
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Increasing the bioactive space of peptide macrocycles by thioamide substitution†
Hitesh
Verma
 a,
Bhavesh
Khatri
a,
Sohini
Chakraborti
a and
Jayanta
Chatterjee
*ab
a,
Bhavesh
Khatri
a,
Sohini
Chakraborti
a and
Jayanta
Chatterjee
*ab
aMolecular Biophysics Unit, Indian Institute of Science, Bangalore 560012, India. E-mail: jayanta@iisc.ac.in
bNMR Research Centre, Indian Institute of Science, Bangalore 560012, India
First published on 22nd January 2018
Abstract
We show that substituting a single atom, O to S (amide to thioamide), in a peptide bond results in global restriction of the conformational flexibility in peptide macrocycles with minimal perturbation of the parent conformation. The van der Waals interactions between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group and the surrounding atoms are the major driving force in inducing the conformational restriction, resulting in well-defined structures of these cyclic peptides with static 3-D presentation of the pharmacophores. Utilizing this property of thioamides, we report the development of a superactive antagonist of pro-angiogenic αvβ3, αvβ5 and α5β1 integrins, which are responsible for cancer cell proliferation and survival. Using simple thio-scanning and spatial screening of a non-efficacious and conformationally flexible cyclic peptide, we could achieve a more than 105 fold enhancement in its efficacy in cellulo via a single O to S substitution. The developed peptide shows better efficacy in inhibiting the pro-angiogenic integrins than the drug candidate cilengitide, with a significantly enhanced serum half-life of 36 h compared to that of cilengitide (12 h). The long shelf-life, absence of non-specific toxicity and resistance to degradation of the thioamidated macrocyclic peptides in human serum suggest the promise of thioamides in markedly improving the affinity, efficacy and pharmacology of peptide macrocycles.
S group and the surrounding atoms are the major driving force in inducing the conformational restriction, resulting in well-defined structures of these cyclic peptides with static 3-D presentation of the pharmacophores. Utilizing this property of thioamides, we report the development of a superactive antagonist of pro-angiogenic αvβ3, αvβ5 and α5β1 integrins, which are responsible for cancer cell proliferation and survival. Using simple thio-scanning and spatial screening of a non-efficacious and conformationally flexible cyclic peptide, we could achieve a more than 105 fold enhancement in its efficacy in cellulo via a single O to S substitution. The developed peptide shows better efficacy in inhibiting the pro-angiogenic integrins than the drug candidate cilengitide, with a significantly enhanced serum half-life of 36 h compared to that of cilengitide (12 h). The long shelf-life, absence of non-specific toxicity and resistance to degradation of the thioamidated macrocyclic peptides in human serum suggest the promise of thioamides in markedly improving the affinity, efficacy and pharmacology of peptide macrocycles.
Introduction
In recent years, peptide macrocycles, as opposed to small molecules, have emerged as an extremely useful scaffold to target various extracellular and intracellular proteins.1 The complex structures of peptide macrocycles give rise to a unique combination of ϕ, ψ, and χ space aiding in molecular recognition towards target binding.2 Nevertheless, macrocyclic structures are often associated with conformational flexibility, introducing an entropic penalty to target binding that severely compromises their affinity and selectivity against a biological target.3 Thus, covalent or noncovalent constraints that minimize the conformational entropy in peptides are highly desirable.3c,4 In particular, constraints that minimally alter the bioactive peptide sequence are of paramount interest. In this context, peptide bond surrogates like peptoids,5 retro-inverso peptides,6 depsipeptides,7 azapeptides8 and N-methylated peptides9 have found use in delivering bioactive analogs with increased affinity, selectivity and metabolic stability. However, the strictest isostere of a peptide bond, the thioamide,10 attracted our attention. Thioamides show longer CS bonds of 1.65 Å compared to 1.23 Å for CO bonds, and a larger van der Waals radius of S of 1.80 Å compared to 1.52 Å for O in oxoamides.10 In addition, the increased rotational barrier about the C–N bond, and the reduced hydrogen bond acceptor but stronger hydrogen bond donor capacity of thioamides compared to regular amides,10 encouraged us to understand the conformational behavior of thiosubstituted macrocyclic peptides. Thioamides have found great utility both in biophysics and medicinal chemistry. They have been utilized as minimal fluorescence quenchers, protease sensors, photoswitchable elements in peptides, and probes to interrogate the dynamics of hydrogen bonds in α-helices and β-sheets.11 Furthermore, thioamides have also shown improved bioactivity and metabolic stability compared to linear bioactive peptides.12 Despite the versatile utility of thioamides, their potential to deliver selective and high-affinity macrocyclic ligands has not been systematically addressed.13
Thus, we set out to investigate the role of thioamides in peptide macrocycles, with special emphasis on the effect of thionation in the secondary structure motifs commonly occurring in bioactive macrocycles viz. βII, βII′, βVI and γ-turns, as observed in cyclosporine A, gramicidin S, somatostatin agonists, integrin antagonists etc., which have found enormous utility in treating pathological conditions.3c This report details the conformational impact of thioamide substitution on cyclic peptide backbones that leads to reduced flexibility of the peptide backbone and allows for the static presentation of the pharmacophores, resulting in a dramatically increased binding affinity and enhanced metabolic stability of the thioamidated macrocyclic peptides.
Results and discussion
Conformational impact of thioamides on a cyclic peptide backbone
To initially determine the conformational impact of thioamide substitution in peptide macrocycles, we synthesized a regioisomeric library of monothionated analogs of the parent cyclic peptide cyclo(–D-Ala–Ala4–), which is known to adopt a βII′-type and γ-type turn (Fig. 1A).14 The 1H NMR spectra of all the thionated cyclic peptides in DMSO-d6 were well dispersed indicating the presence of a well-defined conformation (ESI†). Utilizing the distance restraints obtained from the ROESY spectrum, we derived the solution conformations of the cyclic peptides (Fig. 1A). The introduction of the CS groups did not introduce large perturbation to the global conformations of the cyclic peptides, however, we observed that unlike the CO groups, none of the thioamides were internally oriented, indicating a lack of intramolecular hydrogen bonding with the CS groups. The high-resolution structures of the thionated cyclic pentapeptides indicated the coexistence of βII′-type and γ-type turns. The γ-type turn is in equilibrium between an open (indicated by the NOE between the Ala4HN–Ala5HN protons) and a closed form (suggested by the low temperature coefficient of Ala5HN, which is indicative of intramolecular hydrogen bonding (Fig. 1B)) as demonstrated by Kessler and Marshall et al.15 The thionation of D-Ala1, Ala2, Ala4 and Ala5 resulted in cyclic pentapeptides (1, 2, 4 and 5) with similar global conformations but different ϕ and ψ values (ESI†), where the βII′-type turn is centered about D-Ala1–Ala2 and the γ-turn is centered about Ala4.15 However, the thionation of Ala3 resulted in a conformation with the βII′-type turn centered about Ala2–Ala3 and the γ-turn centered about Ala5 (3).
