
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple organocatalyst component system for asymmetric hetero Diels–Alder reaction of isatins with enones†
Perumalsamy Parasuramana,
Zubeda Beguma,
Madhu Chennapurama,
Chigusa Sekia,
Yuko Okuyamab,
Eunsang Kwonc,
Koji Uwaia,
Michio Tokiwad,
Suguru Tokiwad,
Mitsuhiro Takeshitad and
Hiroto Nakano *a
*a
aDivision of Sustainable and Environmental Engineering, Graduate School of Engineering, Muroran Institute of Technology, 27-1 Mizumoto-cho, Muroran 050-8585, Japan. E-mail: catanaka@mmm.muroran-it.ac.jp
bTohoku Medical and Pharmaceutical University, 4-4-1 Komatsushima, Aoba-Ku, Sendai 981-8558, Japan
cResearch and Analytical Center for Giant Molecules, Graduate School of Sciences, Tohoku University, 6-3 Aoba, Aramaki, Aoba-Ku, Sendai 980-8578, Japan
dTokiwakai Group, 62 Numajiri Tsuduri-Chou Uchigo, Iwaki 973-8053, Japan
First published on 5th May 2020
Abstract
A simple two catalyst component system consisting of primary β-amino alcohols as a catalyst and amino acids as a co-catalyst put together works as an efficient organocatalyst system in the hetero Diels–Alder reaction of isatins with enones to afford the chiral spirooxindole–tetrahydropyranones in good chemical yields and stereoselectivities (up to 86%, up to 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 dr., up to 95% ee).
:15 dr., up to 95% ee).
1. Introduction
Spirooxindoles A are considered to be promising scaffolds in drug discovery.1 The structure of A is contained in many compounds having pharmacological activities such as contraceptive,2 anti-HIV,3 anticancer,4 antituberculosis,5 antimalarial,6 and antiproliferative drugs.9 Therefore, the development of an effective strategy for the preparation of highly optically pure spirooxindole Z is a significantly challenging task in research.1 The hetero Diels–Alder (HDA) reaction is a versatile tool for effectively forming heterocyclic compounds.7 Especially, the catalytic asymmetric version of this reaction is the most efficient and convenient method for constructing a chiral heterocyclic skeleton, which acts as a precursor for many biologically active compounds and drugs.8 In this class of HDA reactions, the reaction of isatins X with enones Y is one of the superior organic transformations for providing unique chiral spirooxindole-tetrahydropyranones Z containing quaternary chiral carbon center on the structure (Scheme 1).9 Most recently, Tanaka and co-workers have reported an efficient organocatalyzed asymmetric HDA reaction of X with Y using three catalysts component system being composed with chiral amine as a catalyst, amino acid and thiourea as co-catalysts for affording Spirooxindole Z with satisfactory chemical yield and stereoselectivity (Scheme 1).9 However, the favourable geometric combination of three catalysts system of complex chiral cinchona alkaloid A as a catalyst and both the prepared complex chiral amino acids B and thioureas C as co-catalysts require time and effort for controlling the enantioselective reaction course for obtaining satisfactory chemical yield and stereoselectivity. Therefore, the development of a more convenient and easier catalytic component system for this versatile reaction is deeply required significantly.