Functional conjugated microporous polymers: from 1,3,5-benzene to 1,3,5-triazine†
Shijie
Ren
,
Robert
Dawson
,
Andrea
Laybourn
,
Jia-xing
Jiang
,
Yaroslav
Khimyak
,
Dave J.
Adams
and
Andrew I.
Cooper
*
Department of Chemistry and Centre for Materials Discovery, University of Liverpool, Crown Street. Liverpool, L69 3BX, U.K. E-mail: aicooper@liverpool.ac.uk; Fax: (+44) 151-794-2304
First published on 30th January 2012
Abstract
Conjugated microporous polymers (CMPs) based on the electron-withdrawing 1,3,5-triazine node (TCMPs) were synthesized by palladium-catalyzed Sonogashira-Hagihara cross-coupling. The porosity in these polymers was found to be comparable to the analogous 1,3,5-connected benzene CMP systems that we reported previously, demonstrating that nodes can be substituted in these amorphous materials in a rational manner, much as for certain crystalline porous metal–organic frameworks. The CO2 adsorption properties of the TCMPs were measured and compared with the corresponding CMPs, and it was found that the TCMP networks adsorbed more CO2 than CMP analogues with comparable BET surface areas. Network TNCMP-2 showed the highest surface area (995 m2 g−1) and a CO2 uptake of 1.45 mmol g−1 at 1 bar at 298 K. The band gap in these triazine-based CMPs could also be engineered through copolymerization with other functional monomers.
Introduction
Microporous organic polymers (MOPs) are attracting increasing attention due to their potential applications in areas such as gas adsorption, separation, and heterogeneous catalysis.1–4 Compared with their inorganic or hybrid counterparts, such as silica, metal oxides, zeolites, and metal–organic frameworks (MOFs), MOPs have possible advantages in terms of the combining wide synthetic diversity with reasonable chemical and thermal stability.5 A variety of MOPs have been developed including polymers of intrinsic microporosity (PIMs),6–10 hypercrosslinked polymers (HCPs),11–14 porous aromatic frameworks (PAFs),15–17 covalent triazine-based frameworks (CTFs),18,19 and covalent organic frameworks (COFs).20,21 The latter two classes of materials (CTFs and COFs) show a degree of crystalline order, while the other porous networks are amorphous.In the last few years, there has been significant interest in a new class of materials described as ‘conjugated microporous polymers’ (CMPs).22,23 CMPs are usually prepared by well-established transition metal catalyzed coupling chemistry. The distinguishing feature of CMP materials is that they combine microporosity and high surface areas with extended conjugation, suggesting a range of new applications. Various chemical functionalities have been incorporated into CMPs24–30 and we have shown that pore size and surface area in CMPs can be tailored through synthetic control.31–33 CMPs are currently being investigated for various applications in areas such as heterogeneous catalysis,34–37 light-harvesting networks,38 metal nanoparticle composites,39 supercapacitors,40 and metal-doped materials with remarkable H2 sorption properties.41
To cater for these various applications, there is a need to further broaden the range of chemical functionalities that can be incorporated into CMPs and particularly to enable applications that can exploit the conjugated nature of the materials. 1,3,5-Triazine containing materials have been widely used in industry because of their high thermal stability, derived from the structural symmetry of 1,3,5-triazine units.42–44 Recently, much attention has been paid to 1,3,5-triazine containing π-conjugated systems because of unique properties such as high electron deficiency and spatial coplanarity.45–56 Introduction of these stable and electron-withdrawing triazine units into CMP systems could be advantageous for stability and also in terms of electronic state manipulation of the polymers. In this context, we report the synthesis and characterization of a series of 1,3,5-triazine based CMPs (TCMPs), and compare the porosity of these materials with the analogous benzene-linked CMPs that we reported previously.25,31
One important potential application for porous materials is the capture and separation of CO2, for example from flue gas. Efficient CO2 adsorption depends not only on the surface area of the material, but also on its chemical functionality57,58 – moreover, physical stability would be important in many real-life applications. It is known that the introduction of functional groups such as amines or carboxylic acids can improve CO2 adsorption properties in porous polymer networks. Potentially, nitrogen-rich triazine units could be advantageous for CO2 adsorption in CMPs, and this was also studied here.
