
Recent developments in Ti-based nanocatalysts for electrochemical nitrate-to-ammonia conversion
Wenda
Chen
a,
Yuan
Xu
a,
Jiaxin
Liu
a,
Huiqun
Cao
a,
Yongliang
Li
 a,
Xiangzhong
Ren
a,
Shenghua
Ye
*ab,
Jianhong
Liu
*ab and
Qianling
Zhang
*a
a,
Xiangzhong
Ren
a,
Shenghua
Ye
*ab,
Jianhong
Liu
*ab and
Qianling
Zhang
*a
aGraphene Composite Research Center, College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, 518060, P. R. China. E-mail: yeshh@szu.edu.cn; liujh@szu.edu.cn; zhql@szu.edu.cn
bShenzhen Eigen-Equation Graphene Technology Co. Ltd, Shenzhen, 518000, PR China
First published on 7th July 2023
Abstract
Recently, electrochemical NO3−-to-NH3 conversion via the nitrate reduction reaction (NO3−RR) has received much attention because it is regarded as an available option for sewage treatment and ammonia synthesis under mild conditions. Exploring promising electrocatalysts with low cost, reduced overpotential, high yield rate and faradaic efficiency toward NH3, and sufficient stability is the most crucial factor for NO3−-to-NH3 conversion and energy efficiencies. Considering the advantages of being nontoxic and having wide availability, outstanding stability, hydrogen evolution inertness, and mature fabrication techniques, titanium-based (mainly metallic Ti- and TiO2-based) nanomaterials have emerged as potential candidates for NO3−-to-NH3 conversion over a wide pH range. This review summarizes the overview of the NO3−RR and fundamental insights into metallic Ti and TiO2, and clarifies the relationship among the design strategy, material structure, and performance enhancement. Furthermore, the recent progress in next-generation Ti-based nanomaterials is discussed, including Ti-based MXene and single atomic catalysts. Finally, the challenges and future directions of the NO3−RR and Ti-based nanocatalysts are elucidated. This review aims to provide some inspiration for developing effective electrocatalysts for electrochemical NO3−-to-NH3 conversion.
1. Introduction
Ammonia (NH3) plays an important role in the manufacturing industries such as fertilizers, plastics, pharmaceuticals, etc.1–3 Recently, the emerging “hydrogen economy” and “carbon neutralization” have caused NH3 to be regarded as a carbon-free fuel and a portable energy carrier owing to its high energy density (4.32 kW h L−1); in addition, NH3 can be used as a “green fuel” to produce electricity via direct ammonia fuel cells.4,5 Importantly, NH3 is a natural hydrogen carrier with a high H2 storage capacity of 17.75%,6 making liquid ammonia a safer hydrogen storage medium. Thus, the primary innovation directions of “ammonia = hydrogen 2.0” are derived,7–10 and a new market for NH3 is estimated to be created soon.However, NH3 is still produced by an energy-intensive Haber–Bosch process which requires harsh conditions of high temperature (400–600 °C) and pressure (200–350 atm), resulting in large amounts of global energy consumption and carbon dioxide emission (450 million metric tons).11–15 Exploring alternative NH3 synthetic techniques driven by sustainable energies at room temperature and under an air atmosphere is significant for developing “green ammonia”.16,17 On the other hand, N2, NH3, and NO3− are the key inorganic species for the global nitrogen cycle. Humans utilize the Haber–Bosch process for nitrogen fixation, and as-produced NH3 is used for chemical fertilizer manufacture or provides nitrates via the Ostwald process for the chemical and ammunition industries. Fertilizer-intensive agriculture generates NO3−-containing sewages,18 and NO3− is also a major waste byproduct stream of industrial production (for instance, the effluent produced from the ammunition industries contains 65% ammonium nitrate and 20% amine nitrates19). Extensive nitrogen fixation eventually results in the accumulation of NO3−, excessive NO3− cannot be converted naturally in time, interferes with the global nitrogen cycle and pollutes the surface and ground waters.20 Therefore, it's necessary to develop a denitrification technique to establish a closed nitrogen cycle. The electrochemical NO3− reduction reaction (NO3−RR) for NO3−-to-NH3 conversion (NO3− + 6H2O + 8e− → NH3 + 9OH−) driven by sustainable energies is regarded as an alternative for “green ammonia” production and an efficient artificial denitrification technique to repair the disturbed global nitrogen cycle, which has received great attention in recent years.21
Electrochemical NO3−-to-NH3 conversion is an aqueous-based electrochemical strategy for ammonia synthesis using water as a hydrogen source, which gets rid of H2 that is needed for NH3 synthesis. Moreover, the high theoretical potential (0.69 V vs. RHE) of NO3−-to-NH3 conversion and the high solubility of NO3− make the efficiency of NH3 synthesis superior to that of the electrochemical nitrogen reduction reaction (NRR) – a traditional electrochemical nitrogen fixation technique.4,8,22 Nevertheless, electrochemical NO3−-to-NH3 conversion still suffers from the following issues: although electrochemical NO3−-to-NH3 conversion presents an ideal theoretical potential, it requires a large overpotential (>400 mV) to trigger the NO3−RR in reality,23,24 due to which the energy conversion efficiency is seriously retarded; the NO3−RR is a complicated multi-electron coupled proton transfer process, in which a series of nitrogen-containing products such as NO2, NO2−, NO, N2O, N2, NH2OH, and NH3 could be generated, and toxic NO2− and low added-value N2 are the major competitive products relative to NH3.25,26 Studying the reactive mechanisms of the NO3−RR and exploring efficient electrocatalysts are the keys to solving the above bottlenecks.
Recently, Cu- and Co-based electrocatalysts for electrochemical NO3−-to-NH3 conversion have been intensively developed.27–35 However, most of them only work in neutral or alkaline electrolytes, as NO3− exhibits strong oxidability in acid solutions. Although noble metal-based electrocatalysts exhibit a strong resistance to acid corrosion, their NO3−RR activity is severely compromised by hydrogen evolution. It is desirable to develop efficient electrocatalysts for NO3−-to-NH3 conversion to treat sewage containing NO3− over a wide pH range and potential range. Ti-based materials have the inherent advantages of being non-toxic and cost-effective, and having superior stability over a wide pH range and HER inertness, making them ideal candidates. Currently, increasing studies reveal that Ti-based electrocatalysts, typically metal Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36–38 and TiO2 composite materials,39–42 exhibit comparable electrochemical NO3−-to-NH3 conversion activity. Furthermore, there are a series of low-cost fabrication techniques that enable large-scale fabrication of Ti-based materials with various nanostructures under mild conditions. The above superiorities indicate that exploring Ti-based nanomaterials is a crucial development direction for electrocatalysts for NO3−-to-NH3 conversion. Although some exciting results of Ti-based electrocatalysts have been reported in the literature, there is still a lack of comprehensive evaluation of Ti-based electrocatalysts for NO3−-to-NH3 conversion, and the fundamentals of compositional and structural features associated with electrochemical NO3−-to-NH3 conversion performances have not been thoroughly summarized.
36–38 and TiO2 composite materials,39–42 exhibit comparable electrochemical NO3−-to-NH3 conversion activity. Furthermore, there are a series of low-cost fabrication techniques that enable large-scale fabrication of Ti-based materials with various nanostructures under mild conditions. The above superiorities indicate that exploring Ti-based nanomaterials is a crucial development direction for electrocatalysts for NO3−-to-NH3 conversion. Although some exciting results of Ti-based electrocatalysts have been reported in the literature, there is still a lack of comprehensive evaluation of Ti-based electrocatalysts for NO3−-to-NH3 conversion, and the fundamentals of compositional and structural features associated with electrochemical NO3−-to-NH3 conversion performances have not been thoroughly summarized.
Herein, brief summaries about the fundamental insights of the NO3−RR and inorganic Ti-based materials are proposed. Ti-based electrocatalysts with various structures and their catalytic performances for NO3−-to-NH3 conversion have been reviewed in detail. Finally, the challenges and opportunities for further research are also discussed. This review aims to give some new inspiration to develop highly efficient Ti-based nanocatalysts for electrochemical NO3−-to-NH3 conversion.
2. Brief overview of the NO3−RR
2.1. Reactive pathway
The NO3−RR is a complicated process involving multi-electron coupled multi-proton transfer (Fig. 1a).43 Different NO3− concentrations, potential ranges and the pH of electrolytes may trigger diverse possible reactive pathways and result in various nitrogen-containing products, including NO2−, NOx, N2, NH3, etc. Moreover, the exact reactive pathway of the NO3−RR is still under debate. Niu et al. proposed five possible reactive pathways of the NO3−RR. They can be classified into O-end, O-side, N-end, N-side and NO-dimer pathways based on the adsorption configuration of *NO (* denotes the state of being adsorbed), as depicted in Fig. 1b.44 However, there is still a lack of sufficient experimental evidence to verify the exact reactive pathway. Some of the studies used the N-end pathway of NO3− → *NO3 → *NO2 → *NO → *NOH → *NHOH → *NH → *NH2 → *NH3 → NH3(g) to perform the density functional theory (DFT) study, while the O-end pathway of NO3− → *NO3 → *NO2 → *HNO2 → *NO → *HNO → *H2NO → *HNO → *O → *OH was often used for Ti-based electrocatalysts.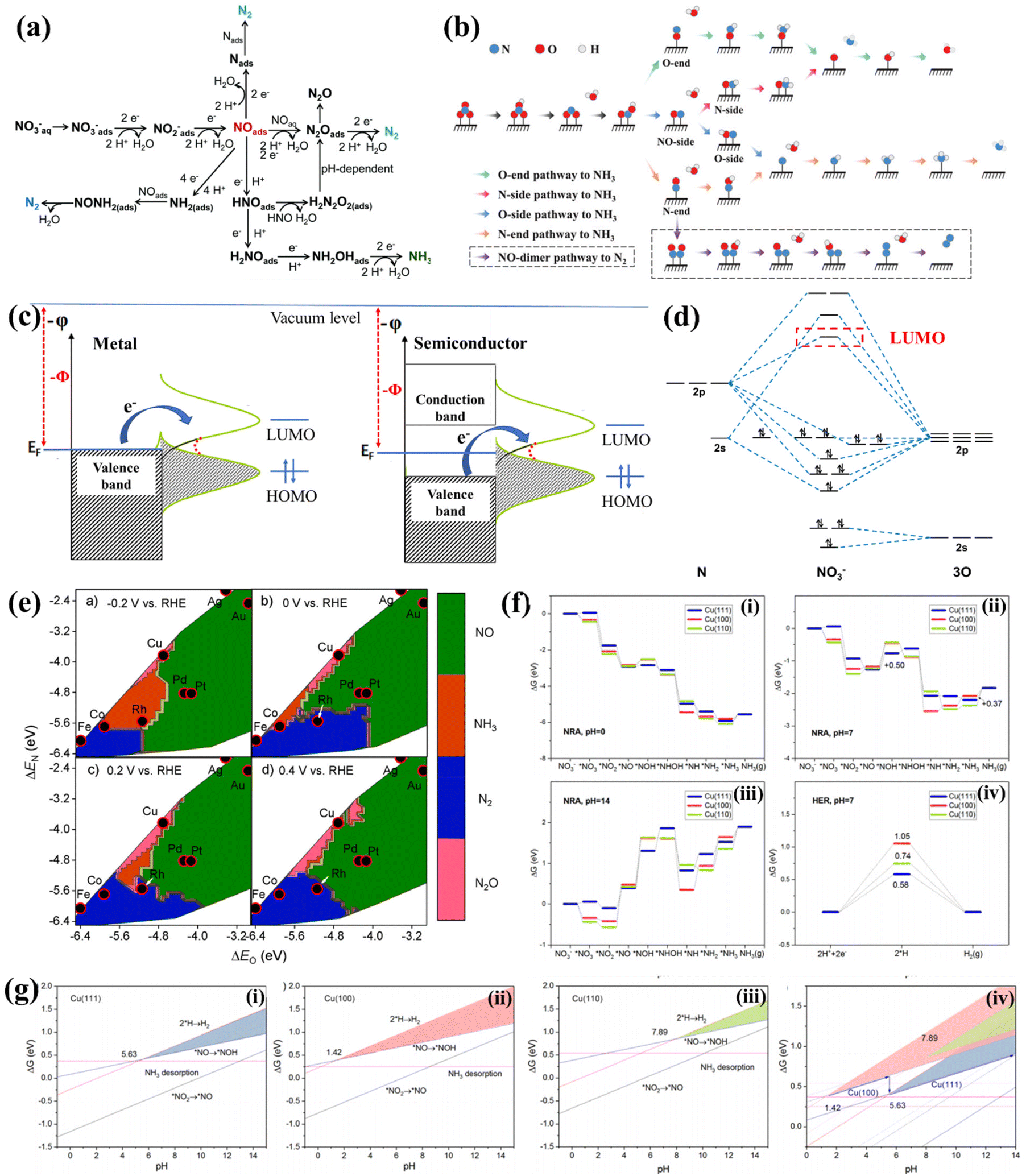 | ||
| Fig. 1 (a) The possible pathways of the NO3−RR.43 Reproduced from ref. 43 with permission from The Royal Society of Chemistry, copyright 2021. (b) Detailed pathways of the NO3−RR, including O-end, O-side, N-end, and N-side pathways to NH3, as well as the NO-dimer pathway to N2.44 Reproduced from ref. 44 with permission from Wiley-VCH, copyright 2020. (c) Electronic band structures of metal- and semiconductor-based electrodes, and the electron transfer from the electrode to the frontier orbitals of the adsorbate. (d) Molecular orbital diagram and the LUMO of NO3−. (e) Theoretical selectivity maps to NO, N2O, N2 or NH3 products from electrocatalytic NO3− reduction as a function of O and N adsorption energy under different applied voltages.49 Reproduced from ref. 49 with permission from American Chemical Society, copyright 2019. (f) NO3−RR pathways at Cu(111), Cu(100), and Cu(110) at (i) pH = 0, (ii) pH = 7, (iii) pH = 14, and (iv) the HER pathway at pH = 7. (g) Competing relationship between the NO3−RR and the HER on (i) Cu(111), (ii) Cu(100), and (iii) Cu(110), and (iv) comparison between different facets. The shaded zones are predominated by the NO3−RR.56 Reproduced from ref. 56 with permission from American Chemical Society, copyright 2021. | ||