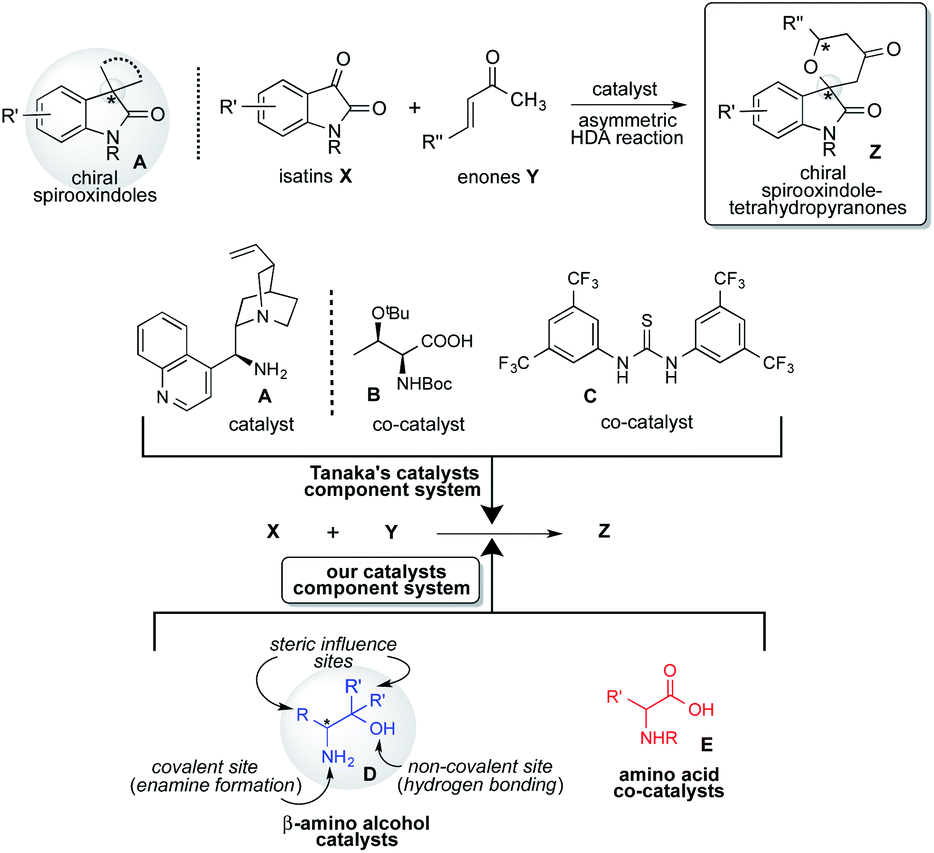 | ||
| Scheme 1 Asymmetric HDA reaction of isatins with enones using catalysts component system. | ||
Based on these backgrounds, we have designed a simple two catalysts component system for this reaction (Scheme 1). About the catalysts system, we focused on a concept of the combination of simple β-amino alcohol D as an organocatalyst for the generation of a diene species and common simple amino acid E as a co-catalyst for the activation of isatin substrate acting as a dienophile comparatively to the complex catalyst system of Tanaka and co-workers having one catalyst and two co-catalysts. Recently, we have reported that simple β-amino alcohols D and their derivatives work as an efficient organocatalyst in various asymmetric reactions.10 As an advantage of catalyst D, it can be easily prepared from commercially available amino acids in a single step and also contains the primary amino group as covalent site, hydroxyl group as a non-covalent site and steric influence site in the single molecule (Scheme 2). Furthermore, simple amino acids as co-catalyst are commercially available. Therefore, combined these properties of amino alcohols as a catalyst and amino acids as a co-catalyst may enable the formation of a simple catalytic component system. This organocatalysed asymmetric HDA reaction might proceed via transition state II (comparing to Tanaka's proposed reaction course I)9 in which the diene species D′ is formed by the reaction of primary amino group on catalyst D with enones Y, and then isatin dienophile X is activated by amino acids co-catalyst E by the two points of hydrogen bonding interactions (Scheme 2). In this transition state II, diene species D′ might attack stereoselectively from less sterically hindered site of the incoming generated dienes to afford the chiral spirooxindoles Z.
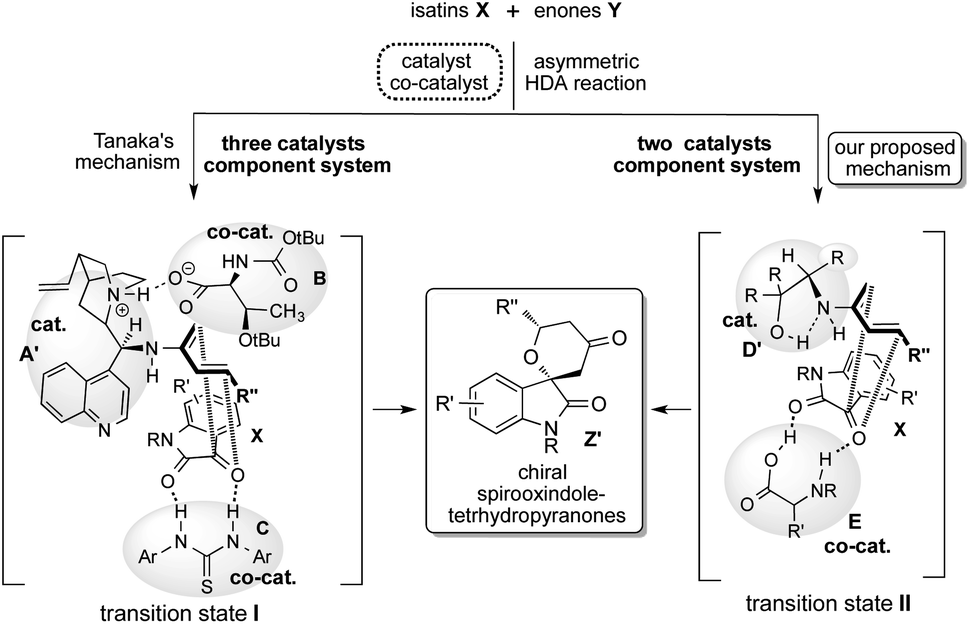 | ||
| Scheme 2 Concept of our two catalysts component system. | ||
Herein, we describe a simple two catalysts component system, primary β-amino alcohols D having only one chiral carbon center on the molecule as a catalyst and simple non-chiral N-protected amino acids E as a co-catalyst, together acts as an efficient component organocatalysts system in the HDA reaction of X with Y to afford the chiral Z in good chemical yields (up to 86%) and with excellent stereoselectivities (up to 85:15 dr., 95% ee).
