
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven rotary molecular motors without point chirality: a minimal design†
Jun
Wang‡
,
Baswanth
Oruganti‡
and
Bo
Durbeej
 *
*
Division of Theoretical Chemistry, IFM, Linköping University, SE-581 83 Linköping, Sweden. E-mail: bodur@ifm.liu.se
First published on 20th February 2017
Abstract
A fundamental requirement for achieving photoinduced unidirectional rotary motion about an olefinic bond in a molecular motor is that the potential energy surface of the excited state is asymmetric with respect to clockwise and counterclockwise rotations. In most available light-driven rotary molecular motors, such asymmetry is guaranteed by the presence of a stereocenter. Here, we present non-adiabatic molecular dynamics simulations based on multiconfigurational quantum chemistry to demonstrate that this chiral feature is not essential for inducing unidirectional rotary motion in molecules that incorporate a cyclohexenylidene moiety into a protonated Schiff-base framework. Rather, the simulations show that it is possible to exploit the intrinsic asymmetry of the puckered cyclohexenylidene to control the direction of photoinduced rotation.
Photoisomerization around an olefinic bond in the retinal protonated Schiff-base chromophore of the visual pigment rhodopsin is the primary event of vision.1,2 Mimicking this reaction in vitro to extract useful mechanical work involves exerting control of the direction – clockwise (CW) or counterclockwise (CCW) – of the photoinduced rotation, usually by means of molecular chirality.3–5 Indeed, by making the potential energy surface (PES) of the excited state asymmetric with respect to the two directions, chirality ensures that the photoinduced rotation prefers one direction over the other.6 Molecules that are able to accomplish a full 360° rotation through the absorption of UV or visible light are commonly known as light-driven rotary molecular motors.
Ever since their introduction in the late 1990s,3 synthetic light-driven rotary molecular motors developed from sterically overcrowded alkenes have attracted considerable interest.4,7–15 In these motors, the asymmetry in the excited-state PES is achieved by the presence of one or two stereocenters and one or two helical motifs that result from a substantial pretwist (ca. 10–25°)8 of the isomerizing olefinic bond. In this work, this is referred to as a “helical” pretwist. The unidirectional rotary motion is produced by consecutive photoisomerization and thermal isomerization steps, where the direction of rotation is controlled by the configuration(s) of the stereocenter(s) (R or S).7,9 In two recent studies, however, it has been demonstrated that permanent point chirality is not an absolute requirement for achieving unidirectional rotary motion by an overcrowded alkene-based motor that exhibits a chiral-like feature in the form of a pseudo-asymmetric carbon,16 or incorporates an axially chiral trityl moiety.17 Interestingly, unidirectional rotary motion has also been realized or conceived for systems that contain a plane of symmetry, either by exploiting mechanically interlocked architectures18 or using polarized laser pulses19,20 or oscillating electric fields21 as the input energy source.
Although the overcrowded alkene-based motor design has shown potential for a variety of applications in nanotechnology,13–15 the performance of these motors is restricted in two different ways. First, under ambient conditions, the rotational frequencies are limited by the fact that the thermal isomerizations of the rotary cycles are much slower than the photoisomerizations.7,10 Second, the quantum yields (QYs) of the photoisomerizations are limited by the pyramidalization of one of the olefinic carbons during these processes.10,22,23 While much effort has been invested to improve the performance of the thermal isomerizations of overcrowded alkenes4,7,12,24–26 or to suggest alternative motor designs that avoid thermal steps altogether,6,27,28 the second limitation has only recently started to be addressed.6,10,11,23,29 Particularly, it has been demonstrated that a new motor design incorporating a protonated or alkylated Schiff base is able to reduce the negative influence of pyramidalization on the photoisomerization QYs.23,29 Moreover, for one such design by Olivucci, Léonard, Gindensperger and their co-workers,6 it has been found that unidirectional rotary motion around an olefinic bond can be achieved from two photoisomerization steps alone, and that a sizable helical pretwist in the associated E and Z motor isomers is not essential.6 However, point chirality is deemed critical for this capability.6
In this work, we attempt to generalize the Schiff-base motor design by demonstrating, through minimum energy path (MEP) calculations and non-adiabatic molecular dynamics (NAMD) simulations30–33 based on multiconfigurational quantum chemistry,34 that this design can also produce unidirectional rotary motion without point chirality. To this end, we present a minimal model of a Schiff-base motor lacking a stereocenter by incorporating a puckered cyclohexenylidene moiety into a pyrrolinium motif. This system is shown in Scheme 1 and is hereafter referred to as motor 1. Because of the intrinsic asymmetry of the cyclohexenylidene attributable to its puckered ring structure, both the E and Z isomers of 1 have two possible orientations of the 5′ carbon relative to the approximate plane formed by the other carbons of the ring – outward (out) and inward (in). Thus, there are four different isomers of 1 that can be labeled 1-Eout, 1-Ein, 1-Zout and 1-Zin, and which form two pairs of axially chiral enantiomers: 1-Eout/1-Ein and 1-Zout/1-Zin.