2.2. Overpotential
Overpotential is a crucial parameter for evaluating the energy conversion efficiency of the NO3−RR. The theoretical potential for NO3−-to-NH3 conversion is 0.69 V vs. RHE.8 In reality, a large overpotential (usually more than 400 mV) is needed for triggering the NO3−-to-NH3 conversion. The ideal electrocatalyst is still to be developed, and the origin of overpotential should be further clarified. The NO3−RR is a cathodic reduction reaction in which NO3− is transferred to a series of nitrogen-containing products by accepting electrons on the electrode surface. The rate-limiting step in the entire reaction is usually the reduction of NO3− to the intermediate product NO2* due to the difficulty of breaking the N–O bond.21,45 It is the process of electron transition from electrodes to the lowest occupied molecular orbital (LUMO) of NO3−, according to the standpoint of frontier orbital theory. This process can be vividly expressed in Fig. 1c for metal and semiconductor electrodes. The molecular orbital diagram of NO3− shown in Fig. 1d suggests that the high-energy LUMO of the NO3− ion and the d orbitals of transition metals (except Hg) are hard to match with the LUMO of NO3−, thus extra energy is needed for reducing NO3− to the intermediate product NO2*.46 Therefore, exploring electrocatalysts with high energy in the highest occupied state (HOS) is essential for reducing the overpotential.47,482.3. Product selectivity
For NO3−-to-NH3 conversion, NH3 is the target product of the NO3−RR. Thus, the selectivity and faradaic efficiency (FE) of NH3 are crucial parameters. However, NO3−-to-NH3 conversion involves eight electrons coupled with the transfer of nine protons. Many intermediates with the valence of nitrogen between +5 and −3 may desorb from electrocatalysts to generate various by-products, and the coupling reaction between neighboring *N leads to the generation of N2, N2H4 or N2O as by-products. Liu et al. revealed that the adsorption strengths of O and N atoms (ΔEO and ΔEN) could be used as descriptors of selectivity on transition metals. As depicted in Fig. 1e, moderate adsorption of O and N prefers NH3 generation, strong adsorption of O and N results in N2 formation, and the weak adsorption intensity of N and O tends to produce NO. They proposed that Co and Rh are ideal candidates for electrochemical NO3−-to-NH3 conversion.49Furthermore, the d-band model is commonly used for describing and anticipating the selectivity of the NO3−RR. The d-band center (Ed) is related to the adsorption strengths of intermediates, which increase with the Ed of active sites approaching the Fermi level. NO2− is regarded as a major byproduct of NO3−-to-NH3 conversion. A more negative Δ(GNO* − GNO2*) is desired for increasing the preference toward nitrite reduction to nitric oxide. As Ed approaches the Fermi level, Δ(GNO* − GNO2*) becomes increasingly negative, and *NO2 tends to be reduced to *NO.50,51 Subsequently, the adsorbed nitric oxide (*NO) serves as a critical intermediate in determining the selectivity between nitrogen/oxides and ammonium or hydroxylamine.52 NH3 could be generated from *NO via two pathways: one is the so-called Eley–Rideal-like proton-coupled electron transfer, in which *NO was reduced to hydroxylamine (*NH2OH),53 and the other involves the dissociation of *NO into *N and *O and the Langmuir–Hinshelwood-like hydrogenation of *N by *H to NH3.54,55 The latter has been proposed to favor NH3 production, namely, dissociative adsorption of nitric oxide is desired for NO3−to-NH3 conversion. Theoretical studies revealed that the dissociation activation barriers of *NO decrease with increasing its adsorption strength, suggesting that the Ed of active sites should be close to the Fermi level to activate the dissociation of nitric oxide for NH3 formation.50
On the other hand, the HER is the major competitive reaction of the NO3−RR, which also retards the selectivity and FE of NO3−-to-NH3 conversion. Hu et al. found that the pH influences the competition between the HER and the NO3−RR on the (100), (111), and (110) facets of Cu. As depicted in Fig. 1f, the Gibbs free energies (ΔG) of intermediates along pathways on Cu(111), Cu(100), and Cu(110) are dependent on the pH of the electrolyte, and the ΔG–pH plots of the potential rate-determining step of the NO3−RR and HER are shown in Fig. 1g(i–iii). The pH range corresponding to the triangle zone is suitable for the NO3−RR, and the critical pH values of the NO3−RR are 5.63, 1.42 and 7.89 for Cu(111), Cu(100) and Cu(110), respectively. Fig. 1g(iv) suggests that Cu(100) works at pH ranging from 1.42 to 5.63 while Cu(111) works at pH ranging from 5.63 to 14, indicating that Cu(111) works more effectively. Moreover, the product selectivity is highly dependent on the applied potential, and a negative potential is needed for NH3 generation while the HER tends to retard the FE and selectivity of NH3.56 The above results verify that the NO3−RR is potential-dependent and pH-sensitive; thus, seeking HER inert materials for constructing NO3−RR electrocatalysts which can work in a wide pH and potential range is highly desired.
3. Fundamental insights of metallic titanium and titanium oxides
Titanium (Ti) is not only a HER inert element but also one of the most abundant elements in the Earth's crust. Metallic Ti or its alloys and oxides have been widely used in aerospace, photocatalysis, energy storage, and other fields due to their advantages of low cost, superior mechanical strength, unique electronic structure, excellent corrosion resistance and stability, etc.57 Some studies revealed that Ti-based inorganic nanomaterials, majorly, metallic Ti- and TiO2-based nanomaterials exhibited impressive performances of electrochemical NO3−-to-NH3 conversion. The exploration of low-cost and highly-efficient Ti-based electrocatalysts for the conversion of NO3−-to-NH3 has attracted much attention. In this part, we systematically review the structure of metallic Ti and TiO2 to help rationalize the design of Ti-based nanomaterials for electrochemical NO3−-to-NH3 conversion.3.1. Metallic Ti
Metallic Ti is a silver-white transition metal with a high melting point of 1660 ± 10 °C, a low density of 4.506 g cm−3, and excellent corrosion resistance. The valence electronic configuration of Ti is [Ar]3d24s2. There are two isomers of titanium: α-Ti and β-Ti; their crystallographic structures are depicted in Fig. 2a and b, respectively. α-Ti belongs to the hexagonal crystal system with an atomic space utilization rate of 74%, while β-Ti presents body-centered cubic dense packing with a utilization rate of 68%. The transition temperature of the above isomers is 882.5 °C, α-Ti is stable below 882 °C, and β-Ti is stable between 882 °C and the melting point; thus α-Ti is the common form in reality.58,59 The work function (Φ shown in Fig. 1c) of metallic Ti is 4.33 eV, lower than those of Co (5 eV), Cu (4.65 eV), Fe (4.5 eV), and Ru (4.71 eV) usually regarded as electrocatalysts for the NO3−RR,60 suggesting that Ti presents a high-energy Fermi level and may be an ideal candidate with a lower overpotential of the NO3−RR. However, titanium usually forms a stable oxide layer on its surface when exposed to air, and the active sites should be carefully identified while using metallic Ti-based materials as catalysts.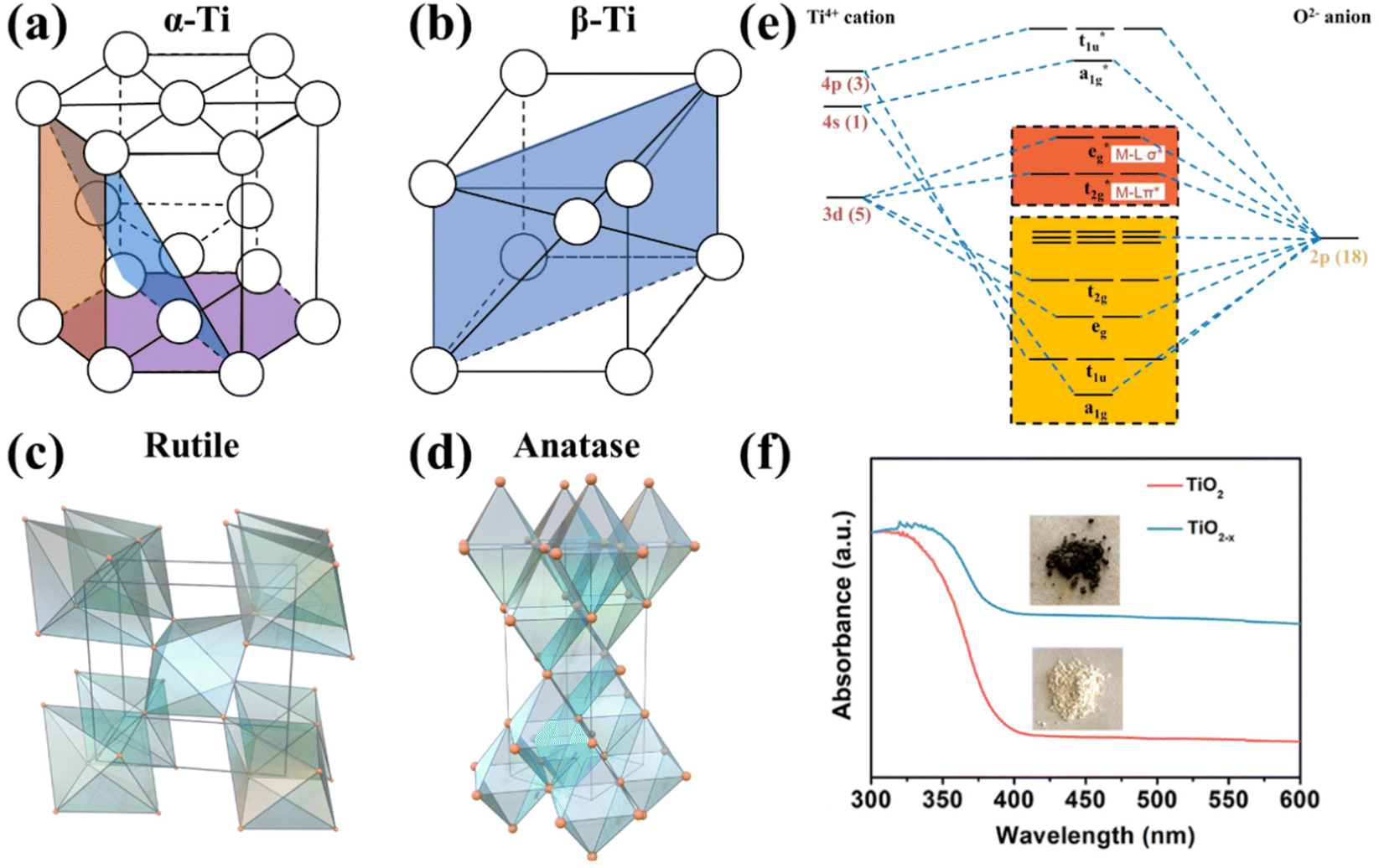 | ||
| Fig. 2 Unit cells of (a) α-Ti, (b) β-Ti, (c) rutile TiO2 and (d) anatase TiO2.61 Reproduced from ref. 61 with permission from American Chemical Society, copyright 2014. (e) Molecular orbitals of the octahedral TiO6 unit. (f) UV–vis absorption spectra (the insets are optical photographs) of TiO2 and TiO2−x.65 Reproduced from ref. 65 with permission from American Chemical Society, copyright 2020. | ||
3.2. Titanium oxides
TiO2 belongs to the category of typical semiconductors.61 There are many kinds of crystallographic structures, including rutile, anatase, brookite and TiO2(B), and several metastable polymorphs, such as TiO2 (H), TiO2 II, and perovskites, have been artificially synthesized.61 Metastable phases are rarely observed as stable electrocatalysts, and the TiO2 (B) phase is less common. Thus, they will not be discussed in this review. Anatase and rutile have more comprehensive applications because they are more stable than brookite,57 hence they are emphatically discussed. Generally, rutile is the most stable phase and is usually synthesized by high-temperature deposition or annealing. Anatase-phased nanomaterials are commonly obtained by solution-based or low-temperature vapor deposition systems.61 Anatase and rutile phases present tetragonal structures (as depicted in Fig. 2c and d) with slightly distorted TiO6 octahedra as a fundamental building block,57,62,63 and have bandgaps of 3.2 and 2.96 eV, respectively.57,64 A large bandgap results in poor electron conductivity of TiO2. Based on the ligand-field theory, the orbital diagram of the TiO6 octahedral unit is shown in Fig. 2e. Since the valence electronic configuration of Ti4+ is [Ar]3d04s0, the bonding orbitals (a1g, t1u, eg and t2g) are mainly contributed by the 2p orbitals of O2−, and the unoccupied anti-bonding orbitals (a1g*, t1u*, eg* and t2g*) are mainly contributed by the 3d, 4s and 4p orbitals of Ti4+. Therefore, the valence band maximum and the conduction band minimum originate from O 2p and Ti 3d, respectively.Oxygen vacancies (OVs) are easily created on the surface of TiO2, thereupon leading to the formation of unsaturated Ti3+ sites and nonstoichiometric TiO2−x (0 < x < 1). According to the stoichiometry theory for semiconductors, TiO2−x belongs to an n-type semiconductor with a narrow bandgap compared to TiO2,57,66 and the electrons in TiO2−x are easily excited from the valence band to the conduction band to form holes and carriers. Therefore, TiO2−x can absorb visible light to show color (Fig. 2f), and the electron conductivity of TiO2−x is strengthened compared to that of intrinsic TiO2.65 Furthermore, the Fermi level could be upshifted by transforming the intrinsic semiconductor to an n-type semiconductor, which is desired for reducing the overpotential of the NO3−RR, as discussed in section 2.2. On the other hand, the electron conductivity of TiO2 can also be modified by heteroatom doping. By controlling the doping pattern, intrinsic TiO2 can be transformed to an n- or p-type semiconductor.57,67 The bandgap, electron conductivity, and the Fermi level could be regulated subsequently.
