
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Same size, same support, same spectator? Selective acetylene hydrogenation on supported Pd nanoparticles†
Marian D.
Rötzer‡
,
Maximilian
Krause‡
,
Tobias
Hinke
 ,
Kevin
Bertrang
,
Florian F.
Schweinberger
,
Andrew S.
Crampton
and
Ueli
Heiz
*
,
Kevin
Bertrang
,
Florian F.
Schweinberger
,
Andrew S.
Crampton
and
Ueli
Heiz
*
Technical University of Munich, TUM School of Natural Sciences, Chair of Physical Chemistry, Catalysis Research Center, Lichtenbergstrasse 4, Garching bei München, Germany. E-mail: marian.roetzer@gmail.com; ulrich.heiz@mytum.de; Tel: +233 89 28954215
First published on 24th April 2024
Abstract
The selective hydrogenation of acetylene catalyzed by Pd nanoparticles is industrially used to increase the purity of ethylene. Despite the implementation of Pd based catalysts on an industrial scale, little is known about metal-support interactions on a fundamental level due to the complexity of these systems. In this study, the influence of metal-support interactions between Pd nanoparticles and two electronically modified a-SiO2 thin films on acetylene hydrogenation is investigated under ultra-high vacuum (UHV) conditions. The hydrogenation is performed under isothermal reaction conditions using a pulsed molecular beam reactive scattering (pMBRS) technique. Besides the activity and selectivity of clean Pd particles also the impact of dehydrogenated species intentionally introduced a priori is elucidated, whereas the active phase of the catalyst is additionally characterized by CO infrared reflection-absorption spectroscopy (IRRAS) and post-mortem temperature-programmed reaction (TPR). Metal-support interactions are found to influence the catalytic properties of Pd particles by charge-transfer, where positive charging leads to increased activity for acetylene hydrogenation. However, the increased activity is accompanied by formation of undesired byproducts. The active sites for acetylene and ethylene hydrogenation are shown to be different as previously proposed by the A and E model. The availability of the two different active sites on the Pd nanoparticles is determined by dehydrogenated species, whose nature and stability can be tuned by metal-support interactions. Based on these findings an electronic model is proposed how selectivity for acetylene hydrogenation can be steered solely by metal-support interactions leading to blocking of unselective sites in situ.
1 Introduction
The selective hydrogenation of acetylene to ethylene is of industrial importance during purification of ethylene feedstocks.1,2 Ethylene as a commodity chemical is produced by non-selective thermal or catalytic cracking of naphtha fractions and typically contains 0.2 to 2% of acetylene. Before further processing, the amount of acetylene has to be lowered (preferentially below 5 ppm), since acetylene acts as a catalyst poison in the downstream processes. In order to achieve these requirements, acetylene is removed by partial hydrogenation to ethylene avoiding fully saturating to ethane.Due to its intrinsic high activity and selectivity, palladium in alloy form is the metal of choice for this process and much effort has been put into understanding the underlying phenomena determining catalytic performance.3–12 However, investigations on these catalytic systems are complicated by the numerous reaction pathways and the rich chemistry of the Pd metal itself. Although ethylene is the only desired product, complete hydrogenation to ethane can occur either by direct conversion of acetylene, or from excess ethylene and hence needs to be avoided in this process.13,14 Further complicating matters, CC-coupling reactions lead to the formation of green oil, effectively poisoning the catalyst.15,16 The oligomerization is believed to initially proceed via 1,3-butadiene, which can either polymerize further or form benzene.17 Even single Pd atoms supported on MgO(100) have been shown to catalyze this intermolecular cyclotrimerization.18,19
The product distribution is decisively controlled by the physical state of the metal catalyst.20–22 The oligomerization products residing on the metal surface block active sites and compete with smaller dehydrogenated species formed upon adsorption of unsaturated hydrocarbons on Pd. The most prominent example of the latter is ethylidyne formed by either acetylene or ethylene,23,24 although various other species have been proposed.16 Besides these surface species, the subsurface of the Pd metal is believed to be crucial for overall catalyst performance as Pd tends to form hydrides and carbides depending on reaction conditions and particle size.25–28
All of these factors combined hamper investigations on these systems since they are inter-correlated and minor changes to one experimental parameter, e.g. particle size, might also alter others in an unforeseen way as it was already shown for the acetylene cyclotrimerization.18,19
Our group has shown that catalytic hydrogenation can be investigated with model catalyst systems. We investigated the reactivity of distinct cluster materials (Ni, Pt, Pd) supported on MgO towards ethylene hydrogenation.29 The particle size of clusters was proven to play a crucial role in the hydrogenation reactions of ethylene and acetylene.30,31 We demonstrated the structural sensitivity and the influence on the overall activity of supported subnanometer particles compared to larger particles.32 Investigation of the catalytic model was widened to support distinct acidity, i.e. SiO2 compared to MgO. The major finding consists of the charging of supported Pt and Pd clusters and the resulting alteration in selectivity towards ethylene hydrogenation by inhibiting undesired side reactions and catalyst deactivation.33,34 Stoichiometry,30 film thickness35 and chemical composition of thin metal oxide films were shown to be crucial for the activity of supported clusters on thin SiO2 or MgO films, comprising less than 5 ML.31 Consequently, structure–activity relationships on a molecular level are elusive insights for acetylene hydrogenation on Pd. For instance, in our previous works, we have shown that nano-engineering of the support material can be used to (de)-charge Pd nanoparticles in the size range of 0.9–1.4 nm supported on amorphous silica films (a-SiO2).33,34 The underlying effect is similar to doping the support material in industrial chemistry. It can be used to increase ethylene hydrogenation activity by one order of magnitude and, at the same time, decrease the amount of carbonaceous deposits. We recently demonstrated the validity of the A-E-model for small Pd clusters on thin MgO films grown on Mo(100).36 We emphasized the application of model catalyst systems under ultra-high vacuum (UHV) conditions, which allow for the investigation of single parameters while keeping others constant. On the basis of these results, the scope of this work was inspired by two main questions: can (de)-charging of Pd nanoparticles by metal-support interactions also be used to steer catalytic activity for acetylene hydrogenation, and if this is the case, what is the impact on overall selectivity?