 | ||
| Scheme 1 The Eout, Zin, Ein and Zout isomers of motor 1 and relevant dihedral angles. | ||
The ground-state (S0) equilibrium geometries of all isomers of 1 were obtained using the complete active space self-consistent field (CASSCF)35 method in combination with the ANO-L-VDZP basis set, employing an active space of six π-electrons in six orbitals (this basis set and this active space were used for all CASSCF-based calculations of this work; see further the ESI†). The helical pretwist shown by the resulting geometries can be characterized by the Δω dihedral angle, where ω is the mean of the θ and θ′ dihedral angles (see Scheme 1) and Δω is the signed deviation of ω from the completely untwisted ω values of 0 or ±180°. Notably, with absolute Δω values of around 3° only, all isomers exhibit a small helical pretwist, which is also predicted by other methods than CASSCF (see Table S3 of the ESI†).
Starting from the vertical Franck–Condon (FC) points in the optically bright first excited singlet ππ* state (S1) of the different isomers of 1, the E → Z and Z → E photoisomerizations were first modeled by performing MEP calculations using state-averaged CASSCF (SA-CASSCF). For each geometry obtained along the respective MEP, singlepoint calculations were carried out with complete active space second-order perturbation theory (CASPT2)36 to account for dynamic electron correlation effects. The resulting MEPs are presented in Fig. 1.
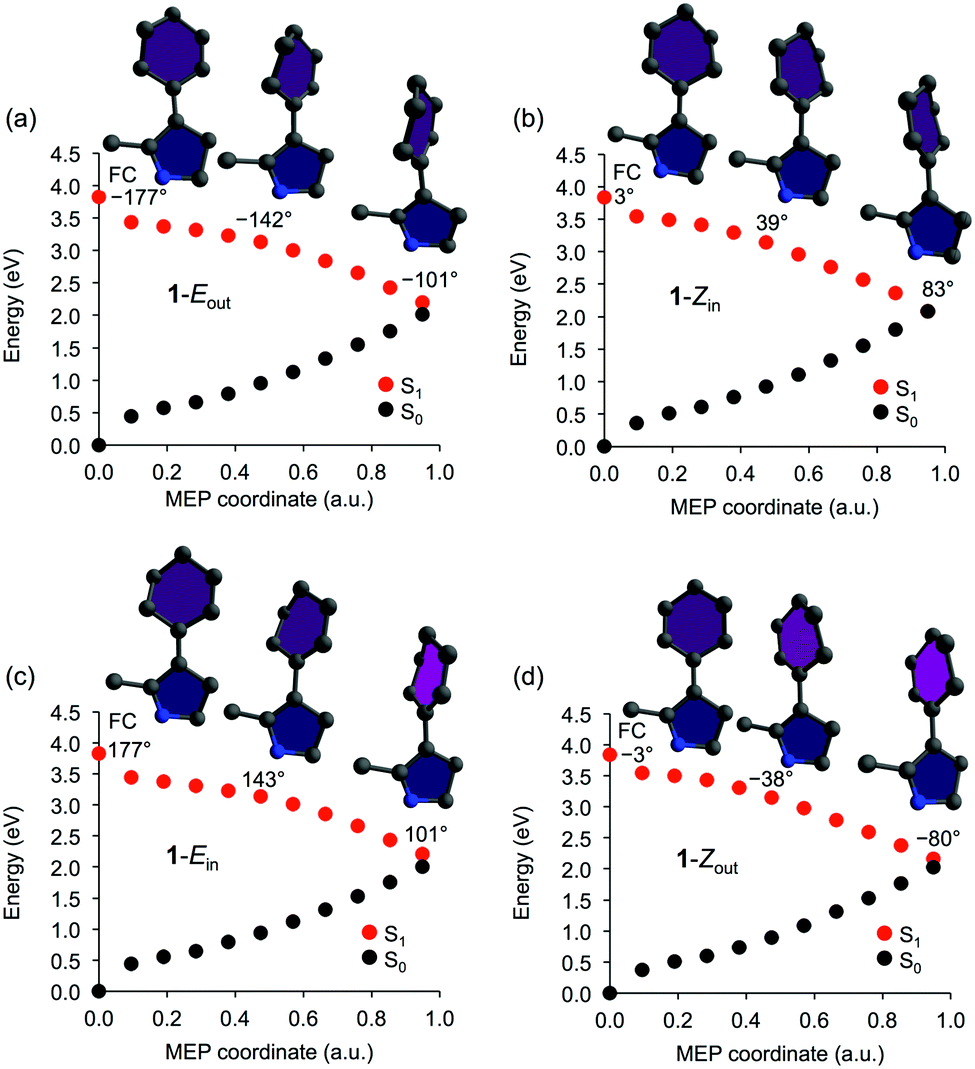 | ||
| Fig. 1 Minimum energy paths (MEPs) from the S1 Franck–Condon (FC) points of the Eout (a), Zin (b), Ein (c) and Zout (d) isomers of motor 1. Shown are also the molecular geometries at the FC point and at two additional points along the paths, as well as the corresponding ω dihedral angles. | ||
As each system evolves from the FC point populated by a near-UV photon of energy ∼3.8 eV (see Table S4 of the ESI†), the MEPs in Fig. 1 show pronounced and barrierless torsional motion around the central olefinic bond toward the S1/S0 conical intersection (CI) region where the system is expected to decay to the S0 state. For the 1-Eout