Experimental section
Chemicals
4-Bromobenzonitrile, trifluoromethanesulfonic acid, 1,4-diethynylbenzene, tetrakis(triphenylphosphine)palladium, copper(I) iodide and anhydrous N,N′-dimethylformamide (DMF) and chloroform were all purchased from Aldrich and used as received. 1,3,5-Triethynylbenzene was purchased from ABCR. 4,4′-Diethynyl-1,1′-biphenyl33 and tris(4-ethynylphenyl)amine25 were synthesized according to the reported procedures.Synthesis of 2,4,6-tris(4-bromophenyl)-1,3,5-triazine (1)
Monomer 1 was synthesized using literature procedures.591H NMR (CDCl3) δ (ppm): 8.63 (d, 6H), 8.73 (d, 6H). MALDI-TOF MS m/z: 542.9 M+. Anal. Calcd for C21H12Br3N3: C, 46.19; H, 2.22; N, 7.70. Found: C, 45.94; H, 2.03; N, 7.59.Synthesis of polymer networks by Sonogashira-Hagihara cross-coupling chemistry
All polymerization reactions were carried out with similar monomer concentration and a fixed reaction temperature and reaction time (100 °C/72 h). The molar ratio of ethynyl to bromo functionalities in the monomer feed was set at 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 based on our previous findings for CMP networks.23,31 A representative experimental procedure for TCMP-3 is given as an example.
:1 based on our previous findings for CMP networks.23,31 A representative experimental procedure for TCMP-3 is given as an example.
TCMP-3
1,4-Diethynylbenzene (142 mg, 1.125mmol), monomer 1 (273 mg, 0.5 mmol), tetrakis(triphenylphosphine)palladium (60 mg), and copper(I) iodide (30 mg) were dissolved in a mixture of anhydrous DMF (5 mL) and Et3N (5 mL). The reaction mixture was heated to 100 °C and stirred for 72 h under a nitrogen atmosphere. The mixture was cooled to room temperature, and the insoluble precipitated network polymer was filtered and washed four times with chloroform, water, methanol, and acetone (100 mL each) to remove any un-reacted monomers or catalyst residues. Further purification of the polymer was carried out by Soxhlet extraction with methanol and CHCl3 successively for 24 h each. The product was then dried under vacuum for 24 h at 60 °C to give brown powder (yield: 226 mg, 92%). FT-IR (KBr, cm−1): 3034, 2207, 1505, 1364, 812. Anal. Calcd for C36H18N3: C, 87.79; H, 3.68; N, 8.53. Found: C, 77.92; H, 3.77; N, 2.87. The deviation of the elemental analysis results from the theoretical value can be due to remaining end groups and incomplete combustion and is consistent with our previous observations.31TCMP-0
1,3,5-Triethynylbenzene (113 mg, 0.75 mmol), monomer 1 (273 mg, 0.5 mmol), tetrakis(triphenylphosphine)palladium (60 mg), copper(I) iodide (30 mg), DMF (5 mL) and Et3N (5 mL) were used this polymerization. TCMP-0 was obtained as brown power (yield: 214 mg, 94%). FT-IR (KBr, cm−1): 3055, 2205, 1516, 1369, 814. Anal. Calcd for C33H15N3: C, 87.40; H, 3.33; N, 9.27. Found: C, 77.80; H, 3.46; N, 6.41.TNCMP-2
Tris(4-ethynylphenyl)amine (238 mg, 0.75 mmol), monomer 1 (273 mg, 0.5 mmol), tetrakis(triphenylphosphine)palladium (60 mg), copper(I) iodide (30 mg), DMF (5 mL) and Et3N (5 mL) were used this polymerization. TNCMP-2 was obtained as dark-brown power (yield: 305 mg, 98%). FT-IR (KBr, cm−1): 3036, 2206, 1505, 1368, 816. Anal. Calcd for C45H24N4: C, 87.08; H, 3.90; N, 9.03. Found: C, 82.24; H, 4.12; N, 7.12.TCMP-5