2 Experimental
All experiments were performed in an UHV chamber with a base pressure of 2 × 10−10 mbar described in detail elsewhere.372.1 Catalyst preparation and film synthesis
Two model systems were synthesized in order to investigate metal-support interactions between Pd nanoparticles and amorphous silica (further denoted as a-SiO2) as support material. The metal component, which was produced with a laser-ablation source, was kept constant for both systems and is identical to our previous work.34 The Pd nanoparticles have a minimum size of 20 atoms and the maximum of the size distribution is found for Pd26. Consequently, the particles are in the size regime of 0.9 to 1.4 nm as evidenced by previous TEM studies.38,39 A coverage of 4.5 × 1012 nanoparticles on the 0.785 cm2 single crystals was used for all experiments.The two thin amorphous silica films were grown either on a Pt(111), or Mo(211), single crystal. Further characterization and information about a-SiO2/Pt(111) can be found in the ESI† and in refs. 30 and 35, whereas synthesis of a-SiO2/Mo(211), which represents a modified route of Goodman et al.,40 is described in ref. 33. Both support materials consist of stoichiometric SiO2 and possess a nominal thickness of 6 ± 2 Å. The usage of different underlying metals below the films lead to positive charging of supported Pd nanoparticles on a-SiO2/Pt(111) relative to a-SiO2/Mo(211) due to changes in the local work function on the support surface. For a detailed discussion of the underlying physical phenomena, see ref. 41 and for these two particular systems.33,34
2.2 Activity measurement and model
The activity of the supported Pd nanoparticles was measured using a pulsed molecular beam technique (pMBRS) described in detail elsewhere.32,42 Briefly, a background pressure of 2 × 10−6 mbar D2 (100% purity, Westfalen, Germany) was established in the UHV chamber. A piezo driven valve (600 μs pulse width, 0.1 Hz) was used to pulse well-defined amounts of acetylene onto the sample, which was placed in front of a skimmer leading to a QMS (Balzers QMA 430, Liechtenstein). The local pressure during the measured quasi steady-state region for one pulse was determined to be 3 × 10−7 mbar for acetylene (99.6%, solvent free, Linde AG, Germany) at 250 K. Consequently, all catalytic measurements were performed in excess of deuterium.To investigate the effect of dehydrogenated C2Hx species on the nanoparticles on catalytic activity, all experiments were performed twice, once with clean particles (sample (i)) and once with C2Hx precovered particles. Hydrogenation of ethylene was performed at 300 K prior to acetylene hydrogenation, resulting in sample (iii), as described in our previous publications.29,34 This reaction leads to formation of C2Hx species on the particle surface (most likely ethylidyne, ethylidene and others) and possibly of a carbide. Several m/z ratios were monitored in order to disentangle the formation of different reaction products. Due to the limited quasi steady-state region within one pulse (≈20 ms, see Fig. 1) and detection by quadrupole mass spectrometry, each m/z ratio had to be measured individually requiring highly reproducible film preparation and particle synthesis enabled by the use of the cluster ablation source. Six different m/z ratios were measured: 30, 31, 33, 34, 54 and 78. The latter m/z ratio is the molecular mass of benzene formed by cyclotrimerization of acetylene. The m/z = 54 ratio was used to monitor the desorption of C4-molecules with 1,3-butadiene being the most abundant molecule in that group. Due to the low amounts of benzene and C4-molecules being formed during reaction, their contribution to lower m/z ratios by overlapping cracking patterns is negligible. It should be noted that due to the high exchange rate between hydrogen and deuterium the given yields of benzene and C4-molecules is rather qualitative than quantitative. The m/z ratios of 30, 31, 33 and 34 have been used to discriminate between ethylene and ethane, which additionally underwent HD-exchange besides the stoichiometric reaction. It was recently shown that during the hydrogenation of ethylene on Pt(111) using D2 not only the stoichiometric product ethane C2H4D2 is formed, but rather a statistical distribution of deuterium content within the ethane molecule.43 This implies the possible presence of over 16 different deuterated ethylenes and ethanes during the hydrogenation of acetylene.44 Since the number of accessible mass to charge ratios is limited for this reaction, the whole system of possible desorbing species is classically under-defined in this case. However, careful data analysis allows at least general trends to be established for the four m/z ratios mentioned above, included in a MatLab-based correlation matrix – see ESI.† Two of these four molecules are attributed to the stoichiometric reaction products, ethylene-D2 and ethane-D4, which are the most abundant reaction products. The cracking ratios of stereoisomers (e.g. Z- and E-CHDCHD) are typically in the range of few percent and the error made by excluding these isomers is small compared to the uncertainty of the experiment.45 The same line of reasoning allows to neglect contribution of 13C. Further molecules included are ethylene-D3 and ethane-D5, which additionally exchanged H by D. Although beneficial for the data evaluation, we did not include m/z-ratios of 32 and 28 in the interpretation of the reaction network, as the used cluster loadings are very low (to guarantee monodisperse clusters). This leads to very small ion currents that are not discernable from the background, even for m/z = 32. The cracking patterns of these molecules are transferred from references45,46 and this approach was additionally validated in situ by use of ethylene-H4, ethylene-D4 and ethane-H6 (below 5% deviation). The contribution of each molecule to one particular m/z ratio can be solved analytically. The recorded voltage, which is now corrected for different molecules, can be correlated to a partial pressure within the quasi steady-state region, see ESI.† Normalization to the amount of used nanoparticles allows to determine TOFs and the errors presented in the following are based on multiple measurements of each particular m/z ratio. The data evaluation is summarized in Fig. 1.
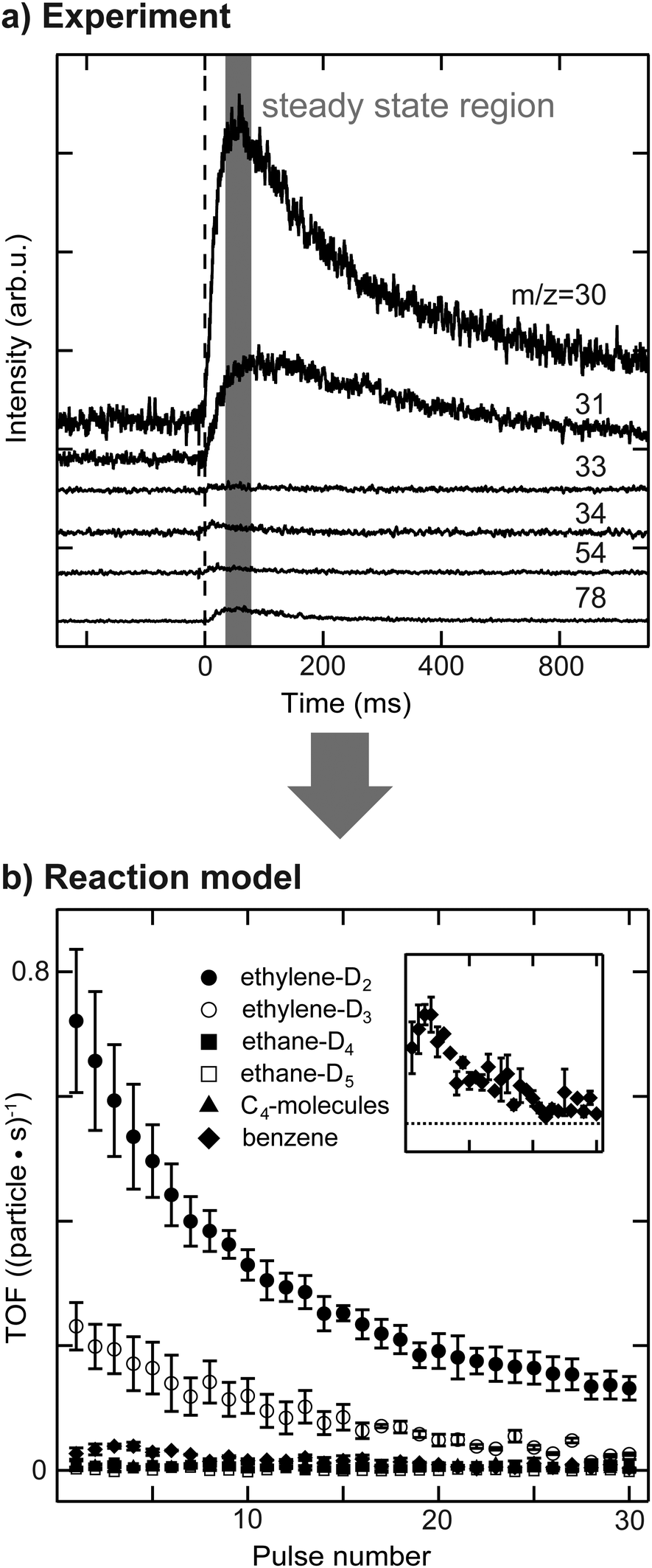 | ||
| Fig. 1 Experimental data acquisition and evaluation: a pulsed molecular beam reactive scattering technique is used to measure a quasi steady-state region for different m/z ratios during acetylene hydrogenation: (a) average m/z ratios of 30 pulses are shown for a typical experiment, where the acetylene pulse starts at 0 ms indicated by the dashed line. (b) The contribution of each assumed molecule to one m/z ratio can be calculated based upon cracking pattern analysis and normalization to the number of particles allows to determine TOFs as a function of pulse number. The inset exhibits a magnification of benzene formation. | ||
Fully deuterated product molecules are not considered analytically due to very low intensities, resulting from scrambling effects. Thus, the quantitative investigation of the ion-fragments only relies on partially deuterated products with known m/z-ratio. In this course, the MatLab source code for evaluating the QMS signals is given in the ESI.†
2.3 Additional catalyst characterization
To exclude reactivity arising from incorporated underlying metal single crystal atom i.e. from Pt(111) or Mo(211) a series of blank measurements was performed using IRRAS and TPD. The corresponding D2O, CO and D2 TPDs are depicted in Fig. S1b and S2 (ESI†). No discernible desorption signals are yielded from both D2O and CO, but physisorption <150 K. The corresponding D2 trace shows no signal at all. The IRRA spectra were recorded after exposing the film to CO, see Fig. S1a (ESI†). The characteristic vibrational Si–O bands at 1238 and 1252 cm−1 and a broad shoulder down to 1100 cm−1 associated with asymmetric longitudinal phonon vibrations for both support materials were observed. In contrast to the Pt-based system, a chemical bond between oxygen and Mo yielding Mo–O–Si vibrations can be found spectroscopically around 1051 cm−1.35 In both cases, no stretch frequencies related to CO adsorption were identified. Consequently, no indications for uncovered metal substrate or metal-atom incorporation in the film are found, and the film is assumed to be fully closed.For post-mortem temperature programmed reaction (TPR), the samples were cooled to 100 K after acetylene hydrogenation at 250 K and 40 L of D2 were dosed (1 L = 1.33 × 10−6 mbar s). The same experimental setup described for pulsed molecular beam reactive scattering (pMBRS) measurements was used, with a linear heating ramp of 2.5 K s−1 (Eurotherm 2408). For Pd nanoparticles supported on a-SiO2/Pt(111) this experiment was also conducted for ethylene hydrogenation at 300 K followed by acetylene hydrogenation at 250 K.
