
Novel properties and applications of carbon nanodots
Lian
Xiao
a and
Handong
Sun
 *abc
*abc
aDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: hdsun@ntu.edu.sg
bCentre for Disruptive Photonic Technologies (CDPT), School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore
cMajuLab, CNRS-UCA-SU-NUS-NTU International Joint Research Unit, Singapore
First published on 17th July 2018
Abstract
In the most recent decade, carbon dots have drawn intensive attention and triggered substantial investigation. Carbon dots manifest superior merits, including excellent biocompatibility both in vitro and in vivo, resistance to photobleaching, easy surface functionalization and bio-conjugation, outstanding colloidal stability, eco-friendly synthesis, and low cost. All of these endow them with the great potential to replace conventional unsatisfactory fluorescent heavy metal-containing semiconductor quantum dots or organic dyes. Even though the understanding of their photoluminescence mechanism is still controversial, carbon dots have already exhibited many versatile applications. In this article, we summarize and review the recent progress achieved in the field of carbon dots, and provide a comprehensive summary and discussion on their synthesis methods and emission mechanisms. We also present the applications of carbon dots in bioimaging, drug delivery, microfluidics, light emitting diode (LED), sensing, logic gates, and chiral photonics, etc. Some unaddressed issues, challenges, and future prospects of carbon dots are also discussed. We envision that carbon dots will eventually have great commercial utilization and will become a strong competitor to some currently used fluorescent materials. It is our hope that this review will provide insights into both the fundamental research and practical applications of carbon dots.
 Lian Xiao | Lian Xiao received his Bachelor's degree in Physics from Sichuan University, China, in 2014. He is currently a year 4 PhD student in the School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore. His research interests include nanomaterials, optical spectroscopy, semiconductor physics, and micro-fluidics. |
 Handong Sun | Handong Sun is currently an associate professor in the School of Physical and Mathematical Science, Nanyang Technological University. He received his Bachelor's degree in Physics from Dalian University of Technology, and his Master's and PhD degrees from Huazhong University of Science and Technology, and Hong Kong University of Science and Technology, respectively. He was elected as a Fellow of the American Physical Society in 2016. His research interests cover optoelectronic materials and devices, semiconductor physics, optical spectroscopy, nanomaterials, and applications in microfluidics. |
1 Introduction
Carbon dots have become a hot topic of numerous scientific studies since their discovery1,2 due to their obvious advantages over conventional semiconductor quantum dots whose notable toxicity and environmental hazards are well documented.3–6 Both in vitro and in vivo toxicity evaluation have demonstrated that carbon dots exhibit excellent biocompatibility, which, along with their photostability, chemical inertness, ease of surface functional group modification, and low cost, endow carbon dots with vast potential scope to be applied in various fields.2,7,8 The exploration of a stable, fast, low-cost synthesis approach is a prerequisite to make use of carbon dots, and fortunately numerous breakthroughs have been achieved over the past decade. Consequently, carbon dots have become the center of significant research efforts to develop an alternative to replace the unsatisfactory traditional fluorescent materials, and the publications about carbon dots have continuously increased since 2006, as seen in Fig. 1 (the dates are generated from the Web of Science up to April 16, 2018, with the search criteria: topic “carbon dots” or “C-dots” or “carbon nanodots” or “graphene quantum dots”, database: Web of Science Core collection). Carbon dots synthesized from different approaches share some similar features, such as less than 10 nm size, bright fluorescent emission, rich hydrophilic surface groups which endow carbon dots with excellent water solubility, and biocompatibility. However, some contradictory behaviors, like non-photoblinking2,9 and photoblinking,10–12 pH-stable13 and pH-dependent PL,14,15 carbon dots size-dependent13,16–18 and size-independent PL,19,20 excitation-dependent20 and independent PL,13,21 have been reported by different research groups. All of these attributes indicate that carbon dots represent a much complex system than expected. It seems that the diverse behaviors of carbon dots result from the different synthesis approaches, that is, different synthesis methods will result in different types of carbon dots.21 Therefore it is necessary to provide the synthesis method when talking about carbon dots. Thus prior to the discussion about the optical properties and applications, we first present the synthesis approaches of carbon dots adopted during the past decades in Section 2 of this review. After more than 10 years of efforts, numerous synthesis methods, including several intriguing one-step approaches, have been discovered by researchers. Generally, the carbon dots synthesis methods can be divided into two categories: top down and bottom up, and each of these contains many sub-categories, which will be presented in detail in Section 2. What is exciting for carbon dots is that researchers have successfully explored highly luminescent carbon dots with a quantum yield up to 80%,22 which is comparable to conventional semiconductor quantum dots, and further, the emission range can be tuned from deep UV to near IR by careful selection of the synthesis approach and precursors.13,17,23–28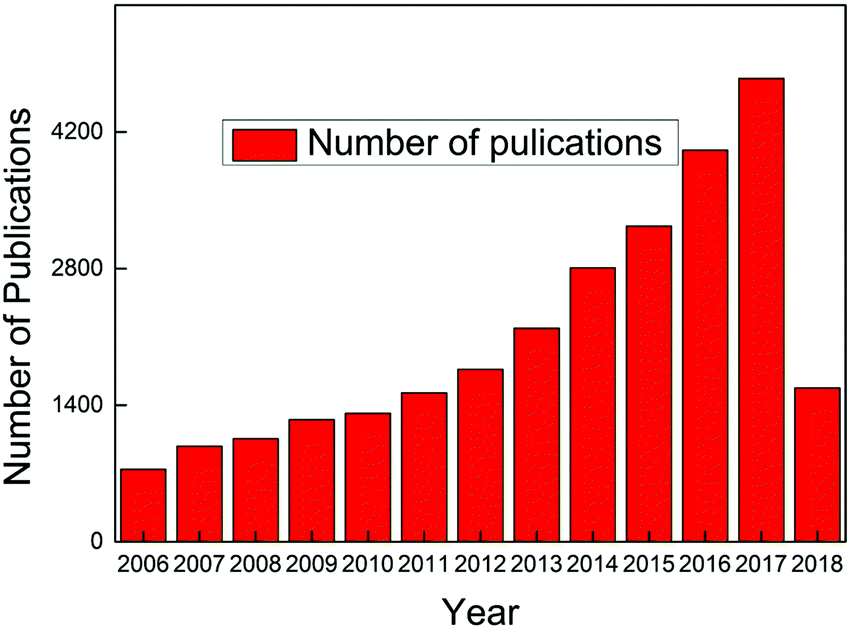 | ||
| Fig. 1 Number of publications about carbon dots since 2006. Dates are generated from the Web of Science up to April 16, 2018; search criteria: topic “carbon dots” or “C-dots” or “carbon nanodots” or “graphene quantum dots”; database: Web of Science Core collection. | ||
To make better use of carbon dots and to tune their optical properties, a full understanding of carbon dots emission is essential. Ironically, the actual emission mechanisms of carbon dots are still controversial even though some breakthroughs have been obtained in recent years. In Section 3, we summarize and display the emission models proposed by different authors and also provide the key evidences for each emission mechanism. Some of the mechanisms are mutually complementary, while some are in sharp conflict. We speculate that different emission mechanisms may be applied to one or more kinds of carbon dots, but these may not be valid to all kinds of carbon dots.
The applications of carbon dots are presented in Section 4. In this review, we focus on the optical and optoelectronic applications, including bioimaging, drug delivery, microfluidic applications, light emitting diodes (LEDs), sensing, especially for high-resolution ratiometric sensing, chemical logic gates, and chiral photonics. Carbon dots have demonstrated good performances in all of the above applications. In the last section, we provide a brief summary and discuss the challenges and future prospects of carbon dots in further exploration. This article is not intended to be a comprehensive review (or a complete list of the published results) in the research field of carbon dots, but rather aims to provide some selective helpful insights, as chosen from our direct experience as well as the recent literature of the subject. We hope that this review article can present a clear picture of carbon dots, from their synthesis, to their characterization and applications, and give valuable insights into both their fundamental physics and practical applications. In addition, we also hope to offer some perspectives and clues to further improve the optical properties and to aid the exploration of more far-reaching applications of carbon dots.
2 Synthesis approaches
Since they were first discovered fortuitously by Xu et al. in 2004,1 and later named by Sun et al. in 2006,2 researchers have paid intense attention to carbon dots because of their striking advantages. However, a stable, economic, easy, and fast synthesis method is a prerequisite to further explore the properties and applications of carbon dots. Overall, carbon dots synthesis methods can be divided into two categories, either top down or bottom up, based on the starting carbon source. Top-down methods are realized by directly exfoliating a bulk carbon source, such as graphite, graphene, or carbon fiber, into nano-scale carbon dots, followed by surface treatment. Opposite to direct exfoliation, bottom-up methods synthesize carbon dots from a carbon element containing small molecules, such as glucose or citric acid. In this part, we review the main progress in the synthesis of carbon dots achieved by researchers in both top-down and bottom-up approaches. To improve and tune the optical properties, such as the quantum yield, emission wavelength, and photostability, surface passivation and hetero-atom doping are employed during the carbon dots synthesis process. It is worth emphasizing that it's not reasonable to evaluate which method is the “best” synthesis approach. As we discussed in Section 1, different kinds of carbon dots exhibit different and even opposite properties, thus we suggest that the choice of the synthesis method depends on the intended application and its requirements.2.1 Top-down approaches for fabricating carbon dots
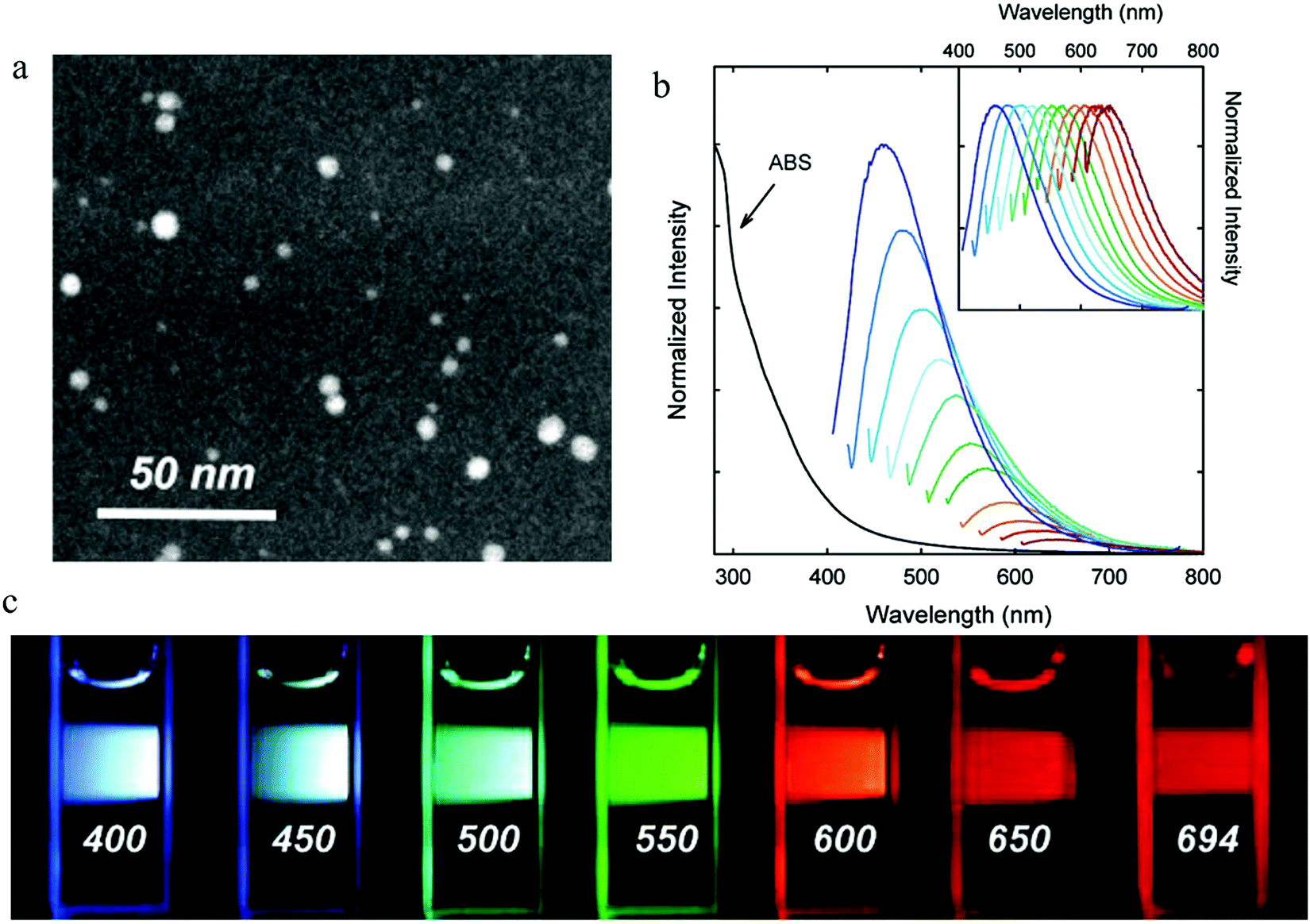 | ||
| Fig. 2 (a) STEM imagine of PEG1500N surface-passivated carbon dots. (b) Absorption and emission spectra for different excitation wavelengths from 400 nm to 600 nm with an increase step of 20 nm. (c) Photographs following excitation by different excitation wavelengths as indicated in the figures. (a–c) Reproduced with permission from ref. 2. Copyright 2006, American Chemical Society. | ||
Later, amended approaches involving combining the laser ablation and surface modification processes into one procedure have also been reported.7,20,29,30 A Nd:YAG pulsed laser (wavelength of 1.064 μm and power density 6.0 × 106 W cm−2) was used to irradiate a mixture of graphite and organic solvent, resulting in the carbon dots synthesis and surface modification being accomplished simultaneously. Besides, researchers have also proposed that the emission properties of carbon dots could be tuned by properly choosing the organic solvent, such as, PEG2000N, diamine hydrate, diethanolamine.
Li and co-workers32 also employed an electrochemical approach to synthesize carbon dots, using the graphite as both the anode and cathode and a current intensity in the range of 10–200 mA cm−2. They demonstrated that the electrolyte environment played an important role in the carbon dots formation. When the electrolyte was alkaline (for example, NaOH/EtOH), carbon dots could be synthesized successfully, while an acid electrolyte (for example, H2SO4/EtOH) could not form the carbon dots. (HR)TEM analysis showed that the synthesized carbon dots had a diameter less than 4 nm with a lattice spacing of around 0.32 nm.
In addition to graphite and carbon nanotubes, other bulk carbon sources, such as graphene film, carbon fiber,33–35 have also been employed to synthesize carbon dots by means of electrochemical approaches.
In addition to candle soot, carbon dots synthesized from the chemical exfoliation of carbon fibers were reported by Ajayan's group.18 A mixture of concentrated H2SO4 and HNO3 was prepared to exfoliate the carbon fiber. The carbon fiber was added to the concentrated acid followed by 2 h sonic treatment. After that, the mixture was heated to a certain temperature for 24 h with vigorous stirring. The synthesized carbon dots exhibited a relatively narrow size distribution (diameter between 1–4 nm) with a lattice parameter of 0.242 nm. The authors also demonstrated that the optical properties (absorption and PL) of the carbon dots could be tuned by simply controlling the reaction temperature to yield different sizes of carbon dots. Later Ye and co-workers36 also employed concentrated H2SO4 and HNO3 to exfoliate coal to synthesize carbon dots.
2.2 Bottom-up approaches for fabricating carbon dots
Later Emmanuel P. Giannelis's group38 did a comprehensive investigation of the carbon dots pyrolysis process. Citric acid monohydrate (C6H8O7) and ethanolamine (C2H7ON) were employed to synthesize carbon dots in different pyrolysis temperatures in the absence of solvent and under reflux in air. The authors demonstrated that, under the condition of low temperature, such as pyrolysis at 180 °C for 30 min (termed as CNP180), the pyrolysis resulted in carbon dots precursors, and a strong PL emission originating from the organic fluorophores. There were several pieces of evidence that supported the molecular assumptions: (1) no carbon dot particles could be observed by either DLS or TEM, and (2) the sample showed excitation-independent PL with a remarkable quantum yield of 50%. More specifically, the authors suggested38 the amide-related molecules contributed to the emission of carbon dots molecular precursors, which was verified by FTIR, XPS, and 1H & 13C NMR. To further confirm the amide-related functional groups, the amide was also synthesized by an alternative approach using citric acid monohydrate (C6H8O7) and ethanolamine (C2H7ON) as the raw materials, and the results showed that the amide and the carbon dots precursors exhibited similar optical behaviors, which validated the mechanism.
Higher temperature (e.g., at 230 °C for 30 min, CNP230) pyrolysis triggered the carbon dots formation, which was verified by TEM analysis, whereupon the carbon dots showed a spherical morphology with an average size of 19 nm. The authors claimed38 that the carbon dots formation was the result of extensive cross-linking reactions and the appearance of interchain imide bonds, as substantiated by the FTIR spectrum. Element analysis showed that the hydrogen component of CNP230 (44.85% C, 5.75% H, 10.85% N) decreased compared to CNP180 (41% C, 7.8% H, 11.7% N). The PL quantum yield of CNP230 decreased to 15%, while the excitation-dependent PL started to appear.
Increasing the temperature to 300 °C resulted in a darkening of the sample and synthesized carbon dots with an average size of 8 nm. TGA-MA analysis uncovered38 that some CO2 emanated from the mixture during the pyrolysis, which contributed to the decrease in size of the carbon dots. Element analysis exhibited an increased C component (50.5% C, 3.7% H, and 13.1% N), which indicated the further carbonization. Different from CNP180 and CNP230, CNP300 displayed distinct excitation-dependent PL with the quantum yield of about 4%. Further increasing the pyrolysis temperature to 400 °C resulted in clusters and nonuniform particles with the size reaching to several hundred nanometers, caused by the aggregation. Even though the resultant carbon dots could still emit light, the intensity was very low.
Later, Fu Wang et al.39 synthesized carbon dots by means of pyrolyzing the citric acid in molten LiNO3 with argon as the protecting gas. Compared to solid LiNO3, the molted alkali metal nitrates could endow the nitrate anion with the ability to form an anion O2 or an oxygen atom O, which could serve as the oxidizing condition. Also, the surface passivation could strongly improve the emission intensity. In a typical experiment, a mixture of carbon dots and PEG1500N were dissolved into a mixture of water and toluene. The surface passivation was started by separating the water from the mixture through a water segregator, and the reaction finished when the water was totally removed from the mixture. The passivated carbon dots had an increased diameter of about 5–8 nm with the PL quantum efficiency reaching 10%.
Beside the citric acid salt, other small molecules, such as ethylenediamine-tetraacetic acid salts, synthetic polymer, e.g., epoxy-enriched polystyrene (PS), and natural materials, such as hair, green tea, konjac flour, paprika,40–47 have also been employed to synthesize carbon dots by means of a thermal pyrolysis approach.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C by FTIR spectroscopy in both carbon dots implied the occurrence of carbonization48 during the hydrothermal process. In addition, the surface passivation occurred by means of the oxidation of –OH groups.
C by FTIR spectroscopy in both carbon dots implied the occurrence of carbonization48 during the hydrothermal process. In addition, the surface passivation occurred by means of the oxidation of –OH groups.
In addition to glucose, natural bioresources,14e.g., orange juice, have also been utilized to synthesize carbon dots by means of a hydrothermal treatment at a relatively low temperature of about 150 °C. The authors14 proposed that the carbon dots emission was the result of the hydrothermal carbonization of the organic components in the orange juice, such as sucrose, glucose, fructose, and citric acid. TEM images showed that the carbon dots had a spherical morphology with a narrow diameter distribution between 1.5 and 4.5 nm with a lattice spacing of 0.18 nm. XPS and FTIR analysis disclosed the existence of many hydrophilic functional groups, such as –OH, –COOH, which endowed the carbon dots with excellent solubility in water. The intriguing features of this approach is that neither post-surface-passivation treatment nor expensive chemicals are needed; thus this work triggered continuous exploration about bioresources-based carbon dots synthesis in a one-step manner.49,50
Later on, Pin-Che Hsu and co-workers51 proposed that organic molecules containing amino and carboxylic acid groups, such as 2-amino-2-hydroxymethyl-propane-1,3-diol (TRIS), ethylenediaminetetraacetic acid (EDTA), and glycine, could benefit the carbon dots formation. They claimed that carbon dots formation underwent four steps: dehydration, polymerization, carbonization, and passivation. Hydrogen bonds played a key role during the molecular assembly, where heating leads to polymerization occurring, resulting in a single burst of nucleation. Finally, the solutes reached to the surface of the nuclei through diffusion, resulting in the growth of carbon dots.
Until now, by choosing proper organic molecules and adjusting the reaction environment (such as alkaline or acidic) with controlled surface function groups by means of hydrothermal method, it is possible to synthesize carbon dots with emission wavelength covering the whole visible range.17,48,52–55
This novel approach opened a new avenue to synthesize carbon dots,57e.g., Sourov et al.58 and Qu et al.59 separately fabricated green emission carbon dots with a relatively high quantum yield, while blue emission carbon dots with a high quantum yield of 44.9% (excited by 360 nm) were reported by Jiang et al.60 Furthermore, Tang et al.26 and Sun et al.61 even achieved deep UV and near IR emission from carbon dots by using a microwave approach.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600; EO = ethylene oxide, PO = propylene oxide). The surface treatment of silica by F127 surfactant was the prerequisite to synthesize carbon dots. The surfactant F127 triggered the micellization-like self-assembly to occur on the sphere surface. After interaction with water, the copolymer chains of F127 were extended on the surface of the sphere, resulting in a core–shell silica-F127 surfactant structure. The opened F127 shell contributed to the carbon source entering into the shell layer, which benefited the molecular loading and polymerization. More specifically, the hydrophilic part of F127 (also called PEO blocks) offered more anchors for resol molecules and for strong hydrogen bond interaction, which contributed to the polymerization of resol molecules, resulting in the construction of the polymer/F127 surfactant/silica sphere composites. After the surface treatment of silica, prepared resols were added as the carbon source to the suspension for the polymerization.
600; EO = ethylene oxide, PO = propylene oxide). The surface treatment of silica by F127 surfactant was the prerequisite to synthesize carbon dots. The surfactant F127 triggered the micellization-like self-assembly to occur on the sphere surface. After interaction with water, the copolymer chains of F127 were extended on the surface of the sphere, resulting in a core–shell silica-F127 surfactant structure. The opened F127 shell contributed to the carbon source entering into the shell layer, which benefited the molecular loading and polymerization. More specifically, the hydrophilic part of F127 (also called PEO blocks) offered more anchors for resol molecules and for strong hydrogen bond interaction, which contributed to the polymerization of resol molecules, resulting in the construction of the polymer/F127 surfactant/silica sphere composites. After the surface treatment of silica, prepared resols were added as the carbon source to the suspension for the polymerization.
To further facilitate the polymerization and dehydrogenation of the resols, calcination of these polymer/F127/silica structure was performed at a temperature of 900 °C for 2 h with Ar as the protection gas. In addition, the surfactant F127 was also totally released during the calcination process. After that, the carbon dots were released from the silica sphere by etching with NaOH solution. The surface passivation was still crucial for achieving bright photoluminescence. The post surface treatment was carried out by heating the mixture of carbon dots solution and diamine-terminated oligomeric poly(ethylene glycol), H2NCH2(CH2CH2O)nCH2CH2CH2NH2 (PEG1500N) at 120 °C for 72 h. Element analysis exposed that the carbon dots contained C 64.65 wt%, H 7.67 wt%, N 1.13 wt%, and O (calculated) 26.55 wt%. The average size of carbon dots was in the range of 1.5–2.5 nm. The absence of a discernible lattice structure indicated that these kinds of carbon dots were amorphous. The carbon dots could emit bright PL with a quantum yield 14.7% when excited by a 360 nm light source.
In addition to silica, NaY zeolite have also been employed as a support to synthesize carbon dots.63,64 It was demonstrated that the anchor-assisted carbon dots showed similar PL behaviors with the non-support-approach synthesized carbon dots.
:1). The different weight of HKUST-1 MOF samples before (1.00 g) and after (1.13 g) loading confirmed the successful loading of glucose. Glucose started to decompose when the loaded MOF power was heated to 200 °C, while the MOF template was still stable even when temperature was as high as 280 °C, which was confirmed by the thermogravimetric (TG) analysis. PXRD analysis demonstrated that the structure of the MOF was not destroyed during calcination. The prominent color changed from blue to green, which signified the carbon dots formation, while the considerable decrease in N2 adsorption ability from 130 cm3 g−1 for pristine MOF to 5 cm3 g−1 for the calcinated MOF indicated the successful filling of the MOF pore by carbon dots.
To release the carbon dots from the MOF temperate, aqueous KOH solution was utilized by the authors65 to dissolve the template’ the whole procedure is illustrated in Fig. 3. After purification, the carbon dots exhibited a rather narrow size distribution with an average diameter of 1.5 nm. Actually, this size was close to the large pore size inside the HKUST-1 MOF (HKUST-1 contains 3 kinds of pores: 1.35 nm, 1.1 nm, 0.5 nm); this was a result of the preference to form carbon dots nuclei in the largest pores, as proposed by the authors.65 By choosing different MOFs as templates with different pore sizes, the authors synthesized different sizes of carbon dots, such as HKUST-1 MOF for 1.5 nm carbon dots with the largest pore size of 1.35 nm, ZIF-8 MOF for 2.0 nm carbon dots with the largest pore size of 1.9 nm, and MIL-101 MOF for 3.2 nm carbon dots with the largest pore size 3.4 nm. However, carbon dots synthesized from the same method without the MOF template showed a nonregular size distribution with an average diameter of 4.5 nm. All of these results suggested the size and morphology controllability offered by the MOF template. The PL of the carbon dots showed a red-shift as the size increased from 1.5 nm to 4.5 nm when excited by UV light.