4,4′-Diethynyl-1,1′-biphenyl (228 mg, 1.125 mmol), monomer 1 (273 mg, 0.5 mmol), tetrakis(triphenylphosphine)palladium (60 mg), copper(I) iodide (30 mg), DMF (5 mL) and Et3N (5 mL) were used this polymerization. TCMP-5 was obtained as brown power (yield: 289 mg, 95%). FT-IR (KBr, cm−1): 3031, 2208, 1508, 1367, 814. Anal. Calcd for C45H24N3: C, 89.09; H, 3.99; N, 6.93. Found: C, 82.08; H, 4.01; N, 4.78.Infrared spectroscopy
IR spectra were collected as KBr disks using a Bruker Tensor 27.Thermogravimetric analysis (TGA)
TGA analysis was carried out using a Q5000IR analyzer (TA Instruments) with an automated vertical overhead thermobalance. The samples were heated at the rate of 20 °C min−1 under a nitrogen atmosphere.Solid-state NMR
Solid-state NMR spectra were measured on a Bruker Avance 400 DSX spectrometer operating at 100.61 MHz for 13C and 400.13 MHz for 1H. 1H-13C cross-polarization magic angle spinning (CP/MAS) NMR experiments were carried out at an MAS rate of 10.0 kHz using zirconia rotors 4 mm in diameter. The 1H π/2 pulse was 3.4 μs, and two-pulse phase modulation (TPPM) decoupling60 was used during the acquisition. The Hartmann–Hahn condition was set using hexamethylbenzene. The spectra were measured using a contact time of 2.0 ms and a relaxation delay of 10.0 s. Typically 984 scans were accumulated. The 13C {1H} MAS NMR spectra were measured at an MAS rate of 10.0 kHz using TPPM decoupling. The 13C π/2 pulse was 2.6 μs. The spectra were measured using a recycle delay of 10.0 s. Typically, 6144 scans were accumulated. The values of the chemical shifts are referred to that of TMS. The analysis of the spectra (deconvolution) was carried out using Bruker TOPSPIN software.UV-vis spectra
UV-vis Spectra were recorded by a SHIMADZU UV-2550 spectrophotometer using powder samples of the networks.Gas sorption analysis
Polymer surface areas and pore size distributions were measured by nitrogen adsorption and desorption at 77.3 K using either a Micromeritics ASAP 2420 or ASAP 2020 volumetric sorption analyzer. The surface areas were calculated in the relative pressure (P/P0) range from 0.01 to 0.05. Pore size distributions and pore volumes were derived from the adsorption branches of the isotherms using the nonlocal density functional theory (NL-DFT) pore model for pillared clay with cylindrical pore geometry. Samples were degassed at 120 °C for 15 h under vacuum (10−5 bar) before analysis. CO2 isotherms were measured at 273 and 298 K on a Micromeritics ASAP 2050. The temperature was controlled using a chiller circulator.Results and discussion
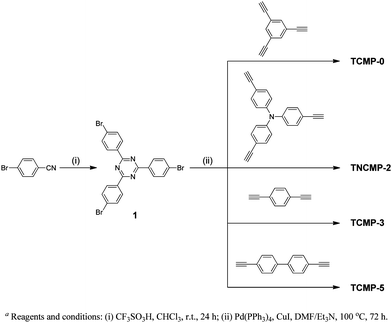
As shown in Scheme 1, the network structures of the triazine based networks TCMP-0, TNCMP-2, TCMP-3 and TCMP-5 are, in principle, isomorphous with respect to our previously reported CMPs, CMP-0,31NCMP-2,25CMP-331 and CMP-5.31 In the TCMP materials, the benzene node is replaced by a 1,3,5-triazine unit. Synthetic routs to the triazine containing monomers and polymers are shown in Scheme 2. 