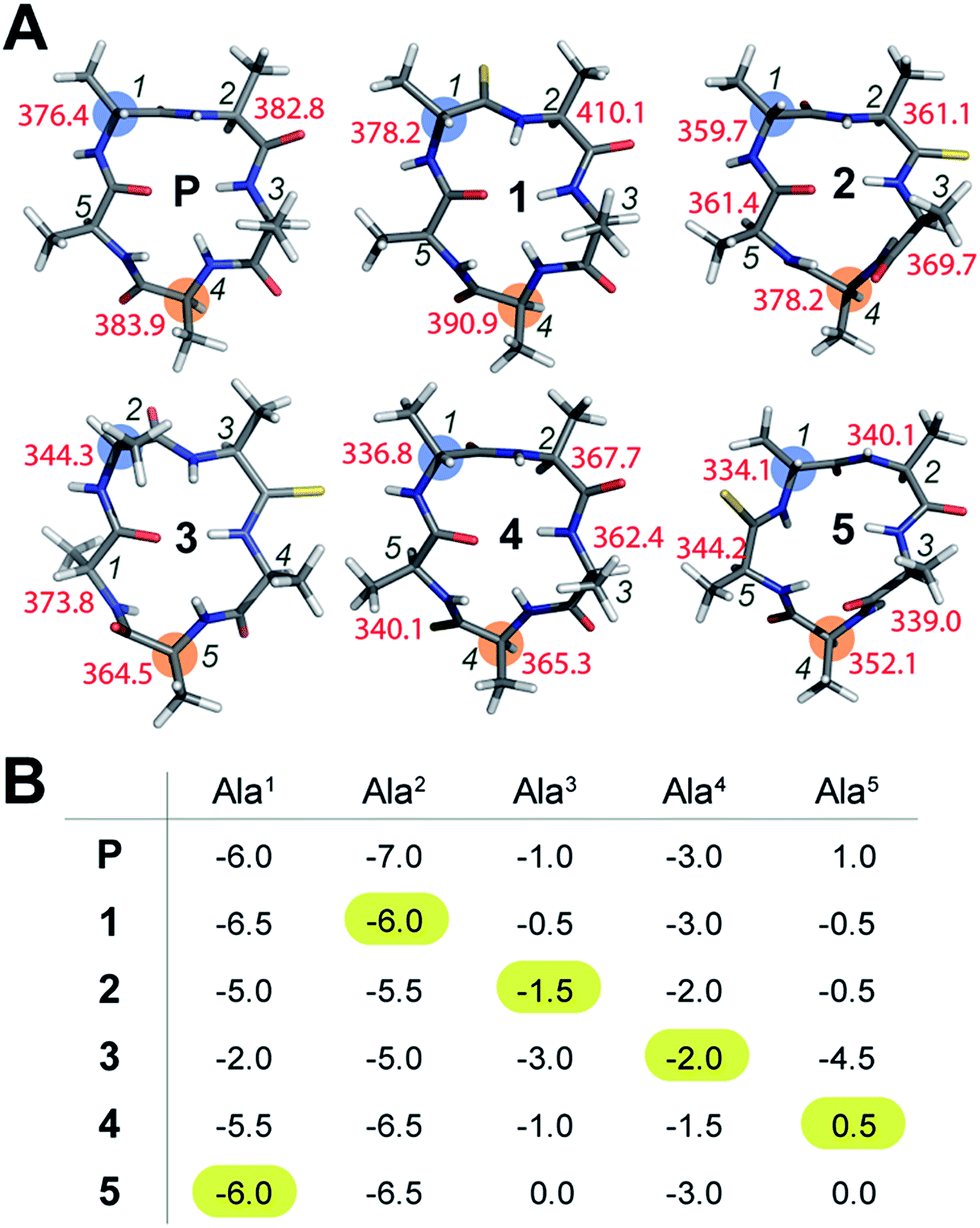 | ||
| Fig. 1 (A) Solution conformation of cyclo(aA4) (P) and its thio analogs (1–5). The T1 values of the resolved Cα are indicated adjacent to the residues. The i + 1 residue in the β-turn, and the residue about which the γ-turn is centered, are highlighted. (B) The amide proton temperature coefficients (in ppb K−1) determined from the 1H NMR spectra acquired between 298 K and 323 K. The value for the thioamide NH is highlighted. | ||
The true externally oriented (solvent exposed) CO groups in the cyclic pentapeptide are at D-Ala1, Ala2 and Ala4, and upon thionation did not perturb the conformation of the parent peptide. However, thionation at the internally oriented CO groups, in the case of Ala3 (3), completely changes the conformation of the peptide; this is also observed for Ala5 (5), albeit to a lesser extent. These results indicate that thioamide substitution at a site where the CO group does not engage in intramolecular hydrogen bonding can retain the conformation of the parent macrocyclic peptide. Thus, we note that thioamides can be valuable probes for identifying the hydrogen bond involved in stabilizing the conformation of a macrocycle.11f However, we were intrigued by the “frame-shifted” conformation of 3 and wondered whether this was a result of the enhanced hydrogen bond (H-bond) donating properties of the thioamide, as noted by Kessler et al.13d To understand this behavior, we analysed the temperature coefficients of the amide protons in these peptides (Fig. 1B) along with the 1H chemical shifts. The upfield shift of the amide resonances in Ala3HN and Ala5HN indicate their involvement in intramolecular hydrogen bonding, as opposed to D-Ala1HN, Ala2HN, and Ala4HN in all the peptides except 3 (ESI†).
It was interesting to note that despite the possibility of Ala2HN being involved in a γ-turn with Ala5CO in 1,16 there was no enhancement in the thioamide H-bond donation and structural alteration. Similarly, the temperature coefficients of the thioamide HN in 2 and 4 did not indicate a significant increase in the thioamide H-bond donation. This suggests that the increased thioamide H-bond donor propensity17 is not the driving force for the structural alteration in 3 and 5, instead, the van der Waals interactions between S (due to the increased size of sulfur and the longer CS bond) and the surrounding atoms are crucial in dictating the conformation of the peptide, as predicted from the computational studies of thioamides containing dipeptides by Lipton et al.11a Thus, the internally oriented Ala3CO completely flips outward on thio substitution in 3, presumably due to the 1,2 and 1,3-diaxial strain between S and the flanking Ala3 and Ala4Cβ. However, owing to the flanking heterochiral residues, thio substitution of Ala5CO allows for a partial flip of the thioamide bond.
Preliminary indication of the structural rigidity in cyclic peptides
To understand whether the thioamide substitution results in reduced flexibility of the cyclic peptide backbone, we determined the 13C spin-lattice relaxation time (T1) of the backbone Cα in these cyclic peptides, which provides qualitative information on the segmental motion of amino acids in peptides.18 It was intriguing to note that in all the peptides, the i + 1 residue in the β-turn displayed the lowest T1 compared to the other residues in the same macrocycle indicating its segmental rigidity (Fig. 1A) (comparison of the intermolecular T1 is not feasible due to differences in the concentration and viscosity of the samples).18a Subsequently, we compared the T1 of the i + 1 residue with the T1 of the alanine about which the flexible γ-turn is centered (highlighted in Fig. 1A). Curiously, we observed that this difference in T1 was smallest in 1 compared to all other thio analogs. Additionally, the difference in T1 within the residues in P was also less than that in the thio analogs, indicating comparable flexibility in the different segments of P. This led us to speculate that thioamide substitution presumably assists in increasing the (segmental) structural rigidity in cyclic peptides.Evidence of enhanced structural rigidity in aqueous solution
To validate our observation of enhanced structural rigidity in peptide macrocycles through thionation in aqueous solution (due to the hydrogen bonding properties of water, it effectively solvates the intramolecular H-bonds in the peptides, leading to greater flexibility), we resorted to a moderate affinity cyclic peptide integrin antagonist cyclo(RGDfV) (6),16b which is also known to adopt a βII′-type turn about D-Phe–Val and a γ-turn about Gly as observed in 1. Interestingly, we noted that 6 displays two conformations in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 on the NMR time scale (Fig. 2A and ESI†) at 25 °C, making it a suitable model to test our hypothesis. Thus, we synthesized a thio-scan library of 6 and assessed the conformations by NMR. Since thioamides undergo sulfur to oxygen exchange under acidic conditions, the orthogonally protected cyclic peptides were initially purified and subjected to global deprotection using an optimized cleavage cocktail of TFA/DCM/TIPS/H2O (62.5:32.5:2.5:2.5).19 Although we were successful in obtaining all the orthogonally protected thioamidated peptides, c(RGDfVt) (8) underwent spontaneous acid catalyzed degradation.19 To analyze whether the flanking amino acids of the thioamide bond have a role to play in the degradation, we synthesized three additional cyclic peptides with identical chirality but different functional groups: c(RGDfAt), c(KGDfVt) and c(NleGDfVt) (Nle: norleucine). It was surprising to note that all of these protected cyclic peptides underwent spontaneous acid catalyzed degradation as observed in 8 (ESI†). This suggested that the backbone conformation of the peptide has a key role in degradation, rather than the amino acid side chains flanking the thioamide bond. It is worth noting at this point that the model cyclic peptides (1–5) did not show any trace of thio to oxo conversion even after 24 months at 25 °C in DMSO solution, indicating the stability of the thiopeptides in an oxidizing environment.