and 1-Zin isomers, the torsional motion is toward increasing values of ω (here defined as CCW motion), whereas it is toward decreasing values of ω for the 1-Ein and 1-Zout isomers (CW motion). Furthermore, S0 geometry optimizations started from the end points of the MEPs continue the torsional motion set forth in S1 and yield 1-Zin and 1-Eout as the photoproducts of the 1-Eout and 1-Zin paths, respectively, and 1-Zout and 1-Ein as the photoproducts of the 1-Ein and 1-Zout paths, respectively. These results suggest that 1, despite lacking a stereocenter, produces 360° unidirectional rotary motion. Furthermore, reminiscent of how the direction of rotation of overcrowded alkenes is dictated by the R/S configuration(s) of the stereocenter(s),7,9 the direction of rotation of 1 is controlled by the out/in orientation of the 5′ carbon, in such a way that the [1-Eout, 1-Zin] pair of isomers complete a 360° CCW rotation and the [1-Ein, 1-Zout] pair a 360° CW rotation.
Besides achieving rotary motion without a stereocenter, it is also interesting to note from Table S5 of the ESI† that 1 shows very little pyramidalization of the central olefinic carbons along the MEPs. For each MEP, the α and α′ dihedral angles (see Scheme 1) remain smaller than 5° for all geometries between the FC point and the CI region. This indicates that 1 produces the type of axial rotation associated with the high QYs of Schiff-base motors.23,29
Having thus obtained promising results from MEP calculations, the photoisomerizations of the four isomers of 1 were then modeled by performing NAMD simulations, as further detailed in the ESI.† Owing to the cost associated with calculating CASPT2 energy gradients, the simulations were carried out at the SA-CASSCF/ANO-L-VDZP level, without inclusion of dynamic electron correlation effects. Importantly, in light of the recent observation that such effects can be quite important in photoisomerization dynamics,33 Fig. S2 of the ESI† suggests that their influence on 1 is minor, insofar that the photoisomerization MEPs obtained with and without CASPT2 singlepoint calculations are very similarly shaped.
