
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in the synthesis of nanostructured magnesium hydroxide
Giulia
Balducci
,
Laura
Bravo Diaz
 and
Duncan H.
Gregory
*
and
Duncan H.
Gregory
*
WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Duncan.Gregory@Glasgow.ac.uk
First published on 3rd October 2017
Abstract
This review highlights synthetic routes for producing nanostructured magnesium hydroxide and focuses on how these various preparative approaches can produce Mg(OH)2 nanoparticles with controlled size and morphology. Mg(OH)2 nanocrystals with rod-, needle-, hollow tube- or platelet-like morphology can be synthesised by the modification of chemical and physical experimental parameters such as the selection of magnesium precursor, solvent and temperature or by employing surface modifiers and templates. Techniques based on hydrothermal/solvothermal treatments, microwave heating and (co-)precipitation are dominant in the production of Mg(OH)2 at the nanoscale, but other materials design approaches are now emerging. Bulk Mg(OH)2 has been extensively studied over decades and finds use in a wide range of applications. Moreover, the hydroxide can also serve as a precursor for other commercially important materials such as MgO. Nanostructuring the material has proven extremely useful in modifying some of its most important properties – not least enhancing the performance of Mg(OH)2 as a non-toxic flame retardant – but equally it is creating new avenues of applied research. We evaluate herein the latest efforts to design novel synthesis routes to nano-Mg(OH)2, to understand the mechanisms of crystallite growth and to tailor microstructure towards specific properties and applications.
 Giulia Balducci | Dr Giulia Balducci graduated from the University of Bologna with an MSc degree in chemistry in 2009. Following the award of two consecutive research bursaries to perform research at the University of Bologna, she moved to the University of Glasgow, where she graduated with a PhD in 2015 under the supervision of Professor Duncan H. Gregory. She worked as a postdoctoral researcher at Glasgow until April 2016. Her research focuses on energy storage materials, with particular emphasis on nanostructured hydrogen and ammonia storage solutions. |
 Laura Bravo Diaz | Laura Bravo Diaz received an MEng degree in chemical engineering from the University of Cantabria, Spain, in 2010. From 2010–2012 she worked as a junior engineer in the renewable energy research sector before joining the European Commission DG Joint Research Centre (DG JRC) as an early-stage-researcher at SolTeF, the EU reference laboratory on hydrogen storage capacity measurements in solid-state materials. Since 2013 she has undertaken PhD studies in Chemistry at the University of Glasgow under the supervision of Professor Duncan H. Gregory while based also at the DG JRC as a research fellow in hydrogen storage technologies. Her research interests include the synthesis and characterisation of advanced nanomaterials and their applications in energy conversion and storage. |
 Duncan H. Gregory | Duncan H. Gregory studied at the University of Southampton completing a PhD in solid state chemistry in 1993 under Prof. Mark Weller. He was an EPSRC advanced fellow, lecturer and reader in materials chemistry at the University of Nottingham until 2006. He then took the WestCHEM chair in inorganic materials in the School of Chemistry at the University of Glasgow. His research interests centre on the synthesis, structure and properties of sustainable energy materials, functional materials and nanomaterials. |
Introduction
Magnesium hydroxide (MH), Mg(OH)2, naturally occurring as the mineral brucite, has attracted much attention over the past decades. Mg(OH)2 is a white, odourless solid characterised by an extremely low solubility in water (0.009 g L−1 at 18 °C) and a refractive index nD = 1.559.1 MH crystallises in the tetragonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group, with lattice parameters of a = 3.148 Å and c = 4.779 Å (Fig. 1).2,3 In the Mg(OH)2 crystal structure, each magnesium, Mg2+, cation is coordinated by 6 hydroxide, OH− anions to form Mg–(OH) octahedra, which share faces in two dimensions resulting in a layered structure. Conversely, each OH− anion is surrounded by three Mg2+ cations in a pyramidal geometry. The oxygen atoms in the OH− anions are located in planes above and below the planes of Mg2+ cations with O–H bonds perpendicular to these metal-containing planes. The extended crystal structure is completed in the third dimension by hydrogen bonding, which weakly binds the hexagonal close packed (HCP) anion layers together, with a shortest H⋯H interlayer distance of 1.97 Å. The ⋯ABABA⋯ HCP layers are spatially distributed such that the hydroxide hydrogens within one layer point towards the centre of the triangular plane (an Mg–OH octahedral face) formed by 3 OH bonds in the next layer.
m1 space group, with lattice parameters of a = 3.148 Å and c = 4.779 Å (Fig. 1).2,3 In the Mg(OH)2 crystal structure, each magnesium, Mg2+, cation is coordinated by 6 hydroxide, OH− anions to form Mg–(OH) octahedra, which share faces in two dimensions resulting in a layered structure. Conversely, each OH− anion is surrounded by three Mg2+ cations in a pyramidal geometry. The oxygen atoms in the OH− anions are located in planes above and below the planes of Mg2+ cations with O–H bonds perpendicular to these metal-containing planes. The extended crystal structure is completed in the third dimension by hydrogen bonding, which weakly binds the hexagonal close packed (HCP) anion layers together, with a shortest H⋯H interlayer distance of 1.97 Å. The ⋯ABABA⋯ HCP layers are spatially distributed such that the hydroxide hydrogens within one layer point towards the centre of the triangular plane (an Mg–OH octahedral face) formed by 3 OH bonds in the next layer.
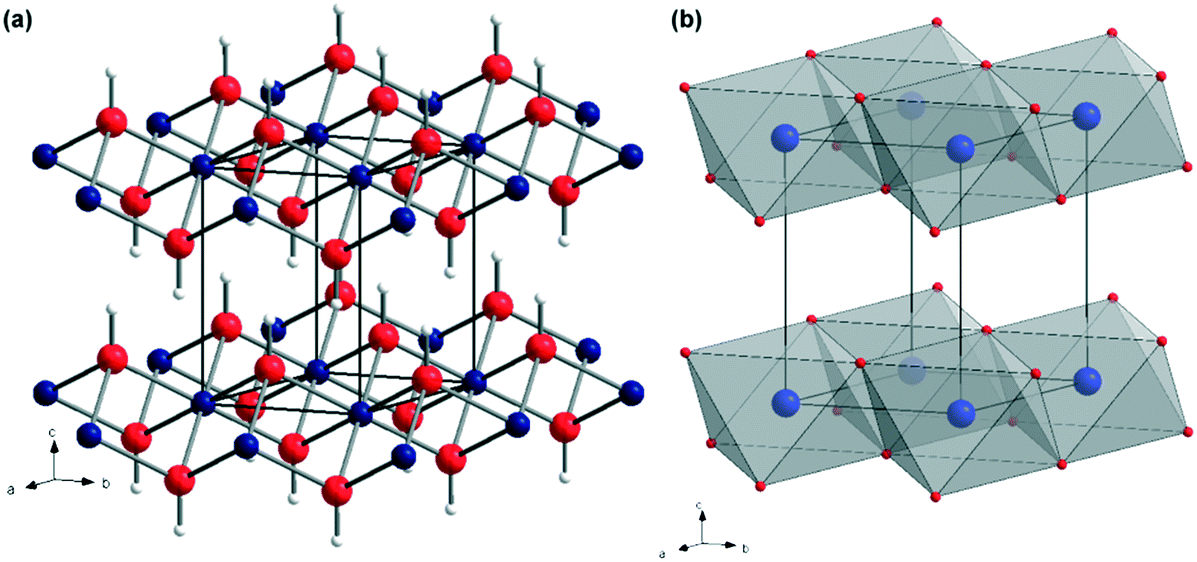 | ||
| Fig. 1 Crystal structure of Mg(OH)2: (a) as a ball-and-stick representation with Mg as blue spheres, O as red spheres and H as white spheres, respectively; (b) as a polyhedral representation showing layers of face-sharing Mg(OH)6 octahedra connected along the <001> direction via hydrogen bonding (adapted from ref. 4). | ||
When heated to temperatures above 350 °C, bulk Mg(OH)2 undergoes a dehydration process resulting in the formation of MgO (cubic, Fmm)5 and the evolution of water (eqn (1)).6,7
| Mg(OH)2 → MgO + H2O ΔH°(298 K) = 98.4 kJ mol−1 H2O | (1) |
The ability of the hydroxide to decompose endothermically releasing water and forming MgO without the production of corrosive or toxic by-products, accounts for its major importance as a commercial non-toxic flame retardant. Magnesium hydroxide also finds application as an acidic waste neutraliser, as a pharmaceutical excipient, in paper conservation, as a component in ethanol chemical sensors and as the most important precursor for the preparation of (nanostructured) magnesium oxide which, in turn, finds application in catalysis.6,8–16
A variety of methods for yielding nanostructured Mg(OH)2 have been reported. These include hydrothermal/solvothermal techniques, precipitation routes and microwave-assisted methods. Further, the use of surfactants and templating agents has been explored as a means to obtain Mg(OH)2 nanocrystals with different morphologies, from hexagonal nanoplates through nanotubes, nanorods and nanosheets to mixture of nanosheets and nanoparticles. However, due to the fact that surfactants are not environmentally sustainable and that their employment results in an additional cost to processing, efforts have been made in order to remove them from the synthetic procedure. Similar environmental concerns arise for synthetic procedures involving non-aqueous solvents, which are commonly used in solvothermal reactions. Ultimately, the goal for the production of Mg(OH)2 is to find novel synthetic routes that are fast, simple, energy-efficient and allow fine control over particle size and morphology. Below we discuss and highlight the latest progress made in terms of synthetic routes to yield nanostructured magnesium hydroxide.
Synthetic routes to nanostructured Mg(OH)2
Conventional hydrothermal/solvothermal synthesis
Hydrothermal and solvothermal methods have been widely employed to produce nanostructured Mg(OH)2. These methods often involve the use of surfactants such as poly(ethylene glycol) (PEG) or ethylendiamine (en), which are considered to play an important role in the mechanism of the nanostructure formation, acting either as templates or growth inhibitors. One of the major drawbacks of these synthetic procedures however is the relatively long reaction time, which is typically of 6 to 24 h or more. Nonetheless, hydro/solvothermal treatments have been extensively explored in terms of morphology and size control for the synthesis of nano-MH.The synthesis of Mg(OH)2 nanorods by solvothermal treatment was first reported in 2000 by Li et al.17 In the experimental procedure Mg metal was used as magnesium source and en as a templating agent. The hydrothermal reaction (eqn (2)) leads to the production of rod-like nanoparticles in a process described as “soft templating”.
| Mg + 2H2O → Mg(OH)2 + H2 | (2) |
The key influence in the crystal growth mechanism is believed to be the presence of the en molecules, which are likely to act as bidentate ligands to form a complex with Mg2+ cations, thereafter controlling the nucleation and growth of the nanorods. The stability of such a complex is expected to decrease as temperature and pressure are increased, coordinating the OH− groups present in solution to the complex and causing the 1D nanorod structures to condense. Ultimately, the Mg–N bonds to the donor ligands become weaker whilst Mg–O bonds form gradually until the point is reached where Mg and N are separated from each other and Mg cations are connected to OH− anions, forming Mg(OH)2. However, in 2001, Ding et al. published a seminal paper in terms of a directed hydrothermal synthesis of Mg(OH)2. They reported the preparation of nanostructured MH in which one could control size and morphology selectively. Rod-, tube-, needle- or lamellar-like nanoparticles could be produced by varying the aqueous solvent composition as well as the magnesium source (Mg, MgSO4 and Mg(NO3)2) and tuning the hydrothermal reaction conditions.18 Ethylendiamine solution, aqueous ammonia or dilute sodium hydroxide solution were investigated as possible solvents and the results obtained are summarised in Table 1.