1,3,5-Triazine monomer 1 was synthesized by the trimerization reaction of 4-bromobenzonitrile, catalyzed by trifluoromethanesulfonic acid at room temperature. As for our previous CMP networks,25,31 the TCMPs were prepared by using (A3 + B2) or (A3 + B3) Sonogashira-Hagihara cross-coupling reactions using a 1.5:1 molar ratio of ethynyl to bromo functionalities, and employing DMF as a solvent. We have shown previously that these conditions give rise to higher surface area materials.24,31,61 The polymer networks were recovered as brown powders in yields of greater than 90%. As for the CMP networks,25,31 the newly synthesized TCMP networks were insoluble in all solvents tested such as THF, toluene, DMF, and chloroform. They were also found to be chemically stable in various aqueous conditions. Thermogravimetric analysis (TGA) performed under a nitrogen atmosphere showed good thermal stability of these polymers (Figure S1, Electronic Supporting Information), with 5% weight loss temperatures at 458 °C, 612 °C, 345 °C and 478 °C, respectively, for TCMP-0, TNCMP-2, TCMP-3 and TCMP-5. TNCMP-2 is therefore one of the most thermally stable polymer networks reported so far in this class. Powder X-ray diffraction measurements (Figure S2) showed that the TCMP networks were amorphous, and no evidence for characteristic reflections from a crystalline phase or layered sheets was observed.
 | ||
| Scheme 1 Representative molecular structures for TCMP networks and the corresponding CMPs. | ||
 | ||
| Scheme 2 Synthesis of the triazine monomer and TCMP networks. | ||
Infrared spectra for these polymers are shown in Fig. 1. The characteristic terminal C–C triple bond vibration peaks at about 3300 cm−1 have very low intensity while the peaks at around 2200 cm−1, which are characteristic of bis-substituted acetylene, are easily detected. This indicates, qualitatively, a high degree of polymerization. All polymers were further characterised at the molecular level by 1H-13C CP/MAS NMR. The assignment of resonances for these networks were straightforward based upon our previous work,23–26 as shown in Fig. 2. The aryleneethynylene units in the struts show peaks at ca. 132 ppm, which correspond to non-protonated CAr–CAr groups, and peaks at ca. 124 ppm are observed for protonated CAr–CAr groups. Ethynylene CAr–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CAr units are observed at 91–92 ppm. Resonances observed at 147 ppm can be ascribed to the tris(phenylethynylene)amine unit in TNCMP-2, which is also consistent with our previous work.25 The triazine functionality displays peaks in the range 170–171 ppm.62 To assess the content of end groups, we have previously calculated the quaternary-to-terminal alkyne ratios (CAr–CC–CAr to CAr–CC–H).26 However, in the case of TCMPs, all of the networks showed negligible amounts of terminal alkyne. No peaks were visible above the level of noise at 70–80 ppm, suggesting high levels of polycondensation for these networks, in accordance with the IR data.
C–CAr units are observed at 91–92 ppm. Resonances observed at 147 ppm can be ascribed to the tris(phenylethynylene)amine unit in TNCMP-2, which is also consistent with our previous work.25 The triazine functionality displays peaks in the range 170–171 ppm.62 To assess the content of end groups, we have previously calculated the quaternary-to-terminal alkyne ratios (CAr–CC–CAr to CAr–CC–H).26 However, in the case of TCMPs, all of the networks showed negligible amounts of terminal alkyne. No peaks were visible above the level of noise at 70–80 ppm, suggesting high levels of polycondensation for these networks, in accordance with the IR data.