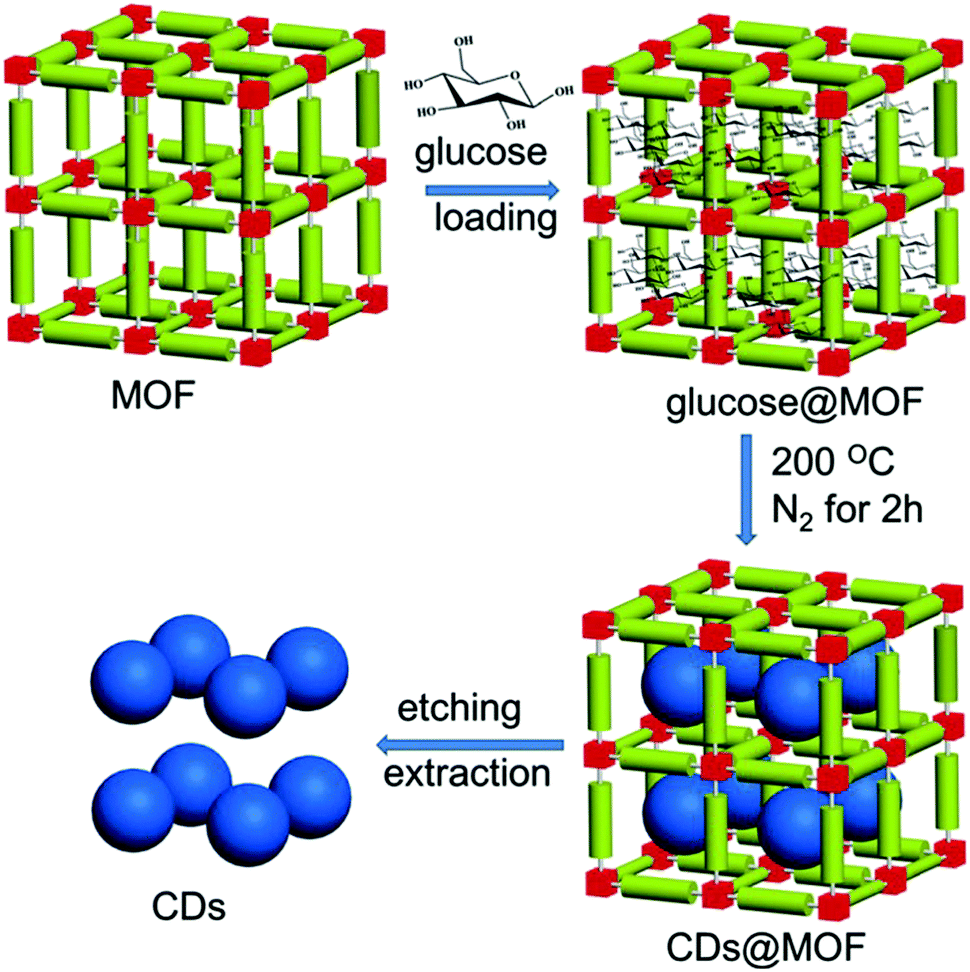 | ||
| Fig. 3 Illustration of carbon dots prepared by using the MOF pores as templates. Reproduced with permission from ref. 65. Copyright 2017, Wiley-VCH. | ||
Due to the intriguing controllability of the size and morphology,66,67 other templates, such as a mixed soft template (copolymer Pluronic P123) and hard template (ordered mesoporous silica (OMS) SBA-15), were also employed by Yang et al.68 to develop carbon dots with a uniform morphology.
The above-mentioned approaches are the most general bottom-up methods widely utilized by researchers to synthesize carbon dots. The discovery of versatile bottom-up approaches, especially fast and simple one-step synthesis methods, ensure the prerequisite of pushing carbon dots into practical applications. Furthermore, in addition to the general bottom-up methods, researchers also proposed some unusual approaches;69,70 for example, Liu et al.70 demonstrated a pathway using a catalase enzyme to decompose graphitic carbon nitride to synthesize carbon dots at room temperature.
3 Emission mechanism
3.1 Band gap emission and quantum confinement effect
By properly controlling the reaction precursor and time, Yuan et al.13 claimed to be able to synthesize band gap edge emission carbon dots, where a different color emission originated from the different size effect. All of the different emission carbon dots were synthesized by a hydrothermal method. Here, hydrothermal treatment of a mixture of CA (citric acid) and 2,3-diaminonaphthalene ethanol solution for 4 h and 9 h resulted in blue and green emission carbon dots, while hydrothermal treatment of CA and 1,5-diaminonaphthalene ethanol solution for 4 h and 9 h formed yellow and orange carbon dots, and red emission carbon dots could be achieved by hydrothermal treatment of a mixture of CA, 1,5-diaminonaphthalene and concentrated sulfuric acid. The wavelength of the absorption peaks of the blue (B), green (G), yellow (Y), orange (O), and red (R) carbon dots were located at 350 nm, 390 nm, 415 nm, 480 nm, and 500 nm respectively as illustrated in Fig. 4a, which displays a continuous increase. The corresponding PL peaks were located at 430 nm, 513 nm, 535 nm, 565 nm, and 604 nm for B, G, Y, O, and R, respectively, as seen in Fig. 4b. Different from the previous reported carbon dots, all of the carbon dots here exhibited excitation-independent PL, and the PLE peaks coinciding with the absorption peaks, both of which indicated that these carbon dots emission originated from the band emission. TEM analysis showed that the size of the carbons dots were 1.95 nm (B-), 2.41 nm (G-), 3.78 nm (Y-), 4.90 nm (O-), and 6.68 nm (R-), respectively, and in addition, the high degree of the crystallinity features for all the carbon dots were exposed by HRTEM. Thus, the authors claimed13 that the carbon dots emission originated from the band edge emission, and that the different color emission was the result of different effective band gaps: 3.02 eV (B-), 2.61 eV (G-), 2.49 eV (Y-), 2.30 eV (O-) and 2.12 eV (R-), where the band gaps were obtained from the absorption peaks (see Fig. 4d). These band gap emission results were also confirmed by UPS spectroscopy, which measured the HOMO and HOMO values of the carbon dots, as presented in Fig. 4c. Combined with the TEM analysis, the authors claimed13 that the different band gaps resulted from the different quantum confinement/size effects. All of the carbon dots exhibited pH-stable (pH value varying from 2 to 13) emission peaks, which, combined with the monoexponential decay of PL lifetimes, further confirmed that the carbon dots’ emission originated from the band emission, instead of surface states emission. In addition, all the excitation-independent emission carbon dots possessed relatively high quantum yield: 75% (B), 73% (G), 58% (Y), 53% (O), and 12% (R).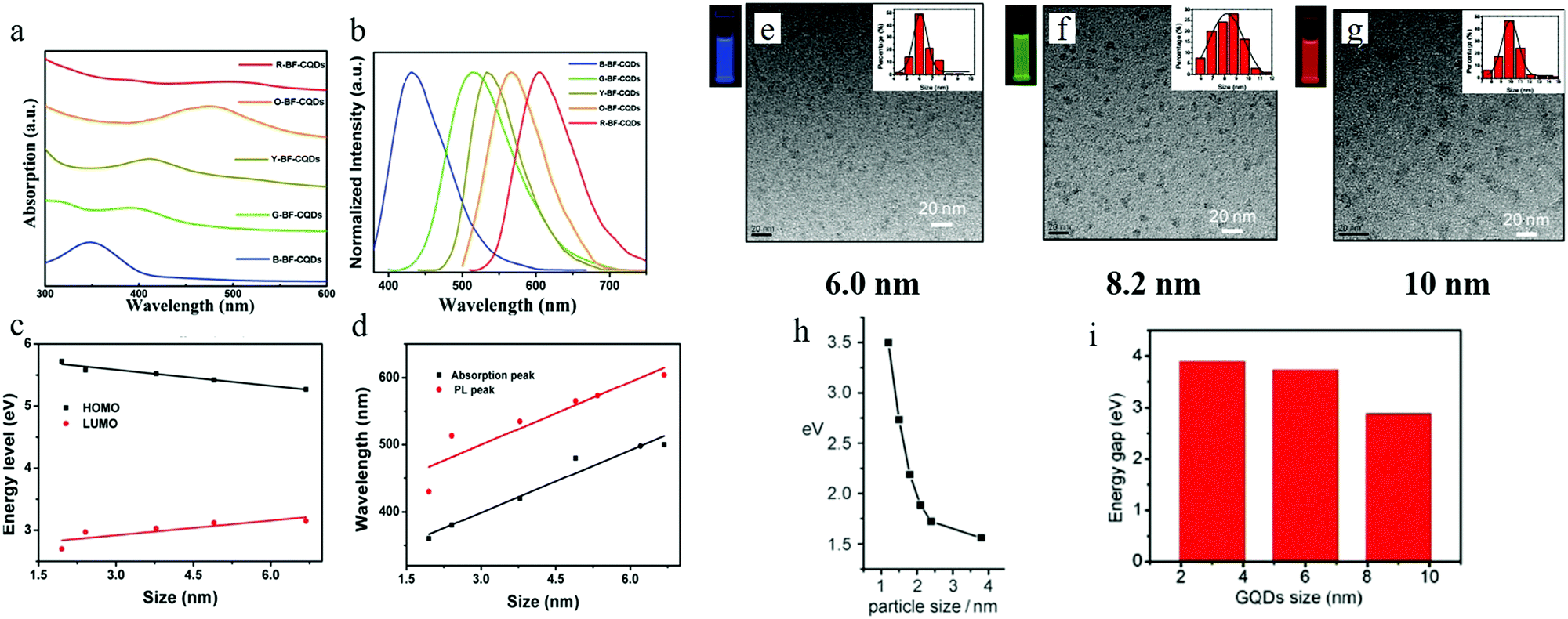 | ||
| Fig. 4 (a and b) The absorption and PL spectra of B-, G-, Y-, O-, R- emission carbon dots. (c) HOMO and LUMO energy for different sizes of carbon dots, obtained from UPS spectroscopy. (d) The corresponding absorption and emission peaks for different sizes of carbon dots. (e–g) 6 nm, 8.2 nm, and 10 nm sized carbon dots (synthesized by Jiang et al.) emitted blue, green, and red light respectively, excitation wavelength 365 nm. (h and i) Relationship between carbon dots size and band gap value for Li et al.'s sample and Peng et al.'s samples, respectively. (a–d) Reproduced with permission from ref. 13. Copyright 2016, Wiley-VCH. (e–g) Reproduced with permission from ref. 17. Copyright 2015, Wiley-VCH. (h) Reproduced with permission from ref. 32. Copyright 2010, Wiley-VCH. (i) Reproduced with permission from ref. 18. Copyright 2012, American Chemical Society. | ||
A quantum-confinement-induced PL spectral shift of carbon dots was also claimed by several other groups. Jiang et al.17 synthesized 6.0 nm, 8.2 nm, and 10.0 nm sized carbon dots by a hydrothermal treatment method, which showed blue, green, and red emissions, respectively (Fig. 4e–g). In addition to the hydrothermal method, carbon dots synthesized by other approaches16,17 can also exhibit size-dependent PL. Li et al.32 and Peng et al.18 separately synthesized carbon dots by utilizing the alkali-assisted electrochemical and chemical exfoliation, and both of them observed a size-dependent PL spectral shift from their carbon dots, as can be seen in Fig. 4h and i.
3.2 Surface states emission
To explore the emission mechanism of carbon dots, the purification of carbon dots is a prerequisite. Ding et al.25 employed the silica column chromatography technique to purify carbon dots synthesized by the hydrothermal treatment of urea and p-phenylenediamine at 160 °C for 10 h. The purified carbon dots emitted blue, green, yellow, and red light, respectively, with these labeled as A, B, C, and D in Fig. 5. The UV-visible absorption spectra of the purified carbon dots displayed distinct absorption peaks in the longer wavelength region located at 383, 410, 488, and 528 nm respectively, and the PLE spectra of each carbon dot samples also exhibited only one peak close to the corresponding absorption bands. Different from the ensemble carbon dots, which presented excitation-dependent PL, the purified carbon dots disclosed an excitation-independent PL behavior, as can be seen in Fig. 5a–d. Furthermore, time-resolved spectroscopy showed that all the carbon dots samples underwent an analogous monoexponential decay process with a lifetime of about 9 ns. All of these suggested that the purified different carbon dots samples manifested a uniformity in their optical properties, while (HR) TEM analysis unveiled that all the carbon dots samples also showed a similar size distribution with an average diameter of 2.6 nm. In addition to the size, the lattice constant of all the carbon dots also showed the same values of about 0.21 nm, as can be seen in Fig. 5a–d in the lower parts; thus it became clear that the different color emission of carbon dots did not originate from the size effect or quantum confinement.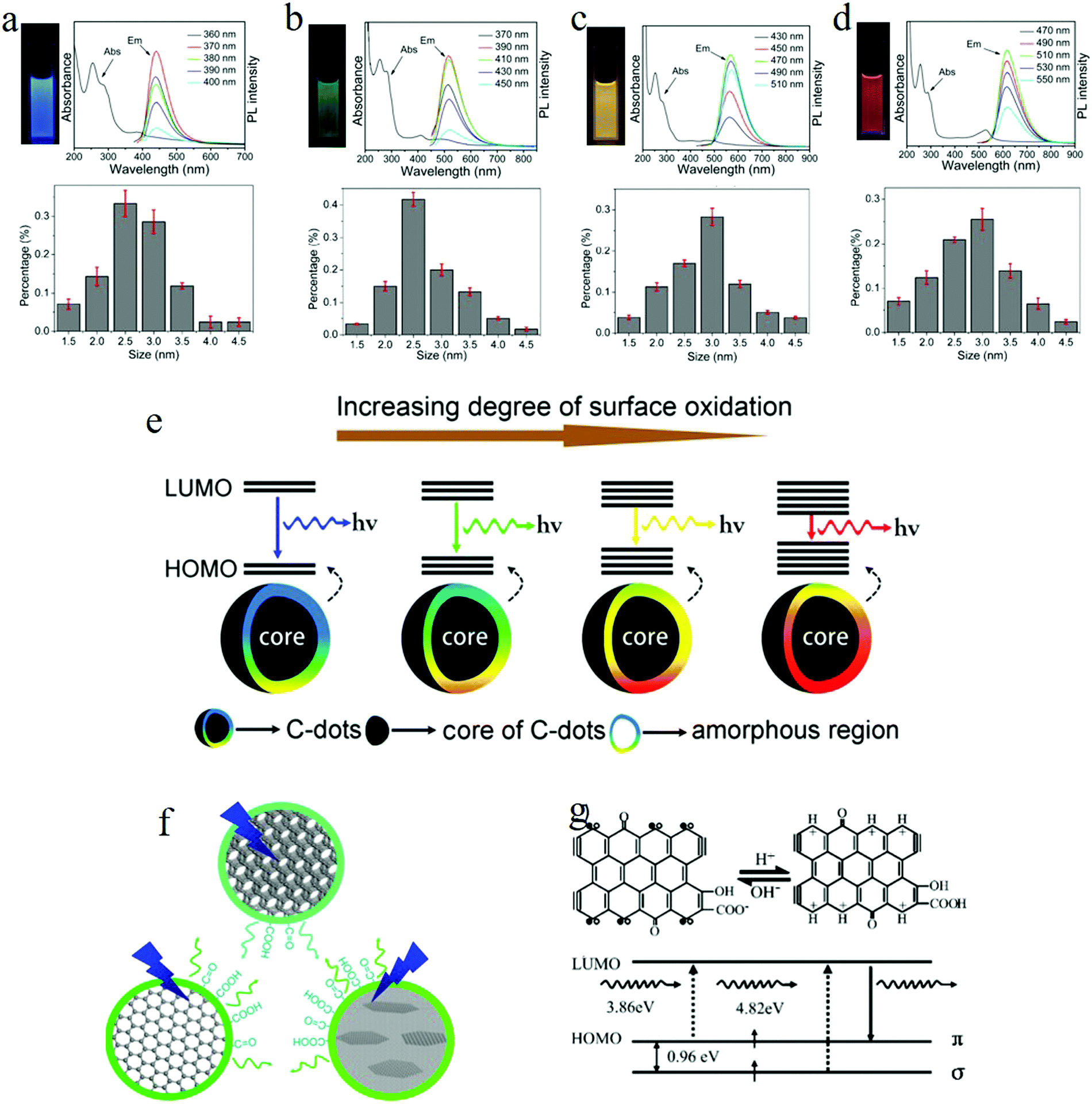 | ||
| Fig. 5 (a–d) Photographs under UV light, absorption and PL spectra, and the size distribution for blue, green, yellow, and red emission carbon dots, respectively. All the purified carbon dots exhibited excitation-independent PL and showed a similar size distribution with an average diameter of 2.6 nm. (e) Model for the tunable PL of carbon dots. The band gap decreased with the increasing degrees of surface oxidation, resulting in a red-shift of carbon dots emission. (f) Wang et al.72 suggested that the green emission of carbon dots originated from hydroxyl and carboxyl surface functional groups. (g) Pan et al. proposed that the blue emission of carbon dots originated from surface zigzag sites, which possess a carbene-like triplet ground state, denoted as s1p1. (a–g) Reproduced with permission from ref. 25. Copyright 2016, American Chemical Society. (f) Reproduced with permission from ref. 72. Copyright 2014, American Chemical Society. (g) Reproduced with permission from ref. 52. Copyright 2010, Wiley-VCH. | ||
FTIR spectroscopy analysis signified that, as the carbon dots emission changed from blue to red (sample A to sample D), the degree of the oxidation continued to increase, which was substantiated by the enhanced COOH bond signal located at 1759 cm−1. Besides, compared to the blue and green carbon dots, the yellow and red carbon dots appeared to hold a more broad O–H band distribution, which suggested a higher polarity, consistent with the column chromatography results. The increased degree of oxidation was also verified by XPS, where both the component of the carboxyl groups (represented by the intensity of the 289.0 eV signal) and the O element continuously increased from blue to red emission carbon dots.
Based on the theory proposed by Chen et al.71 that the COOH groups on the sp2-hybridized carbons will generate local distortions, leading to a decrease of the energy gap, the authors25 recommended the following emission mechanism: the emissive center is located at the surface of the carbon dots, mainly constituted by the conjugated carbon atoms and the bonded oxygen atoms; thus the energy difference between the HOMO and LUMO of the carbon dots is intensely related to the degree of oxidation. More specifically, the increased oxidation or O component leads to the decreased band gap, so the red-shift of the PL of carbon dots originates from the increased surface oxidation, as represented in Fig. 5e. The PL spectra of red emission carbon dots exhibiting a red-shift in acidic conditions also support the surface states emission. The authors attributed25 the pH-triggered PL intensity decrease and peak shift to the protonation–deprotonation-induced surface-charge modification.
The mechanism of surface states emission was also adapted by Wang et al.72 to interpret the carbon dots emission in their samples. They even assigned the surface states emission to the common origin of green light emission. They employed three kinds of carbon dots to explore the green emission of carbon dots, namely carbon dots synthesized by: the electrochemical treatment of a graphite rod, the microwave heating of citric acid and urea, and the hydrothermal treatment of graphene oxide DMF solution. The three kinds of carbon dots displayed similar green emission spectra when excited by 400 nm, even though the absorption spectra were different. By combining transient-absorption and time-resolved spectroscopy, the authors suggested72 that the photoexcited hot carriers were first located at the carbon core, followed by leaking fast to the emissive center and non-radiative traps. In other words, the graphitic carbon core did not contribute to the carbon dot emission but played a role as a temporary hot carrier reservoir. To identity the emission center on the surface, NaBH4 was utilized to achieve the surface chemical reduction for all three kinds of carbon dots, which resulted in different emission spectra. FTIR spectra analysis implied that the CO functional groups, especially the hydroxyl group and carboxyl group, were the main functional groups for both the before and after surface-reduced carbon dots; thus they attributed72 the carbon dots green emission to the hydroxyl and carboxyl surface functional groups, whereby the different quantity of carboxyl surface functional groups before and after the surface reduction resulted in the different green PL spectra of the carbon dots. In addition, the authors also claimed72 that the hybridization/interaction between the carbon core structure and CO functional groups was a prerequisite for the carbon dots emission as the pure CO could not contribute to the green emission.
The surface states emission origin of the carbon dots has been accepted by many researchers,12,20,24,26,52,73–75 although the actual emission fine structures on the surfaces of carbon dots are still controversial; e.g., Pan et al.52 argued that the blue carbon dots emission originated from the surface zigzag sites possessing a carbene-like triplet ground state (Fig. 5g), denoted as s1p1.
3.3 Molecular fluorophores and carbogenic core
Emmanuel P. Giannelis's group38 proposed that, for the thermal pyrolysis approach for obtaining synthesized carbon dots, the emission mechanism was related to the pyrolysis temperature. Briefly speaking, a mixture of citric acid monohydrate (C6H8O7) and ethanolamine (C2H7ON) with a molar ratio of 1:3 was heated at a certain temperature for 30 min under reflux in air. The authors propounded that when the pyrolysis temperature was low (e.g., 180 °C), it could not form carbon dots, but instead formed carbon dots precursors. The precursor contained some organic molecular fluorophores, which contributed to the emission, with a PL quantum efficiency of 50%. The claim of the molecular fluorophore emission was based on the following experimental observations: (1) no carbon dots particles were observed by either DLS or TEM. (2) The sample showed excitation-independent PL. To further identify the molecular structure, the authors employed38 FTIR, XPS, 1H and 13C NMR spectroscopy to study the chemical bond of the carbon dots precursor. The results suggested that the amide-related fluorophore molecules were accountable for the precursor emission. Moreover, amide synthesized by an alternative approach using citric acid monohydrate (C6H8O7) and ethanolamine (C2H7ON) as the raw materials demonstrated similar optical behaviors to the carbon dots precursor, which further validated the mechanism.
Increasing the pyrolysis temperature to 230 °C resulted in the PL quantum yield decreasing to 15% and the appearance of excitation-dependent PL. Further increasing the temperature to 300 °C or even 400 °C also resulted in a different PL quantum yield. Element analysis showed that the carbon component continued to increase with a concomitant decrease in the hydrogen component. Thus the authors claimed38 that the carbon dots underwent further carbonization as the temperature increased, which generated carbogenic cores at the cost of the carbon dots precursor or molecular fluorophores. Both the carbon core and amide-containing molecular fluorophores could contribute to the light emission. This model is illustrated in Fig. 6a.38 When the temperature was low, the emission was from molecular fluorophores, while the carbogenic cores became the main emission mechanism when the temperature was higher (e.g., 400 °C). Both molecular fluorophores and carbogenic cores contributed to the carbon dots emission when the pyrolysis temperature was moderate.
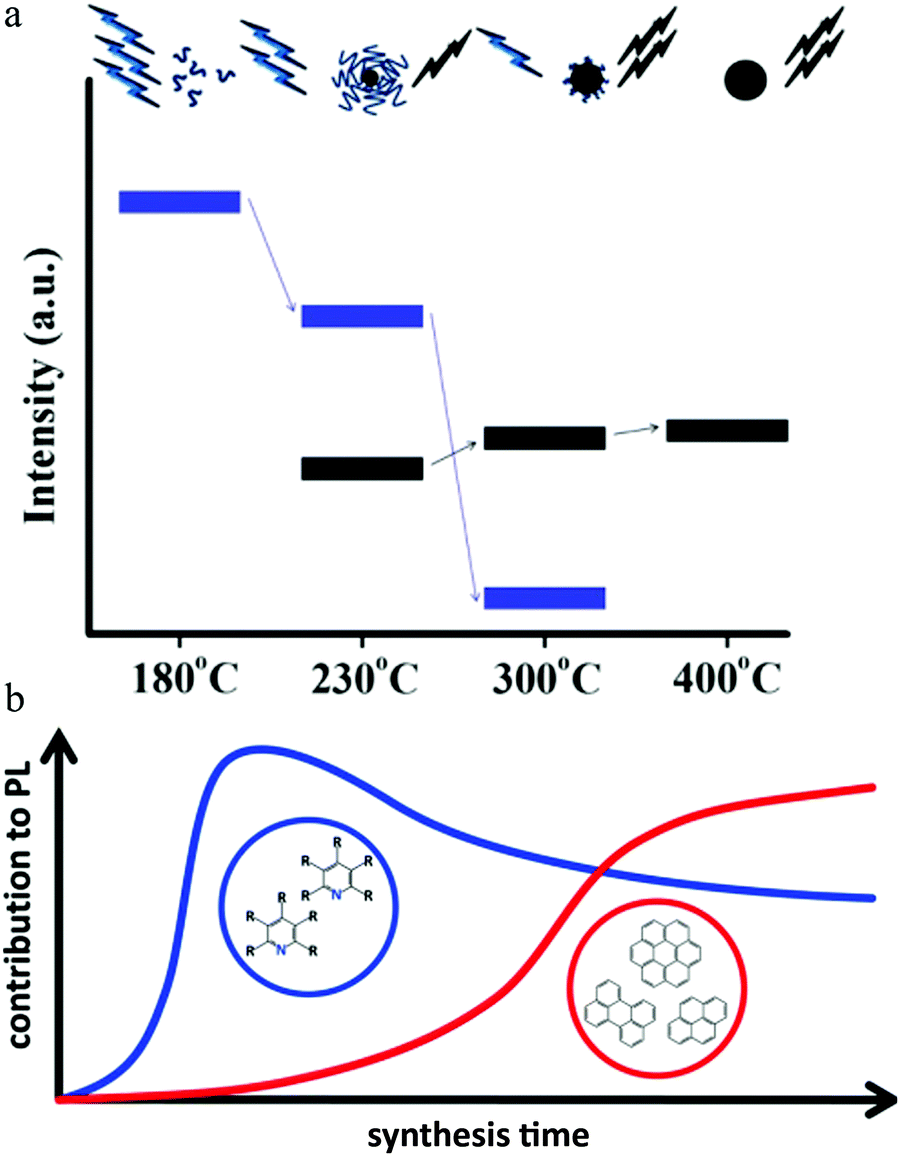 | ||
| Fig. 6 (a) During pyrolysis, the molecular fluorophores (blue groups) were expended to generate a carbogenic core (black sphere), as seen in the upper illustration. So as the temperature increased, the PL intensity corresponding to the carbon core (bold black line, lower part) increased at the cost of the molecular fluorophores (blue line), along with the decrease in the precursor emission (bold blue line, lower part). Reproduced with permission from ref. 38. Copyright 2012, American Chemical Society. (b) When the hydrothermal reaction time was short (e.g., less than 30 min), the emission center was mainly molecular fluorophores, e.g., 2-pyridone derivatives, while as the reaction time increased, the carbon core, which contained an aromatic domain, appeared and played a more important role for carbon dots emission. Reproduced with permission from ref. 76. Copyright 2017, American Chemical Society. | ||
Different from the high-temperature pyrolysis method, Ehrat et al.76 synthesized carbon dots from citric acid and ethylenediamine by means of a hydrothermal treatment. However, they proposed a similar molecular fluorophores and carbogenic core model to interpret the carbon dots emission (Fig. 6b). When the hydrothermal reaction time was short (e.g., less than 30 min), the emission center was mainly molecular fluorophores, e.g., 2-pyridone derivatives, while as the reaction time increased, the carbon core, which contained an aromatic domain, appeared and played a more important role for carbon dots emission.