Starting in the S1 state but allowing a trajectory to hop to the S0 state if the non-adiabatic coupling between the two states is sufficiently large30,34 as described in the ESI,† the simulations were run for maximally 700 fs and with 200 different initial nuclear configurations and velocities for each isomer. Importantly, following an assessment presented in the ESI† of the possible role played by multiple hopping events in the simulations, only one hop between the S1 and S0 states was allowed for each trajectory. The requisite nuclear forces were calculated “on the fly”. Defining successful trajectories as those that isomerize 1-Eout into 1-Zin, 1-Zin into 1-Eout, 1-Ein into 1-Zout, and 1-Zout into 1-Ein by first decaying to the S0 state and subsequently completing a net 180° rotation (in terms of the ω dihedral angle) around the central olefinic bond relative to the starting configuration within 700 fs, the distributions of S1 lifetimes and photoisomerization times (PITs) are presented in Fig. 2. Thus, the PIT is the time it takes to complete one such rotation. Shown also in Fig. 2 are the percentages of trajectories that hop to the S0 state and the percentages of trajectories that are indeed successful.
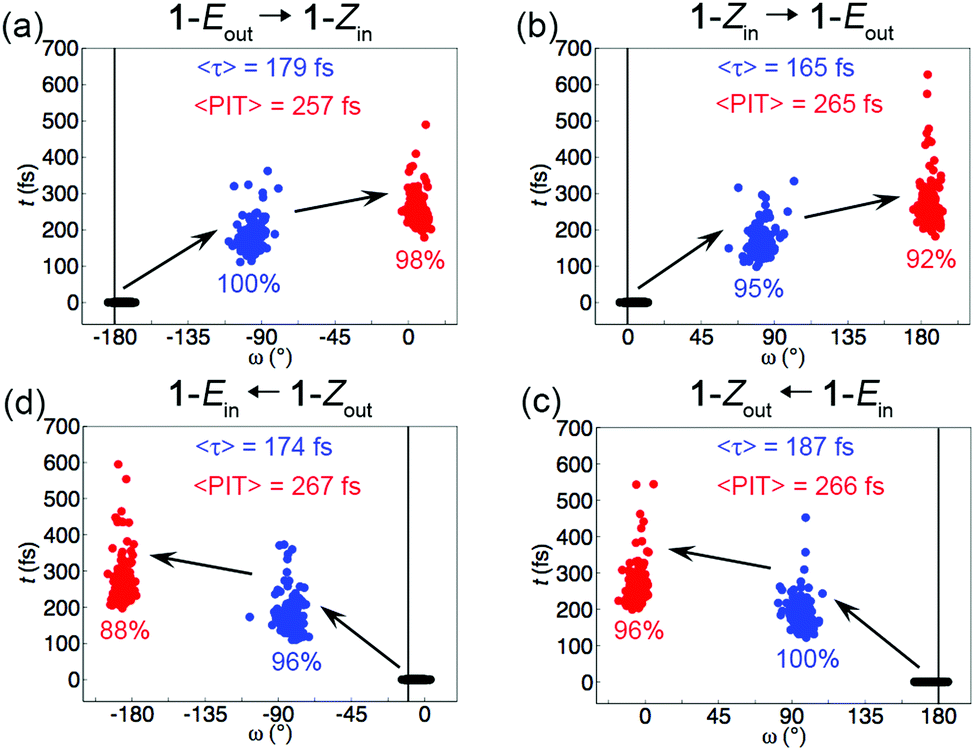 | ||
| Fig. 2 Distributions of S1 lifetimes (τ, blue circles) and photoisomerization times (PITs, red circles) for the 1-Eout → 1-Zin (a), 1-Zin → 1-Eout (b), 1-Ein → 1-Zout (c) and 1-Zout → 1-Ein (d) trajectories and the corresponding changes in the ω dihedral angle relative to the starting configurations (black circles). Displayed for each reaction are also the average values of τ and PIT, as well as the percentages of trajectories that hop to the S0 state and the percentages of trajectories that are successful. | ||
Interestingly, Fig. 2 reveals that the percentage of successful trajectories is very high (≥88%) for each photoisomerization. Furthermore, and surprisingly, each photoisomerization is perfectly directional in that all successful trajectories for the reaction in question proceed in one direction only. Specifically, the direction of rotation is CCW for the 1-Eout and 1-Zin isomers, but CW for the 1-Ein and 1-Zout species. These results corroborate the MEP results that the [1-Eout, 1-Zin] and [1-Ein, 1-Zout] pairs of isomers sustain 360° unidirectional rotary motion through sequential absorption of two photons, despite lacking a stereocenter. The results also reaffirm that the direction of rotation is controlled by the out/in orientation of the 5′ carbon: CCW for [1-Eout, 1-Zin] and CW for [1-Ein, 1-Zout]. Moreover, it is notable that the 〈PIT〉 values are consistently small, only ∼260–270 fs, which suggest that both the S1 dynamics from the FC point to the S1/S0 CI region and the S0 dynamics following the S1 → S0 decay are very fast.