2. Results and discussion
2.1. Preparations of catalysts 2a–e and 4a–e
β-Amino alcohol organocatalysts 2a–e and 4a–e were easily prepared by the reductions of the corresponding amino acids 1a–e and Grignard reactions of the corresponding amino esters 3a–e, respectively (Table 1).10a Furthermore, N-Cbz- and N-Boc-amino acids 5b–g as co-catalyst were also easily derived from the corresponding commercially available non-protected amino acids.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Enone 7a, (eq.) | Cat. 2a–e, 4a–e (mol%) | Co-cat. 5a–k (mol%) | Temp. (°C) | Yielda (%) | drb | Eec (%) |
| a Isolated yield.b Diastereoselectivity (dr) was determined by 1HNMR of the crude reaction mixture (major diastereomer: 8a).c The ee value were determined by HPLC (Daicel chiralpak IB column). | |||||||
| 1 | 4 | 2a (20) | — | rt | 15 | 8515 |
92 |
| 2 | 4 | 4a (20) | — | rt | trace | — | — |
| 3 | 4 | 1a (20) | — | rt | — | — | — |
| 4 | 4 | 2a (20) | a (40) | rt | 14 | 7525 |
95 |
| 5 | 4 | 2a (20) | b (40) | rt | 80 | 7921 |
91 |
| 6 | 4 | 2a (20) | c (40) | rt | 86 | 8020 |
92 |
| 7 | 4 | 2a (20) | d (40) | rt | 61 | 8218 |
88 |
| 8 | 4 | 2a (20) | e (40) | rt | 87 | 8119 |
87 |
| 9 | 4 | 2a (20) | f (40) | rt | 90 | 8218 |
88 |
| 10 | 4 | 2a (20) | g (40) | rt | 97 | 7525 |
84 |
| 11 | 4 | 2a (20) | h (40) | rt | 68 | 7525 |
86 |
| 12 | 4 | 2a (20) | i (40) | rt | 68 | 8416 |
87 |
| 13 | 4 | 2a (20) | j (40) | rt | tra | — | — |
| 14 | 4 | 2a (20) | k (40) | rt | 19 | 7327 |
75 |
| 15 | 4 | 2b (20) | c (40) | rt | 16 | 7525 |
86 |
| 16 | 4 | 2c (20) | c (40) | rt | 66 | 5545 |
72 |
| 17 | 4 | 2d (20) | c (40) | rt | 78 | 6436 |
81 |
| 18 | 4 | 2e (20) | c (40) | rt | 61 | 5050 |
88 |
| 19 | 4 | 4b (20) | c (40) | rt | 14 | 7525 |
24 |
| 20 | 4 | 4c (20) | c (40) | rt | 18 | 7426 |
41 |
| 21 | 4 | 4d (20) | c (40) | rt | 28 | 8317 |
14 |
| 22 | 4 | 4e (20) | c (40) | rt | 24 | 7822 |
6 |
| 23 | 2 | 2a (20) | c (40) | rt | 47 | 7723 |
90 |
| 24 | 1 | 2a (20) | c (40) | rt | 17 | 7327 |
89 |
| 25 | 4 | 2a (20) | c (40) | 0 | 56 | 8119 |
93 |
| 26 | 4 | 2a (20) | c (20) | rt | 54 | 7822 |
89 |
| 27 | 4 | 2a (20) | c (10) | rt | 52 | 7921 |
87 |
| 28 | 4 | 2a (10) | c (10) | rt | 54 | 77822 |
89 |
| 29 | 4 | 2a (10) | c (20) | rt | 60 | 88119 |
89 |
| 30 | 4 | 2a (10) | c (5) | rt | 52 | 7921 |
87 |
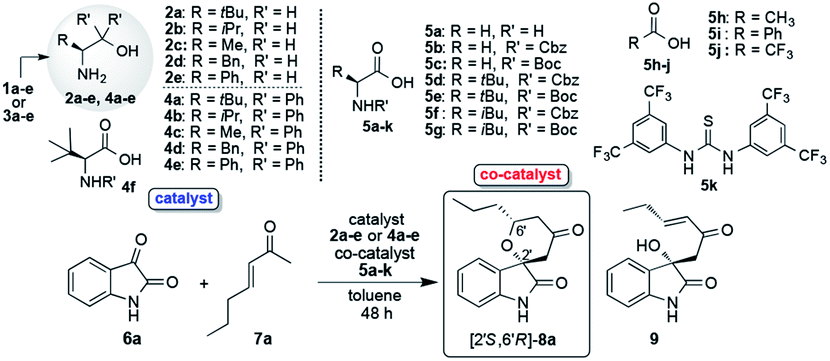
2.2. Screening of catalysts 2a–e and 4a–e
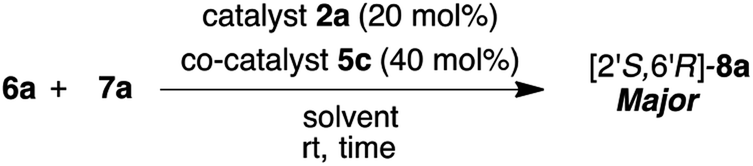
Firstly, we examined the HDA reaction of isatin 6a as a dienophile with heptene-2-one 7a as a diene source, using only amino alcohol organocatalysts 2a with primary hydroxyl methyl or 4a with bulkier hydroxyl diphenylmethyl groups (entries 1 and 2, Table 1). The reaction was carried out with catalysts 2a or 4a in toluene at room temperature for 48 h for comparison with the catalytic efficiency of three catalysts component system by Tanaka and co-workers.9 Simple amino alcohol 2a showed good catalytic activity in this reaction and the corresponding HDA adduct [2′S,6′R]-8a was obtained in excellent enantioselectivity (92% ee) and with good diastereoselectivity (85:15), although the chemical yield was extremely low (15%) (entry 1). On the other hand, the use of bulkier amino alcohol catalyst 4a did not show catalytic activity in this reaction condition (entry 2). These results deeply suggested the necessity of co-catalyst for the activation of isatin dienophile, and also the structure of amino alcohol catalyst may be important for showing a good catalytic activity.