| Mg source | Solvent | Temperature (T)/K | Time/h | Morphology | Dimensions/nm |
|---|---|---|---|---|---|
| Mg | en–H2O (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
453 | 20 | Rod-like | Diameter: 20, length: 200 |
| Mg | en–H2O (1:6) |
453 | 20 | Lamellar | Diameter: 50–100, thickness: 10 |
| Mg | NH3·H2O (pH 10) | 453 | 20 | Lamellar; tube-like | Diameter: 25–200; outer diameter: 40, length: 60 |
| MgSO4 | en–H2O (4:1) |
453 | 20 | Needle-like | Diameter:10–20, length: 50–100 |
| MgSO4 | en–H2O (1:1) |
453 | 20 | Lamellar | Diameter: 100–150 |
| MgSO4 | NH3·H2O (pH 11) | 453 | 20 | Lamellar | Diameter: 150 |
| Mg(NO3)2·6H2O | en | 453 | 20 | Lamellar | Diameter: 80–100 |
| Mg(NO3)2·6H2O | NH3·H2O (pH 10) | 453 | 20 | Lamellar | Diameter: 100–200 |
| Mg(NO3)2·6H2O | NaOH (0.1 M) | 353 | 2 | Lamellar | Diameter: 50 |
Two years after Ding et al.'s paper, Fan et al. reported the development of the above concept by employing en itself as a suitable solvent.19 The synthesis used nanowires of Mg10(OH)18Cl2·5H2O as a magnesium source to yield hollow Mg(OH)2 nanotubes. The synthesis of the hydrated hydroxide chloride precursor was previously reported by Christensen et al.20
Following a synthetic procedure analogous to the one reported by Ding et al. (discussed above),18 it was possible to produce nanotubes defined by outer diameters of 80–150 nm, wall thicknesses of 30–50 nm and lengths of up to 5–10 μm. A growth mechanism has been proposed that involves the exchange of Cl− and OH− anions during solvothermal treatment. The mechanism of crystal growth is consistent with the one originally proposed by Li et al.,17 in which en is also believed to play a pivotal role in controlling the product morphology by acting as a bidentate ligand leading to the production of the one dimensional structure. However, the fibre-like morphology of the magnesium precursor is also believed to be very important, as one might anticipate.19 In the following year (2004), many of the same researchers published further work providing additional insight into the above-mentioned system together with the study of two other possible solvents: 1,6-diaminohexane and pyridine.21 It was found that size and morphology of the hydroxide product were greatly influenced by the solvent used and by the reaction temperature during the solvothermal process. In particular, both en and diaminohexane lead to the formation of nanotubes with outer diameters of 80–300 nm, wall thicknesses of 30–80 nm and lengths of several microns, whilst the use of pyridine resulted in a rod-like morphology. This difference in morphology is attributed to the coordination behaviour of the respective ligands; both en and diaminohexane act as bidentate ligands whereas pyridine is a monodentate ligand. Nonetheless, the mechanism of crystal growth for all structures is believed to be the fundamentally identical to that reported previously (in 2003).19 Interestingly several years later, high aspect ratio nanowires of “Mgx(OH)yClz·nH2O” were used in the templated pseudomorphic synthesis of MH nanowires (by reaction with NaOH in 3:1 ethanol:water) at 25–70 °C and ambient pressure without the need of additional chelating species.22
Subsequent to the original oxychloride nanowire template experiments above, a solvothermal process was developed for yielding hollow MH nanotubes without the requirement of such a template or for chelating ligands such as en and diaminohexane. In this case, magnesium chloride was used as the Mg source and aqueous ammonia was employed as a solvent.23 By stopping the progress of the reaction at different times and characterising the reaction mixture, it was possible to propose a mechanism of nanotube formation. Evidence suggested that the process involved anisotropic growth-dissolution–reorganization. Under solvothermal conditions, the precursor dissolves as the temperature increases and as the concentration of the Mg precursor reaches and exceeds saturation, the chloride recrystallises and grows into nanoplates. These plates are then proposed to dissolve again to form smaller growth nuclei, which subsequently reorganize to yield hollow nanotubes of Mg(OH)2.
An ammonia-hydrothermal method has also been tested in the presence of established surfactants and templating agents. Magnesium hydroxide hexagonal nanoplates can be prepared by hydrothermally reacting a solution of MgCl2, citric acid and aqueous ammonia in the presence of monoethanolamine (MEA), diethanoamine (DEA) or triethanolamine (TEA).24 MEA, DEA and TEA were believed to promote the dissolution–recrystallisation process of the nanostructured MH taking place during the hydrothermal treatment. Just as with the use of ligating amines, such as en, the ethanolic amines were proposed to act by forming complexes with Mg. The mechanism requires dissolution of the precursor, so the complexing is apparently crucial in governing solubility. It was found that MEA exhibits the best complexing effect towards Mg cations, hence promoting the dissolution process and contributing to the recrystallisation of hexagonal MH nanoplates. Cetyltetramethylammonium bromide (CTAB) has also been investigated as a possible cationic surfactant for obtaining ultrafine, highly-dispersed magnesium hydroxide lamellar nanoparticles in aqueous ammonia.25 Using magnesium chloride hexahydrate as an Mg precursor, Yan et al. evaluated the significance of the hydrothermal treatment by performing the synthesis in solution at room temperature with or without an additional final hydrothermal step. Important differences were observed in the obtained morphologies; with no hydrothermal treatment, flower-like Mg(OH)2 nanoparticles formed from the intergrowth of platelets while with an additional hydrothermal step it was possible to synthesise dispersed hexagonal nanoplates of MH. In this sense CTAB does not prevent agglomeration when the reaction is carried out at ambient temperature, but it appears to inhibit the intergrowth of the particles during hydrothermal treatment leading to a much higher dispersion of primary nanoparticles (nanoplates). However, it would appear that the individual (nano)plates also grow larger when synthesised in the presence of CTAB. Its effect on the crystal growth may be explained by invoking a dissolution–precipitation mechanism as proposed previously by Wu et al. for the hydrothermal synthesis of nanostructured Mg(OH)2 in the presence of CaCl2.26 According to this mechanism, the first step is the dissolution of Mg(OH)2, followed by the dissociation to “[MgOH]+”, Mg2+ and OH−, with the last step being the combination of the ions to yield Mg(OH)2. It is believed that [MgOH]+ is the rate controlling component of the process, as its concentration is lower than that of Mg2+ and OH−. The CTAB, being a cationic surfactant, is believed to combine with OH− groups (as CTA+), promoting the formation of Mg2+ as well as [MgOH]+ and accelerating the dissolution of small crystals. CTAB was again employed for the preparation of Mg(OH)2 nanostructures via a hydrothermal method in 2011. In this case, uniform nanorods with a length up to 300 nm and a diameter of 10–40 nm were obtained using magnesium oxide as a source of Mg, which was dissolved in an aqueous solution of CTAB.27 For comparison, the synthesis was performed with and without CTAB and as a result, interesting differences in morphologies were observed. In the absence of CTAB, hexagonal nanoplates were observed. By contrast the synthesis including CTAB was found to produce nanorods. Nanorod formation is again speculated to be attributed to the interaction of the positively charged CTA+ cations and OH− groups. This time the interaction leads to an adsorption of CTAB on the nanocrystal faces which are parallel to the c-axis, which prevents solute diffusion to these faces. The growth along the lateral facet is constrained which favours 1D growth from the crystal nuclei, ultimately producing rods rather than hexagonal plates.
Nanostructured MH materials originating from hydrothermal syntheses can also possess high porosity. The production of mesoporous nano-Mg(OH)2 obtained hydrothermally using only MgO and H2O was originally reported towards the start of the 21st century.11 The synthetic procedure does not involve the use of any surfactants or templating agents and was found to produce single phase Mg(OH)2 nanoparticles characterised by an hexagonal platelet-like morphology with a thickness of 50–110 nm and lateral dimensions of several μm (Fig. 2). Plates were found to agglomerate to form larger almost spherical particles with typical diameters over tens of microns. The particles exhibited a bimodal distribution of mesopores, measuring 3.6 nm and 36 nm in diameter, to give a BET specific surface area of 100 m2 g−1. It was hypothesised that the growth process starts with the dissolution of bulk MgO in water, leading to the formation of primary nanoparticles. The porosity of the hydroxide originates during the aggregation of these primary particles resulting in the production of the wormhole-like mesoporous nanoplates.
 | ||
| Fig. 2 High magnification SEM images of hexagonal mesoporous Mg(OH)2 nanoplates obtained via surfactant-free hydrothermal synthesis. Reprinted with permission from J. C. Yu, A. Xu, L. Zhang, R. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64–70. Copyright 2004 American Chemical Society.11 | ||
Sodium hydroxide solutions using different solvents have been explored as components of solvothermal synthesis of nanostructured Mg(OH)2. Ethanolic and aqueous solutions of sodium hydroxide were used in the solvothermal treatment of 5 Mg(OH)2·MgSO4·3H2O (“513 MOS”) nanowire precursors in order to obtain different one-dimensional Mg(OH)2 nanostructures.28 The 513 MOS precursor was initially prepared by hydrothermal treatment of a slurry containing MgSO4 solution and ammonia at 150 °C (giving nanowires of 100–200 μm in length and of 80–200 nm in diameter). Using 1 mol L−1 NaOH aqueous solution yielded hexagonal Mg(OH)2 plates (with a diameter of ca. 200–600 nm) but the morphology of the MH could be modified by changing the sodium hydroxide concentration and solvent.
Reducing the concentration of the NaOH (aq) solution to 0.2 mol L−1 led to predominantly rectangular MH particles (length ∼600 nm–2.0 μm; width ∼150–300 nm) whereas by changing solvent from water to ethanol (maintaining a concentration of 0.2 mol L−1), it was possible to grow 1D MH nanowires (length ∼10–20 μm; diameter ∼100–200 nm) selectively. This difference in morphology was attributed to the different solubility of MgSO4 (rather than that of the product Mg(OH)2) in aqueous and ethanolic NaOH. The reduced solubility and slower dissolution of MgSO4 in ethanol is believed to be responsible for the slower release of SO42−, favouring the formation of the 1D wire morphology. Related to this work, Gao and Jia used aqueous NaOH in the synthesis of nano-MH from magnesium acetate in the presence of citrate. In this case, the product morphology depended primarily on the hydrothermal treatment time; nano-MH was transformed from nanosheets to nanodiscs by prolonging the heating duration.29 In fact, under these conditions, a continuous evolution of morphology was observed as a function of time. On heating at 180 °C for 6 h both nanosheets (<5 nm thick) and nano-needles (3–5 nm in diameter and 50–100 nm in length) were produced. It was suggested that the latter formed from the vertical aggregation of nanosheets, where local stresses lead to a decrease of the surface-free energy. On heating for 10 h, nanosquares of 150–250 nm in size were fabricated in addition to sheets. These nanosquares were each composed of several sheets. On further heating (20 h), disc-like nanocrystals were the only product. The overall transformation was proposed to occur in two steps: first, nanosquares form by the vertical aggregation of nanosheets and second, the oriented aggregation of nanoparticles on the edge of the nanosquares form nanodiscs at longer reaction times. In 2015 a concept of post-synthesis surface modification for nano-Mg(OH)2 was introduced.30 Magnesium acetate and aqueous NaOH were employed in synthesis via a continuous flow hydrothermal (supercritical) process. Poly(ethylene-co-acrylic acid) in toluene was used as a surface coating agent and injected downstream from the hydrothermal reactor. MH nanorods with a diameter of ca. 20 nm and a length of ca. 120 nm were produced without the addition of modifier whereas smaller, more porous hydrophobic Mg(OH)2 nanorods (∼15 nm diameter; length ∼70 nm) were produced in the surface-modified process (Fig. 3).
 | ||
| Fig. 3 TEM images of (a) unmodified and (b) poly(ethylene-co-acrylic acid)-modified Mg(OH)2 nanorods grown by continuous flow hydrothermal processing. Reprinted from Powder Technology, 278, S. Elbasuney and S. F. Mostafa, Continuous flow formulation and functionalization of magnesium di-hydroxide nanorods as a clean nano-fire extinguisher, 72–83, copyright 2015, with permission of Elsevier.30 | ||
Although ostensibly the morphologies appear very similar by transmission electron microscopy (TEM), the specific surface area of the higher porosity poly(ethylene-co-acrylic acid)–Mg(OH)2 nanorods is greater by an order of magnitude (31.104 m2 g−1vs. 2.803 m2 g−1).