4. Nitrate-to-ammonia conversion performance of Ti-based nanocatalysts
4.1. Metallic Ti-based electrocatalysts
Considering that titanium exhibits a higher Fermi level, poor HER activity and high corrosion resistance, as discussed in section 3, some researchers tried to explore metallic Ti-based electrocatalysts for NO3−-to-NH3 conversion.Fajardo et al. studied the NO3−RR on a series of transition metals Ti, Fe, Co, Ni, Cu, Zn, and Sn by electroreduction of 100 mg L−1 NO3−-N in 50 mM Na2SO4 at 20 mA cm−2 and 360 min of treatment time. They found that the NO3− degradation on the above materials conformed to the pseudo-first-order characteristic. The selectivity of N2 for Ti is almost negligible, the FENH3 of Ti is close to those of Co and Fe and higher than those of Ni, Cu, Zn and Sn, and the reaction kinetics of Ti is even comparable to that of Pt. The above results suggest that NH3 generation on Ti is electrocatalytically preferential over N2 evolution.36 To further reveal the rule of the NO3−RR on Ti, McEnaney et al. systematically studied the effect of pH, nitrate concentration, and applied potential on the FENH3 of the NO3−RR on the Ti electrode. The heatmaps constructed by an entire grid of electrolyte conditions and FENH3 shown in Fig. 3a suggest that (1) more extreme pH values give significantly higher total current densities than those of moderate pH, (2) lower pH generally corresponds to higher FENH3, and (3) in moderate base and moderate acid electrolytes, moderate nitrate concentrations (between 0.025 and 0.2 M NO3−) generate higher FENH3. According to the above results, the authors used an electrolytic cell constructed by using a Nafion membrane divider with 0.1 M HNO3/0.3 M KNO3 electrolyte for the NO3−RR, and achieved the highest FENH3 of 82%. Importantly, the authors first revealed that titanium hydride (TiHx) is generated on the Ti electrode after the reaction.13 Subsequently, Liu et al. focused on this issue and utilized highly surface-sensitive techniques such as ex situ grazing-incidence X-ray diffraction (GIXRD) and total electron yield X-ray absorption spectroscopy (TEY XAS) to deeply elucidate the self-reconstruction of metallic Ti foil. The GIXRD result suggests that α-Ti was the primary phase of unamended Ti foil; the diffraction patterns for TiHx appeared after the NO3−RR was performed at −0.6 V vs. RHE in 0.1 M HClO4 + 0.8 mM KNO3. The quantitative analysis of Ti K-edge TEY XAS measurements further suggested that more negative applied potential and longer applied durations promoted near-surface TiH2 enrichment, as depicted in Fig. 3b. The electrochemical NO3−RR performance of unamended Ti and preformed TiH2/Ti electrodes was assessed with 30 min chronoamperometry experiments at −0.4, −0.6, −0.8, and −1.0 V vs. RHE in 1 M NaClO4 + 10 mM HNO3, GIXRD of unamended Ti electrodes after such a short-time chronoamperometry showed that self-reconstruction could not be observed, and the NO3−RR performance of unamended Ti could be attributed to the intrinsic Ti foil. Surprisingly, the FENH3, FENO2− and FEH2 of unamended Ti and preformed TiH2/Ti electrodes depicted in Fig. 3c followed a similar trend with almost identical partial current densities of NH3.38 Thus, the mystery of whether the active species of the metallic Ti electrode for the NO3−RR is Ti, Ti hydride, or a combination of species remains under debate, and the above two studies remind us that the self-reconstruction during the NO3−RR cannot be neglected, even if Ti-based materials are usually regarded as stable components. It should be highlighted that the near-surface structure is important for the NO3−RR.
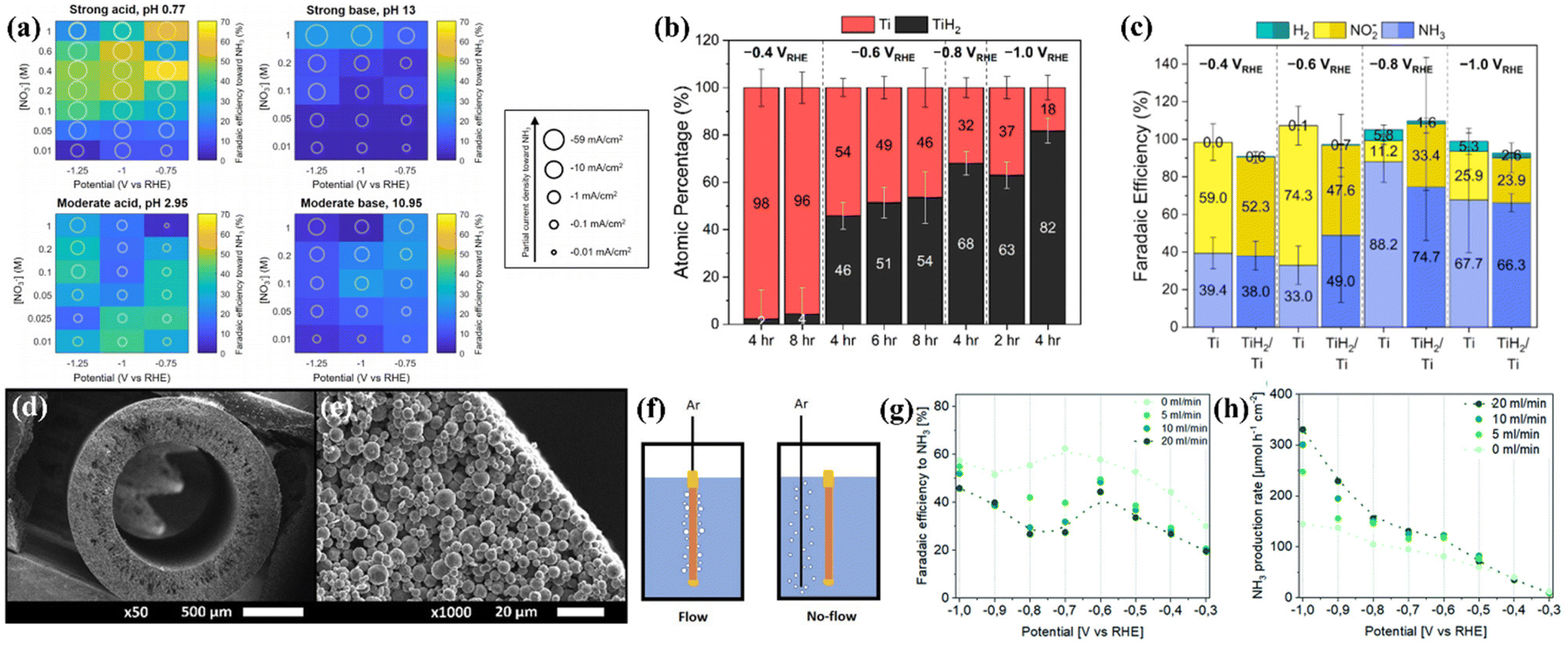 | ||
| Fig. 3 (a) Heatmap plots at four distinct pH values showing faradaic efficiency to NH3 by varying applied potentials and nitrate concentrations. Each grid block displays the data from a 30 min potentiostatic experiment performed at the indicated pH, nitrate concentration, and applied potential.13 Reproduced from ref. 13 with permission from American Chemical Society, copyright 2020. (b) Ti atomic percentages of the Ti foil cathode after the NO3−RR obtained from the EXAFS modeling. (c) Faradaic efficiencies toward the production of ammonia, nitrite, and hydrogen gas on unamended Ti and TiH2/Ti electrodes.38 Reproduced from ref. 38 with permission from American Chemical Society, copyright 2022. (d) SEM images the cross section of a Ti hollow fiber electrode at different magnifications; (e) gas flow configurations of the working electrode compartment; (f) performance of Ti hollow fiber electrodes for the electrochemical reduction of NO3− to NH3 in 0.1 M HClO4 using 50 mM KNO3; potential- and Ar flow rate-dependent (g) faradaic efficiency and (h) production rate toward ammonia.37 Reproduced from ref. 37 with permission from the Royal Society of Chemistry, copyright 2022. | ||
A representative study about electrochemical NO3−-to-NH3 conversion on metallic Ti-based nanomaterials reported by Krzywda et al. suggested that a tubular porous Ti electrode prepared by dry-wet spinning exhibited interesting catalytic behaviors. The as-prepared Ti electrode exhibited uniform pore distribution over the entire length of the fiber (Fig. 3d and e). They introduced a flow of inert gas exiting the wall of the hollow fiber electrode (Ar flow through the hollow fiber electrode from the inside to the outside, denoted as flow-through conditions), as illustrated in Fig. 3f. The current density of tubular porous Ti electrodes with flow-through conditions is higher than that with no-flow (Ar was introduced through an external sparging line next to a hollow fiber electrode) and vigorous magnetic stirring conditions. This result excluded the possibility that enhanced current density originated from the bubbling induced convective flow. Although the flow-through conditions decreased the FENH3 (Fig. 3g), the yield rates and partial current densities of NH3 eventually improved (Fig. 3h). The authors suggested that the flow-through conditions promote the transport of protons and nitrate towards the electrode, thus increasing the catalytic current density. However, the flow-through conditions take NO and N2O away from the solution, promote the homogeneous reaction of NO2− and NH2OH to NO and N2O, hinder the consecutive electrochemical reduction of NO2− and NH2OH towards NH3, and eventually result in a decrease of FENH3.37 Furthermore, Tarpeh et al. constructed a representative flow-cell configuration with a polycrystalline titanium electrode for the NO3−RR and revealed that NO2− and NH3 accounted for almost all NO3−RR products, the selectivity was flow rate dependent, and NH3 was favored at the lowest flow rate. The above results suggested that the NO3−RR was subject to mass transport limitations.68
4.2. TiO2-based electrocatalysts
TiO2 is suitable as a substrate for loading active materials due to its low cost, non-toxic nature and corrosion resistance. The electrons in TiO2 can be excited from the valence band to the conduction band, and the electrons and holes are able to diffuse to the semiconductor surface for participating in the reaction; thus, modulating the valence electronic structure is an alternative for narrowing the bandgap and improving the electron conductivity of intrinsic TiO2. In this section, three common strategies for modulating the NO3−-to-NH3 conversion performances of TiO2-based nanomaterials including heteroatom doping, creating oxygen vacancies and heterostructures will be introduced.Firstly, the electrochemical NO3−-to-NH3 conversion behavior of intrinsic TiO2 must be clarified. Xu et al. synthesized anatase TiO2 and systematically studied its NO3−RR behavior affected by electrolytic conditions, including pH values, nitrate concentration, and the type of electrolyte. The heatmap in Fig. 4a shows that the relationship between FENH3 and the NO3− concentration exhibits an approximate volcano shape, and the relationship between FENH3 and the pH of the electrolyte also obeys a similar tendency. According to the heatmap, higher FENH3 could be achieved in neutral media of 0.4 M KNO3 + 1.0 M PBS (pH = 6.6), unlike metallic Ti, which is more inclined to produce NH3 in acidic electrolytes (Fig. 3e). This phenomenon demonstrates that NO3−RR behaviors on both metallic Ti and TiO2 are pH-sensitive. To further understand the insightful effect of the electrolyte, the electrolyte was switched into another neutral solution of 0.4 M KNO3 + 1.0 M Na2SO4 (pH = 6.7). Fig. 4b and c suggest that TiO2 displayed a much lower catalytic current density, FENH3 and partial current density of NH3 in an electrolyte of 0.4 M KNO3 + 1.0 M Na2SO4. To reveal the essence of this phenomenon, the authors tested the pH values of two electrolytes after 30 min electrolysis. Consequently, PBS only increased 0.3 pH units while in Na2SO4 solution the pH drastically increased from 7 to 13. Therefore, the pH of the electrolyte could be maintained in a suitable range for NH3 formation in the buffer. However, the pH of Na2SO4 quickly ascended to an inferior area shown in Fig. 4a, which inhibited the NH3 formation. Moreover, the authors proposed that faster proton transport in PBS is responsible for the larger catalytic current density. This effect could increase the proton concentration on the electrode surface and facilitate the hydrogenation of NO3− to NH3.39 This study preliminarily revealed the NO3−RR behavior on TiO2 and demonstrated optimal electrochemical conditions for electrochemical NO3−-to-NH3 conversion using TiO2 as an electrocatalyst.