Just in case, the catalytic activity of amino acid 1a (L-tert-leucine) with the primary amino group for generating diene species was also examined under the same reaction condition (entry 3). However, its catalytic activity was not confirmed at all, for a reason that neutral amino acids exist in betaine form which might not work for the generation of the diene species. The most curious thing is that enantioselectivity was controlled almost completely (92% ee) to afford the HDA adduct 8a using simple small β-amino alcohol molecules independently. Thus, amino alcohol alone worked as a catalyst for almost completely shielding one side of the enantiotopic face when diene attack to dienophile. These results indicated the necessity of our two catalysts component system comprising of amino alcohol catalyst for generating diene species and for controlling stereoselective reaction course and amino acid co-catalyst for activating isatin dienophile. Based on the results in entries 1 and 3, we next examined this reaction using the combinations of catalyst 2a (20 mol%) with amino acids 5a–g or common organic acids 5h–j as co-catalysts (40 mol%) at room temperature for 48 h (entries 4–13). First, the reaction using the simplest amino acid 5a having free amino group as a co-catalyst was carried out in the presence of catalyst 2a (entry 4). Contrary to expectation, neutral acid 5a, which hardly worked as co-catalyst for activating of isatin dienophile 7a, showed excellent enantioselectivity (95% ee) with good diastereoselectivity, although chemical yield was quite low (14% ee). Interestingly, the use of 2a and 5a combined together increased the enantioselectivity (95% ee) then the result (92% ee) of the independently use of amino alcohol 2a (entry 1). Amino acid 5a might act as steric factor for controlling the attacking direction of diene to afford 8a with superior enantioselectivity. Next, we tried the combinations of superior catalyst 2a with other N-protected amino acids 5b–g or common organic acids 5h–j as co-catalysts in this reaction condition (entries 5–13). All of co-catalysts 5b–g assisted the progress of the reaction for affording chiral 8a with moderate to good results. Especially, highly satisfactory results for chemical yields and stereoselectivities were obtained when the reactions were carried out in the presence of simple non-chiral amino acids, N-Cbz-protected 5b and N-Boc-protected 5c with good chemical yields and stereoselectivities (5b: 80%, 79:21 dr., 91% ee, 5c: 86%, 80:20, 92% ee) (entries 5 and 6). On the other hand, the uses of common organic acids 5h, i brought about the decrease of chemical yield, even though good stereoselectivities were obtained (entries 11 and 12). Furthermore, strongest trifluoro acetic acid (TFA) 5j did not work as a co-catalyst in this reaction condition (entry 13). Moreover, thioureas 5k that was used as co-catalyst in Tanaka's three catalysts component system9 was also applied with amino alcohol organocatalyst 2a in this reaction. However, this component system of 2a and 5k did not work effectively in this reaction (19%, 73:27 dr., 75% ee) (entry 14). In addition, three catalysts component system of catalyst 2a and co-catalysts of both amino acid 5c and thiourea 5k also did not show better catalytic activity (85%, 75:25 dr., 82% ee) than two catalysts component system of 2a and 5c (86%, 80:20 dr., 92% ee). We next examined the reaction of 6a with 7a in the presence of β-amino alcohols 2b–e (20 mol%) as catalysts along with superior simple non-chiral N-Boc-amino acid 5c, as a co-catalyst (40 mol%) in this reaction condition (entries 15–18). Although, all catalysts combination systems, of catalysts 2b–e and co-catalyst 5c showed good catalytic activities to afford the HDA adduct 8a with moderate to good chemical yields, diastereoselectivities and enantioselectivities, but showed inferior results compared to catalyst 2a and co-catalyst 5c (entry 6). Moreover, the utility of combination of the catalysts bulkier amino alcohol catalysts 4a–e and superior simplest non-chiral N-Boc-amino acid co-catalyst 5c were also examined in this reaction condition (entries 19–22). However, better catalytic activities were not confirmed at all than that of the combination of simple catalysts 2a–e with aprimary hydroxyl group and 5c (entry 6). From these results, it was revealed that the best catalyst combination was β-amino alcohols 2a with primary hydroxyl group as a catalyst and non-chiral N-Boc-amino acid as a co-catalyst 5c. Next, the ratio of substrate amounts 6a and 7a (6a:7a = 1:2 and 6a:7a = 1:1) were examined in the presence of optimised 2a and co-catalyst 5c under same reaction condition (entries 23 and 24). However, these results displayed considerable decrease in chemical yields and the reaction temperature performed at 0 °C also showed a large decrease in chemical yield up to 56% (entry 25). Next, we examined the molar ratio of catalyst 2a and co-catalyst 5c in this reaction of 6a with 7a (4 equiv.) at room temperature (entries 26–30). Satisfactory enantioselectivities and diastereoselectivities were confirmed under all of the molar ratios of 2a and 5c. However, chemical yields comparatively decreased when the reaction was carried out under the molar ratio of 20 mol% of catalyst 2a and 40 mol% of co-catalyst 5c (entry 6).