The Pd nanoparticles were further characterized by IRRAS (Thermo Electron Corp. Nicolet FT-6700) using CO as a probe molecule before and after reaction. This method has often been applied to investigate the presence of carbon deposits on Pd and Pt.47,48 These measurements were performed in single reflection mode with an external MCT-detector at 100 K. After background measurement, 10 L CO were dosed and a spectrum recorded (256 scans, 4 cm−1 resolution). The base-line corrected spectra after reaction were recorded directly after the 250 K reaction step and after five additional pulses at 300 K. In addition, IRRAS signals are employed to exclude cluster sintering, since an increase in particle size is expected with a concomitant shift of the CO frequency. Moreover, an alteration of the clusters due to the reaction conditions can be neglected to the considerably low reaction temperatures and the strong interaction of the clusters with the support, according to ref. 33.
3 Results
Activity results of acetylene hydrogenation catalyzed by Pd nanoparticles are shown in Fig. 2 with (a) a-SiO2/Pt(111) and (b) a-SiO2/Mo(211) as support material. The reactivity of the same particles is measured either as initially clean (sample (i)) or after previous introduction of C2Hx species and carbides, which is labelled as “after ethylene hydrogenation” or sample (iii).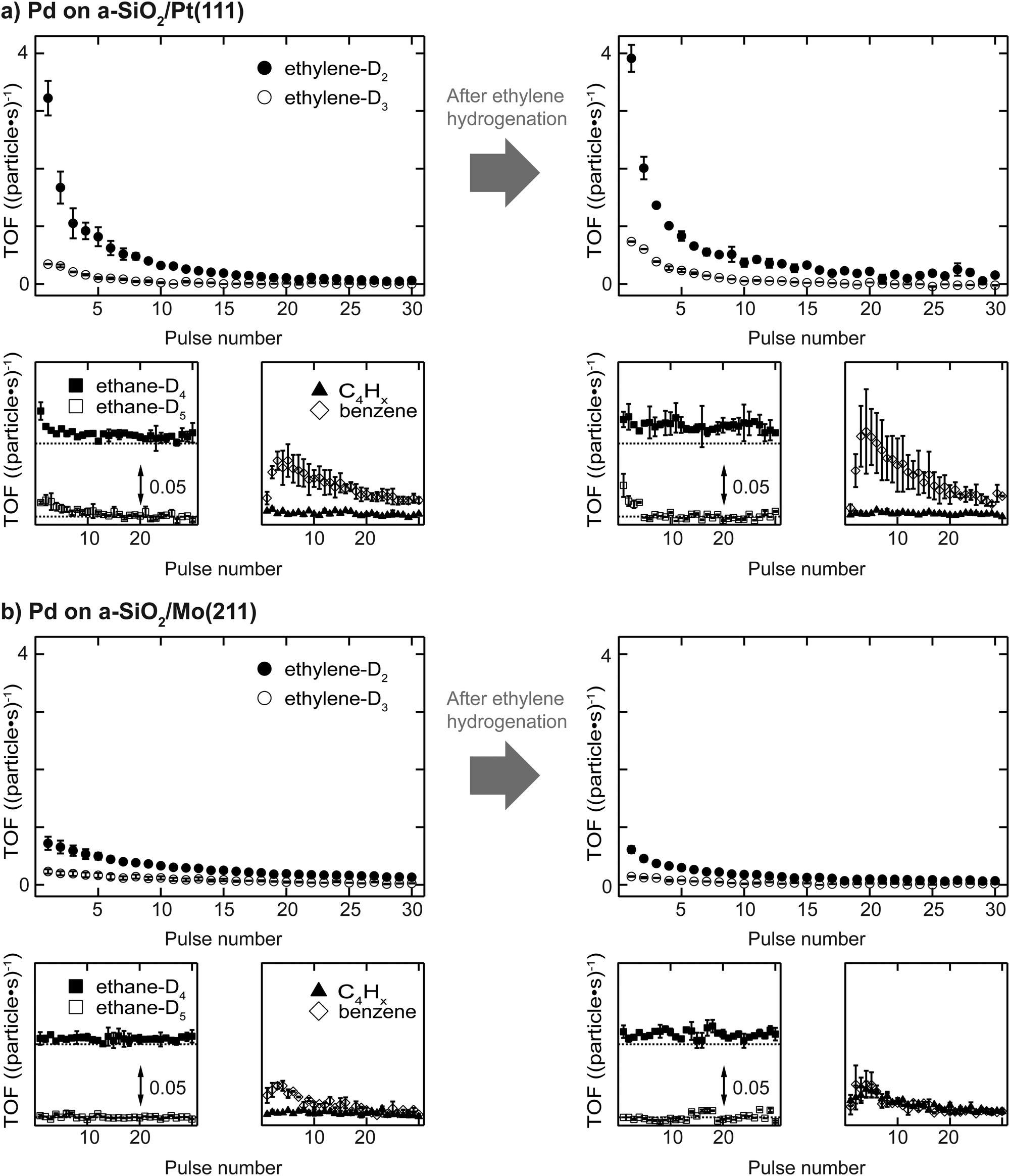 | ||
| Fig. 2 Activity results for acetylene hydrogenation catalyzed by Pd nanoparticles supported on a-SiO2/Pt(111) (a) and a-SiO2/Mo(211) (b). The reaction is performed at 250 K using a pMBRS technique and the TOFs for different reaction products are given as a function of pulse number. Each support system exhibits three subfigures devoted to ethylene as the main product, ethane and oligomerization. Note that ethane-D4 exhibits an offset in ethane subfigures indicated by the dotted line for better visibility. For each support the activity of initially clean and poisoned metal particles is given, where the poisoning is achieved by previous ethylene hydrogenation (see Experimental for details). The y-scale of each of the smaller graphs is the same for all. | ||
Clean particles deactivate over the course of the experiment irrespective of support material due to the formation of dehydrogenated species and limited desorption of acetylene. At the beginning, Pd nanoparticles supported on a-SiO2/Pt(111) show higher ethylene production compared to the same particles supported on a-SiO2/Mo(211). In both cases not only the stoichiometric reaction product ethylene-D2 is formed, but also significant amounts of ethylene-D3, which additionally exchanged hydrogen with deuterium. However, the higher acetylene hydrogenation activity of Pd particles supported on a-SiO2/Pt(111) is accompanied by additional overhydrogenation to ethane as evidenced by initial desorption of ethane-D4 and ethane-D5, which is not observed for a-SiO2/Mo(211) as support. No desorption of C4-molecules can be detected for clean particles, whereas higher benzene formation is shown by particles on a-SiO2/Pt(111) having a maximum after several pulses of acetylene. After performing ethylene hydrogenation, a higher initial catalytic activity is observed for Pd particles supported on a-SiO2/Pt(111) compared to clean particles. More ethylene-D2 as well as ethylene-D3 is formed. No desorption of C4-moieties can be detected, whereas an increase in benzene formation is observed. The ethane-D4 background is increased and small amounts of ethane-D5 desorb during the first pulses. In contrast to a-SiO2/Pt(111), Pd nanoparticles on a-SiO2/Mo(211) show slightly decreased catalytic activity for ethylene and ethane after ethylene hydrogenation. Production of C4-moieties follows the one of benzene, which is not observed for clean particles.
More information about the physical state of the metal nanoparticles is necessary in order to understand the activity results shown in Fig. 2. To this end the Pd nanoparticles are further characterized by post-mortem TPR and CO IRRAS to gain insight into species residing on the particle. The corresponding post-mortem TPR spectra are measured after p-MBRS hydrogenation experiments at 250 K, by using the p-MBRS stated in the Experimental section. For the following samples the desorption of deuterium, as evidenced by m/z = 4, was monitored: (i) clean Pd particles samples directly after deposition, (ii) Pd samples after an ethylene hydrogenation step, (iii) Pd samples after an ethylene hydrogenation step and a subsequent acetylene hydrogenation step at 250 K and (iv) Pd samples after acetylene hydrogenation at 250 K or 300 K. For this purpose D2 is dosed at 100 K for all three samples. The resulting TPR spectra after reaction are summarized in Fig. 3 for particles supported on a-SiO2/Pt(111) (a) and a-SiO2/Mo(211) (b). No deuterium desorption (m/z = 4) is observed for initially clean particles on a-SiO2/Pt(111) after acetylene hydrogenation (sample (iv)). Several desorption features of C4-species (m/z = 54, 56) and benzene (m/z = 78) can be detected above 250 K. Subsequent performance of ethylene and acetylene hydrogenation leads to a broad desorption signal of deuterium as evidenced by m/z = 4, for sample (iii). Contrary to the observation of two D2 desorption features for sample (ii), post-mortem TPR spectra of sample (iii) only yields one feature at around 350 K. In addition to the dehydrogenation of the overlayer at 350 K present for sample (iii), two low temperature features below 200 K are observed in Fig. 3, which might be indicative of partial carbide formation, which is further discussed later. Particles supported on a-SiO2/Mo(211) show D2 desorption at 350 K and higher hydrocarbons desorb above 300 K. The amount of benzene desorption is significantly lower than for particles on a-SiO2/Pt(111).