:2 on the NMR time scale (Fig. 2A and ESI†) at 25 °C, making it a suitable model to test our hypothesis. Thus, we synthesized a thio-scan library of 6 and assessed the conformations by NMR. Since thioamides undergo sulfur to oxygen exchange under acidic conditions, the orthogonally protected cyclic peptides were initially purified and subjected to global deprotection using an optimized cleavage cocktail of TFA/DCM/TIPS/H2O (62.5:32.5:2.5:2.5).19 Although we were successful in obtaining all the orthogonally protected thioamidated peptides, c(RGDfVt) (8) underwent spontaneous acid catalyzed degradation.19 To analyze whether the flanking amino acids of the thioamide bond have a role to play in the degradation, we synthesized three additional cyclic peptides with identical chirality but different functional groups: c(RGDfAt), c(KGDfVt) and c(NleGDfVt) (Nle: norleucine). It was surprising to note that all of these protected cyclic peptides underwent spontaneous acid catalyzed degradation as observed in 8 (ESI†). This suggested that the backbone conformation of the peptide has a key role in degradation, rather than the amino acid side chains flanking the thioamide bond. It is worth noting at this point that the model cyclic peptides (1–5) did not show any trace of thio to oxo conversion even after 24 months at 25 °C in DMSO solution, indicating the stability of the thiopeptides in an oxidizing environment.
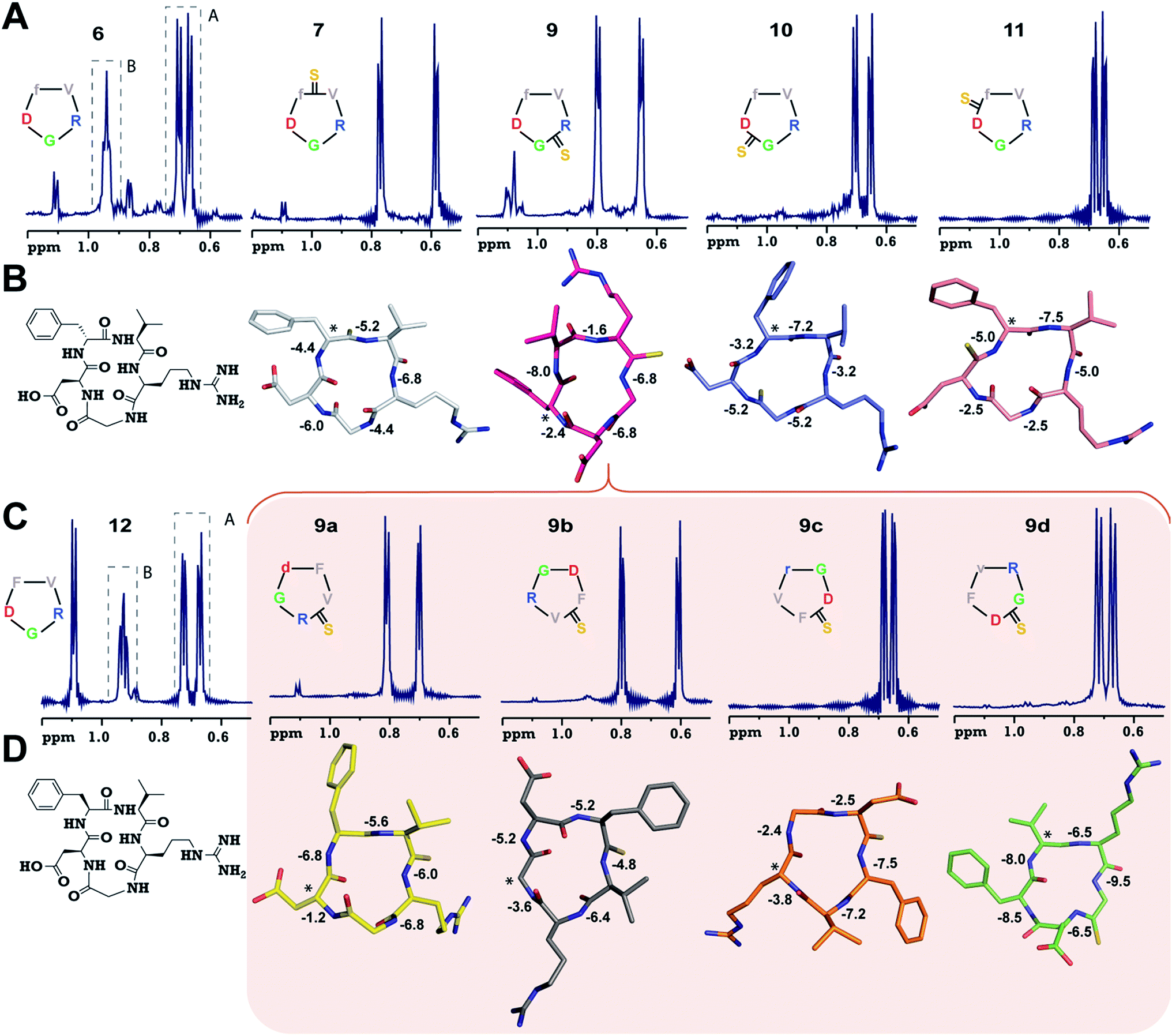 | ||
| Fig. 2 (A) 1H NMR spectra of cyclo(RGDfV) (6) and its thio-analogs 7–11. The γ-CH3 of Val in both the conformers (A and B) of 6 is shown. (B) The solution structures of 7–11 deduced from 2D 1H NMR spectroscopy in aqueous conditions. (C) 1H NMR spectra of cyclo(RGDFV) (12) showing two conformers. (D) Solution structures of spatial screen analogs of 9 (9a–9d). The temperature coefficients of the amide protons (ppb K−1) are shown and the asterisk denotes the site of the D-amino acid (Gly in 9b). Significant resonance overlap precluded the structure determination of 6 and 12. The hydrogen atoms are omitted for the sake of clarity. | ||
Adding to our excitement, all the thio-variants of 6 displayed a single conformation on the NMR time scale at room temperature, unlike the all-amide 6 (Fig. 2A). Variable temperature 1H NMR, recorded from 5 °C to 50 °C, also did not reveal the existence of any minor conformers in slow exchange in 7–11, (ESI†) directly demonstrating the rigidifying role of the thioamides in the peptide macrocycles, irrespective of the substitution site.