It is also interesting to note from Fig. 2 that virtually all (≥95%) trajectories for each isomer decay to S0 and that the average S1 lifetime falls in the ∼170–190 fs range (see also Table S6 of the ESI†). Furthermore, Fig. S3 of the ESI† shows that 80% (1-Eout), 89% (1-Zin), 74% (1-Ein) and 81% (1-Zout) of the trajectories that do decay, do so within 200 fs. These results underline one advantage of the Schiff-base motor design6,23,29 over overcrowded alkene-based motors, which typically have S1 lifetimes of ∼1 ps or more.10,22 As for mechanistic photoisomerizations details, relevant information from the NAMD simulations is summarized in Fig. S4 and S5 of the ESI.† For example, Fig. S5 (ESI†) presents the distributions of central olefinic bond lengths and ω values in the starting and S1 → S0 hopping configurations for each photoisomerization. As expected, the S1 → S0 decay occurs at ω values close to ±90°.
From Fig. 2, it can be seen that the 200 sampled starting configurations have Δω values (i.e., ω values relative to the completely untwisted ω values of 0 or ±180°), or helical pretwists, that are not evenly distributed around 0 or ±180°; rather, the distributions are somewhat skewed toward positive Δω values for 1-Eout and 1-Zin and toward negative Δω values for 1-Ein and 1-Zout. Through the sampling, this skewness arises because the S0 equilibrium geometries of the isomers also have Δω values that, albeit small, are positive for 1-Eout and 1-Zin and negative for 1-Ein and 1-Zout (see Table S3, ESI†). However, since it was found above that all successful photoisomerization trajectories for 1-Eout and 1-Zin proceed in the direction of increasing ω and those for 1-Ein and 1-Zout proceed in the direction of decreasing ω, it is of interest to assess whether the tendency to isomerize in a particular direction depends on the sign of Δω at the starting configuration. This is done in Fig. 3 and Fig. S6 and S7 of the ESI.†
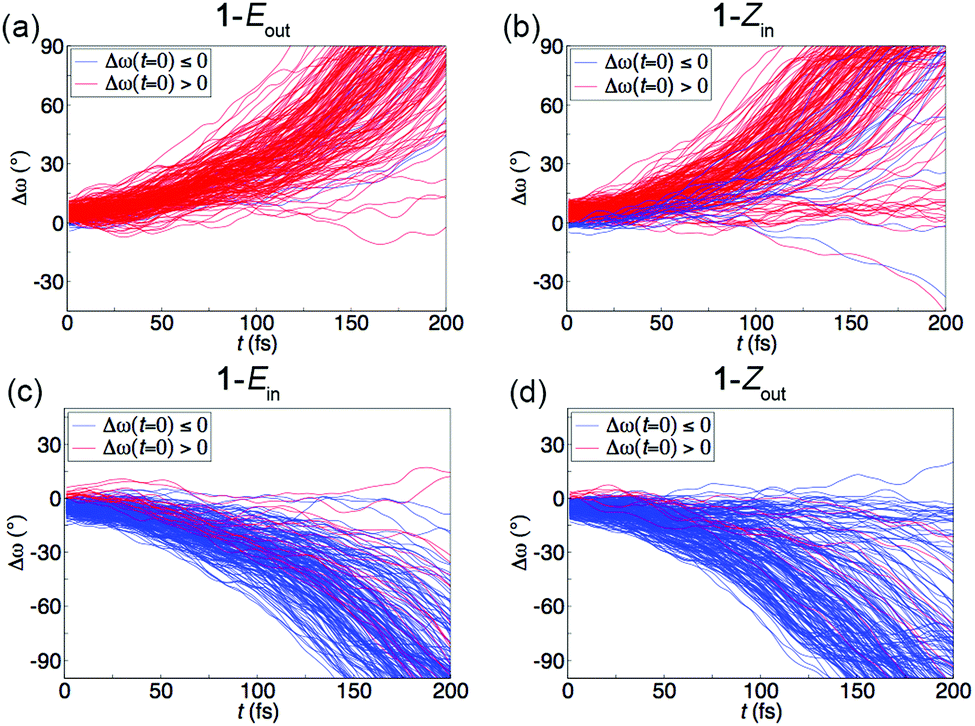 | ||
| Fig. 3 Influence of the sign of the initial (at t = 0) Δω value on the direction of photoinduced torsional motion around the central olefinic bond during the first 200 fs of all 200 trajectories run for the Eout (a), Zin (b), Ein (c) and Zout (d) isomers of motor 1. | ||