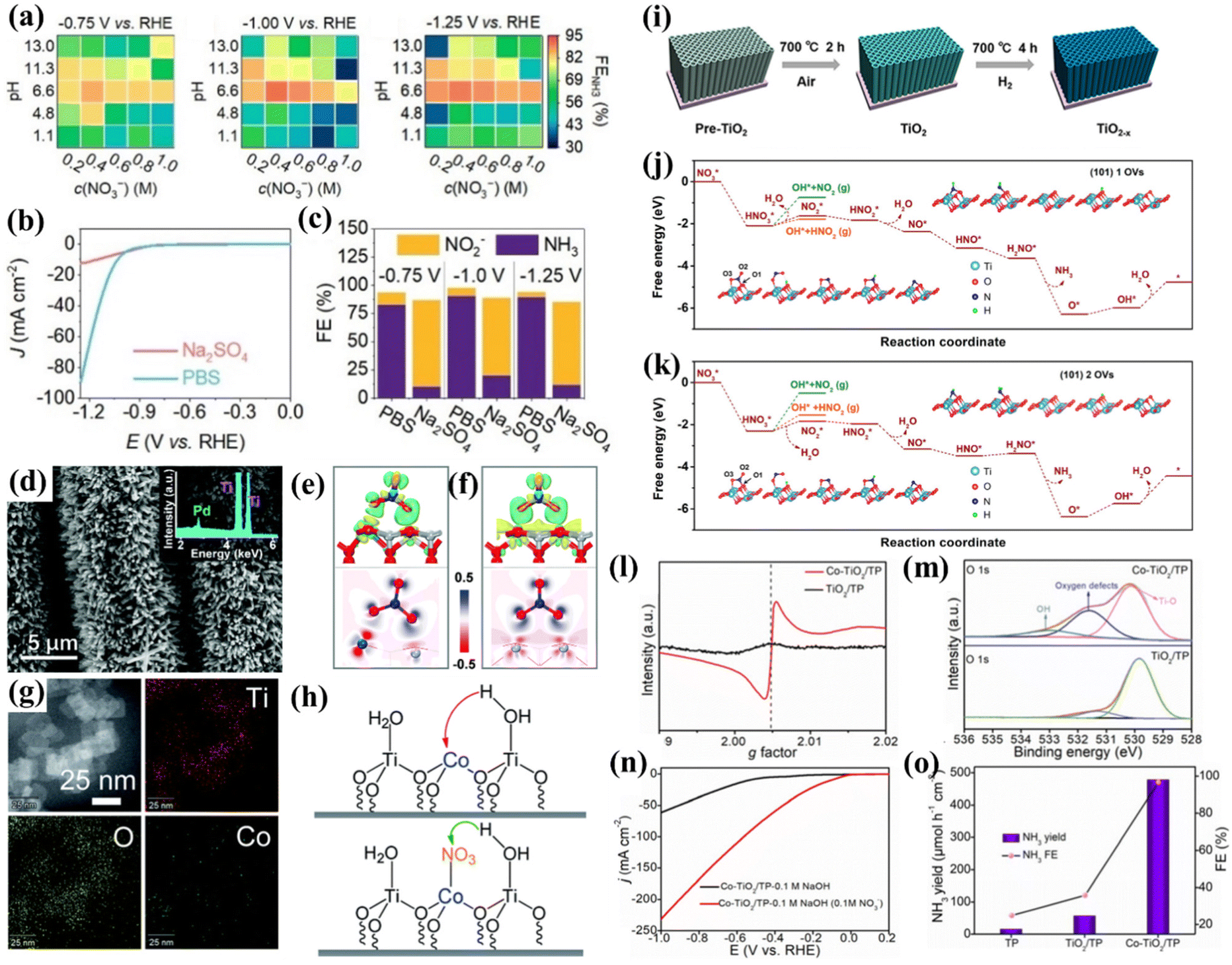 | ||
| Fig. 4 (a) Heatmaps of FENH3 in electrolyte solution with different NO3− concentrations and pH values under three applied potentials; comparison of NO3−RR activities in 1.0M PBS and 1.0M Na2SO4 with 0.4M KNO3, (b) LSV curves, (c) FENH3;39 reproduced from ref. 39 with permission from Wiley-VCH, copyright 2022; (d) SEM images and the EDX spectrum (inset) of Pd/TiO2 nanoarrays; (e and f) 3D and 2D electron density difference mappings for the optimized (e) Pd/TiO2-NO3− and (f) TiO2-NO3− structures;40 reproduced from ref. 40 with permission from the Royal Society of Chemistry, copyright 2021; (g) EDS elemental mapping images of Co/TiO2 NSs; (h) proposed hydrogen-activated routes and the corresponding hydrogen acceptors in the HER and NO3−RR processes;41 reproduced from ref. 41 with permission from the Royal Society of Chemistry, copyright 2022; (i) schematic illustration for TiO2−x synthesis; calculated free energy changes of the nitrate reduction reaction on the TiO2 (101) surface with (j) one, and (k) two oxygen vacancies in one 1 × 3 slab at 0 V vs. RHE;65 reproduced from ref. 65 with permission from American Chemical Society, copyright 2020; (l) EPR spectra and (m) O 1s regions of Co-TiO2/TP and TiO2/TP; (n) LSV curves of Co-TiO2/TP in 0.1 M NaOH with and without 0.1 M NO3−; (o) NH3 yields and FEs of Co-TiO2/TP, TiO2/TP, and bare TP at −0.5 V vs. RHE.42 Reproduced from ref. 42 with permission from The Royal Society of Chemistry, copyright 2022. | ||
A representative study of heteroatom doping is presented by Guo et al., who successfully doped Pd atoms into TiO2 (Fig. 4d) for NO3−-to-NH3 conversion. They proved that the lattice stress caused by doping Pd atoms into the TiO2 phase creates dislocations and distortions forming the active sites of the catalytic reactions. DFT studies revealed that the HOMO of NO3− is located at the O atoms. Fig. 4(e and f) further illustrate that the introduction of Pd atoms makes the electrons of TiO2 transfer to Pd atoms, forming an electron-rich accumulation on Pd, promoting electron transfer to the oxygen atoms of nitrate ions, and eventually accelerating the dissociation of the N–O bond. Meanwhile, a novel Zn-nitrate cell system was assembled for the first time based on the Pd/TiO2 catalyst as the cathode and metal Zn as the anode. The battery delivers remarkable dual functions, i.e., it generates electricity and produces NH3 at the same time with a peak power density of 0.87 mW cm−2 and a high FENH3 of 81.3%. This study demonstrated the feasibility of the galvanic nitrate-based cell, which broadens the field of Zn-based batteries.40 Xu et al. synthesized Co(II)-decorated TiO2 nanosheets (Fig. 4g) exhibiting excellent nitrate performance with an FE and NH3 yield of 97.4% and 0.348 mmol cm−2 h−1 respectively in 1.0 M phosphate buffer solution (PBS, pH = 6.53). The inhibited HER mechanism was also investigated. They demonstrated a proton pumping mechanism for thermodynamically facilitating proton transfer in the presence of nitrate as follows: in the HER, the hydrogen/proton acceptor should be the Co atoms on Co/TiO2 NSs, while the hydrogen/proton acceptor switches into the N/O atoms of nitrate adsorbed on the Co sites in the NO3−RR, and Ti serves as active sites for water dissociation, as depicted in Fig. 4h. Due to the negative charge and delocalized conjugated π electron cloud on NO3−, the barrier of proton transportation from H2O to NO3− (0.508 eV) is much lower than that to Co2+ (0.805 eV), thus the HER could spontaneously switch to the NO3−RR and the energy input for the NO3−RR could be lowered simultaneously.41
Jia et al. synthesized oxygen vacancy-enriched TiO2−x nanotubes by hydrogen reduction (Fig. 4i) and the as-prepared TiO2−x nanotubes were regarded as an efficient electrocatalyst for NO3−-to-NH3 conversion. DFT calculations suggested that introducing OVs on the surface of TiO2 resulted in the occupation of excess 3d electrons of Ti, and the Fermi level could be moved into the conduction band minimum, eventually giving rise to the metallic behavior of TiO2−x and then improving the electron conductivity. This study revealed that OVs are the adsorption sites filled by the oxygen atoms in nitrate, which weaken the N–O bonding. By comparing the Gibbs free energy diagram of TiO2 (101) with one and two vacancies, the latter needed a higher reaction barrier for the formation of HNO2 (Fig. 4j and k), thus suppressing the generation of byproducts.65
The enhancement of oxygen vacancies by metal doping has also received much attention. Song et al. utilized electrochemical anodic oxidation and electrodeposition to synthesize a highly dispersed Cu-doped TiO2 nanotube array (Cu/TNTA) cathode with a high electrocatalytic NO3−RR efficiency and long-term stability. They discovered that just doping trace amounts of Cu into TiO2 could activate more OVs during the NO3−RR process. The OVs highly promoted charge transfer between the NO3− and the electrocatalyst, thus reducing the energy barrier of the NO3−RR, eventually reducing the overpotential required for triggering the NO3−RR and improving the yield rate of NH3 and FENH3.69 Similarly, Yu's group synthesized a unique structure of Cu clusters homogeneously supported on TiO2 nanosheets with abundant OVs (10Cu/TiO2−x). DFT calculations suggested that OVs should be the strong adsorption sites of NO3−. The N–O bond of NO3− breaks automatically during the adsorption process, and the by-products of NO2 or HNO2 are suppressed by OVs with higher reaction barriers, suggesting that the positive effect of interfacial OVs optimized the NO3−RR on 10Cu/TiO2−x.70 Zhao et al. skillfully utilized Na2Ti2O5 grown on a Ti plate as a precursor to prepare CoTi2O5 by an ion exchange strategy, and then the Co-doped TiO2 nanoribbon array grown on the Ti plate (Co-TiO2/TP) was achieved by Ar-annealing of CoTi2O5. XRD patterns suggested that Co in the lattice of TiO2 reduced the crystal quality. Moreover, the EPR and XPS results verified that OVs were introduced into TiO2 after Co doping (Fig. 4l and m). DFT calculations revealed that Co-TiO2 shows a higher impurity level near the Fermi level compared to TiO2, which facilitates charge transfer at the interface. Co doping and OVs also reduced the Gibbs free energy barrier from *NO to *N, which was regarded as a potential determining step in these systems. These advantages enabled Co-TiO2/TP to attain an improved onset potential compared to TiO2/TP, a large NH3 yield of 1127 μmol h−1 cm−2 at −0.9 V and a high FENH3 of 98.2% at −0.5 V vs. RHE (Fig. 4n and o).42
The above Cu/Co induced OVs generated in TiO2 could be attributed to the charge difference between Cu2+/Co2+ and Ti4+. Doping Cu2+/Co2+ into the TiO2 lattice must create OVs to compensate for the missing positive charge while remaining charge neutral.71
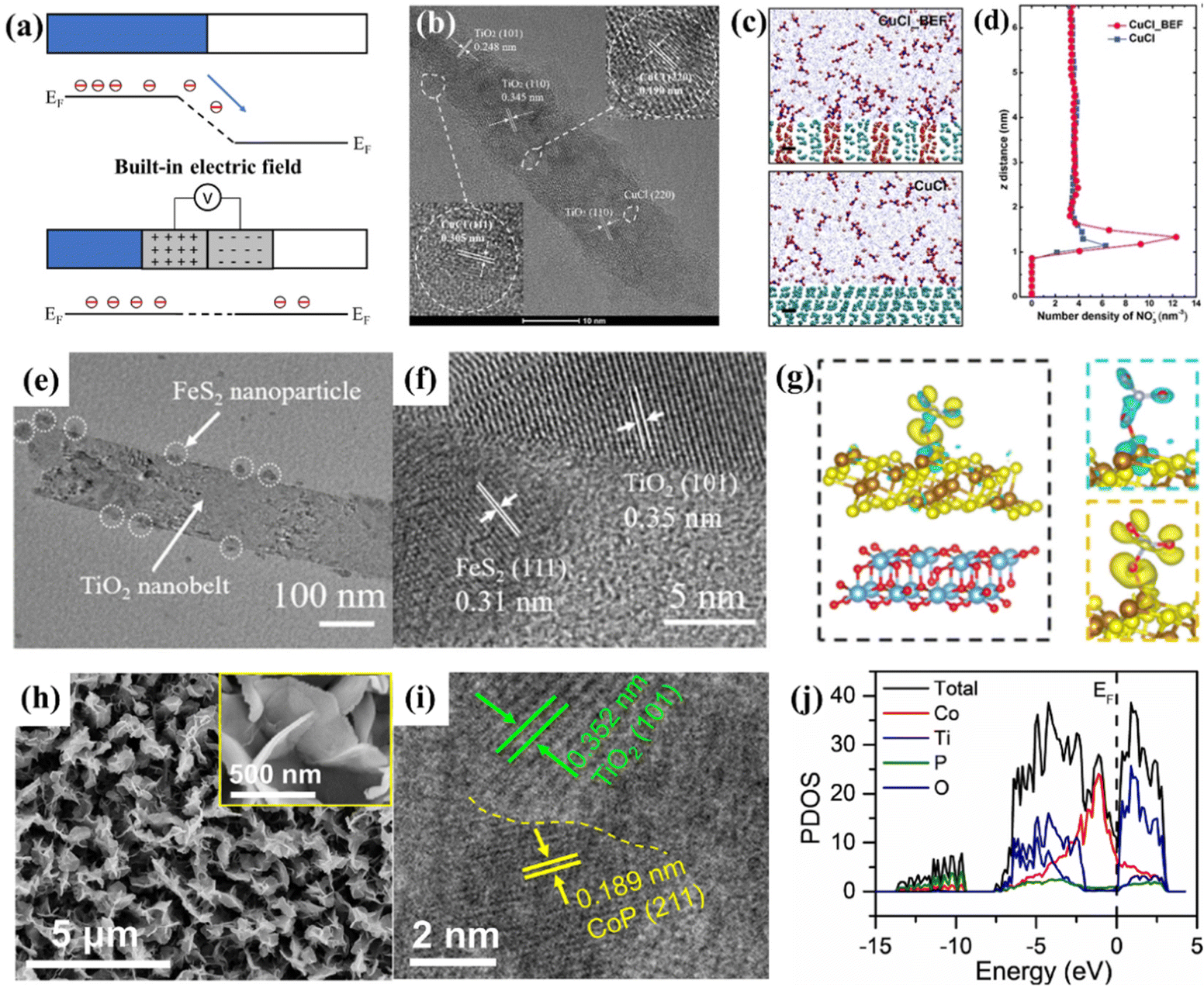 | ||
| Fig. 5 (a) The formation of a built-in electric field at the heterostructure composed of two components with different Fermi levels. (b) Representative HRTEM image of CuCl_BEF. (c) Molecular dynamics simulation of CuCl and CuCl_BEF in KNO3 (100 mg L−1) solution. Scale bar, 0.5 nm. (d) Distribution of NO3− along the z-axis electrode distance based on the molecular dynamics simulation.72 Reproduced from ref. 72 with permission from Wiley-VCH, copyright 2022. (e) TEM and (f) HRTEM images of FeS2@TiO2/TP. (g) Charge density difference of FeS2@TiO2 with adsorbed NO3−, where yellow and cyan color indicate electron accumulation and depletion, respectively, and the isosurface values are set to 0.000213 Å−3.73 Reproduced from ref. 73 with permission from The Royal Society of Chemistry, copyright 2022. (h) SEM images and (i) HRTEM images of CoP/TiO2. (j) Calculated PDOS of CoP/TiO2.76 Reproduced from ref. 76 with permission from Elsevier, copyright 2022. | ||