To demonstrate the uniform behavior of the thioamides in dictating the conformation of the cyclic peptide, we analyzed the solution conformations of 7–11 (Fig. 2B). It was encouraging to note that the conformation of these thionated RGD peptides reflects the conformation of the model alanine peptides with identical patterns of thioamide substitution. Furthermore, as observed in 3, 9 displayed a “frame-shifted” conformation due to the steric bulk associated with the CS group and not the improved thioamide H-bonding ability.17 Despite the involvement of the GlyHN thioamide in a β-type turn in 9, its temperature coefficient is −6.8 ppb K−1, indicating the absence of intramolecular hydrogen bonds, as opposed to D-PheHN (−2.4 ppb K−1) and ArgHN (−1.6 ppb K−1), which are involved in γ-turns (Fig. 2B). Likewise, the thioamide in 11 (D-PheHN) is also solvent exposed (−5.0 ppb K−1) and was not involved in stabilizing the γ-turn about Asp with GlyCO.
We were quite excited to observe that unlike N-methylation, which introduces severe conformational heterogeneity into macrocyclic peptides via cis–trans isomerism (due to the reduced cis–trans rotational barrier and the pseudoallylic strain between the N-methyl and the flanking Cβ groups within two homochiral residues),20 thioamides lower the conformational heterogeneity by reducing the conformational space of the cyclic peptide, which is guided by the van der Waals repulsion between S and the surrounding atoms and the increased cis–trans rotational barrier of the thioamide bond.21 Furthermore, despite structural alteration by thionation of the internally oriented amide bonds (3 and 9), the structural rearrangement results in a parent-like conformation, but with an altered side chain identity. These two observations suggested that (i) thioamides are valuable tools to reduce the conformational entropy in peptide macrocycles and (ii) thioamide substitution is an ideal modification for spatial screening22 of peptide sequences in search of bioactive conformations.
Thioamide incorporation yields superactive pro-angiogenic integrin antagonists
To validate our hypothesis and demonstrate the potential of thioamides in delivering selective and high-affinity ligands, we chose to target the cell-adhesion receptors, integrins.23 Integrins mediate a diverse set of cellular functions by binding to extracellular matrix components through a set of 24 distinct integrin heterodimers, formed by a combination of 18α-subunits and 8β-subunits. We were keen to simultaneously target the pro-angiogenic αvβ3, αvβ5 and α5β1 integrins, which have partially overlapping ligand specificity and are associated with migration, proliferation and survival in tumor cells.23b We sought to develop subtype selective integrin antagonists because: (a) these integrins are validated drug-targets for treating cancer, (b) pre-organization of the pharmacophore (–RGD– sequence) is known to modulate the integrin subtype selectivity, and (c) the N-methylated cyclic peptide, cilengitide (Cilen), unfortunately failed in phase III clinical trials against glioblastoma.24The inhibitory potency of the peptides to block the association between the extracellular matrix and the integrins overexpressed on cell-surfaces was determined using an in vitro competitive solid phase binding assay.25 We assessed the affinity of these peptides against the pro-angiogenic αvβ3, αvβ5 and α5β1 integrins, and their selectivity against their αIIbβ3 integrin, which plays an important role in platelet aggregation (Table 1).26 The parent peptide, 6, showed a moderate affinity towards the pro-angiogenic integrins due to the conformational flexibility, which does not allow the static presentation of the pharmacophores responsible for tight binding. On the contrary, the conformationally rigid thio analogs yielded very interesting results. 7, which can be directly compared with Cilen, where an N-methylated amide bond is substituted with a thioamide bond, displayed a higher IC50 than 6 against αvβ3. However, remarkably it showed a better IC50 than Cilen against αvβ5 and α5β1 in the in vitro assays with purified integrins.
| Compound | In vitro | In cellulo | αvβ3Xαvβ5a | ||||
|---|---|---|---|---|---|---|---|
| αvβ3 | αvβ5 | α5β1 | αIIbβ3 | MDA-MB-231 | U-87 MG | ||
| a Multiplication of in vitro IC50 against these integrins. | |||||||
| 6 | 140 ± 95 | 516 ± 117 | 164 ± 134 | >105 | >105 | >105 | >105 |
| 7 | 372 ± 52 | 0.7 ± 0.2 | 1.3 ± 0.6 | >104 | 4 ± 2 | 326 ± 97 | 260.4 |
| 9 | >105 | >104 | 53 ± 42 | >104 | >105 | >105 | >105 |
| 10 | 252 ± 200 | >105 | >105 | >105 | >105 | >105 | >105 |
| 11 | 72 ± 63 | 1.3 ± 1 | 62 ± 45 | >105 | 4.2 ± 4 | 96 ± 77 | 93.6 |
| 9a | 169 ± 100 | 0.5 ± 0.3 | 23 ± 12 | >105 | 11 ± 9 | 40 ± 28 | 84.5 |
| 9b | 0.2 ± 0.1 | 1.6 ± 0.3 | 2 ± 0.4 | >105 | 1 ± 0.1 | 1.6 ± 0.6 | 0.32 |
| 9c | >105 | >104 | 461 ± 246 | >105 | >105 | >105 | >105 |
| 9d | 584 ± 504 | >104 | 104 ± 95 | >105 | >105 | >105 | >105 |
| 12 | 166 ± 98 | >104 | 48 ± 20 | >105 | >105 | >105 | >105 |
| Cilen | 0.6 ± 0.4 | 3 ± 2 | 6 ± 1 | >105 | 1.2 ± 1 | 3 ± 2 | 1.8 |
To validate our in vitro data and determine the efficacy of the peptides against the pro-angiogenic integrins in a complex cellular milieu, we subjected the peptides to a competitive cell-adhesion assay (Table 1) utilizing MDA-MB-231 human breast cancer cells expressing αvβ5 integrins, and U-87 MG human glioblastoma cells expressing αvβ3, αvβ5 and α5β1 integrins.27 Despite the moderate affinity of 6 towards the pro-angiogenic integrins in vitro, it failed to show any efficacy at all in the cell-based assay, which could be attributed to the off-target binding associated with the conformational flexibility in 6. On the other hand, we observed a remarkable correlation between the in vitro affinity of the thioamidated peptides against the purified integrins and their efficacy in cellulo (αvβ5 with MDA-MB-231, and αvβ3 and αvβ5 with U-87 MG). This suggests the high specificity and selectivity of the thioamidated analogs towards the pro-angiogenic integrins in the presence of complex cellular components, which can otherwise sequester the peptides, reducing their efficacy.
We were quite excited to note that as detected in the solid phase assay, 7 strongly competed with vitronectin (Vn) in inhibiting the adhesion of MDA-MB-231 cells, suggesting its strong affinity towards αvβ5. A relatively lower potency of 7 was observed when inhibiting U-87 MG cell-adhesion to Vn (Vn binds to αvβ3 and αvβ5),23a which is in accord with the lower affinity of 7 towards αvβ3. 11 was the other member of the thio-scan library that showed better affinity than 6 towards the pro-angiogenic integrins in vitro, which was also reflected by its efficacy in the cell-adhesion assay.
The lowest affinity towards the pro-angiogenic integrin αvβ3 was obtained for 9 with thionation at Arg, which led to a complete reshuffling of the residues about which both the turns in 9 were centered (Fig. 2B), resulting in a total loss of its efficacy. However, since 9 displayed a βII′- and γ-type turn, we hypothesized that a subsequent spatial screening by altering the spatial position of the pharmacophore on the cyclic peptide backbone might improve the affinity and efficacy of the resulting peptides.