We also examined the effects of various solvents and the reaction times to this reaction with an optimized catalyst combination of 2a (20 mol%) and 5c (40 mol%) at room temperature (Table 2). As a result, aromatic solvents performed better giving satisfactory chemical yields and stereoselectivities (entries 1–3). Particularly, toluene was found to be effective in this reaction (entry 1). Furthermore, no significant improvement in chemical yields and stereoselectivities was observed when the reaction times were shortened for 24 h and prolonged for 72 h and 96 h, respectively (entries 14–16). From these results, it was revealed that the catalyst combination of simple catalyst 2a (20 mol%) and simple non-chiral N-Boc-glycine 5c (40 mol%), toluene as solvent, room temperature and 48 h reaction time was best reaction condition for this reaction. This reaction using three catalysts component system by Tanaka and co-workers mainly afforded HDA adduct 8a which was obtained by concerted HDA cycloaddition, while this reaction also slightly afforded aldol product 9 which is obtained by aldol reaction as a by-product. Similarly, our catalysts component system also slightly afforded similar aldol product 9 in low chemical yield (12%) and stereoselectivities (72:28 dr, 16% ee) like Tanaka and co-workers.9
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time (h) | Yielda (%) | drb | Eec (%) |
| a Isolated yield.b Diastereoselectivity (dr) was determined by 1HNMR of the crude reaction mixture (major diastereomer: 8a).c The ee value were determined by HPLC (Daicel chiralpak IB column). | |||||
| 1 | Toluene | 48 | 86 | 8020 |
92 |
| 2 | Benzene | 48 | 60 | 7822 |
90 |
| 3 | Xylene | 48 | 73 | 7723 |
88 |
| 4 | Cyclohexane | 48 | 66 | 7426 |
89 |
| 5 | Hexane | 48 | trace | — | — |
| 6 | Et2O | 48 | 55 | 7822 |
90 |
| 7 | iPr2O | 48 | 68 | 7723 |
89 |
| 8 | THF | 48 | 40 | 7921 |
82 |
| 9 | CH2Cl2 | 48 | 74 | 7921 |
90 |
| 10 | CHCl3 | 48 | 34 | 8416 |
92 |
| 11 | C2H4Cl2 | 48 | 75 | 7723 |
88 |
| 12 | CH3CN | 48 | 70 | 7525 |
88 |
| 13 | MeOH | 48 | 38 | 6832 |
83 |
| 14 | Toluene | 24 | 73 | 7921 |
90 |
| 15 | Toluene | 72 | 86 | 7822 |
86 |
| 16 | Toluene | 96 | 78 | 7822 |
86 |
| 17 | Neat | 24 | 87 | 7129 |
86 |
| 18 | Neat | 48 | 75 | 6832 |
82 |
2.3. Substrate scope