Sun et al. stacked CuCl (111) and rutile TiO2 (110) layers together (Fig. 5b), a BEF could be induced by electron transfer from TiO2 to CuCl due to the difference in their Fermi levels, and the BEF strength was roughly estimated to be 8 × 108 V m−1. Molecular dynamics simulation and finite element analysis suggested that the BEF accumulated NO3− in the diffusion layer near the surface of the electrocatalyst (Fig. 5c and d). This study demonstrated that the BEF increased the free energy of *ON, resulting in a great decrease of ΔG of the potential determining step. This phenomenon could be elucidated as the BEF resulted in electron richer Cu(I), which suppressed the electron donation from *NO but facilitated the back donation to the π* anti-bonding orbital of *NO, thus, destabilizing the N–O bond of *NO. The above advantages endowed the heterostructure with an NH3 selectivity of 98.6% and a yield rate of 1.82 mg h−1 cm−2 at −1.0 V vs. RHE in 100 mg L−1 NO3− + 0.5 M Na2SO4.72
Wang et al. reported a heterostructure of an FeS2 nanoparticle-decorated TiO2 nanobelt array supported on a titanium plate (FeS2@TiO2/TP), as depicted in Fig. 5e and f. They proved that the abundant heterostructures endowed FeS2@TiO2/TP with excellent electrocatalytic performance with an NH3 yield of 860.3 mmol h−1 cm−2 and a FENH3 of 97% at −0.4 V vs. RHE in 0.1 M NaOH + 0.1 M NO3−. Furthermore, they elucidated the positive effect of the FeS2@TiO2 heterostructure by DFT studies. The charge density difference and partial density of states suggested that the electrons were spontaneously transferred from TiO2 to FeS2 (Fig. 5g). The corresponding charge accumulation and depletion endowed the FeS2@TiO2 interface with local nucleophilic and electrophilic regions, thus favoring the adsorption of targeted species. Moreover, such a heterostructure achieves the targeted adsorption of molecules by electrostatic interaction, promoting the charge transfer between the active site and the O atom of NO3−, resulting in the activation of the N–O bond.73 Similar results are obtained for the CoS2@TiO2/TP heterostructure synthesized by Zhao et al. They utilized XPS to prove the electron transfer from TiO2 to CoS2, and the heterostructure eventually resulted in an improved electrocatalytic performance of CoS2@TiO2/TP with a high FENH3 and NH3 yield rate of 92.80% and 538.21 mmol h−1 cm−2, respectively.74
Based on the conclusion that CoP is proven to be an effective and stable electrocatalyst for NO3−-to-NH3 conversion,75 Deng et al. designed a heterostructure of CoP/TiO2@TP, as depicted in Fig. 5h and i. Interestingly, XPS proved that the BEF of the CoP/TiO2 heterostructure was established by the electrons transferring from CoP to TiO2, which is reversed for the heterostructures mentioned above. This phenomenon should be attributed to the fact that CoP exhibits the metallic character of a continuous electron occupation state at the Fermi level, and the work function of CoP should be lower than that of TiO2 with semiconductor properties. Moreover, the heterostructure displayed an impurity level near the Fermi level compared with CoP and TiO2, indicating that more charge carriers were created (Fig. 5j), thereby enhancing the electron conductivity of CoP/TiO2. The Gibbs free energy diagram suggested that the potential-determining step of conversion of NO3− to *NO3 presented a lower energy barrier in the CoP/TiO2 heterostructure. Therefore, such CoP/TiO2@TP attained an excellent FENH3 of 95.0% with a large NH3 of 499.8 μmol h−1 cm−2.76
4.3. New generation Ti-based MXene and single atomic catalysts
In addition to the traditional metallic Ti- and TiO2-based nanomaterials, MXene attracts much attention in electrocatalysis. Moreover, single atomic catalysts (SACs) are emerging as fantastic materials due to their ultra-high active site utilization and outstanding catalytic activity. Therefore, developing Ti-based MXene and SACs for electrochemical NO3−-to-NH3 conversion could be a prevailing trend, although related studies are still rarely reported. In this section, we summarized some preliminary experimental and theoretical achievements of Ti-based MXene and SACs for the NO3−RR and hope to provide inspiration for designing novel Ti-based electrocatalysts.Li's group investigated the possible mechanism and catalytic activity of electrochemical NO3−-to-NH3 conversion on Ti3C2 MXene by theoretical calculations. They proposed that the reactive pathway of NO3− → *NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3 → NH3(g) is thermodynamically preferable to that of NO3− → *NO3 → *NO2 → *NO → *NOH → *NHOH → *NH → *NH2 → *NH3 → NH3(g). *NO3 and *NO2 prefer to adsorb on Ti3C2 MXene in parallel adsorption modes due to the O atoms of *NO3 and *NO2 being inclined to bond with Ti, resulting in drastic distortion of *NO3 and *NO2 (Fig. 6a). Moreover, the Gibbs free energy suggests that the NO3−RR prefers to occur on the basal plane rather than the edge plane of Ti3C2 MXene because basal plane sites present lower energy barriers for the potential determining step (Fig. 6b). The termination effects of Ti3C2T2 (T = O, OH, H, Cl, F, representing the terminal atoms located at the surface of Ti3C2 MXene) for NO3−-to-NH3 conversion were also elucidated in detail. As illustrated in Fig. 6c, the NO3−RR is more competitive than the HER on the O-terminated Ti3C2O2 with OVs (denoted as Ti3C2O2-Ov), and all free energy changes of NO3−-to-NH3 on the Ti3C2O2-Ov are negative, signifying that the termination effect of oxygen promotes the hydrogenation of nitrogen-containing intermediates.79 Another study utilized Ti3C2Tx nanosheets as a promising molecular catalyst substrate. CuPc can be spontaneously dispersed on delaminated Ti3C2Tx to overcome their large π-conjugated stacking. The prepared CuPc@MXene exhibited a high yield rate and selectivity for NH3, which is superior to other counterparts of FePc@MXene, NiPc@MXene, and CoPc@MXene. DFT calculations suggested that NO3−-to-NH3 conversion tends to obey the ONH pathway because the Gibbs free energy required only 0.27 eV to form *ONH.80
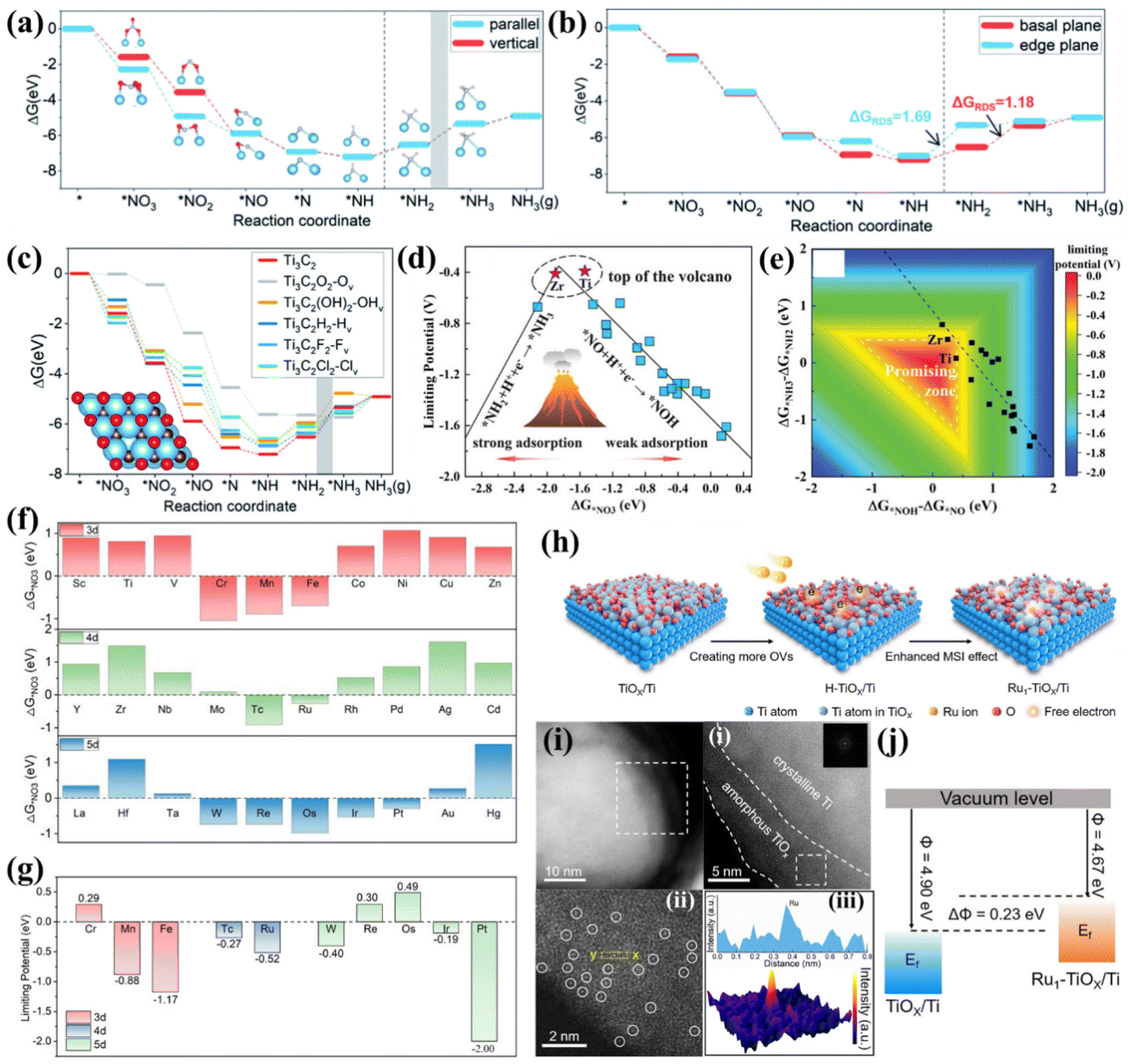 | ||
| Fig. 6 (a) Gibbs free energy diagram of NO3−-to-NH3 on Ti3C2 in vertical and parallel *NO3 adsorption modes. The grey zone means that *NH2 → *NH3 is the RDS. (b) Gibbs free energy diagram of NO3−-to-NH3 on the basal plane and edge plane of Ti3C2. (c) Gibbs free energy diagram of the reaction pathway of Ti3C2T2-Tv, and the grey zone highlights the step *NH2 → *NH3 as the RDS.79 Reproduced from ref. 79 with permission from The Royal Society of Chemistry, copyright 2022. (d) NO3−RR volcano plot of TM/g-CN with a descriptor of ΔG*NO3. (e) Contour plot of limiting potential as a function of two potential-determining steps (*NO + H+ + e− → *NOH and *NH2 + H+ + e− → *NH3). The promising zone is highlighted.44 Reproduced from ref. 44 with permission from Wiley-VCH, copyright 2020. (f) Gibbs free energy change of NO3− absorption on Ti3C2O2-TMSA. (g) Summary of limiting potentials on Ti3C2O2-TMSA.81 Reproduced from ref. 81 with permission from Springer Nature, copyright 2023. (h) Schematic illustration of preparing the Ru1-TiOx/Ti electrode via an inherent surface oxide anchoring strategy. (i) (i and ii) Low-magnified HAADF-STEM images of Ru1-TiOx/Ti. (iii) Atomic-resolution HAADF-STEM image of Ru1-TiOx/Ti; Ru single atoms are indicated by white circles. (iv) Line intensity profile (inset) taken along the yellow dashed rectangle in HAADF-STEM and the corresponding 3D surface intensity profile. (j) Schematic illustration of the work functions of Ru1-TiOx/Ti and TiOx/Ti.82 Reproduced from ref. 82 with permission from Wiley-VCH, copyright 2022. | ||