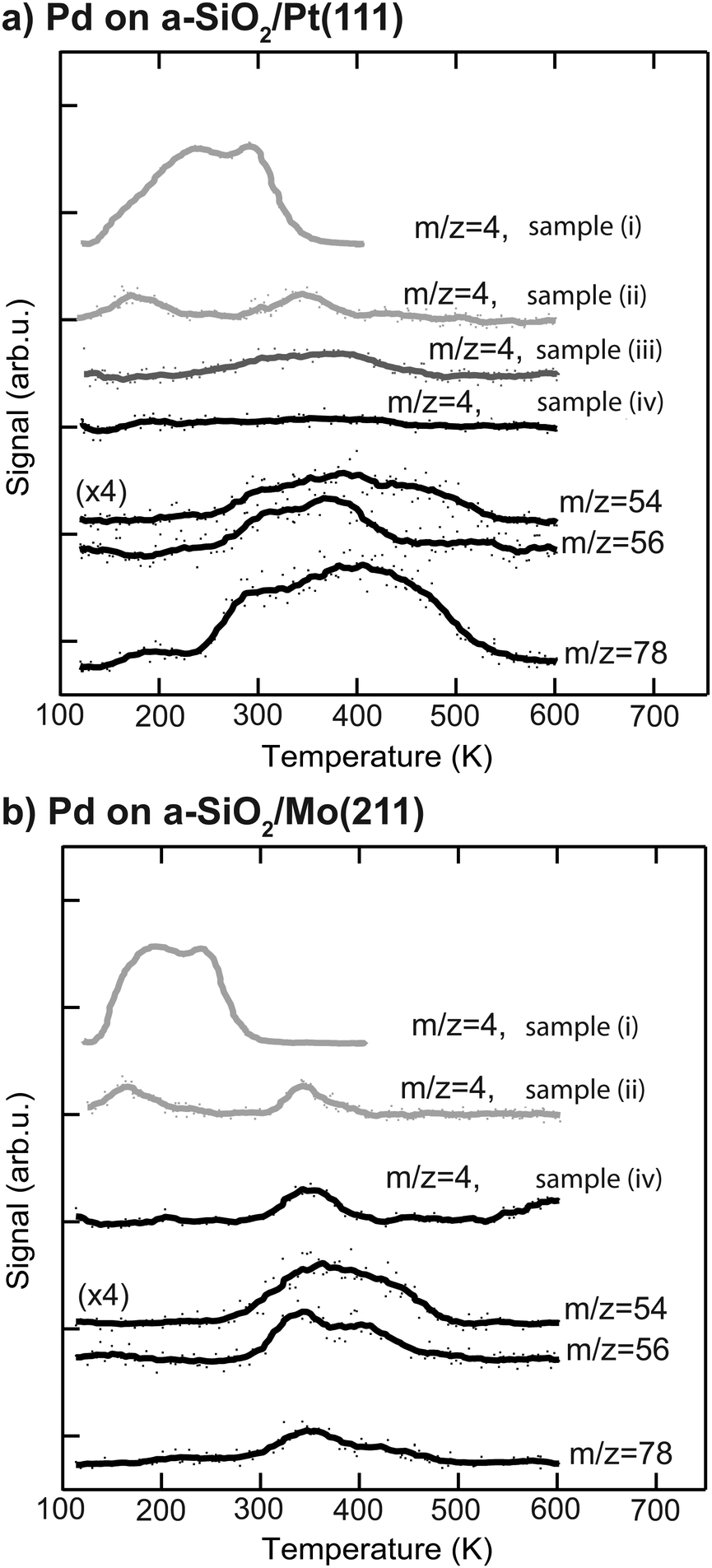 | ||
| Fig. 3 Post-mortem TPR spectra of Pd nanoparticles supported on a-SiO2/Pt(111) (a) and a-SiO2/Mo(211) (b) after acetylene hydrogenation. (sample (iv)) shown in black for m/z = 4,56 and 78. Additional D2 is dosed at 100 K after reaction of C2–H2 and D2 at 250 K (sample (iv)) and a TPR spectrum is recorded, shown in black for m/z = 4, 56 and 78. D2-TPR spectra of particles after deposition (sample (i)) without hydrocarbon hydrogenation and after exclusive ethylene hydrogenation (sample (ii)) are given in grey. In the case of Pd particles supported on a-SiO2/Pt(111) an additional trace m/z = 4 is given for ethylene hydrogenation followed by subsequent acetylene hydrogenation denoted as sample (iii). | ||
IRRA spectra of CO adsorbed on Pd nanoparticles before and after reaction are displayed in Fig. 4 for (a) a-SiO2/Pt(111) and (b) a-SiO2/Mo(211), respectively. For both support systems linear (2100 cm−1) and bridge bonded (1920 to 2000 cm−1) CO species can be observed for the clean particles (sample (i)). After ethylene hydrogenation (sample (ii)) the linear species are red-shifted due to carbonaceous deposits on the metal particles, which weaken the CO bond by increased metal back-donation into the 2p orbital.34,48,49 After acetylene hydrogenation the linear CO stretch frequency is further red-shifted for particles supported on a-SiO2/Pt(111) depending on reaction temperature (sample (iv)) or pretreatment with ethylene (denoted as sample (iii) in Fig. 4). In the case of particles supported on a-SiO2/Mo(211) no CO bands can be detected after acetylene hydrogenation regardless of reaction temperature (sample (iv)) and preconditioning (sample (iii)).