3.4 Polycyclic aromatic hydrocarbon molecular emission
Following the report on molecular emission,38 Alexander's group9 proposed the polycyclic aromatic hydrocarbon fluorescent molecular emission. The carbon dots utilized in the experiment were synthesized by the hydrothermal treatment of citric acid and ethylenediamine, and showed a diameter between 2 and 5 nm. The authors claimed that the carbon dots were a kind of organic molecular nano-crystal based on the following evidence: (1) XPS spectroscopy indicated that there existed sp2 (e.g., CO bond) and sp3 (e.g. C–C bond) hybrid bonds. (2) C 1s XPS spectra showed a dramatic change after treatment by Ar-ion sputtering. Specifically, the authors declared that the carbon dots contained many sp2 carbon domains embedded in a sp3 hybridized carbon matrix. Single particle measurements on the carbon dots still exhibited excitation-dependent PL, indicating that there existed multi-chromophores in one carbon dot. As a result, the authors employed9 three basic polycyclic aromatic hydrocarbons (PAHs), namely anthracene (3 rings), pyrene (4 rings), and perylene (5 rings), to mimic the emission nano-domain, with poly(methyl methacrylate) (PMMA) chosen as the sp3 hybridized matrix. The authors stated that they chose these PAHs because: (1) the absorption and PL spectra of these PAHs overlapped with those of the carbon dots, see Fig. 7a and b and (2) their structures were relatively simple. After optimization, the fixed molecular ratio of 10:10:1:20 for anthracene/pyrene/perylene/PMMA was utilized to mimic the carbon dots. The results are reproduced in Fig. 7c and d, where it can be seen that the absorption spectra of the PAH films and carbon dots displayed a similar shape, A red-shift was observed for the PL spectra when excited at 340 nm, while the PL line shape and width were similar. Increasing the excitation wavelength resulted in the PL spectra changing, but under the conditions of 420 nm, 440 nm and 480 nm excitation, the PAH films PL spectra matched very well with the carbon dots emission. Based on this model, the authors9 gave the following picture: when excited by short wavelengths (<400 nm), the PAHs that have the largest bandgap (e.g., anthracene and pyrene) were mainly excited (the authors claimed that the smaller band gap PAHs, such as perylene, did not absorb strongly at shorter wavelengths), and could emit light directly contributing to the main peak emission, or transfer energy to the smaller bandgap PAHs (e.g., perylene), resulting in the longer wavelength emission. Whereas, when the excitation wavelength was greater than 400 nm, both kinds of PAHs (large and small band gap PAHs) could be exited directly, resulting in a red-shift of the carbon dots emission. Continuously increasing the excitation wavelength led to the small band gap PAHs absorption increasing and the large PAHs band gap decreasing, contributing to the PL spectra of carbon dots showing a continual red-shift. Finally, the authors also stated9 that there most likely existed other PAHs that were involved in the carbon dots optical process in addition to the above-mentioned three PAHs.
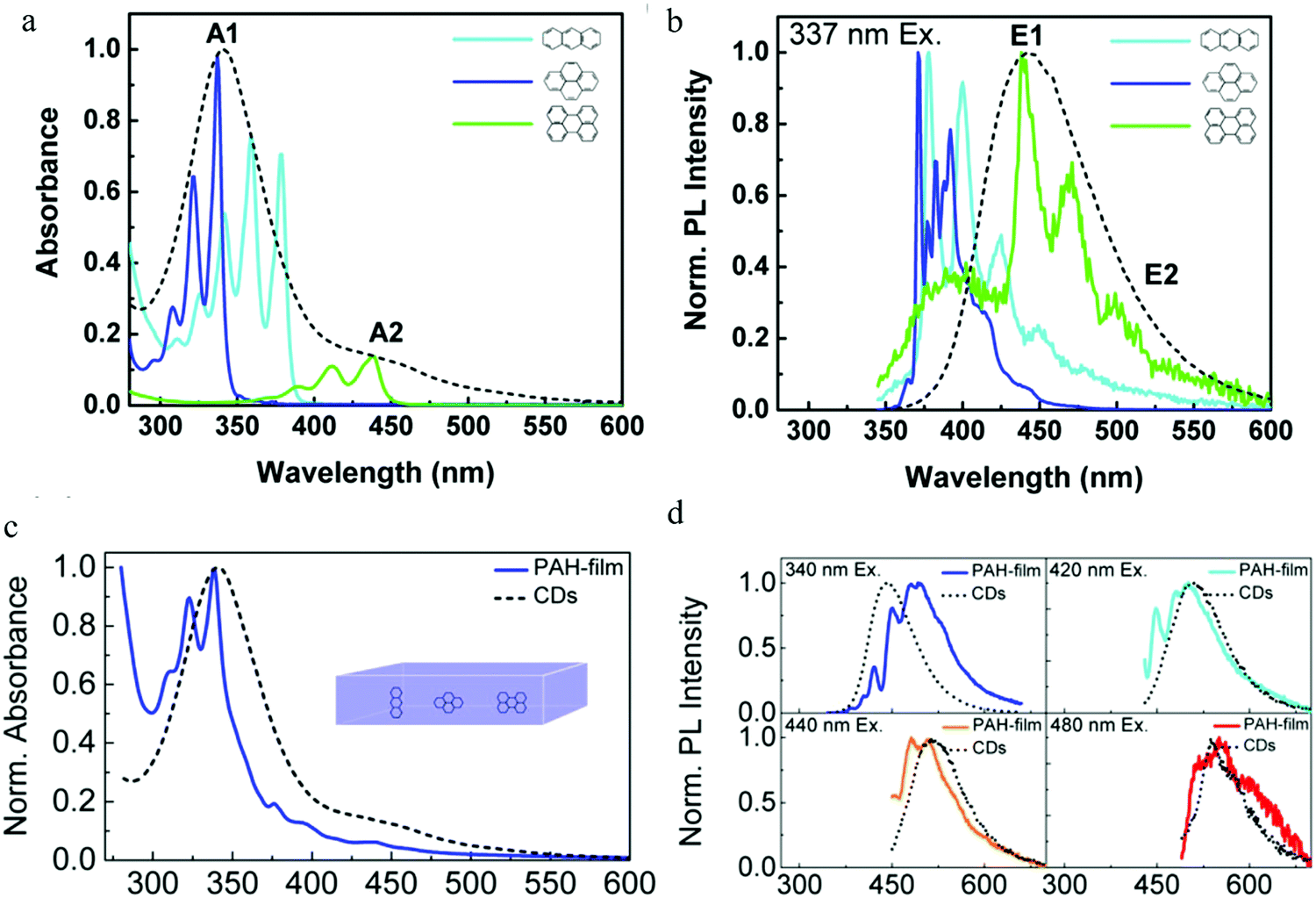 | ||
| Fig. 7 (a and b) Absorption and normalized PL spectra of anthracene (light blue), pyrene (dark blue), and perylene (green) in a PMMA matrix (concentration: 0.01 mol%) and CDs aqueous solution (black dashed line). (c) Absorption spectra of a PMMA film containing three PAHs (blue) (molecular ratio: anthracene/pyrene/perylene/PMMA 10:10:1:20) and CDs aqueous solution (black dashed line), (d) PL spectra of the film stimulated by different wavelengths, CD PL spectrum (black dashed line) also given for reference. (a–d) Reproduced with permission from ref. 9. Copyright 2015, American Chemical Society. | ||
3.5 Slowed solvent relaxation or solvatochromic shift
Apart from the electronic structure of the carbon dots itself, the effect of the surrounding solvent molecular environment of the carbon dots was also considered by Khan et al.,77 as illustrated in Fig. 8a. Different from the solid sample, the carbon dots aqueous solution has two kinds of decay processes after photoexcitation (time scale of about 10−15 s) occurring simultaneously: (1) hot carriers (e.g., elections, holes) vibrational relaxation, which results in the well-known Stokes red-shift, and (2) solvent relaxation or dipolar reorganization, which takes place at the band edge, leading to a further red-shift of the emission spectra, commonly termed as the solvatochromic shift.77 According to the Lippert–Mataga theory,78,79 a greater polarity of solvent will lead to a larger red-shift of the emission spectra. The authors77 assumed that the hot carrier variation time was much less than the fluorescent lifetimeτf, thus only the solvent relaxation effect (solvent relaxation lifetime: τr) was considered. When τf ≫ τr, which means fast solvent relaxation, the solvent relaxation is fully finished before the light emission, resulting in the solvatochromic shift being red-shifted, also called homogeneous broadening; when τf ≪ τr, which is the case in a much slower solvent relaxation, there is no solvent relaxation effect during the PL; while when the fluorescent lifetime and solvent relaxation time are the same order of magnitude e.g., τf ≅ τr, a set of sub-states will be formed, where all of the sub-states can contribute to the emission maximum of the carbon dots. This condition will lead to a broadened band emission, called inhomogeneous broadening. The temperature, solvent dipole moment, and fluorophore dipole moment all can affect the inhomogeneous broadening, which is governed by the following equation:80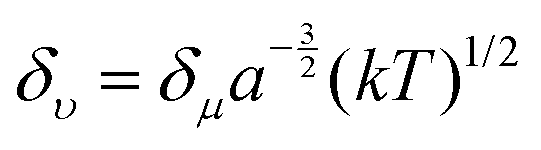 , where a is the Onsagar sphere radius. Considering the dynamics of the relative slow solvent relaxation, e.g., τf ≅ τr, process, the initial energy sates will be migrated to lower energy states during the PL lifetime, which results in two effects: (1) the time-resolved emission spectra will red-shift as time increases, and (2) the time-integrated PL spectra will red-shift as the excitation wavelength increases. More specifically, the center of mass of the time-integrated spectra caused by the solvent relaxation is determined by the following equation:81υ = υt=∞ − (υt=0 − υt=∞)τr/(τf + τr), where υt=∞ is the PL peak when the time is long enough, and υt=0 represents the instant that the PL occurs. It is obvious that υ = υt=∞ for τf ≫ τr, υ = υt=0 for τf ≪ τr, and in both two cases, the emission spectra are independent on the detection time. Only when the two time constants are of the same order do the time-dependent emission spectra appear.
, where a is the Onsagar sphere radius. Considering the dynamics of the relative slow solvent relaxation, e.g., τf ≅ τr, process, the initial energy sates will be migrated to lower energy states during the PL lifetime, which results in two effects: (1) the time-resolved emission spectra will red-shift as time increases, and (2) the time-integrated PL spectra will red-shift as the excitation wavelength increases. More specifically, the center of mass of the time-integrated spectra caused by the solvent relaxation is determined by the following equation:81υ = υt=∞ − (υt=0 − υt=∞)τr/(τf + τr), where υt=∞ is the PL peak when the time is long enough, and υt=0 represents the instant that the PL occurs. It is obvious that υ = υt=∞ for τf ≫ τr, υ = υt=0 for τf ≪ τr, and in both two cases, the emission spectra are independent on the detection time. Only when the two time constants are of the same order do the time-dependent emission spectra appear.
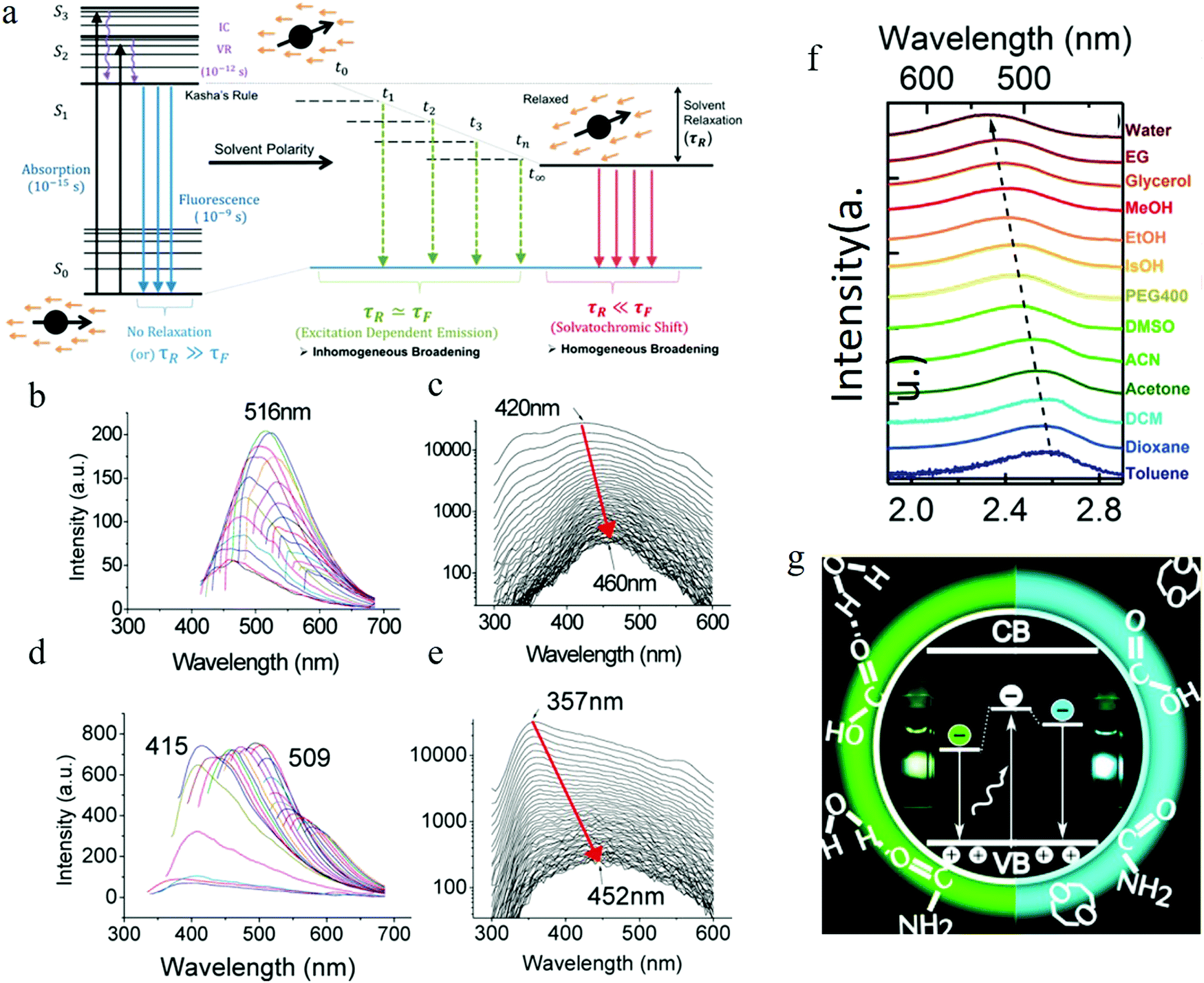 | ||
| Fig. 8 (a) Illustrating solvent relaxation. (Fluorescent lifetime τf, solvent relaxation lifetime: τr). (b–e) Function of solvent dipole moment. (b and c) Time-integrated PL spectra and time-resolved PL spectra as a function of time of the carbon dots in water. (d and e) Time-integrated PL spectra and time-resolved PL spectra as a function of time of the carbon dots in dimethylformamide (DMF). The red arrow indicated that as the time increased, the emission spectra red-shifted. The wavelength migration during the lifetime was 40 nm for water, while it reached to 100 nm for DMF. (f) Solvent-dependent spectral shift. (g) Recombination of the electrons located at the surface with the valence band holes in the carbon dots crystalline core contributing to the carbon dots emission. (a–e) Reproduced with permission from ref. 77. Copyright 2015, American Chemical Society. (f and g) Reproduced with permission from ref. 82. Copyright 2016, American Chemical Society. | ||
The carbon dots employed by the authors77 were synthesized by means of a microwave-assisted method. They synthesized different carbon dots from different starting materials (carbon dots 1 from chitosan and PEG, carbon dots 2 from adenine sulfate, carbon dots 3 from sodium citrate and sodium thiosulfate, carbon dots 4 from potato-dextrose agar) but all exhibited similar behaviors, thus the authors carried out the analysis by using carbon dots 1 as a representative sample. Further purification of carbon dots 1 was achieved by column chromatography, which resulted in two kinds of carbon dots with different polarity: lower polarity carbon dots, which presented blue light emission, and the higher polarity carbon dots, which showed longer wavelength emission. Water and dimethylformamide (DMF), which have different dipole moments, were adopted by the authors77 to dissolve the carbon dots to explore the solvent polar effect. Both the water and DMF carbon dots solutions demonstrated time-integrated excitation-dependent PL and time-resolved PL spectra migration, as described in Fig. 8b–e. The results indicated that the solvent relaxation time was the same order as the PL lifetime, and the excitation-dependent PL was the result of solvent relaxation. Compared to water, DMF has a higher dipole moment, which led to more populated sub-states, resulting in the larger time-resolved emission spectra migration, reaching 100 nm (see Fig. 8e) (but only 40 nm emission migration for the carbon dots aqueous solution).
The two kinds of carbon dots purified from column chromatography also displayed different time-resolved emission spectra migration due to the different polarity properties, which was consistent with the author's model.77 The less oxidized and polar fraction carbon dots demonstrated a lower or no time-resolved emission spectra red-shift, which could be interpreted by the faster photon emission before significant relaxation occurs. Whereas, the highly oxidized and high polarity carbon dots fractions were considered to hold a higher lifetime value and showed clear time-resolved PL spectra migration, as the high polarity carbon dots sustained significant solvent relaxation during the PL lifetime, which resulted in a population of the sub-states, contributing to the red-shift edge effect and time-resolved spectra migration.
Time-resolved anisotropic analysis estimated77 the rotational time of the dipole in the carbon dots to be in the range of 0.61 to 1.51 ns (for pH = 7, although the values varied for different emission wavelengths), i.e., the same order as the PL lifetime (several ns) of carbon dots, consistent with the relatively slowed solvent relaxation assumption. Combined with the pH-dependent measurement, carbon-dots-concentration-dependent measurements, and single step photobleaching, the authors77 claimed that carbon dots emission was neither from the quantum confinement nor particle heterogeneity, instead they proposed that the carbon dots were a kind of single emitter, and the excitation-dependent PL was from the slowed solvent relaxation or solvatochromic red-shift. Furthermore, based on the observation of the blue-shift of carbon dots at high pH (>10), the authors claimed77 that the emission center was located at the surface of the carbon dots.
Even though the polar solvent effect has been observed by some other groups,82,83 the slow relaxation of polar solvents mechanism has been argued by some researchers.82 The appearance of PL tunability even in apolar solvents combined with the disappearance of the spectral migration during the PL lifetime of carbon dots (synthesized by the thermal pyrolysis of citric acid monohydrate and urea) in all solvents contradicted the slow relaxation hypothesis in the polar solvents mechanism. Instead, some researchers82 have attributed the solvation-dependent PL to the surface functional groups of carbon dots. Observation of the solvent effect being more efficient for excited sates than ground states combined with the fact that the amide and carboxylic surface groups are assumed not to undergo any transitions at the 2–3 eV energy range have guided researchers to propose that the carbon dots emission is a result of the recombination of the electrons located at the surface with the valence band holes in the carbon dots crystalline core,82 as illustrated in Fig. 8f and g. The inhomogeneous broadening of the PL spectra is consider a the result of the size distribution, specifically, the size distribution affects the core band gap, resulting in the different efficient “energy band gap” between the valence band and the surface energy states.
3.6 Self-trapped exciton emission
The PL spectra of carbon dots synthesized by the microwave heating of glucose and urea manifested two distinct features:84 (1) when exited by 442 nm, the emission wavelength started nearly from the excitation wavelength 450 nm with a broadened emission spectra over 1 eV, and (2) the emission intensities of the whole spectra were the same as the magnitude, which was contrary to band edge emission, where the majority of the emission intensity was located at the band edge. Based on these observations, it's reasonable to speculate that the carbon dots emission originates from the localized emission. To verify this assumption, Xiao et al.84 employed polarization spectroscopy for a comprehensive investigation and uncovered the emission mechanism of carbon dots. Polarization spectroscopy can provide information on the energy and momentum relaxation dynamics, which are closely related to the electronic structure and emission mechanism.85,86 Linear polarized light will align the momentum of photogenerated hot carriers (hot electrons and holes), with the corresponding principles ruled by the transition density matrix combined with the angular momentum distribution under dipole approximation.87 The radiative recombination of carriers with the aligned momentum will emit linear polarized light as the selection rules are the same for photoexcitation and emission. However, the decay process of the hot carriers will randomize the aligned momentum, resulting in an isotropic momentum distribution of the ensemble carriers by means of collision and/or phonon emission.85 As a result, the linearly polarized fluorescence can only be obtained from hot carrier recombination, where the momentum alignment/anisotropy excited by the linear excitation light is totally or partially conserved, but on the contrary, the thermalized carriers, which exhibit an isotropic momentum distribution, can only emit un-polarized light. Thus, the appearance of linearly polarized light unambiguously indicates the momentum alignment, which is the result of the hot carrier property. The linearly polarized emission of carbon dots84 are depicted in Fig. 9. The carbon dots show distinct emission anisotropy, as seen in Fig. 9a (The vertical axis is the angle between the polarization of the excitation and emission light with the excitation polarization fixed.) and linear polarized emission. To quantitatively analyze the emission anisotropy, the degree of linear polarization is defined as P = (I‖ − I⊥)/(I‖ + I⊥), where I‖ and I⊥ are the parallel (0°) and perpendicular (90°) emission compared to the excitation light, respectively. The carbon dots exhibited extremely high linear polarization at the instant of excitation, reaching to 0.5, and then continuously decreased as the emission energy decreased, as seen in Fig. 9b. However, there was still a large polarization value for the carbon dots (about 0.25 for 690 nm) even when the energy difference between the excitation and emission light was more than 1 eV, which unambiguously -implied the hot carrier properties. Based on these observations, the authors claimed84 that the carbon dots emission originated from the localized emission, where the momentum, energy, and vibrational relaxation were strongly suppressed due to the existence of a strong local potential field. As a result, the momentum relaxation time became comparable or even longer that the fluorescent lifetime, contributing to the highly polarized emission; therefore the carbon dots emission was totally different from the band edge emission, where the fluorescence lifetime of the band edge emission is much longer than the momentum and energy relaxation time (several orders of magnitude longer), and consequently the emission is from the isotropic momentum distrusted and thermalized carriers.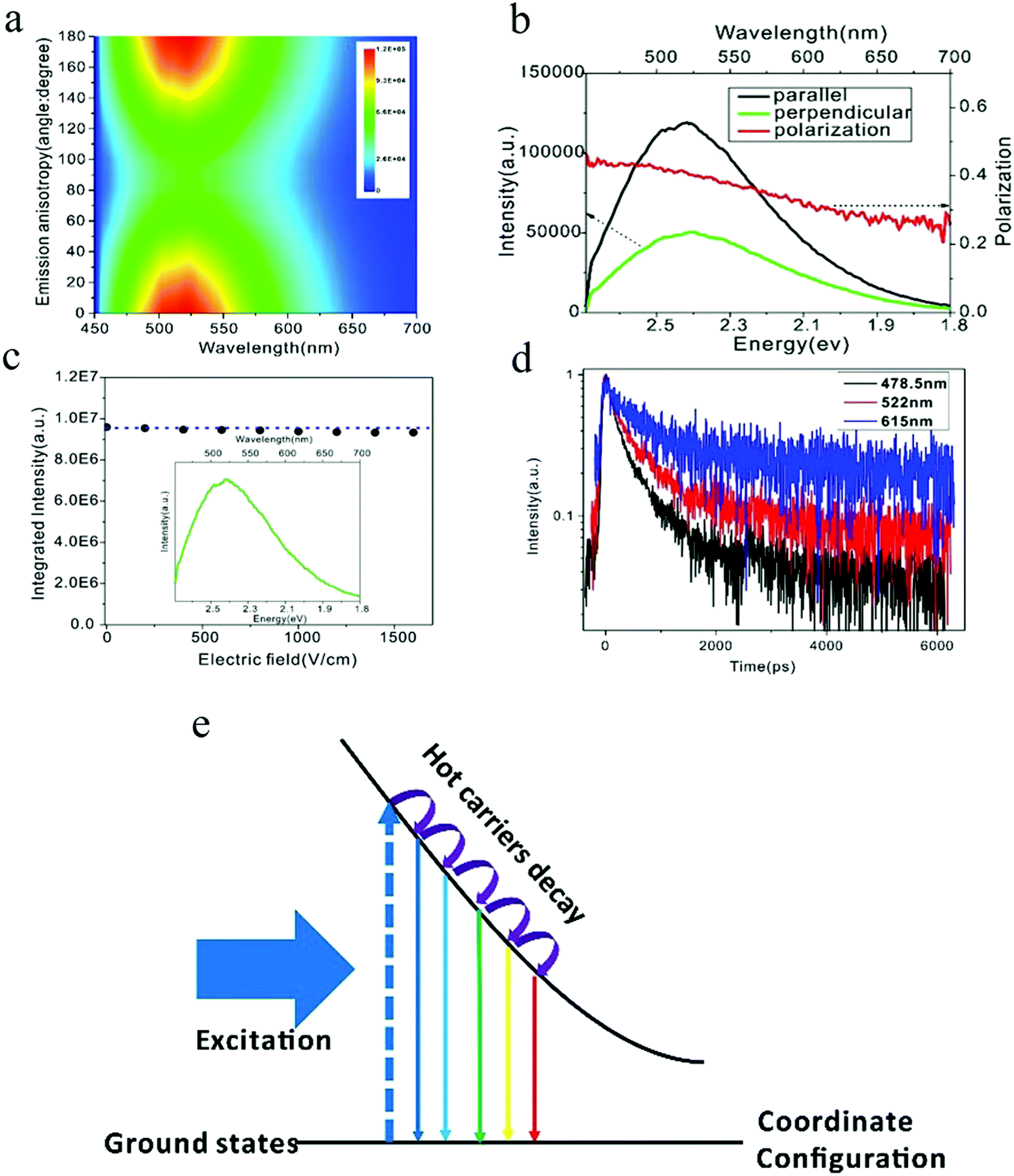 | ||
| Fig. 9 (a and b) Emission anisotropy and degree of linear polarization of carbon dots. (c) Electric-field modulation of carbon dots emission. (d) Time-resolved spectroscopy for different emission wavelengths. (e) Electronic structure of self-trapped excitons, which contribute to carbon dots emission. Excitation wavelength: 442 nm. (a–e) Reproduced with permission from ref. 84. Copyright 2017, Royal Society of Chemistry. | ||
The localized emission was further confirmed by electric-field-modulation experiments,84 as can be seen in Fig. 9c. Both the line shape and the intensity of the carbon dots PL were stable even when the electric field was as high as 1500 V cm−1, which was consistent with the localized emission assumption, whereby the strong local potential field in carbon dots protects the hot carriers against the quenching/tuning effect by the high external electric field.88,89 Power-dependent PL experiments showed that the emission intensity of carbon dots exhibited a linear relationship to the excitation power, even when the excitation power density was up to 1800 mW cm−2, which implied that the carbon dots emission originated from self-trapped excitons rather than defects.90,91 The self-trapped exciton property was further validated by time-resolved spectroscopy.84 For self-trapped excitons, a smaller charge distance r possesses a larger wave function overlap with the transition matrix, and they recombine first.92 As a consequence, the emission shifts to a longer wavelength as the time increases, so the increased lifetime for the longer emission wavelength can be predicted. The time-resolved experiment results demonstrated that, as the emission wavelength increased from 478 to 615 nm, as seen in Fig. 9d, the PL lifetime also continuously increased, which corroborated the self-trapped exciton emission. Combining photoexcitation, relaxation, and polarization emission, an energy diagram contributing to the carbon dots emission was given by the authors,84 as illustrated here in Fig. 9e, where the horizontal axis is the coordinate distance, which represent the relative distance r between the hot carrier pairs. With time evolution after the photoexcitation, the larger distance self-trapped exciton states become activated and emit longer wavelength because they undergo a relaxation process. The excitation-dependent PL of carbon dots could be also interpreted by the self-trapped exciton model, whereby a monochromatic light source excites the self-trapped exciton states with energy equal to the excitation photon energy. The subdued decay process results in the emission starting from the excitation wavelength. When the excitation wavelength changes, the photons excite different self-trapped exciton states as determined by the excitation photon energy, and the suppressed decay process is then performed in the same way as shown before. As a result, the emission starts from the new excitation wavelength and the spectra also shift, as observed by the experiment.84 Another predicted phenomenon was the reduced linear polarization in the same fluorescence range as the excitation energy increases. As reported by the authors,84 compared to 442 nm excitation, the linear polarization decreased for the 325 nm excitation in the 460–690 nm range, which was a result of the further relaxation and momentum randomization for the 325 nm excitation.
