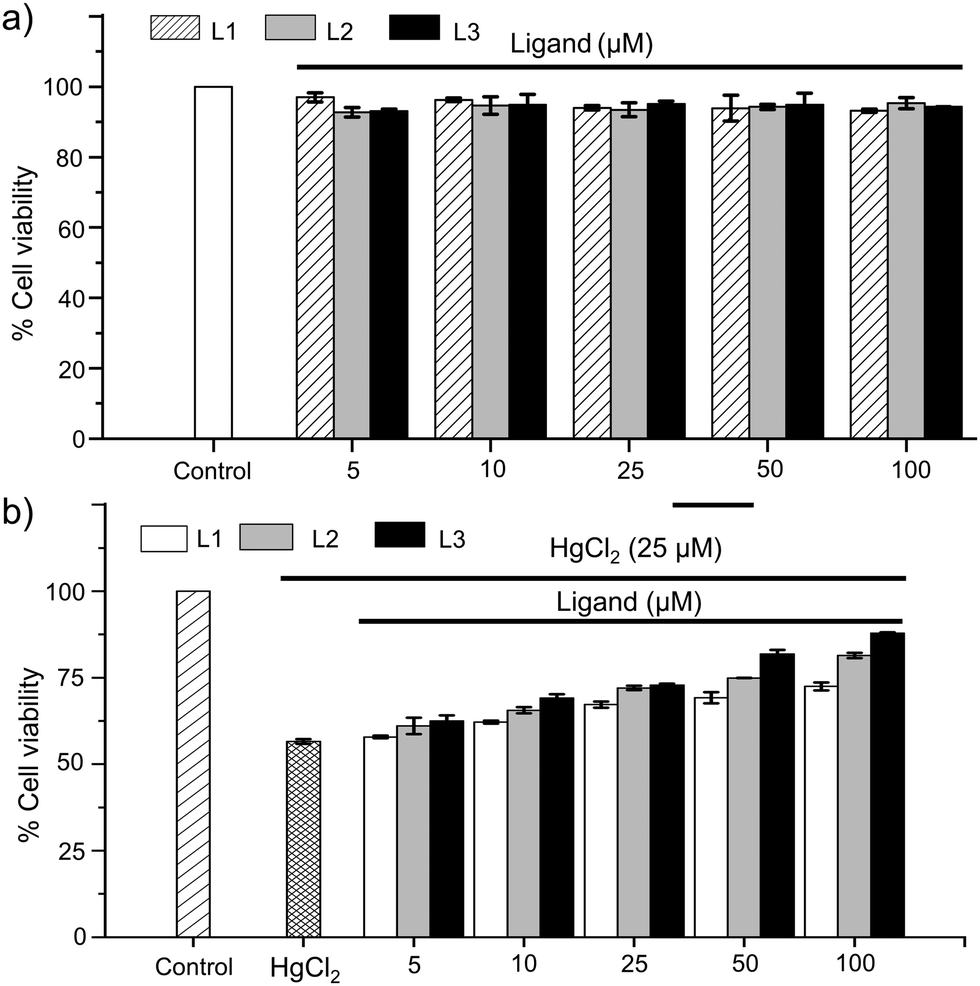
After detailed studies to understand the nature of coordination of [S1] and [S2]-donor ligands to the mercury center of various Hg(II) salts in both solution and solid states, we investigated the protective effect of these imidazole-based thiones against Hg(II) induced toxicity in a cellular system. To examine the protective effect of the imidazole-based thiones against Hg(II)-induced toxicity in hepatocytes we have also employed another new N,N-disubstituted imidazole-based [S2]-donor ligand L3 with –N(CH2)3CO2H substituents. The synthetic procedure of L3 is mentioned in the supporting information and the crystal structure is shown in Fig. 6e. First, we determined the cytotoxicity of these imidazole-based thiones in human HepG2 cells. To investigate the cytotoxic effect, cells (1.0 × 104) were seeded in 96 well-plates and incubated with various concentrations of ligands (0–100 μM) for 24 h and their toxic effects were analysed using standard MTT assays.32a To our delight, we found that these thione based ligands are not cytotoxic to HepG2 cells even up to 100 μM concentrations, and more than 90% of cell viability was observed in the presence of 100 μM ligands, as illustrated in Fig. 8a, suggesting that these thiones can be used safely even up to 100 μM concentration to study their protective effect against Hg(II) toxicity. Next we investigated the cytotoxic effect of HgCl2 in a dose dependent manner in HepG2 cells. As mentioned above, cells (1.0 × 104) were seeded into 96 well-plates and incubated with various concentrations of HgCl2 (0–50 μM) for 24 h to analyse its cytotoxic effect.32b Almost 90% cell death was observed at 50 μM and ∼50% cell survival was observed at 25 μM concentration of HgCl2 (IC50 = 25 μM) and, thus, 25 μM concentration of HgCl2 was used to study the protective effect of thiones (L1, L2 or L3) against HgCl2 toxicity (Fig. S28, ESI†). Cells were co-treated with HgCl2 (25 μM) and various concentrations of thiones (0–100 μM) and incubated for 24 h. In the case of [S1]-donor ligand L1, we have observed only 10% and 15% protection of HepG2 cells at 50 μM and 100 μM concentrations of L1, respectively, in comparison to the cells treated with 25 μM HgCl2 only. Whereas in the case of [S2]-donor ligands, we have observed up to 25% and 35% protection at 100 μM concentration of L2 and L3, respectively, as illustrated in Fig. 8b. These observations clearly suggest that [S2]-donor ligands L2 and L3 have greater protective effects in comparison to the [S1]-donor ligand L1. Again, the among [S2]-donor ligands, L3 has more protective effect than L2 against HgCl2 toxicity in liver cells, indicating that L3 possibly coordinates effectively with Hg2+ ions in a cellular system and protects enzymes, proteins and GSH from Hg2+.
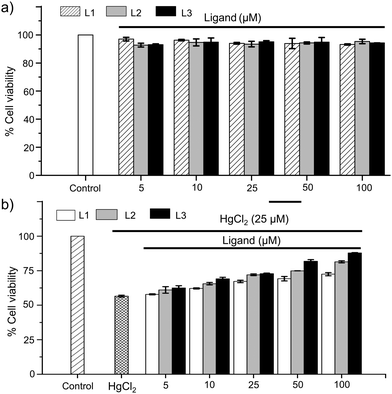 |
| | Fig. 8 (a) Effect of L1, L2 and L3 (5–100 μM) on cell viability in HepG2 cells. (b) Percentage of cell viability of HepG2 cells treated with HgCl2 (25 μM) and co-treated with HgCl2 (25 μM) and various amounts of ligands (5–100 μM). | |
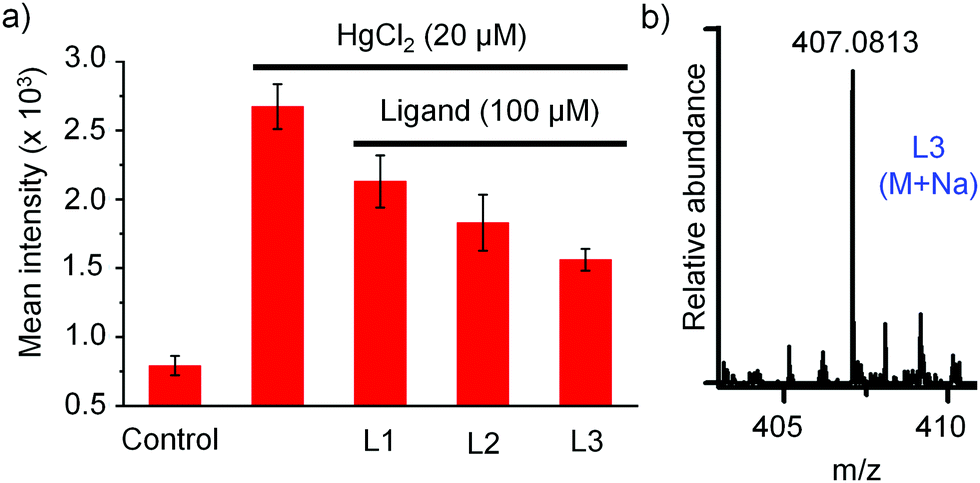
Intracellular ROS estimation.