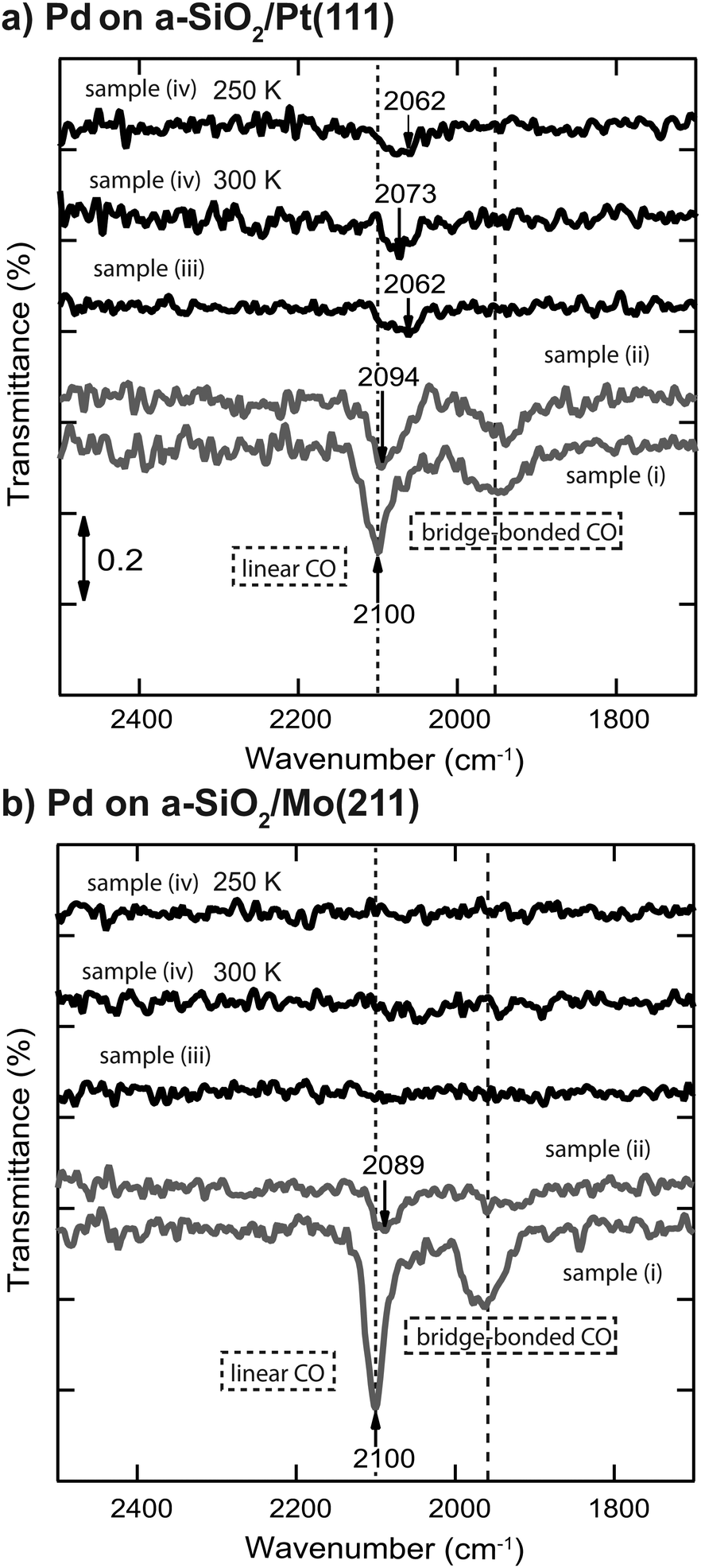 | ||
| Fig. 4 CO IRRAS data of Pd nanoparticles supported on a-SiO2/Pt(111) (a) and a-SiO2/Mo(211) (b): Clean particles (sample (i)) exhibit CO-frequencies of linear (2100 cm−1) and bridge bonded (1920 to 2000 cm−1) CO species. CO is adsorbed at the nanoparticles at 100 K after performing the reaction at the temperature indicated. Spectra after preconditioning with ethylene at 300 K followed by acetylene hydrogenation at 250 K are denoted as sample (iii). IRRA spectra of clean particles (sample (i)) and after ethylene hydrogenation (sample (ii)) are shown for comparison (grey). | ||
4 Discussion
The Discussion section is divided into two parts. The first part deals with the activity and selectivity of clean particles and the influence of support material on overall catalytic performance. In the second section the impact of preconditioning on the supported Pd nanoparticles is discussed and the effect of the dehydrogenated overlayer is elucidated.4.1 Metal-support interactions on clean particles
Electron-poor particles supported on a-SiO2/Pt(111) show higher acetylene hydrogenation activity compared to electron-rich particles supported on a-SiO2/Mo(211) as can be seen in Fig. 2. The same effect, although more pronounced, has been observed for ethylene hydrogenation on the same particles in our previous work,34 as well as for similarly supported Pt clusters.30,33 The difference in ethylene activity has been rationalized by charging arguments based on the d-band center (εD) in comparison to the Fermi energy (EF) when looking at the particle as an entity or varied localized electron densities for an active ensemble of metal atoms.50 Here, a large distance between εD and EF favors hydrogenation, whereas decreased distance leads to dehydrogenation by electron-backdonation of the metal to the σCH* orbital of ethylene or ethyl adsorbed on the surface. This ethylene hydrogenation model was shown to be applicable to small metal nanoparticles in the nanometer size range comprised of either Pt, Pd or Ni supported on MgO(100).29 Modification of the electronic structure of the d-band of a Pt catalyst has shown promising results in the application to selective acetylene hydrogenation.51,52During ethylene hydrogenation, the adsorption of the olefin is described by the Dewar–Chatt–Duncanson model,53 which was also applied theoretically to the interactions between alkynes and metals.54,55 Following these descriptions of bonding, the increased activity of Pd nanoparticles on a-SiO2/Pt(111) can be attributed to an electronic effect, where decreased electron density at the metal leads to weaker back-donation and consequently higher hydrogenation activity.
The mechanism of acetylene hydrogenation is believed to follow a Horiuti-Polanyi mechanism analogous to ethylene,56 in which sequential addition of atomic hydrogen to adsorbed acetylene leads first to a vinyl-intermediate followed by ethylene formation. Subsequently, ethylene can either desorb or be further hydrogenated to undesired ethane. This overhydrogenation to the alkane involves an alkyl-intermediate on the metal surface. During ethylene hydrogenation on the same system, we have shown that the backreaction from adsorbed alkyl to ethylene is negligible, since this reaction would involve significant HD-exchange not observed in the desorbing products.34 The limited HD-exchange was attributed to a large activation barrier for the backreaction leading to accumulation of alkyl-species on the surface.57 However, in the case of acetylene hydrogenation shown in Fig. 2, significant HD-exchange is observed for desorbing ethylene leading to ethylene-D3 for both support systems. Consequently, the relative stability of both intermediates vinyl and ethyl on the surface has to be different. This discrepancy for acetylene was accounted for by the assumption that the normal vinyl group is in equilibrium with a free radical state, which was used to explain the formation of E- and asym-C2H2D2 isomers and might also be applicable to the exchange of hydrogen by deuterium.58
This free radical form is also believed to play a role during oligomerization of acetylene via formation of adsorbed 1,3-butadiene,59 which can further polymerize or trimerize to yield benzene, although also a mechanism based on coupling between acetylene and vinylidene to form a C4 intermediate has been experimentally verified.60 In Fig. 2 no desorption of C4-species is observed for clean particles on both supports. However, benzene desorption, which is the consecutive reaction product of C4-molecules, can be detected having a maximum after several pulses in both cases. This shows, that C4-species are built up over the course of the experiment and remain on the surface, until the final product of cyclotrimerization benzene desorbs. Pd particles supported on a-SiO2/Pt(111) show higher benzene yield than on a-SiO2/Mo(211) during pMBRS. Whether this higher yield is caused by greater catalytic production or facilitated desorption can only be clarified if the residues on the metal particles are known.
To answer this question post-mortem TPR spectra shown in Fig. 3 can be used. After acetylene hydrogenation at 250 K and subsequent TPR, benzene desorption (m/z = 78) starts at 250 K in the case of Pd particles on a-SiO2/Pt(111), whereas the first desorption feature is located at 350 K for Pd on a-SiO2/Mo(211). At the same time a significantly higher amount of benzene desorbs during TPR on the Pt based system. The lower desorption temperature in the case of a-SiO2/Pt(111) suggest that the production of benzene is enhanced on electron-poor particles by a lower activation barrier, whereas the higher overall yield might also be caused residual acetylene on the surface, which undergoes cyclotrimerization upon heating. In addition to benzene (m/z = 78), two m/z ratios of 54 and 56 are shown, which are characteristic for C4-entities. Assuming only one desorbing C4-molecule, e.g. 1,3-butadiene, these ratios should have identical features, which is clearly not observed for both systems in Fig. 3. A recent theoretical work evaluated C–C bond formation leading to green oil on Pd based catalysts.61 In their vast reaction network the authors have shown, that there exist several pathways for CC-coupling of low activation energies. The appearance of several desorption features and non-overlapping traces reflect the presence of several oligomerization products, which may have underwent different degrees of hydrogenation as well as HD-exchange as observed for ethylene in Fig. 2. The discussion on the absence of deuterium desorption (m/z = 4) of Pd particles on a-SiO2/Pt(111) after acetylene hydrogenation as well as the signal at 350 K for a-SiO2/Mo(211) is postponed to the next section, as these involve dehydrogenated C2Hx species on the surface.