3.7 Surface dipole emission center
To reveal the single particle emission mechanism and its structure information, a combined spectroscopic technique was adapted by Ghosh et al.93 By combining high-resolution transmission electron microscopy (HRTEM) with PL imaging spectroscopy, they achieved the correlative measurement of PL, the crystal structure, and the shape of a single carbon dot. The carbon dots were synthesized by a microwave-assisted method with sucrose as the carbon source and PEG as the passivation agent. The combined HRTEM disclosed that there existed two kinds of carbon particles: one with an onion-like structure with an amorphous carbon shell, as seen in the left part of Fig. 10a. EDX analysis indicated that the amorphous shell mainly contained oxygen and carbon elements. The diameter of the onion-like carbon dots varied from 7 to 20 nm, but they had the same lattice constant of about 0.35 to 0.36 nm. The other structure was a kind of fully crystalline carbon dots with diameters ranging from 2 to 5 nm and a lattice constant of 0.24 nm, as presented in the right part of Fig. 10a. All the individual single carbon dots exhibited much narrower PL spectra compared to the ensemble carbon dots and displayed a longer wavelength tail, thus the author suggested that there existed two splitting bands in the carbon dots. By using two Gaussian functions to fit the emission spectra of single carbon dots, the authors claimed93 that the splitting energy was in the range of 70 to 150 meV and showed no relationship to the emission peak. Based on the crystal structures of single carbon dots provided by HRTEM and the energy splitting information, the authors calculated the optical phonons contributing to the carbon dots emission by using a density-functional tight-binding method. Three optical phonons were proposed93 to be involved in the carbon dots emission: 98 meV phonon for onion-shaped structure carbon dots, and 112 and 113 meV for fully crystallized carbon dots; thus the longer wavelength PL peak was the result of the coupling between the radiative carriers with the optical phonons. The interaction between the electrons and lattice vibrations (phonons) led to coupled states that possessed lower energy.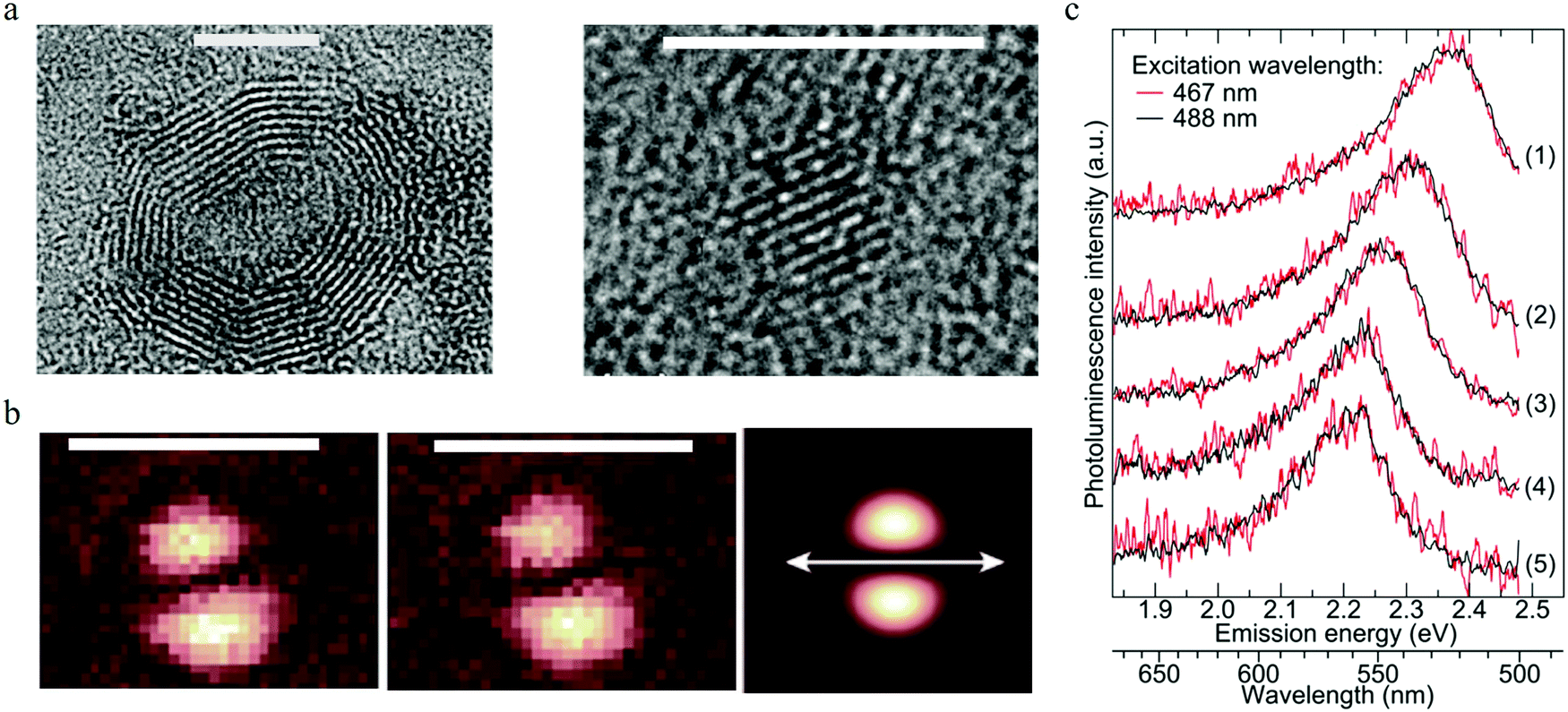 | ||
| Fig. 10 (a) Two types of carbon dots. Left: The onion-like shaped structure, right: full crystalline structure. Scale bar 5 nm. (b) Consecutive imagines of the excitation TDM for the same carbon dots particle, the third one presents a theoretically calculated excitation pattern, where the orientation of the dipole is indicated by the white double arrow. Scale bar 1 μm. (c) Normalized PL spectra of different single CNDs (5 single carbon dots) on the surface of a glass cover slide stimulated by 467 nm (red) and 488 nm (black) laser lines. The results show that the emission spectra varied for different single carbon dots, while each single carbon dot presented the same PL spectra, even when the excitation wavelengths were different. (a–c) Reproduced with permission from ref. 93. Copyright 2014, American Chemical Society. | ||
Focused or defocused imaging with an azimuthally polarized laser beam (APLB), which can measure and provide information on the excitation or emission transition dipole moment (TDM), respectively,94–96 was employed in one study93 to uncover the emission mechanism of carbon dots. The results are illustrated in Fig. 10b. Both the excitation and emission TDM patterns exhibited two nearby bright spots with an elliptical shape, indicating the fixed linear excitation and emission TDM. The TDM projection to the surface of a glass cover slide is shown by the white arrows, which clearly show that the excitation and emission TDM were orientated parallel to each other93 for the same carbon dots, while a projection perpendicular to the surface could not be detected. The fixed excitation and emission TDM suggested that the carbon dots behaved as a dipole for both excitation/absorption and emission. Single step PL intermittency and bleaching between the on–off states and monoexponential decay of the lifetime were observed for a single carbon dots, which indicated that the carbon dots were a kind of a single quantum emitter. To further confirm the single emitter property of single carbon dots, the authors checked93 whether the PL spectra of the carbon dots changed when excited by different wavelengths. The results are displayed in Fig. 10c. There was no difference in PL spectral shape for all the five individual single carbon dots when excited by 467 nm (red curve) and 488 nm (black curve) lasers. The results indicated that individual carbon dots were a kind of single emitter, and the excitation-dependent PL of carbon dots was the result of heterogeneity. The authors also measured93 the PL quantum yield for different single carbon dots by means of a nanocavity-based method, and the results demonstrated that different single carbon dots possessed different quantum yields within the main range of 0–0.3, which was consistent with the single quantum emitter assumption. Considering that the different crystal structures did not present distinct PL properties, the authors speculated93 that the defect emission center was located at the surface of the carbon dots rather than its core. The surface emission center notion was also supported by the ion-quenching experiments. In addition, the homogeneous distributed PL peak and lifetime values indicated that the all the PL spectra originated from similar defect emission centers.
Based on the all the observations, the following emission mechanism was suggested by the authors.93 A single carbon dot is a single emitter with the defect emission center located at the surface of the carbon dot, where strong coupling between the charged carriers with the lattice vibrational (phonon modes) results in longer wavelength emission, while excitation-dependent PL in carbon dot ensembles originates from the different carbon dots heterogeneity.
3.8 Aggregate emission center
Following the model of a single surface dipole emission center,93 researchers have continued to explore the details of the structure aspects that contribute to carbon dots emission.97 As the absorption of light occurs by the coupling between the photons and aromatic species in carbon dots, two general energy coupling approaches have been considered: excitonic coupling between the π electronic systems in carbon dots, which is efficient in the condition of a near contact, and dipole–dipole resonance (also called Förster resonance energy transfer (FRET)), which possesses a longer interaction distance. The observed parallel orientated excitation and emission transition dipole moment (TDM) in single carbon dots93 demonstrate that it's the excitonic coupling rather than the dipole–dipole interaction that is responsible, as the dipole–dipole interaction will rotate the orientation of the emission TDM from the absorption TDM direction.97According to the theory proposed by Kasha,98 only two kinds of aggregate conditions can contribute to the excitonic coupling effect for molecular complexes, namely J- and H-aggregates. Parallel-aligned molecular dipoles can constitute J-aggregates, which have distinct absorption and PL spectra features.99 Compared to molecular monomers, both PL and the absorption spectra of J-aggregates are narrow and shifted to longer wavelength. Furthermore, the Stokes shift between the excitation and emission is small, and all the properties of J-aggregates are contradictory to the observations from single carbon dots. The other type of π–π stacked H-aggregates possess opposite properties with large energy shifted absorption spectra compared to molecular monomers and a large Stokes shift between the excitation and PL, consistent with experimental observation. The energy diagram was given by Alexander P. Demchenko97 and is illustrated in Fig. 11a, where it can be seen that the π–π-stacked aggregates lead to the dimer excited state splitting into two energy states, consisting of a higher energy excitonic state and a lower energy excitonic state. According to the classic theory proposed by Kasha,98 the relaxation to the lower excitonic states is forbidden, which means that the pure π–π-stacked H-aggregates are non-emissive centers, which also contradicts the observed bright emission from carbon dots. Considering the recently observed bright emission from H-aggregates from other materials,100,101 the authors rationalized97 the optical spectroscopic observations from the carbon dots within the concept of excitation theory. Different from J-aggregates, excitonic coupling in H-aggregates could occur by a cofacial stacking alignment determined by weak van der Waals interactions. As a result, a slight rotation of the coupled monomer in H-aggregates could result in a non-zero transition probability from the excited states to the lower excitonic states, which represents the emission center; thus even a small structure disorder could lead to transformation of non-emissive H-aggregates to highly emissive H-aggregates. H-aggregates formed by different mono-chromophores (e.g., number, size, shape, surface) exhibited different absorption and emission properties, which interpreted the heterogeneity of the lifetimes and PL spectra for different single carbon dots, while the energy decay process led to decay of the hot carriers to the lower excitonic states, which contributed to the large Stokes shift.
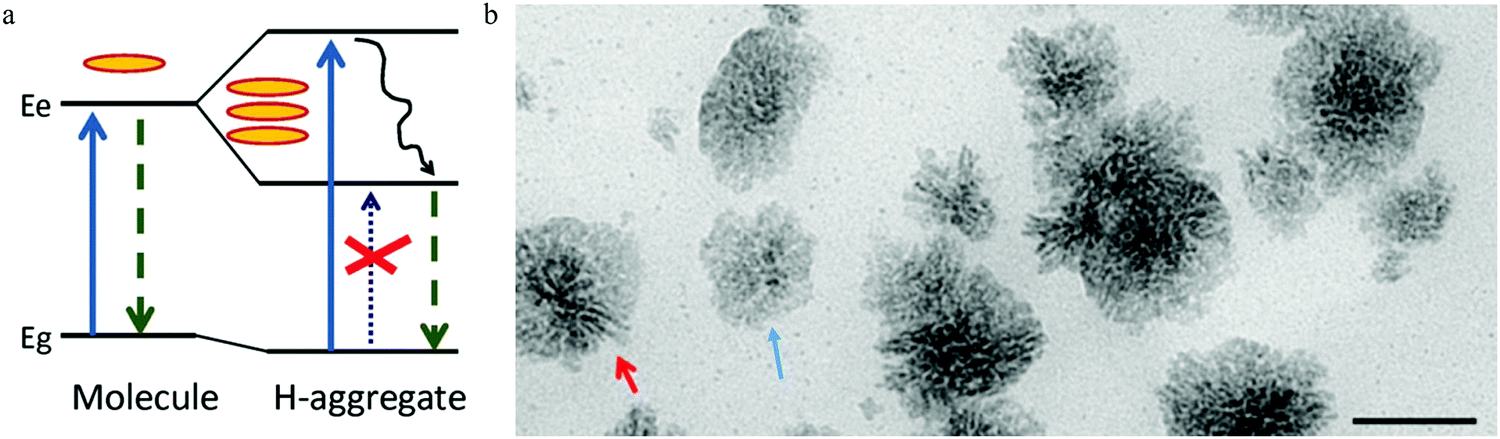 | ||
| Fig. 11 (a) The H-type aggregates formed by π–π stacking or side-to-side stacking of the chromophores, then the resulting dimer excited states split into higher energy and lower energy excitonic states. The disorder in the H-aggregates leads to the non-emissive lower excitonic states transferring to the emissive excitonic states, which contributes to the carbon dots emission and large Stokes shift. Reproduced with permission from ref. 97. Copyright 2016, Royal Society of Chemistry. (b) The authors103 also proposed that both the H (rod-like, as indicated by red arrow) and J (flake-like, as indicated by blue arrow) aggregates contribute to carbon dots emission. Reproduced with permission from ref. 103. Copyright 2016, American Chemical Society. | ||
Different from the dyes’ molecular or supramolecular H-aggregates, the authors suggested97 that the H-aggregates in carbon dots were assembled during the synthesis process by means of a regular packing of graphene sheets. Accepting the assumption proposed by Siddharth Ghosh et al.,93 the authors suggested97 that the cofacial positions of chromophores were located at the surface of carbon dots, and that surface functional polar groups, such as CO and CN, could modify the optical properties of H-aggregates containing carbon dots.
The aggregates-triggered emission was also adopted by several research groups,102–106 but with some modifications. Arjun Sharma et al.103 synthesized carbon dots by directly heating citric acid and urea. However, unlike the pure H-aggregates emission center, they proposed that the carbon dots emission originated from the discrete multiple electronic states, and they ascribed the PL excitation band of 350 nm, 450 nm, and 520 nm to the carbon dots monomers, H-aggregates (see Fig. 11b, red arrow), and J-aggregates (see Fig. 11b, blue arrow), respectively. The corresponding emission bands were 460 nm, 540 nm, and 600 nm, which suggested that both H and J-aggregates contributed to the carbon dots emission. Furthermore, H- and J-aggregates manifested different PL responses with increased temperature, whereby the PL intensity of H-aggregates increase with the temperature increase as the thermal activated higher excitonic states are emissive, while the J-aggregates display decreased PL intensity as the thermal activated states return to the vibrational states, which are non-emissive. Therefore, the authors also carried out temperature-dependent PL analysis,103 and the results were as follows: the H aggregated band (450 nm/540 nm excitation/emission band) presented an increased emission intensity as the temperature increased, while the J aggregated band (520 nm/600 nm excitation/emission band) exhibited a decreased PL intensity as the temperature increased, which was consistent with the authors’ claims.
4 Applications
4.1 Bioimaging
Compared to fluorescent organic dyes and genetically engineered fluorescent proteins, carbon dots have significant advantages, like a high PL quantum yield, photostability, and resistance to metabolic degradation, which endows them with huge potential for use in bioapplications. While the toxicity evaluation of carbon dots is necessary and a prerequisite for exploring the bioapplications of carbon dots, human breast cancer MCF-7 cells and human colorectal adenocarcinoma HT-29 cells were employed by Yang et al.7 to assess the in vitro toxicity of carbon dots synthesized by the laser ablation of graphite powder and cement using PEG1500N as a surface passivation agent. All the observations of cell proliferation, mortality, and viability from both cell lines indicated that the carbon dots exhibited superior biocompatibility, even when the concentration of carbon dots was up to 50 μg ml−1, which is much higher than the practical application demand, for example in living cell imaging. Additionally, in vivo toxic evaluation was also carried out by the authors using male CD-1 mice;7 whereby, two groups of mice were intravenously injected with carbon dots at a concentration of 8 mg kg−1 and 40 mg kg−1 separately, while a third group of mice was injected with 0.9% NaCl aqueous solution as the nontoxic control. During the four weeks of the experiment, the behaviors of all the mice were normal, whereby neither violent nor lethargic behavior was observed. In addition, all the experimental mice exhibited no clinical anorexia or symptoms such as hair loss or scabs. Serum biochemistry assays uncovered that both the hepatic injury indicators alanine amino transferase (ALT) and aspartate amino transferase (AST) and kidney injury indicators uric acid (UA), blood urea nitrogen (BUN), and creatinine (Cr) displayed similar levels in the carbon dots-injected mice and in the control group mice, which indicated that the carbon dots were nontoxic to the mice. Histopathological analyses showed that the carbon dots did not cause a structure change of the hepatic, splenic, and kidneys sections. All of the above observations unambiguously demonstrated the nontoxicity of carbon dots both in vitro and in vivo. Besides the mice evaluation, similar results were also observed from an experiment with zebrafish,107 which also exhibited the excellent biocompatibility of carbon dots.Moreover, the plane cell toxicity of the carbon dots (synthesized by the microwave heating of citric acid and urea) was also verified by Qu et al.59 A carbon dots aqueous solution with a concentration of 1.5 mg ml−1 was adopted to grow bean sprouts, and the results indicated that the carbon dots could be ingested by the plant cells, thus resulting in bright emissive bean sprouts, and also no obvious toxicity or plant growth inhibition were observed.
Due to the excellent biocompatibility and bright fluorescence, carbon dots are naturally considered for use in bio- and clinical applications. Ya-Ping Sun et al. performed the pioneering work on carbon dots bioimaging.2 They first employed carbon dots (synthesized by laser ablation) to label the E. coli ATCC 25922 cells, as depicted in Fig. 12a–d, and the cells displayed different colors when the excitation wavelength was changed. Later, the same group also demonstrated the in vivo imaging of carbon dots,108 as can be seen in Fig. 12e–i. Female DBA/1 mice (∼25 g) were injected with 30 μg carbon dots (dissolved in 30 μl aqueous solution) via subcutaneous injection. As seen from Fig. 12f and h, the subcutaneously injected mice showed bright green emission when stimulated by 470 nm, and red light emission for 525 nm excitation. Following the author's in vitro and in vivo imaging demonstrations, various cell lines7,14,17,30,62,109–113 (such as HeLa cells, MG-63 cells) and animals8,107,114,115 (such as mice, zebra-fish) have been also imaged using diverse fluorescent carbon dots.