Hydrazine was first proposed as a reagent in the hydrothermal synthesis of Mg(OH)2 in 2008. Well-defined hexagonal Mg(OH)2 nanoflakes (thickness ∼40 nm) result from simply reacting magnesium nitrate and hydrazine hydrate (N2H4·H2O) hydrothermally (Fig. 4).31 A mechanism of crystal growth was proposed, again involving ligation at the Mg centre as in the case of previous experiments with en as a ligand. In this case, the formation of the complex cation [Mg(N2H4)6]2+ is proposed to form in solution prior to the hydrothermal treatment. The hydrazine hydrate molecule has lone pair electrons and so could readily act as an electron pair-donating hard Lewis base in combination with Mg2+ as an electron pair-accepting hard Lewis acid. During the hydrothermal process the hydrazine is believed to dissociate into NH3 and N2, with the former generating OH− anions in aqueous solution, ultimately leading to the formation of Mg(OH)2. In comparison to other alkalis added to MH syntheses, such as sodium hydroxide and aqueous ammonia as discussed above, hydrazine hydrate first has to undertake a decomposition process that leads to a relatively slow release of OH− under solvothermal conditions. The slower reaction and growth should favour a more measured crystallization process resulting in the symmetrical, regular hexagonal morphology of the single nanocrystallites. The authors also suggested that steric effects from the NO3− anions plane could restrict growth in the third dimension and favour formation of the lamellar structure, although it is not clear why this should be the case.
 | ||
| Fig. 4 TEM image and selected area electron diffraction (SAED) pattern (inset) of a regular hexagonal Mg(OH)2 nanoflake synthesised from Mg(NO3)2 and N2H4·H2O. Reprinted from Materials Chemistry and Physics, 112, D. Jin, X. Gu, X. Yu, G. Ding, H. Zhu and K. Yao, Hydrothermal synthesis and characterization of hexagonal Mg(OH)2 nano-flake as a flame retardant, 962–965, copyright 2008, with permission of Elsevier.31 | ||
More recently, well-dispersed, discrete hexagonal MH nanoflakes were obtained hydrothermally using magnesium nitrate hexahydrate in the presence of the polymeric templating agent, PEG 20000.32 As shown in Fig. 5, the synthesised magnesium hydroxide nanoparticles present a hexagonal flake-like morphology with a thickness of only ca. 10 nm, a lateral size of ca. 100 nm and a specific surface area of ∼69 m2 g−1. In the absence of PEG 20000, similarly prepared MH particles exhibited a higher polydispersity and a slightly reduced specific surface area (∼42 m2 g−1). There is no firm evidence to indicate the exact role played by PEG in the MH nanoflake formation mechanism and the authors speculate that its main functions are to prevent the agglomeration of the hexagonal Mg(OH)2 nanoflakes and to control the growth rate. The morphology of the synthesised nanocrystals is proposed to originate from the formation of “PEG–Mg2+ pairs”. The authors proposed that the “PEG–Mg2+ pairs” develop initially via complex formation, with PEG coordinating to Mg2+ ions in Mg(NO3)2 through oxygen donors prior to the hydrothermal treatment. These “PEG–Mg2+ pairs” are then proposed to combine during stirring and aging to form a skeleton of PEG chains that are twisted and coiled with each other to form a series of continuous hexagonal pores. When NaOH was added to the system, the OH− ions are proposed to react preferentially with Mg2+ ions located in the hexagonal holes of the entangled PEG skeleton to form small crystal nuclei of magnesium hydroxide. With increasing temperature and time over the period of the hydrothermal treatment, these Mg(OH)2 crystal nuclei grow until finally they detach from the hexagonal pores (which act as a template). Compared to synthesis and hydroxide growth without PEG, the initial coordination process and the subsequent PEG templating process is predicted to be slow in kinetic terms. It is evident that further syntheses using varying molecular weight polymeric additives and closer scrutiny of the ensuing crystallisation processes are required in order to understand the templated MH growth in such systems more fully and a fuller comparison with PEG-templated coprecipitation methods performed at ambient pressure (see below) would be useful.
 | ||
| Fig. 5 SEM images of Mg(OH)2 nanoflakes (a) synthesised with PEG20000 (at low magnification); (b) synthesised with PEG20000 (at higher magnification) and (c) synthesised without PEG 20000. Reprinted from Materials Research Bulletin, 51, Q. Wang, C. Li, M. Guo, L. Sun and C. Hu, Hydrothermal synthesis of hexagonal magnesium hydroxide nanoflakes, 35–39, copyright 2014, with permission of Elsevier.32 | ||
From all the examples in this section, it can be seen that hydrothermal/solvothermal methods can be widely utilised to obtain Mg(OH)2 nanostructures with degrees of control over size and morphology.
The main advantages lie with a high level of reproducibility and generally the method encourages production of 2D plate-like nanostructures with dimensions and porosity that can be modified by appropriate selection of experimental parameters (such as temperature, time and pH) and choice of reagents. Such syntheses typically lead to a low degree of agglomeration and opportunities to produce monodisperse MH particles of high surface area. By contrast, the growth of 1D nanostructures is rarer and more difficult, requiring more “forcing” growth conditions such as the use of hard templates (e.g. nanowire precursors). Nevertheless, solvothermal methods allow one to tailor the crystal growth to different morphologies for different applications.
Given the range of chemical variables applied in solvothermal syntheses – such as solvent, magnesium source, molecular (ligating) templating agents (e.g. en, en–H2O, diaminohexane, pyridine or hydrazine hydrate) or other soft/hard templates – it is difficult to appreciate the precise role of each of these variables in a single set of experiments and even more complex to consider their combinations across multiple synthesis regimes. The nucleation and crystal growth is often initiated by the ability of molecular additives (acting as ligands) to form coordination complexes with Mg2+ cations (typically present in a salt precursor or oxidised from Mg metal). To some extent the nanorod, nanoneedle, nanotube or various lamellar morphologies should depend on the coordination behaviour of the respective ligands to the metal centre as well as on the magnesium precursor employed and the morphology of the precursor itself. Syntheses involving aqueous ammonia (for example, in the formation of MH nanotubes23) are interesting in that coordination complex formation apparently need not be an initial step in the nucleation and subsequent growth process and the proposed mechanisms involve anisotropic growth-dissolution–reorganization processes. In such cases, the precursor dissolves and as the temperature/time increases recrystallization is induced and the use of ethanolic amines with aqueous ammonia, for example, was thought to promote this dissolution–recrystallisation process.24
Synthesis via microwave-assisted methods
The use of microwaves as an alternative to conventional heating for material synthesis in the solid state as well as in solution has attracted considerable attention.33–37 In fact, using microwave (MW) heating can enable reaction times to be dramatically decreased from days to even a matter of minutes when compared to conventional heating approaches. Great progress has been made over the past decade in terms of the MW preparation of nanostructured Mg(OH)2 (Table 2).| Mg source | Solvent/additives | Power/W | Time/h | Morphology | Dimensions | Ref. |
|---|---|---|---|---|---|---|
| a All performed at 2.45 GHz. b Reagents directed through semi-permeable collodion membrane. c MWs applied to form plasma. d To prepare 60 g. e Hydrothermal conditions. | ||||||
| Mg(NO3)2 | Water/NaOHb | 20 | 120 | Fibrous | Diameter: 20–40 nm, length: 100–150 nm | 38 |
| MgCl2·6H2O | Water/NaOH/urea | 1000 | 0.5 | Spherical/lamellar | Sphere diameter: 30 μm; plate thickness: 95 nm, plate diameter: 1–10 μm | 13 |
| MgSO4 | Water/NaOH/CTAB/ethanol | 266 | 0.25 | Lamellar | Thickness: 5 nm | 39 |
| Mg (rod) | Water | 160c | 1d | Lamellar | Diameter: 25–125 nm | 40 |
| Mg (powder) | Water | 180 | 0.13 | Lamellar | Diameter: ca. 100–1000 nm thickness: 36–45 nm | 41 |
| MgO | Watere | 800 | 0.03 | Lamellar | Diameter: 100–600; thickness: 10–60 nm | 6 |
In 2004, Wu et al. reported the first synthetic route to yield fibre-like Mg(OH)2 nanoparticles exploiting microwave heating.38 Magnesium hydroxide nanoparticles were obtained by reacting magnesium nitrate and sodium hydroxide in aqueous solution. The reagents were directed though a semi-permeable membrane made from collodion (nitrocellulose in ether/alcohol) while irradiating them with low power microwaves (20 W, 2.45 GHz) for 5 days. This resulted in the production of fibre-like nanoparticles (diameter ∼20–40 nm; length ∼100–150 nm). An equivalent experiment performed without irradiation produced MH lamellae (diameter ∼300 nm) together with agglomerates of fibres. The authors hypothesised that both microwave irradiation and the use of the membrane (and additionally the concentration of NaOH and hence the pH) were important factors in the formation of the fibre-like morphology of the hydroxide nanoparticles rather than the lamellar morphology otherwise observed. An unconfirmed mechanism for fibre formation was proposed by which local microwave-induced hot spots provoke Mg(OH)2 plates into curling into fibre-like nanoparticles. It was also observed that when the concentration of NaOH > 0.1 M, the reaction proceeded at a high rate such that magnesium hydroxide nanoparticles aggregated. A more complete understanding of the rationale for nanofibre formation in the absence of hard or soft templates has not yet been forthcoming.
More recently, other magnesium salts have been studied as precursors in the MW synthesis of nanostructured magnesium hydroxide. In 2012, Al-Hazmi et al. employed a microwave-assisted solvothermal route to induce the rapid growth of hydroxide nanosheets, which can find application in the fabrication of ethanol sensors.13 Magnesium chloride hexahydrate, sodium hydroxide and urea were irradiated with microwaves at 1000 W with a temperature limit of 220 °C. The reaction reached completion after 30 min and produced hierarchical structures with a spherical morphology (diameter ∼30 ± 3 μm). SEM revealed the ball-like structures were agglomerates of nanosheets, each with a thickness of 95 ± 10 nm and lateral dimensions of up to several μm (Fig. 6).
 | ||
| Fig. 6 Typical (a and b) low-magnification and high-resolution (c–f) FESEM images of Mg(OH)2 nanosheet networks as synthesised via microwave hydrothermal processing. Reprinted from Journal of Alloys and Compounds, 519, F. Al-Hazmi, A. Umar, G. N. Dar, A. A. Al-Ghamdi, S. A. Al-Sayari, A. Al-Hajry, S. H. Kim, R. M. Al-Tuwirqi, F. Alnowaiserb and F. El-Tantawy, Microwave assisted rapid growth of Mg(OH)2 nanosheet networks for ethanol chemical sensor application, 4–8, copyright 2012, with permission of Elsevier.13 | ||
Magnesium sulfate has also proved to be an effective precursor for Mg(OH)2 nanosheets, with a structure of highly porous sheets made of particles smaller than 5 nm and a surface area of 80.27 m2 g−1.39 The synthesis is based on the reduction of magnesium sulfate by sodium hydroxide in a microwave reactor (266 W; 10–15 min), using cetylmethylammonium bromide (CTAB) as dispersant. After precipitation, the synthesised particles were subjected to thermal treatment at 300 °C to remove the CTAB and obtain pure phase Mg(OH)2.