It is well-known that the inorganic mercury toxicity is mainly due to its strong affinity toward thiol and selenol containing proteins including many vital antioxidant enzymes like thioredoxin (Trx), thioredoxin reductase (TrxR) and glutathione peroxidase (GPx), glutathione reductase (GR), and endogenous thiols including GSH and L-CysH and thereby it reduces the concentration of GSH, an important antioxidant, thiol containing tripeptide present in living cells, leading to the production of reactive oxygen species (ROS) in various tissues which causes DNA damage, protein oxidation, and lipid peroxidation. Overproduction of ROS within the cells leads to a situation called oxidative stress, which ultimately leads to cell death.6,11,12 When cells were treated with 20 μM HgCl2 for 2 h followed by the treatment with DCFH-DA for 0.5 h, we observed remarkably strong fluorescence signal of DCF due to the production of huge amounts of ROS in HgCl2-treated HepG2 cells in comparison to that observed in untreated cells (UT), as shown in Fig. 9a and b. In accordance, the bright field image of HgCl2-treated HepG2 cells confirmed that the cells were completely stressed in the presence of 20 μM HgCl2. However, co-treatment of cells with 20 μM of HgCl2 and 100 μM of ligand (HgCl2/Ln hereafter) decreased the production of ROS in the cells and as a result we observed a significant amount of decrease of fluorescence intensity in co-treated cells, Fig. 9c–e and 10a. The mean intensity profile showed ∼30% decrease of ROS production in cells co-treated with HgCl2/L1 in comparison to the cells treated with HgCl2 only, as indicated by weaker fluorescence signal of DCF. However, to our delight, in the case of [S2]-donor ligands, we observed ∼45% and ∼60% decrease of ROS production in cells co-treated with HgCl2/L2 and HgCl2/L3, respectively, in comparison to the cells treated with HgCl2 only, as indicated by much weaker fluorescence signal of DCF. These observations strongly suggest that [S2]-donor ligands L2 and L3 have more cytoprotective effect against HgCl2 induced toxicity, 1.5 and 2 times, respectively, in comparison to L1. Interestingly, in accordance with the ROS results, the bright field images confirmed that the cells are not in stress and, moreover, the shapes of the HepG2 cells remained intact when they were co-treated with HgCl2 and 100 μM of L2 or L3.
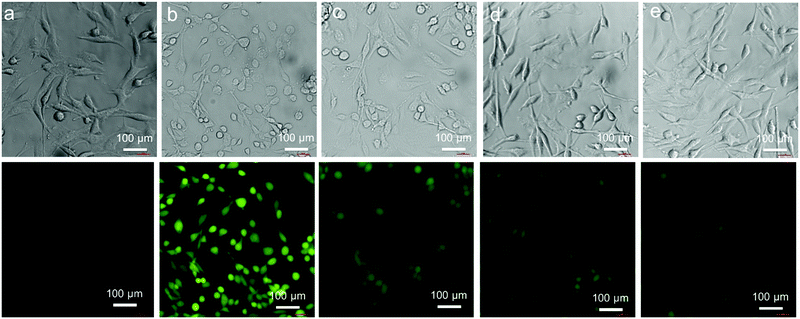 |
| | Fig. 9 Bright field (top) and the corresponding fluorescence images (bottom) of untreated (a), 20 μM HgCl2 (b), 20 μM HgCl2 + 100 μM L1 (c), 20 μM HgCl2 + 100 μM L2 (d), and 20 μM HgCl2 + 100 μM L3 (e) treated HepG2 cells. | |
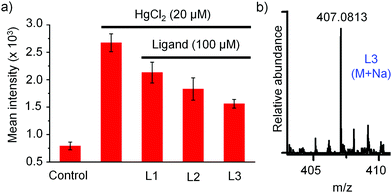 |
| | Fig. 10 (a) The relative mean intensity profile of ROS production in HeG2 cells treated with HgCl2 and co-treated with HgCl2 plus ligands (L1, L2 or L3). (b) HRMS of L3 detected in cell lysates. | |
In order to investigate the internalization of the active compound L3 into the HepG2 cells we have incubated cells with L3 (100 μM) for 4 h and performed HRMS-QTOF analysis with cell lysates after thorough washing of cells with PBS buffer. A detailed experimental procedure is mentioned in the ESI.† Identification of L3 in cell lysates by mass spectrometry [L3: m/z for (M + Na) = 407.0813], Fig. 10b, confirmed internalization of L3 into the HepG2 cell. Next we investigated the protecting effect of L3 due to the intracellular complexation with mercury. For this, at first, cells were incubated in the absence (control experiment) and presence of L3 (100 μM) for various times (4 h, 12 h, and 24 h) in 6-well plates. After incubation of cells with L3 at various times, the medium was completely removed, the cells were washed with PBS buffer and then fresh medium was added in 6-well plates to treat the cells further with HgCl2. Cells were incubated with HgCl2 (20 μM) for 2 h and then the production of ROS in the cells was measured using DCFH-DA as mentioned and compared with that observed in untreated cells, as shown in Fig. S29 (ESI†). The mean fluorescence intensity of the DCF profile, Fig. S30 (ESI†), showed 15%, 28%, and 36% reduction of ROS level in cells pre-treated with L3 for 4 h, 12 h and 24 h, respectively, in comparison to the untreated cells.