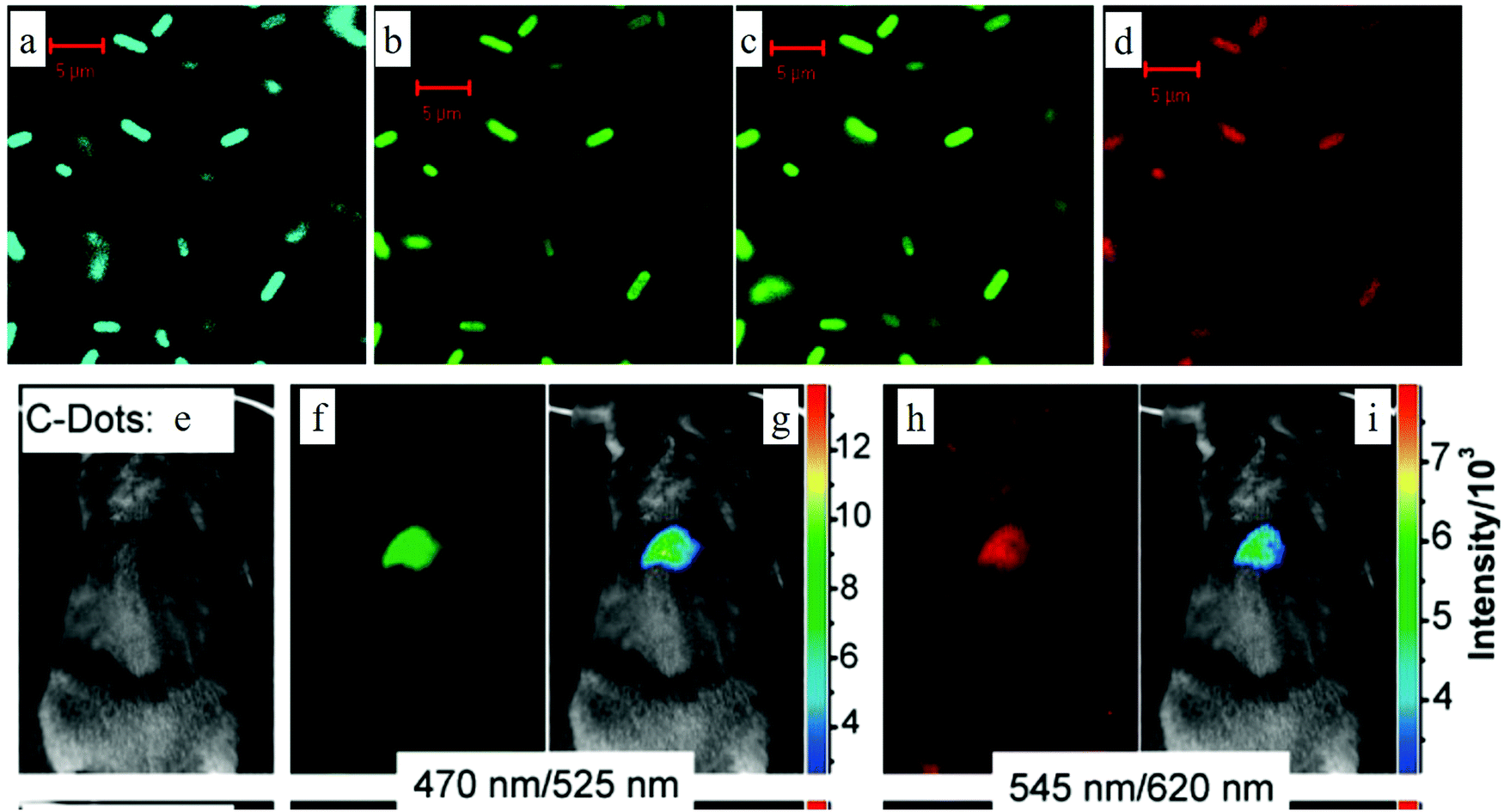 | ||
| Fig. 12 (a–d) Confocal microscopy images of E. coli ATCC 25922 cell lines using carbon dots as the fluorescent material. (a) 465 nm excitation, (b) 477 nm excitation, (c) 488 nm excitation, (d) 514 nm excitation. (e–i) In vivo imaging. (e) Bright field image, (f and h) in vivo fluorescent imaging for 470 nm excitation/525 nm emission and 545 nm excitation/620 nm emission, respectively. (g and i) Color-coded images for (b and d), respectively. The carbon dots were subcutaneously injected into the mice. (a–d) Reproduced with permission from ref. 2. Copyright 2006, American Chemical Society. (e–i) Reproduced with permission from ref. 108. Copyright 2009, American Chemical Society. | ||
For further preclinical and potential clinical applications, carbon dots must meet the requirements of the U.S. Food and Drug Administration (FDA), where agents injected into the human body must be removed entirely in a reasonable period of time;116 thus the injected agents cannot accumulate in the body and their circulation time should be optimized. The bright emissive carbon dots could be used in in vivo bioimaging, thus they also enable the possibility to explore the biodistribution and clearance in animals. A main challenge for carbon dots8 (synthesized by laser ablation of a carbon target, followed by PEG1500N surface passivation) in vivo bioimaging is the relative low quantum yield in the near-infrared (NIR) wavelength range of 650–900 nm. In vivo imaging possesses high quality in the near-infrared (NIR) region as both the tissue absorption and scattering are suppressed in this region. To overcome this issue, a near-infrared dye ZW800 (emission wavelength about 795 nm) was employed by Huang and co-workers8 to link to the carbon dots by means of conjugating the dye NHS ester with amino groups on the C-dots. The resulted ZW800-carbon dots showed two absorption peaks at 420 nm and 770 nm and two emission peaks at 510 nm and 800 nm, respectively, corresponding to the carbon dots and dye ZW800. Various biological fluids (fetal bovine serum (FBS), blood, urine, and tissue lysate) tests demonstrated that the ZW800-carbon dots exhibited photostability, chemical stability, and a low emission background.
The carbon dots in vivo dynamics was explored by means of three different injection routes: intravenous (iv), subcutaneous (sc), and intramuscular (im). The blood circulation behaviors of carbon dots were revealed by the relative PL intensity in the venous blood, which was analyzed using a Maestro all-optical imaging system. The results demonstrated that different injection routes resulted in opposed carbon dots concentration dynamics. For the iv injection approach, the carbon dots concentration in blood continued to decrease post-injection. Quantitative analysis disclosed that the PL intensity of the blood after 1 min of post-injection was about 17.3 times higher than at 1 h. While for the subcutaneous (sc) and intramuscular (im) injection routes, the carbon dots concentration gradually increased and reached the maximum value 30 and 20 min post-injection, respectively, as indicated by the increased blood PL intensity. These blood circulation behaviors were observed with both the carbon dots part (420 nm excitation) and the ZW800 component (770 nm excitation) emission. In addition to the blood circulation, the clearance or accumulation of carbon dots was evaluated by the authors.8 Generally, nano-materials are cleared from the body via two major approaches, namely the liver, which convert them into bile, and the kidneys, which convert them into urine. Ex vivo imaging of the organs analysis demonstrated that for all three types of injection, most of the carbon dots were gathered by the kidneys after 1 h post-injection. Also, the accumulation rate displayed the following sequence: im > sc > iv, whereas, on the contrary, only a small amount of carbon dots were collected by the liver. However, when the time increased to 24 h, no obvious PL signal could be detected regardless of the injection approaches, as illustrated in Fig. 13a. All of these observations ambiguously implied that the carbon dots were totally removed from the body. Beside the ex vivo imaging, the authors8 also employed the positron emission tomography (PET) imaging technique, which showed similar results, which thus further confirmed the low accumulation of carbon dots in the body. Further urine clearance evaluation was analyzed by in vivo bladder imaging analysis. The bladder red emission imaging demonstrated that the intravenous (iv) injection exhibited the fastest clearance and reached its maximum value after 10 min of injection, while the subcutaneous (sc) and intramuscular (im) injections displayed a much lower urine clearance rate within 10 min, as indicated by the relatively low emission intensity. More specifically, the urine clearance rate of the three injection routes followed the sequence of iv > im > sc, which was in accordance with the carbon dots ZW800 clearance rate from the blood. The carbon dots ZW800 clearance mechanism was also confirmed by the carbon dots emission, while the strong background of urine in the visible range caused the contrast to be much lower. Besides, the positron emission tomography (PET) imaging results also suggested the same urine clearance mechanism. We want to emphasize here that different carbon dots samples may show different in vivo dynamics. Hong Bi's group117 demonstrated that carbon dots synthesized from the thermal pyrolysis of konjac flour accumulated in both the kidney and liver after 4 h of intravenous injection. While 24 h after the injection, all the carbon dots were cleared from the mice; thus no PL signal can be detected from the organs.
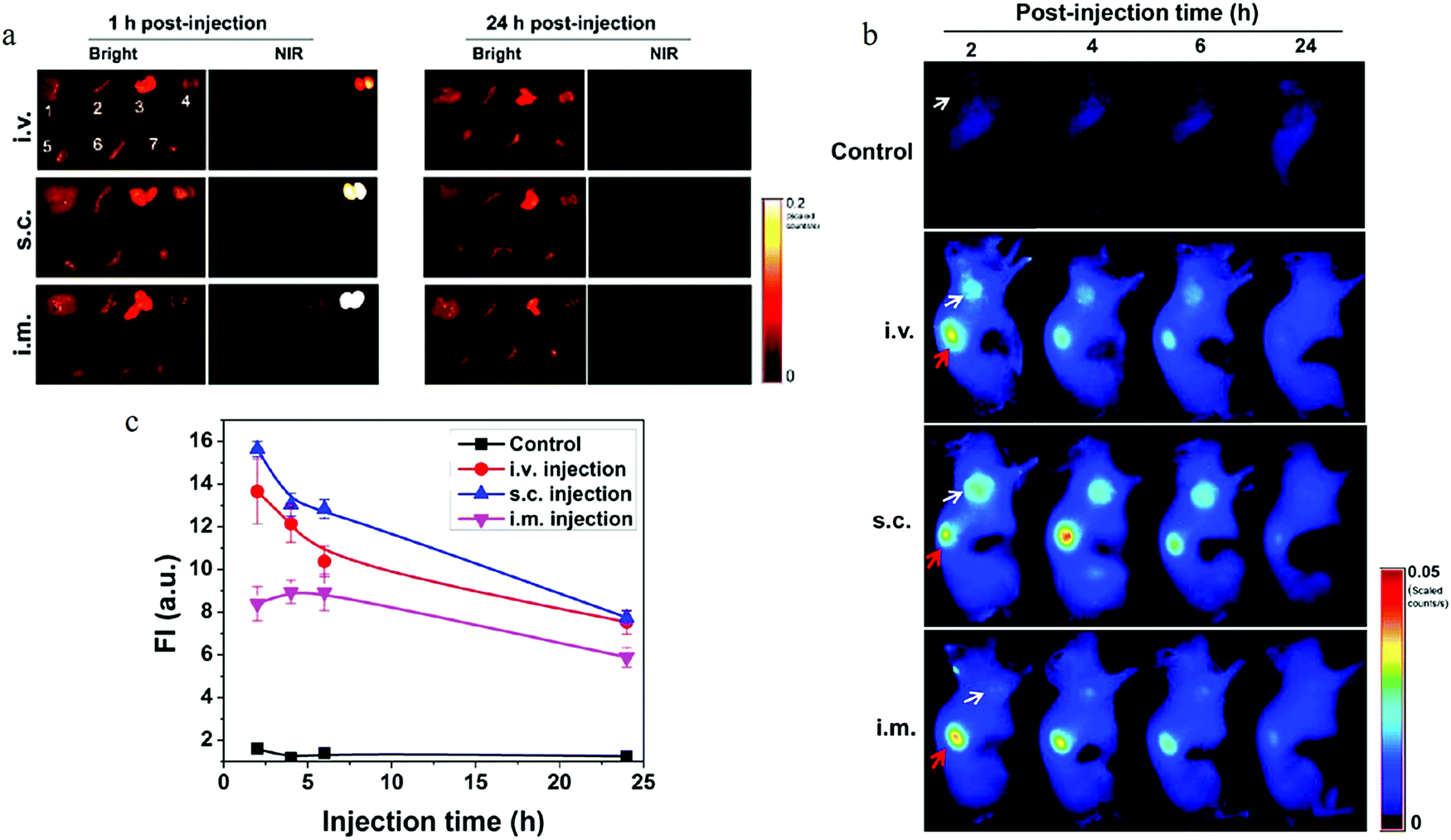 | ||
| Fig. 13 (a) Ex vivo imaging of the biodistribution of C-dot particles. After the injection of ZW800-carbon dots, the major organs and tissues were collected from Balb/C mice at 1 h (left) and 24 h (right), and subsequently, the bright field and NIR imaging were obtained using a Maestro imaging system. Top, iv injection; middle, sc injection; bottom, im injection; bright field numbers: 1, liver; 2, spleen; 3, lung; 4, kidneys; 5, muscle; 6, intestine; 7, heart. (b) Tumor accumulation of ZW800-carbon dots for different injection approaches. In vivo fluorescence images of SCC-7 tumor-bearing mice acquired after 2, 4, 6, and 24 h injection: control (without injection); iv; sc; im (white arrow labels tumor; red arrow labels kidney). (c) Tumor fluorescence intensity analysis. (a–c) Reproduced with permission from ref. 8. Copyright 2013, American Chemical Society. | ||
Another fascinating discovery by the authors8 was that the prominent and prolonged ZW800-carbon dots were uptaken by the tumor. Athymic nude mice bearing a subcutaneous SCC7 tumor were employed as the tumor model. All the ZW800-carbon dots injected by the three routes manifested accumulation around the tumor, resulting in bright red light emission, thus the tumor became distinguishable from the surrounding normal tissue. In addition to the tumor accumulation, the remaining carbon dots exhibited no accumulation for the other normal organs and were totally removed via the kidneys (red arrow), which was uncovered by the in vivo imaging analysis using the Maestro all-optical imaging system. The carbon dots accumulation ability at the tumor site also displayed an injection route dependence. The subcutaneous (sc) injection route gave the highest accumulation ability, and thus exhibited the brightest emission. The intravenous (iv) group displayed a lower accumulation, but the light emission was still clear from the tumor. The lowest accumulation ability was observed with the intramuscular (im) injection, which showed the minimum PL intensity, as seen in Fig. 13b and c.8
All the observations have great significance for clinical applications. Carbon dots displayed no accumulation over time for all the normal organs in body and could be entirely removed from the body via the kidneys,8 which meets the requirement of the U.S. Food and Drug Administration (FDA). In addition, the unique tumor accumulation ability implied that the carbon dots-carried drug may exhibit better therapeutic effect, as discussed in the next sub-section. Moreover, the in vivo performance such as tumor targeting, blood circulation time, could be controlled by properly choosing the injection route.
4.2 Drug delivery
The excellent biocompatibility and clearance from the body of carbon dots desirably meet the prerequisite for in vivo applications. Rich and tunable function groups, such as amino, carboxyl, or hydroxyl, can endow carbon dots with the ability to carry therapeutic agents, generating theranostic nanomedicines.118–122 The bright emission of carbon dots provide the opportunity to dynamically and in real-time monitor the drug distribution and response. Carbon dots synthesized by the thermal pyrolysis of citric acid and polyene polyamine were employed by Zheng and co-workers to carry oxaliplatin,123 a platinum-based drug, as platinum-based drugs are the most efficient anticancer drugs and are used in more than 50% of chemotherapeutic treatments for clinical cancer patients. One of the oxaliplatin(IV) derivatives, Oxa(IV)-COOH, was employed to link with the carbon dots, forming CD-Oxa, and the linkage was finished by the reaction between the amino groups on the carbon dots surface and the activated COOH group of Oxa(IV)-COOH, where the COOH group was activated by using EDC/sulfo-NHS, as shown in Fig. 14a. A newly appeared Pt 4f XPS signal confirmed the successful bonding of Oxa(IV) to the carbon dots. Inductively coupled plasma mass spectrometry (ICP-MS) analysis unveiled that the platinum element of carbon dots-Oxa was 1.5 wt%, corresponding to a 4.2 wt% oxaliplatin(IV). In addition, the TEM data indicated that the size of carbon dots-Oxa was 2.71 ± 0.43 nm, thus larger than the diameter of pure carbon dots (2.28 ± 0.42 nm).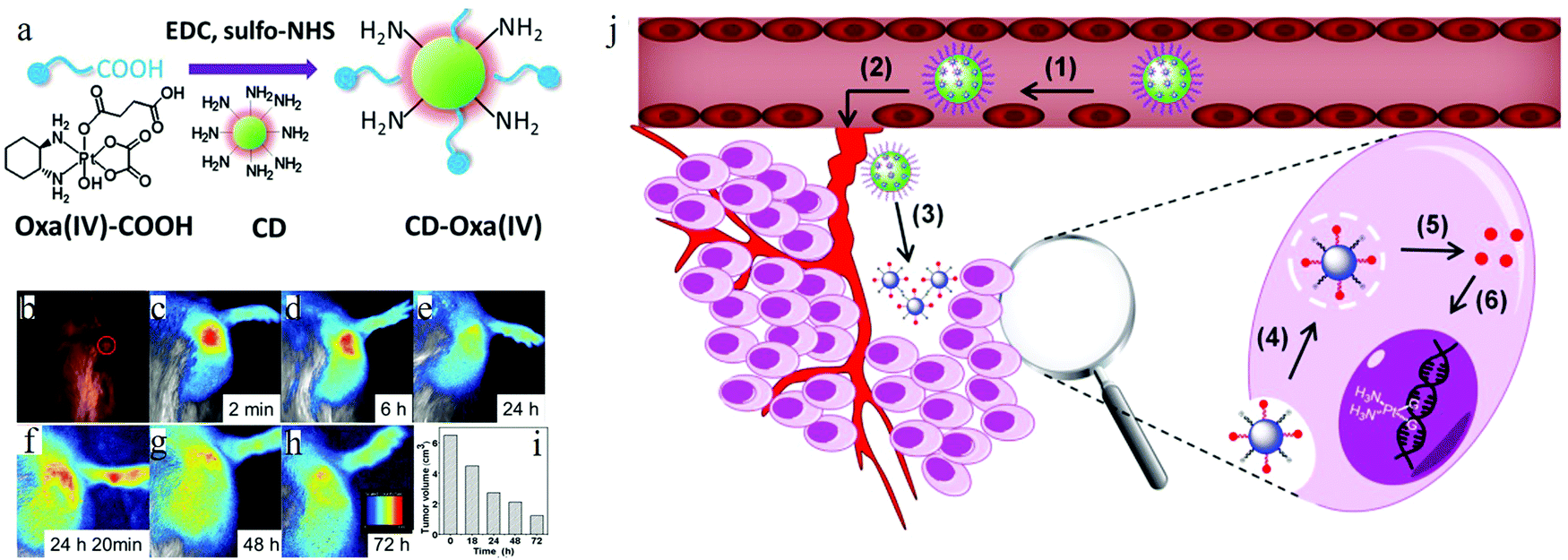 | ||
| Fig. 14 (a) Linkage illustration between the Oxa(IV)-COOH and carbon dots. (b–h) In vivo imaging of H22 xenograft model mice for different periods of time after intralesional injection. (b) Injection time; red circle means the tumor site (white light view). (c–e) Corresponding to the times of 2 min, 6 h and 24 h. (f) Second injection. (f–g) The in vivo imaging for 48 h and 72 h. (i) Tumor size of H22 xenograft model, where it can be clearly seen that the tumor size continuously decreases. (j) Illustration of the charge-convertible carbon dots-based drug-delivery process: (1) negatively charged carbon dots extend the circulation time in blood. (2) The drug accumulates at the tumor position via the enhanced permeability and retention effect. (3) Drug response in the tumor microenvironment. (4) Drug uptake by the cancer cells. (5) Drug release from carbon dots carrier through a proton sponge effect. (6) Cisplatin attaching with DNA to display cancer cell cytotoxicity. (a–i) Reproduced with permission from ref. 123. Copyright 2014, Wiley-VCH. (j) Reproduced with permission from ref. 124. Copyright 2016, American Chemical Society. | ||
The synthesized carbon dots-Oxa was injected into the hepatocarcinoma 22 cell line (H22) liver cancer of Chinese Kun Ming (KM) mice by means of intralesional injection,123 and the dynamical distribution of the drug was observed with an in vivo optical imaging system using blue light as the excitation source. At the instant of exposure123 (Fig. 14b), a highlight point was observed from the in vivo imaging due to the high concentration of carbon dots, and the following delivery of carbon dots-Oxa resulted in a fluorescent intensity decrease from the initial position and a broadened intensity distribution. After 24 h post-injection (Fig. 14e), the PL signal became much weak, which implied a decrease of the oxaliplatin(IV) drug, thus a second injection was carried out by the authors123 and the PL intensity became strong again. Furthermore, the PL intensity declined over another 24 h (Fig. 14g), and then the PL signal totally disappeared when the time reached 72 h (Fig. 14h), which implied that the carbon dots were cleared from the body entirely. The tumor size of the mice continued to decrease with the time of therapy, from 6.5 cm3 at the initial exposure time to 1.2 cm3 within 2 times injection, as observed by the authors123 (Fig. 14i). The dynamic drug distribution in the tumor and body during the curing process were obtained by the fluorescence of the carbon dots, suggesting it would be possible to achieve “personalized medicine”, which means that the injection doses, time, etc. could be controlled for individual patients.
Later, charge-convertible carbon dots were also fabricated for drug delivery by Feng et al.124 The carbon dots surface was negatively charged at pH = 7.4, thus corresponding to normal physiological conditions, while the surface was reversed to positively charged when the pH decreased to 6.8, mimicking the tumor microenvironment. The negatively charged carbon dots surface in the normal cell microenvironment suppressed the drug absorption of normal cells by means of electrostatic repulsion between the carbon dots surface and the negatively charged cell membrane, thus reducing the side effects. On the contrary, the positively charged carbon dots surface in the tumor cell microenvironment could promote the internalization and cancer drug absorption because of the electrostatic interaction with the negatively cell membrane (Fig. 14g). Both the in vitro and in vivo demonstration indicated124 that carbon dots could be used as an effective imaging-guided drug carrier with increased therapeutic efficiency and deceased side effects.
4.3 Microfluidics marker
Microfluidic systems have become a superior platform for investigating fluidic physics at the micro-scale.125,126 Their dramatically increased surface-to-volume ratio results in surface tension and viscosity being dominant over the inertia effects, thus making the fluid easier to manipulate. The typical micro-fluidic cases are static laminar flows and dynamic droplet formation, with both possessing various merits, such as less reagents consumption, high sensitivity, high output, which makes them find numerous applications in bioassays, chemical reactions, drug delivery, etc.127–129 Most of the applications are based on fluid flow visualization in the microfluidic circuit. However, the currently employed fluorescent materials cannot balance the biocompatibility and low cost, which is an urgent issue for microfluidic applications, especially for bioapplications. To address this issue, carbon dots (synthesized from the microwave heating of glucose and urea) were, for the first time, employed to visualize micro fluid flows by Sun's group.130 The authors explored the mix dynamics of DI water and glycerol with carbon dots dissolved DI water as the fluorescent marker. At t = 0 s (Fig. 15a), the flat flow was determined by the hydrodynamic driving forces, where an applied electric field (136.36 kV m−1) then ruptured the interface and resulted in the complete mixing at t = 3.76 s, as seen in Fig. 15a. The instability process was balanced by the pressure gradient, viscous dissipation, and electric-field induced body-force ruled by the ion species conversion law in each fluid. An electric field higher than a threshold ruptures the interface and leads to rapid mixing at the micro-scale.131 In addition to the laminar flow, mono-dispersed droplets were also achieved by the authors130 using a flow focusing configuration, where the carbon dots aqueous solution was the dispersed phase and the mineral oil acted as the continuous phase. The diameter of the droplets followed an inverse relationship to the capillary numbers (Ca), defined as Ca = μν/γ, where μ and ν are the dynamic viscosity and characteristic velocity of the mineral phase, respectively, and γ represents the interfacial tension between the dispersed (carbon dots aqueous solution) and mineral oil continuous phases. A larger capillary number means a higher interfacial shear force, thus the diameter of the droplets will decrease. The results are shown in Fig. 15b and c, where the generated droplets changed from squeezing to dripping and finally reached jetting along with the increasing capillary number (Ca). Besides, the multiple component droplet, merged droplet and double emulsion, which possess unique core–shell structures, were also demonstrated successfully by the authors130 by making use of superior carbon dots. To further quantitatively measure the velocity of the flow field, carbon dots fluorescent seeding particles were also fabricated by stir-mixing 3 μm porous polystyrene microparticles with a carbon dots aqueous solution. The velocity field measured by the micro-particle image velocimetry (μ-PIV) technique using carbon dots seeding particles is exhibited in Fig. 15d and e, and matches very well with the simulation results (Fig. 15f).130 All of the static and dynamic, qualitative and quantitative measurements unambiguously exhibited the high potential of applying carbon dots in microfluidics.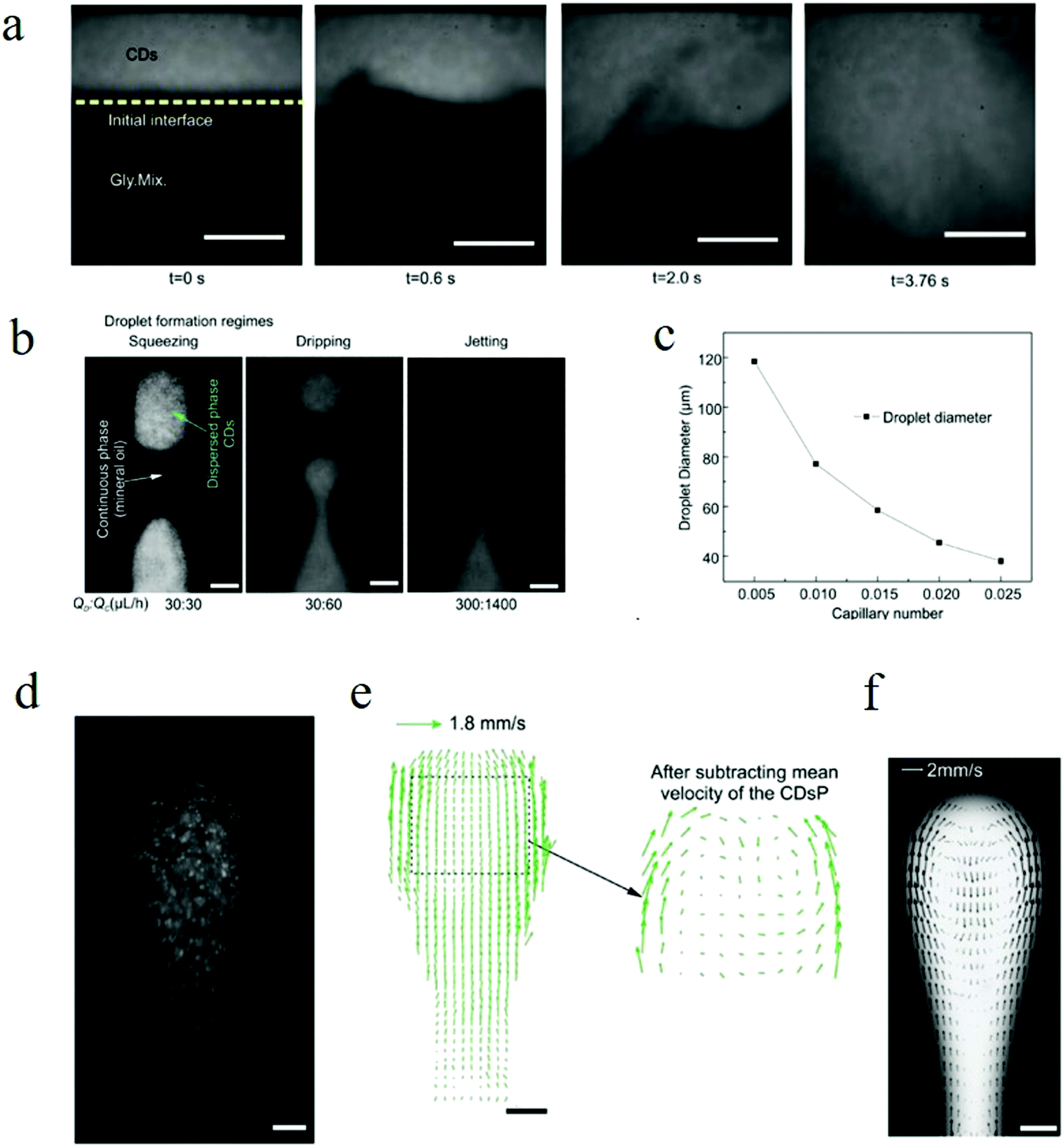 | ||
| Fig. 15 (a) Mixing evolution of carbon dots aqueous solution and glycerol at an applied electric field of 136.36 kV m−1; scale bar 100 μm. (b) Droplet formations in three typical regimes: squeezing, dripping, and jetting, which correspond to the different capillary number; scale bar 50 μm. (c) Relationship between the capillary number and droplet diameter. (d–f) Flow velocity field measurement using a carbon dots-based seeding particle. (d) Fluorescent imaging of a droplet generated with the carbon dots seeding particle distrusted in the dispersing phase. (e) The measured velocity field by using the micro-PIV technique; scale bar 50 μm. (f) The simulation results of the velocity field of the droplet, which match very well with the measurements. For all experiments and simulations, the flow rate of the dispersed phase and continues phase was 20:100 μl h−1. Reproduced with permission from ref. 130. Copyright 2017, Wiley-VCH. | ||
4.4 LEDs
In 2010, Wang et al.39 achieved direct white light emission from carbon dots, which offered the possibility to fabricate carbon dots-based white light emitting devices.132–135 Later, they carried out tests on white light emitting devices using white light carbon dots as the emissive layer.136 Briefly, the carbon dots were synthesized by a thermal pyrolysis method using citric acid as the carbon source and 1-hexadecylamine as the surface passivation agent, which showed a relative high quantum yield of 60% with an average diameter of 5 nm.The device structure, energy alignment, and organic molecular structure adopted by the authors136 are illustrated in Fig. 16a–c. The architecture of the device comprised three layers. The buffer layer consisted of 40 nm poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) in the anode, which had two functions: (1) increase the work-function from 4.7 eV (ITO) to 5.0 eV. (2) Increase the smoothness of the anode. The carbon dots emissive layer was fabricated by a spin-casting method with an optimized thickness of 20 nm. A thicker film, e.g., 35 nm, would lead to bad charge transport in the carbon dot film, while a thinner film, e.g., 10 nm, would increase the density of voids, grain boundaries, etc., both of which would decrease the device EL efficiency. AFM measurements indicated that the surface roughness of the carbon dot film was less than 3 nm, which implied good compatibility between the carbon dots and the buffer layer. Finally, the electron transport layer was employed through 40 nm thick 1,3,5-tris(N-phenylbenzimidazol-2-yl) benzene (TPBI) due to its good electron transport ability. The electrode consisted of 1 nm LiF and 120 nm Al fabricated by thermal evaporation.