Niu et al. established several transition metal atoms from Ti to Au supported on graphitic carbon nitrides (g-CN) as representatives (TM/g-CN) to study the NO3−RR rule of SACs. They found that the adsorption energies of NO3− (ΔG*NO3), protons (ΔG*H), and the N2 molecule (ΔG*N2) on TM/g-CN exhibited periodic regulations. Furthermore, they proposed that the N-end pathway is the most reasonable on TM/g-CN because the N-end adsorption of NO presents the lowest energy. Based on the above reactive pathway, the performance criteria of limiting potential on TM/g-CN were evaluated. A volcano plot of limiting potential vs. ΔG*NO3 on TM/g-CN was proposed. As shown in Fig. 6d, too strong adsorption of NO3− results in a larger energy barrier in the step from *NH2 to *NH3. In contrast, too weak adsorption leads to a sluggish step from *NO to *NOH, and Ti/g-CN and Zr/g-CN stand exactly near the top of the volcano. Regarding selectivity, the large energy barriers prevented the formation of NO2, NO, N2O, and N2 on Ti/g-CN, guaranteeing a high FENH3 of Ti/g-CN. In addition, the linear scaling relationship between (ΔG*NH3 − ΔG*NH2) and (ΔG*NOH − ΔG*NO) hinders the exploration of more effective electrocatalysts (Fig. 6e), hence, a critical strategy to break such a scaling relationship is desired.44 Another study that predicts TM/g-C3N4 (TM = Sc to Au, except Tc, Cd, and Hg) for the NO3−RR also proposes a similar volcano plot and suggests that Ti/g-C3N4 should be an ideal candidate with outstanding limiting potential and FENH3.90
Recently, Ti-based MXene and oxides are also predicted to be suitable substrates for anchoring SAs. Wang et al. screened out 30 single transition metal atoms (3d: Sc–Zn, 4d: Y–Cd, and 5d: La–Hg) anchored on Ti3C2O2 by means of first principles calculations. They found that SAs with half-filled d orbitals (around d5) are favorable for NO3− activation due to high electronic states at the Fermi level (Fig. 6f). Further screening suggested that Cr, Re and Os SAs anchored on Ti3C2O2 present negative free energy changes in the whole NO3−-to-NH3 conversion process and thus exhibit positive limiting potentials (Fig. 6g); hence, they are recognized as the most efficient candidates.66
An experimental study utilized Ti foil as a substrate to prepare binder-free monolithic single-atom electrodes (MSAEs). Metallic Ti inevitably undergoes oxidation in air to form a thin metal oxide layer (TiOx) with abundant dangling bonds and defects, thereby providing unique on-site hosts for the dispersion of SACs. A two-step annealing approach depicted in Fig. 6h was employed to successfully anchor Ru atoms into the TiOx layer (Fig. 6i). The as-prepared MSAEs were named Ru1-TiOx/Ti. Anchoring of Ru atoms upshifted the Fermi level, as proven by the lower work function of Ru1-TiOx/Ti compared to TiOx/Ti, as shown in Fig. 6j. This characteristic endowed Ru1-TiOx/Ti with much improved catalytic current density and FENH3 at various applied potentials. The highest FENH3 and NH3 yield rate of 87.6% and 22.2 mol g−1 were respectively achieved at −0.3 V vs. RHE for Ru1-TiOx/Ti.82
Although DFT studies predicted Ti-based SACs to be outstanding candidates as electrocatalysts for NO3−-to-NH3 conversion, there is still a lack of experimental achievement to fulfill the above results. Further synthesis strategies and performance studies should be conducted.
5. Conclusion and perspective
Emerging “hydrogen economy” and “carbon neutralization” have caused NH3 to be regarded as a carbon-free fuel and a portable energy carrier, endowing the electrochemical nitrate reduction reaction for NO3−-to-NH3 conversion with a new opportunity for “green ammonia” synthesis. Compared to noble metal-based electrocatalysts, Ti-based electrocatalysts mainly composed of metallic Ti- and TiO2-based nanomaterials are attractive due to their inherent advantages of being nontoxic and cost-effective and having outstanding stability and HER inertness. Fundamental studies revealed that metallic Ti and TiO2 are more suitable for NO3−-to-NH3 conversion over a wide pH range. Many strategies including morphological design, heteroatom doping, creation of OVs, and the construction of heterostructures have been developed for reducing the overpotential while improving the FENH3. To sum up, regulating the work function (for metallic Ti) or ionizing energy (for TiO2) to reduce the gap between the HOS and the LUMO of nitrogen-containing intermediates is essential for reducing the overpotential, modulating the d-band structure to optimize the binding energy of O- and N-containing intermediates is crucial for regulating the reactive route and faradaic efficiency of NH3, and designing secondary active sites for splitting H2O on Ti-based nanomaterials is also crucial for the hydrogenation of NO3− and consequent intermediates in neutral and alkaline electrolytes. In particular, since potential induced self-reconstruction exists for Ti-based electrocatalysts, the use of in situ characterization techniques is encouraged to reveal the real active sites for the NO3−RR.Finally, the difficulties and opportunities of Ti-based nanocatalysts for electrochemical NO3−-to-NH3 conversion are discussed.
The large bandgap of TiO2 (∼3 eV) results in poor electronic conductivity which is the major reason for low electrochemical activity. Narrowing the bandgap of TiO2 is an essential strategy to solve the problem of electronic conductivity. Therefore, characterizing the bandgap by UV-Vis spectroscopy, UPS, or DFT calculations is suggested to be performed for TiO2-based nanomaterials. Moreover, constructing metal/TiO2 heterostructures (such as Ti/TiO2) and in situ compositing the TiO2-based nanomaterials with electronic conductive additives such as graphene, carbon nanotubes or acetylene black are effective strategies for solving the electronic conductivity issue. Recently, novel Ti-based MXenes with a large specific area and outstanding electron conductivity have been proposed as ideal candidates for nitrate-to-ammonia conversion; thus developing Ti-based MXene electrocatalysts is an important development direction in the future.
The effect of lattice facets is rarely considered for Ti-based nanomaterials for the NO3−RR. Different lattice facets present unique atomic arrangements, signifying that the nanostructure with different exposed lattice facets exhibits disparate catalytic performance. Typically, high-index facets represent better catalytic activity. Therefore, exploring the effect of the lattice facets of Ti-based nanomaterials on the catalytic performance of the NO3−RR is important for designing Ti-based nanomaterials with desired exposed lattice facets, reducing the overpotential and improving the FENH3.
Although heteroatom doping is a common modification strategy for inorganic materials, OVs in TiO2 could also be simultaneously formed by doping metallic atoms with lower valence than Ti4+. Therefore, doping-induced OVs should be considered but unfortunately, they are often neglected.
In addition to NH3, producing high value-added organic molecules with deeply reduced nitrogen functional groups (such as urea,91,92 methylamine,93 formamide,94 glycine,95etc.) by electrochemical reduction of NO3− is also an emerging direction. Since TiO2-based nanomaterials exhibit profound reduction ability of NO3−, some preliminary studies successfully coupled the CO2 reduction reaction and the NO3−RR on TiO2-based nanomaterials to produce urea.91 Zhang et al. revealed that constructing secondary active sites on TiO2 for adsorbing carbon-containing intermediates is a feasible strategy for the gC–N coupled reaction.92
We hope that this review will shed light on the exploration of Ti-based nanocatalysts and pave the way for low-cost and efficient Ti-based nanocatalysts to realize large-scale industrial applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We appreciate the financial support from the National Key Research and Development Program of China (No. 2020YFC1909604), the Shenzhen Key Projects of Technological Research (JSGG20200925145800001) and the Shenzhen Science and Technology Program (CJGJZD20210408092801005).References
- R. F. Service, Chemistry. New recipe produces ammonia from air, water, and sunlight, Science, 2014, 345, 610 CrossRef CAS PubMed.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- C. Tang and S. Z. Qiao, How to explore ambient electrocatalytic nitrogen reduction reliably and insightfully, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- Y. Zhao, B. P. Setzler, J. Wang, J. Nash, T. Wang, B. Xu and Y. Yan, An Efficient Direct Ammonia Fuel Cell for Affordable Carbon-Neutral Transportation, Joule, 2019, 3, 2472–2484 CrossRef CAS.