3. Conclusions
In summary, we demonstrated the coordination behaviour of [S1] and [S2]-donor ligands such as L1, L2 or L3 with mercury(II) halides in solution and the solid state by NMR, UV-Vis, and single crystal X-ray diffraction studies. NMR studies revealed that L1 or L2 ligand coordinated rapidly and reversibly to the mercury center of HgX2 (X = Cl, Br, or I) at room temperature. A significant chemical shift of the 1H and 13C resonance of the ligands was observed upon coordination to the mercury center of HgX2 in comparison to the free ligand, indicating strong interaction between [S1] or [S2]-donor ligands with Hg2+ in solution. The UV-Vis titration and Job's plot confirmed that [S1]-donor ligand L1 formed a 1:1 complex with HgI2 in solution when both were mixed in a 1:1 molar ratio at room temperature. Single crystal X-ray studies also confirmed the formation of a 1:1 bimetallic complex of [L1HgI2]2 in the solid state when L1 and HgI2 reacted in a 1:1 molar ratio. However, addition of excess L1 into the solution of HgI2 afforded (L1)3Hg2I4, a 3:2 bimetallic complex. In contrast, treatment of [S2]-donor ligand L2 to the solution of HgI2, in a 1:1 molar ratio, afforded a polymeric structure of k1-[L2HgI2]n. Interestingly, treatment of L2 with HgX2 (X = Cl or Br) yielded stable sixteen-membered neutral binuclear metallacycle k1-(L2)2Hg2Cl4 or k1-(L2)2Hg2Br4 where two Cl or Br atoms are sandwiched in between the two five-membered imidazole rings of the metallacycle. However, possibly due to the large size of iodine, the mixture of L2 and HgI2 afforded a stable polymeric structure of k1-[L2HgI2]n. In vitro investigation of the cellular toxicity of ligands L1, L2 and L3 in HepG2 cells suggests that these ligands are not cytotoxic to human liver cells. Water-soluble [S2]-donor ligand L3 with N-(CH2)3CO2H substituents showed excellent cytoprotective effects against HgCl2 induced toxicity in hepatocytes. In MTT assay, 35% more cell survival was observed when HepG2 cells were co-treated with HgCl2 (25 μM) and L3 (100 μM) in comparison to that in the case of cells treated with HgCl2 (25 μM) alone. Fluorescence imaging study, for the estimation of ROS production induced by HgCl2, in HepG2 cells demonstrated that L3 has an excellent property to reduce the oxidative stress in a cellular system, mostly through binding to HgCl2.
4. Experimental section
4.1 General experimental
Methyl imidazole, diiodomethane, sulfur powder, 4-bromo butyrate MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], DCFH-DA (2′,7′-dichlorofluorescin diacetate) and DMSO-d6 were purchased from Sigma-Aldrich. Mercuric chloride, mercuric bromide, and mercuric iodide were purchased from CDH chemicals and other chemicals were purchased from local companies. The L1 and L2 ligands were prepared by following the literature procedure,33–35 and L3 was synthesized in a new synthetic method (ESI†). All the synthetic experiments were carried out under anhydrous and anaerobic conditions using standard Schlenk techniques for the synthesis. Mass spectrometric analysis was carried out using an Agilent 6540 accurate mass Q-TOF HRLC/MS equipped with an electrospray ionisation source (ESI). 1H (400 MHz), 13C (100 MHz), and 199Hg (71.6 MHz) NMR spectra were obtained on a Bruker Advance 400 MHz NMR spectrometer using the solvent as an internal standard for 1H and 13C. Chemical shifts (1H, 13C) are cited with respect to tetramethylsilane (TMS). The 199Hg NMR spectra are reported in ppm relative to neat Me2Hg (δ = 0 ppm) and HgCl2 (δ = −1501 ppm for 1 M solution in DMSO-d6 at 21 °C) was used as an external standard. Electronic spectra were recorded on a UV-1800 Shimadzu UV-Vis spectrophotometer at 298 K in acetonitrile.
4.2 Synthesis
Synthesis of [L1HgI2]2.
To a stirred solution of HgI2 (100 mg, 0.21 mmol) in acetone a solution of L1 (27 mg, 0.21 mmol) in acetone was added and the reaction mixture was stirred at room temperature for 0.5 h. Upon slow evaporation of the reaction solution, a pale yellow coloured crystalline product was obtained. Yield: 89.2 g (73%). 1H NMR (DMSO-d6) δ = 3.68 (s, 12H), 7.51 (s, 4H), 13C NMR (DMSO-d6) δ = 35.8, 121.5, 151.7. HR-ESIMS (m/z): calcd for [M]+ C10H16N4S2Hg2I4 = 1165.638, observed value: [M − HgI3]+ = 584.9590.
Synthesis of (L1)3Hg2I4.
To a stirred solution of HgI2 (100 mg, 0.21 mmol) in acetone a solution of excess L1 (108 mg, 0.84 mmol) in methanol was added and reaction mixture was stirred at room temperature for 0.5 h. The reaction solution was removed and washed with dichloromethane to get a pale yellow coloured crude product. The complex was crystallized from hot DMSO solvent as cube shaped crystals. Yield: 84.2 g (66%). 1H NMR (DMSO-d6) δ = 3.57 (s, 18H), 7.31 (s, 4H), 13C NMR (DMSO-d6) δ = 35.2, 119.9, 156.5.
Synthesis of (L1)2HgBr2.
To a solution of L1 (50 mg, 0.39 mmol) in dichloromethane one equivalent of HgBr2 (140.4 mg, 0.39 mmol) dissolved in acetonitrile was added and immediate formation of a white precipitate was observed. The suspension was stirred for another 0.5 h at room temperature. The white precipitate was filtered and washed with dichloromethane and dried under high vacuum. The obtained product was crystallized from hot DMF solvent as colourless needle-shaped crystals. Yield: 103.2 g (86%). 1H NMR (DMSO-d6) δ = 3.68 (s, 12H), 7.50 (s, 4H), 13C NMR (DMSO-d6) δ = 35.5, 121.1, 151.6. HR-ESIMS (m/z): calcd for [M]+ C10H16N4S2HgBr2 = 617.8854, observed value: [M − Br]+ = 536.9713.
Synthesis of k1-(L2)2Hg2Cl4.
To a 5 mL solution of L2 (50 mg, 0.21 mmol) in dichloromethane one equivalent of mercury(II) chloride (56.47 mg, 0.21 mmol) dissolved in acetonitrile was added. The immediate formation of white precipitates occurred and the suspension was stirred for another 0.5 h at 30 °C. A white solid of (L2)2Hg2Cl4 was filtered from the reaction mixture and suitable single crystals were obtained from a DMSO solution of the complex. Yield: 89.2 g (83%). 1H NMR (DMSO-d6) δ = 3.66 (s, 12H), 6.57 (s, 4H), 7.59–7.59 (d, J = 1.6 Hz, 4H), 7.86–7.87 (d, J = 1.6 Hz, 4H), 13C NMR (DMSO-d6) δ = 35.5, 57.3, 120.3, 122.1, 154.1. HR-ESIMS (m/z): calcd for [M]+ C18H24N8S4Hg2Cl4 = 1023.95, observed value: [M − HgCl3]+ = 717.0430, [M-C9H12N4S2HgCl3]+ = 476.9911.
Synthesis of k1-(L2)2Hg2Br4.
k
1-(L2)2Hg2Br4 was synthesized following a procedure similar to that for k1-(L2)2Hg2Cl4, except mercury(II) bromide (75 mg, 0.21 mmol) was added in place of mercury(II) chloride. Over time, block shaped single crystals of the complex were settled from the saturated solution of the complex in DMSO at room temperature. Yield: 100 mg (76%). 1H NMR (DMSO-d6) δ = 3.60 (s, 12H), 6.57 (s, 4H), 7.53–7.54 (d, J = 1.6 Hz, 4H), 7.89–7.890 (d, J = 1.6 Hz, 4H), 13C NMR (DMSO-d6) δ = 36.0, 57.7, 121.2, 122.4, 152.3. HR-ESIMS (m/z): calcd for [M]+ C18H24N8S4Hg2Br4 = 1201.7082, observed value: [M − HgBr3]+ = 760.9913, [M − C9H12N4S2HgBr3]+ = 520.9402.