After optimizing the reaction conditions, we examined the generality of the developed superior two catalysts component system of 2a and 5c in the reactions of different isatins 6a–f with enones 7a–e (Scheme 3). This system also showed better catalytic activity in the reactions and afforded the corresponding chiral spirooxindole-tetrahydropyranones 8b–j in good to excellent stereoselectivities with moderate to good chemical yields, except the result from the reaction of 6a with 7e to did not afford the adduct 8j. From the results, it is strongly indicated that our simple two catalysts component system works effectively in this reaction using variety of substrates.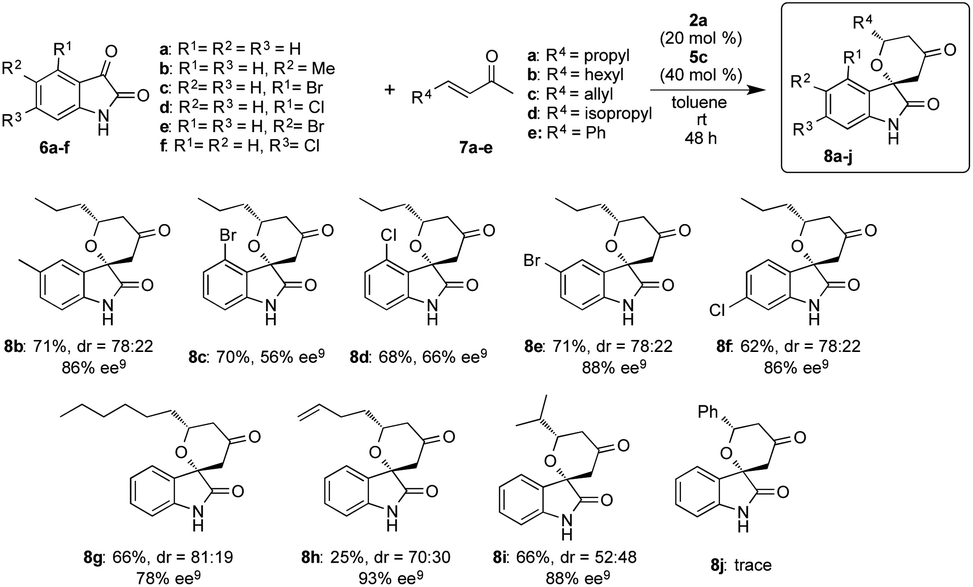 | ||
| Scheme 3 Substrate scope for asymmetric HDA reaction. | ||
We also examined this reaction using a large amount of substrate (6a: 1 g, 7a: 3.05 g) to demonstrate the practically utility of the two component system in best reaction condition. As a result, the HDA adduct 8a was successfully obtained with 87% chemical yield with good stereoselectivites (dr = 80:20, 85% ee), although a slight decrease of ee was observed. From this result, it is expected that this HDA reaction using our two catalyst components system may be useful for practical aspect.
2.4. Reaction mechanism
Based on the observed highly enantiopurity of the obtained HDA adduct [2S,6R]-8a (rt: 92% ee, 0 °C: 93% ee, entries 6 and 25, Table 1), the model of the enantioselective reaction course was proposed as shown in Scheme 4. First, the reaction of β-amino alcohol catalyst 2a with enone 7a forms the diene intermediate I-1 that has less steric interaction of between amino alcohol that is fixed by intramolecular hydrogen bonding and substituted diene parts on generated diene I-1 than that of intermediate I-2. Furthermore, isatin 6a is activated by the two points of hydrogen bonding interactions with N-Boc amino acid co-catalyst 5c. Then, the reaction might proceed through TS-1 to afford 8a that has a less steric interaction between I-1 and dienophile 6a than those of TS-2-4 to afford 8a′–8a′′ that have more steric interaction between I-1 and 6a. Thus, diene I-1 might attack stereoselectively from less sterically hindered site of the incoming activated isatin dienophile 6a to afford [2S,6R]-8a with excellent optically purity (93% ee). On the other hand, it is also expected that the formation of adduct 8a via aldol reaction followed by oxa-Michael addition may be minor pathway based on the chemical yield and enantioselectivity of the obtained aldol product 9 and 8a was quite low (9: 12%, 72:28 dr., 16% ee, 8a: 8%, 80:20 dr., 86% ee).