 | ||
| Fig. 1 Infrared spectra of the TCMP networks. The spectra were recorded using KBr pellets. | ||
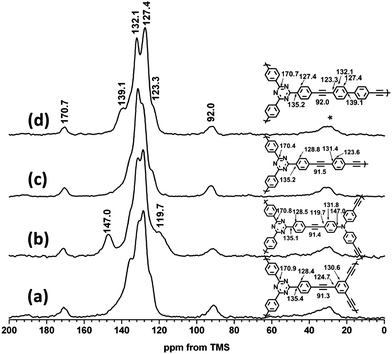 | ||
| Fig. 2 1H-13C CP/MAS NMR spectra of triazine containing networks. (a) TCMP-0, (b) TNCMP-2, (c) TCMP-3, and (d) TCMP-5. Spectra were collected at an MAS rate of 10 kHz. Asterisks denote spinning sidebands. | ||
UV-vis spectra of the TCMP networks were recorded in the solid state and are shown in Fig. 3. All four polymers showed a broad absorption across the wavelength range 200–600 nm, which is consistent with other reported networks.23,63 Although the shapes of the spectra were similar, they did show differences in terms of the onset of the absorption, which correlates with the band gap of the conjugated system. The highly branched A3 + B3 type polymer, TCMP-0, showed an absorption onset at around 540 nm, while the two less branched A3 + B2 polymers, TCMP-3 and TCMP-5, showed absorption onsets at 625 nm and 590 nm, respectively. Compared with TCMP-0, the other A3 + B3 network, TNCMP-2, synthesized from both an electron-donating unit (triphenylamine) and an electron-withdrawing unit (1,3,5-triazine), showed a significantly red-shifted absorption onset at 625 nm. These results indicate clearly that the electronic states of the CMP systems can be adjusted and tuned by judicious choice of monomers. The results suggest that both lower levels of branching and donor–acceptor approaches can achieve narrower band gaps.
 | ||
| Fig. 3 UV-vis spectra of the TCMP networks. The spectra were recorded using powder samples of the polymers. | ||
The porosity in the TCMP networks was investigated by nitrogen adsorption/desorption experiments (Fig. 4), from which the Brunauer-Emmet-Teller (BET) surface areas of the polymers were calculated. Detailed porosity data for the TCMPs in this study and the analogous CMPs are compiled in Table 1. As shown in Fig. 4, networks TNCMP-2 and TCMP-3 gave rise to typical Type I physisorption isotherms according to IUPAC classifications,64 indicating a predominantly microporous structure. While still mainly showing Type I isotherms, networks TCMP-0 and TCMP-5 gave some H3 hysteresis loop character, indicative of the existence of mesopores and/or small macropores. Pore size distribution curves for the four TCMPs are shown in Fig. 5, as calculated using nonlocal density functional theory (NL-DFT). All the polymer networks exhibit a micropore diameter centering around 1.1 nm with a shoulder peak at about 1.6 nm, similar to the corresponding CMPs.25,31 An apparent peak at about 5 nm for TCMP-5 shows there are some mesopores in the network, in agreement with the shape of the N2 isotherms (Fig. 4).
 | ||
| Fig. 4 Nitrogen adsorption and desorption isotherms measured at 77 K (the adsorption and desorption branches are labelled with filled and empty symbols, respectively). | ||
| Polymer | SA BET (m2 g−1)a | V 0.1 (cm3 g−1)b | V tot (cm3 g−1)c | V 0.1/Vtot | CO2 uptake (mmol g−1)d | N2 uptake (mmol g−1)e | CO2/N2f | |
|---|---|---|---|---|---|---|---|---|
| 273 K | 298 K | |||||||
| a BET surface area calculated over the pressure range 0.01–0.05 P/P0. b V 0.1 = pore volume at P/P0 = 0.1. c V tot, total pore volume calculated at P/P0 = 0.99. d CO2 uptake measured at 1 bar. e N2 uptake measured at 1 bar at 298 K. f Selectivity of CO2 and N2 uptake at 1 bar at 298 K. g Data from ref. 31. h Data from ref. 25. | ||||||||