Synthesis of k1-[L2HgI2]n.
To 10 mL of L2 (50 mg, 0.21 mmol) in MeOH, one equivalent of mercuric iodide (98.7 mg, 0.21 mmol) was added and the reaction mixture was stirred for another 30 minutes at 30 °C. After completion, the reaction solution was concentrated and dried under vacuum to obtain a white solid. Diamond shaped single crystals of k1-[L2HgI2]n were obtained from the solution of acetone/DMSO solution. Yield: 84 mg. 1H NMR (DMSO-d6) δ = 3.59 (s, 6H), 6.58 (s, 2H), 7.53–7.54 (d, J = 2 Hz, 2H), 7.91–7.92 (d, J = 2 Hz, 2H), 13C NMR (DMSO-d6) δ = 36.1, 57.4, 120.6, 122.1, 154.7.
4.3 NMR titration experiments of L1 and L2 with HgX2 (X = Cl, Br or I)
A solution of HgX2 (0.05 mmol) in DMSO-d6 (0.6 mL) was titrated with various equivalents (0–11 equiv.) of L1 (or L2) in DMSO-d6 and the 1H NMR spectra were recorded at room temperature.26 The variation of the chemical shifts of the proton (1H) and carbon (13C) resonances of the N-CH3 group, olefinic-C and olefinic-H of L1, and bridged-methylene (N-CH2-N), N-CH3 group, olefinic-H or olefinic-C of L2 were reported with respect to solvent the residual peak (DMSO-d6, 1H δ = 2.5 ppm), as shown in Fig. S1–S8 (ESI†). Full titration spectra are presented in the ESI.†
4.4 UV-Visible spectroscopic analysis
UV-Visible titration studies were performed using a UV-1800 Shimadzu UV-Vis spectrophotometer at 298 K in acetonitrile. 50 μM of L1 (or L2) solution in 1 mL acetonitrile was transferred into a 1 cm path length UV cuvette and various amounts of HgX2 were added (0.1–2 or 3 equiv.) to get a titration profile. For determining the complex composition in solution by Job's method we used 5 × 10−5 M of HgX2 and 5 × 10−5 M of ligands (L1 or L2 or L3) in acetonitrile. In total, nine mixtures of HgX2 and ligands were prepared. The volumes of ligand solution varied from 9 to 1 mL and those of HgX2 solution from 1 to 9 mL to obtain 1 to 9 mole fractions. The total volume was always kept at 10 mL. The complex inflection point in the Job's plot was calculated with respect to the LMCT wavelength of the corresponding complex.
4.5 Protection of HepG2 cells
Cell viability.
Cell culture experiment and cell viability assay were performed following the general procedure. HepG2 cells were continuously grown in a C25-mL cultural flask in RPMI 1640 supplemented with 5% FBS, 100 U mL−1 penicillin and 100 U mL−1 streptomycin in a CO2 incubator (5%) at 37 °C. MTT assay was performed to quantify the viability of the cells treated with ligands L1, L2, L3 and HgCl2 as mentioned below. In brief, 1 × 104 HepG2 cells were seeded into each well of a 96-well cell culture plate. After being grown for another 24 h, the cells were treated with various concentrations of ligands L1, L2 and L3 (1 to 100 μM) to determine the cytotoxicity of each ligand. After incubation of the cells with different concentrations of ligand for 24 h, the medium was removed and the cells were treated with 100 μL of 500 μg mL−1 of MTT (thiazolyl blue tetrazolium bromide) and incubated for another 4 h. Finally, MTT solution was removed and the formazan product was dissolved by adding 100 μL DMSO, and allowing for vibration for 10 min. Absorbance was read on an ELISA plate reader (Thermo Scientific, USA) at 590 nm. Likewise, the cytotoxicity of HgCl2 was also determined using the MTT assay, as mentioned above. To determine the IC50 value, the cells were treated with various concentrations of HgCl2 (5–50 μM). To investigate the protective effect of the ligands against HgCl2 toxicity, the cells were co-treated with ligand of various concentrations (5–100 μM) and HgCl2 (25 μM) and a similar procedure was followed, as mentioned above.
4.5 ROS estimation
The intracellular oxidative stress, induced by HgCl2, was detected by fluorescent imaging study. DCFH-DA was used as a fluorescent dye to measure the intracellular ROS level following the literature procedure.36 In brief, HepG2 cells (5 × 105) were seeded in 12-well plates and grown in a complete medium in a CO2 incubator to 80% confluence. Cells were exposed to 20 μM HgCl2 (alone) and co-treated with 100 μM concentration of the ligands (L1, L2, or L3) for 2 h. After 2 h incubation, the cells were washed with PBS and labelled with 10 μM DFCH–DA and incubated for 0.5 h at 37 °C. After being washed with PBS (3 times), the intracellular ROS level was determined using a Nikon eclipse Ti-u fluorescence microscope with the excitation wavelength set at 488 nm and emission wavelength at 530 nm.
4.6 Single crystal X-ray analysis
Single crystal X-ray diffraction data were collected on a D8 Venture Bruker AXS single crystal X-ray diffractometer equipped with a CMOS PHOTON 100 detector having monochromatized microfocus sources (Mo-Kα = 0.71073 Å). Single crystals of L3 (CCDC 1857559), (L1)2HgBr2 (CCDC 1486402), [L1HgI2]2 (CCDC 1857557), (L1)3Hg2I4 (CCDC 1857558), k1-(L2)2Hg2Cl4·DMSO (CCDC 1534013), k1-(L2)2Hg2Br4 (CCDC 1857556) and k1-[L2Hg2I2]n (CCDC 1857560) suitable for X-ray diffraction study were obtained from a slow evaporation process as described in the Experimental section.† Crystallographic parameters of L3 and the complexes are mentioned in the ESI† (Tables S1 and S2).37 All crystal data were collected at room temperature and solved using the SHELX program implemented in APEX3.38–41 The non-H atoms were located in successive difference fourier syntheses and refined with anisotropic thermal parameters. All the hydrogen atoms were placed at the calculated positions and refined using a riding model with appropriate HFIX commands. The program “Mercury” was used for molecular packing analysis.42 The crystal structure of [L2HgI2]n was disordered at the Hg2 atom, and the disordered mercury atom was treated using the PART command with occupancy (74:26)% (Hg2A:Hg2B). Similarly, k1-(L2)2Hg2Cl4·DMSO having solvent molecule disorder at the O1 atom was split by the PART command with occupancy (63:37)% (O1A:O1B).43 The crystal structure of k1-(L2)2Hg2Br4 has disorder in the solvent molecule which was removed by SQUEEZE option using PLATON software.44
Funding source
This work was supported by Shiv Nadar University (SNU), Shiv Nadar Foundation (SNF), and SERB (SB/S1/IC-44/2013), Govt. of India.
Caution
Mercuric salts are highly toxic to humans, and thus appropriate safety precautions must be taken in handling these toxic chemicals.