4.2 The role of dehydrogenated species during acetylene hydrogenation
The hydrogenation of ethylene performed at 300 K leads to preconditioning of the Pd nanoparticles on both supports. We have shown in our previous study that in both cases an identical dehydrogenated overlayer C2Hx is formed on the metal particles during that process.34 This overlayer exchanged hydrogen completely by deuterium (and will still be denoted as “C2Hx” for simplicity) and a single desorption feature at 350 K of these species is observed during post-mortem TPR on the m/z = 4 curve in Fig. 3. The amount of dehydrogenated species depends on the electron density of the particles with less electron density leading to less dehydrogenated species as observed by a smaller red-shift of the CO frequency in Fig. 4 after ethylene hydrogenation. The formation of C2Hx species (mainly consisting of ethylidyne and ethylidene) is assumed to be responsible for the decreasing hydrogenation activity within the first 20 pulses of each scattering/p-MBRS experiment. Despite this fact, ethylene pre-conditioning of the sample leads to slightly decreased activity with electron-rich particles on SiO2/Mo(112) contrary to a slightly increased activity for electron-poor species on a Pt-based system. Conclusively, increased dehydrogenation on SiO2/Mo(112) reduces the overall activity in comparison to SiO2/Pt(111), see Fig. 2. In turn, overhydrogenation is less pronounced on the Mo-based system compared to the Pt support. Furthermore, C2Hx seems to alter the dehydrogenation activity of differently supported Pd particles, with respect to their electron deficiency, since less higher hydrocarbon products, e.g. C4 and benzene are observed, see Fig. 2. In addition to dehydrogenation of the overlayer at 350 K two low temperature features below 200 K are observed in Fig. 3, which might be indicative of partial carbide formation.In our previous work we have shown that the formation of dehydrogenated species leads to decreased ethylene hydrogenation activity between 50 and 25% for Pd particles on a-SiO2/Pt(111) and a-SiO2/Mo(211), respectively.34 Assuming identical active sites for acetylene and ethylene, the same decreased activity would be expected when ethylene hydrogenation is used to precondition the metal particles. However, although a small decline in activity can be observed for particles on a-SiO2/Mo(211) in Fig. 2, this decrease is significantly smaller than expected for identical active sites. Particles on a-SiO2/Pt(111) even show higher activity after ethylene hydrogenation than starting with clean particles. This leads to the conclusion that the active sites for acetylene hydrogenation have to be different than those for ethylene. This observation is in line with the A and E model developed by Borodziński.62,63 Here, at least two active sites exist on the Pd surface for acetylene (A) and ethylene (E), whereas ethylene is unable to adsorb on A sites due to geometric hindrance. On E sites, both molecules can be hydrogenated and therefore this site is made responsible for unselective overhydrogenation. This model has been developed for ambient conditions and particle sizes between 4.2 and 26.2 nm.64
By introduction of the dehydrogenated overlayer, it is evident that this model also holds true for particles as small as twenty atoms and can be successfully mimicked under UHV conditions as we could show previously.36 The reactivity data gained on Pd particles of the same size as featured here and supported on MgO/Mo(100) under the same conditions shows very similar activities and characteristics as for Pd/a-SiO2/Mo(112). This loosely suggests comparable surface chemistry as well as the validity of the A–E-model for the present system, as was found over MgO. The similar basicities of the SiO2/Mo(112) and the MgO/Mo(100) systems would lead to expect a similar influence on supported catalysts, at least concerning effects resulting from different electron densities.31 Gaining control over these sites, e.g. by structural modification65 or as realized in this work, by electronic influence of the support, could be a keyway to optimize selectivity in a future application of this system in selective acetylene hydrogenation.
Preconditioning of electron-rich particles on a-SiO2/Mo(211) leads to slightly decreased acetylene hydrogenation activity as can be seen in Fig. 2(b). This observation is in accordance with the C2Hx species being stable spectator species, which effectively block active sites of the metal nanoparticles. Interestingly, the same dehydrogenation signal at 350 K is observed during post-mortem TPR in Fig. 3(b) for pure acetylene and ethylene hydrogenation, which indicates, that the formed dehydrogenated overlayer is partially the same for both molecules. Unfortunately, further elaboration of the spectator species is not feasible, e.g. by IRRAS due to the low dipole moment of ethylidyne in comparison to CO and the low cluster coverage of <1%/ML.
The analogy between acetylene and ethylene with respect to surface chemistry on clean transition metals, has also been observed for Pd single crystals.24 However, no CO vibration can be detected in the IRRAS data of Fig. 4(b) after acetylene hydrogenation regardless of reaction temperature and pretreatment. This complete poisoning of the metal surface can be explained by reaction pathways, which are available for acetylene, but not for ethylene. The products of oligomerization leading either to polymers or to cyclotrimerization, remain partially on the surface and alter the availability of active sites. If these active sites are modified by stable C2Hx spectator species on electron-rich Pd particles, the adsorption of C4-molecules, which would otherwise remain on the surface, is weakened leading to desorption identical to benzene as can be seen in Fig. 2(b) after ethylene hydrogenation.
The role of C2Hx species on electron-deficient Pd particles supported on a-SiO2/Pt(111) is more complex. After ethylene hydrogenation a single dehydrogenation feature at 350 K is observed in post-mortem TPR, which is identical to electron-rich particles regardless of ethylene or acetylene. However, after ethylene followed by acetylene hydrogenation this signal becomes broader in Fig. 3(a) indicating a dynamic change in the dehydrogenated overlayer. After sole acetylene hydrogenation no D2-desorption can be detected at all, which evidences the absence of significant amounts of C2Hx on the particles. At the same time higher desorption of ethylene as well as ethane is observed during pMBRS in Fig. 2(a) after ethylene pretreatment leading to the conclusion that these species are replaced during acetylene hydrogenation and desorb. Especially the higher amount of ethane-D5 after ethylene preconditioning in Fig. 2(b) supports the displacement of the spectator species as it was shown, that these species contain almost exclusively deuterium instead of hydrogen.34 The displacement of spectator species should lead to identical electronic structure of the supported Pd nanoparticles during acetylene hydrogenation regardless of pretreatment and is indeed observed in the CO IRRAS of Fig. 4. In both cases, after reaction starting with clean particles (sample (i)) or after ethylene pretreatment (sample (iii)), a single CO vibration is detected at 2062 cm−1, which shifts to 2073 cm−1 after increasing temperature to 300 K (sample (iv) 300 K). This blue-shift at 300 K is caused by desorption of benzene shown in Fig. 3(a) decreasing the amount of dehydrogenated species on the metal surface. As a consequence, metal-support interactions not only determine the nature and amount of dehydrogenated species on nanoparticles, but also their relative stability. A similar effect has been observed for a platinum single crystal, where the exact nature of the dehydrogenated overlayer determined the stability in a hydrogenation atmosphere.66 A closer look at the nature of the carbon-rich overlayer under different conditions could yield a more detailed picture of the involved species and might be a future point of attack to understand the exact interaction between catalyst and these species, but goes in another direction than the scope of this work.67
One of the key goals for Pd based acetylene hydrogenation catalysts is to achieve high ethylene selectivity, whereas overall activity is of minor importance. So far, two key observations have been achieved in this work: First, the active sites for acetylene and ethylene are different for nanoparticles in the nm size regime. The overall activity can be steered by controlling the charge state of the particles by metal-support interactions. However, by increasing reactivity also undesired byproducts are produced. Second, one major part of the dehydrogenated adlayer, C2Hx species, can be tuned by the same interactions either to be stable in the case of electron-rich particles or to be displaced upon formation for electron-poor particles. Here, the formation of these species follows charging arguments based on the local electron density. In order to achieve higher selectivity these two factors have to be combined as simplified shown in Fig. 5. The main aim is to minimize the amount of unselective E sites in situ by blocking them with dehydrogenated species. A high electron density at the E site favors ethylene dehydrogenation leading to stable C2Hx species as realized by a-SiO2/Mo(211) as support system. The same site on electron-deficient particles leads to undesired overhydrogenation, and even if dehydrogenated species are formed, they are unstable during acetylene hydrogenation. This self-poisoning should have a similar effect on selectivity as an external poison, e.g. lead. By increasing electron density not only unselective sites are poisoned, but also the kinetic factor should favor acetylene hydrogenation, as it was shown that the gain in acetylene activity by decreased electron density in this work is considerably lower than observed for ethylene hydrogenation on the same system.34 These considerations neglect the influence of higher hydrocarbons on E sites, which has yet to be determined.