The spatial screening of 9 resulted in 9a–9d (Fig. 2C), of which 9c and 9d did not show any appreciable change in affinity and efficacy against the pro-angiogenic integrins. However, 9a displayed a remarkable improvement in affinity, and the most exciting observation was the dramatically enhanced (∼105 fold) affinity and efficacy of 9b in comparison to 9, by simple alteration in the spatial positioning of the pharmacophores on the thioamide backbone. To emphasize the effect of thio substitution, we synthesized compound 12 (a non-thio analog of 9b) (Fig. 2C), which showed moderate affinity only towards αvβ3 and α5β1 integrins with no efficacy in the cell-adhesion assay. Additionally, we noted that 12 also displayed two conformers (1:2) on the NMR time scale, as observed in 6, whereas 9b (along with 9a, 9c and 9d) showed a single conformation, further reinforcing the rigidifying role of thioamides in peptide macrocycles.
Interestingly, the backbone conformations of 9 and the spatial screen analogs (9a–9c) were comparable, except for 9d, which did not undergo the conformational switch (Fig. 2D). This is due to the absence of Cβ in glycine resulting in its ability to adopt a left-handed α-helical conformation,28 allowing the conformation of 9d. This result, along with the high temperature coefficient value of the thioamide in 9d, re-emphasizes the fact that the van der Waals interactions between the CS group and the surrounding atoms are critical in dictating the conformation of thioamidated macrocycles.
Taking into consideration the in vitro and in cellulo activity of the synthesized peptides (Table 1), we then performed docking simulations to understand the structure activity relationships of the thioamidated peptides with respect to the αvβ3 integrin, for which the crystal structure in complex with Cilen is already known (Fig. 3A).29 The most potent thioamidated peptide 9b and Cilen display a similar bound pose, the similarity being closest at the –RGD– motif (Fig. 3D) (ESI†). The important electrostatic interactions mediated by Arg and Asp of the –RGD– motif and the hydrophobic contacts mediated by Gly and the aromatic ring of Phe are also maintained in 9b.29 Additionally, 9b shows an aromatic hydrogen bond (C–H⋯O interaction) between Hε of Phe and one of the carboxylate oxygens of (β)-Asp126 (Fig. 3B). The C–H⋯O interactions are very weak but play an important role in protein–ligand binding events.30 This extra C–H⋯O interaction, together with the improved van der Waals interactions (mediated by a S atom as discussed later), possibly contributes to the slightly improved inhibitory potency of 9b.
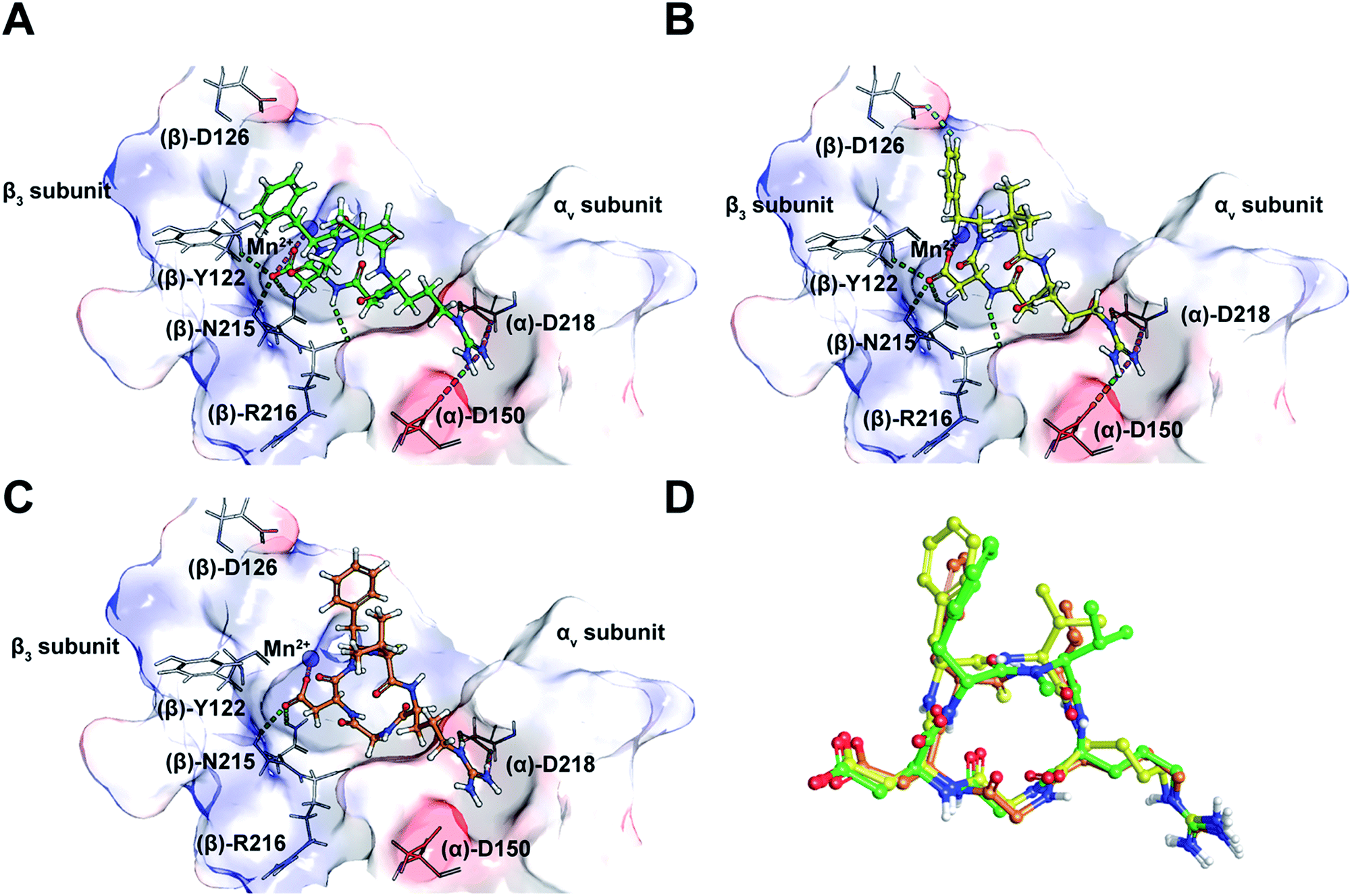 | ||
| Fig. 3 (A) The interaction of cilengitide (shown as a green ball and stick model) bound to the αvβ3 integrin in the PDB entry 1L5G. (B) The interaction of docked 9b (shown as a yellow ball and stick model) with the αvβ3 integrin. (C) The interaction of docked 7 (shown as an orange ball and stick model) with the αvβ3 integrin. (D) An overlay of the docked poses of 9b and 7 onto that of Cilen co-crystallized with the αvβ3 integrin in 1L5G. The green, magenta and cyan dashed lines represent hydrogen bonds, salt bridges and aromatic hydrogen bonds, respectively. The binding site of the protein is shown as a surface representation and the carbon atoms of the protein residues are shown in grey. Oxygen, nitrogen, hydrogen and sulphur atoms are shown in red, blue, white and yellow, respectively. Please see the ESI† for a detailed description of the interactions between the various ligands and the αvβ3 integrin. | ||
The docked poses of all the other thioamidated peptides do not show such a C–H⋯O interaction, except for 11. However, the lower potency of 11 stems from the loss of some crucial interactions with the receptor mediated by the –RGD– motif (ESI†). Likewise, all other analogs except 9b show the loss of one or multiple electrostatic interactions with the receptor. The substitution of O with S provides a greater contact surface area between the peptide and the receptor, which leads to improved van der Waals interactions mediated by the CS group compared to the CO group.10 Although we did not notice any direct hydrogen bonds between the protein and S atom,31 we cannot fully negate the possibility of any water mediated hydrogen bonding between the S atom and the receptor.32
Interestingly, 9b and 7, which differ only in the chirality about the phenylalanine, show comparable affinity for αvβ5 and α5β1, but >103 fold difference in αvβ3 affinity. Docking of 7 revealed that this difference is due to the smaller distance between Arg and Asp Cβ compared to Cilen and 9b, leading to the loss of end-on hydrogen bonding between Arg and (α)-Asp150 (Fig. 3C) and the side chain mediated interaction of Asp with the backbone of (β)-Tyr122. Additionally, the backbone mediated interaction of Asp and (β)-Arg216 is also lost due to a flip in the Gly–Asp amide bond (Fig. 3D). This result demonstrates how an inversion of chirality drastically affects the overall efficacy of a macrocyclic peptide.26 Lastly, our docking results were nicely validated by 9, which did not yield any meaningful binding mode (ESI†). The complete loss in its affinity could be rationalized by the absence of biologically relevant interactions mediated by the Asp and Arg side chains with αvβ3. Thus, our in silico analysis corroborates the in vitro and in cellulo results.