One of the first studies to use magnesium metal as a precursor for the microwave synthesis of nano-Mg(OH)2 in fact employed an indirect method. In 2011, Hattori et al. used microwave-generated plasma in water in the continuous synthesis of MH from Mg rod (at 60 g h−1); a method that could also be used to prepare Zn(OH)2.40 Characteristic hexagonal nanoplates of Mg(OH)2 were produced (alongside (truncated) triangular crystallites) using an incident power of only 160 W. More recently direct MW synthesis was demonstrated by simply heating magnesium powder and water. Al-Gaashani et al. detailed an additive-free MW synthesis of nano-MH achieved by irradiation in a domestic microwave oven for 8 minutes without the addition of surfactants or templating agents.41 However, the reaction resulted in the presence of two different Mg(OH)2 morphologies: a mixture of nanoparticles and nanosheets of 36–45 nm in thickness were obtained. The proposed reaction mechanism invokes the combination of Mg2+ cations with hydroxide ions as follows (eqn (3)):
| Mg2+(aq) + 2OH− ↔ MgOH+ + OH− → Mg(OH)2 | (3) |
The MgOH+ ions (magnesium hydroxo ions) are believed to act as the precursor in producing the Mg(OH)2 nuclei that initiate the growth of both particles and sheets (Fig. 7). Only the monomeric MgOH+ cation (of several hypothetical [Mgp(OH)q](2p–q)+ species) has been previously identified with confidence as forming prior to magnesium hydroxide (brucite) precipitation (typically at high pH) and at high temperature the cation's existence is fleeting at best.42,43 Al-Gaashani et al. also suggested (with reference to Zn(OH)2/ZnO formation) that the exposure of the reactant solution to MW irradiation without stirring could promote differences in the temperature distribution and to the viscosity of the solution, leading ultimately to different morphologies.44 In fact, if one considers this non-uniform temperature profile and the relative stability of the cations, it is perhaps not unreasonable to suppose that the formation of [MgOH]+ is localised and crystallite formation could proceed either directly from Mg2+ or via the hydroxo cation.
 | ||
| Fig. 7 Mechanism of crystal growth hypothesises by Al-Gaaneshi et al. reprinted from Journal of Alloys and Compounds, 521, R. Al-Gaashani, S. Radiman, Y. Al-Douri, N. Tabet and A. R. Daud, Investigation of the optical properties of Mg(OH)2 and MgO nanostructures obtained by microwave-assisted methods, 71–76, copyright 2012, with permission of Elsevier.41 | ||
In 2015, a surfactant-free hydrothermal MW synthesis inside a multimode cavity MW reactor was proposed, which could yield gram-quantities of single-phase nano-Mg(OH)2 from only MgO and without the use of additives.6 The synthesis was performed hydrothermally in a Teflon-lined autoclave and the reaction time could be decreased from 6 to 2 minutes by increasing the incident power from 750 to 800 W. The hexagonal nanoplates so-produced were 100–600 nm across and with a typical thickness of 10–60 nm (Fig. 8). The experimental hydrothermal procedure followed is broadly the microwave analogue to the conventional one proposed by Yu et al.11 The mechanism of crystal growth proposed for the MW–HT synthesis contrasts slightly with the one suggested by Yu et al. and it consists of dissolution–precipitation steps followed by crystallite growth (eqn (3), Fig. 9).
 | ||
| Fig. 8 (a) SEM micrograph of hexagonal nanoplates of Mg(OH)2 obtained in 4 minutes at 800 W; (b) SEM micrograph showing the thickness of individual nanoplates from the same sample. Reproduced from ref. 6 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 9 Proposed growth processes for (a) conventional hydrothermal synthesis proposed by Yu et al.11 and (b) MW-hydrothermal synthesis proposed by Hanlon et al.6 reproduced from ref. 6 with permission from the Royal Society of Chemistry. | ||
The use of MWs results in a much higher rate of both heating and cooling and this could lead to an extremely fast initial MgO dissolution step (the solubility of MgO and Mg(OH)2 increases with increasing temperature45,46). This is believed to be followed by the formation of magnesium hydroxide on the oxide surface via intermediate Mg(OH)+ species and then by the removal of Mg(OH)2 from the MgO surface (eqn (4)).47,48
| MgO(s) + H2O(l) → MgOH+(ads) + OH−(aq) → Mg(OH)2(s) | (4) |
The possible agglomeration of the particles is believed to occur on cooling. The mechanism (Fig. 9b) shows that the use of templating agents or surfactants is not essential for the synthesis of nano-MH, but that their use could help in suppressing the agglomeration of the nanoparticles, resulting in an increased surface area. Yu et al. had previously proposed that in conventional hydrothermal synthesis the agglomeration of small primary nanoparticles into nanoplates leads to the formation of a bimodal distribution of mesopores.11
Precipitation methods
Solution based precipitation and co-precipitation methods are attractive ways to prepare MH under relatively mild conditions. Routes with and without the use of surfactants and surface modifiers have been explored as means to obtain nanostructures with controlled size and morphology (Table 3). In 1993, Láska et al. started studying the influence of experimental parameters on the size distribution of magnesium hydroxide prepared by hydrating magnesium oxide, concluding that increasing the pH of the solutions resulted in a smaller crystal size, whereas raising the reaction temperature resulted in the opposite effect.49 Ten years later, Henrist et al. conducted a systematic and wide-ranging investigation of the influence of a host of synthesis parameters including the chemical nature of magnesium precursors with different counter-ions (MgCl2 and Mg(NO3)2), the chemical nature of the basic precipitating agent (NaOH, NH4OH) and the variation in reaction temperature. The ambient pressure precipitation process was also compared to hydrothermal syntheses.50 It was reported that the use of sodium hydroxide results in the preparation of “cauliflower-like” agglomerates, whilst the use of aqueous ammonia leads to the formation of hexagonal platelets. This contrast was attributed to the influences of pH and cation structure in solution. Also, as one might expect, the temperature has a strong effect, affecting the degree of agglomeration of the precipitated nanoparticles. Notably, particles tend to intergrow (agglomerate) at 60 °C and above, whereas at lower temperature, single, pseudo-circular platelets are obtained. A comparatively mild (180 °C; 14 h) hydrothermal treatment results in an increased mean particle size and decreased specific surface area.| Mg source | Additives | pH | T/K | Morphology | Dimensions | Ref. |
|---|---|---|---|---|---|---|
| a Aggregates of spherical particles from NaOH, lamellae from NH4OH. b Not reported. c Rods formed on addition of CuSO4·5H2O. d Dispersed best with PEG 200. e Agglomerated. f Dispersed. | ||||||
| MgO | NaOH/NH3, TEA | 10.2–13.0 | 393–413 | Lamellar | Ave. diameter: 1.5–3.4 μme | 49 |
| MgCl2/Mg(NO3)2 | NaOH/NH4OH | ca. 10–13 | 283–313 | “Cauliflower” agglomerates /lamellara | Sphere diameter: ca. 300 nm;e plate diameter ca. 200–450 nmf | 50 |
| Mg(NO3)2·6H2O | NaOH, urea/EtOH | 333 | Lamellar | Diameter: 50–200 nmf | 52 | |
| MgSO4 | NaOH, MgSA, CuSO4·5H2O | 273–353 | Lamellar/rod-likec | Plate diameter: ∼50–100 nm; rod length: ∼100–300 nm, rod diameter: ∼10–30 nmf | 54 | |
| MgCl2·6H2O | NaOH, ODP | Lamellar | Diameter: ∼100 nm; thickness: ∼35 nmf | 55 | ||
| Mg(NO3)2·6H2O | NaOH, SDS, MAP | 358 | Lamellar | Diameter: ∼100 nmf | 56 | |
| MgCl2·6H2O | NH4OH, PEG 400 | 323 | Lamellar | Diameter: ∼100 nm; thickness: 10–20 nmf | 12 | |
| MgCl2·6H2O | NH4OH, PEG 12000 | 293–353 | Needle-like/lamellar | Needle length: ∼1 μm; plate diameter: ∼200 nmf | 57 | |
| MgSO4 | NaOH, PEG 200/PEG 8000/PEG 20000 | 353 | Lamellar | Diameter: ∼30 nm-5 μmd,f | 58 | |
| MgSO4 | NaOH, OA, PMMA | 343 | Lamellar | Diameter: ∼100 nmf | 60 | |
In 2007, Zou et al. reported the synthesis of lamellar MH nanostructures obtained from the oxidation of magnesium metal in a mixture of formamide and water.51 The proposed one-step synthesis produces densely packed layers of agglomerated Mg(OH)2 particles as a result of simply immersing magnesium ribbons in a 6% formamide/water mixture at 80 °C for 12 h. Further, as shown in Fig. 10, the growth of the lamellar structures could be moderated as a function of reaction time when using a 4% formamide/water mixture at 80 °C. Initial Mg oxidation was very slow, limiting the Mg concentration and inducing heterogeneous nucleation preferentially on the metal substrate. As the reaction time increased, Mg species originating from the thermal decomposition of Mg–formamide complexes were continuously supplied for subsequent crystal growth on heterogeneous nuclei. Fig. 10(a) shows that at 1 h rod-like nuclei grow on the substrate from heterogeneous nucleation of MH. At 2 h, the nuclei start to branch on the surface, gradually forming the beginnings of a 3D porous skeleton for further growth (Fig. 10(b)). By 6 h into the nucleation and growth process, the lamellar MH structures develop into a continuous porous network (Fig. 10(d)).
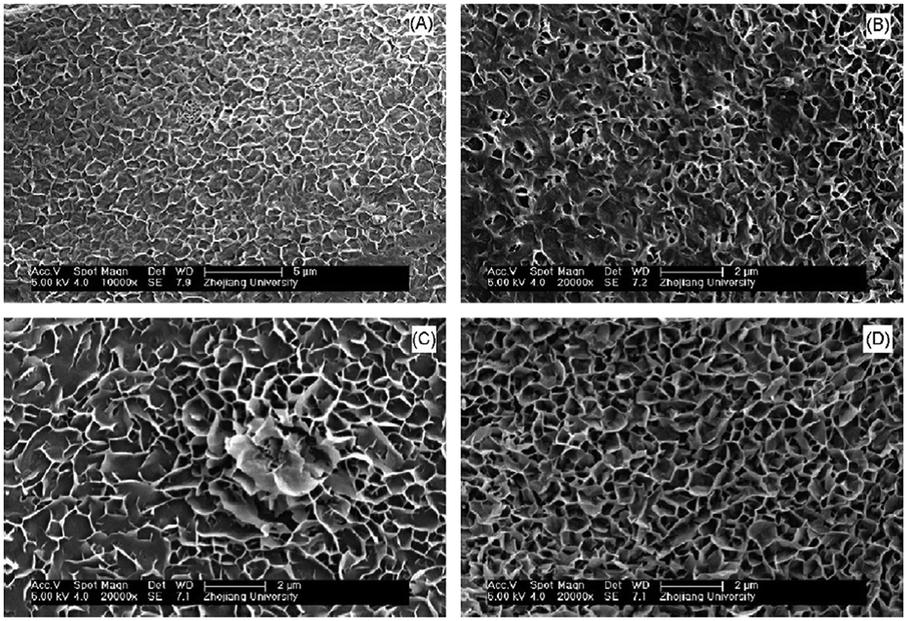 | ||
| Fig. 10 Time-dependent evolution of magnesium hydroxide particle crystal morphology at different stages for Mg metal immersed in a 4% formamide/water solution at 80 °C: (A) 1 h, (B) 2 h, (C) 3 h, (D) 6 h, respectively. Reprinted from Materials Research Bulletin, 42, G. Zou, R. Liu, W. Chen, Z. Xu, Preparation and characterization of lamellar-like Mg(OH)2 nanostructures via natural oxidation of Mg metal in formamide/water mixture, 1153–1158, copyright 2017, with permission of Elsevier.51 | ||
An alternative surfactant-free precipitation route to lamellar MH nanoparticles is possible via reaction of aqueous solutions of magnesium chloride hexahydrate and sodium hydroxide in the presence/absence of urea and/or ethanol.52 Addition of ethanol apparently produced more regular hexagonal lamellae. Although no surfactants were employed, urea evidently prevented agglomeration presumably by coordinating to Mg2+ in solution. Moreover, chloride impurities were reduced on urea addition. In fact, an earlier study showed similar nanostructured MH lamellae could be obtained from lower concentrations of the same reactants without the use of urea (or other structure directing additives).15
Although surfactant-free syntheses are environmentally favourable, there is no doubt that a very high level of control over size and morphology of nanomaterials can be achieved when employing surfactants, dispersants and surface modifiers in precipitation processes. Over a decade ago, Lv et al. were able to produce MH nanoparticles with three different morphologies exploiting the precipitation process of a magnesium precursor species (MgCl2·6H2O) in the presence of different complex dispersants and surfactants.53 In their comprehensive study, they also studied the influence of synthesis parameters such as temperature, concentration and type of precipitating reagent (NH4OH, NaOH). A multitude of additives were investigated; gelatin, lauryl sodium sulfate, polyvinylpirrolidone, polyglycol ether, polysorbate 80, polyvinyl alcohol, sodium polymethacrylate and polyacrylamide. By tuning the various reaction conditions, it was possible to obtain needle-, lamellar- and rod-like nanoparticles selectively. Although the selection of complex dispersant/surfactant was instrumental in checking growth and controlling size distribution, the alkali solution concentration appeared to be more important in controlling morphology. The use of lower concentration aqueous ammonia (5 wt%) promoted the formation of 1D structures, whereas a higher concentration (25 wt%) promoted the formation of lamellae. It was proposed that the polymer dispersants exert more structural control at the lower NH3(aq) concentration, anisotropically limiting the growth of crystal nuclei resulting in needle-like particles which became rods when ammonia was added at a much slower rate. At the higher NH3(aq) concentration, the structure directing effect of the dispersants was less significant and crystal nuclei agglomerate and form lamellar-like particles.