| TCMP-0 | 963 | 0.38 | 0.98 | 0.39 | 2.38 | 1.34 | 0.14 | 9.6 |
| CMP-0 g | 1018 | 0.38 | 0.56 | 0.68 | 2.10 | 1.21 | 0.08 | 15.1 |
| TNCMP-2 | 995 | 0.40 | 0.55 | 0.73 | 2.62 | 1.45 | 0.19 | 7.6 |
| NCMP-2 h | 900 | 0.32 | 0.55 | 0.58 | 2.10 | 1.15 | 0.11 | 10.5 |
| TCMP-3 | 691 | 0.28 | 0.36 | 0.78 | 2.25 | 1.26 | 0.05 | 25.2 |
| CMP-3 g | 522 | 0.18 | 0.26 | 0.69 | 2.18 | 0.81 | 0.06 | 13.5 |
| TCMP-5 | 494 | 0.19 | 0.51 | 0.37 | 1.22 | 0.68 | 0.04 | 17.0 |
| CMP-5 g | 512 | 0.16 | 0.47 | 0.34 | 1.11 | 0.63 | 0.07 | 9.0 |
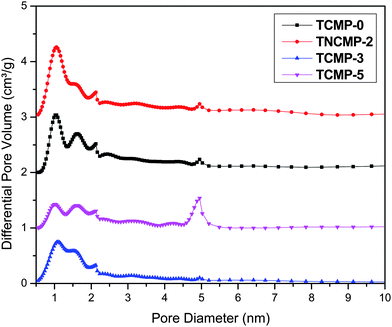 | ||
| Fig. 5 Pore size distribution curves of the TCMP networks (for clarity, the curves of TCMP-5, TCMP-0 and TNCMP-2 are shifted vertically by 1, 2 and 3 cm3 g−1, respectively. | ||
The apparent BET surface areas for these networks range from 494 m2 g−1 (TCMP-5) to 995 m2 g−1 (TNCMP-2), as shown in Table 1. Interestingly, the BET surface areas of the TCMPs correlate well with those of the analogous CMPs (to within less than 10%), showing that the replacement of benzene for a triazine unit does not greatly affect the porous character in the systems. In line with our previous work,25,31 networks formed by A3 + B3 copolymerization (TCMP-0 and TNCMP-2) have higher surface areas than those obtained from A3 + B2 reactions (TCMP-3 and TNCMP-5); we believe this is due to polymers made by the A3 + B3 methods having shorter rigid connecting struts between branching nodes, as depicted in Scheme 1. The pore volume in the materials was also calculated from the nitrogen isotherms. The total pore volume at P/P0 = 0.1 gives a good approximation of the micropore volume (V0.1), as described previously.26 The micropore volume divided by the total pore volume (V0.1/Vtot) gives an indication of the percentage microporosity in the material. In this study, TCMPs have comparable micropore volumes to the corresponding CMPs. The V0.1/Vtot values of TNCMP-2 and TCMP-3 were both greater than 0.70, which means that micropores are dominant in the networks, while the V0.1/Vtot values of TCMP-0 and TCMP-5 were less than 0.40, indicating the coexistence of mesopores or interparticulate porosity.
The CO2 gas sorption for the four TCMP networks was measured at 273 K and 298 K, and compared with that of the corresponding benzene-centred CMPs (Fig. 6, Table 1 and Figure S5–S12). Among these networks, TNCMP-2, which was synthesized from the triazine and triphenylamine monomers, showed the highest volumetric CO2 uptake at both 273 K (2.62 mmol g−1) and 298 K (1.45 mmol g−1). These values are comparable or better than those reported for other polymer networks with similar surface areas synthesized by Sonogashira-Hagihara coupling reactions.57 As far as the TCMP polymers are concerned, networks with larger surface areas lead to higher CO2 uptakes; however, the correlations between CO2 uptake and the surface area or pore volume are not proportional, suggesting that chemical functionality, and perhaps pore size, also plays a role. For example, TCMP-0 and TCMP-3 show comparable CO2 sorption properties although there are apparent differences in their levels of porosity.