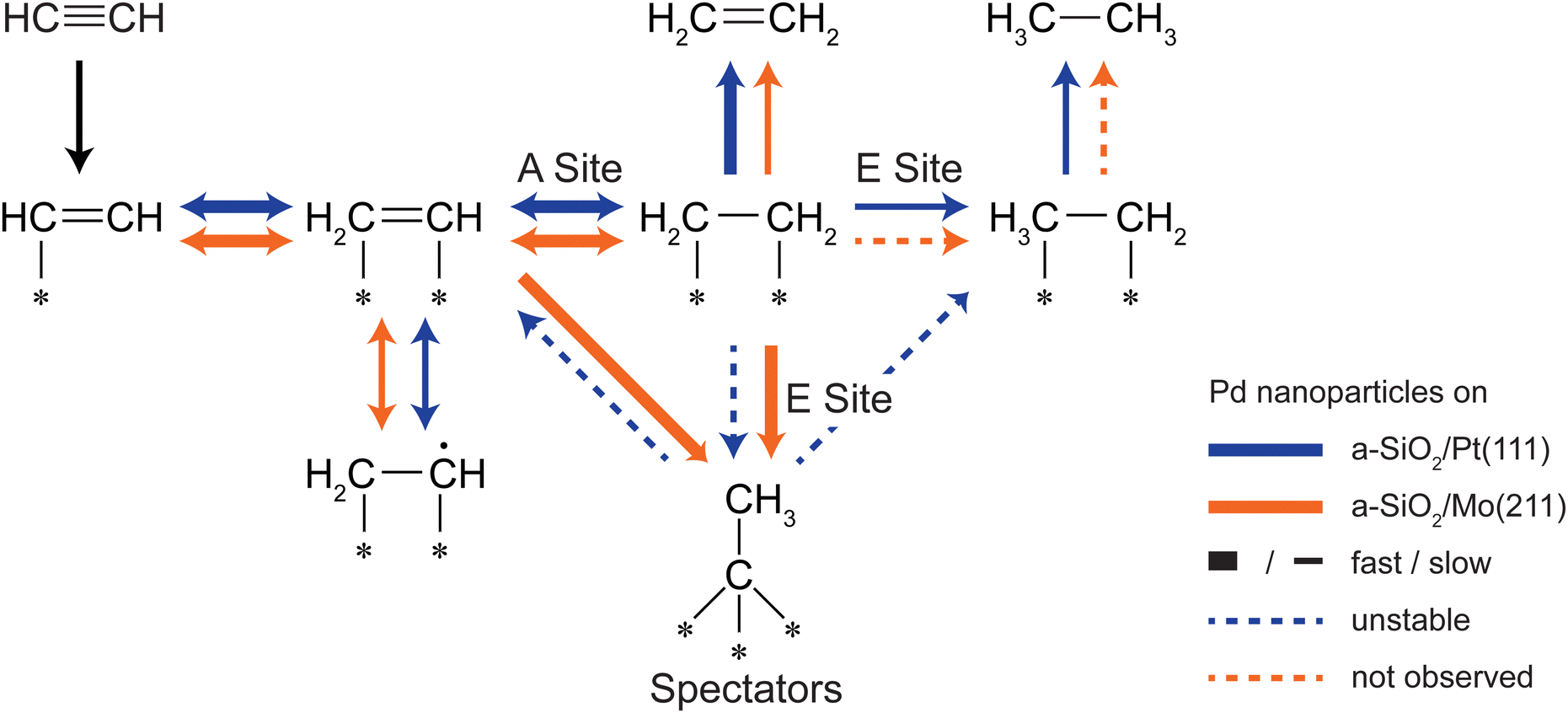 | ||
| Fig. 5 Available C2 reaction pathways of acetylene and ethylene on supported Pd nanoparticles: a-SiO2/Pt(111) (orange) and a-SiO2/Mo(211) (blue). On electron-rich particles stable spectator species C2Hx are formed either by acetylene or ethylene, whereas on electron-deficient particles the formation of these species is hindered in the case of acetylene or unstable in the case of ethylene pretreatment. Due to the formation of stable spectator on E sites, overhydrogenation to ethane can be effectively suppressed. Further reaction pathways, e.g. oligomerization, are not shown for simplicity. | ||
5 Conclusions
The influence of metal-support interactions on catalytic properties of supported Pd nanoparticles for selective acetylene hydrogenation is provided in this work. Two different a-SiO2 samples have been prepared in order to (de-)charge identical Pd nanoparticles in the size regime of 0.9 to 1.4 nm. Here, particles supported on a-SiO2/Pt(111) have less electron density as identical particles supported on a-SiO2/Mo(211) caused by differences in the local work function of the support. The following main conclusions were reached:1. Electron-poor particles have higher initial activity for acetylene hydrogenation than electron-rich particles. This metal-support induced increase in catalytic reactivity follows the trend observed for ethylene hydrogenation, although less pronounced, and can be rationalized by simple charging arguments. However, the increase in ethylene formation is accompanied by production of undesired byproducts, e.g. ethane, at the same time.
2. By introduction of carbonaceous deposits it was shown, that the active site for acetylene and ethylene have to be different for particles as small as twenty atoms. The occurrence of at least two distinct active sites is in line with the A and E model developed by Borodziński under applied reaction conditions.
3. The metal-support interactions have direct influence on the nature of the dehydrogenated species on the metal nanoparticles. For particles with increased electron density, identical dehydrogenation features are observed between ethylene and acetylene showing their similar surface chemistry. In contrast to this, adsorption of acetylene leads to different surface species than ethylene for positively charged Pd nanoparticles. Preconditioning with species formed by ethylene under hydrogenation conditions and performing acetylene hydrogenation afterwards leads to replacement of initially adsorbed dehydrogenated species in contrast to their spectator character typically applied in mechanistic discussions. This shows, that metal-support interactions not only determine the amount of different species on the particles, but also their relative stability. We refer to this atypical behavior as support-dictated stability of spectator species.
4. Given the different catalytic active sites and stability of dehydrogenated species, an increase in selectivity should be accessible by increasing electron-density at the E site or reversed d-band engineering. According to the d-band model a large distance between EF and d-band center leads to increased ethylene hydrogenation activity and the same goal can be achieved by decreased local electron density. Since this reaction is undesired in the case acetylene hydrogenation, the local electron density has to be increased at the E site leading to an in situ blockade of unselective sites by formation of stable spectator species. Again, this self-poisoning of unselective sites is only feasible, if the reaction takes place at different active sites as shown in the case of acetylene and ethylene in this work.
Author contributions
Marian D. Rötzer and Maximilian Krause contributed equally to this publication and were responsible for investigation, data curation, formal analysis validation and writing – original draft. Florian F. Schweinberger and Andrew S. Crampton were responsible for conceptualization and investigation. Tobias Hinke and Kevin Bertrang were responsible for validation and writing – review & editing. Ueli Heiz was responsible for project administration, supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by Clariant in the framework of MuniCat – Munich Catalysis Alliance of Clariant and Technical University Munich. We also acknowledge financial support of the Deutsche Forschungsgemeinschaft (DFG, project HE5434 23-1). M. K. acknowledges financial support of the Studienstiftung des Deutschen Volkes and K. B. the Fond Nationale de la Recherche Luxembourg for financial support, grant ID 14559324. M. D. R. is grateful to Richard Fischer for helpful discussions and Normen Szesni for initializing the project.References
- A. Borodziński and G. C. Bond, Catal. Rev., 2006, 48, 91–144 CrossRef
.
- A. Borodziński and G. C. Bond, Catal. Rev., 2008, 50, 379–469 CrossRef
- A. J. McCue, A. Guerrero-Ruiz, C. Ramirez-Barria, I. Rodríguez-Ramos and J. A. Anderson, J. Catal., 2017, 355, 40–52 CrossRef CAS
- X. Huang, Y. Xia, Y. Cao, X. Zheng, H. Pan, J. Zhu, C. Ma, H. Wang, J. Li, R. You, S. Wei, W. Huang and J. Lu, Nano Res., 2017, 10, 1302–1312 CrossRef CAS
- Y. Cao, Z. Sui, Y. Zhu, X. Zhou and D. Chen, ACS Catal., 2017, 7, 7835–7846 CrossRef CAS
- A. J. McCue and J. A. Anderson, Front. Chem. Sci. Eng., 2015, 9, 142–153 CrossRef CAS
- P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. Matthijs de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582–587 CrossRef CAS
- D. Mei, M. Neurock and C. M. Smith, J. Catal., 2009, 268, 181–195 CrossRef CAS
- N. López, B. Bridier and J. Pérez-Ramírez, J. Phys. Chem. C, 2008, 112, 9346–9350 CrossRef
- N. A. Khan, S. Shaikhutdinov and H.-J. Freund, Catal. Lett., 2006, 108, 159–164 CrossRef CAS
- A. A. Ibrahim, A. Lin, M. S. Adly and M. S. El-Shall, J. Catal., 2020, 385, 194–203 CrossRef CAS
- M. R. Ball, K. R. Rivera-Dones, E. B. Gilcher, S. F. Ausman, C. W. Hullfish, E. A. Lebrón and J. A. Dumesic, ACS Catal., 2020, 10, 8567–8581 CrossRef CAS
- Y. Guo, Y. Li, X. Du, L. Li, Q. Jiang and B. Qiao, Nano Res., 2022, 15, 10037–10043 CrossRef CAS