Starting with Fig. 3, virtually all 200 trajectories of each isomer are propagating in the same direction, irrespective of the sign of the initial Δω value. Furthermore, plotting the S1 lifetimes and PITs as functions of the initial Δω values in Fig. S6 and S7 (ESI†), these entities do not seem to depend on whether Δω is positive or negative. Altogether, then, the sign of the initial Δω appears to have no bearing on the directionality (or efficiency) of the rotary motion produced by the [1-Eout, 1-Zin] and [1-Ein, 1-Zout] pairs of isomers. The reason why each isomer has such a clear propensity to photoisomerize in one particular direction only is, we believe, that the S1 PES is markedly asymmetric with respect to the CW and CCW directions (see Fig. S8 of the ESI†). Thereby, the ability of 1 to achieve unidirectional rotary motion is not a consequence of the small positive and negative helical pretwists in the different isomers of 1, but rather a manifestation of how the out and in orientations of the cyclohexenylidene 5′ carbon influence the S1 PES.
In order for experimental realizations of 1 or its structural analogues to work, it is important that the thermal free-energy barriers separating the Eout and Ein and the Zout and Zin enantiomers are not too small. Otherwise, it would be difficult to synthesize or isolate an enantiopure out/in isomer, as the isomers have identical energies (see Table S3, ESI†), and similarly challenging to control the direction of photoinduced rotation through the out/in orientation of the 5′ carbon. In this light, it is pleasing that neither 1-Eout ↔ 1-Ein nor 1-Zout ↔ 1-Zin enantiomerizations were observed during the NAMD simulations (a more rigorous assessment of the possible occurrence of such processes would require that quantum dynamics effects are accounted for, which is beyond the scope of this work).
However, to investigate this issue in further detail, the enantiomerization barriers were calculated using both density functional theory (DFT) and second-order Møller–Plesset perturbation theory (MP2) as described in the ESI.† From these calculations, room-temperature barriers of around 25 kJ mol−1 were predicted (see Table S7 of the ESI†), whereby efficient control of the rotary cycles of 1 would seem to require low temperatures. Alternatively, it may be possible to increase the enantiomerization barriers by introducing other substituents than hydrogens at the 5′ position and/or other positions of the cyclohexenylidene moiety. To test this possibility, all four hydrogens at the 4′ and 5′ positions were replaced with bulkier methyl groups, with the idea that these could slow down the enantiomerizations on steric grounds. The resulting species, labeled 2-Eout, 2-Zin, 2-Ein and 2-Zout, are shown in Scheme S1 of the ESI.† Importantly, these species remain axially chiral (i.e., lack a stereocenter).