Thionated macrocyclic peptides show minimal non-specific cytotoxicity and enhanced serum half-life
Since integrin antagonists are known to inhibit the proliferation of cancer cells,33 we assessed the effect of a subset of these analogs on the proliferation of MDA-MB-231 and U-87 MG cells and simultaneously determined whether the incorporation of thioamides in peptide macrocycles leads to unspecific cytotoxicity, which is of paramount importance in lead design (Fig. 4). Although in both the cell types we observed a moderate dose-dependent decrease in cell viability at 48 h, a very high concentration of the thio-analogs and Cilen was required to achieve the half maximal inhibitory concentration (IC50). This clearly suggested that incorporation of thioamides into macrocyclic peptides does not result in unspecific cytotoxicity even after 48 h, which is an important concern for the use of thioamides in lead design. We also noted that glioblastoma cells were more sensitive towards these antagonists (Cilen, 9b and 7) than breast cancer cells, presumably due to the expression of all the pro-angiogenic integrins, despite the known resistance of U-87 MG cells against detachment-induced apoptosis.27 Furthermore, as expected the least efficacious flexible cyclic peptides 6 and 12 displayed minimal detachment-induced apoptosis in breast cancer cells compared to the potent thioanalogs. | ||
| Fig. 4 Proliferation of (A) breast cancer cells and (B) glioblastoma cells at 48 h; n = 4 ± SEM. Note the slow dose-dependent decrease in proliferation and minimal cytotoxicity even with a 2 × 104 fold increase in initial concentration. Please refer to the ESI† for the entire dose-dependent study. | ||
One major drawback to translating peptides as drugs is their inherent metabolic instability due to the action of endo- and exoproteases.4b Thus, we determined the stability of the unmodified peptide macrocycles, Cilen (N-methylated peptide) and the most potent thioanalogs in human serum ex vivo for 72 h (Fig. 5). As expected, 6 showed slightly better metabolic stability (t1/2: 9 h) than 12 (t1/2: 7 h) due to the presence of D-Phe. It is interesting to note the significant improvement in metabolic stability of all the thioamidated analogs throughout the course of the experiment as observed previously in linear thionated peptides.12a,b,e,f Since these analogs have thionation at different amino acids, we speculate that thioamidation has the potential to significantly improve the metabolic stability of macrocyclic peptides irrespective of the site and amino acid being substituted. It was also quite striking to note that 11 and 9b showed a significantly improved serum half-life (t1/2: 36 h each) compared to that of the clinical candidate Cilen (t1/2: 12 h). These data collectively suggest that thioamidation could have a stronger influence in improving the metabolic stability of macrocyclic peptides than D-amino acid incorporation.
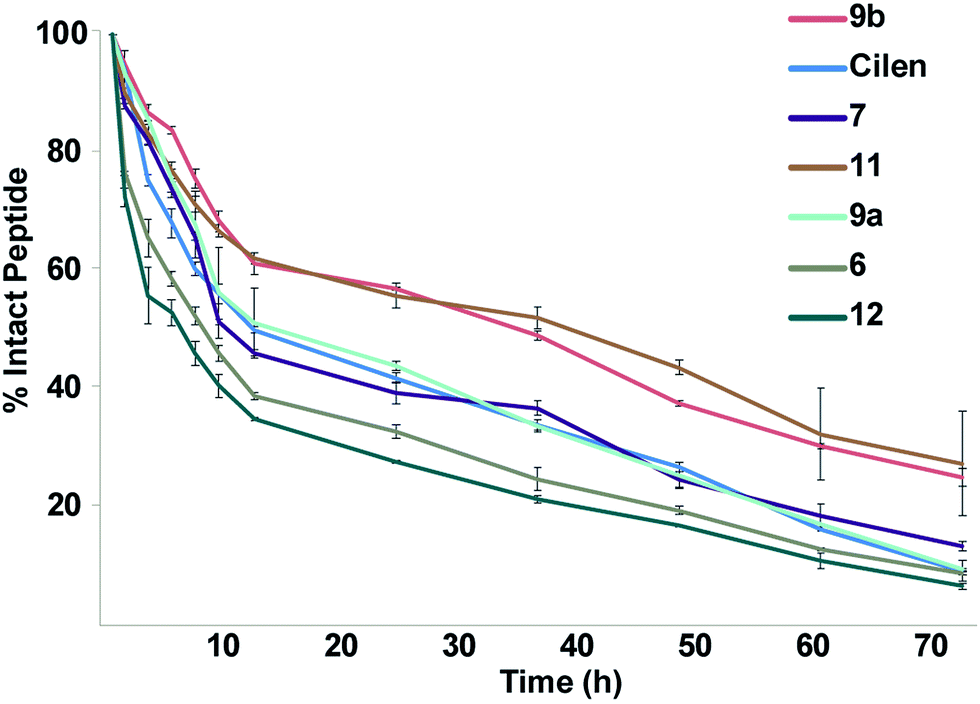 | ||
| Fig. 5 The ex vivo metabolic stability of the macrocyclic peptides in freshly isolated human serum for 72 h; n = 6 ± SEM. Note the rapid degradation of the unmodified cyclic peptides 6 and 12. Even at 72 h, 1/4 of the thioamidated peptides 9b and 11 are intact. | ||
Conclusions
In summary, we have demonstrated that macrocyclic peptides exhibit conformational flexibility in both apolar (model alanine peptides) and polar environments (integrin antagonists), which tends to reduce their bioactivity.3a,b,4c We observed that CO to CS substitution in peptide macrocycles dramatically reduces the conformational flexibility while minimally perturbing the global conformation as observed in the model alanine, thio-scan and spatial screen integrin antagonists. Thus, thioamide incorporation leads to predictable changes in the conformation of the peptide backbone, unlike other amide bond modifications. Thioamide incorporation fine tunes the 3D-pharmacophore orientation, which qualifies thioamides as important bioisosteric mimics of peptide bonds.34 The remarkable correlation between the in vitro and in cellulo data against the pro-angiogenic integrins demonstrates the specificity of these thioamidated macrocyclic peptides against their endogenous target, which can be extended to other bioactive peptides as well. Thioamidated macrocyclic peptides are non-cytotoxic and, presumably due to their marked conformational rigidity, display enhanced stability against degradation by proteases,35 leading to their increased therapeutic efficacy. Due to the adoption of well-defined structures (turns) caused by increased conformational rigidity, thioamides might have the potential to increase the membrane permeability of cyclic peptides.36
It is curious to note that few thioamide containing peptide natural products, such as thioviridamide, thioholgamides, and their related analogs,37 which are ribosomally synthesized and post-translationally modified (RiPP), or are of non-ribosomal origin (closthioamide),38 show potent antibacterial and antineoplastic activity compared to their all-oxo-amide variant.38a Thus, the potent biological activity in these compounds possibly arises from a combination of conformational restriction, increased metabolic stability and enhanced cellular permeability by thio substitution. Furthermore, as observed in 10, the conformational restriction imparted by the thioamidation of glycine is also predicted to stabilize the local conformation in the vicinity of the active site in methyl-coenzyme M reductase, leading to the increased half-life of the protein.39 Therefore, we strongly believe that this single atom substitution, by successfully overcoming the two major drawbacks of peptides i.e. conformational flexibility and metabolic instability, will accelerate the discovery of peptide-based drugs and antibacterials.40 All the efficacious thioamidated macrocyclic peptides reported here have the potential to block angiogenesis in vivo and are currently being investigated.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the DBT through the project 6242/P-17/2015 and the DST through the project SB/YS/LS-220/2013 for JC, the DBT-IISc partnership program, the DST-FIST and the Indian Institute of Science (MHRD) for funding this work. We thank the DST & DBT funded NMR and proteomics facility at the IISc. We are grateful to Prof. R. Sowdhamini for her generous support in providing access to the Schrödinger suite of programs, Prof. S. P. Sarma for help with NMR relaxation experiments and Prof. D. Pal for access to Discovery Studio. Prof. A. Rangarajan and Prof. K. Somasundaram are acknowledged for their kind gift of MDA-MB-231, MCF7 and U-87 MG cell lines.References
- (a) C. Heinis, Nat. Chem. Biol., 2014, 10, 696 CrossRef CAS PubMed; (b) A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC; (c) L. J. Walport, R. Obexer and H. Suga, Curr. Opin. Biotechnol., 2017, 48, 242 CrossRef CAS PubMed; (d) A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed.