Conflicts of interest
The authors declare no competing financial interest and have no conflicts to declare.
Acknowledgements
We thank Shiv Nadar University (SNU), Shiv Nadar Foundation (SNF) and SERB (SB/S1/IC-44/2013), Govt. of India for funding. RK, and AC thank the SNU for fellowships.
References
-
(a) D. Mergler, H. A. Anderson, L. H. M. Chan, K. R. Mahaffey, M. Murray, M. Sakamoto and A. H. Stern, Methylmercury exposure and health effects in humans: a worldwide concern, Ambio, 2007, 36, 3–11 CrossRef CAS
 ;
(b) T. W. Clarkson, The toxicology of mercury, Crit. Rev. Clin. Lab. Sci., 1997, 34, 369–403 CrossRef CAS ;
(c) P. B. Tchounwou, C. G. Yedjou, A. K. Patlolla and D. J. Sutton, Heavy Metals Toxicity and the Environment, EXS, 2012, 101, 133–164 Search PubMed .
;
(b) T. W. Clarkson, The toxicology of mercury, Crit. Rev. Clin. Lab. Sci., 1997, 34, 369–403 CrossRef CAS ;
(c) P. B. Tchounwou, C. G. Yedjou, A. K. Patlolla and D. J. Sutton, Heavy Metals Toxicity and the Environment, EXS, 2012, 101, 133–164 Search PubMed .
-
(a) T. W. Clarkson, The three modern faces of mercury, Environ. Health Perspect., 2002, 110, 11–23 CrossRef CAS ;
(b) M. Korbas, T. C. MacDonald, I. J. Pickering, G. N. George and P. H. Krone, Chemical form matters: differential accumulation of mercury following inorganic and organic mercury exposures in zebrafish larvae, ACS Chem. Biol., 2012, 7, 411–420 CrossRef CAS ;
(c) H. H. Harris, I. J. Pickering and G. N. George, The chemical form of mercury in fish, Science, 2003, 301, 1203 CrossRef CAS .
-
(a) I. Bando, M. I. S. Reus, D. Andrés and M. Cascales, Endogenous antioxidant defence system in rat liver following mercury chloride oral intoxication, J. Biochem. Mol. Toxicol., 2005, 19, 154–161 CrossRef CAS ;
(b) C. M. L. Carvalho, E. H. Chew, S. I. Hashemy, J. Lu and A. Holmgren, Inhibition of the human thioredoxin system a molecular mechanism of mercury toxicity, J. Biol. Chem., 2008, 283, 11913–11923 CrossRef CAS ;
(c) M. Aschner and J. L. Aschner, Mercury neurotoxicity: mechanisms of blood–brain barrier transport, Neurosci. Biobehav. Rev., 1990, 14, 169–176 CrossRef CAS .
-
(a) T. W. Clarkson and L. Magos, The toxicology of mercury and its chemical compounds, Crit. Rev. Toxicol., 2006, 36, 609–662 CrossRef CAS ;
(b) P. J. Gallagher and R. L. Lee, The role of biotransformation in organic mercury neurotoxicity, Toxicology, 1980, 15, 129–134 CrossRef CAS .
- M. A. Khan and F. Wang, Mercury–selenium compounds and their toxicological significance: toward a molecular understanding of the mercury–selenium antagonism, Environ. Toxicol. Chem., 2009, 28, 1567–1577 CrossRef CAS PubMed .
- C. Chen, H. Yu, J. Zhao, B. Li, L. Qu, S. Liu, P. Zhang and Z. Chai, The roles of serum selenium and selenoproteins on mercury toxicity in environmental and occupational exposure, Environ. Health Perspect., 2006, 114, 297–301 CrossRef CAS .
-
(a) C. M. Carvalho, J. Lu, X. Zhang, E. S. Arnér and A. Holmgren, Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: implications for treatment of mercury poisoning, FASEB J., 2011, 25, 370–381 CrossRef CAS ;
(b) J. L. Franco, T. Posser, P. R. Dunkley, P. W. Dickson, J. J. Mattos, R. Martins, A. C. D. Bainy, M. R. Marques, A. L. Dafre and M. Farina, Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase, Free Radical Biol. Med., 2009, 47, 449–457 CrossRef CAS ;
(c) V. Branco, J. Canário, J. Lu, A. Holmgren and C. Carvalho, Mercury and selenium interaction in vivo: effects on thioredoxin reductase and glutathione peroxidase, Free Radical Biol. Med., 2012, 52, 781–793 CrossRef CAS ;
(d) V. Chatziargyriou and S. Dailianis, The role of selenium-dependent glutathione peroxidase (Se-GPx) against oxidative and genotoxic effects of mercury in haemocytes of mussel Mytilus galloprovincialis (Lmk.), Toxicol. In Vitro, 2010, 24, 1363–1372 CrossRef CAS .
-
(a) G. Carrier, M. Bouchard, R. C. Brunet and M. Caza, A toxicokinetic model for predicting the tissue distribution and elimination of organic and inorganic mercury following exposure to methyl mercury in animals and humans. II. Application and validation of the model in humans, Toxicol. Appl. Pharmacol., 2001, 171, 50–60 CrossRef CAS ;
(b) C. C. Bridges and R. K. Zalups, Transport of inorganic mercury and methylmercury in target tissues and organs, J. Toxicol. Environ. Health, Part B, 2010, 13, 385–410 CrossRef CAS ;
(c) W. L. Hughes, A physicochenical rationale for the biological activity of mercury and its compounds, Ann. N. Y. Acad. Sci., 1957, 65, 454–460 CrossRef CAS .
-
(a) R. K. Zalups and D. W. Barfuss, Intrarenal distribution of inorganic mercury and albumin after co-administration, J. Toxicol. Environ. Health, 1993, 40, 77–103 CrossRef CAS ;
(b) R. K. Zalups and D. W. Barfuss, Nephrotoxicity of inorganic mercury co-administered with L-cysteine, Toxicology, 1996, 109, 15–29 CrossRef CAS .
-
(a) D. H. Roos, R. L. Puntel, T. H. Lugokenski, R. P. Ineu, D. Bohrer, M. E. Burger, J. L. Franco, M. Farina, M. Aschner, J. B. T. Rocha, N. B. de and V. Barbosa, Complex methylmercury–cysteine alters mercury accumulation in different tissues of mice, Basic Clin. Pharmacol. Toxicol., 2010, 107, 789–792 CrossRef CAS ;
(b) Z. Yin, H. Jiang, T. Syversen, J. B. T. Rocha and M. Farina, The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter, J. Neurochem., 2008, 107, 1083–1090 CAS ;
(c) M. K. Donais, B. Duncan, D. George and C. Bizzarri, Dynamic accumulation and redistribution of methylmercury in the lens of developing zebrafish embryos and larvae, X-Ray Spectrom., 2010, 39, 146–153 CrossRef CAS ;
(d) T. A. Simmons-Willis, A. S. Koh, T. W. Clarkson and N. Ballatori, Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2, Biochem. J., 2002, 367, 239–246 CrossRef CAS .