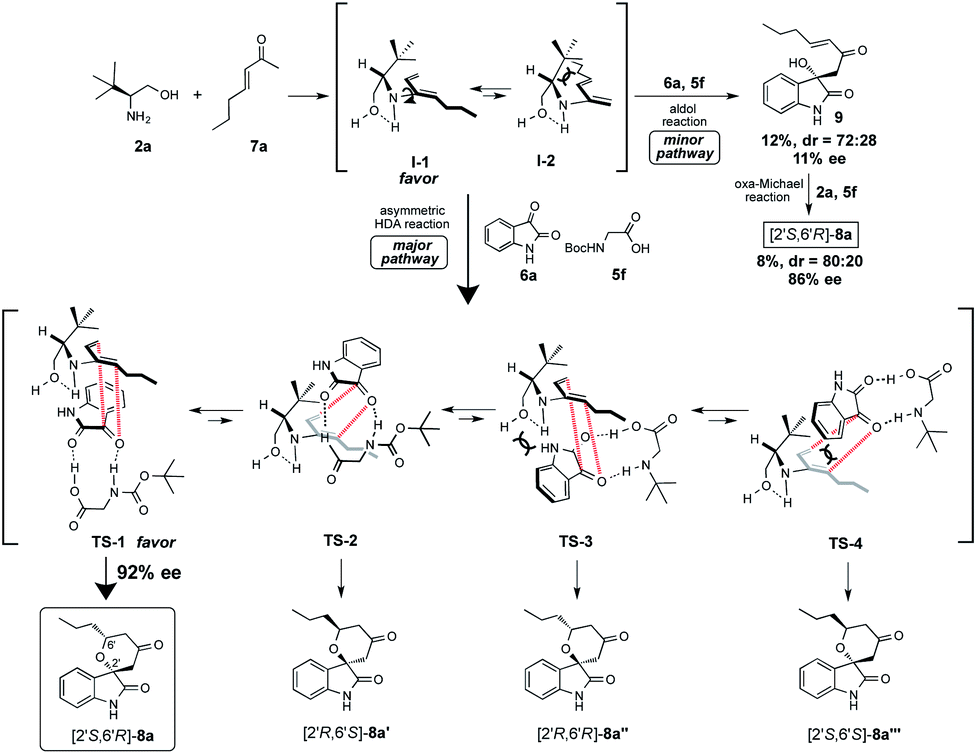 | ||
| Scheme 4 Plausible reaction course for asymmetric HDA reaction. | ||
3. Conclusion
We have developed a simple two catalysts component system consisting of primary β-amino alcohol 2a as a catalyst and N-protected amino acid 5c as a co-catalyst for the asymmetric HDA reaction of isatins with enones for the first time. This dual component system showed efficient catalytic activity to afford the chiral spirooxindole-tetrahydropyranones 8a–j that are efficient synthetic intermediates for many biologically active compounds and drug discovery, in good chemical yields (up to 86%) and with enough stereoselectivities (up to 85:15 dr, 95% ee). In addition, the independent use of simple β-amino alcohol catalyst 2a also showed good catalytic activity for affording 8a with an excellent enantioselectivity (92% ee), although chemical yield was low. The modification of the combination of amino alcohols and detailed mechanistic study of this reaction using our catalysts system are in progress.
4. Experimental
4.1. General information
All reagents and dry solvents were purchased from commercial vendors and used directly without further purification. All reactions were placed in dried sample vials inserted with magnetic beads. Thin-layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates and the analytes were identified under UV light. Flash column chromatography was performed using silica gel pore size 60N (40–100 μm). Melting points were recorded with a micro-melting point apparatus. IR spectra were recorded with a JASCO-4100 Fourier transform infrared spectrophotometer. 1H and 13C NMR spectroscopic data were recorded using a JEOL JNM-ECA500 instrument with tetramethyl silane as the internal standard. HPLC data were collected using the TOSOH instrument equipped with (UV-8020, DP-8020, and SD-8022) detectors using CHIRALPAK IB column. Optical rotations were recorded using a JASCO DIP-360 digital polarimeter. High-resolution mass spectrometry (HRMS) data were collected by electron impact (EI) modes using Hitachi RMG-GMG and JEOL JNX-DX303 sector instruments.4.2. General procedure for the hetero Diels–Alder (HDA) reaction of isatins (6a–f) with enones (7a–e)
To a solution of the corresponding isatins 6a–f (0.2 mmol, 1 eq.) and enones 7a–e (0.8 mmol, 4 eq.) in anhydrous toluene (0.3 mL) were added catalysts 2a–e or 4a–e (0.04 mmol, 20 mol%) and co-catalysts 5a–k (0.08 mmol, 40 mol%) at room temperature and the mixture were stirred at that temperature for 48 h. The mixture was purified by flash column chromatography (SiO2: hexane/ethyl acetate, 7:3) to afford the corresponding major HDA adducts 8a–j.9 The diastereoselectivity (dr) of the obtained HDA adducts were determined by the crude reaction mixture by 1H-NMR.9 The enantiomeric excess (ee) of 8a–j were determined by HPLC (CHIRALPAK-IB, hexane/i-PrOH = 70:30, 90:10 and 95:5, 1.0 mL and 0.6 mL min−1, λ = 245 nm).9
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. Tan, N. R. Candeias and C. F. Barbas, Nat. Chem., 2011, 3, 473 CrossRef CAS PubMed; (b) B. D. Horning and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 6442 CrossRef CAS PubMed; (c) T.-P. Gao, J.-B. Lin, X.-Q. Hu and P.-F. Xu, Chem. Commun., 2014, 50, 8934 RSC; (d) H. Huang, M. Bihani and C.-G. Zhao, Org. Biomol. Chem., 2016, 14, 1755 RSC; (e) D. G. Hall, T. Rybak and T. Verdelet, Acc. Chem. Res., 2016, 49, 2489 CrossRef CAS PubMed; (f) M. Uroos, P. Pitt, L. M. Harwood, W. Lewis, A. J. Blake and C. J. Hayes, Org. Biomol. Chem., 2017, 15, 8523 RSC; (g) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926 RSC; (h) G.-L. Mei and F. Shi, Chem. Commun., 2018, 54, 6607 RSC.