 | ||
| Fig. 6 CO2 adsorption and desorption isotherms of TCMP networks (red) and the corresponding CMPs (black) measured at 1 bar at 298 K (the adsorption and desorption branches are labelled with filled and empty symbols, respectively). | ||
In general, the triazine networks adsorbed more CO2 at 298 K than the corresponding benzene analogues despite having similar surface areas (Fig. 6). For example, at 1 bar, TCMP-3 shows 50% higher CO2 uptake (1.26 mmol g−1) than the corresponding polymer CMP-3 (0.81 mmol g−1). Similar findings were made at lower sorption temperatures (Figure S5–S12). This again indicates that not only the surface area and pore volume are important for the CO2 adsorption, but also the chemical functionalities in the porous materials.57,58 CO2/N2 absorption selectivity is another important index for the practical application of the gas adsorbents. The ideal CO2/N2 absorption selectivity for these TCMPs and the analogous CMPs were calculated directly from their individual uptakes at 1 bar at room temperature. As listed in Table 1, networks with lower surface areas showed better CO2/N2 selectivity. Network TCMP-3 showed the highest CO2/N2 selectivity of 25.2:1.
Conclusions
A series of 1,3,5-triazine based CMP networks was prepared using Sonogashira-Hagihara cross-coupling chemistry. These TCMP networks are structurally analogous with benzene-linked CMPs we reported previously, and their porous properties can be well fine-tuned using the strategies previously developed in our group. UV-vis spectra measurements showed that the band gap of the networks could be engineered by judicious choice of monomers. Although the TCMPs have surface areas that are similar to the corresponding CMPs, they show better CO2 absorption capacities. Of the TCMP networks, TNCMP-2 is particularly noteworthy. This network is very thermally stable, highly microporous, and is efficient in terms of CO2 adsorption. Also, TNCMP-2 is a polymer constructed with both an electron acceptor (1,3,5-triazine) and an electron donor (triphenylamine), and is hence potentially able to afford interesting optoelectronic properties. CMPs comprising such monomer combinations could be useful in applications such as photovoltaics or photocatalysis. Research on this subject and development of new methods to synthesize 1,3,5-triazine containing porous networks are ongoing.Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council (EPSRC) for funding (EP/H000925/1), and both EPSRC and E.ON for funding (EP/G061785/1) through the E.ON-EPSRC strategic call on CCS. A.I.C. is a Royal Society Wolfson Merit Award holder.References
- J. Germain, J. M. J. Fréchet and F. Svec, Small, 2009, 5, 1098–1111 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2011 DOI:10.1016/j.progpolymsci.2011.09.002.
- J.-X. Jiang and A. Cooper, Top. Curr. Chem., 2010, 293, 1–33 CrossRef CAS.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780–2781 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782–2783 RSC.
- N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804–1807 CrossRef CAS.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC.
- C. R. Mason, L. Maynard-Atem, N. M. Al-Harbi, P. M. Budd, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Macromolecules, 2011, 44, 6471–6479 CrossRef CAS.
- J. Germain, J. Hradil, J. M. J. Fréchet and F. Svec, Chem. Mater., 2006, 18, 4430–4435 CrossRef CAS.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- M. G. Schwab, A. Lennert, J. Pahnke, G. Jonschker, M. Koch, I. Senkovska, M. Rehahn and S. Kaskel, J. Mater. Chem., 2011, 21, 2131–2135 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291–293 RSC.
- D. Yuan, W. Lu, D. Zhao and H.-C. Zhou, Adv. Mater., 2011, 23, 3723–3725 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2008, 42, 319–326 CrossRef.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS.
- J.-X. Jiang, A. Laybourn, R. Clowes, Y. Z. Khimyak, J. Bacsa, S. J. Higgins, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 7577–7582 CrossRef CAS.
- J.-X. Jiang, A. Trewin, F. Su, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
- R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42(22), 8809–8816 CrossRef CAS.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS.
- R. Dawson, F. Su, H. Niu, C. D. Wood, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2008, 41, 1591–1593 CrossRef CAS.