- (a) V. J. Hruby, Life Sci., 1982, 31, 189 CrossRef CAS PubMed; (b) S. Jackson, W. Degrado, A. Dwivedi, A. Parthasarathy, A. Higley, J. Krywko, A. Rockwell, J. Markwalder, G. Wells, R. Wexler, S. Mousa and R. Harlow, J. Am. Chem. Soc., 1994, 116, 3220 CrossRef CAS; (c) T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed.
- (a) M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901 CrossRef CAS PubMed; (b) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136 CrossRef CAS PubMed; (c) G. R. Marshall, Tetrahedron, 1993, 49, 3547 CrossRef CAS; (d) C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef CAS PubMed.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg, C. K. Marlowe, D. C. Spellmeyer, R. Y. Tan, A. D. Frankel, D. V. Santi, F. E. Cohen and P. A. Bartlett, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367 CrossRef CAS.
- G. Guichard, N. Benkirane, G. Zederlutz, M. H. V. Vanregenmortel, J. P. Briand and S. Muller, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 9765 CrossRef CAS.
- T. Cupido, J. Spengler, J. Ruiz-Rodriguez, J. Adan, F. Mitjans, J. Piulats and F. Albericio, Angew. Chem., Int. Ed., 2010, 49, 2732 CrossRef CAS PubMed.
- J. Zhang, M. Mulumba, H. Ong and W. D. Lubell, Angew. Chem., Int. Ed., 2017, 56, 6284 CrossRef CAS PubMed.
- E. Biron, J. Chatterjee, O. Ovadia, D. Langenegger, J. Brueggen, D. Hoyer, H. A. Schmid, R. Jelinek, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2008, 47, 2595 CrossRef CAS PubMed.
- A. Choudhary and R. T. Raines, ChemBioChem, 2011, 12, 1801 CrossRef CAS PubMed.
- (a) D. R. Artis and M. A. Lipton, J. Am. Chem. Soc., 1998, 120, 12200 CrossRef CAS; (b) R. Frank, M. Jakob, F. Thunecke, G. Fischer and M. Schutkowski, Angew. Chem., Int. Ed., 2000, 39, 1120 CrossRef CAS; (c) J. H. Miwa, A. K. Patel, N. Vivatrat, S. M. Popek and A. M. Meyer, Org. Lett., 2001, 3, 3373 CrossRef CAS PubMed; (d) J. H. Miwa, L. Pallivathucal, S. Gowda and K. E. Lee, Org. Lett., 2002, 4, 4655 CrossRef CAS PubMed; (e) A. Reiner, D. Wildemann, G. Fischer and T. Kiefhaber, J. Am. Chem. Soc., 2008, 130, 8079 CrossRef CAS PubMed; (f) R. M. Culik, H. Jo, W. F. DeGrado and F. Gai, J. Am. Chem. Soc., 2012, 134, 8026 CrossRef CAS PubMed; (g) J. M. Goldberg, S. Batjargal, B. S. Chen and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 18651 CrossRef CAS PubMed; (h) J. M. Goldberg, X. Chen, N. Meinhardt, D. C. Greenbaum and E. J. Petersson, J. Am. Chem. Soc., 2014, 136, 2086 CrossRef CAS PubMed; (i) C. R. Walters, D. M. Szantai-Kis, Y. Zhang, Z. E. Reinert, W. S. Horne, D. M. Chenoweth and E. J. Petersson, Chem. Sci., 2017, 8, 2868 RSC.
- (a) P. A. Bartlett, K. L. Spear and N. E. Jacobsen, Biochemistry, 1982, 21, 1608 CrossRef CAS PubMed; (b) P. Campbell and N. T. Nashed, J. Am. Chem. Soc., 1982, 104, 5221 CrossRef CAS; (c) K. Clausen, A. F. Spatola, C. Lemieux, P. W. Schiller and S. O. Lawesson, Biochem. Biophys. Res. Commun., 1984, 120, 305 CrossRef CAS PubMed; (d) M. Schutkowski, S. Wollner and G. Fischer, Biochemistry, 1995, 34, 13016 CrossRef CAS PubMed; (e) B. Zacharie, M. Lagraoui, M. Dimarco, C. L. Penney and L. Gagnon, J. Med. Chem., 1999, 42, 2046 CrossRef CAS PubMed; (f) A. Bach, J. N. N. Eildal, N. Stuhr-Hansen, R. Deeskamp, M. Gottschalk, S. W. Pedersen, A. S. Kristensen and K. Stromgaard, J. Med. Chem., 2011, 54, 1333 CrossRef CAS PubMed; (g) X. Chen, E. G. Mietlicki-Baase, T. M. Barrett, L. E. McGrath, K. Koch-Laskowski, J. J. Ferrie, M. R. Hayes and E. J. Petersson, J. Am. Chem. Soc., 2017, 139, 16688 CrossRef CAS PubMed.