- M. A. Mushtaq, M. Arif, G. Yasin, M. Tabish, A. Kumar, S. Ibraheem, W. Ye, S. Ajmal, J. Zhao, P. Li, J. Liu, A. Saad, X. Fang, X. Cai, S. Ji and D. Yan, Recent developments in heterogeneous electrocatalysts for ambient nitrogen reduction to ammonia: Activity, challenges, and future perspectives, Renewable Sustainable Energy Rev., 2023, 176, 113197 CrossRef CAS.
- L. Jiang and X. Fu, An Ammonia–Hydrogen Energy Roadmap for Carbon Neutrality: Opportunity and Challenges in China, Engineering, 2021, 7, 1688–1691 CrossRef.
- Y. Ren, C. Yu, L. Wang, X. Tan, Z. Wang, Q. Wei, Y. Zhang and J. Qiu, Microscopic-Level Insights into the Mechanism of Enhanced NH3 Synthesis in Plasma-Enabled Cascade N2 Oxidation-Electroreduction System, J. Am. Chem. Soc., 2022, 144, 10193–10200 CrossRef CAS PubMed.
- N. Salmon and R. Bañares-Alcántara, Green ammonia as a spatial energy vector: a review, Sustainable Energy Fuels, 2021, 5, 2814–2839 RSC.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Ammonia as a possible element in an energy infrastructure: catalysts for ammonia decomposition, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Beyond fossil fuel-driven nitrogen transformations, Science, 2018, 360, 6391 Search PubMed.
- Y. Song, D. Johnson, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, J. Huang, F. Yang, F. Zhang, R. Qiao, A. P. Baddorf, T. J. Tschaplinski, N. L. Engle, M. C. Hatzell, Z. Wu, D. A. Cullen, H. M. Meyer III, B. G. Sumpter and A. J. Rondinone, A physical catalyst for the electrolysis of nitrogen to ammonia, Sci. Adv., 2018, 4, e1700336 CrossRef PubMed.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, Electrolyte Engineering for Efficient Electrochemical Nitrate Reduction to Ammonia on a Titanium Electrode, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- M. A. Mushtaq, A. Kumar, G. Yasin, M. Arif, M. Tabish, S. Ibraheem, X. Cai, W. Ye, X. Fang, A. Saad, J. Zhao, S. Ji and D. Yan, 3D interconnected porous Mo-doped WO3@CdS hierarchical hollow heterostructures for efficient photoelectrochemical nitrogen reduction to ammonia, Appl. Catal., B, 2022, 317, 121711 CrossRef CAS.
- M. A. Mushtaq, M. Arif, X. Fang, G. Yasin, W. Ye, M. Basharat, B. Zhou, S. Yang, S. Ji and D. Yan, Photoelectrochemical reduction of N2 to NH3 under ambient conditions through hierarchical MoSe2@g-C3N4 heterojunctions, J. Mater. Chem. A, 2021, 9, 2742–2753 RSC.
- W. Ye, M. Arif, X. Fang, M. A. Mushtaq, X. Chen and D. Yan, Efficient Photoelectrochemical Route for the Ambient Reduction of N2 to NH3 Based on Nanojunctions Assembled from MoS2 Nanosheets and TiO2, ACS Appl. Mater. Interfaces, 2019, 11, 28809–28817 CrossRef CAS PubMed.
- S. Yang, W. Ye, D. Zhang, X. Fang and D. Yan, Layered double hydroxide derived bimetallic nickel–iron selenide as an active electrocatalyst for nitrogen fixation under ambient conditions, Inorg. Chem. Front., 2021, 8, 1762–1770 RSC.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a century of ammonia synthesis changed the world, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- N. C. Kani, N. H. L. Nguyen, K. Markel, R. R. Bhawnani, B. Shindel, K. Sharma, S. Kim, V. P. Dravid, V. Berry, J. A. Gauthier and M. R. Singh, Electrochemical Reduction of Nitrates on CoO Nanoclusters–Functionalized Graphene with Highest Mass Activity and Nearly 100% Selectivity to Ammonia, Adv. Energy Mater., 2023, 13, 2204236 CrossRef CAS.
- N. Gruber and J. N. Galloway, An Earth-system perspective of the global nitrogen cycle, Nature, 2008, 451, 293–296 CrossRef CAS PubMed.
- C. Wang, Y. Zhang, H. Luo, H. Zhang, W. Li, W. X. Zhang and J. Yang, Iron-Based Nanocatalysts for Electrochemical Nitrate Reduction, Small Methods, 2022, 6, 2200790 CrossRef CAS PubMed.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, Electrochemical Ammonia Synthesis—The Selectivity Challenge, ACS Catal., 2016, 7, 706–709 CrossRef.
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S. X. Dou, L. Zhang and J. C. Yu, Efficient Ammonia Electrosynthesis from Nitrate on Strained Ruthenium Nanoclusters, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed.
- W. He, J. Zhang, S. Dieckhofer, S. Varhade, A. C. Brix, A. Lielpetere, S. Seisel, J. R. C. Junqueira and W. Schuhmann, Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia, Nat. Commun., 2022, 13, 1129 CrossRef CAS PubMed.
- Q. Hu, Y. Qin, X. Wang, Z. Wang, X. Huang, H. Zheng, K. Gao, H. Yang, P. Zhang, M. Shao and C. He, Reaction intermediate-mediated electrocatalyst synthesis favors specified facet and defect exposure for efficient nitrate–ammonia conversion, Energy Environ. Sci., 2021, 14, 4989–4997 RSC.
- X. Liang, H. Zhu, X. Yang, S. Xue, Z. Liang, X. Ren, A. Liu and G. Wu, Recent Advances in Designing Efficient Electrocatalysts for Electrochemical Nitrate Reduction to Ammonia, Small Struct., 2023, 4, 2200202 CrossRef CAS.
- J. Y. Fang, Q. Z. Zheng, Y. Y. Lou, K. M. Zhao, S. N. Hu, G. Li, O. Akdim, X. Y. Huang and S. G. Sun, Ampere-level current density ammonia electrochemical synthesis using CuCo nanosheets simulating nitrite reductase bifunctional nature, Nat. Commun., 2022, 13, 7899 CrossRef CAS PubMed.
- W. Wen, P. Yan, W. Sun, Y. Zhou and X. Y. Yu, Metastable Phase Cu with Optimized Local Electronic State for Efficient Electrocatalytic Production of Ammonia from Nitrate, Adv. Funct. Mater., 2022, 33, 2212233 Search PubMed.
- W. Zhu, X. Zhang, F. Yao, R. Huang, Y. Chen, C. Chen, J. Fei, Y. Chen, Z. Wang and H. Liang, A Hydrazine-Nitrate Flow Battery Catalyzed by a Bimetallic RuCo Precatalyst for Wastewater Purification along with Simultaneous Generation of Ammonia and Electricity, Angew. Chem., Int. Ed., 2023, 62, e2023003 CrossRef PubMed.
- H. Liu, X. Lang, C. Zhu, J. Timoshenko, M. Ruscher, L. Bai, N. Guijarro, H. Yin, Y. Peng, J. Li, Z. Liu, W. Wang, B. R. Cuenya and J. Luo, Efficient Electrochemical Nitrate Reduction to Ammonia with Copper-Supported Rhodium Cluster and Single-Atom Catalysts, Angew. Chem., Int. Ed., 2022, 61, e202202556 CAS.
- W. Gao, K. Xie, J. Xie, X. Wang, H. Zhang, S. Chen, H. Wang, Z. Li and C. Li, Alloying of Cu with Ru Enabling the Relay Catalysis for Reduction of Nitrate to Ammonia, Adv. Mater., 2023, 35, 2202952 CrossRef CAS PubMed.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J. F. Li, Z. Q. Tian, W. Liu, A. Wang and T. Zhang, Potential-Driven Restructuring of Cu Single Atoms to Nanoparticles for Boosting the Electrochemical Reduction of Nitrate to Ammonia, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS.
- J. Wang, S. Zhang, C. Wang, K. Li, Y. Zha, M. Liu, H. Zhang and T. Shi, Ambient ammonia production via electrocatalytic nitrate reduction catalyzed by a flower-like CuCo2O4 electrocatalyst, Inorg. Chem. Front., 2022, 9, 2374–2378 RSC.
- J. Geng, S. Ji, H. Xu, C. Zhao, S. Zhang and H. Zhang, Electrochemical reduction of nitrate to ammonia in a fluidized electrocatalysis system with oxygen vacancy-rich CuOx nanoparticles, Inorg. Chem. Front., 2021, 8, 5209–5213 RSC.
- R. Zhang, S. Zhang, Y. Guo, C. Li, J. Liu, Z. Huang, Y. Zhao, Y. Li and C. Zhi, A Zn–nitrite battery as an energy-output electrocatalytic system for high-efficiency ammonia synthesis using carbon-doped cobalt oxide nanotubes, Energy Environ. Sci., 2022, 15, 3024–3032 RSC.
- A. S. Fajardo, P. Westerhoff, C. M. Sanchez-Sanchez and S. Garcia-Segura, Earth-abundant elements a sustainable solution for electrocatalytic reduction of nitrate, Appl. Catal., B, 2021, 281, 119465 CrossRef CAS.
- P. M. Krzywda, A. Paradelo Rodríguez, L. Cino, N. E. Benes, B. T. Mei and G. Mul, Electroreduction of NO3− on tubular porous Ti electrodes, Catal. Sci. Technol., 2022, 12, 3281–3288 RSC.
- M. J. Liu, J. Guo, A. S. Hoffman, J. H. Stenlid, M. T. Tang, E. R. Corson, K. H. Stone, F. Abild-Pedersen, S. R. Bare and W. A. Tarpeh, Catalytic Performance and Near-Surface X-ray Characterization of Titanium Hydride Electrodes for the Electrochemical Nitrate Reduction Reaction, J. Am. Chem. Soc., 2022, 144, 5739–5744 CrossRef CAS PubMed.
- Y. T. Xu, Z. Peng, Y. Han, H. Zhong, J. Yang and Y. Cao, Insight into Hydrogenation Selectivity of the Electrocatalytic Nitrate-to-Ammonia Reduction Reaction via Enhancing the Proton Transport, ChemSusChem, 2022, 15, e202102450 CAS.
- Y. Guo, R. Zhang, S. Zhang, Y. Zhao, Q. Yang, Z. Huang, B. Dong and C. Zhi, Pd doping-weakened intermediate adsorption to promote electrocatalytic nitrate reduction on TiO2 nanoarrays for ammonia production and energy supply with zinc–nitrate batteries, Energy Environ. Sci., 2021, 14, 3938–3944 RSC.
- Y.-T. Xu, Y. Han, D. K. Sam and Y. Cao, The selective electrocatalytic reduction of nitrate to ammonia using Co(ii)-decorated TiO2 nanosheets, J. Mater. Chem. A, 2022, 10, 22390–22398 RSC.
- D. Zhao, C. Ma, J. Li, R. Li, X. Fan, L. Zhang, K. Dong, Y. Luo, D. Zheng, S. Sun, Q. Liu, Q. Li, Q. Lu and X. Sun, Direct eight-electron NO3−-to-NH3 conversion: using a Co-doped TiO2 nanoribbon array as a high-efficiency electrocatalyst, Inorg. Chem. Front., 2022, 9, 6412–6417 RSC.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Nitrate electroreduction: mechanism insight, in situ characterization, performance evaluation, and challenges, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Theoretical Insights into the Mechanism of Selective Nitrate–to–Ammonia Electroreduction on Single–Atom Catalysts, Adv. Funct. Mater., 2020, 31, 2008533 CrossRef.
- D. He, Y. Li, H. Ooka, Y. K. Go, F. Jin, S. H. Kim and R. Nakamura, Selective Electrocatalytic Reduction of Nitrite to Dinitrogen Based on Decoupled Proton-Electron Transfer, J. Am. Chem. Soc., 2018, 140, 2012–2015 CrossRef CAS PubMed.
- J. Wang, T. Feng, J. Chen, V. Ramalingam, Z. Li, D. M. Kabtamu, J.-H. He and X. Fang, Electrocatalytic nitrate/nitrite reduction to ammonia synthesis using metal nanocatalysts and bio-inspired metalloenzymes, Nano Energy, 2021, 86, 106088 CrossRef CAS.
- W. Chen, X. Yang, Z. Chen, Z. Ou, J. Hu, Y. Xu, Y. Li, X. Ren, S. Ye, J. Qiu, J. Liu and Q. Zhang, Emerging Applications, Developments, Prospects, and Challenges of Electrochemical Nitrate–to–Ammonia Conversion, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202300512.