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- Q. Chen, M. Luo, T. Wang, J.-X. Wang, D. Zhou, Y. Han, C.-S. Zhang, C.-G. Yan and B.-H. Han, Macromolecules, 2011, 44, 5573–5577 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- S. Yuan, B. Dorney, D. White, S. Kirklin, P. Zapol, L. Yu and D.-J. Liu, Chem. Commun., 2010, 46, 4547–4549 RSC.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS.
- X. Du, Y. Sun, B. Tan, Q. Teng, X. Yao, C. Su and W. Wang, Chem. Commun., 2010, 46, 970–972 RSC.
- J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS.
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS.
- T. Hasell, C. D. Wood, R. Clowes, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Mater., 2009, 22, 557–564 CrossRef.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS.
- A. Li, R.-F. Lu, Y. Wang, X. Wang, K.-L. Han and W.-Q. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330–3333 CAS.
- T. Fang and D. A. Shimp, Prog. Polym. Sci., 1995, 20, 61–118 CrossRef CAS.
- D. R. Anderson and J. M. Holovka, J. Polym. Sci., Part A-1, 1966, 4, 1689–1702 CrossRef CAS.
- T. Nishikubo, A. Kameyama and C. Saito, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3604–3611 CrossRef CAS.
- I. Nenner and G. J. Schulz, J. Chem. Phys., 1975, 62, 1747–1758 CrossRef CAS.
- R. Fink, C. Frenz, M. Thelakkat and H.-W. Schmidt, Macromolecules, 1997, 30, 8177–8181 CrossRef CAS.
- H. Meier, E. Karpuk and H. Christof Holst, Eur. J. Org. Chem., 2006, 2609–2617 CrossRef CAS.
- S. Ren, Q. Fang, Y. Lei, H. Fu, X. Chen, J. Du and A. Cao, Macromol. Rapid Commun., 2005, 26, 998–1001 CrossRef CAS.
- S. Ren, Q. Fang, F. Yu and D. Bu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6554–6561 CrossRef CAS.
- T. Yamamoto, S. Watanabe, H. Fukumoto, M. Sato and T. Tanaka, Macromol. Rapid Commun., 2006, 27, 317–321 CrossRef CAS.
- S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian and Q. Fang, J. Phys. Chem. B, 2010, 114, 10374–10383 CrossRef CAS.
- L. Zou, Y. Fu, X. Yan, X. Chen and J. Qin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 702–712 CrossRef CAS.
- K. M. Omer, S.-Y. Ku, Y.-C. Chen, K.-T. Wong and A. J. Bard, J. Am. Chem. Soc., 2010, 132, 10944–10952 CrossRef CAS.
- R. Kannan, G. S. He, T.-C. Lin, P. N. Prasad, R. A. Vaia and L.-S. Tan, Chem. Mater., 2003, 16, 185–194 CrossRef.
- L. Zou, Y. Liu, N. Ma, E. Macoas, J. M. G. Martinho, M. Pettersson, X. Chen and J. Qin, Phys. Chem. Chem. Phys., 2011, 13, 8838–8846 RSC.
- L. Zou, Z. Liu, X. Yan, Y. Liu, Y. Fu, J. Liu, Z. Huang, X. Chen and J. Qin, Eur. J. Org. Chem., 2009, 5587–5593 CrossRef CAS.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- R. Dawson, E. Stockel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 CAS.
- A. Ranganathan, B. C. Heisen, I. Dix and F. Meyer, Chem. Commun., 2007, 3637–3639 RSC.
- A. E. Bennett, R., C. M., M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef CAS.
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS.
- M. J. Bojdys, S. A. Wohlgemuth, A. Thomas and M. Antonietti, Macromolecules, 2010, 43, 6639–6645 CrossRef CAS.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011 DOI:10.1021/ja208284t , in press.
- K. S. W. Sing, E., D. H., R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures for solid state NMR, powder X-ray diffraction measurements, BET plots of the N2 adsorption isotherms, CO2 adsorption at different temperatures and CO2/N2 adsorption selectivity. See DOI: 10.1039/c2py00585a |
| This journal is © The Royal Society of Chemistry 2012 |