- A. Fensome, W. R. Adams, A. L. Adams, T. J. Berrodin, J. Cohen, C. Huselton, A. Illenberger, J. C. Kern, V. A. Hudak, M. A. Marella, E. G. Melenski, C. C. McComas, C. A. Mugford, O. D. Slayden, M. Yudt, Z. Zhang, P. Zhang, Y. Zhu, R. C. Winneker and J. E. Wrobel, J. Med. Chem., 2008, 51, 1861 CrossRef CAS PubMed.
- G. Kumari, Nutan, M. Modi, S. K. Gupta and R. K. Singh, Eur. J. Med. Chem., 2011, 46, 1181 CrossRef CAS PubMed.
- (a) J.-J. Liu and Z. Zhang, US Pat., US2008114013A1, 2008; (b) H.-L. Cui and F. Tanaka, US Pat., US9309261B2, 2014; (c) K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130 CrossRef CAS PubMed; (d) K. Ding, Y. Lu, Z. Nikolovska- Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432 CrossRef CAS PubMed; (e) Q. Ding, J.-J. Liu and Z. Zhang, WO2007104714A1, 2007.
- V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hubel, D. Rauh and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed.
- (a) M. Rottmann, C. McNamara, B. K. Yeung, M. C. Lee, B. Zou, B. Russell, P. Seitz, D. M. Plouffe, N. V. Dharia, J. Tan, S. B. Cohen, K. R. Spencer, G. E. Gonzalez-Paez, S. B. Lakshminarayana, A. Goh, R. Suwanarusk, T. Jegla, E. K. Schmitt, H. P. Beck, R. Brun, F. Nosten, L. Renia, V. Dartois, T. H. Keller, D. A. Fidock, E. A. Winzeler and T. T. Diagana, Science, 2010, 329, 1175 CrossRef CAS PubMed; (b) B. K. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, S. Keller-Maerki, C. Fischli, A. Goh, E. K. Schmitt, P. Krastel, E. Francotte, K. Kuhen, D. Plouffe, K. Henson, T. Wagner, E. A. Winzeler, F. Petersen, R. Brun, V. Dartois, T. T. Diagana and T. H. Keller, J. Med. Chem., 2010, 53, 5155 CrossRef CAS PubMed.
- (a) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS PubMed; (b) L.-L. Wang, L. Peng, J.-F. Bai, Q.-C. Huang, X.-Y. Xu and K.-X. Wang, Chem. Commun., 2010, 46, 8064 RSC; (c) Y.-B. Lan, H. Zhao, Z.-M. Liu, J.-C. Tao and X.-W. Wang, Org. Lett., 2011, 13, 4866 CrossRef CAS PubMed; (d) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104 CrossRef CAS PubMed.
- (a) V. Gouverneur and M. Reiter, Chem.–Eur. J., 2005, 11, 5806 CrossRef CAS PubMed; (b) H. Pellissier, Tetrahedron, 2009, 65, 2839–2877 CrossRef CAS; (c) V. E-Lux, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2014, 53, 2 CrossRef; (d) M. M. Heravi, T. Ahmadi, M. Ghavidel, B. Heidari and H. Hamidi, RSC Adv., 2015, 5, 101999 RSC; (e) S. Jayakumar, K. Louven, C. Strohmann and K. Kumar, Angew. Chem., Int. Ed., 2017, 56, 15945 CrossRef CAS PubMed; (f) D. Zhang and F. Tanaka, RSC Adv., 2016, 6, 61454 RSC.
- (a) H.-L. Cui and F. Tanaka, Chem. - Eur. J., 2013, 19, 6213 CrossRef CAS PubMed; (b) H.-L. Cui, P. V. Chouthaiwale, F. Yin and F. Tanaka, Asian J. Org. Chem., 2016, 5, 153 CrossRef CAS; (c) H.-L. Cui, P. V. Chouthaiwale, F. Yin and F. Tanaka, Org. Biomol. Chem., 2016, 14, 1777 RSC.
- (a) Y. Kohari, Y. Okuyama, E. Kwon, T. Furuyama, N. Kobayashi, T. Otuki, J. Kumagai, C. Seki, K. Uwai, G. Dai, T. Iwasa and H. Nakano, J. Org. Chem., 2014, 79, 9500 CrossRef CAS PubMed; (b) T. Takahashi, U. V. Subba Reddy, Y. Kohari, C. Seki, T. Furuyama, N. Kobayashi, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Tetrahedron Lett., 2016, 57, 5771 CrossRef CAS; (c) U. V. Subba Reddy, M. Chennapuram, C. Seki, E. Kwon, Y. Okuyama and H. Nakano, Eur. J. Org. Chem., 2016, 24, 4124 CrossRef; (d) H. Nakano, I. A. Owolabi, M. Chennapuram, Y. Okuyama, E. Kwon, C. Seki, M. Tokiwa and M. Takeshita, Heterocycles, 2018, 97, 647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and HPLC data. See DOI: 10.1039/d0ra03006f |
| This journal is © The Royal Society of Chemistry 2020 |