-
(a) N. Ballatori and T. W. Clarkson, Biliary secretion of glutathione and of glutathione–metal complexes, Fundam. Appl. Toxicol., 1985, 5, 816–831 CrossRef CAS ;
(b) N. Ballatori and T. W. Clarkson, Dependence of biliary secretion of inorganic mercury on the biliary transport of glutathione, Biochem. Pharmacol., 1984, 33, 1093–1098 CrossRef CAS ;
(c) N. Ballatori and T. W. Clarkson, Inorganic mercury secretion into bile as a low molecular weight complex, Biochem. Pharmacol., 1984, 33, 1087–1092 CrossRef CAS ;
(d) W. Wang, T. W. Clarkson and N. Ballatori, Gamma-glutamyl transpeptidase and L-cysteine regulate methylmercury uptake by HepG2 cells, a human hepatoma cell line, Toxicol. Appl. Pharmacol., 2000, 168, 72–78 CrossRef CAS .
-
(a) G. Gstraunthaler, W. Pfaller and P. Kotanko, Glutathione depletion and in vitro lipid peroxidation in mercury or maleate induced acute renal failure, Biochem. Pharmacol., 1983, 32, 2969–2972 CrossRef CAS ;
(b) M. Farina, J. B. T. Rocha and M. Aschner, Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies, Life Sci., 2011, 89, 555–563 CrossRef CAS ;
(c) M. Farina, M. Aschner and J. B. T. Rocha, Oxidative stress in MeHg-induced neurotoxicity, Toxicol. Appl. Pharmcol., 2011, 256, 405–417 CrossRef CAS PubMed .
-
(a) L. V. Papp, J. Lu, A. Holmgren and K. K. Khanna, From selenium to selenoproteins: synthesis, identity, and their role in human health, Antioxid. Redox Signaling, 2007, 9, 775–806 CrossRef CAS ;
(b) N. H. Stacey and H. Kappas, Cellular toxicity and lipid peroxidation in response to mercury, Toxicol. Appl. Pharmacol., 1982, 63, 29–35 CrossRef CAS .
-
(a) A. O. Summers, Organization, expression, and evolution of genes for mercury resistance, Annu. Rev. Microbiol., 1986, 40, 607–634 CrossRef CAS ;
(b) J. L. Schottel, The mercuric and organomercurial detoxifying enzymes from a plasmid-bearing strain of Escherichia coli, J. Biol. Chem., 1978, 253, 4341–4349 CAS ;
(c) A. O. Summers and L. I. Sugarman, Cell-free mercury(II)-reducing activity in a plasmid-bearing strain of Escherichia coli, J. Bacteriol., 1974, 119, 242–249 CAS ;
(d) L. A. Rojas, C. Yáñez, M. González, S. Lobos, K. Smalla and M. Seeger, Characterization of the metabolically modified heavy metal-resistant Cupriavidus metallidurans strain MSR33 generated for mercury bioremediation, PLoS One, 2011, 6, 17555 CrossRef .
-
(a) J. Lafrance-Vanasse, M. Lefebvre, P. D. Lello, J. Sygusch and J. Omichinski, Crystal structures of the organomercurial lyase merb in its free and mercury-bound forms insights into the mechanism of methylmercury degradation, J. Biol. Chem., 2009, 284, 938–944 CrossRef CAS ;
(b) J. M. Parks, H. Guo, C. Momany, L. Liang, S. M. Miller, A. O. Summers and J. C. Smith, Mechanism of Hg–C Protonolysis in the Organomercurial Lyase MerB, J. Am. Chem. Soc., 2009, 131, 13278–13285 CrossRef CAS PubMed .
-
(a) M. Blanusa, V. M. Varnai, M. Piasek and K. Kostial, Chelators as antidotes of metal toxicity: therapeutic and experimental aspects, Curr. Med. Chem., 2005, 12, 2771–2794 CrossRef CAS ;
(b)
W. Martindale, The Extra Pharmacopoeia, Royal Pharmaceutical Society, London, 31st edn, 1996, pp. 989–992 Search PubMed .
-
(a) V. E. Petrunki, Synthesis and properties of mercapto derivatives of alkysulfonic acids, Ukr. Khim. Zh., 1956, 22, 603–607 Search PubMed ;
(b) E. Friedheim, J. R. Dasilva and A. V. Martins, Treatment of Schistosomiasis mansoni with antimony a-dimercapto-potassium succinate (TWSb), Am. J. Trop. Med. Hyg., 1954, 3, 714–727 CrossRef CAS ;
(c) H. V. Aposhian and M. M. Aposhian,
Meso-2,3-dimercaptosuccinic acid: chemical, pharmacological and toxicological properties of an orally effective metal chelating agent, Annu. Rev. Pharmacol. Toxicol., 1990, 30, 279–306 CrossRef CAS ;
(d) R. A. Peters and L. Stocken, A. British anti-lewisite (BAL), Nature, 1945, 156, 616–619 CrossRef CAS ;
(e) J. P. Rooney, The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury, Toxicology, 2007, 234, 145–156 CrossRef CAS .
-
(a) R. A. Steele and S. J. Opella, Structures of the Reduced and Mercury-Bound Forms of MerP, the Periplasmic Protein from the Bacterial Mercury Detoxification System, Biochemistry, 1997, 36, 6885–6895 CrossRef CAS ;
(b) R. Ledwidge, B. Patel, A. Dong, D. Fiedler, M. Falkowski, J. Zelikova, A. O. Summers, E. F. Pai and S. M. Miller, NmerA, the Metal Binding Domain of Mercuric Ion Reductase, Removes Hg2+ from Proteins, Delivers It to the Catalytic Core, and Protects Cells under Glutathione-Depleted Conditions, Biochemistry, 2005, 44, 11402–11416 CrossRef CAS ;
(c) E. Rossy, O. Sénèque, D. Lascoux, D. Lemaire, S. Crouzy, P. Delangle and J. Covès, Is the cytoplasmic loop of MerT, the mercuric ion transport protein, involved in mercury transfer to the mercuric reductase?, FEBS Lett., 2004, 575, 86–90 CrossRef CAS ;
(d) E. Mesterházy, S. Crouzy, A. Jancsó and P. Delangle, Short oligopeptides with three cystein residues as models of sulphur-rich Cu(I)-and Hg(II)-binding sites in proteins, Metallomics, 2018, 10, 1232–1244 RSC .
-
(a) V. Mah and F. Jalilehvand, Mercury(II) Complex Formation with Glutathione in Alkaline Aqueous Solution, JBIC, J. Biol. Inorg. Chem., 2008, 13, 541–553 CrossRef CAS ;
(b) F. Jalilehvand, B. O. Leung, M. Izadifard and E. Damian, Mercury(II) Cysteine Complexes in Alkaline Aqueous Solution, Inorg. Chem., 2006, 45, 66–73 CrossRef CAS ;
(c) V. Mah and F. Jalilehvand, Glutathione Complex Formation with Mercury(II) in Aqueous Solution at Physiological pH, Chem. Res. Toxicol., 2010, 23, 1815–1823 Search PubMed ;
(d) T. Warner and F. Jalilehvand, Formation of Hg(II) Tetrathiolate Complexes with Cysteine at Neutral pH, Can. J. Chem., 2016, 94, 373–379 CrossRef CAS .