By subjecting 2 to the same type of DFT and MP2 calculations performed for 1, it was then found that the added methyl groups raise the enantiomerization barriers by about 20 kJ mol−1 (see Table S7, ESI†). Thus, the potential problem with facile enantiomerizations appears circumventable by introducing substituents that could raise the barriers even further. For example, complementary calculations on a tetra-brominated (at the 4′ and 5′ positions) analogue of motor 1, labeled 3 in Table S7 (ESI†), predict that the barriers can actually be raised by 50 kJ mol−1 relative to 1. This approach to increase the barriers by such magnitudes can be exploited in established asymmetric synthesis techniques, like dynamic kinetic resolution,37,38 to obtain an enantiopure analogue of 1 from a racemic mixture of 1, using a chiral catalyst.37,38
Finally, by subjecting 2 to MEP calculations and NAMD simulations, results presented in Fig. S9, S10 and Table S8 of the ESI† suggest that producing 360° unidirectional rotary motion through sequential absorption of two photons is not a behavior unique to 1, but is a behavior that can be attributed to any structural analogue of 1 containing a puckered cyclohexenylidene moiety.
Conclusions
We have demonstrated that light-driven unidirectional rotary motion is achievable by axially chiral molecules that incorporate a cyclohexenylidene moiety into a protonated Schiff-base framework, and of which 1 is a minimal model. Despite lacking a stereocenter, the direction of photoinduced rotation of 1 is controllable by the orientation of the 5′ carbon relative to the plane formed by the other carbons of the cyclohexenylidene. Furthermore, the photoisomerizations retain key favorable features, such as short S1 lifetimes,6,23,29 observed in previous studies of Schiff-base motors. It is our hope that the results of this work will stimulate the further development of improved rotary motors by having brought molecules that have no point chirality into play.Acknowledgements
We acknowledge financial support from the Swedish Research Council (grant 621–2011–4353), the Olle Engkvist Foundation (grant 2014/734), the Carl Trygger Foundation (grant CTS 15:134) and Linköping University, as well as grants of computing time at the National Supercomputer Centre (NSC) in Linköping.References
- G. Wald, Nature, 1968, 219, 800–807 CrossRef CAS PubMed.
- H. G. Khorana, J. Biol. Chem., 1992, 267, 1–4 CAS.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS PubMed.
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Nat. Commun., 2015, 6, 8406 CrossRef CAS PubMed.
- G. Marchand, J. Eng, I. Schapiro, A. Valentini, L. M. Frutos, E. Pieri, M. Olivucci, J. Léonard and E. Gindensperger, J. Phys. Chem. Lett., 2015, 6, 599–604 CrossRef CAS PubMed.
- M. M. Pollard, M. Klok, D. Pijper and B. L. Feringa, Adv. Funct. Mater., 2007, 17, 718–729 CrossRef CAS.
- A. Kazaryan, J. C. M. Kistemaker, L. V. Schäfer, W. R. Browne, B. L. Feringa and M. Filatov, J. Phys. Chem. A, 2010, 114, 5058–5067 CrossRef CAS PubMed.
- N. Ruangsupapichat, M. M. Pollard, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 53–60 CrossRef CAS PubMed.
- J. Conyard, K. Addison, I. A. Heisler, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, Nat. Chem., 2012, 4, 547–551 CrossRef CAS PubMed.
- J. Conyard, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 9692–9700 CrossRef CAS PubMed.
- J. Vachon, G. T. Carroll, M. M. Pollard, E. M. Mes, A. M. Brouwer and B. L. Feringa, Photochem. Photobiol. Sci., 2014, 13, 241–246 CAS.