Morphological selectivity was also achieved in the presence of magnesium stereate (MgSA) as a surfactant, although as in the example above, the two different observed morphologies of MH (lamellar and rod-like particles) were more likely determined by other factors.54 Magnesium(II) sulfate and NaOH solution were used to coprecipitate MH in the presence of MgSA. Notably, it was only possible to move from growth of MH lamellae to rod-like MH crystals when copper(II) salts were added. The premise was presented that similarly sized Cu2+ could partly replace Mg2+ within individual octahedra in the MH structure during crystallization. It was proposed that adding Cu2+ (0.4 mol%) into magnesium salt solution moderates growth in specific directions to form rod-like nuclei, which evolve into rod-like crystals. NaOH concentration, reaction time and temperature were also deemed significant (by increasing the temperature from close to 0 °C through 50 °C to 80 °C, the morphology switched from lamellae to rods back to lamellae) mediating the kinetics of Cu2+ diffusion. Ultimately, the role of Cu(II) salts in the growth mechanism is unresolved and whether Cu2+ is included in the MH crystal structure, for example, is unclear.
Octadecyl dihydrogen phosphate (ODP) was proposed to check crystal growth by surface modification.55 In a simple one-step wet precipitation process, Mg(OH)2 nanoparticles were prepared from magnesium chloride hexahydrate, sodium hydroxide and ODP. ODP was proposed to engender hydrophobic (001) surfaces and thus restrict the crystal growth in the c direction. Lamellar nano-Mg(OH)2 plates synthesised in the presence of ODP grew with an average lateral dimension of 60 nm and an average thickness of ca. 35 nm whereas an average lateral dimension of 130 nm and an average thickness of ca. 20 nm predominated when no surface modifier was employed. Similarly sodium dodecyl sulfate (SDS) and monoalcohol ether phosphate (MAP) (2:1 by weight) were applied as surface-modifiers in an MH co-precipitation starting from magnesium nitrate hexahydrate and NaOH.56 Addition of SDS/MAP in different amounts from 0.0 to 1.0 wt% had positive effects on the MH dispersion properties in liquid paraffin with an optimal suspension volume obtained for 0.2 wt% of SDS/MAP. All Mg(OH)2 particles formed hexagonal plates, but the surface-modified nanocrystals were notably smaller in lateral dimensions (across the face of each plate).
Polymeric surfactants might be expected to have an even more pronounced effect on constraining crystal growth. PEG is a widely-used surfactant with properties that can be tuned via the molecular weight of the polymer. Wang et al. utilised PEG 400 in the synthesis of MH nanoplates (to be used as a precursor to nanostructured MgO). MgCl2·6H2O was combined with ammonium hydroxide as a precipitating agent and PEG 400 as a non-ionic surfactant.12 Plates of 10–20 nm in thickness could be produced but were found to agglomerate in large clusters and it was postulated that this was due to the relatively low molecular weight of the PEG, which was not very effective in capping the MH particles. A later study explored the effect of using higher molecular weight PEG 12000 in the synthesis of MH nanoparticles (also using magnesium chloride hexahydrate as the magnesium precursor and aqueous ammonia as the precipitating agent).57 It was found that unlike PEG 400 in the earlier study, PEG 12000 influences both the size and morphology of the synthesised nanocrystals. Close to room temperature (20 °C, 40 °C) needle-like MH nanocrystallites are formed whereas once the reaction temperature is raised to 60 °C or 80 °C, lamellae form selectively. It was postulated that the binding of the PEG 12000 to the MH particles changes with temperature favouring either 1D or 2D growth depending on to what crystal surfaces the polymer preferentially adsorbs. In 2013, Pilarska et al. further investigated the effect of PEG molecular weight on the synthesis of nano-MH (from MgSO4 and NaOH), comparing PEG 200, 8000 and 20000 respectively at concentrations of 1, 2 and 5 wt% each.58 Modification of the PEG molecular weight and concentration had no obvious effect on the shape of the crystallites; platelets were obtained in all cases. Modifying the surfactant concentration did affect the dispersity however, and an optimum concentration of 2% wt of PEG 200 produced hydrophobic nano-MH plates 28–79 nm across with a BET surface area of 131 m2 g−1. The degree of coverage of the MH surface with PEG was suggested to decrease with increasing PEG molecular weight. However, it is worth noting that all the syntheses in this study were performed at 80 °C and so consistent with previous work,12,57 if PEG is adsorbed on (001) faces then Mg(OH)2 nanoplates are preferentially formed. Intriguingly, the use of PEG-1000 was also argued as a means to obtain Mg(OH)2 nanotubes from magnesium chloride hexahydrate and ammonium hydroxide.59 In the absence of HRTEM images and further characterisation, the MH nanotubes (with an estimated inner diameter of 6–34 nm and lengths of 63–200 nm) are difficult to discern, but were synthesised only when 2.5 wt% of PEG 1000 was used in the liquid phase reaction system. Lower concentrations of PEG 1000 led to lamellae (2 wt%) whereas needles were obtained at much higher concentration (3.5 and 4 wt%). Overall, there appears to be a fine balance between 2D and 1D growth with the use of PEG and the preferred morphology is likely very sensitive to PEG molecular weight and reaction temperature (which could, in turn, fragment PEG when increased significantly above ambient).
In a variation on the above methodology, polymers have been successfully employed post-synthesis to improve dispersiblilty of nano-MH still further.60 Magnesium hydroxide nanoparticles with a mean particle size of 100 nm were first obtained from MgSO4 and NaOH using oleic acid (OA) as a structural modifier and to make the MH surface lipophillic. Polymethyl metacrylate (PMMA) (prepared in situ using poly(N-vinyl-2-pyrrolidone) (PVP) and monomeric methyl metacrylate (MMA) with azoisobutyronitrile (AIBN) as an initiator) was grafted onto the OA-rich surface of the nanoplates to form hydrophobic nano-MH lamellae. Hence in this two-step process, initially –OH groups on the MH surface are replaced by the acidic COO– functionality of OA and subsequently the alkyl chains from the affixed OA is incorporated into the PMMA backbone. The MH dispersibility in water is improved considerably as a result; untreated MH phase-separated instantly while PMMA-grafted MH remained dispersed in water for 30 days.
Other synthetic routes
Aside from solvothermal and precipitation methods, a number of other synthetic techniques have been explored for producing nanostructured magnesium hydroxide of controlled size and morphology. Although some of these techniques might lack the simplicity of the above solution-based methods, in many cases experiments have led to unexpected and intriguing outcomes and can provide further insight into nano-MH growth mechanisms. Among these alternative approaches are electrodeposition, liquid–solid arc discharge and pulsed-laser ablation. Progress has also been made in demonstrating how solution-based flow processes can be scaled up towards commercial production of nano-MH.Deposition of metal hydroxides such as Ni(OH)2 and Mg(OH)2 by cathodic reduction of chlorides, for example (in the hydrogen evolution reaction; HER) has been known for some time.61,62 Micron-thick coatings of Mg(OH)2, for example, can be deposited on stainless steel electrodes under basic conditions from reduction of aqueous Mg(NO3)2.63 The films become thicker with increasing deposition time, current density and nitrate concentration and consist of characteristic MH nanoplates (ca. 13 nm thick) that grow with their hexagonal faces approximately perpendicular to the stainless-steel substrate. Following a very similar method, porous films composed of MH nanoplates could also be deposited on to indium tin oxide (ITO)/glass cathodic substrates.64 Interestingly and perhaps counter-intuitively, if a structure-directing agent (chelating species; sodium acetate) is added to the electrochemical system, deposition of clusters of “hierarchical” structures are formed.65 These flower-like objects are formed from the agglomeration of individual MH nanoplates and grow on layers of existing deposited – more aligned – MH films. The number of these clusters increases with deposition time. For a fixed time, increasing the acetate concentration also encourages cluster growth and leads to less uniform films.
Fascinatingly liquid–solid arc discharge produces 1D MH nanostructures, yielding nanorods with a diameter of ca. 10 nm and lengths up to 250 nm.66 The method is based on the discharge between two magnesium metal strips, immersed in a NaCl solution, to which a voltage between 50 and 200 V was applied for set periods of time. The heat generated in the process melts the magnesium strip such that the MH product is distributed throughout the NaCl solution. The proposed reaction pathway involves the formation of a colloidal Mg solution from the metal, followed by oxidation to Mg2+ cations and finally the growth of magnesium hydroxide nanorods. Although the length of the rods (and therefore the aspect ratio), can be increased by extending the discharge time, the underpinning 1D morphology of the synthesised Mg(OH)2 nanorods does not change. This would suggest that the MH morphology is determined in the very early stages of the reaction (and is quite possibly stimulated by the structure of the rapidly solidifying Mg colloidal particles).
In a similar vein, Liang et al. took magnesium metal plate immersed in distilled water (with or without the presence of SDS as a surfactant) as the basis of a synthesis configuration, but in this case pulsed-laser ablated the plate.67 By varying the reaction conditions and amount of surfactant, it proved feasible to obtain wormhole-, tube-, rod- or platelet-like nanoparticles selectively. Further, the authors proposed a multi-step mechanism of crystal growth which starts with the formation of Mg species induced by the laser (initially as a plasma plume above the surface of the metal), followed by their almost instantaneous reaction with water to form Mg(OH)2. The growth of the nanostructured crystals is then principally directed by the surfactant and the selective formation of 1D vs. 2D structures is extremely sensitive to the concentration of SDS; low concentration favours growth of nanorods while increasing the proportion of SDS leads to nanoplates.
Finally, Li et al. demonstrated how a series of flow reactors can be designed to produce hexagonal nanoplates of nano magnesium hydroxide using cheap raw materials.68 Crude calcined magnesia (MgO) was reacted with nitrogen fertiliser ((NH4)2SO4) in the presence of two different surfactants, gelatin and polyvinyl alcohol. The surfactants were added primarily to avoid aggregation of the nanoparticles in micrometric clusters. The reaction proceeds in 2 steps whereby ultimately gaseous ammonia is bubbled through an aqueous solution of magnesium sulfate (eqn (5) and (6)):
| MgO + (NH4)2SO4 → MgSO4 + 2NH3 + H2O | (5) |
| MgSO4 + 2NH3 + 2H2O → Mg(OH)2 + (NH4)2SO4 | (6) |
Both pH and the surfactant concentration had an impact on the morphology of the synthesised MH: surfactant-free reactions at high pH resulted in the formation of flower-like clusters, whilst at lower pH and in the presence of surfactant, discrete hexagonal platelets were obtained. It was highlighted that aside from the direct influence of the surfactant, the pH is extremely important in the process; by exceeding the isoelectric point of Mg(OH)2 in water (pH 12) it was possible to positively charge the surface of the nano-MH, facilitating the adsorption of the negatively charged surfactant and thus preventing aggregation.