- (a) D. B. Sherman and A. F. Spatola, J. Am. Chem. Soc., 1990, 112, 433 CrossRef CAS; (b) D. Seebach, S. Y. Ko, H. Kessler, M. Kock, M. Reggelin, P. Schmieder, M. D. Walkinshaw, J. J. Bolsterli and D. Bevec, Helv. Chim. Acta, 1991, 74, 1953 CrossRef CAS; (c) H. Kessler, A. Geyer, H. Matter and M. Kock, Int. J. Pept. Protein Res., 1992, 40, 25 CrossRef CAS PubMed; (d) H. Kessler, H. Matter, A. Geyer, H. J. Diehl, M. Kock, G. Kurz, F. R. Opperdoes, M. Callens and R. K. Wierenga, Angew. Chem., Int. Ed., 1992, 31, 328 CrossRef; (e) Y. Hitotsuyanagi, J. Suzuki, Y. Matsumoto, K. Takeya and H. Itokawa, J. Chem. Soc., Perkin Trans. 1, 1994, 1887 RSC; (f) H. Morita, Y. S. Yun, K. Takeya, H. Itokawa and O. Shirota, Bioorg. Med. Chem., 1997, 5, 631 CrossRef CAS PubMed; (g) P. Jeschke, A. Harder, W. Etzel, W. Gau, G. Thielking, G. Bonse and K. Iinuma, Pest Manage. Sci., 2001, 57, 1000 CrossRef CAS PubMed; (h) W. L. Lin, F. Erdmann, A. Quintero, G. Fischer and Y. X. Zhang, Bioorg. Med. Chem. Lett., 2016, 26, 5754 CrossRef CAS PubMed.
- J. Chatterjee, D. Mierke and H. Kessler, J. Am. Chem. Soc., 2006, 128, 15164 CrossRef CAS PubMed.
- (a) D. F. Mierke, M. Kurz and H. Kessler, J. Am. Chem. Soc., 1994, 116, 1042 CrossRef CAS; (b) X. M. Zhang, G. V. Nikiforovich and G. R. Marshall, J. Med. Chem., 2007, 50, 2921 CrossRef CAS PubMed.
- (a) Z. P. Liu and L. M. Gierasch, Biopolymers, 1992, 32, 1727 CrossRef CAS PubMed; (b) R. Haubner, R. Gratias, B. Diefenbach, S. L. Goodman, A. Jonczyk and H. Kessler, J. Am. Chem. Soc., 1996, 118, 7461 CrossRef CAS.
- H. J. Lee, Y. S. Choi, K. B. Lee, J. Park and C. J. Yoon, J. Phys. Chem. A, 2002, 106, 7010 CrossRef CAS.
- (a) J. P. Meraldi, V. J. Hruby and A. I. R. Brewster, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1373 CrossRef CAS PubMed; (b) C. H. Niu, L. G. Pease and E. R. Blout, Biopolymers, 1978, 17, 115 CrossRef CAS.
- S. Mukherjee, H. Verma and J. Chatterjee, Org. Lett., 2015, 17, 3150 CrossRef CAS PubMed.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, Acc. Chem. Res., 2008, 41, 1331 CrossRef CAS PubMed.
- K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1995, 117, 2201 CrossRef CAS.
- T. Weide, A. Modlinger and H. Kessler, Top. Curr. Chem., 2007, 272, 1 CrossRef CAS.
- (a) M. Barczyk, S. Carracedo and D. Gullberg, Cell Tissue Res., 2010, 339, 269 CrossRef CAS PubMed; (b) J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9 CrossRef CAS PubMed.
- R. Stupp, M. E. Hegi, T. Gorlia, S. C. Erridge, J. Perry, Y. K. Hong, K. D. Aldape, B. Lhermitte, T. Pietsch, D. Grujicic, J. P. Steinbach, W. Wick, R. Tarnawski, D. H. Nam, P. Hau, A. Weyerbrock, M. J. B. Taphoorn, C. C. Shen, N. Rao, L. Thurzo, U. Herrlinger, T. Gupta, R. D. Kortmann, K. Adamska, C. McBain, A. A. Brandes, J. C. Tonn, O. Schnell, T. Wiegel, C. Y. Kim, L. B. Nabors, D. A. Reardon, M. J. van den Bent, C. Hicking, A. Markivskyy, M. Picard, M. Weller, E. O. R. T. Canc, C. B. T. Consortium and C. S. Team, Lancet Oncol., 2014, 15, 1100 CrossRef CAS PubMed.
- C. Mas-Moruno, J. G. Beck, L. Doedens, A. O. Frank, L. Marinelli, S. Cosconati, E. Novellino and H. Kessler, Angew. Chem., Int. Ed., 2011, 50, 9496 CrossRef CAS PubMed.
- C. Mas-Moruno, F. Rechenmacher and H. Kessler, Anti-Cancer Agents Med. Chem., 2010, 10, 753 CrossRef CAS PubMed.
- J. W. Kim, F. V. Cochran and J. R. Cochran, J. Am. Chem. Soc., 2015, 137, 6 CrossRef CAS PubMed.
- J. S. Richardson, Adv. Protein Chem., 1981, 34, 167 CrossRef CAS PubMed.
- J. P. Xiong, T. Stehle, R. G. Zhang, A. Joachimiak, M. Frech, S. L. Goodman and M. A. Arnaout, Science, 2002, 296, 151 CrossRef CAS PubMed.
- A. C. Pierce, K. L. Sandretto and G. W. Bemis, Proteins: Struct., Funct., Genet., 2002, 49, 567 CrossRef CAS PubMed.
- V. R. Mundlapati, S. Gautam, D. K. Sahoo, A. Ghosh and H. S. Biswal, J. Phys. Chem. Lett., 2017, 8, 4573 CrossRef CAS PubMed.
- M. S. Bodnarchuk, Drug Discovery Today, 2016, 21, 1139 CrossRef CAS PubMed.
- A. Abdollahi, D. W. Griggs, H. Zieher, A. Roth, K. E. Lipson, R. Saffrich, H. J. Grone, D. E. Hallahan, R. A. Reisfeld, J. Debus, A. G. Niethammerl and P. E. Huber, Clin. Cancer Res., 2005, 11, 6270 CrossRef CAS PubMed.
- J. Vagner, H. C. Qu and V. J. Hruby, Curr. Opin. Chem. Biol., 2008, 12, 292 CrossRef CAS PubMed.
- K. V. Lawson, T. E. Rose and P. G. Harran, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3753 CrossRef PubMed.
- (a) G. Lattig-Tunnemann, M. Prinz, D. Hoffmann, J. Behlke, C. Palm-Apergi, I. Morano, H. D. Herce and M. C. Cardoso, Nat. Commun., 2011, 2, 453 CrossRef PubMed; (b) J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488 CrossRef CAS PubMed.
- (a) Y. Hayakawa, K. Sasaki, H. Adachi, K. Furihata, K. Nagai and K. Shin-ya, J. Antibiot., 2006, 59, 1 CrossRef CAS PubMed; (b) L. Frattaruolo, R. Lacret, A. R. Cappello and A. W. Truman, ACS Chem. Biol., 2017, 12, 2815 CrossRef CAS PubMed; (c) L. Kjaerulff, A. Sikandar, N. Zaburannyi, S. Adam, J. Herrmann, J. Koehnke and R. Muller, ACS Chem. Biol., 2017, 12, 2837 CrossRef CAS PubMed.
- (a) T. Lincke, S. Behnken, K. Ishida, M. Roth and C. Hertweck, Angew. Chem., Int. Ed., 2010, 49, 2011 CrossRef CAS PubMed; (b) V. F. Miari, P. Solanki, Y. Hieba, R. A. Stabler and J. T. Heap, Antimicrob. Agents Chemother., 2017, 61, 929 CrossRef PubMed.
- D. D. Nayak, N. Mahanta, D. A. Mitchell and W. W. Metcalf, eLife, 2017, 6, 29218 Search PubMed.
- D. W. Carney, K. R. Schmitz, J. V. Truong, R. T. Sauer and J. K. Sello, J. Am. Chem. Soc., 2014, 136, 1922 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04671e |
| This journal is © The Royal Society of Chemistry 2018 |