- P. T. Chiang, J. Mielke, J. Godoy, J. M. Guerrero, L. B. Alemany, C. J. Villagómez, A. Saywell, L. Grill and J. M. Tour, ACS Nano, 2012, 6, 592–597 CrossRef CAS PubMed.
- D. J. van Dijken, J. Chen, M. C. A. Stuart, L. Hou and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 660–669 CrossRef CAS PubMed.
- S. J. Wezenberg, C. M. Croisetu, M. C. A. Stuart and B. L. Feringa, Chem. Sci., 2016, 7, 4341–4346 RSC.
- J. C. M. Kistemaker, P. Štacko, J. Visser and B. L. Feringa, Nat. Chem., 2015, 7, 890–896 CrossRef CAS PubMed.
- S. C. Everhart, U. K. Jayasundara, H. Kim, R. Procúpez-Schtirbu, W. A. Stanbery, C. H. Mishler, B. J. Frost, J. I. Cline and T. W. Bell, Chem. – Eur. J., 2016, 22, 11291–11302 CrossRef CAS PubMed.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed.
- G. Pérez-Hernández, A. Pelzer, L. González and T. Seideman, New J. Phys., 2010, 12, 075007 CrossRef.
- J. Zhang, A. P. Sergeeva, M. Sparta and A. N. Alexandrova, Angew. Chem., Int. Ed., 2012, 51, 8512–8515 CrossRef CAS PubMed.
- D. Horinek and J. Michl, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14175–14180 CrossRef CAS PubMed.
- A. Kazaryan, Z. Lan, L. V. Schäfer, W. Thiel and M. Filatov, J. Chem. Theory Comput., 2011, 7, 2189–2199 CrossRef CAS PubMed.
- M. Filatov and M. Olivucci, J. Org. Chem., 2014, 79, 3587–3600 CrossRef CAS PubMed.
- G. Pérez-Hernández and L. González, Phys. Chem. Chem. Phys., 2010, 12, 12279–12289 RSC.
- B. Oruganti, C. Fang and B. Durbeej, Phys. Chem. Chem. Phys., 2015, 17, 21740–21751 RSC.
- B. Oruganti, J. Wang and B. Durbeej, ChemPhysChem, 2016, 17, 3399–3408 CrossRef CAS PubMed.
- C. García-Iriepa, M. Marazzi, F. Zapata, A. Valentini, D. Sampedro and L. M. Frutos, J. Phys. Chem. Lett., 2013, 4, 1389–1396 CrossRef PubMed.
- Y. Amatatsu, J. Phys. Chem. A, 2012, 116, 10182–10193 CrossRef CAS PubMed.
- A. Nikiforov, J. A. Gamez, W. Thiel and M. Filatov, J. Phys. Chem. Lett., 2016, 7, 105–110 CrossRef CAS PubMed.
- G. Groenhof, M. Bouxin-Cademartory, B. Hess, S. P. de Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark and M. A. Robb, J. Am. Chem. Soc., 2004, 126, 4228–4233 CrossRef CAS PubMed.
- M. Barbatti, WIREs Comput. Mol. Sci., 2011, 1, 620–633 CrossRef CAS.
- I. Fdez. Galván, M. G. Delcey, T. B. Pedersen, F. Aquilante and R. Lindh, J. Chem. Theory Comput., 2016, 12, 3636–3653 CrossRef PubMed.
- L. Liu, J. Liu and T. J. Martinez, J. Phys. Chem. B, 2016, 120, 1940–1949 CrossRef CAS PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. L. Manni, H. Lischka, D. Ma, P.-Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS.
- J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251–1255 CrossRef CAS PubMed.
- H. Pellissier, Tetrahedron, 2011, 67, 3769–3802 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Computational details, Scheme S1, Fig. S1–S10, Tables S1–S8, description of multimedia files, and Cartesian coordinates for equilibrium geometries. See DOI: 10.1039/c6cp08484b |
| ‡ These two authors contributed equally to this work and are co-first authors. |
| This journal is © the Owner Societies 2017 |