Applications of nanostructured Mg(OH)2
The alkalinity, hydrophilicity, thermal stability and low toxicity of magnesium hydroxide has facilitated its application in numerous fields. Traditionally, MH has been widely applied as a non-toxic flame retardant, as an acidic waste neutraliser, as a pharmaceutical excipient and as a precursor to magnesium oxide, MgO, which is widely used in catalysis and as an adsorbent.8–10,12,14 A comprehensive review of the properties of magnesium hydroxide has been published recently and the reader is directed to this text for a more complete description of the many applications of the bulk material.69 As has been discussed in previous sections, advances in the controlled synthesis of nanostructured magnesium hydroxide and in the techniques required to characterise them offer new possibilities in terms of applications, particularly when one considers the range of available nanoscale morphologies (from platelets through needles, rods and tubes to hierarchical structures and films). In this section, we focus both on how specific nano-morphologies can enhance the “traditional” properties of MH and on how such nanomaterials can be exploited in new directions.The low flammability, non-toxicity, low cost and high abundance of MH have long been qualities that have assured its dominance as a flame-retardant filler. In a nanostructured form, the material becomes even more attractive given the potential of enhanced surface area and excellent dispersion. One of the first examples of using nano-MH in composites as a flame retardant was reported in 2003.70 Needle-like and lamellar nanostructures were synthesised via a surfactant-mediated solution method (using PVP as a surfactant) and blended with ethylene-vinyl acetate (EVA) copolymer by milling and hot pressing as 3 mm-thick sheets. The replacement of micrometric (2–5 μm) inorganic material in an MH/EVA composite with nanometric MH (as needle-like nanoparticles with 3–6 nm in diameter and 50–100 nm in length) showed a significant improvement in the flame-retardant properties as evinced by measuring the limiting oxygen index (LOI; a value of 38.3 for the nano-MH/EVA as opposed to 24 for the composite containing micron-sized MH). This enhancement was attributed to the high dispersion of MH nanoparticles in EVA matrix, leading to a more compact char after burning, which could block the flow of gas molecules during combustion. A similar example of compact char formation was observed when lamellar MH nanoparticles (ca. 350 nm in diameter and ca. 50 nm in thickness) were embedded in an EVA matrix (and to which small concentrations of microcapsulated red phosphorus were also added).71 Composites with MH nanoparticles again demonstrated considerably better LOI values than micrometer size MH for all loading levels. Moreover, the tensile strength of the nano-MH/EVA composite was also improved.
More complex, novel flame-retardant ternary nanocomposites in which nano-MH was combined with various polymers (EVA or polyamide 6; PA) and crosslinked rubbers (acrylonitrile butadiene latex or carboxyl acrylonitrile butadiene latex) were investigated by Gui et al. in 2007.72 The flame retardancy was measured in terms of the heat release rate (HRR) and the time to ignition (TTI) and the new ternary composites outperformed composites without nano-MH in terms of HRR, TTI and thermal stability. As with bulk Mg(OH)2, it was postulated that the nano-MH restricts the combustion of the polymers (normally generating highly flammable decomposition products) due to its endothermic decomposition (cooling the condensed phase) and the concurrent release of water. The disperse distribution of nano-MH within the ternary composite expedites this process. It was also proposed that the resulting MgO crust that forms after combustion can protect the underlying polymer from external heat.
By a slight extrapolation, nano-MH particles can also be embedded in an epoxy matrix and there are several examples of the flame-retardant performance of these types of composites and how they compare to equivalents using micro particles of MH. One such example involves the preparation of a composite containing epoxy resin (a diglycidyl ether of bisphenol-A) and nano-MH that had been surface-modified post-synthesis with γ-aminopropyltrietoxysilane (γ-APS) to impart hydrophobicity.73 Both heat release rates and the total heat released were improved compared to composites formed with micro-MH, as also were the mechanical properties. Similar flame-retardant nanocomposites can be produced using surface-modified MH nanorods synthesised in a supercritical hydrothermal flow process, for example.30
In an environmental context, magnesium oxide, magnesium hydroxide and various magnesium salts have been used for many years as acidic waste neutralisers, particularly for the removal of liquid pollutants and as adsorbents for water purification. Among recent examples, MH has been used with kaolin as a coagulant agent to remove reactive dyes from wastewater, as an adsorbent supported on granular activated carbon (GAC) to remove cadmium(II) ions from aqueous solution, with bentonite as an adsorbent to remove phosphate ions and as a supported adsorbent for defluoridation of groundwater.74–77 The addition of MH to the clays above, for example, appears to increase the specific surface area and incorporating Mg(OH)2 in GAC is an improvement over GAC alone in terms of Cd(II) removal from aqueous solution. However, the effect of nanostructured MH in comparison to micron-scale or bulk MH particles for water purification has not been studied in detail. Indeed, in some cases, such the defluoridation example above, MgO appears to outperform MH.
Development of new chemical sensors is an important area of current research and MH nanosheets have recently been proposed for the fabrication of highly efficient chemical sensors for ethanol.13,78 MH nanosheets were deposited as a slurry (with butyl carbitol acetate) on a glassy carbon electrode and used in a 2-electrode system with Pd wire as a counter electrode, 0.1 M phosphate buffer solution and 0.1 μM to 1.0 M concentrations of ethanol. The fabricated sensor is proposed to function in a similar way to a semiconducting device such as a nanocrystalline Sm-doped Co3O4-coated silver electrode reported previously.79 Oxygen atoms adsorbed on the nano-MH surface are proposed to transiently convert into anions (O2− and O−) after extracting electrons from the surface (while also generating holes which participate in the ethanol sensing mechanism). Initially, this process leads to a decrease in the conductivity of the MH nanodiscs and a reduced current. Subsequently, however, ethanol present in solution is converted to CO2 and H2O in the presence of the adsorbed O− species with the generation of electrons. These electrons, it is suggested, enter the empty conduction band of the Mg(OH)2, increasing the conductivity of the electrode and increasing the current. Given the dependence of the conduction mechanism on the adsorbed species, the surface area and dispersity of the deposited Mg(OH)2 is likely to be influential. Although the validity of the mechanism for Mg(OH)2 requires some further corroboration, measurements indicate that a sensor based on MH nanosheets has a superior sensitivity (6.89 ± 0.01 μA cm−2 μM−1) to equivalent devices fabricated from ceria, copper(II) oxide or Ni-doped SnO2 and a competitive lower detection limit (73 nM).
One emerging application that very much relies on the properties of magnesium hydroxide at the nanoscale is its use as a non-toxic antibacterial agent. MH nanoparticles were found to be effective against Escherichia coli (E. coli) and Burkholderia phytofirmans in suspension.80 However, neither the OH− nor the Mg2+ ions in the MH-water suspension were found to be directly responsible for killing the bacteria. Two possible mechanisms were proposed: in the first it was claimed that direct penetration of MH nanoparticles into the cell wall occurred, destroying the structure of proteins and leading to membrane damage and consequent cell death. In the second proposed mechanism, it was contended that adsorption of moisture occurred on the surface of the MH nanoparticles, forming a meniscus around the particles due to capillary condensation. The pH of this thin water meniscus could be much higher than its equilibrium solution and once in contact with the bacteria, this meniscus could damage the membrane leading to the death of the cells. In 2013, Pan et al. studied the antibacterial action of specific MH nanostructures on E. coli.16 Three slurries were obtained using different magnesium precursors: MgCl2 was coprecipitated with NaOH under ambient conditions; MgSO4 was reacted with ammonia at room temperature; while MgO was reacted with water at 600 °C for 2 h, quenched and aged for 24 hours while stirring. The SEM images of the samples show clear differences in the resulting morphology that are dependent on the magnesium precursor (Fig. 11). Although all each hydroxide sample was composed of crystalline plates, their size and distribution was synthesis-route dependent.
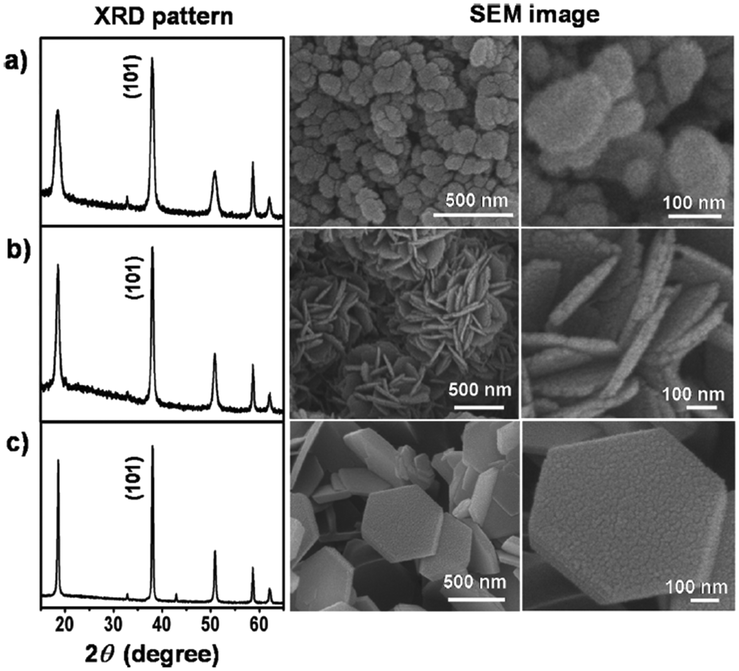 | ||
| Fig. 11 PXD patterns and SEM images for nano-MH synthesised from: (a) MgCl2 (b) MgSO4 and (c) MgO precursors respectively. Reprinted with permission from X. Pan, Y. Wang, Z. Chen, D. Pan, Y. Cheng, Z. Liu, Z. Lin and X. Guan, ACS Applied Materials and Interfaces, 2013, 5, 1137–1142. Copyright 2013 American Chemical Society.16 | ||
The coprecipitation using MgCl2 with NaOH led to agglomerated nano-flakes, while ammonia treatment of MgSO4 produced flower-like clusters of platelets and hydrothermal synthesis with MgO created larger hexagonal nanoplates respectively. The antibacterial mechanism of nano-MH was proposed to comprise 2 steps: 1) positive charged nano-MH particles adsorb on the negative charged bacterial surface by charge attraction; 2) the adsorbed MH nanoparticles destroy the integrity of cell walls, resulting in the subsequent death of the bacteria. One of the most interesting conclusions from this study concerned the morphology-dependence of the nano-MH antibacterial activity (Fig. 12). SEM and zeta potential analysis revealed that the highest adherence ability was observed for MH synthesised from MgCl2 (“Mg(OH)2_MgCl2”) with a morphology characterised by clusters of nano-flakes, each <100 nm across and with positive surface charges. Conversely, MH synthesised by hydrothermal treatment of MgO (“Mg(OH)2_MgO”), which formed as discrete hexagonal nanoplates several hundred nm across with a negatively charged surface, did not demonstrate any antibacterial efficacy since only very limited adsorption on the bacterial surface was possible. Intermediate between these two extremes, MH synthesised from MgSO4 (“Mg(OH)2_MgSO4”) which existed as flower-like clusters of plates with positively charged surfaces, demonstrated some limited ability to adsorb at the E. coli cell surface and to damage the bacterial cell membrane. In 2014, the antibacterial behaviour of MH nanoparticles against E. coli was revisited and a novel possible antibacterial mechanism of MH nanoparticles was suggested by Dong et al.81 They proposed that MH nanoparticles would directly enter into the cell through endocytosis and accumulate in vivo upon contact with a bacterial cell. Then, the MH nanoparticles would unavoidably dissolve in the water content inside the cell (70%) leading to OH− release until an equilibrium is reached (pH of 10). Thus, the intracellular high pH would lead to cell death.