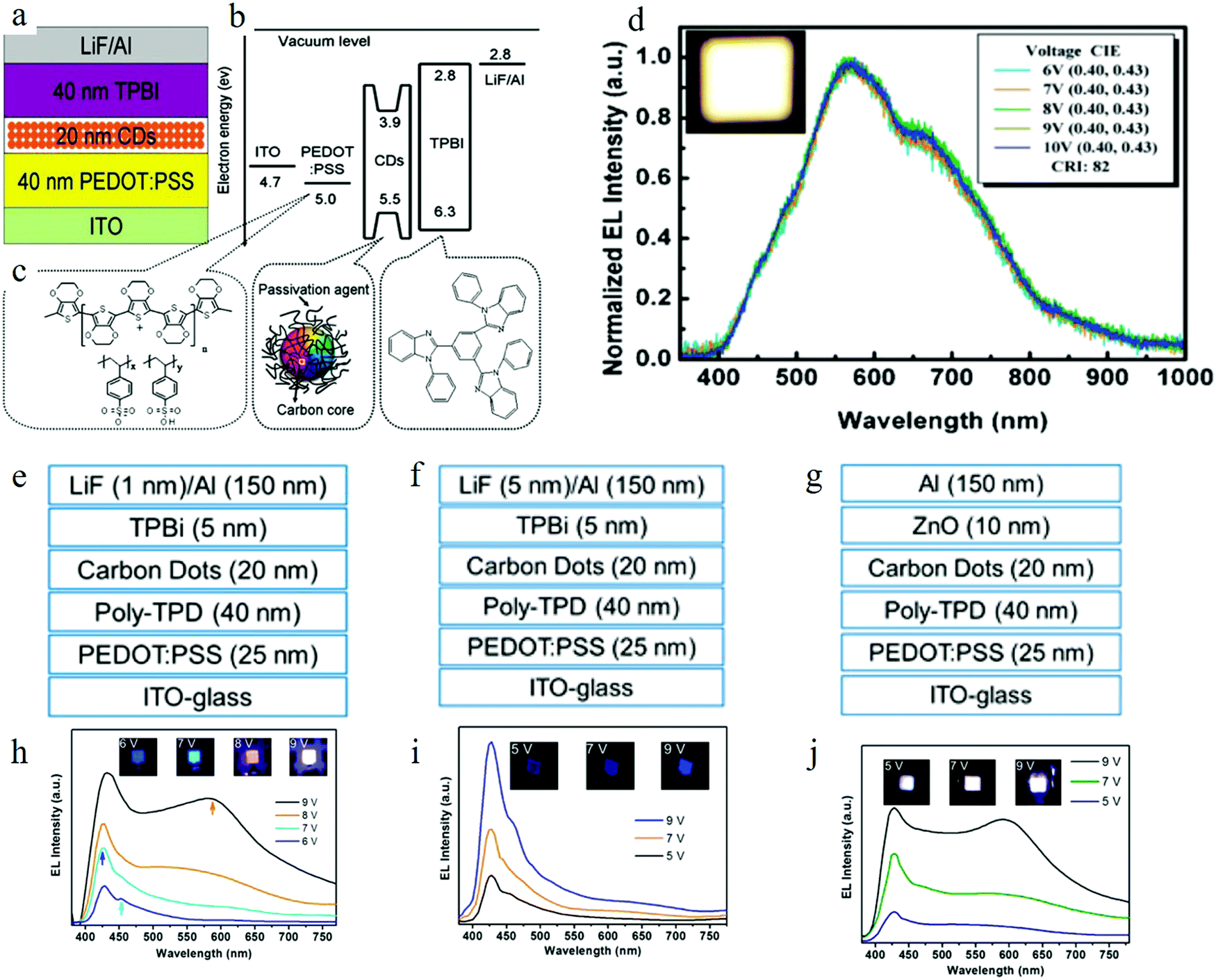 | ||
| Fig. 16 (a–c) White light LED device structure illustration, energy alignment, and organic molecular structures. (d) Normalized electroluminescence spectra of the carbon dots white light LED; inset is the photograph of the white light emission from the LED with a voltage of 9 V. (e–g) Schematic diagrams of the device structure for tunable EL light, blue light, and white light emission LEDs respectively. (h–j) The corresponding EL spectra for the tunable, blue and white carbon dots LEDs; insets are the photographs of the devices at different applied voltages. (a–d) Reproduced with permission form ref. 136. Copyright 2011, Royal Society of Chemistry. (e–g) Reproduced with permission from ref. 137. Copyright 2013, American Chemical Society. | ||
The pure carbon dots exhibited broad PL with an excitation-dependent emission feature, while the LED device displayed stable emission even when the voltage was increased to 10 V, as shown in Fig. 16d.136 The Commission International d’Eclairage (CIE) coordinates of the emitted light were (0.40, 0.43) ((0.33, 0.33) for pure white light), with a color-rendering index CRI of 82. The WLED had a turn-on voltage of 6 V, with the maximum external quantum efficiency (EQE) reaching 0.083% at a current density of 5 mA cm−2, and a maximum brightness output of 35 cd m−2 with a current density of 160 mA cm−2 and voltage of 9 V.
By modifying the device structure, color-switchable CD LEDs were demonstrated using the same carbon dots by Zhang's group.137 According to the excitation-dependent PL and time-resolved measurement, the authors claimed that there existed three emission channels, whose lifetimes were 2 ns, 5–6 ns and 14–15 ns, contributing to 420 nm, 460 nm, and 580 nm band emissions, respectively. In addition, the authors proposed137 that the relaxation process for high energy emission was faster. Adopting this assumption to fabricate the carbon dot LED, different current injections would lead to the following conclusion: when the injection current density is low, the hot carries will relax by means of the fast decay channel, resulting in a high energy emission, e.g., 412 nm band or blue emission, while a high injected current density could stimulate all three emission bands. In addition, the highly populated excited states could feed the lower emission band, e.g., the 480 nm and 580 nm states, thus the lower band emission becomes dominant in the case of a high current injection. So by adjusting the injection current, the authors achieved137 tunable color emission based on one kind of carbon dots.
The structure illustration of the CD LED device adopted by the authors is shown in Fig. 16e–g.137 Briefly, the LED consisted of six layers: an ITO anode, 25 nm hole-injection layer, 40 nm hole-transport layer (HTL), 20 nm carbon dots layer, 5 nm electron transport layer (ETL), and top cathode layer. Poly(ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) was utilized as the hole-injection layer. Poly-(N,N′-bis(4-butylphenyl)-N,N′-bis(phenyl)benzidine) (poly-TPD) was employed as the HTL because of its superior hole-transport capability, the matched energy alignment between poly-TPD and the PEDOT:PSS layer, and as it was easy to synthesize a uniform thin film. The ETL and the cathode layer were designed for a certain purpose. When choosing 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI) as the ETL and 1 nm LiF and 150 nm aluminum as the cathode double layer, the turn-on voltage was 5 V and the injection current density could be tuned from relatively low to relatively high (reaching 600 mA cm−2), while, as discussed above, different injection current densities (or the corresponding applied voltage) resulted in different color emissions, as can be seen in Fig. 16h. Blue, cyan, magenta, and white light emissions occurred at voltages of 6 V, 7 V, 8 V, 9 V, respectively, while the Commission Internationale de l’Eclairage (CIE) coordinates for the four emission colors were (0.198, 0.151), (0.212,0.162), (0.260, 0.221), and (0.318, 0.320).137
To obtain pure blue emission CD LED, a low current injection density was necessary; thus the authors137 increased the thickness of the LiF layer of the cathode layer from 1 nm to 5 nm, and as a result, the current density decreased to 150 mA cm−2, resulting in a pure blue CD LED with a turn-on voltage 5 V and a maximum luminance of 24 cd m−2, as illustrated in Fig. 16i. Similarly, a high current density injection could result in white light emission. Thus, using ZnO nanoparticles as the ETL and with the cathode layer only containing 150 nm Al, the high electron mobility of ZnO nanoparticles (2 × 10−3 cm2 V−1 s−1 for ZnO, one order of magnitude higher than TPBI 1 × 10−4 cm2 V−1 s−1) endowed a high injection current density to the emissive layer, resulting in white light emission, as depicted in Fig. 16j. The white light CD LED possessed the maximum luminance of 90 cd m−2 with a turn-on voltage of about 4.6 V.
Different from controlling the injection current density, carbon dots with excitation-independent emission were also proposed by Yang's group to fabricate mono-color light emission LEDs.13 The carbon dots were synthesized by means of hydrothermal methods. By controlling the carbon source (citric acid), nitrogen source (2,3-diaminonaphthalene or 1,5-diaminonaphthalene), surface passivation/carbonization (concentrated sulfuric acid), and reaction time, all blue, green, yellow, orange, and red emission carbon dots were achieved. All the carbon dots exhibited relatively high quantum yields: 75% (B), 73% (G), 58% (Y), 53% (O), and 12% (R). The EL spectra of all the carbon dots were consistent with the PL spectra with the peaks located at 455 nm (blue), 536 nm (green), 555 nm (yellow), 585 nm (orange), and 628 nm (red) and all displayed voltage-independent EL colors. The authors13 attributed the tunable PL emission and voltage stable EL emission to the band emission of carbon dots with the quantum confinement effect. The CIE coordinates, maximum luminance, and current efficiency for the monochrome LEDs were13 ((0.19, 0.20), 136 cd m−2, 0.084 cd A−1), ((0.31, 0.47), 93 cd m−2, 0.045 cd A−1), ((0.37, 0.52), 60 cd m−2, 0.02 cd A−1), ((0.46, 0.48), 65 cd m−2, 0.027 cd A−1), ((0.55, 0.41), 12 cd m−2, 0.0028 cd A−1) for the blue, green, yellow, orange, and red LEDs, respectively.
4.5 Sensing
The fluorescence intensity of carbon dots is closely related to the surrounding environment. The interaction between carbon dots and chemicals results in the quenching/enhancement of carbon dots emission. Thus carbon dots can be used as a kind of fluorescent probe to detect the quantity of items. Various sensors based on the fluorescence of carbon dots have been reported and can be generally categorized into the following groups: ion sensing,138–141 such as Eu3+, Fe3+, Fe2+, F−, Tl+ (thallium); pH-value sensing;142–145 biomaterial sensing,145–148 such as intracellular lysine, ascorbic acid, guanosine 3′-diphosphate-5′-diphosphate (ppGpp); enzyme (e.g., thioredoxin reductase (TrxR)); DNA; and temperature sensing.149–151 However, single fluorescent wavelength intensity change-related sensing requires strict conditions. The fluctuation of the light source, concentration of fluorescence probes, different optical paths, and even the aggregation of the probes could devalue the reliability of the sensing results. Thus ratiometric sensing as an alternative has been proposed,143,145,152,153 as it can simultaneously collect information on the intensity change of two separated emission bands. The intensity ratio of the two wavelengths is the indicator of the sensing item and is considered as the output. Compared to a single emission sensor, ratiometric sensing provides for self-calibration for both the light source and sensing environment, and thus can vastly improve the sensing accuracy and reliability.Carbon dots-based ratiometric pH sensing was first proposed by Wen and co-workers,152 using carbon dots synthesized by the thermal pyrolysis of citric acid with 4,7,10-trioxa-1,13-tridecanediamine (TTDDA) as the surface passivation agent. Both pH-sensitive fluorescent fluoresceinisothiocyanate (FITC) and pH-insensitive rhodamine B isothiocyanate (RBITC) were utilized to post-treat the carbon dots, endowing the carbon dots with two emission bands located at 515 nm and 575 nm (excited by 488 nm), respectively. The author's demonstration152 indicated that, as the pH value changed from 5 to 9, which covered the full range of the physiological environment of the human body, the intensity of the 515 nm emission band increased substantially, while that for 575 nm only increased a little, thus it was clearly shown that the carbon dots exhibited a linear ratiometric response to the pH input (see Fig. 17a and b). In addition, the authors also demonstrated152 that both the intracellular genres (such as ions, saccharides, and proteins) and redox substances (e.g., H2O2, ClO, reduced glutathione (GSH), GSH inhibitor (NEM), and GSH precursor (NAC)) have a neglectful effect on the carbon dots ratiometric response, which verified the selectivity of the carbon dots-based ratiometric sensing.
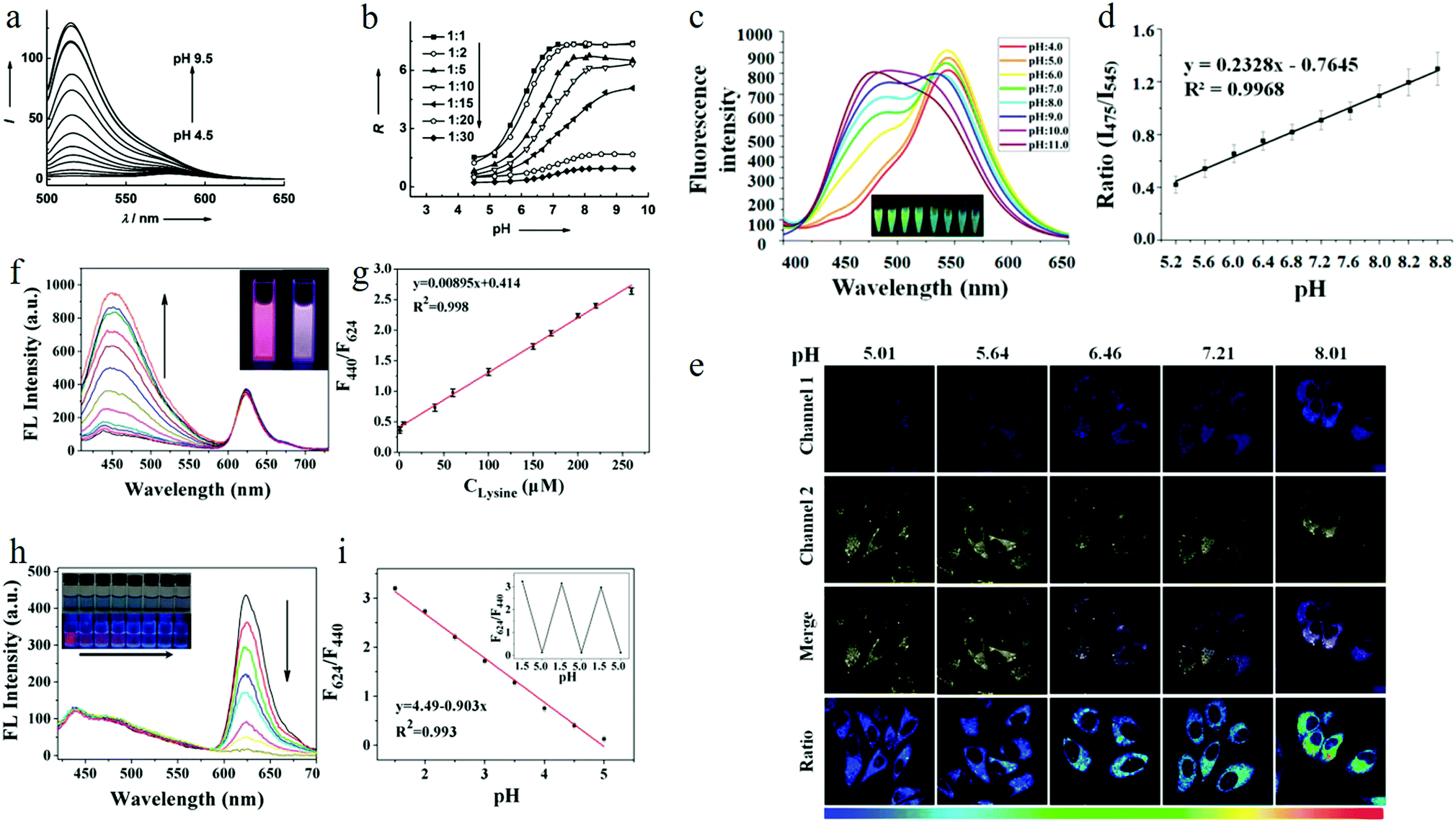 | ||
| Fig. 17 (a) PL emission spectra of carbon dots in the pH range of 4.5–9. (b) The relationship between the intensity ratio (I515/I575) R and pH values. The carbon dots were surface passivated by FITC and RBITC with different molar ratios from 1:1 to 1:30. (c) PL spectra of carbon dots in PBS buffer solution with different pH values (stimulated by 380 nm); inset shows the corresponding color change under 365 nm UV. (d) Linear relationship between the intensity ratio and pH value. (e) Confocal fluorescence images of HeLa cells. Channel 1: 465–495 nm band pass filter. Channel 2: 535–565 nm band pass filter. The ratio of the two band emission was obtained by imaging processing. All the samples were stimulated by 380 nm. (f) PL emission spectra of carbon dots for different concentrations of lysine: from 0.5–260 μM in 100 mM buffer solution with the pH value of 2. (g) Linearly relationship between the intensity ratio (F440/F624) and the lysine concentration. (h) PL emission spectra of carbon dots for buffer solutions at different pH values (in the range of 1.5–5.0); inset shows photographs of the buffer solutions under white light (upper) and UV light (lower) excitation, respectively, left to right: pH value 1.5 to 5.0. (i) The linearly relationship between the intensity ratio (F624/F440) and the pH values. (a and b) Reproduced with permission form ref. 152. Copyright 2012, Wiley-VCH. (c–e) Reproduced with permission from ref. 154. Copyright 2016, American Chemical Society. (f–i) Reproduced with permission from ref. 145. Copyright 2017, American Chemical Society. | ||
Small molecule linked carbon dot ratiometric sensors possess high accuracy, although their ease of photobleaching, and the leaching of small molecules from the carbon dots limit their practical applications. To overcome these issues, a label-free carbon dots ratiometric pH sensor was proposed by Huang's group154 using carbon dots synthesized by the hydrothermal treatment of citric acid and basic fuchsin, where the fuchsin had a pH-sensitive property. The carbon dots displayed two emission peaks located at 475 nm and 545 nm, respectively, when excited by 380 nm. The pH-dependent experiments uncovered that, as the pH values increased from 4.0 to 11.0, the intensity of the 475 nm emission band continuously increased, while the intensity of the 545 nm emission band manifested a different performance which slightly increased from acid to neutral condition, while further increasing the pH from 7 (neutral condition) to 14 (alkaline condition) resulted in the decrease of the 545 nm emission band, as shown in Fig. 17c. The intensity ratio of 457 nm and 545 nm exhibited a linear relationship to the pH values in the range of 5.1 to 8.8, as can be seen in Fig. 17d,154 which implied that the carbon dots could be used as a ratiometric pH sensor. The reproducibility experiments demonstrated that carbon dots emission could totally recover between the strong acidic environment (pH = 4.0) and strong alkaline condition (pH = 11.0). In addition, the authors also substantiated154 that both the metal ions (such as Fe3+, Cu2+, Hg2+), biothiol (e.g., GSH, Cys), and reactive oxygen species (ROS) (e.g., H2O2, HClO) had negligible influence on the ratiometric pH sensing, thus verifying the selectivity of the sensing. Living HeLa cells were employed by the authors154 to inspect the function and applicability of the carbon dots sensor. The HeLa cells were treated by various buffered solutions with different pH values (pH = 5.01, 5.64, 6.46, 7.21, and 8.01). The results are displayed in Fig. 17e. As the pH values increased from 5.0 to 8.0, the blue emission corresponding to 475 nm band (465–495 nm, channel 1) increased, but the 545 nm yellow light (535–565 nm, channel 2) showed a slight decrease. The merged graphs of the blue emission and yellow emission bands exhibited a pronounced color change154 as the pH value continued to increase, which could be clearly seen by the naked eye. The fluorescence ratio imaging was accomplished by using imaging processing, as illustrated in Fig. 17e. The calibrated ratio-pH function was also obtained154 by calculating the average PL intensity of the blue channel and yellow channel. Later, Song et al.145 also proposed another kind of dual emissive carbon dots (synthesized by the hydrothermal treatment of o-phenylenediamine and phosphoric acid). The carbon dots exhibited two emission bands at 400 nm and 624 nm, where the 400 nm blue emission could be enhanced by lysine, while the 624 nm red emission band was inert to lysine. However, when the input was changed to different pH values, the results were different: as the pH value increased from 1.5–5.0, the 400 nm blue band became stable, while the 624 red band emission decreased. In both cases, the intensity ratio of the two emission bands manifested a linear relationship to the input items (lysine or pH), thus the authors advised145 that the carbon dots could be used to sense both lysine (linear performance in the range of 0.5–260 μM with a detection limit of 98 nM) and the pH value (linear performance in the range of 1.5–5) in a ratiometric manner, as seen in Fig. 17f–i.
4.6 Logic gates
The development of molecular devices can help to achieve multifunctional “lab-on-a-molecule”,155 and triggers the applications of molecular information processing, communication, molecular based biosensing, etc.156 All of these intriguing applications are based on basic logic operations, and the PL signal is regarded as a desirable logic gate readout due to its fast response, increased signal-to-noise ratio, and high resolution. All these good merits of fluorescent carbon dots make them an effective competitor in logic gates.157–162The prerequisite to achieve a carbon dots chemical logic gate is to generate a variable PL output, which could be realized by heavy metal ions causing PL quenching. A carbon dots aqueous solution synthesized by the thermal pyrolysis of lemon juice was adopted by Pal's group,15 and displayed two absorption bands located at 268 and 320 nm, respectively. The authors15 also discovered that increasing the pH value to 10.2 resulted in an enhanced 268 nm absorption and the appearance of a new broad band at longer wavelength. The pH-dependent PL of carbon dots was also evaluated by the authors.15 Exited by 360 nm, the PL intensity increased when the pH value increased from 1 to 7, with the PL peak nearly remaining the same (the absorption difference of 360 nm was eliminated by the authors15via normalizing the PL intensity to the relative absorption). On the contrary, continuously increasing the pH value from 7 to 10 led to a decrease of the PL intensity, along with an intriguing PL spectral red-shift. The authors15 also discovered that different excitation wavelengths in the range of 320–380 nm resulted in the same emission peak located at 445 nm for both acidic and basic conditions. However, when the excitation wavelength was increased to 400 nm, the PL spectra of the carbon dots in alkaline conditions showed a red-shift with the peak shifted to 465 nm, while nearly no PL signal could be observed from acidic carbon dots. Both the pH-dependent absorption and emission spectra were completely reversible, which implied that neither the acidic or basic conditions degenerated the carbon dots.