- Y. Guo, Y. Huang, B. Zeng, B. Han, M. Akri, M. Shi, Y. Zhao, Q. Li, Y. Su, L. Li, Q. Jiang, Y.-T. Cui, L. Li, R. Li, B. Qiao and T. Zhang, Nat. Commun., 2022, 13, 2648 CrossRef CAS PubMed
- I. Y. Ahn, J. H. Lee, S. S. Kum and S. H. Moon, Catal. Today, 2007, 123, 151–157 CrossRef CAS
- X. Deng, J. Wang, N. Guan and L. Li, Cell Rep. Phys. Sci., 2022, 3, 101017 CrossRef CAS
- D. Stacchiola, G. Wu, H. Molero and W. T. Tysoe, Catal. Lett., 2001, 71, 1–4 CrossRef CAS
- S. Abbet, A. Sanchez, U. Heiz and W.-D. Schneider, J. Catal., 2001, 198, 122–127 CrossRef CAS
- S. Abbet, A. Sanchez, U. Heiz, W. D. Schneider, A. M. Ferrari, G. Pacchioni and N. Rösch, Surf. Sci., 2000, 454, 984–989 CrossRef
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86–89 CrossRef CAS PubMed
- D. Teschner, J. Borsodi, Z. Kis, L. Szentmiklósi, Z. Révay, A. Knop-Gericke, R. Schlögl, D. Torres and P. Sautet, J. Phys. Chem. C, 2010, 114, 2293–2299 CrossRef CAS
- D. Teschner, E. Vass, M. Hävecker, S. Zafeiratos, P. Schnörch, H. Sauer, A. Knop-Gericke, R. Schlögl, M. Chamam, A. Wootsch, A. S. Canning, J. J. Gamman, S. D. Jackson, J. McGregor and L. F. Gladden, J. Catal., 2006, 242, 26–37 CrossRef CAS
- I. Jungwirthová and L. L. Kesmodel, J. Phys. Chem. B, 2001, 105, 674–680 CrossRef
- J. A. Gates and L. L. Kesmodel, Surf. Sci., 1983, 124, 68–86 CrossRef CAS
- M. W. Tew, M. Janousch, T. Huthwelker and J. A. van Bokhoven, J. Catal., 2011, 283, 45–54 CrossRef CAS
- M. García-Mota, B. Bridier, J. Pérez-Ramírez and N. López, J. Catal., 2010, 273, 92–102 CrossRef
- D. Teschner, A. Pestryakov, E. Kleimenov, M. Hävecker, H. Bluhm, H. Sauer, A. Knop-Gericke and R. Schlögl, J. Catal., 2005, 230, 186–194 CrossRef CAS
- W. Ludwig, A. Savara, R. J. Madix, S. Schauermann and H.-J. Freund, J. Phys. Chem. C, 2012, 116, 3539–3544 CrossRef CAS
- A. S. Crampton, M. D. Rötzer, F. F. Schweinberger, B. Yoon, U. Landman and U. Heiz, J. Catal., 2016, 333, 51–58 CrossRef CAS
- A. S. Crampton, M. D. Rötzer, F. F. Schweinberger, B. Yoon, U. Landman and U. Heiz, Angew. Chem., Int. Ed., 2016, 55, 8953–8957 CrossRef CAS PubMed
- A. S. Crampton, M. D. Rötzer, U. Landman and U. Heiz, ACS Catal., 2017, 7, 6738–6744 CrossRef CAS
- A. S. Crampton, M. D. Rötzer, C. J. Ridge, F. F. Schweinberger, U. Heiz, B. Yoon and U. Landman, Nat. Commun., 2016, 7, 10389 CrossRef PubMed
- M. D. Rötzer, M. Krause, A. S. Crampton, U. Heiz, B. Yoon and U. Landman, ACS Catal., 2020, 10, 1799–1809 CrossRef
- M. D. Rötzer, M. Krause, M. Huber, F. F. Schweinberger, A. S. Crampton and U. Heiz, J. Catal., 2021, 397, 90–97 CrossRef
- A. S. Crampton, C. J. Ridge, M. D. Rötzer, G. Zwaschka, T. Braun, V. D'Elia, J. M. Basset, F. F. Schweinberger, S. Günther and U. Heiz, J. Phys. Chem. C, 2015, 119, 13665–13669 CrossRef CAS
- M. Krause, M. D. Rötzer, A. S. Crampton, M. Huber and U. Heiz, J. Chem. Phys., 2019, 151, 244304 CrossRef PubMed
- U. Heiz, F. Vanolli, L. Trento and W.-D. Schneider, Rev. Sci. Instrum., 1997, 68, 1986–1994 CrossRef CAS
- S. Kunz, K. Hartl, M. Nesselberger, F. F. Schweinberger, G. Kwon, M. Hanzlik, K. J. J. Mayrhofer, U. Heiz and M. Arenz, Phys. Chem. Chem. Phys., 2010, 12, 10288–10291 RSC
- K. Wettergren, F. F. Schweinberger, D. Deiana, C. J. Ridge, A. S. Crampton, M. D. Rötzer, T. W. Hansen, V. P. Zhdanov, U. Heiz and C. Langhammer, Nano Lett., 2014, 14, 5803–5809 CrossRef CAS PubMed
- S. Wendt, E. Ozensoy, T. Wei, M. Frerichs, Y. Cai, M. S. Chen and D. W. Goodman, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 115409 CrossRef
- L. Giordano, F. Cinquini and G. Pacchioni, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045414 CrossRef
- C. J. Harding, S. Kunz, V. Habibpour, V. Teslenko, M. Arenz and U. Heiz, J. Catal., 2008, 255, 234–240 CrossRef CAS
- Y. Dong, M. Ebrahimi, A. Tillekaratne, J. P. Simonovis and F. Zaera, Phys. Chem. Chem. Phys., 2016, 18, 19248–19258 RSC
- D. H. Lenz and W. C. Conner, Anal. Chim. Acta, 1985, 173, 227–238 CrossRef CAS
- F. L. Dibeler, V. H. abd Mohler and M. deHemptinne, J. Res. Natl. Bur. Stand, 1954, 53, 107–111 CrossRef
- Y. Amenomiya and R. F. Pottie, Can. J. Chem., 1968, 46, 1735–1739 CrossRef CAS
- T. P. Beebe and J. T. Yates, Surf. Sci., 1986, 173, L606–L612 CrossRef CAS
- R. M. Rioux, J. D. Hoefelmeyer, M. Grass, H. Song, K. Niesz, P. Yang and G. A. Somorjai, Langmuir, 2008, 24, 198–207 CrossRef CAS PubMed
- M. J. Lundwall, S. M. McClure and D. W. Goodman, J. Phys. Chem. C, 2010, 114, 7904–7912 CrossRef CAS
- V. Pallassana and M. Neurock, J. Catal., 2000, 191, 301–317 CrossRef CAS
- Z. Wang, A. Garg, L. Wang, H. He, A. Dasgupta, D. Zanchet, M. J. Janik, R. M. Rioux and Y. Román-Leshkov, ACS Catal., 2020, 10, 6763–6770 CrossRef CAS
- K. Yan, Y. Cao, X. Ge, Y. Li, J. Zhang, X. Gong, G. Qian, X. Zhou and X. Duan, AIChE J., 2023, 69(3), e17922 CrossRef CAS
- J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC
- A. Clotet and G. Pacchioni, Surf. Sci., 1996, 346, 91–107 CrossRef CAS
- A. Fahmi and R. A. van Santen, Surf. Sci., 1997, 371, 53–62 CrossRef CAS
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC
- C. J. Heard, C. Hu, M. Skoglundh, D. Creaser and H. Grönbeck, ACS Catal., 2016, 6, 3277–3286 CrossRef CAS
- G. C. Bond and P. B. Wells, J. Catal., 1966, 6, 397–410 CrossRef CAS
- G. C. Bond and P. B. Wells, J. Catal., 1966, 5, 65–73 CrossRef CAS
- M. Kaltchev, D. Stacchiola, H. Molero, G. Wu, A. Blumenfeld and W. T. Tysoe, Catal. Lett., 1999, 60, 11–14 CrossRef CAS
- E. Vignola, S. N. Steinmann, A. Al Farra, B. D. Vandegehuchte, D. Curulla and P. Sautet, ACS Catal., 2018, 1662–1671 CrossRef CAS
- A. Borodziński and A. Gołebiowski, Langmuir, 1997, 13, 883–887 CrossRef
- A. Borodziński, Catal. Lett., 1999, 63, 35–42 CrossRef
- A. Borodziński, Catal. Lett., 2001, 71, 169–175 CrossRef
- Y. Liu, A. J. McCue, C. Miao, J. Feng, D. Li and J. A. Anderson, J. Catal., 2018, 364, 406–414 CrossRef CAS
- J. Simonovis, A. Tillekaratne and F. Zaera, J. Phys. Chem. C, 2017, 121, 2285–2293 CrossRef CAS
- H. Yin, L.-Q. Zheng, W. Fang, Y.-H. Lai, N. Porenta, G. Goubert, H. Zhang, H.-S. Su, B. Ren, J. O. Richardson, J.-F. Li and R. Zenobi, Nat. Catal., 2020, 3, 834–842 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00719k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2024 |