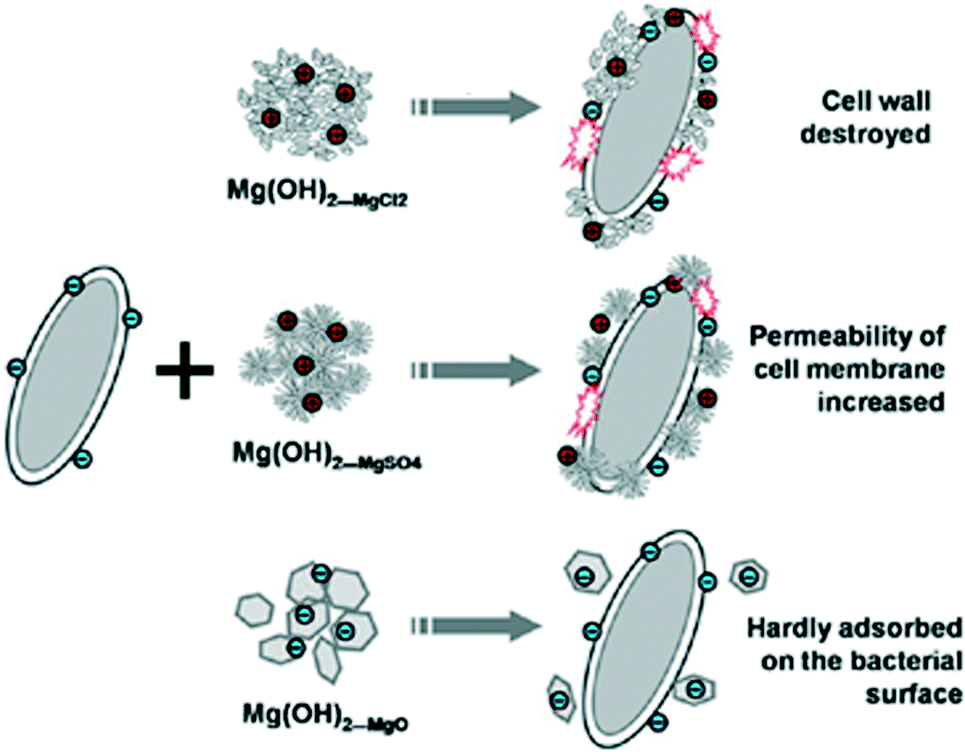 | ||
| Fig. 12 Schematic depicting how the varying size and morphology of nano-MH materials affects their interactions with bacterial cells. Reprinted with permission from X. Pan, Y. Wang, Z. Chen, D. Pan, Y. Cheng, Z. Liu, Z. Lin and X. Guan, ACS Applied Materials and Interfaces, 2013, 5, 1137–1142. Copyright 2013 American Chemical Society.16 | ||
Related also to its alkalinity, MH has found application in paper conservation since its nanoparticles were found effective in the deacidification treatment and protection against cellulose aging.15 The performance of MH nanoparticles synthesised from magnesium salts by precipitation was compared with established deacidification methods (such as the Wei t'O method based on the use of alkoxides which are hydrolysed in situ and in a second step react with CO2 to form protective carbonate). MH nanoparticles reported in this study were prepared simply by heating an aqueous solution of Mg salt (either chloride, nitrate, sulfate or perchlorate) with NaOH and the counter-anion of the respective salts was discovered to be significant in governing the size of the nanoplates so-produced. In fact, it was commented by the authors that the particle sizes obtained from the respective anions appear to evoke the Hofmeister series; sulfate < chloride < nitrate < perchlorate, where perchlorate increases the solubility of the solid (Mg(OH)2 in this case) by weakening the hydrophobic effect and leads to the largest particles. Although the mechanism for growth control was not investigated in detail, parameters such as the surface charge of the particles and the ionic adsorption on the particle surface were thought to play a crucial role. Numerous advantages were observed for the use of MH nanoparticles over previous paper deacidification approaches; most significantly, impregnation of nano-MH in paper results in a higher efficacy in the deacidification treatment, which follows from a higher reactivity. The nano-MH method is also evaluated to be easier, more economic and less aggressive than the Wei t'O process and leads to paper with a higher tensile strength.
As alluded to previously, one of the main applications of MH is in its use as precursor to MgO which itself is used for a wide variety of purposes due to its catalytic activity, flame resistance, mechanical strength and dielectric resistance. Nanostructured MgO presents a range of improved properties compared to that of the bulk.12,18,69,82 Nanostructured MgO has been used in optical materials and phase plasma display technologies,83–85 heterogeneous catalysis,86 environmental remediation77,87 and medical products as bactericide.88 A clear advantage in using nano-MH as a precursor for the synthesis of nanostructured MgO is the pseudomorphic nature of the reaction since the oxide retains the nanostructure of the MH after dehydration.6
Among the most exciting applications of nano-MH are those that have emerged directly from the nanostructuring of the hydroxide and relate to its use in the production of advanced materials with enhanced properties. One example is MH applied to polyurethane foams for sound absorption purposes, where the MH particles are orders of magnitude smaller than the majority of the foam pores.89 In fact, the nano-MH was proposed to improve the sound absorption coefficient relative to the unfilled foam as a result of the creation of partially open pores and due to “synergistic mechanisms of wave damping by fillers and wave collisions through various pores”. Another example is the use of nano-MH as a filler in combination with starch as a matrix to form polymeric bionanocomposites applicable as non-toxic and environmentally-friendly food packing materials.90 The use of the nano-MH filler could considerably improve the tensile strength and elastic modulus of the matrix as well as also improving the thermal stability. Related is the rise of nano-MH in biomedical applications such as biodegradable implants.91 The biodegradable and biocompatible nature of MH is an obvious advantage in such applications and with surface modification of MH by oligolactide (OLA), it is possible to prepare extremely effective biocomposites in combination with poly(L-lactide) (PLLA). The MH–OLA–PLLA composite carries two main advantages: First it can nullify the body's inflammatory response to PLLA by neutralising the acids generated from the hydrolysis of ester bonds and second it can improve the mechanical properties of the proposed implant. Polymeric composites have also proved important for water purification. Given a superior chemical and oxidation resistance coupled with good mechanical and thermal stability, poly(vinylidene fluoride) (PVDF) has been widely exploited as a membrane for various types of filtration. PVDF, however, is hydrophobic and susceptible to membrane fouling, which can result in considerable flux losses. Inorganic additives have shown the ability to counter these fouling effects and nanostructured Mg(OH)2 was demonstrated to reduce fouling without a reduction in flux, principally by changing the membrane from hydrophobic to hydrophilic.92 The porosity of the membrane could be maintained as long as the MH filler was effectively dispersed and surfactants (such as PEG) were found to be important in ensuring a good level of dispersion. Finally and looking forward to future possibilities, as can be seen from the above, most existing applications of nano-MH rely on its structural or chemical properties. Its functional (e.g. electronic) properties remain essentially under-explored and unexploited. One new computational study suggests that if one could develop synthetic methods to make particles of MH small enough, then it should be possible to isolate mechanically stable nanoclusters (with shear moduli similar to the bulk) with reduced interlayer binding energies and band gaps approximately 3 eV smaller than the bulk material.93
Conclusions and final remarks
In summary, a vast range of synthetic approaches for yielding nanostructured magnesium hydroxide have been reported based on a much smaller number of dominant underpinning reaction procedures (which can be summarised as hydrothermal/solvothermal treatments, microwave heating and precipitation methods). Mg(OH)2 (brucite) crystals tend to form as large, hexagonal plates by default. However, the size and morphology of the crystalline material from 2D nanostructures such as hexagonal platelets and sheets to 1D configurations such as rods, needles and hollow tubes can be modified by accessing and selecting an array of experimental variables including the source of magnesium, solvent, temperature, pressure, use of templating agents/surface modifiers, aging conditions and pH. To some extent, the relative importance of these variables depends on the synthesis technique employed, but there are nonetheless some underlying principles that are likely to influence the nano-MH growth mechanism more generally.In the case of hydrothermal/solvothermal methods, the solvent/solution itself unsurprisingly plays a major role in the determination of the nature of the crystalline products. By employing solvents/templating agents such as en, en–H2O, diaminohexane, pyridine or hydrazine hydrate, the crystal nucleation and growth are governed by the ability of these molecules to act as ligands and form complexes with the Mg2+ cations supplied by the starting material. The type of morphology obtained should therefore depend on the coordination behaviour of the respective ligands as well as the magnesium salt/source employed (and indeed the morphology of the source). However, the morphology of the product is often dominated by the relatively harsh physical conditions of the hydro/solvothermal process (temperature, reaction time, pH of the solution) and when adding bases such as aqueous ammonia, anisotropic growth-dissolution–reorganization processes can be observed during solvothermal synthesis. Surfactants undoubtedly have a major influence on the solvothermal crystallisation process (discussed further below for precipitation methods). Most commonly they dictate size and dispersity rather than morphology, but in addition to inhibiting the intergrowth of the particles they can also inhibit growth of individual particles selectively by adsorbing to specific crystal planes. MW-assisted syntheses are typically variants of the conventional solvothermal approach, although the different (dielectric) heating mechanism evoked in the presence of polar solvents can drastically reduce reaction times when compared to conventional solvothermal approaches. The mechanism of crystal growth proposed for MW-assisted solvothermal synthesis follows a characteristic dissolution–precipitation pathway followed by crystallite growth. Principally, the use of MWs leads to much higher rates of both heating and cooling. Magnesium hydroxide nanoparticles are believed to form via intermediate Mg(OH)+ species.
In the case of solution-based precipitation and co-precipitation methods performed at or close to room temperature, the single most important factor in steering crystallisation away from the “default brucite process” (to hexagonal plates) is the employment of surfactants or other surface modifiers/templates. In all cases, the morphology of the (nano-)MH is extremely sensitive to subtle changes in surfactant/precipitation agent concentration. Other factors such as temperature and reaction time can in turn influence growth probably as a result of the effects of these physical variables on the structure directing agent. From the body of evidence, it would appear that at low concentrations, surfactant preferentially adsorbs to the edges of a hexagonal MH nucleus (seed). The ensuing growth is therefore one dimensional (parallel to the longer crystallographic c-axis of the hexagonal crystal structure) resulting in rods and needles, for example. At higher concentrations (and/or in the presence of polymeric surfactants with higher MW), surfactant predominantly adsorbs to the hexagonal faces (the ab plane of the hexagonal MH structure) giving rise to 2D growth and platelets that can exhibit large lateral dimensions, but that are often thinner than those grown at lower surfactant concentration (since growth along the c-axis in the <001> direction is now inhibited).
The last two decades has seen considerable advances in the study of the properties of magnesium hydroxide and the implementation of the material for new applications. The ability to modify MH at the nanoscale has been instrumental in this renaissance. Control of particle size and dispersity has had wide implications for performance in applications as diverse as flame retardants and antibacterial activity. That MH is non-toxic, abundant and inexpensive is a major advantage in terms of the commercial viability of materials. Ensuring that production methods are economic while also being energy-efficient and environmentally sustainable are fundamental to the adoption of nano-MH based products. If continuing momentum can be maintained in terms of reducing MH particle size and prescribing precise MH morphology, then even more opportunities await, not least in developing a generation of new (multi)functional hydroxide materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of Glasgow for a studentship for LDB and the EPSRC (EP/I0225701/1) for a studentship for GB. Associated research received funding from the European Union's Seventh Framework 610 Programme (FP7/2007–2013) for the Fuel Cells and Hydrogen Joint Technology Initiative under Grant 611 agreement number 303447.Notes and references
- P. Patnaik, Handbook of Inorganic Chemicals, McGraw-Hill, New York, 2003 Search PubMed.
- L. Desgranges, G. Calvarin and G. Chevrier, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 82–86 CrossRef.
- V. Y. Kazimirov, M. B. Smirnov, L. Bourgeois, L. Guerlou-Demourgues, L. Servant, A. M. Balagurov, I. Natkaniec, N. R. Khasanova and E. V. Antipov, Solid State Ionics, 2010, 181, 1764–1770 CrossRef CAS.
- G. Balducci, PhD Thesis, University of Glasgow, 2015 Search PubMed.
- V. Tsirelson, A. Avilov, Y. A. Abramov, E. Belokoneva, R. Kitaneh and D. Feil, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 8–17 CrossRef.
- J. M. Hanlon, L. Bravo Diaz, G. Balducci, B. A. Stobbs, M. Bielewski, P. Chung, I. MacLaren and D. H. Gregory, CrystEngComm, 2015, 17, 5672–5679 RSC.
- R. C. Turner, I. Hoffman and D. Chen, Can. J. Chem., 1963, 41, 243–251 CrossRef CAS.
- R. N. Rothon and P. R. Hornsby, Polym. Degrad. Stab., 1996, 54, 383–385 CrossRef CAS.
- P. R. Hornsby, J. Wang, R. Rothon, G. Jackson, G. Wilkinson and K. Cossick, Polym. Degrad. Stab., 1996, 51, 235–249 CrossRef CAS.
- J. Kang and S. P. Schwendeman, Biomaterials, 2002, 23, 239–245 CrossRef CAS PubMed.
- J. C. Yu, A. Xu, L. Zhang, R. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64–70 CrossRef CAS.
- W. Wang, X. Qiao, J. Chen and H. Li, Mater. Lett., 2007, 61, 3218–3220 CrossRef CAS.
- F. Al-Hazmi, A. Umar, G. N. Dar, A. A. Al-Ghamdi, S. A. Al-Sayari, A. Al-Hajry, S. H. Kim, R. M. Al-Tuwirqi, F. Alnowaiserb and F. El-Tantawy, J. Alloys Compd., 2012, 519, 4–8 CrossRef CAS.
- Z. P. Wang, C. H. Li, Y. Mu, Z. Lin, A. J. Yi, Q. Zhang and B. Yan, J. Hazard. Mater., 2015, 287, 296–305 CrossRef CAS PubMed.