-
(a) F. Jalilehvand, K. Parmar and S. Zielke, Mercury(II) Complex
Formation with N-acetylcysteine, Metallomics, 2013, 5, 1368–1376 RSC ;
(b) B. O. Leung, F. Jalilehvand and V. Mah, Mercury(II) Penicillamine Complex Formation in Alkaline Aqueous Solution, Dalton Trans., 2007, 4666–4674 RSC ;
(c) A. M. Pujol, C. Lebrun, C. Gateau, A. Manceau and P. Delangle, Mercury-Sequestering Pseudopeptides with a Tris (cysteine) Environment in Water, Eur. J. Inorg. Chem., 2012, 3835–3843 CrossRef CAS ;
(d) A. S. Jullien, C. Gateau, C. Lebrun and P. Delangle, Mercury Complexes with Tripodal Pseudopeptides Derived from D-Penicillamine Favour a HgS3 Coordination, Eur. J. Inorg. Chem., 2015, 3674–3680 CrossRef CAS .
-
(a) H. V. Aposhian, Biological chelation: 2,3-dimercaptopropanesulfonic acid and meso-dimercaptosuccionic acid, Adv. Enzyme Regul., 1982, 20, 301–319 CrossRef CAS ;
(b) R. K. Zalups and C. C. Bridges, Relationships between the renal handling of DMPS and DMSA and the renal handling of mercury, Chem. Res. Toxicol., 2012, 25, 1825–1838 Search PubMed .
- R. Karri, M. Banerjee, A. Chalana, K. K. Jha and G. Roy, Activation of the Hg–C Bond of Methylmercury by [S2]-Donor Ligands, Inorg. Chem., 2017, 56, 12102–12115 CrossRef CAS .
-
(a) J. G. Melnick and G. Parkin, Cleaving mercury-alkyl bonds: a functional model for mercury detoxification by MerB, Science, 2007, 317, 225–227 CrossRef CAS ;
(b) J. G. Melnick, K. Yurkerwich and G. Parkin, Synthesis, Structure, and Reactivity of Two-Coordinate Mercury Alkyl Compounds with Sulfur Ligands: Relevance to Mercury Detoxification, Inorg. Chem., 2009, 48, 6763–6772 CrossRef CAS ;
(c) M. Banerjee, R. Karri, K. S. Rawat, K. Muthuvel, B. Pathak and G. Roy, Chemical Detoxification of Organomercurials, Angew. Chem., 2015, 127, 9455–9459 (
Angew. Chem., Int. Ed.
, 2015
, 54
, 9323–9327
) CrossRef ;
(d) M. Banerjee and G. Roy, Cleavage of Hg–C Bonds of Organomercurials Induced by ImOHSe via Two Distinct Pathways, Inorg. Chem., 2017, 56, 12739–12750 CrossRef CAS .
-
(a) H. Wang, B. Chen, S. Zhu, X. Yu, M. He, B. Hu, S. Cuello, L. Goya, Y. Madrid, S. Campuzano, M. Pedrero, L. Bravo, C. Cámara and S. Ramos, Molecular mechanisms of methylmercury-induced cell death in human HepG2 cells, Food Chem. Toxicol., 2010, 48, 796–802 Search PubMed ;
(b) H. Wang, B. Chen, S. Zhu, X. Yu, M. He and B. Hu, Chip-Based Magnetic Solid-Phase Microextraction Online Coupled with MicroHPLC-ICPMS for the Determination of Mercury Species in Cells, Anal. Chem., 2016, 88, 796–802 CrossRef CAS .
-
(a) Z. Popović, D. M. Calogović, Ž. Soldin, G. Pavlović, N. Davidović and D. V. Topić, Mercury(II) compounds with 1,3-imidazole-2-thione and its 1-methyl analogue. Preparative and NMR spectroscopic studies. The crystal structures of di-μ-iodo-bis[iodo(1,3-imidazolium-2-thiolato-S)mercury(II)], bis[bromo(1,3-imidazolium-2-thiolato-S)]mercury(II) and bis[μ-(1-N-methyl-1,3-imidazole-2-thiolato-S)]mercury(II), Inorg. Chim. Acta, 1999, 294, 35–46 CrossRef ;
(b) Z. Popovic, G. Pavlovic, D. Matkovic-Calogovic, Z. Soldin, M. Rajic, D. Vikic-Topic and D. Kovacek, Mercury(II) complexes of heterocyclic thiones: part 1. Preparation of 1:2 complexes of mercury(II) halides and pseudohalides with 3,4,5,6-tetrahydropyrimidine-2-thione. X-ray, thermal analysis and NMR studies, Inorg. Chim. Acta., 2000, 306, 142–152 CrossRef CAS ;
(c) C. Wang, D. Zhang, G. Zhang, J. Xiang and D. Zhu, Chemistry, 2008, 14, 5680–5686 CrossRef CAS ;
(d) M. I. Wazeer and A. A. Isab, Omplexations of Hg(CN)2 with imidazolidine-2-thione and its derivatives: solid state, solution NMR and antimicrobial activity studies, Spectrochim. Acta, Part A, 2007, 68, 1207–1212 CrossRef ;
(e) R. Karri, M. Banerjee, R. Rai and G. Roy, Synthesis and Characterization of 1:2 Complex of Mercury (II) Chloride with 1,3-Dimethyl-1H-Imidazole-2-(3H)-Thione, Proc. Natl. Acad. Sci., India, Sect. A, 2016, 86, 611–617 CrossRef CAS ;
(f) E. Buncel, A. R. Norris, S. E. Taylor and W. J. Racz, Metal ion–biomolecule interactions. IV Methylmercury(II) complexes of L-methylimidazoline-2-thione (methimazole), a potentially useful protective agent in organomercurial intoxication, Can. J. Chem., 1982, 60, 3033–3038 CrossRef CAS ;
(g) A. R. Norris, S. E. Taylor, E. Buncel, F. Belangergariepy and A. L. Beauchamp, Crystal structures of two methylmercury complexes with 1-methylimidazoline-2-thione, Can. J. Chem., 1983, 61, 1536–1541 CrossRef CAS .
- J. P. Palmer and G. Parkin, Protolytic Cleavage of Hg–C Bonds Induced by 1-Methyl-1,3-dihydro-2H-benzimidazole-2-selone: syn-thesis and Structural Characterization of Mercury Complexes, J. Am. Chem. Soc., 2015, 137, 4503–4516 CrossRef CAS .
- N. A. Bell, T. N. Branston, W. Clegg, J. R. Creighton, L. Cucurull-Sanchez, M. R. J. Elsegood and E. S. Raper, Complexes of hetero-cyclic thiones and Group 12 metals Part 3. Preparation and characterisation of 1:2 complexes ofmercury(II) halides with 1-methylimidazoline-2(3H)-thione: the crystal structures of [(HgX2)(1-methylimidazoline-2(3H)-thione)2] (X = Cl, Br, I) at 160 K, Inorg. Chim. Acta, 2000, 303, 220–227 CrossRef CAS .
- A. A. Isab and H. P. Perzanowski,
1H, 13C and 199Hg NMR Studies of the Complexation of HgCl2 by Imidazolidine-2-Thione and its Derivatives, J. Coord. Chem., 1990, 21, 247–252 CrossRef CAS .