- S. Ye, X. Yang, Z. Huang, W. Chen, T. Huang, Z. Ou, W. Xiong, Y. Li, X. Ren, J. Hu, J. Liu and Q. Zhang, The Activity Origin of FeCo Prussian Blue Analogue for Ambient Electrochemical Hydrogenation of Nitrate into Ammonia in Neutral Electrolyte, Sci. China Mater., 2023 DOI:10.1007/s40843-023-2475-6.
- J.-X. Liu, D. Richards, N. Singh and B. R. Goldsmith, Activity and Selectivity Trends in Electrocatalytic Nitrate Reduction on Transition Metals, ACS Catal., 2019, 9, 7052–7064 CrossRef CAS.
- O. Q. Carvalho, R. Marks, H. K. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Arnadottir and K. A. Stoerzinger, Role of Electronic Structure on Nitrate Reduction to Ammonium: A Periodic Journey, J. Am. Chem. Soc., 2022, 144, 14809–14818 CrossRef CAS PubMed.
- S. A. Akhade, R. M. Nidzyn, G. Rostamikia and M. J. Janik, Using Brønsted-Evans-Polanyi relations to predict electrode potential-dependent activation energies, Catal. Today, 2018, 312, 82–91 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, Mechanistic Study on the Electrocatalytic Reduction of Nitric Oxide on Transition-Metal Electrodes, J. Catal., 2001, 202, 387–394 CrossRef CAS.
- S. Kuwabata, S. Uezumi, K. Tanaka and T. Tanaka, Assimilatory and Dissimilatory Reduction of NO3− and NO2− with an (n-Bu4N)3[Mo2Fe6S8(SPh)9] Modified Glassy-Carbon Electrode in Water, Inorg. Chem., 1986, 25, 3018–3022 CrossRef CAS.
- Z. Wang, D. Richards and N. Singh, Recent discoveries in the reaction mechanism of heterogeneous electrocatalytic nitrate reduction, Catal. Sci. Technol., 2021, 11, 705–725 RSC.
- B. H. Ko, B. Hasa, H. Shin, Y. Zhao and F. Jiao, Electrochemical Reduction of Gaseous Nitrogen Oxides on Transition Metals at Ambient Conditions, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- T. Hu, C. Wang, M. Wang, C. M. Li and C. Guo, Theoretical Insights into Superior Nitrate Reduction to Ammonia Performance of Copper Catalysts, ACS Catal., 2021, 11, 14417–14427 CrossRef CAS.
- J. Bai and B. Zhou, Titanium dioxide nanomaterials for sensor applications, Chem. Rev., 2014, 114, 10131–10176 CrossRef CAS PubMed.
- W. W. Xu, S. L. Shang, B. C. Zhou, Y. Wang, L. J. Chen, C. P. Wang, X. J. Liu and Z. K. Liu, A first-principles study of the diffusion coefficients of alloying elements in dilute alpha-Ti alloys, Phys. Chem. Chem. Phys., 2016, 18, 16870–16881 RSC.
- F. Yuan, G. Li, F. Han, Y. Zhang, A. Muhammad, W. Guo, J. Ren, C. Liu, H. Gu and G. Yuan, A new type face-centered cubic zirconium phase in pure zirconium, J. Mater. Sci. Technol., 2021, 81, 236–239 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, CRC Press, 2013, ch. 4, pp. 32–33 Search PubMed.
- X. Wang, Z. Li, J. Shi and Y. Yu, One-dimensional titanium dioxide nanomaterials: nanowires, nanorods, and nanobelts, Chem. Rev., 2014, 114, 9346–9384 CrossRef CAS PubMed.
- X. Chen and S. S. Mao, Titanium Dioxide Nanomaterials: Synthesis, Properties, Modifications, and Applications, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- T. L. Thompson and J. T. Yates Jr., Surface Science Studies of the Photoactivation of TiO2-New Photochemical Processes, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed.
- W. Wunderlich, T. Oekermann and L. Miao, J. Ceram. Process. Res., 2004, 5, 343 Search PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, Boosting Selective Nitrate Electroreduction to Ammonium by Constructing Oxygen Vacancies in TiO2, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- M. Liu, N. Li, S. Cao, X. Wang, X. Lu, L. Kong, Y. Xu and X. H. Bu, A “Pre-Constrained Metal Twins” Strategy to Prepare Efficient Dual-Metal-Atom Catalysts for Cooperative Oxygen Electrocatalysis, Adv. Mater., 2022, 34, 2107421 CrossRef CAS PubMed.
- A. M. Ruiz, G. Sakai, A. Cornet, K. Shimanoe, J. R. Morante and N. Yamazoe, Cr-doped TiO2 gas sensor for exhaust NO2 monitoring, Sens. Actuators, B, 2003, 93, 509–518 CrossRef CAS.
- J. Guo, P. Brimley, M. J. Liu, E. R. Corson, C. Muñoz, W. A. Smith and W. A. Tarpeh, Mass Transport Modifies the Interfacial Electrolyte to Influence Electrochemical Nitrate Reduction, ACS Sustainable Chem. Eng., 2023, 11, 7882–7893 CrossRef CAS.
- Q. Song, S. Zhang, X. Hou, J. Li, L. Yang, X. Liu and M. Li, Efficient electrocatalytic nitrate reduction via boosting oxygen vacancies of TiO2 nanotube array by highly dispersed trace Cu doping, J. Hazard. Mater., 2022, 438, 129455 CrossRef CAS PubMed.
- X. Zhang, C. Wang, Y. Guo, B. Zhang, Y. Wang and Y. Yu, Cu clusters/TiO2−x with abundant oxygen vacancies for enhanced electrocatalytic nitrate reduction to ammonia, J. Mater. Chem. A, 2022, 10, 6448–6453 RSC.
- M. Sahu and P. Biswas, Single-step processing of copper-doped titania nanomaterials in a flame aerosol reactor, Nanoscale Res. Lett., 2011, 6, 441 CrossRef PubMed.
- W. J. Sun, H. Q. Ji, L. X. Li, H. Y. Zhang, Z. K. Wang, J. H. He and J. M. Lu, Built-in Electric Field Triggered Interfacial Accumulation Effect for Efficient Nitrate Removal at Ultra-Low Concentration and Electroreduction to Ammonia, Angew. Chem., Int. Ed., 2021, 60, 22933–22939 CrossRef CAS PubMed.
- H. Wang, D. Zhao, C. Liu, X. Fan, Z. Li, Y. Luo, D. Zheng, S. Sun, J. Chen, J. Zhang, Y. Liu, S. Gao, F. Gong and X. Sun, FeS2@TiO2 nanobelt array enabled high-efficiency electrocatalytic nitrate reduction to ammonia, J. Mater. Chem. A, 2022, 10, 24462–24467 RSC.
- X. E. Zhao, Z. Li, S. Gao, X. Sun and S. Zhu, CoS2@TiO2 nanoarray: a heterostructured electrocatalyst for high-efficiency nitrate reduction to ammonia, Chem. Commun., 2022, 58, 12995–12998 RSC.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- Z. Deng, C. Ma, X. Fan, Z. Li, Y. Luo, S. Sun, D. Zheng, Q. Liu, J. Du, Q. Lu, B. Zheng and X. Sun, Construction of CoP/TiO2 nanoarray for enhanced electrochemical nitrate reduction to ammonia, Mater. Today Phys., 2022, 28, 100854 CrossRef CAS.
- D. Zhang, S. Yang, X. Fang, H. Li, X. Chen and D. Yan, In situ localization of BiVO4 onto two-dimensional MXene promoting photoelectrochemical nitrogen reduction to ammonia, Chin. Chem. Lett., 2022, 33, 4669–4674 CrossRef CAS.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Advances in the Synthesis of 2D MXenes, Adv. Mater., 2021, 33, 2103148 CrossRef CAS PubMed.
- T. Hu, M. Wang, C. Guo and C. M. Li, Functionalized MXenes for efficient electrocatalytic nitrate reduction to ammonia, J. Mater. Chem. A, 2022, 10, 8923–8931 RSC.
- L.-X. Li, W.-J. Sun, H.-Y. Zhang, J.-L. Wei, S.-X. Wang, J.-H. He, N.-J. Li, Q.-F. Xu, D.-Y. Chen, H. Li and J.-M. Lu, Highly efficient and selective nitrate electroreduction to ammonia catalyzed by molecular copper catalyst@Ti3C2Tx MXene, J. Mater. Chem. A, 2021, 9, 21771–21778 RSC.
- M. Wang, T. Hu, C. Wang, F. Du, H. Yang, W. Sun, C. Guo and C. Li, Screening MXene-based Single-Atom Catalysts for Selective Nitrate-to-Ammonia Electroreduction, Sci. China Mater., 2023 DOI:10.1007/s40843-022-2406-5.
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Single Atom Ru Monolithic Electrode for Efficient Chlorine Evolution and Nitrate Reduction, Angew. Chem., Int. Ed., 2022, 61, e202208215 CAS.
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J. L. Luo and X. Sun, Recent Advances in MOF–Derived Single Atom Catalysts for Electrochemical Applications, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- C. Wan, X. Duan and Y. Huang, Molecular Design of Single–Atom Catalysts for Oxygen Reduction Reaction, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS.
- S. Ye, S. Xie, Y. Lei, X. Yang, J. Hu, L. Zheng, Z. Chen, Y. Fu, X. Ren, Y. Li, X. Ouyang, Q. Zhang, J. Liu and X. Sun, Modulating the electronic spin state by constructing dual-metal atomic pairs for activating the dynamic site of oxygen reduction reaction, Nano Res., 2022, 16, 1869–1877 CrossRef.
- Z. Chen, W. Chen, L. Zheng, T. Huang, J. Hu, Y. Lei, Q. Yuan, X. Ren, Y. Li, L. Zhang, S. Huang, S. Ye, Q. Zhang, X. Ouyang, X. Sun and J. Liu, Rational design of Ru species on N-doped graphene promoting water dissociation for boosting hydrogen evolution reaction, Sci. China: Chem., 2022, 65, 521–531 CrossRef CAS.
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han, M. Jaroniec, Y. Zhu, Y. Zheng and S. Z. Qiao, Short-Range Ordered Iridium Single Atoms Integrated into Cobalt Oxide Spinel Structure for Highly Efficient Electrocatalytic Water Oxidation, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed.
- S. Ye, W. Xiong, P. Liao, L. Zheng, X. Ren, C. He, Q. Zhang and J. Liu, Removing the barrier to water dissociation on single-atom Pt sites decorated with a CoP mesoporous nanosheet array to achieve improved hydrogen evolution, J. Mater. Chem. A, 2020, 8, 11246–11254 RSC.
- Z. Lei, W. Cai, Y. Rao, K. Wang, Y. Jiang, Y. Liu, X. Jin, J. Li, Z. Lv, S. Jiao, W. Zhang, P. Yan, S. Zhang and R. Cao, Coordination modulation of iridium single-atom catalyst maximizing water oxidation activity, Nat. Commun., 2022, 13, 24 CrossRef CAS PubMed.
- N. Sathishkumar, S.-Y. Wu and H.-T. Chen, Mechanistic exploring the catalytic activity of single-atom catalysts anchored in graphitic carbon nitride toward electroreduction of nitrate-to-ammonia, Appl. Surf. Sci., 2022, 598, 153829 CrossRef CAS.
- D. Saravanakumar, J. Song, S. Lee, N. H. Hur and W. Shin, Electrocatalytic Conversion of Carbon Dioxide and Nitrate Ions to Urea by a Titania-Nafion Composite Electrode, ChemSusChem, 2017, 10, 3999–4003 CrossRef CAS PubMed.
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, Oxygen vacancies enhanced cooperative electrocatalytic reduction of carbon dioxide and nitrite ions to urea, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed.
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Direct electrosynthesis of methylamine from carbon dioxide and nitrate, Nat. Sustainability, 2021, 4, 725–730 CrossRef.
- C. Guo, W. Zhou, X. Lan, Y. Wang, T. Li, S. Han, Y. Yu and B. Zhang, Electrochemical Upgrading of Formic Acid to Formamide via Coupling Nitrite Co-Reduction, J. Am. Chem. Soc., 2022, 144, 16006–16011 CrossRef CAS PubMed.
- J. E. Kim, J. H. Jang, K. M. Lee, M. Balamurugan, Y. I. Jo, M. Y. Lee, S. Choi, S. W. Im and K. T. Nam, Electrochemical Synthesis of Glycine from Oxalic Acid and Nitrate, Angew. Chem., Int. Ed., 2021, 60, 21943–21951 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2023 |