- R. Giorgi, C. Bozzi, L. Dei, C. Gabbiani, B. W. Ninham and P. Baglioni, Langmuir, 2005, 21, 8495–8501 CrossRef CAS PubMed.
- X. Pan, Y. Wang, Z. Chen, D. Pan, Y. Cheng, Z. Liu, Z. Lin and X. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 1137–1142 CAS.
- Y. Li, M. Sui, Y. Ding, G. Zhang, J. Zhuang and C. Wang, Adv. Mater., 2000, 12, 818–821 CrossRef CAS.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- W. Fan, S. Sun, L. You, G. Cao, X. Song, W. Zhang and H. Yu, J. Mater. Chem., 2003, 13, 3062–3065 RSC.
- A. N. Christensen, P. Norby and J. C. Hanson, J. Solid State Chem., 1995, 114, 556–559 CrossRef CAS.
- W. Fan, S. Sun, X. Song, W. Zhang, H. Yu, X. Tan and G. Cao, J. Solid State Chem., 2004, 177, 2329–2338 CrossRef CAS.
- P. Jeevanandam, R. S. Mulukutla, Z. Yang, H. Kwen and K. J. Klabunde, Chem. Mater., 2007, 19, 5395–5403 CrossRef CAS.
- L. Zhuo, J. Ge, L. Cao and B. Tang, Cryst. Growth Des., 2009, 9, 1–6 CAS.
- Y. Chen, T. Zhou, H. Fang, S. Li, Y. Yao and Y. He, Procedia Eng., 2015, 102, 388–394 CrossRef CAS.
- H. Yan, X. Zhang, J. Wu, L. Wei, X. Liu and B. Xu, Powder Technol., 2008, 188, 128–132 CrossRef CAS.
- Q. L. Wu, L. Xiang and Y. Jin, Powder Technol., 2006, 165, 100–104 CrossRef CAS.
- H. Dhaouadi, H. Chaabane and F. Touati, Nano-Micro Lett., 2011, 3, 153–159 CrossRef CAS.
- X. T. Sun, L. Xiang, C. Zhu and Q. Liu, Cryst. Res. Technol., 2008, 43, 1057–1061 CrossRef CAS.
- B. Jia and L. Gao, J. Am. Ceram. Soc., 2006, 89, 3881–3884 CrossRef CAS.
- S. Elbasuney and S. F. Mostafa, Powder Technol., 2015, 278, 72–83 CrossRef CAS.
- D. Jin, X. Gu, X. Yu, G. Ding, H. Zhu and K. Yao, Mater. Chem. Phys., 2008, 112, 962–965 CrossRef CAS.
- Q. Wang, C. Li, M. Guo, L. Sun and C. Hu, Mater. Res. Bull., 2014, 51, 35–39 CrossRef CAS.
- H. J. Kitchen, S. K. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman and D. H. Gregory, Chem. Rev., 2014, 114, 1170–1206 CrossRef CAS PubMed.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358 RSC.
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed.
- K. L. Harrison and A. Manthiram, Chem. Mater., 2013, 25, 1751–1760 CrossRef CAS.
- J. Zhao and W. Yan, in Modern Inorganic Synthetic Chemistry, ed. R. Xu, W. Pang and Q. Huo, Elsevier, Amsterdam, Editon edn, 2011, pp. 173–195 Search PubMed.
- H. Wu, M. Shao, J. Gu and X. Wei, Mater. Lett., 2004, 58, 2166–2169 CrossRef CAS.
- K. M. Saoud, S. Saeed, R. M. Al-Soubaihi and M. F. Bertino, Am. J. Nanomater., 2014, 2, 21–25 CAS.
- Y. Hattori, S. Mukasa, H. Toyota, T. Inoue and S. Nomura, Mater. Chem. Phys., 2011, 131, 425–430 CrossRef CAS.
- R. Al-Gaashani, S. Radiman, Y. Al-Douri, N. Tabet and A. R. Daud, J. Alloys Compd., 2012, 521, 71–76 CrossRef CAS.
- P. L. Brown, S. E. Drummond Jr. and D. A. Palmer, J. Chem. Soc., Dalton Trans., 1996, 3071–3075 RSC.
- D. A. Palmer and D. J. Wesolowski, J. Solution Chem., 1997, 26, 217–232 CrossRef CAS.
- R. Al-Gaashani, S. Radiman, N. Tabet and A. R. Daud, Mater. Chem. Phys., 2011, 125, 846–852 CrossRef CAS.
- H. Remy and A. Kuhlmann, Z. Anal. Chem., 1924, 65, 1 CrossRef CAS.
- J. K. Gjaldbaek, Z. Anorg. Allg. Chem., 1925, 144, 145 CrossRef.
- G. L. Smithson and N. N. Bakhshi, Can. J. Chem., 1969, 47, 508–513 CrossRef.
- S. D. Rocha, M. B. Mansur and V. S. Ciminelli, J. Chem. Technol. Biotechnol., 2004, 79, 816–821 CrossRef CAS.
- M. Láska, J. Valtýni and P. Fellner, Cryst. Res. Technol., 1993, 28, 931–936 CrossRef.
- C. Henrist, J. P. Mathieu, C. Vogels, A. Rulmont and R. Cloots, J. Cryst. Growth, 2003, 249, 321–330 CrossRef CAS.
- G. Zou, R. Liu, W. Chen and Z. Xu, Mater. Res. Bull., 2007, 42, 1153–1158 CrossRef CAS.
- W. Jiang, X. Hua, Q. Han, X. Yang, L. Lu and X. Wang, Powder Technol., 2009, 191, 227–230 CrossRef CAS.
- J. Lv, L. Qiu and B. Qu, J. Cryst. Growth, 2004, 267, 676–684 CrossRef.
- D. Chen, L. Zhu, H. Zhang, K. Xu and M. Chen, Mater. Chem. Phys., 2008, 109, 224–229 CrossRef CAS.
- D. An, L. Wang, Y. Zheng, S. Guan, X. Gao, Y. Tian, H. Zhang, Z. Wang and Y. Liu, Colloids Surf., A, 2009, 348, 9–13 CrossRef CAS.
- H. Dong, Z. Du, Y. Zhao and D. Zhou, Powder Technol., 2010, 198, 325–329 CrossRef CAS.
- P. Wang, C. Li, H. Gong, H. Wang and J. Liu, Ceram. Int., 2011, 37, 3365–3370 CrossRef CAS.
- A. Pilarska, M. Wysokowski, E. Markiewicz and T. Jesionowski, Powder Technol., 2013, 235, 148–157 CrossRef CAS.
- J. Zheng and W. Zhou, Mater. Lett., 2014, 127, 17–19 CrossRef CAS.
- H. Yan, X. Zhang, L. Wei, X. Liu and B. Xu, Powder Technol., 2009, 193, 125–129 CrossRef CAS.
- E. J. McHenry, Electrochem. Technol., 1967, 5, 275 CAS.
- G. H. A. Therese and P. V. Kamath, J. Appl. Electrochem., 1998, 28, 539–543 CrossRef CAS.
- M. Dinamani and P. V. Kamath, J. Appl. Electrochem., 2004, 34, 899–902 CrossRef CAS.
- G. Zou, R. Liu and W. Chen, Mater. Lett., 2007, 61, 1990–1993 CrossRef CAS.
- G. Zou, W. Chen, R. Liu and Z. Xu, Mater. Chem. Phys., 2008, 107, 85–90 CrossRef CAS.
- L. Hao, C. Zhu, X. Mo, W. Jiang, Y. Hu, Y. Zhu and Z. Chen, Inorg. Chem. Commun., 2003, 6, 229–232 CrossRef CAS.
- C. Liang, T. Sasaki, Y. Shimizu and N. Koshizaki, Chem. Phys. Lett., 2004, 389, 58–63 CrossRef CAS.
- X. Li, C. Ma, J. Zhao, Z. Li, S. Xu and Y. Liu, Powder Technol., 2010, 198, 292–297 CrossRef CAS.
- A. Pilarska, L. Klapiszewski and T. Jesionowski, Powder Technol., 2017, 319, 373–407 CrossRef CAS.
- L. Qiu, R. Xie, P. Ding and B. Qu, Compos. Struct., 2003, 62, 391–395 CrossRef.
- J. Lv and W. Liu, J. Appl. Polym. Sci., 2007, 105, 333–340 CrossRef CAS.
- H. Gui, X. Zhang, Y. Liu, W. Dong, Q. Wang, J. Gao, Z. Song, J. Lai and J. Qiao, Compos. Sci. Technol., 2007, 67, 974–980 CrossRef CAS.
- R. Suihkonen, K. Nevalainen, O. Orell, M. Honkanen, L. Tang, H. Zhang, Z. Zhang and J. Vuorinen, J. Mater. Sci., 2011, 47, 1480–1488 CrossRef.
- H. Li, S. Liu, J. Zhao and N. Feng, Colloids Surf., A, 2016, 494, 222–227 CrossRef CAS.
- K. Wang, J. Zhao, H. Li, X. Zhang and H. Shi, J. Taiwan Inst. Chem. Eng., 2016, 61, 287–291 CrossRef CAS.
- M. El Bouraie and A. A. Masoud, Appl. Clay Sci., 2017, 140, 157–164 CrossRef CAS.
- N. A. Oladoja, S. Chen, J. E. Drewes and B. Helmreich, Chem. Eng. J., 2015, 281, 632–643 CrossRef CAS.
- A. Umar, F. Al-Hazmi, G. N. Dar, S. A. Zaidi, R. M. Al-Tuwirqi, F. Alnowaiserb, A. A. Al-Ghamdi and S. W. Hwang, Sens. Actuators, B, 2012, 166–167, 97–102 CrossRef CAS.
- M. M. Rahman, A. Jamal, S. B. Khan and M. Faisal, J. Phys. Chem. C, 2011, 115, 9503–9510 CAS.
- C. Dong, J. Cairney, Q. Sun, O. L. Maddan, G. He and Y. Deng, J. Nanopart. Res., 2010, 12, 2101–2109 CrossRef CAS.
- C. Dong, G. He, W. Zheng, T. Bian, M. Li and D. Zhang, Mater. Lett., 2014, 134, 286–289 CrossRef CAS.
- J. Liu, W. Wang, Z. Guo, R. Zeng, S. Dou and X. Chen, Chem. Commun., 2010, 46, 3887 RSC.
- A. Kumar and J. Kumar, J. Phys. Chem. Solids, 2008, 69, 2764–2772 CrossRef CAS.
- N. C. S. Selvam, R. T. Kumar, L. J. Kennedy and J. J. Vijaya, J. Alloys Compd., 2011, 509, 9809–9815 CrossRef CAS.
- L. Kumari, W. Z. Li, C. H. Vannoy, R. M. Leblanc and D. Z. Wang, Ceram. Int., 2009, 35, 3355–3364 CrossRef CAS.
- N. M. Julkapli and S. Bagheri, Rev. Inorg. Chem., 2015, 36, 1–41 CrossRef.
- B. Nagappa and G. T. Chandrappa, Microporous Mesoporous Mater., 2007, 106, 212–218 CrossRef CAS.
- F. Luo, J. Lu, W. Wang, F. Tan and X. Qiao, Micro Nano Lett., 2013, 8, 479 CAS.
- G. Sung, J. W. Kim and J. H. Kim, J. Ind. Eng. Chem., 2016, 44, 99–104 CrossRef CAS.
- F. K. V. Moreira, D. C. A. Pedro, G. M. Glenn, J. M. Marconcini and L. H. C. Mattoso, Carbohydr. Polym., 2013, 92, 1743–1751 CrossRef CAS PubMed.
- C. H. Kum, Y. Cho, Y. K. Joung, J. Choi, K. Park, S. H. Seo, Y. S. Park, D. J. Ahn and D. K. Han, J. Mater. Chem. B, 2013, 1, 2764 RSC.
- C. Dong, G. He, H. Li, R. Zhao, Y. Han and Y. Deng, J. Membr. Sci., 2012, 387–388, 40–47 CrossRef CAS.
- S. Jahangiri and N. J. Mosey, Phys. Chem. Chem. Phys., 2017, 19, 1963 RSC.
| This journal is © The Royal Society of Chemistry 2017 |