Based on the opposite zeta potential values measured from acidic (ξ = 0.13 mV for pH = 2.6) and basic (ξ = −7.8 mV for pH = 9) carbon dots, the authors15 attributed the pH-dependent spectral shift and intensity variation to the pH-triggered protonation and deprotonation. FTIR spectroscopy analysis implied that two stretching bonds at the carbon dots surface gradually disappeared as the pH value was increased from 2.5 to 10. One of the bonds was the CO bond of the carboxylic acid with infrared absorption at 1725 cm−1, along with the appearance of two new carboxylate categories at 1575 and 1390 cm−1. The other disappeared functional group was the C–OH stretching of the phenolic category with absorption at 1220 cm−1, which was converted to a phenolate bond at 1278 cm−1.
Generally, carboxylic functional groups possess a lower pKa value in the range of 4–5 due to their relative acidic properties, while more basic phenolic functional groups hold a higher pKa value of about 8–10. Considering that there was only one inflection point for the pH-dependent absorption density and PL spectra shift, the authors15 assumed that only one functional group was involved in the emission of carbon dots. The fact that both the pH-dependent absorption optical density (@400 nm) and the PL peaks manifested a sigmoidal pattern with a similar inflection point at pH = 7.9 led the authors to advocate that it was the phenolic functional groups that contributed to the carbon dots PL red-shift in high pH conditions. More specifically, the phenolic group was strongly coupled with the emissive center located at the surface of the carbon dots, which could modify the energy level of the emissive states. Therefore, Pal et al.15 suggested the following pH-dependent PL mechanism: the high pH value condition leads to the deprotonation equilibrium of phenol ↔ phenolate (where coupling with phenol and phenolate will result in different energy states), namely the transformation between phenol and phenolate, which can tune the PL emission of carbon dots. The carboxylic functional groups cannot affect the PL properties of carbon dots because there is no coupling between the emission center and carboxylic groups, even though the carboxylic ↔ carboxylate deprotonation equilibrium also exists. Thus the authors proposed15 that a tunable PL signal of carbon dots could be achieved by properly controlling the surrounding environment, e.g., the pH and surface chemical modification, e.g., the presence of Hg2+, which can offer the opportunity to produce carbon dots-based chemical logic gates.
As observed by the authors,15 in an acidic environment, e.g., pH = 2.7, both the carboxylic (even if the carboxylic group was not directly coupled with emissive states, it could also affect the PL quenching process by means of electrostatic interaction) and hydroxyl categories were absolutely protonated, thus the Hg2+ could not quench the PL of carbon dots. As a result, the authors suggested15 that the YES logic gate could be realized by regarding the Hg2+ as the input and the PL intensity as the output, as can be seen in the left part of Fig. 18. Increasing the pH value to 8.9 (basic environment) results in the deprotonation of both the carboxylic and phenolic functional groups, and consequently, the surface of the carbon dots is negatively charged, which could interact with the Hg2+, leading to PL quenching. Therefore, the NOT gate was realized by Pal et al.15 in the condition of a basic environment when considering Hg2+ as the input and the PL intensity as the output (middle part of Fig. 18). The authors also discovered that the quenched PL could be recovered by adding extra glutathione (GSH), as the thiol group in glutathione could combine with a Hg2+ ion and release the carbon dots from the Hg2+ ion. But it should be noted that pure glutathione could not affect the PL properties of carbon dots. Therefore, the implication logic gate was fulfilled by Choudhury and co-workers15 by using the Hg2+ ion and glutathione as the chemical input and the PL intensity as the output, as seen in the right part of Fig. 18. If the pH, Hg2+, and GSH were all viewed as the chemical input and the PL intensity as the readout, a more complex logic gate could also be demonstrated.
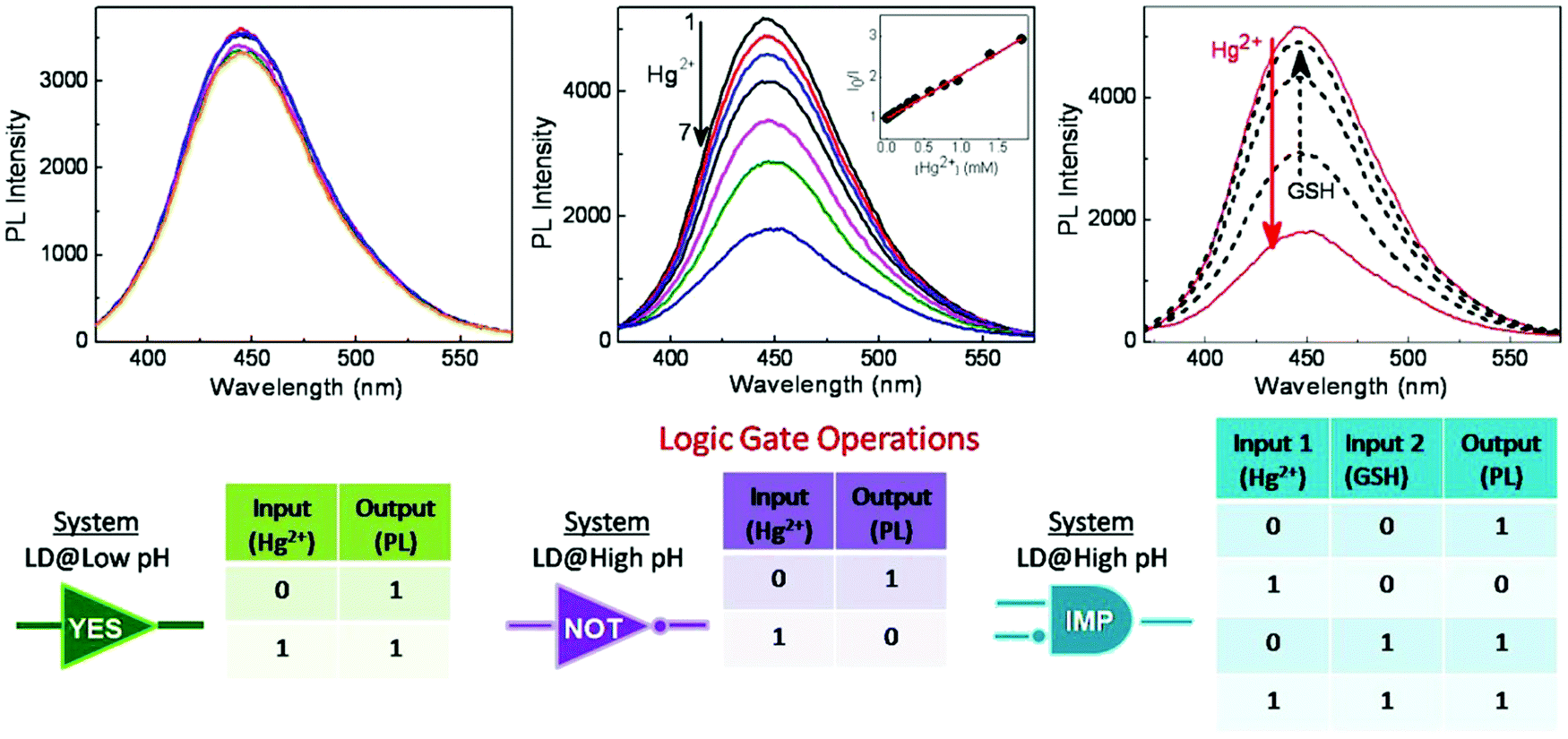 | ||
| Fig. 18 PL spectra, corresponding logic gate, and Truth tables for “YES,” “NOT,” and “IMP” logic gates, respectively. Reproduced with permission from ref. 15. Copyright 2017, American Chemical Society. | ||
Carbon dots synthesized by an induction heater (500 W, 100 °C) of citric acid and ethylenediamine manifested pH-stable PL in the range of 4–11; however, they could be also used to realize a chemical logic gate.157 Chattopadhyay's group uncovered that the PL intensity (excited by 365 nm, detected at 433 nm) of the carbon dots could be quenched by picric acid via static quenching. An additional mixture of acetonitrile, ethyl acetate, and sodium chloride could transfer the picric acid to the organic phase, thus recovering the PL of the carbon dots. Alternatively, the authors also discovered157 that carbon dots could be quenched by Fe3+ through dynamic quenching, accomplished by ultrafast carrier transfer between the carbon dots and Fe3+ ions. A following treatment by ascorbic acid or cysteine could reverse the PL quenching, whereby the ascorbic acid could reduce the Fe3+ ion to Fe2+ ion and cysteine could yield a complex with Fe3+, both of which could consume the Fe3+ ion and release carbon dots emission. Diverse and multifunctional logic gates were reported by the authors157 using the variable PL as the output: the NOT logic gate was realized by using the picric acid (or Fe3+ ion) as the input (input logic 1); for the Fe3+-ion-treated carbon dots system; whereby the input of ascorbic acid (input logic 1) and cysteine (input logic 1) could result in the OR logic, as only when both of the inputs were 0 (i.e., both the ascorbic acid and cysteine were absent), was the output 0 (namely the PL intensity was low). By properly designing the system and chemical component input, even higher complex logical systems were demonstrated by Sk and co-workers.157
4.7 Carbon dots chiral photonics
Chirality plays an important role in various practical application fields, such as chiral drug recognition, chiral molecular biology, and chiral chemistry.163–165 Thus the combination of carbon dots with chirality can give rise to intriguing chiral optics based on carbon dots, as first proposed by M. Vázquez-Nakagawa et al.166 In their pioneering work, the carbon dots were synthesized by means of the chemical exfoliation (concentrated sulfuric acid and nitric acid) of graphite, and thionyl chloride was employed to convert the carbon dots surface carboxylic acid groups into acid chlorides. The simultaneous reaction between the acid chlorides and the (R) or (S)-2-phenyl-1-propanol (chiral molecular) resulted in enantiomerically pure esters and the formation of chiral carbon dots, as shown in Fig. 19a. The formation166 of the enantiomerically esters and chiral carbon dots was confirmed by 13C-NMR and FTIR spectroscopy, where the appearance of 120–150 ppm peak in the 13C-NMR indicated the existence of the phenyl substituents and the 1731–1727 cm−1 (CO stretch vibration), 1300 cm−1 (asymmetric stretching vibrations of C–O–C), and 1200 cm−1 (symmetric stretching vibrations of C–O–C) signals in the FTIR spectra implied the occurrence of ester groups.
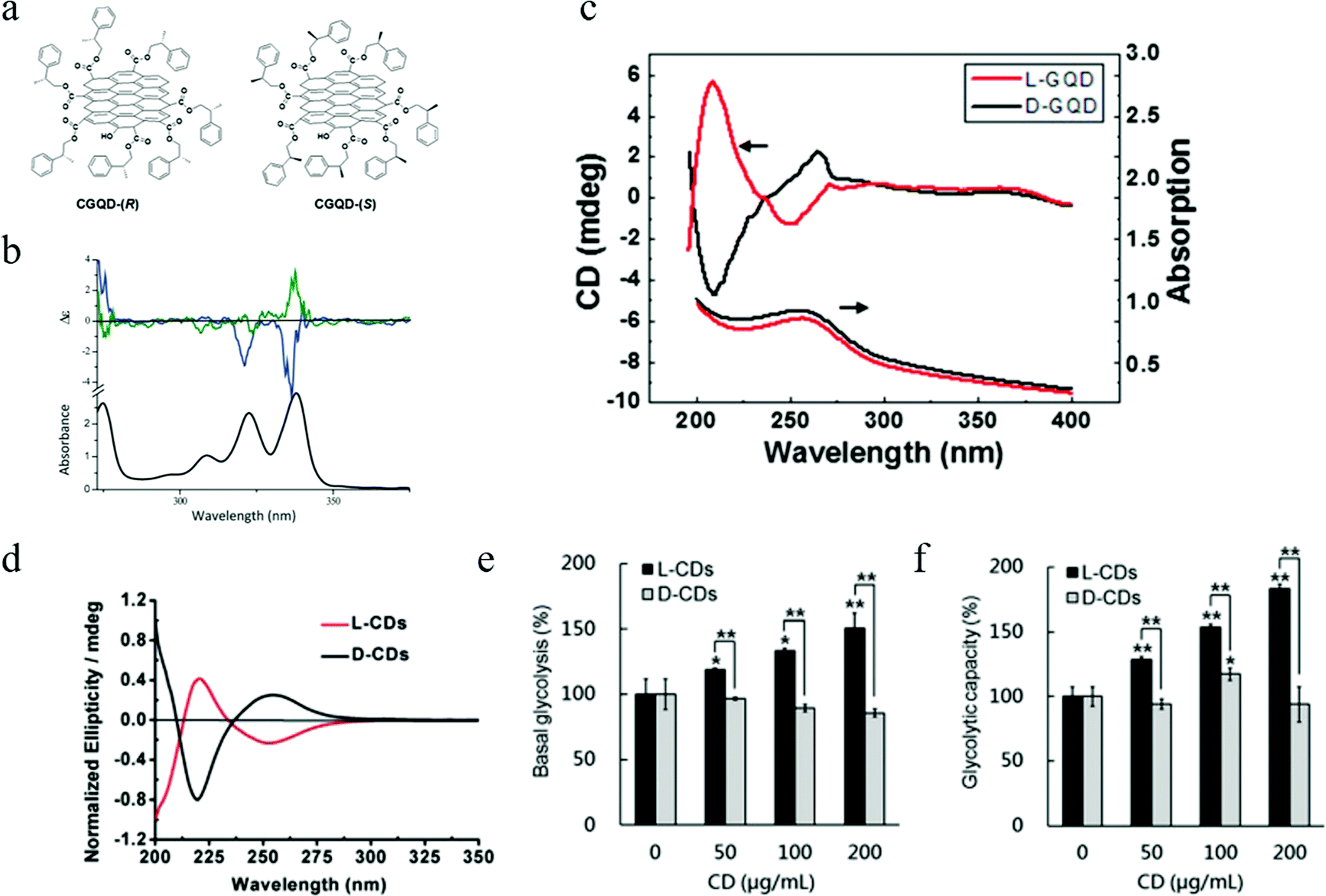 | ||
| Fig. 19 (a and b) Chiral carbon dots synthesized by Martin's group. (a) Structure illustration of chiral carbon dots obtained from the (R) or (S)-2-phenyl-1-propanol. (b) Upper: Circular dichroism spectra of chiral carbon dots-(R)/pyrene (blue) and chiral carbon dots-(S)/pyrene (green) in NMP solution. Bottom: UV-visible spectra of pyrene in NMP. (c) Circular dichroism spectra of carbon dots synthesized by Nicholas A. Kotov's group.170 (d) Circular dichroism spectra of chiral carbon dots synthesized by Guangjun Nie's group.171 (e and f) The tunability of L- and D-carbon dots to ECAR (extracellular acidification rate), where the basal glycolysis and the glycolytic capacity are the indicators of the ECAR. (a and b) Reproduced with permission from ref. 166. Copyright 2016, Royal Society of Chemistry. (c) Reproduced with permission from ref. 170. Copyright 2016, American Chemical Society. (d–f) Reproduced with permission from ref. 171. Copyright 2018, Royal Society of Chemistry. | ||
The chiral carbon dots N-methylpyrrolidone (NMP) solution was prepared for circular dichroism (CD) measurements. However, the authors discovered that166 the chiral moieties at the carbon dots surface did not have any obvious chiral absorption with wavelengths longer than 300 nm, thus the pyrene molecule was introduced to the chiral carbon dots solution to aid forming a supermolecular species, which could trigger the chiral property transfer from the chiral carbon dots to the supermolecular species by means of strong π–π stacking forces, as observed from other carbon nano-forms.167–169 Consequently, the pyrene/chiral carbon dots NMP solution displayed an obvious CD signal, as observed by the authors166 (Fig. 19b). More specifically, the S-chiral carbon dots exhibited a positive Cotton effect in the absorption range of 320–350 nm, while the R-chiral carbon dots showed the opposite negative Cotton effect in the similar absorption range. The authors attributed166 the small wavelength shift of the CD signal for the R- and S-chiral carbon dots to the non-identical surface functionalization and organization.
In addition to the (R) and (S)-2-phenyl-1-propanol chiral molecules, the L/D-cysteine chiral molecule was also adopted by Nozomu Suzuki et al.170 to synthesize chiral carbon dots. Briefly, the carbon dots were synthesized by the chemical exfoliation of carbon fiber in the presence of sulfuric acid and nitric acid, while a following carbodiimide/N-hydroxysuccinimide (EDC/NHS) cross-linking method linked the L/D-cysteine to the surface of the carbon dots by means of covalently bonding the amine group in L/D-cysteine with the carboxylic group in the carbon dots, thus resulting in chiral carbon dots, labeled as L-/D-carbon dots. Both the pristine and chiral carbon dots exhibited a similar size distribution in the range of 2–7 nm and a broadened XRD peak at 2θ = 25°. In addition, both the position and the relative intensity of the A1g D band Raman signal at 1355 cm−1 and the E2g G band Raman signal at 1590 cm−1 were the same for the pristine and chiral carbon dots. All of these indicated that the cysteine surface modification did not change the structure of the center graphene carbon sheets, as suggested by the authors.170 The appearance of the S–H bond (2390 cm−1) in the FTIR spectra implied that the cysteine ligands were linked to the carbon dots successfully, and the linkage was finished via covalent bonding, which was verified by the newly formed C–N bond (830 cm−1) in the chiral carbon dots.
CD spectral measurements demonstrated170 that both the L and D chiral carbon dots exhibited two chiral bands, as seen in Fig. 19c: one located at the high energy band of 210–220 nm, which was close to the chiral band of the pure cysteine at 209 nm, and another at a lower energy chiral band located at 250–265 nm, which had an opposite sign compared to the chirality of cysteine used to modify the carbon dots. In addition, the lower energy (located at 250–265 nm) chiral band manifested an overlap with the absorption peak at 260 nm and displayed a relatively high g factor of 1 × 10−4. By using the combined calculation methods of the semi-empirical ZINDO algorithm, MMFF algorithm, DFT+ZINDO algorithms, and by comparing with the XPS and CD spectral data, the authors suggested170 that the high energy chiral band originated from the hybridization of the atomic orbit of the cysteine chiral molecule and the molecular orbit of the small segment or edge groups of the graphene sheet. The hybridization was caused by the symmetry-breaking triggered perturbation of the graphene surface edge states due to the existence of the chiral ligands on the surface. Such a mechanism was termed as170 “local chirality induction.” However, the authors also pointed out that the local hybridization mechanism was not suitable for interpreting the longer wavelength chiral band (at 250–265 nm) because, based on the theory, for the larger diameter 10–15 nm carbon dots, no chiral band should be observed for wavelengths longer than 220 nm even though local hybridization exists on the surface of the large diameter carbon dots. On the contrary, the authors170 attributed the longer wavelength chiral band to the change of the conformation structure of the carbon dots themselves. The cysteine ligand was linked to the carbon dots by means of noncovalent intermolecular interaction. Intermolecular interaction between the chiral molecule and the large carbon dots results in a buckling deformation, and as a result, the equilibrium conformation of the carbon dots sustains a strong twist; more specifically, L-cysteine and D-cysteine result in a right-hand and left-hand twist, respectively, corresponding to the different twisted excitonic states. The twisted carbon dots could facilitate the electrons and holes to transfer to one of the excitonic states, resulting in the longer wavelength chiral excitonic band. It's clear that the chirality (i.e., rotatory handedness) of the carbon dots was opposite to chirality of the modification chirality molecular, consistent with the authors’ experiment observations.170
In addition to surface chiral molecular modification,172,173 chiral carbon dots could also be directly prepared by the chiral precursor guanosine 5′-monophosphate, (5′-GMP).174 Briefly, the chiral molecular disodium salt of guanosine 5′-monophosphate, Na2(5′-GMP) was heated by microwave at 160 °C for 5 min to synthesize chiral carbon dots. (HR)TEM analysis indicated that the carbon dots had an average diameter of less than 5 nm, with a highly disordered amorphous carbon core. The amorphous carbon dots were also confirmed by Raman spectroscopy due to the absence of D band and G band signals for the carbon dots. The synthesized chiral carbon dots displayed several chiral peaks, including three positive peaks at 218, 270, and 300 nm, and two negative chiral peaks at 230 nm and 260 nm. The similarity of the CD spectra of carbon dots and the 5′-GMP indicated that the chiral structure was converted during the carbon dots synthesis, as suggested by the authors.174
The most exciting news for chiral carbon dots relates to the recent achievements in the chiral control of bioreactions. Xin et al.175 demonstrated that D-glutamic acid (D-Glu)-carbon dots could drastically destroy both the Gram-negative and Gram-positive bacteria cell wall, resulting in the death of bacteria, while their counterpart L-glutamic acid (L-Glu)-carbon dots manifested negligible effect to bacteria, which suggested that chiral carbon dots could be used as chiral antimicrobial nanoagents. L- and D-cysteine-based carbon dots have also been utilized to control the enzyme's chiral activity,176e.g., L-cysteine carbon dots increased the laccase enzyme activity by 20.2%, while D-cysteine carbon dots exhibited the opposite behavior, suppressing the enzyme activity by 10.4%. Very recently, Li et al.171 even discovered that L/D-cysteine-based carbon dots could even affect the cellular energy metabolism. In the future, we believe that more chiral carbon dots-based applications will be explored,177 and chiral carbon dots will become a new but fascinating field due to their huge potential practical applications.
5 Summary and prospects
Carbon dots as a new kind of carbon-based fluorescent material have triggered numerous investigations. Various breakthroughs have been obtained from their fundamental optical-physical properties to potential applications. It's believed that carbon dots will play a more important role in various fields in the future. Considering their excellent biocompatibility, it's reasonable to predict that carbon dots will become an important platform for bioapplications. Thus, exploring high quantum yield NIR emission carbon dots is urgent for bioimaging, as near IR light manifests a high tissue penetration depth and deceased autofluorescent background. Carbon dots linked with proper moieties and therapeutic drugs offer the potential to identify the challenge of cancer heterogeneity and adaptation. However, as pointed out by Gao et al.,178 before the practical clinical applications of carbon dots, researchers should provide more evidence that carbon dots-based drug delivery is better than monoclonal antibody-bonded anticancer drugs179 and biodegradable polymer-linked drugs.180 Consequently, the next step for carbon-based nano-therapy is to explore carbon dots that can hugely reduce the non-specific uptake by normal cells, while increasing the specific targeting capability, especially enhancing the cancer cell specific targeting ability, and elongate blood circulation to increase the therapeutic effect, i.e., to explore carbon dots targeted nano-medicine to achieve cancer-targeted therapy. Such carbon dots-based targeted cancer therapy can vastly increase the therapeutic effect and decrease the side effects, and thus may trigger a revolution in the diagnosis, treatment, and prevention of cancer.178Although carbon dots-based optoelectronic applications have been demonstrated, e.g., in LEDs, their external quantum efficiency and maximum luminance are still low, so further improvement of the efficiency is needed, which requires higher quantum yield carbon dots and proper device structure design. Both of these are related to a clear understanding of the optical-physical properties of carbon dots. As we discussed above, researchers have paid much attention to try to understand the emission essence of carbon dots, and various breakthroughs have been achieved in recent years, but there are still some debates and challenges remaining. Even through different emission models proposed by researchers may contribute to different kinds of carbon dots emission, the fine structure (e.g., for self-trapped exciton emission,84 the bond geometry configuration, bond length, bond angle, and local potential distribution) of the emission center is still a big issue. Thus more theoretical and experiment methods need to be employed to uncover the mystery of carbon dots emission.
The carbon dots-based sensing platform may play a more important role in the future, especially for biosensing. The discovery of ratiometric sensing and the demonstration of in vitro pH mapping imply a high potential for practical sensing applications. However, to compete with commercial sensors, carbon dots-based sensing should further improve the sensitivity, selectivity, and robustness. Also, to achieve in vivo sensing and mapping will also be a part of the future development of carbon dots-based biosensing. Sun's group130 demonstrated the potential to apply carbon dots in a microfluidic platform. Considering the widespread applications, especially the bioapplications of such a microfluidic platform, we believe that bright fluorescent carbon dots will light up the micro-channels and facilitate more applications based on microfluidic systems, such as bioassays, drug delivery, and biosystem mimics.
We want to highlight the discovery of chiral carbon dots. One very exciting piece of recent news is that chiral carbon dots-based applications have started to appear; for example, chirality controlled enzyme activity and cellular energy metabolism.171,175,176 We predict that more and more chiral carbon dots-based applications will appear in biology, catalysis, optic electronic, and sensing. However, at the current stage, the chiral carbon dots only hold UV range CD signals due to the chiral molecule; thus one of the future directions is to explore chiral carbon dots that possess visible and even near IR range CD signals. Another big issue for chiral carbon dots is the circular polarized emission. Circular polarization emission has wide applications, such as in chiral sensing, chiral drug discovery, flat panel display, and quantum communications.163,181–183 While it's still difficult to achieve circular polarized emission from carbon dots, which hugely hinders the applications of chiral carbon dots, future research efforts should be paid to address this issue.
In summary, researchers have achieved gratifying progress with carbon dots in both understanding their fundamental aspects and in their practical applications in recent years, even though there still exist some debates and challenges to overcome. We believe that this kind of novel nanomaterial holds a bright future. Someday, carbon dots may replace some currently used fluorescent materials in various fields, such as bioimaging, drug delivery, nanomedicine, biosensing, and LEDs. Besides these traditional fields, carbon dots may find a place in some current urgent and pressing fields such as green chemistry and clean energy production.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ministry of Education Singapore through the Academic Research Fund under Projects RG105/16 and RG189/17.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef PubMed.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang and H. Wang, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef PubMed.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef PubMed.
- Y. Wang, X. Li, X. Zhao, L. Xiao, H. Zeng and H. Sun, Nano Lett., 2015, 16, 448–453 CrossRef PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165 CrossRef PubMed.