-
(a) S. Meyer, S. Demeshko, S. Dechert and F. Meyer, Synthesis, structure and Mössbauer characterization of polymeric iron(II) complexes with bidentate thiourea ligands, Inorg. Chim. Acta, 2010, 363, 3088–3092 CrossRef CAS ;
(b) B. S. Stadelman, M. M. Kimani, C. A. Bayse, C. D. McMillen and J. L. Brumaghim, Synthesis, characterization, DFT calculations, and electrochemical comparison of novel iron(II) complexes with thione and selone ligands, Dalton Trans., 2016, 45, 4697–4711 RSC ;
(c) M. M. Kimani, H. C. Wang and J. L. Brumaghim, Investigating the copper coordination, electrochemistry, and Cu(II) reduction kinetics of biologically relevant selone and thione compounds, Dalton Trans., 2012, 41, 5248–5259 RSC ;
(d) M. M. Kimani, D. Watts, L. A. Graham, D. Rabinovich, G. P. A. Yap and J. L. Brumaghim, Dinuclear copper(I) complexes with N-heterocyclic thione and selone ligands: synthesis, characterization, and electrochemical studies, Dalton Trans., 2015, 44, 16313–16324 RSC ;
(e) M. M. Kimani, C. A. Bayse and J. L. Brumaghim, Synthesis, characterization, and DFT studies of thione and selone Cu(I) complexes with variable coordination geometries, Dalton Trans., 2011, 40, 3711–3723 RSC .
-
(a) K. Srinivas, A. Sathyanarayana, C. N. Babu and G. Prabhusankar, Bismuth(III) dichalcogenones as highly active catalysts in multiple C–C bond formation reactions, Dalton Trans., 2016, 45, 5196–5209 RSC ;
(b) P. Rousselot-Pailley, O. Sénèque, C. Lebrun, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Model Peptides Based on the Binding Loop of the Copper Metal-lochaperone Atx1: Selectivity of the Consensus Sequence MxCxxC for Metal Ions Hg(II), Cu(I), Cd(II), Pb(II), and Zn(II), Inorg. Chem., 2006, 45, 5510–5520 CrossRef CAS ;
(c) S. Pires, J. Habjanič, M. Sezer, C. M. Soares, L. Hemmingsen and O. Iranzo, Design of a Peptidic Turn with High Affinity for Hg II, Inorg. Chem., 2012, 51, 11339–11348 CrossRef CAS ;
(d) J. S. Casas and M. M. Jones, Mercury(II) complexes with sulfhydryl containing chelating agents: stability constant inconsistencies and their resolution, J. Inorg. Nucl. Chem., 1980, 42, 99–102 CrossRef CAS ;
(e) B. N. Ahamed, M. Arunachalam and P. Ghosh, Thiomethoxychalcone-Functionalized Ferrocene Ligands as Selective Chemodosimeters for Mercury(II): Single-Crystal X-ray Structural Signature of the [Hg8(μ8-S)(SCH3)12]2+ Cluster, Inorg. Chem., 2010, 49, 4447–4457 CrossRef CAS .
- H. Schmidbaur and A. Schier, Mercurophilic Interactions, Organometallics, 2015, 34, 2048–2066 CrossRef CAS .
-
(a) B. T. Mossman,
In vitro approaches for determining mech-anisms of toxicity and carcinogenicity by asbestos in the gastrointestinal and respiratory tracts, Environ. Health Perspect., 1983, 53, 155–161 CrossRef CAS ;
(b) M. Kaivalya, B. N. Rao and B. S. Satish Rao, Mangiferin: a xanthone attenuates mercury chloride induced cytotoxicity and genotoxicity in HepG2 cells, J. Biochem. Mol. Toxicol., 2011, 25, 108–116 CrossRef CAS .
-
(a) B. L. Benac, E. M. Burgess and A. J. Arduengo III, 1,3-Dimethyl-1H-imidazole-2(3H)-thion, Org. Synth., 1986, 64, 92–95 CrossRef CAS ;
(b) G. Roy, D. Das and G. Mugesh, Bioinorganic chemistry aspects of the inhibition of thyroid hormone biosynthesis by anti-hyperthyroid drugs, Inorg. Chim. Acta, 2007, 360, 303–316 CrossRef CAS .
-
(a) R. M. Claramunt, J. Elguero and T. J. Meco,
N-Polyazolylmethanes. III. Synthesis and proton NMR study of 1,1′-methylenediimidazole and 1,1′-methylenedibenzimidazole derivatives, Heterocycl. Chem., 1983, 20, 1245–1249 CrossRef CAS ;
(b) E. Diez-Barra, A. de la Hoz, A. Sanchez-Migallon and J. Tejeda, Phase transfer catalysis without solvent: synthesis of bisazolylalkanes, Heterocycles, 1992, 34, 1365–1372 CrossRef CAS .
-
(a) R. M. Silva, M. D. Smith and J. R. Gardinier, Unexpected New Chemistry of the Bis(thioimidazolyl)methanes, J. Org. Chem., 2005, 70, 8755–8763 CrossRef CAS ;
(b) D. J. Williams, D. Vanderveer, R. L. Jones and D. S. Menaldino, Main group metal halide complexes with sterically hindered thioureas XI. Complexes of antimony(III) and bismuth(III) chlorides with a new bidentate thiourea 1,1′-methylenebis(3-methyl-2H-imidazole-2-thione), Inorg. Chim. Acta, 1989, 165, 173–178 CrossRef CAS ;
(c) K. P. Bhabak, K. Satheeshkumar, S. Jayavelu and G. Mugesh, Inhibition of peroxynitrite- and peroxidase-mediated proteintyrosine nitration by imidazole-based thiourea and selenourea derivatives, Org. Biomol. Chem., 2011, 9, 7343–7350 RSC ;
(d) C. N. Babu, P. Suresh, K. Srinivas, A. Sathyanarayana, N. Sampath and G. Prabusankar, Catalytically active lead(II)–imidazolium coordination assemblies with diversified lead(II) coordination geometries, Dalton Trans., 2016, 45, 8164–8173 RSC .
- X. Chang, W. Lu, T. Dou, X. Wang, D. Lou, X. Sun and Z. Zhou, Paraquat inhibits cell viability via enhanced oxidative stress and apoptosis in human neural progenitor cells, Chem.-Biol. Interact., 2013, 206, 248–255 CrossRef CAS .
- CCDC: 1486402, 1534013 and 1857556–1857560†.
- Bruker Support APEX3, SAINT and SADABS: Software for data reduction, absorption correction and structure solution, Bruker AXS Inc., Madison, Wisconsin, USA, 2015, http://www.brukersupport.com/.
-
G. M. Sheldrick, SHELXTL Version2014/7: Programs for the determination of small and macromolecular crystal structures by single crystal X-ray and neutron diffraction. University of Göttingen Germany, 2014, http://shelx.uni-ac.gwdg.de/SHELX/index.php.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef .
- L. J. Farrugia, ORTEP-3 for Windows – a version of ORTEP-III with a Graphical User Interface (GUI), J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS .
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. V. Streek and P. A. Wood, Mercury CSD 2.0 – New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS .
-
P. Muller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement, Oxford University Press, 2006 Search PubMed .
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*