- J. Geys, A. Nemmar, E. Verbeken, E. Smolders, M. Ratoi, M. F. Hoylaerts, B. Nemery and P. H. Hoet, Environ. Health Perspect., 2008, 116, 1607 CrossRef PubMed.
- S.-T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J.-H. Liu, Y. Liu and M. Chen, J. Phys. Chem. C, 2009, 113, 18110–18114 CrossRef PubMed.
- X. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo, J. Guo, K. Tackett, P. Anilkumar, G. Liu and Q. Quan, ACS Nano, 2013, 7, 5684–5693 CrossRef PubMed.
- M. Fu, F. Ehrat, Y. Wang, K. Z. Milowska, C. Reckmeier, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2015, 15, 6030–6035 CrossRef PubMed.
- R. Schmidt, C. Krasselt, C. Göhler and C. von Borczyskowski, ACS Nano, 2014, 8, 3506–3521 CrossRef PubMed.
- S. K. Das, Y. Liu, S. Yeom, D. Y. Kim and C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef PubMed.
- Y. Choi, B. Kang, J. Lee, S. Kim, G. T. Kim, H. Kang, B. R. Lee, H. Kim, S.-H. Shim and G. Lee, Chem. Mater., 2016, 28, 6840–6847 CrossRef.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- S. Dutta Choudhury, J. M. Chethodil, P. M. Gharat and H. Pal, J. Phys. Lett., 2017, 8, 1389–1395 Search PubMed.
- B. Zhu, S. Sun, Y. Wang, S. Deng, G. Qian, M. Wang and A. Hu, J. Mater. Chem. C, 2013, 1, 580–586 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan and G. Gao, Nano Lett., 2012, 12, 844–849 CrossRef PubMed.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef PubMed.
- S.-L. Hu, K.-Y. Niu, J. Sun, J. Yang, N.-Q. Zhao and X.-W. Du, J. Mater. Chem., 2009, 19, 484–488 RSC.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker and T. Clark, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef PubMed.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., 2013, 125, 4045–4049 CrossRef.
- L. Tang, R. Ji, X. Li, G. Bai, C. P. Liu, J. Hao, J. Lin, H. Jiang, K. S. Teng and Z. Yang, ACS Nano, 2014, 8, 6312–6320 CrossRef PubMed.
- L. Bao, C. Liu, Z. L. Zhang and D. W. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef PubMed.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2015, 10, 484–491 CrossRef PubMed.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng and J. Hao, ACS Nano, 2012, 6, 5102–5110 CrossRef PubMed.
- X. Meng, Q. Chang, C. Xue, J. Yang and S. Hu, Chem. Commun., 2017, 53, 3074–3077 RSC.
- D. Zhou, D. Li, P. Jing, Y. Zhai, D. Shen, S. Qu and A. L. Rogach, Chem. Mater., 2017, 29, 1779–1787 CrossRef.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774–3776 RSC.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca and D. Murray, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef PubMed.
- J. Zhou, C. Booker, R. Li, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef PubMed.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef PubMed.
- Q.-L. Zhao, Z.-L. Zhang, B.-H. Huang, J. Peng, M. Zhang and D.-W. Pang, Chem. Commun., 2008, 5116–5118 RSC.
- L. Bao, Z. L. Zhang, Z. Q. Tian, L. Zhang, C. Liu, Y. Lin, B. Qi and D. W. Pang, Adv. Mater., 2011, 23, 5801–5806 CrossRef PubMed.
- Y. Li, Y. Hu, Y. Zhao, G. Shi, L. Deng, Y. Hou and L. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef PubMed.
- R. Ye, C. Xiang, J. Lin, Z. Peng, K. Huang, Z. Yan, N. P. Cook, E. L. Samuel, C.-C. Hwang and G. Ruan, Nat. Commun., 2013, 4, 2943 CrossRef PubMed.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455–458 CrossRef PubMed.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2011, 134, 747–750 CrossRef PubMed.
- F. Wang, M. Kreiter, B. He, S. Pang and C.-y. Liu, Chem. Commun., 2010, 46, 3309–3311 RSC.
- X. Guo, C.-F. Wang, Z.-Y. Yu, L. Chen and S. Chen, Chem. Commun., 2012, 48, 2692–2694 RSC.
- S.-S. Liu, C.-F. Wang, C.-X. Li, J. Wang, L.-H. Mao and S. Chen, J. Mater. Chem. C, 2014, 2, 6477–6483 RSC.
- H. G. Baldovi, S. Valencia, M. Alvaro, A. M. Asiri and H. Garcia, Nanoscale, 2015, 7, 1744–1752 RSC.
- S. Qu, D. Zhou, D. Li, W. Ji, P. Jing, D. Han, L. Liu, H. Zeng and D. Shen, Adv. Mater., 2016, 28, 3516–3521 CrossRef PubMed.
- D. Pan, J. Zhang, Z. Li, C. Wu, X. Yan and M. Wu, Chem. Commun., 2010, 46, 3681–3683 RSC.
- C. Zhu, M. Yan, X. Shi, J. Fan and H. Bi, RSC Adv., 2016, 6, 38470–38474 RSC.
- X. Teng, C. Ma, C. Ge, M. Yan, J. Yang, Y. Zhang, P. C. Morais and H. Bi, J. Mater. Chem. B, 2014, 2, 4631–4639 RSC.
- J. Chen, X. Zhang, Y. Zhang, W. Wang, S. Li, Y. Wang, M. Hu, L. Liu and H. Bi, Langmuir, 2017, 33, 10259–10270 CrossRef PubMed.
- Z.-C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X.-H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- L. Wang and H. S. Zhou, Anal. Chem., 2014, 86, 8902–8905 CrossRef PubMed.
- P.-C. Hsu and H.-T. Chang, Chem. Commun., 2012, 48, 3984–3986 RSC.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef PubMed.
- K. i. Holá, M. Sudolská, S. Kalytchuk, D. Nachtigallová, A. L. Rogach, M. Otyepka and R. Zbořil, ACS Nano, 2017, 11, 12402–12410 CrossRef PubMed.
- L. Wang, Y. Wang, T. Xu, H. Liao, C. Yao, Y. Liu, Z. Li, Z. Chen, D. Pan and L. Sun, Nat. Commun., 2014, 5, 5357 CrossRef PubMed.
- G. Ren, M. Tang, F. Chai and H. Wu, Eur. J. Inorg. Chem., 2018, 153–158 CrossRef.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- X. Zhai, P. Zhang, C. Liu, T. Bai, W. Li, L. Dai and W. Liu, Chem. Commun., 2012, 48, 7955–7957 RSC.
- S. Chandra, P. Das, S. Bag, D. Laha and P. Pramanik, Nanoscale, 2011, 3, 1533–1540 RSC.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., 2012, 124, 12381–12384 CrossRef.
- J. Jiang, Y. He, S. Li and H. Cui, Chem. Commun., 2012, 48, 9634–9636 RSC.
- S. Sun, L. Zhang, K. Jiang, A. Wu and H. Lin, Chem. Mater., 2016, 28, 8659–8668 CrossRef.
- R. Liu, D. Wu, S. Liu, K. Koynov, W. Knoll and Q. Li, Angew. Chem., 2009, 121, 4668–4671 CrossRef.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539–4541 CrossRef.
- B. Wang, Y. Mu, H. Yin, Z. Yang, Y. Shi and J. Li, Nanoscale, 2018, 10, 10650–10656 RSC.
- Z. G. Gu, D. J. Li, C. Zheng, Y. Kang, C. Wöll and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 6853–6858 CrossRef PubMed.
- H. Xu, S. Zhou, L. Xiao, H. Wang, S. Li and Q. Yuan, J. Mater. Chem. C, 2015, 3, 291–297 RSC.
- A. J. Amali, H. Hoshino, C. Wu, M. Ando and Q. Xu, Chem. – Eur. J., 2014, 20, 8279–8282 CrossRef PubMed.
- Y. Yang, D. Wu, S. Han, P. Hu and R. Liu, Chem. Commun., 2013, 49, 4920–4922 RSC.
- T. Huang, T. Wu, Z. Zhu, L. Zhao, H. Ci, X. Gao, K. Liu, J. Zhao, J. Huang and Y. Yan, Chem. Commun., 2018, 54, 5960–5963 RSC.
- N. Liu, J. Liu, W. Kong, H. Li, H. Huang, Y. Liu and Z. Kang, J. Mater. Chem. B, 2014, 2, 5768–5774 RSC.
- C. T. Chien, S. S. Li, W. J. Lai, Y. C. Yeh, H. A. Chen, I. S. Chen, L. C. Chen, K. H. Chen, T. Nemoto and S. Isoda, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef PubMed.
- L. Wang, S.-J. Zhu, H.-Y. Wang, S.-N. Qu, Y.-L. Zhang, J.-H. Zhang, Q.-D. Chen, H.-L. Xu, W. Han and B. Yang, ACS Nano, 2014, 8, 2541–2547 CrossRef PubMed.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef PubMed.
- Y. Chen, H. Lian, Y. Wei, X. He, Y. Chen, B. Wang, Q. Zeng and J. Lin, Nanoscale, 2018, 10, 6734–6743 RSC.
- S. Zhu, J. Zhang, X. Liu, B. Li, X. Wang, S. Tang, Q. Meng, Y. Li, C. Shi and R. Hu, RSC Adv., 2012, 2, 2717–2720 RSC.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef PubMed.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Nano Lett., 2015, 15, 8300–8305 CrossRef PubMed.
- E. v. Lippert, Z. Elektrochem., 1957, 61, 962–975 Search PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef.
- N. Nemkovich, A. Rubinov and V. Tomin, Top. Fluoresc. Spectrosc., 1991, 2, 367 Search PubMed.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 1999 Search PubMed.
- A. Sciortino, E. Marino, B. v. Dam, P. Schall, M. Cannas and F. Messina, J. Phys. Lett., 2016, 7, 3419–3423 Search PubMed.
- C. J. Reckmeier, Y. Wang, R. Zboril and A. L. Rogach, J. Phys. Chem. C, 2016, 120, 10591–10604 CrossRef.
- L. Xiao, Y. Wang, Y. Huang, T. Wong and H. Sun, Nanoscale, 2017, 9, 12637–12646 RSC.
- B. P. Zakharchenya, D. N. Mirlin, V. Perel' and I. Reshina, Phys.-Usp., 1982, 25, 143–166 CrossRef.
- V. Dymnikov, D. Mirlin, L. Nikitin, V. Perel, I. Reshina and V. Sapega, Zh. Eksp. Teor. Fiz., 1981, 80, 1766–1778 Search PubMed.
- V. Dymnikov, M. D'yakonov and N. Perel, Sov. Phys. - JETP, 1976, 44, 1252 Search PubMed.
- H. Koyama, T. Oguro and N. Koshida, Appl. Phys. Lett., 1993, 62, 3177–3179 CrossRef.
- S. Kazaoui, R. Ross and N. Minami, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R11665 CrossRef.
- C. Itoh, K. Tanimura and N. Itoh, J. Phys. C: Solid State Phys., 1988, 21, 4693 CrossRef.
- M. Matus, H. Kuzmany and E. Sohmen, Phys. Rev. Lett., 1992, 68, 2822 CrossRef PubMed.
- D. Emin, J. Non-Cryst. Solids, 1980, 35, 969–973 CrossRef.
- S. Ghosh, A. M. Chizhik, N. Karedla, M. O. Dekaliuk, I. Gregor, H. Schuhmann, M. Seibt, K. Bodensiek, I. A. Schaap and O. Schulz, Nano Lett., 2014, 14, 5656–5661 CrossRef PubMed.
- A. I. Chizhik, A. M. Chizhik, D. Khoptyar, S. Bär and A. J. Meixner, Nano Lett., 2011, 11, 1131–1135 CrossRef PubMed.
- T. Schmidt, A. I. Chizhik, A. M. Chizhik, K. Potrick, A. J. Meixner and F. Huisken, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125302 CrossRef.
- A. I. Chizhik, A. M. Chizhik, A. Huss, R. Jäger and A. J. Meixner, J. Phys. Lett., 2011, 2, 2152–2157 Search PubMed.
- A. P. Demchenko and M. O. Dekaliuk, Nanoscale, 2016, 8, 14057–14069 RSC.
- M. Kasha, H. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 Search PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- U. Rösch, S. Yao, R. Wortmann and F. Würthner, Angew. Chem., 2006, 118, 7184–7188 CrossRef.
- J. Gierschner, L. Lüer, B. a. Milián-Medina, D. Oelkrug and H.-J. Egelhaaf, J. Phys. Lett., 2013, 4, 2686–2697 Search PubMed.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, J. Phys. Lett., 2016, 7, 3695–3702 Search PubMed.
- A. Sharma, T. Gadly, S. Neogy, S. K. Ghosh and M. Kumbhakar, J. Phys. Lett., 2017, 8, 1044–1052 Search PubMed.
- J. Zhang, L. Yang, Y. Yuan, J. Jiang and S.-H. Yu, Chem. Mater., 2016, 28, 4367–4374 CrossRef.
- Y. Malyukin, O. Viagin, P. Maksimchuk, M. Dekaliuk and A. Demchenko, Nanoscale, 2018, 10, 9320–9328 RSC.
- Y. Xu, Y.-H. Li, Y. Wang, J.-L. Cui, X.-B. Yin, X.-W. He and Y.-K. Zhang, Analyst, 2014, 139, 5134–5139 RSC.
- S.-T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y.-P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef PubMed.
- H. Zhang, B. Zhang, C. Di, M. C. Ali, J. Chen, Z. Li, J. Si, H. Zhang and H. Qiu, Nanoscale, 2018, 10, 5342–5349 RSC.
- F. Jia, S. Lv and S. Xu, RSC Adv., 2017, 7, 53532–53536 RSC.
- Y. Zhang, X. Liu, Y. Fan, X. Guo, L. Zhou, Y. Lv and J. Lin, Nanoscale, 2016, 8, 15281–15287 RSC.
- L. Pan, S. Sun, L. Zhang, K. Jiang and H. Lin, Nanoscale, 2016, 8, 17350–17356 RSC.
- Q. Liu, B. Guo, Z. Rao, B. Zhang and J. R. Gong, Nano Lett., 2013, 13, 2436–2441 CrossRef PubMed.
- J. Liu, D. Li, K. Zhang, M. Yang, H. Sun and B. Yang, Small, 2018, 1703919 CrossRef PubMed.
- D. Li, P. Jing, L. Sun, Y. An, X. Shan, X. Lu, D. Zhou, D. Han, D. Shen, Y. Zhai, S. Qu, R. Zbořil and A. L. Rogach, Adv. Mater., 2018, 30, 1705913 CrossRef PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef PubMed.
- J. Chen, S. Li, Y. Zhang, W. Wang, X. Zhang, Y. Zhao, Y. Wang and H. Bi, Adv. Healthcare Mater., 2017, 6, 1700746 CrossRef PubMed.
- W.-Q. Li, Z. Wang, S. Hao, L. Sun, M. Nisic, G. Cheng, C. Zhu, Y. Wan, L. Ha and S.-Y. Zheng, Nanoscale, 2018, 10, 3744–3752 RSC.
- H. Wang, C. Liu, Z. Liu, J. Ren and X. Qu, Small, 2018, 14, 1703710 CrossRef PubMed.
- H.-J. Jian, R.-S. Wu, T.-Y. Lin, Y.-J. Li, H.-J. Lin, S. G. Harroun, J.-Y. Lai and C.-C. Huang, ACS Nano, 2017, 11, 6703–6716 CrossRef PubMed.
- J. Tang, B. Kong, H. Wu, M. Xu, Y. Wang, Y. Wang, D. Zhao and G. Zheng, Adv. Mater., 2013, 25, 6569–6574 CrossRef PubMed.
- Q. Wang, X. Huang, Y. Long, X. Wang, H. Zhang, R. Zhu, L. Liang, P. Teng and H. Zheng, Carbon, 2013, 59, 192–199 CrossRef.
- M. Zheng, S. Liu, J. Li, D. Qu, H. Zhao, X. Guan, X. Hu, Z. Xie, X. Jing and Z. Sun, Adv. Mater., 2014, 26, 3554–3560 CrossRef PubMed.
- T. Feng, X. Ai, G. An, P. Yang and Y. Zhao, ACS Nano, 2016, 10, 4410–4420 CrossRef PubMed.
- Y. Huang, Y. Wang and T. Wong, Lab Chip, 2017, 17, 2969–2981 RSC.
- Y. Huang, J. Chen, T. Wong and J.-L. Liow, Soft Matter, 2016, 12, 6206–6213 RSC.
- J. Atencia and D. J. Beebe, Nature, 2004, 437, 648 CrossRef PubMed.
- M. P. Stewart, A. Sharei, X. Ding, G. Sahay, R. Langer and K. F. Jensen, Nature, 2016, 538, 183 CrossRef PubMed.
- H. Song, D. L. Chen and R. F. Ismagilov, Angew. Chem., Int. Ed., 2006, 45, 7336–7356 CrossRef PubMed.
- Y. Huang, L. Xiao, T. An, W. Lim, T. Wong and H. Sun, Small, 2017, 13, 1700869 CrossRef PubMed.
- H. Lin, B. D. Storey, M. H. Oddy, C.-H. Chen and J. G. Santiago, Phys. Fluids, 2004, 16, 1922–1935 CrossRef.
- W. Kwon, Y.-H. Kim, C.-L. Lee, M. Lee, H. C. Choi, T.-W. Lee and S.-W. Rhee, Nano Lett., 2014, 14, 1306–1311 CrossRef PubMed.
- Z. Luo, G. Qi, K. Chen, M. Zou, L. Yuwen, X. Zhang, W. Huang and L. Wang, Adv. Funct. Mater., 2016, 26, 2739–2744 CrossRef.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, A. Chen and M. Jin, Nat. Commun., 2018, 9, 2249 CrossRef PubMed.
- Z. Zhou, P. Tian, X. Liu, S. Mei, D. Zhou, D. Li, P. Jing, W. Zhang, R. Guo and S. Qu, Adv. Sci., 2018, 1800369 CrossRef.
- F. Wang, Y.-h. Chen, C.-y. Liu and D.-g. Ma, Chem. Commun., 2011, 47, 3502–3504 RSC.
- X. Zhang, Y. Zhang, Y. Wang, S. Kalytchuk, S. V. Kershaw, Y. Wang, P. Wang, T. Zhang, Y. Zhao and H. Zhang, ACS Nano, 2013, 7, 11234–11241 CrossRef PubMed.
- Y.-L. Zhang, L. Wang, H.-C. Zhang, Y. Liu, H.-Y. Wang, Z.-H. Kang and S.-T. Lee, RSC Adv., 2013, 3, 3733–3738 RSC.
- H. X. Zhao, L. Q. Liu, Z. De Liu, Y. Wang, X. J. Zhao and C. Z. Huang, Chem. Commun., 2011, 47, 2604–2606 RSC.
- X. Lu, J. Zhang, Y.-N. Xie, X. Zhang, X. Jiang, X. Hou and P. Wu, Anal. Chem., 2018, 90, 2939–2945 CrossRef PubMed.
- T.-H. Chen and W.-L. Tseng, Anal. Chem., 2017, 89, 11348–11356 CrossRef PubMed.
- X. Jin, X. Sun, G. Chen, L. Ding, Y. Li, Z. Liu, Z. Wang, W. Pan, C. Hu and J. Wang, Carbon, 2015, 81, 388–395 CrossRef.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X.-A. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef.
- A. Chandra and N. Singh, Chem. Commun., 2018, 54, 1643–1646 RSC.
- W. Song, W. Duan, Y. Liu, Z. Ye, Y. Chen, H. Chen, S. Qi, J. Wu, D. Liu and L. Xiao, Anal. Chem., 2017, 89, 13626–13633 CrossRef PubMed.
- J. S. Sidhu, A. Singh, N. Garg, N. Kaur and N. Singh, Analyst, 2018, 143, 1853–1861 RSC.
- B. B. Chen, M. L. Liu, L. Zhan, C. M. Li and C. Z. Huang, Anal. Chem., 2018, 90, 4003–4009 CrossRef PubMed.
- L.-L. Feng, Y.-X. Wu, D.-L. Zhang, X.-X. Hu, J. Zhang, P. Wang, Z.-L. Song, X.-B. Zhang and W. Tan, Anal. Chem., 2017, 89, 4077–4084 CrossRef PubMed.
- S. Qu, H. Chen, X. Zheng, J. Cao and X. Liu, Nanoscale, 2013, 5, 5514–5518 RSC.
- S. Kalytchuk, K. i. Poláková, Y. Wang, J. P. Froning, K. Cepe, A. L. Rogach and R. Zboril, ACS Nano, 2017, 11, 1432–1442 CrossRef PubMed.
- H. K. Sadhanala, S. Senapati and K. K. Nanda, Carbon, 2018, 133, 200–208 CrossRef.
- W. Shi, X. Li and H. Ma, Angew. Chem., 2012, 124, 6538–6541 CrossRef.
- X. Sun, C. Brückner and Y. Lei, Nanoscale, 2015, 7, 17278–17282 RSC.
- J. Shangguan, D. He, X. He, K. Wang, F. Xu, J. Liu, J. Tang, X. Yang and J. Huang, Anal. Chem., 2016, 88, 7837–7843 CrossRef PubMed.
- P. A. de Silva, N. H. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42 CrossRef.
- L. Feng, A. Zhao, J. Ren and X. Qu, Nucleic Acids Res., 2013, 41, 7987–7996 CrossRef PubMed.
- M. P. Sk, S. K. Sailapu and A. Chattopadhyay, ChemPhysChem, 2015, 16, 723–727 CrossRef PubMed.
- C. Shen, Y. Zhao, H. Liu, Y. Jiang, N. Li, S. Lan, H. Bao, B. Yang and Q. Lin, Polym. Chem., 2018, 9, 2478–2483 RSC.
- N. Dhenadhayalan and K.-C. Lin, Sci. Rep., 2015, 5, 10012 CrossRef PubMed.
- W.-S. Zou, Q.-C. Zhao, W.-L. Kong, X.-F. Wang, X.-M. Chen, J. Zhang and Y.-Q. Wang, Chem. Eng. J., 2018, 337, 471–479 CrossRef.
- M. Li, Z. Wang, J. Liang, H. Yao, L. Shen, H. Liu and L. Fan, Nanoscale, 2018, 10, 7484–7493 RSC.
- J. Deng, Z. Tao, Y. Liu, X. Lin, P. Qian, Y. Lyu, Y. Li, K. Fu and S. Wang, Chem. Commun., 2018, 54, 3110–3113 RSC.
- F. P. Milton, J. Govan, M. V. Mukhina and Y. K. Gun'ko, Nanoscale Horiz., 2016, 1, 14–26 RSC.
- X.-L. Liu, S. Tsunega and R.-H. Jin, Nanoscale Horiz., 2017, 2, 147–155 RSC.
- R. Walker, D. Pociecha, J. Abberley, A. Martinez-Felipe, D. Paterson, E. Forsyth, G. Lawrence, P. Henderson, J. Storey and E. Gorecka, Chem. Commun., 2018, 54, 3383–3386 RSC.
- M. Vázquez-Nakagawa, L. Rodríguez-Pérez, M. Herranz and N. Martín, Chem. Commun., 2016, 52, 665–668 RSC.
- C. B. Kc, G. N. Lim and F. D'Souza, Angew. Chem., Int. Ed., 2015, 54, 5088–5092 CrossRef PubMed.
- R. J. Chen, Y. Zhang, D. Wang and H. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef PubMed.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- N. Suzuki, Y. Wang, P. Elvati, Z.-B. Qu, K. Kim, S. Jiang, E. Baumeister, J. Lee, B. Yeom and J. H. Bahng, ACS Nano, 2016, 10, 1744–1755 CrossRef PubMed.
- F. Li, Y. Li, X. Yang, X. Han, Y. Jiao, T. Wei, D. Yang, H. Xu and G. Nie, Angew. Chem., Int. Ed., 2018, 57, 2377–2382 CrossRef PubMed.
- Y. Zhang, L. Hu, Y. Sun, C. Zhu, R. Li, N. Liu, H. Huang, Y. Liu, C. Huang and Z. Kang, RSC Adv., 2016, 6, 59956–59960 RSC.
- M. J. Deka and D. Chowdhury, RSC Adv., 2017, 7, 53057–53063 RSC.
- A. Ghosh, B. Parasar, T. Bhattacharyya and J. Dash, Chem. Commun., 2016, 52, 11159–11162 RSC.
- Q. Xin, Q. Liu, L. Geng, Q. Fang and J. R. Gong, Adv. Healthcare Mater., 2017, 6, 1601011 CrossRef PubMed.
- L. Hu, H. Li, C. a. Liu, Y. Song, M. Zhang, H. Huang, Y. Liu and Z. Kang, Nanoscale, 2018, 10, 2333 RSC.
- R. Butbul, S. Bhunia, S. Shaham-Niv, S. Kolusheva, E. Gazit and R. Jelinek, Chem. Commun., 2018, 54, 7762–7765 RSC.
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2015, 44, 362–381 RSC.
- T. A. Waldmann, Nat. Med., 2003, 9, 269 CrossRef PubMed.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. Frechet, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef PubMed.
- Y. Wang, X. Li, F. Li, W.-Y. Sun, C. Zhu and Y. Cheng, Chem. Commun., 2017, 53, 7505–7508 RSC.
- E. M. Sánchez-Carnerero, F. Moreno, B. L. Maroto, A. R. Agarrabeitia, M. J. Ortiz, B. G. Vo, G. Muller and S. d. l. Moya, J. Am. Chem. Soc., 2014, 136, 3346–3349 CrossRef PubMed.
- J. Han, P. Duan, X. Li and M. Liu, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |