
Recent advances in the green processing of organic photovoltaic devices from nanoparticle dispersions
Mohammad
Rammal
a,
Patrick
Lévêque
 b,
Guy
Schlatter
a,
Nicolas
Leclerc
*a and
Anne
Hébraud
*a
b,
Guy
Schlatter
a,
Nicolas
Leclerc
*a and
Anne
Hébraud
*a
aInstitut de Chimie et Procédés pour l’Energie, l’Environnement et la Santé (ICPEES), UMR 7515 ECPM Université de Strasbourg – CNRS, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: nicolas.leclerc@unistra.fr; anne.hebraud@unistra.fr
bLaboratoire des Sciences de l’Ingénieur, de l’Informatique et de l’Imagerie (ICube), UMR 7357 Université de Strasbourg – CNRS, 23 rue du Loess, 67037 Strasbourg, France
First published on 15th July 2020
Abstract
Two decades of research have allowed organic solar cells to appear today as an alternative to hybrid and inorganic photovoltaic devices. However, several issues need to be addressed in order to facilitate their production on an industrial scale. Active layer processing is one of them. Indeed, high power conversion efficiency organic semiconductors are poorly soluble and require not only the use of toxic solvents but also moderate temperature (above 80 °C) processing for which the amount of generated vapors is critical. Recently, the use of conjugated polymer nanoparticle (NP) dispersions in water or alcohols has emerged as a possible solution to avoid toxic solvents. Recent studies have demonstrated that it is possible to finely tune the active-layer morphology using NPs and achieve high power conversion efficiencies. In this review, we aim at providing an up-to-date overview of this field by focusing on the different steps that make up the development of an organic photovoltaic device, from the preparation of nanoparticles to the characterization of the device. Finally, we provide perspectives for the future development of organic photovoltaic devices using a NP-based active layer.
 Mohammad Rammal | Mohammad Rammal obtained his Master of molecular and macromolecular chemistry degree from the University of Bordeaux (France) in 2017. The same year, he began his PhD under the supervision of Dr Anne Hébraud at the Institute for Chemistry and Processes for Energy, Environment and Health ICPEES in Strasbourg. His research interests focus on the development of new eco-friendly optoelectronic devices based on organic polymer nanoparticles. |
 Patrick Lévêque | Patrick Lévêque received his PhD in Materials Science from Poitiers University (France) in 1998. After completing his post-doctoral research at KTH-FTE in Bengt G. Svensson's team in Stockholm (Sweden) on defect engineering in inorganic semiconductors, he joined the ICube laboratory of the University of Strasbourg (France) as an associate professor in 2004. His research interests have focused on the charge-carrier dynamics in organic semiconductors and on organic photovoltaics since 2006. |
 Guy Schlatter | Guy Schlatter received his PhD in materials science in 1995 from Mines ParisTech, France. After completing his postdoctoral research at Ecole Polytechnique of Montréal, Canada, he was appointed as an assistant professor at the University of Strasbourg, France. Since 2007, he has been a full professor at the European Engineering School of Chemistry, Polymers and Materials, University of Strasbourg. His research interests focus on the micro and nano-fabrication of polymer systems as well as electrospinning with an emphasis on the understanding and the control of material organization during the process of fabrication. |
 Nicolas Leclerc | Nicolas Leclerc received his PhD from the Pierre and Marie Curie University (Paris, France) in 2003. After completing his post-doctoral research at Laval University in Mario Leclerc's team (Canada), he joined the ICPEES institute of the University of Strasbourg (France) as a CNRS researcher in 2005. His research interests focus on the development of new molecular and macromolecular organic semiconductor materials and their applications in optoelectronics, mainly in the fields of photovoltaics and thermoelectricity. |
 Anne Hébraud | Anne Hébraud obtained her PhD from the Pierre and Marie Curie University (Paris, France) in 2004. After a post-doctoral fellowship in the group of Katharina Landfester at the University of Ulm (Germany), she was appointed assistant Professor at the University of Strasbourg in the ICPEES institute. Her research interests focus on the physicochemistry of polymers and their use in the elaboration of colloids and nanofibers for diverse applications. |
1. Introduction
The development of renewable energy, reducing CO2 emissions and natural resource consumption, is a key challenge to limit global warming. In this context, major efforts are being made to increase the performance of photovoltaic panels and improve their life cycle in terms of energy payback time (EPBT) and environmental loads.1 Among the different photovoltaic technologies, organic photovoltaics (OPVs) look appealing. OPV performances, now reaching 18% at the laboratory scale,2 are still below those of mono- and poly-crystalline based silicon photovoltaics. OPVs have nevertheless several advantages as OPV modules are light weight and flexible, their shape and color can easily be tuned and they can be produced at low cost by solution processes requiring lower energy. In recent years, the increase of the Power Conversion Efficiencies (PCE) of OPV devices has been achieved thanks to the evolution of photoactive material design on the one hand and the improvement of the different device sub-layers on the other hand.Regarding photoactive materials, in order to meet industrial criteria, two key concerns remain to be addressed: (i) their synthesis cost, closely related to the number of steps and reagents used, and (ii) their processing in thin-films. If the first point starts to be well documented with in particular the recent introduction of the following new indexes: the synthetic complexity (SC) and the figure of merit (FOM),3,4 the second point still needs to be addressed as high PCE organic semiconductors (OSCs) are less and less soluble and require not only the use of toxic organic solvents but also moderate temperature (above 80 °C) processing where the amount of generated vapors is critical.5,6 Therefore, the development of alternative eco-friendly processing allowing the use of green solvents appears as a key step towards industrialization of OPV technology. Three main solutions are currently being developed consisting of (i) research on alternative and less toxic solvents,7 (ii) the design of water soluble materials8 and (iii) the dispersion of OSC nanoparticles (NPs) in water or alcohols, resulting in an eco-friendly active layer ink.
Two recent reviews have been published discussing these three different strategies,9,10 but none of them described thoroughly all the aspects of the NP dispersion route. Our review focuses on the NP dispersion strategy and aims at describing its different steps, from the elaboration of the NPs to their assembly in the active layer, the OPV device characterization and its optimization. It also highlights the essential parameters that can be tuned at each step to improve the performance of the final device.
Regarding the device sub-layer optimization, the NP approach also allows one to specifically address the control of the active-layer thin film morphology in an innovative way. Indeed, the photoactive layer of efficient solution-processed OPV devices is made of phase-separated domains of electron-acceptor (A) and electron-donor (D) materials known as bulk heterojunctions (BHJs, Fig. 1). Two different levels of organization should be considered. First, the structuration of the materials in each domain at the molecular scale (crystallinity, self-assembly properties, etc.) is a major parameter to obtain high charge-carrier mobilities. Then, at the mesoscale, the morphology of the phase separation is critical as it simultaneously enables both exciton dissociation and free-charge collection. The ideal structure is an interpenetrating network of the two phases with a typical length scale of the order of the exciton diffusion length, i.e. 10–20 nm. The two levels of organisation of the active-layer depend on the materials’ physico-chemical properties (solubility, planarity, self-assembly properties, etc.) and on the film preparation process. The elaboration of separate electron-donor NPs and electron-acceptor NPs or composite NPs containing both materials is an attractive way to address these issues, because it provides the possibility to control the morphology of the molecules and their phase separation at the scale of the nanoparticles. These NPs are then assembled to form the active layer. Accordingly, the NP approach offers an additional control degree over the active-layer morphology.
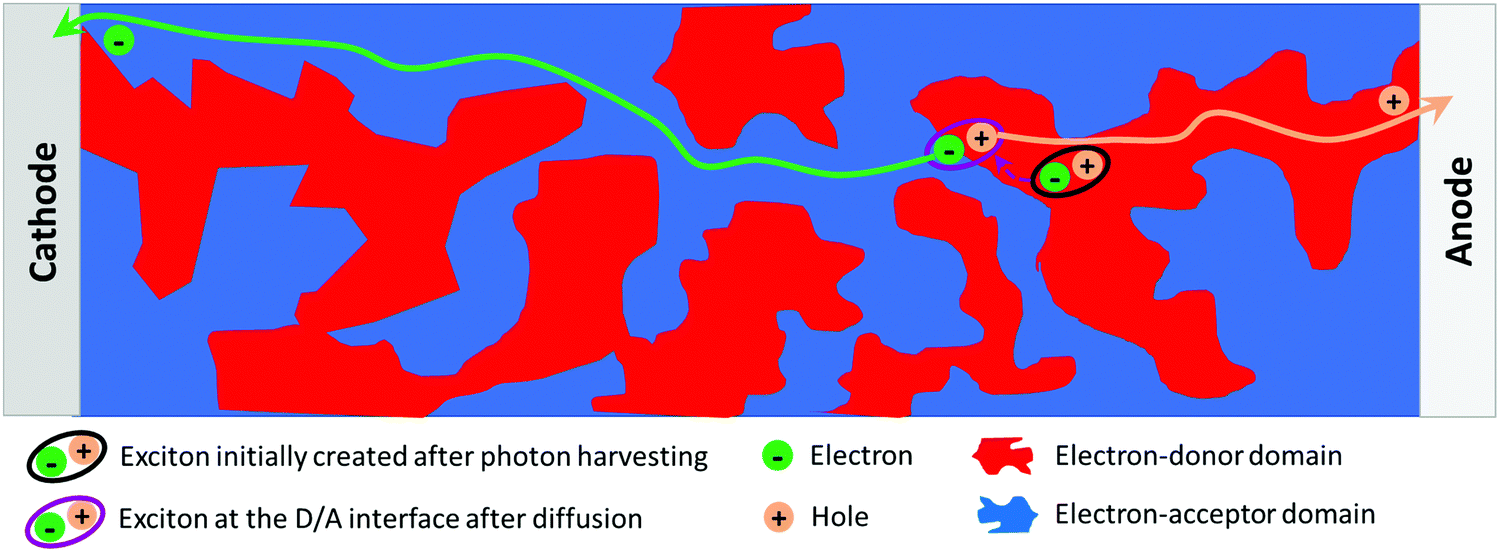 | ||
| Fig. 1 Typical BHJ scheme for a donor:acceptor blend in organic solar cells. | ||
In this review, we aim at providing an up-to-date overview of this field by focusing on the different steps that make up the development of an OPV device, from the preparation of nanoparticles to the characterization of the device (Fig. 2). Thus, in the first part of the review, we focus on the elaboration of separate and composite OSC NPs by the two main post-polymerization processes: miniemulsion and nanoprecipitation. We then discuss the impact of the materials and different processing parameters on the diameter and internal morphology of these NPs. In the second part, we present the different NP deposition processes and their optimization from spin-coating at the lab scale, to roll-to-roll processing allowing the preparation of large-scale devices. The morphology of the active layer obtained from the assembly of the NPs is then described in detail and the influence of thermal annealing on the purity and size of the segregated donor and acceptor domains is discussed. Finally, the last part of the review focuses on the performances of OPV devices obtained by this strategy. The charge-carrier generation and recombination dynamics, which may limit NP OPV devices, as well as the optimization processes, are extensively discussed.
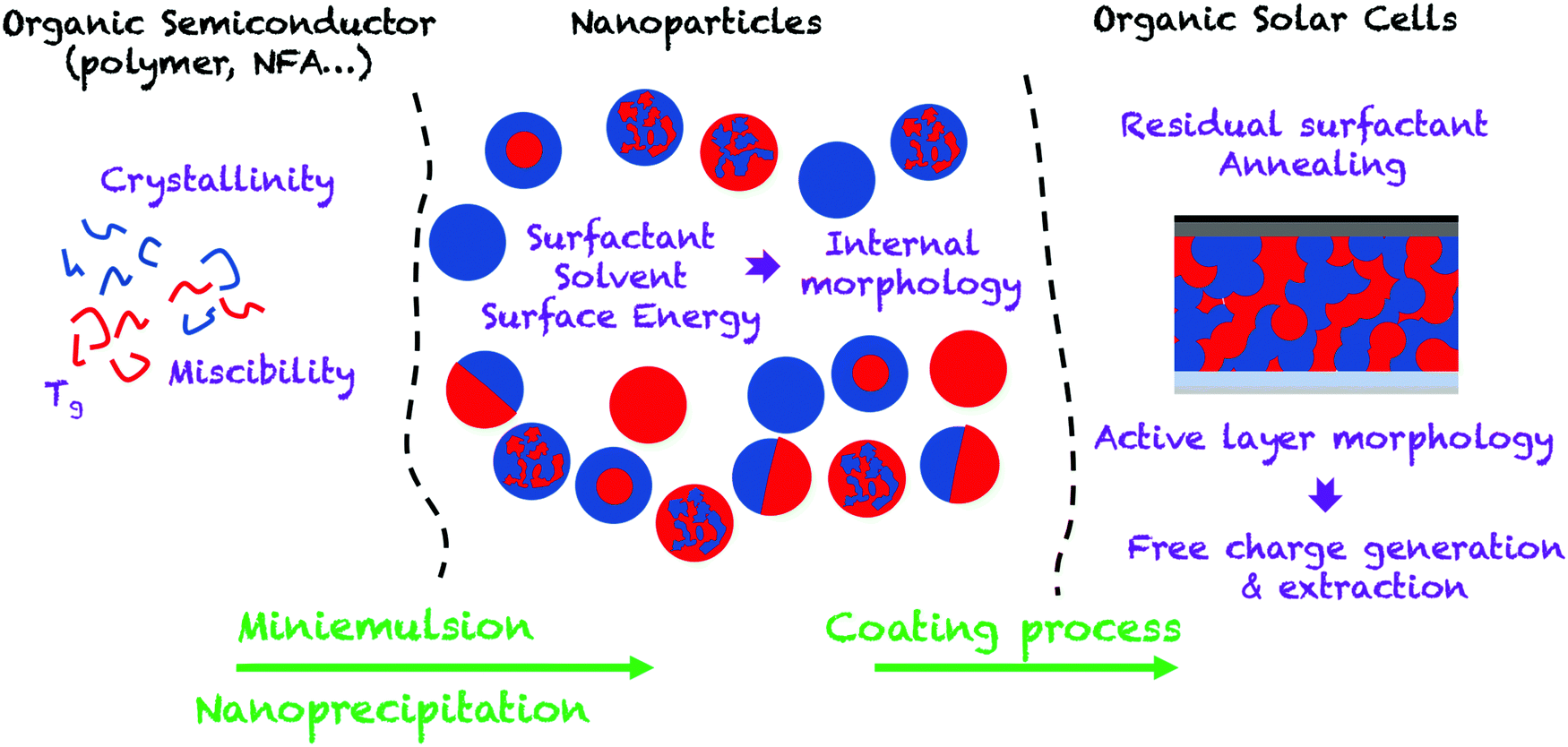 | ||
| Fig. 2 Scheme of the NP dispersion route to OPV cells. | ||
2. Elaboration of nanoparticle dispersions in green solvents
Different ways have been used to synthesize semiconducting polymer NPs by polymerization in heterophase systems or post-polymerization methods.11,12 We focus here on the post-polymerization preparation methods because they are very versatile, allowing the preparation of NPs from most recent organic semiconducting materials. Readers interested in the heterophase polymerization strategy can refer the review of Pecher et al.11Two main post-polymerization processes can be used to prepare semiconducting NPs with diameters in the range of 20 to 200 nm: miniemulsion and nanoprecipitation.
2.1. The miniemulsion technique
Preparation of aqueous dispersions of semiconducting polymer NPs by the miniemulsion process was first demonstrated by Landfester et al.13 In this process, two non-miscible phases are prepared: an organic phase composed of the polymer in a good solvent and an aqueous phase containing a surfactant (Fig. 3). The organic phase is then dispersed in the aqueous phase by ultrasonication, giving a metastable miniemulsion, i.e. a dispersion of very small organic droplets in an aqueous phase. After evaporation of the organic solvent, a polymer dispersion is obtained. The diameter of the obtained NPs, typically between 50 and 250 nm, depends on the surfactant concentration, with an increase of the surfactant concentration resulting in a decrease of the NP size.13 Moreover, as the NPs are obtained after evaporation of the solvent from the miniemulsion droplets, the NP size also increases with the initial polymer concentration in the organic phase.14 If low boiling temperature (Tb) solvents such as chloroform (Tb = 61 °C) are usually preferred for the miniemulsion technique, it was also possible to prepare NPs from high boiling point solvents such as ortho-dichlorobenzene (o-DCB, Tb = 178 °C)15 or o-xylene (Tb = 144 °C),16 thus expanding the range of semi-conducting polymers that can be processed by this technique. In this case, evaporation of the solvent is completed after several hours at 60 or 75 °C and water has to be regularly added into the flask in order to compensate for water loss through evaporation. Using this process, composite NPs containing two different materials in the same particle can also be prepared, by mixing the two materials in the initial organic phase, prior to emulsification.17,18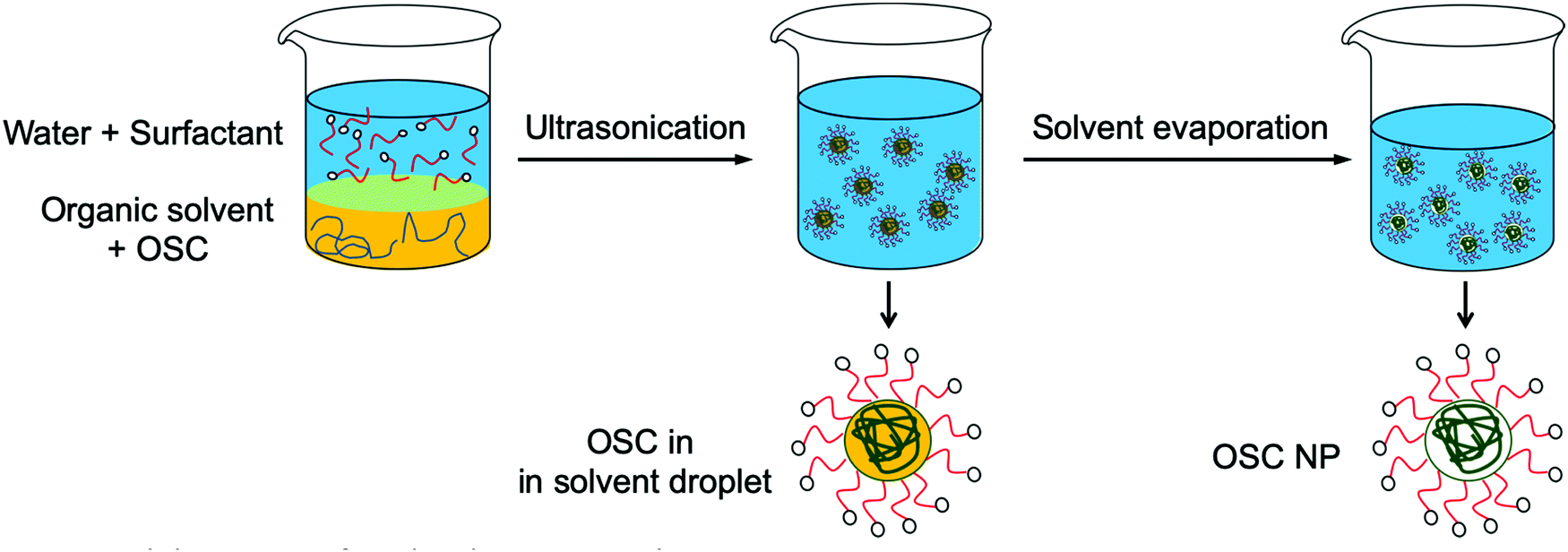 | ||
| Fig. 3 Elaboration of NPs by the miniemulsion process. | ||
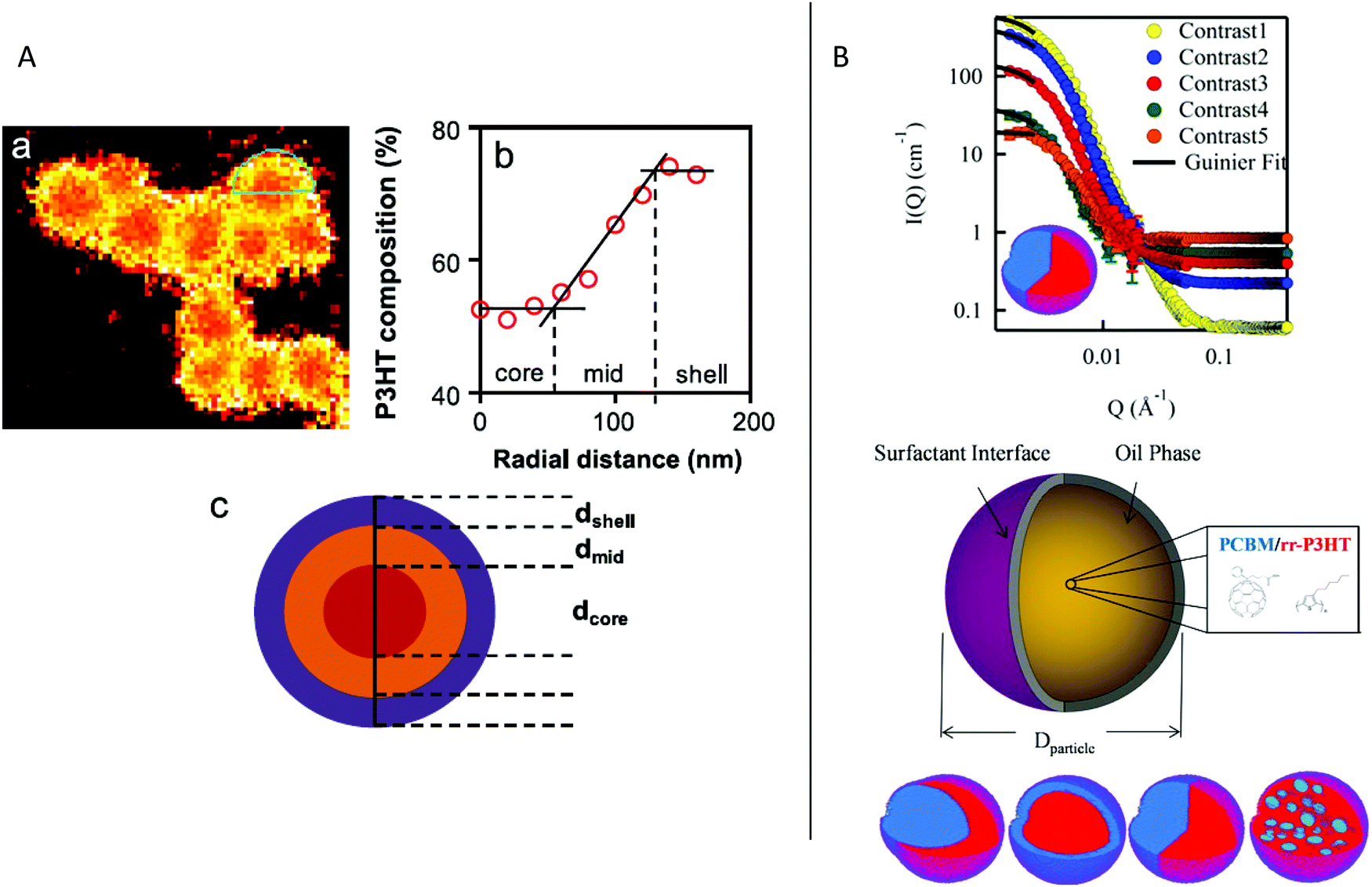 | ||
| Fig. 4 (A) STXM observation of a P3HT:PCBM NP. (a) P3HT composition map, (b) P3HT radial composition calculated from the green area in the STXM image, and (c) scheme of the core–shell NP. Adapted with permission from ref. 19 (Copyright 2013 Elsevier B.V.). (B) Scattering profile obtained by contrast variation SANS for a Janus P3HT:PCBM NP, and scheme of the different possible NP internal morphologies. Adapted with permission from ref. 21 (Copyright 2014 American Chemical Society). | ||
The internal structure of the NPs depends on the surface energy of the materials, but can also vary with the evaporation kinetics or other processing parameters. In general, the materials with higher surface energy are found in the core of the NPs. Thus, when PCBM is used in combination with an electron donor polymer, the NPs obtained by miniemulsion have usually a core–shell morphology with the fullerene derivative in the core and the conjugated polymer in the shell. It is the case of PCDTBT:PC71BM NPs, PC71BM having a higher surface energy (48 mJ m−2) than PCDTBT (37 mJ m−2),14 or of P3HT:PC61BM19 with surface energies of 38.2 mJ m−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and 26.9 mJ m−224 for PC61BM and P3HT, respectively (see the structures in Fig. 5). Moreover, the size and composition of the two segregated phases, usually not pure, depend on different parameters. The molar mass of the polymer has a limited impact on the composition of the two phases after elaboration. However, it changes the fullerene derivative diffusion during annealing as discussed later in Section 3.2. Indeed, for P3HT:PC61BM NPs,19 variation of the molar mass of P3HT between 9 and 72 kg mol−1 did not change significantly the composition of the core and shell, as obtained just after the miniemulsion process. The P3HT-rich shell contained 72 ± 5% P3HT and the PC61BM-rich core contained 73 ± 12% PC61BM. Only for the lowest studied P3HT molar mass of 5 kg mol−1 was the particle fully blended with a composition of 54 ± 6% P3HT and 46 ± 6% PC61BM. The impact of the ratio of the two materials on the morphology of the NPs was demonstrated by Holmes et al.25 Indeed, an increase of the PC61BM content in the P3HT:PC61BM NPs from 1:0.5 to 1:2 slightly increased the radius of the PCBM-rich core, while maintaining its composition at around 70% PC61BM. However, a favorable increase in the amount of PC61BM in the P3HT-rich shell from 18 to 33% before annealing was obtained, reaching 46% after annealing, which is higher than the percolation thresholds in bulk films.26 Moreover, the phase separation and composition of the core and shell depend also logically on the nature of the materials and their miscibility. By STXM, Dam et al.27 have compared the structure and composition of P3HT:PC71BM and PSBTBT:PC71BM NPs prepared by miniemulsion, with the same 1:1 blend ratio and a similar NP radius. PSBTBT is a highly crystalline low band gap polymer (Fig. 5). They showed that while the radius of the core and the composition of the polymer-rich shell were similar, the purity of the PC71BM-rich core was very different with 80% PC71BM in the core of P3HT:PC71BM NPs and only 60% PC71BM in the core of PSBTBT:PC71BM NPs. Finally, in the case of 1:1 P3HT:ICBA NPs, ICBA being a C60-bis-adduct derivative (Fig. 5) highly miscible in P3HT, a core–shell structure was still obtained, due to the difference in the surface energy of the two materials, but with a large core containing 41% ICBA and a thin shell region containing only 23% ICBA. However, after annealing and due to this higher miscibility, the core–shell structure was quickly lost and the NPs became homogeneously blended.28
23 and 26.9 mJ m−224 for PC61BM and P3HT, respectively (see the structures in Fig. 5). Moreover, the size and composition of the two segregated phases, usually not pure, depend on different parameters. The molar mass of the polymer has a limited impact on the composition of the two phases after elaboration. However, it changes the fullerene derivative diffusion during annealing as discussed later in Section 3.2. Indeed, for P3HT:PC61BM NPs,19 variation of the molar mass of P3HT between 9 and 72 kg mol−1 did not change significantly the composition of the core and shell, as obtained just after the miniemulsion process. The P3HT-rich shell contained 72 ± 5% P3HT and the PC61BM-rich core contained 73 ± 12% PC61BM. Only for the lowest studied P3HT molar mass of 5 kg mol−1 was the particle fully blended with a composition of 54 ± 6% P3HT and 46 ± 6% PC61BM. The impact of the ratio of the two materials on the morphology of the NPs was demonstrated by Holmes et al.25 Indeed, an increase of the PC61BM content in the P3HT:PC61BM NPs from 1:0.5 to 1:2 slightly increased the radius of the PCBM-rich core, while maintaining its composition at around 70% PC61BM. However, a favorable increase in the amount of PC61BM in the P3HT-rich shell from 18 to 33% before annealing was obtained, reaching 46% after annealing, which is higher than the percolation thresholds in bulk films.26 Moreover, the phase separation and composition of the core and shell depend also logically on the nature of the materials and their miscibility. By STXM, Dam et al.27 have compared the structure and composition of P3HT:PC71BM and PSBTBT:PC71BM NPs prepared by miniemulsion, with the same 1:1 blend ratio and a similar NP radius. PSBTBT is a highly crystalline low band gap polymer (Fig. 5). They showed that while the radius of the core and the composition of the polymer-rich shell were similar, the purity of the PC71BM-rich core was very different with 80% PC71BM in the core of P3HT:PC71BM NPs and only 60% PC71BM in the core of PSBTBT:PC71BM NPs. Finally, in the case of 1:1 P3HT:ICBA NPs, ICBA being a C60-bis-adduct derivative (Fig. 5) highly miscible in P3HT, a core–shell structure was still obtained, due to the difference in the surface energy of the two materials, but with a large core containing 41% ICBA and a thin shell region containing only 23% ICBA. However, after annealing and due to this higher miscibility, the core–shell structure was quickly lost and the NPs became homogeneously blended.28
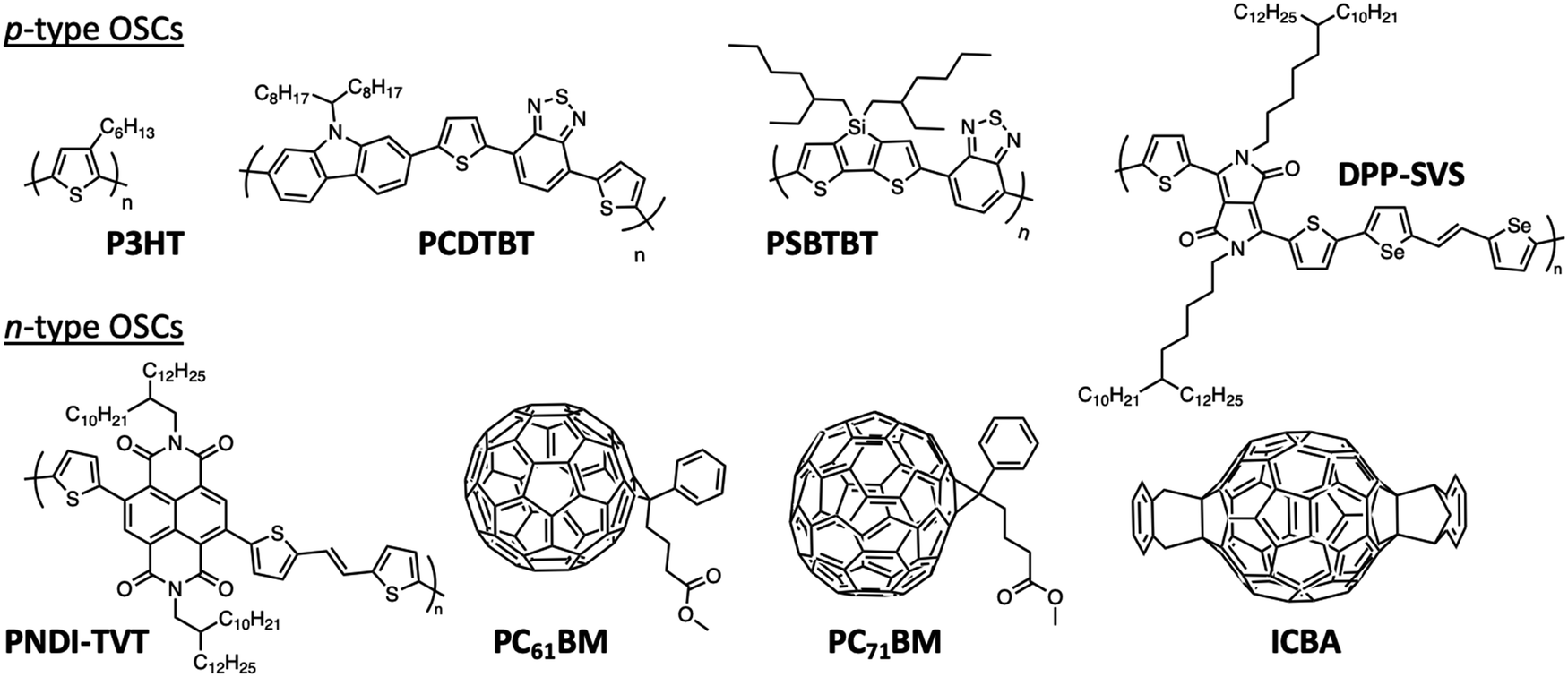 | ||
| Fig. 5 Chemical structures of P3HT, PCDTBT, PSBTBT, DPP-SVS, PNDI-TVT, PC61BM, PC71BM and ICBA. | ||
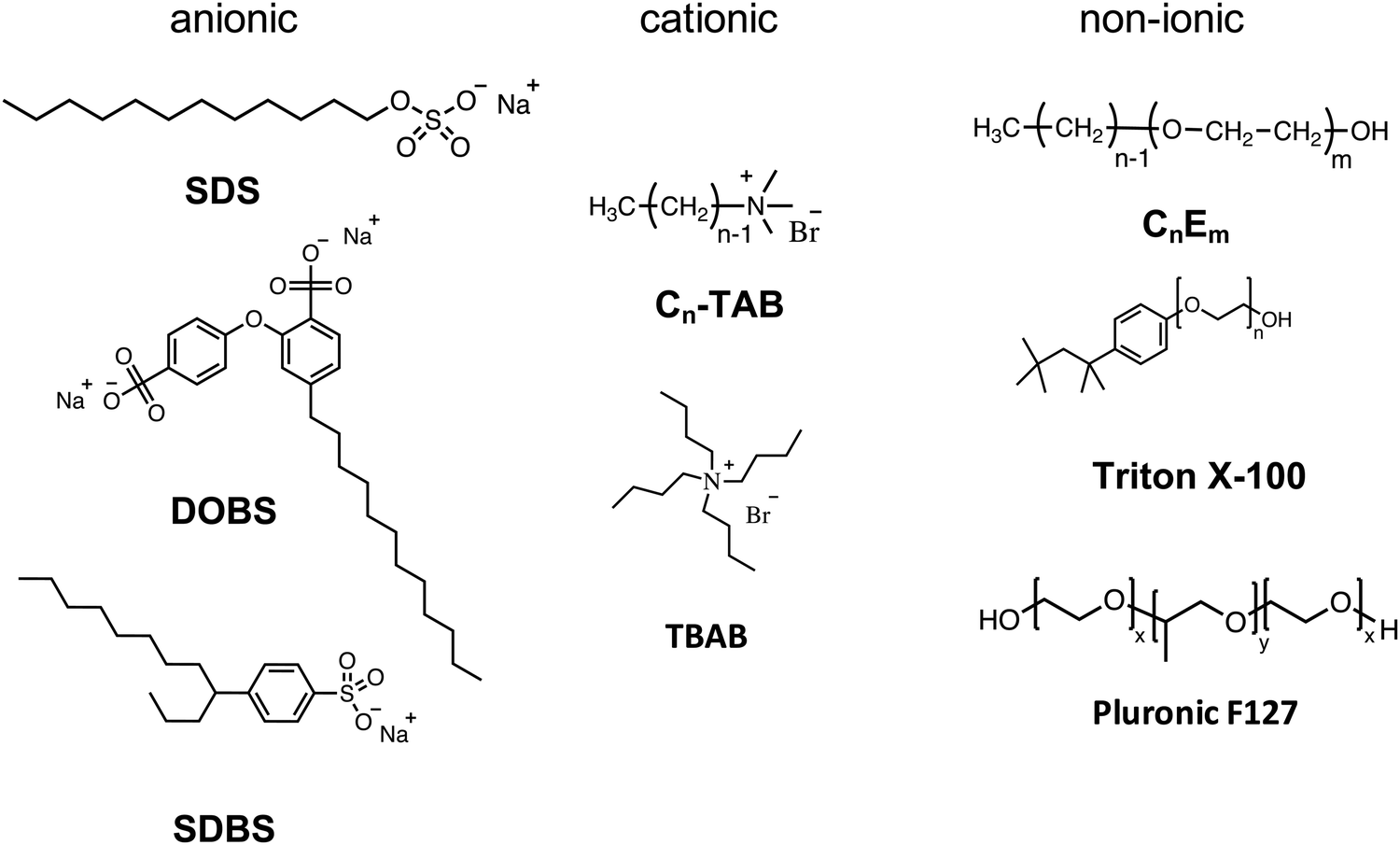 | ||
| Fig. 6 Chemical structures of surfactants used in the miniemulsion process. | ||
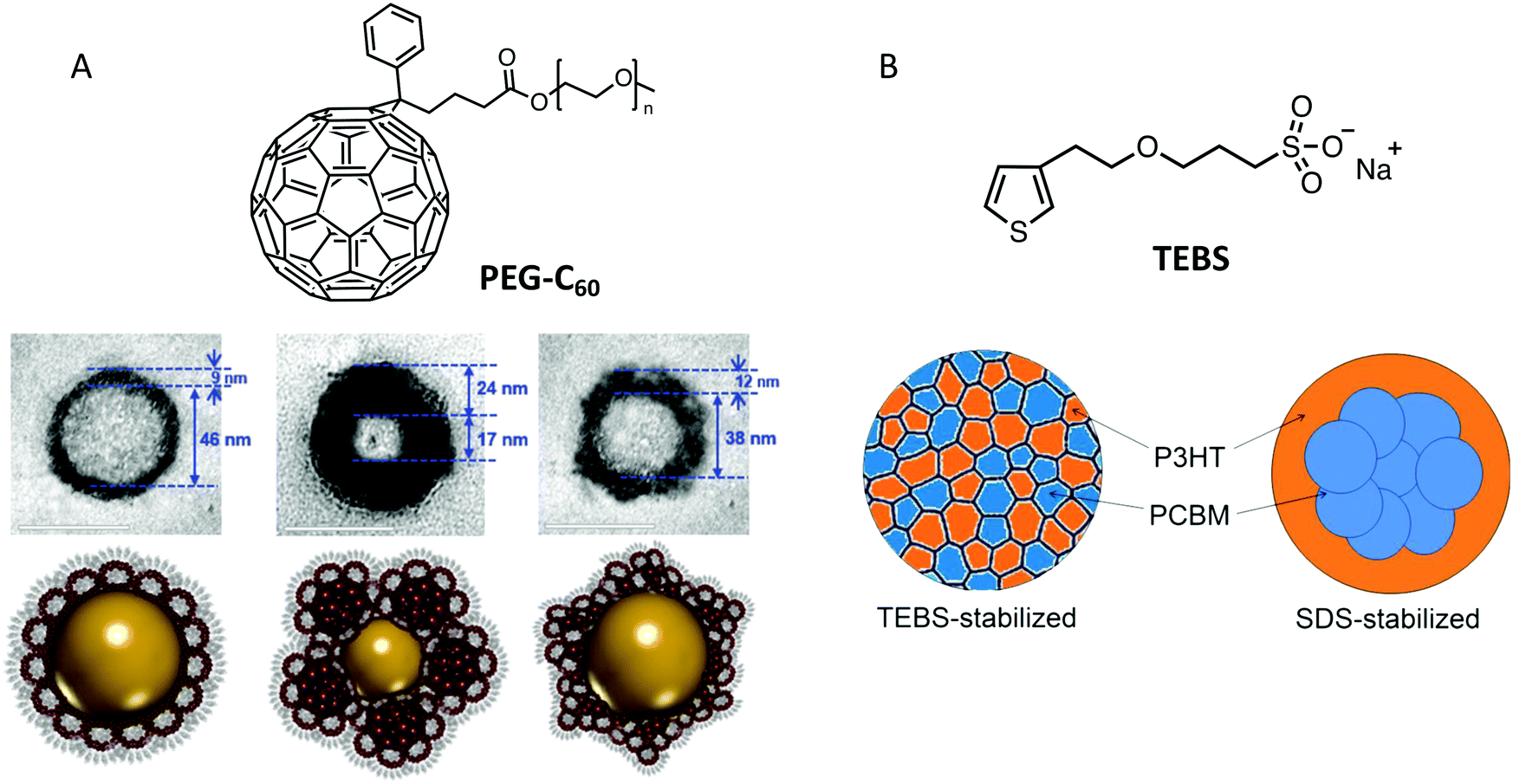 | ||
| Fig. 7 (A) TEM image of a core–shell NP stabilized by PEG-C60, the core is composed of P3HT and the shell of PEG-C60 micelles mixed with various amounts of PC61BM. Adapted with permission from ref. 37 (Copyright 2018 Wiley-VCH Verlag). (B) Scheme of the structure of P3HT:PC61BM stabilized by TEBS. Adapted with permission from ref. 38 (Copyright 2018 American Chemical Society). | ||
2.2. Nanoprecipitation
The nanoprecipitation technique is the second way to prepare OSC NPs in an aqueous or alcohol based non-toxic solvent. A polymer solution in an organic solvent is injected into a non-solvent of the polymer, miscible with the initial solvent (Fig. 8A). The solubility of the polymer decreases and the system becomes supersaturated. At low supersaturation, in the metastable region between the binodal and spinodal lines of the phase diagram, particle formation takes place through nucleation and growth, whereas for high supersaturation in the spinodal region, the systems demixes spontaneously39 (Fig. 8B). The organic solvent is then evaporated. The advantage of this technique, as compared to the miniemulsion evaporation process, is that no surfactant is needed. However, the NP dispersions tend to aggregate during long-term storage and the formation of stable small NPs is usually obtained in the binodal region at very low polymer concentrations, leading to very dilute dispersions. Moreover, to obtain dispersions in water or in alcohol, the choice of suitable solvents is limited to miscible ones such as tetrahydrofuran (THF) or chloroform.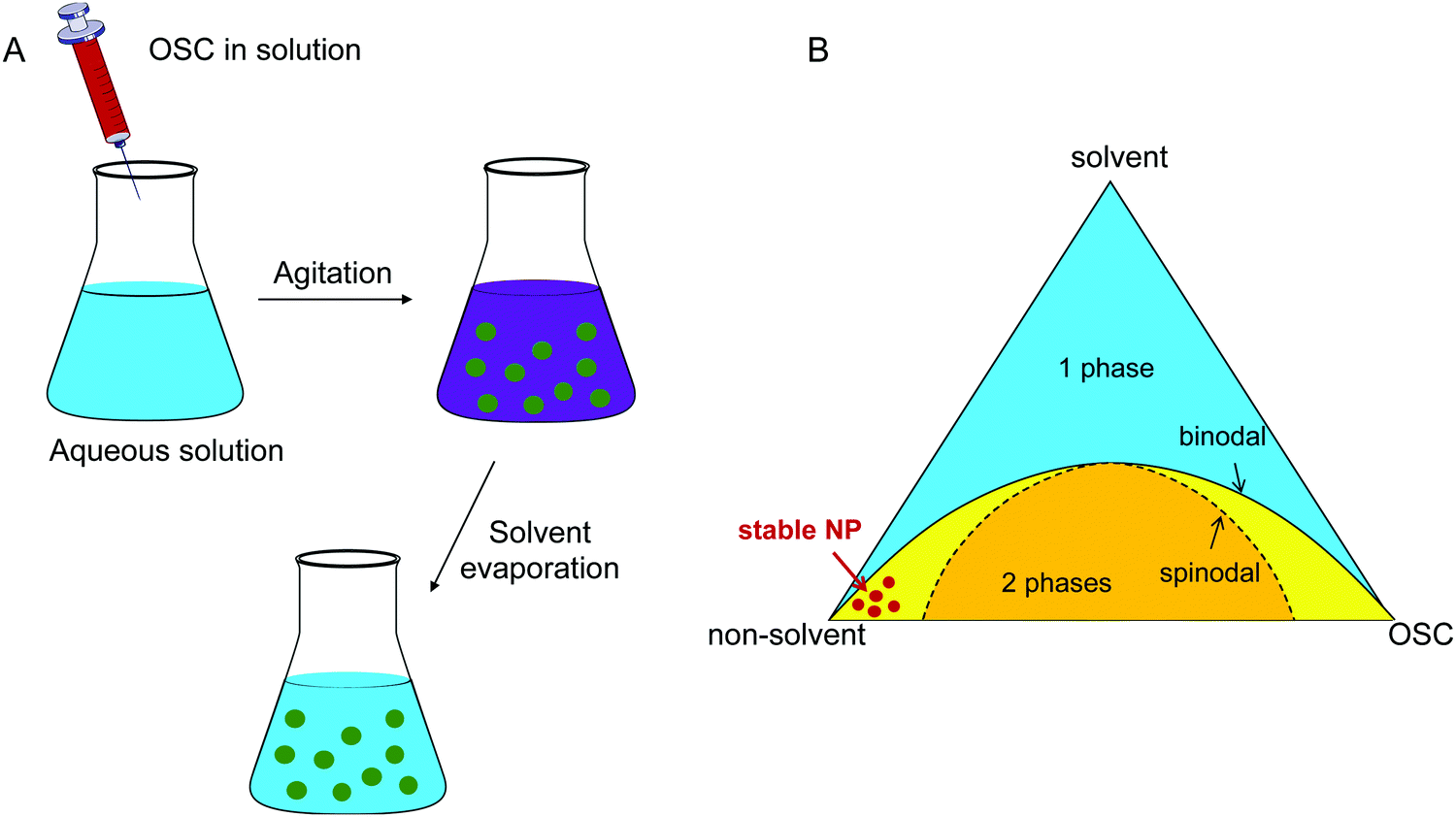 | ||
| Fig. 8 (A) Scheme of the nanoprecipitation process, and (B) phase diagram of the OSC/solvent/non-solvent system showing the concentration zone in which stable NPs are obtained. | ||
Wang et al.40 studied the aggregation-driven growth of the NPs in the first hours after their nucleation, as a function of the structure of the polymer. They compared the aggregation behavior of poly(bithiophene-alt-azulene) bearing either alkyl (alkyl-PTA) or alkoxy side chains (alkoxy-PTA) during its nanoprecipitation from chloroform solution into methanol (Fig. 9a). The diameter of alkyl-PTA NPs increases from 60 nm, 1 min after vigorously mixing the 10−5 M polymer solution in chloroform with an equal volume of methanol, to 400 nm after 4 h, after which the NPs became polydisperse. In the case of alkoxy-PTA, a smaller initial diameter of 30 nm was obtained. The NP diameter increased slightly up to 130 nm after 5 h but kept a very low dispersity (Fig. 9). To explain the difference between these two behaviors, the authors observed the evolution of the UV-visible absorption spectra with time and concluded that the electron donation to the conjugated polymer backbone is enhanced for alkoxy groups as compared to alkyl groups, resulting in stronger π–π stacking of the polymer backbone. Thus, alkoxy side chains favor the nucleation step, leading to an increased number of smaller NPs and faster consumption of the free macromolecules. This example illustrates the mechanism of nucleation and growth of the nanoprecipitate. Moreover, as in the case of miniemulsion, several processing parameters, such as the solvent or the relative concentration of species, may also influence the size of the NPs, as well as their crystallinity. Millstone et al.41 have prepared P3HT NPs by nanoprecipitation from chloroform polymer solutions into ethanol. They have shown that the final diameter of the NPs increases from 30 to 83 nm with the initial concentration of P3HT in chloroform increasing from 0.005% to 0.5%. Moreover, when using monodisperse regioregular P3HT synthesized by Grignard metathesis, they obtained NPs with anisotropic shape due to their high crystallinity. As in the case of the miniemulsion technique, composite NPs can also be prepared by nanoprecipitation, starting from a solution containing both electron donor and acceptor materials further mixed with a non-solvent of the two materials. In this case also, the processing parameters allow one to control the size and stability of the prepared NPs. Using a high-throughput engineering method with a robot, Xie et al.42 investigated systematically the effect of the different processing parameters on the size and stability of P3HT:ICBA NPs precipitated from chloroform solutions into five different alcohols with increasing alkyl chain length from ethanol to cis-3-hexen-1-ol. They also showed that, whatever the used alcohol, increasing the material concentration in chloroform, from 0.1 to 20 mg mL−1, raised the NP diameter from around 20 nm to 100 nm. The nature of the alcohol had a smaller impact on the NP diameter, which decreased slightly with the length of the alkyl chain. However, it had an important impact on the surface charge density of the NPs and, consequently, on the stability of the dispersion over time. Indeed, NPs produced in a more polar solvent, like ethanol, acquired a higher zeta potential, resulting in more electrostatic repulsion and better stability, as compared to NPs dispersed in cis-3-hexen-1-ol, which aggregated within less than 24 h. Finally, they observed a tendency of the diameter of the NPs to decrease with an increasing content of ICBA in the NPs, which could be due to a difference in solubility of the two components leading to a difference in the number of nuclei formed during the nucleation period.
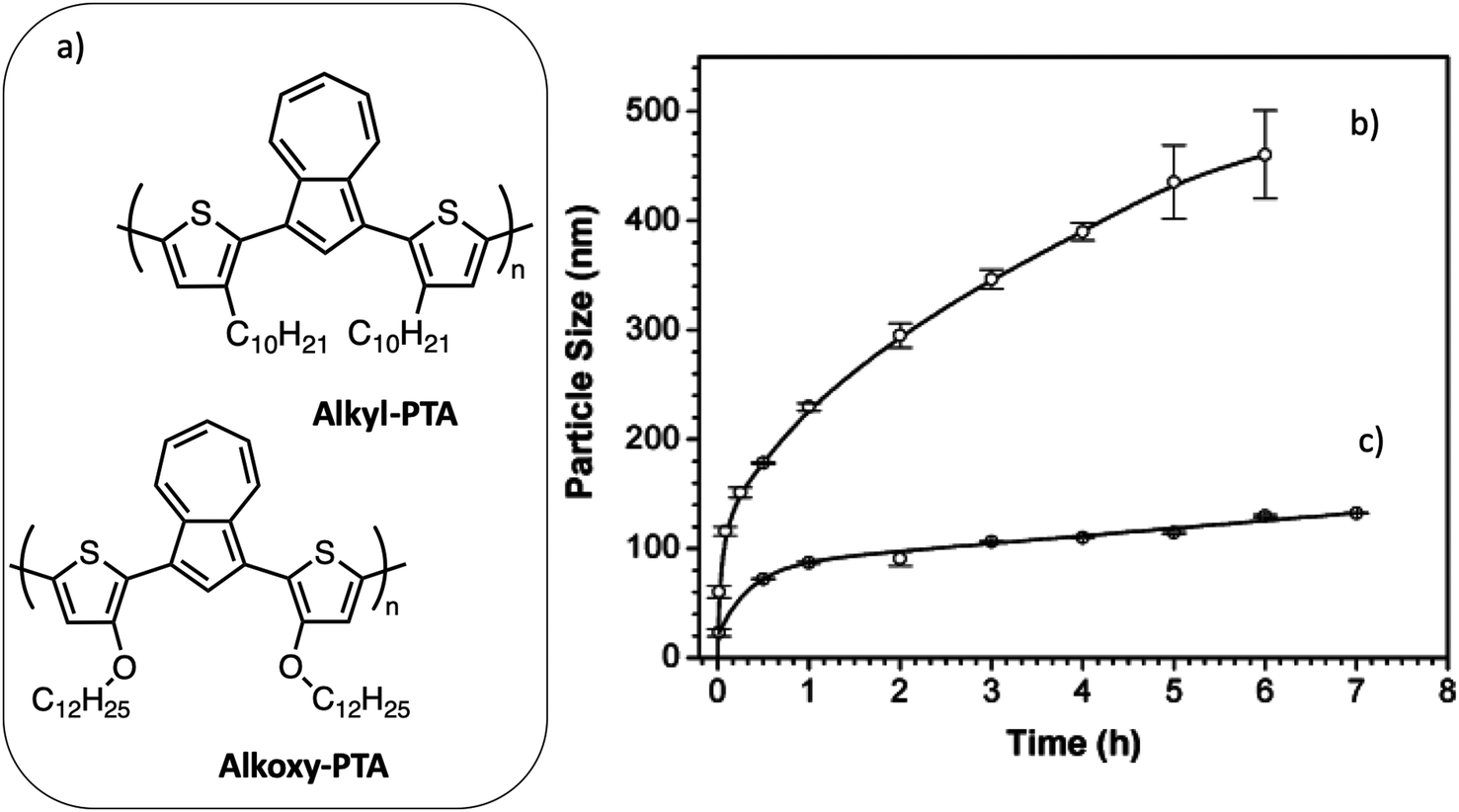 | ||
| Fig. 9 (a) Chemical structures of poly{1,3-bis[2-(3-n-decylthienyl)]azulene} (C10H21-PTA) and poly{1,3-bis[2-(3-n-dodecoxythienyl)]azulene} (C12H25O-PTA). Time-dependent size evolutions of conjugated polymer nanoparticles in 1:1 mixed chloroform/methanol solutions containing (b) 10−5 M C10H21-PTA and (c) 10−5 M C12H25O-PTA. Adapted with permission from ref. 40 (Copyright 2005 Wiley-VCH Verlag). | ||
Compared to the miniemulsion process, simple nanoprecipitation normally allows the formation of more uniform phase separation inside the NPs without the use of a surfactant, which makes it very attractive for OPV applications. However, the production of small NPs with low dispersity in size and good stability over time is a real challenge. Moreover, its success depends on the material/solvent/non-solvent phase diagram, which is different for each molecule, the stable dispersion being obtained in the binodal region at very low polymer concentration, yielding very dilute NP dispersions. Recently, Xie et al.49 developed a surfactant assisted nanoprecipitation method which allows the synthesis of stable dispersions at higher concentrations in the spinodal region and is therefore more versatile regarding the nature of the materials. As a surfactant, they cleverly chose Pluronic F127 (Fig. 6), a thermosensitive block copolymer of ethylene glycol and propylene glycol. It has the advantage of being non-ionic. Moreover, its solubility in water increases at low temperature. F127 could therefore be easily stripped off from the NPs at 4 °C in a few centrifugation dialysis steps, much more efficiently than for SDS removal (Fig. 10). With this technique, they prepared composite NPs of P3HT and a non-fullerene acceptor (NFA) o-IDTBR (Fig. 10a), starting from a 5 mg mL−1 material solution in THF followed by precipitation into water with a high solvent:non-solvent ratio of 1:2, whereas it is usually in the range of 1:8 or lower in standard nanoprecipitation. Such a strategy resulted in a dispersion having a higher final NP concentration than what it is usually achieved in classical nanoprecipitation. Moreover, after surfactant stripping, the NP dispersions remained stable for more than 3 months. Finally, the authors demonstrated the versatility of this technique by preparing dispersions of composite NPs with two LBG polymers PCE10 or PBQ-QF, and two NFAs, o-IDTBR or ITIC (Fig. 10a), which have been used in OPVs to reach a record efficiency for NP based systems of 7.5%.
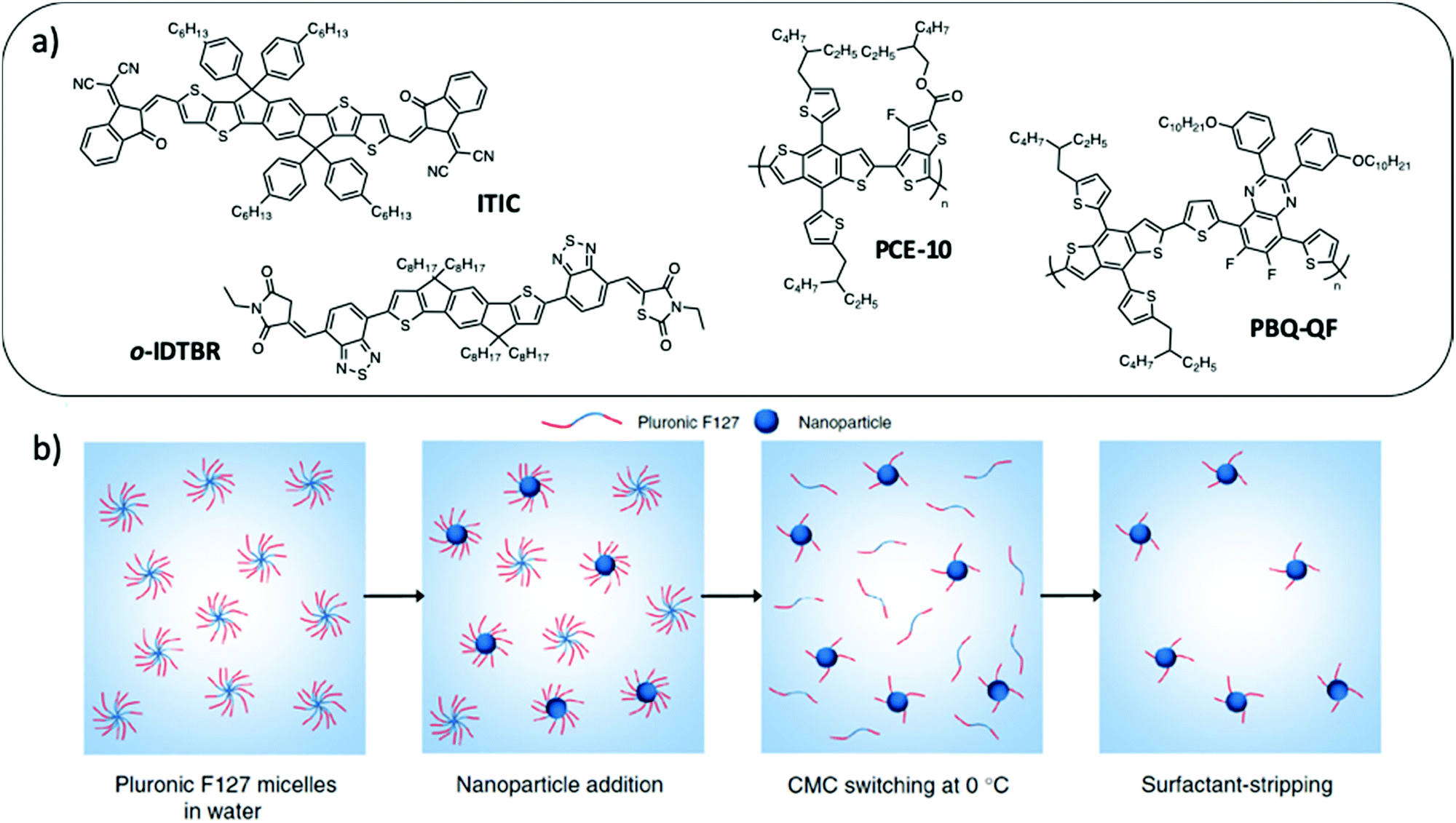 | ||
| Fig. 10 (a) Chemical structures of ITIC, o-IDTBR, PCE-10 and PBQ-QF used to prepare NPs by surfactant-assisted nanoprecipitation. (b) Pluronic F127 is a copolymeric surfactant with a temperature-dependent critical micelle concentration (cmc). After elaboration of the NPs at room temperature, the dispersion is cooled down to 0 °C. At this low temperature, the surfactant becomes more soluble in water, its cmc increases and it desorbs from the NP surface. It can then be easily removed by centrifugal dialysis in a few steps only. Adapted with permission from ref. 49 (Copyright 2018 Springer Nature). | ||
3. Building the active layer by NP assembly, and its resulting morphology
Once synthesized, the NPs have to be assembled into a functional active layer. As already described, the ideal morphology of the film is a two phase bicontinuous structure with percolating pathways allowing the efficient conduction of electrons and holes towards the electrodes (Fig. 1).The elaboration of an active layer from an NP ink is performed in two steps: first, the deposition and assembly of the solid NPs during solvent evaporation and then an annealing step allowing the diffusion of the materials in the interparticle voids, leading to more compact and smooth active layers. Rearrangement of the donor and acceptor phases can also occur during annealing. These two steps can then be repeated several times in order to obtain a thicker and/or a multilayered film.
3.1. Processes used to build an active layer from NP dispersions
Different processes have been used to build a film from the NP dispersions for the preparation of active layers from the laboratory scale to the industrial one. Several issues are encountered. Among them, one can cite the low solid contents of the prepared aqueous dispersions (Table 1) requiring additional concentration steps, and their low viscosity leading to low film thicknesses. It is then often necessary to repeat the deposition step in order to increase the active layer thickness. Another difficulty is the poor wettability of the aqueous dispersions on more or less hydrophobic substrates, resulting in a non-homogeneous layer with large roughness, responsible for current leakages and failure of the device.| OSC system | Solution [mg mL−1] | Dispersion, solid content, mg mL−1 | Ref. | ||
|---|---|---|---|---|---|
| After elaboration | For the coating process | ||||
| P3HT/o-IDTBR | Solution | 24 in CB | 50 | ||
| Surfactant-assisted nanoprecipitation | 2.5 in water | 80 in water | 49 | ||
| P3HT/PC61BM | Solution | ∼40 in o-DCB | 50 | ||
| Surfactant-assisted nanoprecipitation | 2.5 in water | 50 in water | 49 | ||
| Miniemulsion | 3.3 in water | 10 in water | 51 | ||
| P3HT/ICBA or P3HT/ICxBA | Solution | 40 in CB | 52 | ||
| Nanoprecipitation | 0.025 to 5 in ethanol | 10 in ethanol | 42 and 53 | ||
| Miniemulsion | 10 in water | 10 in water | 28 and 54 | ||
| PBDTTPD/PC71BM | Solution | 17.5–20 in CB | 55 | ||
| Miniemulsion | 9.5 to 28.5 in water | 40 in water | 15 | ||
| PCE10(PTB7-Th)/o-IDTBR | Solution | 20–37.5 in o-DCB | 56 | ||
| Surfactant-assisted nanoprecipitation | 2.5 in water | 50 in water | 49 | ||
| PBQ-QF/o-IDTBR or PBQ-QF/ITIC | Solution | 12 in THF | 57 | ||
| Surfactant-assisted nanoprecipitation | 2.5 in water | 50 in water | 49 | ||
At the lab scale, the main used process is the spin-coating of the NP dispersions. Bag et al.51 have optimized the spin-coating conditions of a P3HT:PC61BM blend in order to obtain a reproducible deposited active layer. Optimization was performed both for separate and composite NPs for a direct PV structure of glass/ITO/PEDOT:PSS/active layer/Ca/Al, the direct structure allowing illumination through a transparent hole-collecting electrode (ITO/PEDOT:PSS). Thus, UV-O3 treatment of the PEDOT–PSS layer allowed its water contact angle to be decreased to less than 2°, leading to rapid and uniform spreading of the NP dispersion and a low surface roughness. Better packing of the NPs in the layer was also obtained by spin-coating NPs in a solvent mixture of ethanol:water (20:80) rather than water alone, probably thanks to a decrease of the electrostatic repulsion between the NPs, while increasing van der Waals attraction. Moreover, in order to prevent the PEDOT–PSS sublayer from re-dissolving when in contact with the aqueous dispersion, the evaporation of the solvent was sped up by preheating the substrate with an IR lamp. An ambient atmosphere was also important as a relative humidity below 30% has also been shown to increase the porosity and roughness of the deposited layer. Finally, the addition of a thin PC61BM buffer layer on top of the active layer, before Ca/Al electrode deposition, reduced the surface roughness to 10 nm and prevented leakage current to the electron-collecting electrode. OPV device characterization clearly highlights the positive effect of this buffer layer, especially on the fill factor (FF), which reached 65%. Although modest, the final PCE of 2.15% is clearly in the average of the PCEs obtained for such P3HT-based blends.58
By studying composite NPs made of two fluorene-based polymers, one electron donating derivative, called PFB, and one electron accepting derivative, called F8BT (Fig. 11b), Stapleton et al.59 have shown that the successive deposition of NPs by spin-coating, with rapid annealing at 70 °C between each layer deposition for drying purposes, allowed one to tune the thickness of the final film. In addition, they observed that the roughness of the film decreased with the number of deposited layers (Fig. 11a). The voids left in the underlying film were filled by the freshly deposited NPs, thus increasing the compactness and quality of the film. However, when the film became too thick, cracks appeared on its surface, probably due to negative capillary pressure between NPs resulting from water removal. Hence, an optimum has to be found for the number of NP deposition layers and thickness of the film in order to achieve the best morphology.
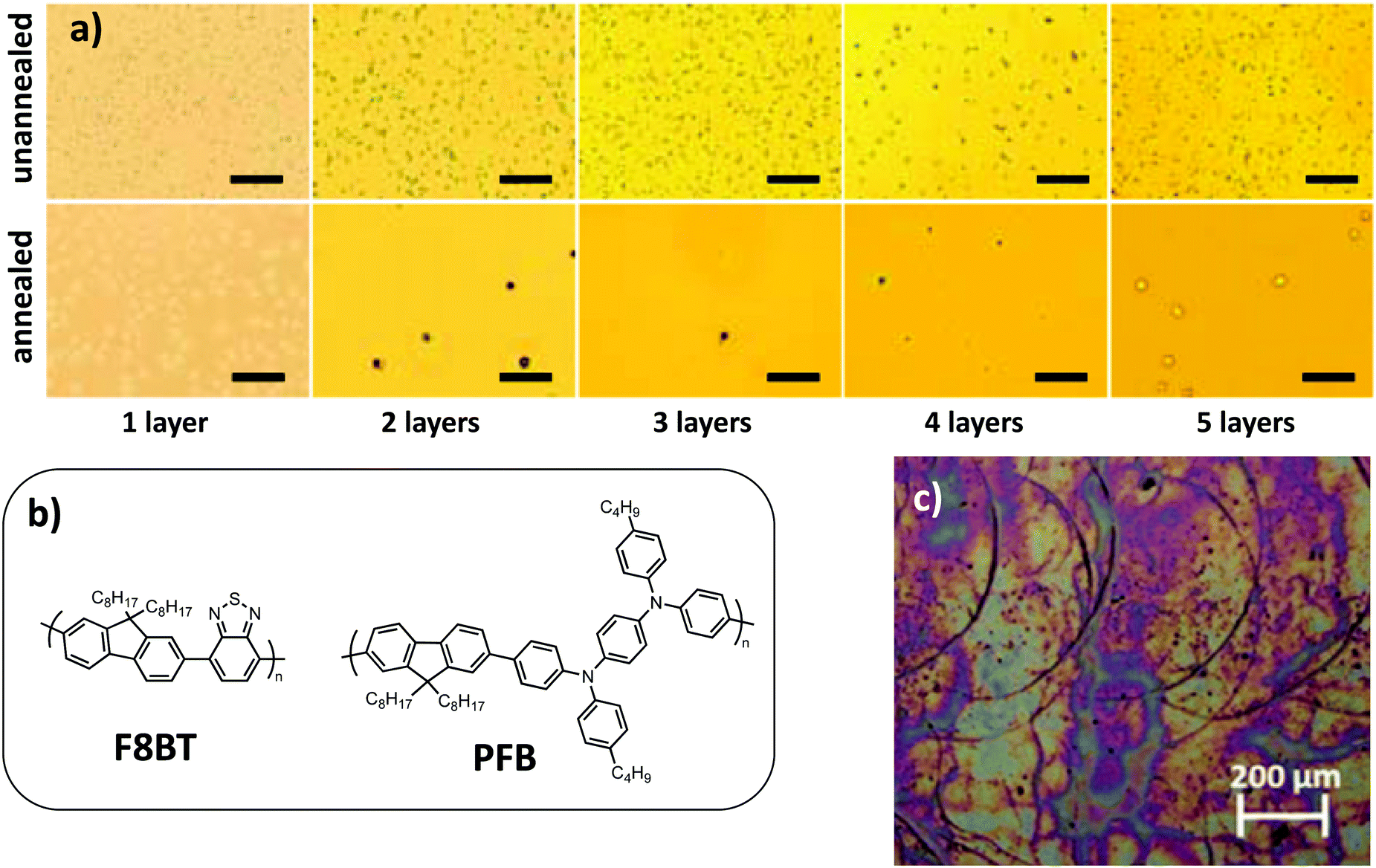 | ||
| Fig. 11 (a) Optical micrographs for the unannealed (upper row) and annealed (lower row) nanoparticulate films consisting of one to five layers. The scale bar is 5 mm in each micrograph. Adapted with permission from ref. 59 (Copyright 2012 Elsevier B.V.). (b) Polymer chemical structures. (c) Optical micrograph of an ink-jet printed P3HT:IC[60]BA nanoparticulate film after annealing, showing the typical signature of droplet edges, originating from the coffee stain effect. Adapted with permission from ref. 60 (Copyright 2015 Elsevier B.V.). | ||
Preparation of large surface devices is not possible by using spin-coating; therefore, other deposition techniques have been studied such as spray coating, ink-jet printing or doctor blading.
P3HT films were prepared by spray coating of aqueous dispersions of P3HT NPs in water leading to NP assemblies with thicknesses up to 1.5 μm.61 The cleaned ITO substrate was heated at 80–85 °C and a UV-O3 pretreatment improved the film quality. The presence of surfactant in the dispersion had an important influence on the packing of NPs in the films, leaving voids in the film due to electrostatic repulsion between the particles. Therefore, removal of the excess surfactant, as discussed for the miniemulsion process (Section 2.1), was necessary to obtain close-packed NP assemblies.
Ink-jet printing is an interesting technique that allows the deposition of solar cells with custom-designed shapes, while doctor blading is limited to large surfaces homogeneously coated.62 The formulation of the dispersion has to be adapted in order to avoid nozzle clogging for ink-jet printing. As an example, in the case of P3HT:ICBA NP dispersions in ethanol, a higher boiling point solvent, butoxyethanol (boiling point 170 °C), often used in paints, had to be added.60 The temperature of the substrate can also be optimized to tune the spreading of the droplets and evaporation rate. If the deposited film is too thin, several layers can also be printed. However, one of the difficulties of this technique is to avoid the coffee stain effect, an accumulation of NPs at the edge of the deposited droplets. Indeed, it leads to the formation of ridges with increased thickness in the final film (Fig. 11c).
Doctor-blading is a deposition technique that closely resembles slot-die coating used in roll-to-roll processes. Sankaran et al.60 have prepared inverted OPV devices with structure glass/ITO/ZnO/composite NPs/PEDOT:PSS/Ag by doctor-blading active layers of surfactant-free P3HT:ICBA NP dispersions in ethanol onto ZnO. Two blading sublayers were applied before thermal annealing at 150 °C, leading to similar performances to spin coated devices. They also demonstrated the possibility to prepare a device active area of 1.1 cm2 with minimal loss of performance from 3.9% for 0.105 cm2 to 3.4%. Xie et al.42 have also prepared inverted OPV devices with structure glass/ITO/ZnO/composite NPs/MoOx/Ag by doctor-blading P3HT:ICBA NPs in different alcohols on ZnO. The best device performance was obtained for ethanol with a PCE of 4.26%. Indeed, NPs in alcohol with increasing alkyl chain length up to hexanol showed stronger aggregation and lower wetting on the substrate. Four sequentially blade-coated layers were necessary to obtain an active layer thickness of 240 nm.
OPV active layers have also been deposited onto flexible PET foil from aqueous dispersions of composite conjugated polymer:fullerene derivative NPs by roll-to-roll processing.54,63 One of the important issues encountered in the case of slot-die coating of an aqueous dispersion onto a ZnO electron transporting layer (ETL) for inverted structures or a PEDOT–PSS hole transporting layer (HTL) for direct structures is the dewetting of the NP ink before complete drying of the film. This phenomenon can be controlled by fine tuning of the ink formulation. In particular, it was shown that leaving an optimized concentration of SDS surfactant in the ink allowed one to control dewetting, but was detrimental to the device performance. Andersen et al.63,64 have added a non-ionic fluorosurfactant (FSO-100) to the ink formulation. However, in excess, this surfactant could be responsible for low film adhesion to the substrate. Another way to control the interfacial surface energy is to use ozone treatment to change the substrate chemistry. Applied to the PEDOT–PSS HTL, this treatment lowered the water contact angle by increasing the PSS fraction at the surface of the film, resulting in good wetting of the NP ink slot-die coated onto it and thus increasing the homogeneity of the active layer film.54 Simultaneous double slot-die coating of the active layer and the PEDOT–PSS HTL onto ZnO was also demonstrated.108 In this case, it was shown by film delamination in NaOH solution, and by Time of Flight-Secondary Ion Mass Spectrometry (TOF-SIMS) analysis, that a double layer had been obtained with only limited inter-diffusion between the active and the hole transporting layers, the diffusion of NPs being too slow for the two dispersions to mix before drying. Even if significant process optimization has still to be done in order to increase the device performances, the possibility to prepare NP OPV flexible devices entirely processed by roll-to-roll has been demonstrated with a PCE up to 0.45%.54 Roll-to-roll processing of aqueous NP dispersions is thus the most promising route to large scale printing of OPV devices from aqueous solvents.
3.2. Morphology of the BHJ film at the mesoscale
In this part, we discuss the morphology of the active layer composed of both electron donor and acceptor materials, formed by the assembly of the NPs, as building blocks. The phase separation of the two materials is discussed, keeping in mind that the ideal morphology is formed by two percolating material networks allowing exciton dissociation at interfaces and charge transport to the electrodes.65 In order to illustrate these features, a correlation with the OPV device performances is also made when it makes sense. In order to make this review as exhaustive and clear as possible, the reader will find in Table 2 the main features associated with the performance of organic photovoltaic devices reported in the literature.| Electron donor/acceptor | NP synthesis technique | Good/bad solvent | Surfactant | AL thermal annealing T °C/time (min) | V oc (mV) | J sc (mA cm−2) | FF (%) | PCE (%) | Device architecture | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| PFB/F8BT | Miniemulsion | CF/water | SDS | n.d. | n.d. | n.d. | n.d. | n.d. | Standard ITO/PEDOT–PSS/AL/Ca/Al | 70 |
| PFB/F8BT | Miniemulsion | CF/water | SDS | 140/2 | 770 | 1.81 | 28 | 0.39 | Standard ITO/PEDOT–PSS/AL/Al | 59 |
| PFB/F8BT | Miniemulsion | CF/water | SDS | n.d. | n.d. | n.d. | n.d. | 1.7 | Standard ITO/AL/Ca/Al | 18 |
| P3HT/PC61BM | Miniemulsion | CF/water | SDS | 80/60 under a vacuum | 470 | 4.89 | 50.5 | 1.16 | Inverted ITO/ZnO/AL/MoO3/Ag | 92 |
| P3HT/PC61BM | Miniemulsion | CF/20% EtOH in water | SDS | RT under a vacuum | 509 | 6.38 | 66.2 | 2.15 | Standard ITO/PEDOT–PSS/AL/Ca/Al | 68 |
| P3HT/PC61BM | Miniemulsion | CF/water | SDS | None | 360 ± 10 (380) | 5.9 ± 0.5 (7.1) | 52 ± 2 (54) | 0.9 ± 0.1 (1.2) | Standard ITO/PEDOT:PSS/AL/ZnO/Al | 30 |
| P3HT/PC61BM | Miniemulsion | CF/water | SDS | 140/4 | 529 ± 4.7 (524) | 4.18 ± 0.27 (4.6) | 41 ± 1 (42) | 0.91 ± 0.07 (1.00) | Standard ITO/PEDOT–PSS/AL/Ca/Al | 25 |
| P3HT/PC61BM | Nanoprecipitation | CF/EtOH | — | 130/4 | 634 ± 37 | 4.84 ± 0.78 | 36 ± 2 | 1.09 ± 0.16 | Standard ITO/PEDOT–PSS/AL/Al | 77 |
| P3HT/PC61BM | Miniemulsion | CF/water | SDS | 150/n.d. | 510 (520) | 5.84 (6.38) | 65.4 (67.9) | 1.94 (2.15) | Standard ITO/PEDOT–PSS/AL/Al | 51 |
| P3HT/PC61BM + PEG-C60 | Miniemulsion | CF/water | PEG-C60 | n.d. | 540 ± 10 | 10.3 ± 0.03 | 55.3 ± 0.06 | 2.94 (3.08) | Inverted ITO/MoO3/AL/LiF/Al | 37 |
| P3HT/PEG-C60 | Miniemulsion | CB/water | None | n.d. | 540 | 9.11 | 53.4 | 2.62 | Inverted ITO/MoO3/AL/LiF/Al | 36 |
| TQ1/PC61BMP3HT/PC61BM | Miniemulsion | CF/water | SDS | 80/— | 910 | 0.37 | 28.2 | 0.1 | Inverted tandem PET/ZnO/AL1/V2O5/ZnO/AL2/PEDOT–PSS/Ag (4 cm2) | 64 |
| P3HT/ICBA | Miniemulsion | CF/water | SDS | 150/10 | 763 ± 17 | 5.85 ± 0.20 | 55 ± 1 | 2.44 ± 0.04 | Standard ITO/PEDOT–PSS/AL/Ca/Al | 28 |
| P3HT/ICBA | Nanoprecipitation | o-DCB/MeOH | — | 200/10 | 781 | 9 | 58 | 4.1 | Inverted ITO/ZnO/AL/MoOx/Ag | 78 |
| P3HT/ICBA | Nanoprecipitation | CF/water | — | 150/10 | 834 ± 2 | 9.0 ± 0.1 | 55 ± 1 | 4.2 ± 0.1 | Inverted ITO/ZnO/AL/PEDOT–PSS/Ag | 53 |
| P3HT/ICBA | Nanoprecipitation | CF/EtOH | — | 150/10 | 797 ± 6 | 9.2 ± 0.2 | 53 ± 1 | 3.9 ± 0.1 | Inverted PET/ITO/ZnO/AL/PEDOT:PSS/Ag | 46 |
| P3HT/ICBA | Nanoprecipitation | o-DCB/EtOH | — | 150/10 | 832 ± 1 | 9.4 ± 0.1 | 55 ± 1 | 4.3 ± 0.1 | Inverted ITO/ZnO/AL/PEDOT–PSS/Ag | 60 |
| P3HT/ICBA | Nanoprecipitation | CF/EtOH | — | 160/10 | 810 ± 10 | 9.74 ± 0.27 | 56.5 ± 0.9 | 4.44 ± 0.15 (4.52) | Inverted ITO/ZnO/AL/MoOx/Ag | 42 |
| P3HT/ICxBA | Miniemulsion | CF/water | SDS | 110/10 | 500 ± 10 (510) | 3.64 ± 0.22 (3.95) | 36 ± 1 (37) | 0.67 (0.73) | Standard ITO/PEDOT–PSS/AL/Ca/Al | 31 |
| P3HT/ICxBA | Miniemulsion | CF/water | SDS | 110/10 | 573 ± 17 (600) | 4.18 ± 0.26 (4.73) | 47 ± 1 (49) | 1.14 ± 0.11 (1.39) | Standard ITO/PEDOT–PSS/AL/Ca/Al (1 cm2) | 54 |
| P3HT/o-IDTBR | Surfactant assisted nanoprecipitation | THF/water | F127 | 150/10 | 760 ± 10 | 10.36 ± 0.39 | 62.9 ± 2.0 | 4.95 ± 0.32 (5.23) | Inverted ITO/ZnO/AL/MoOx/Ag | 49 |
| PCDTBT/PC71BM | Nanoprecipitation | THF/water | — | 160/4 | 480 | 1.65 | 29 | 0.19 | Inverted ITO/ZnO/AL/MoO3/Ag | 43 |
| PSBTBT/PC61BM | Miniemulsion | CF/water | SDS | 80/— | 470 | 3.99 | 29.3 | 0.55 | Inverted PET/ITO/ZnO/AL/PEDOT:PSS/Ag-(printed) (4 cm2) | 63 |
| PSBTBT/PC61BM | Miniemulsion | CF/water | SDS | 140/2 | 620 | 6.2 | 33 | 1.3 | Inverted ITO/ZnO/AL/PEDOT–PSS/Ag | 27 |
| PDPP-TNT/PC71BM | Miniemulsion | CF/water | SDS | 130/10 | 760 | 6.09 | 43 | 1.99 | Standard ITO/PEDOT–PSS/AL/Ca/Al | 75 |
| PDPP5T/PC71BM | Miniemulsion | CF/water | SDS | 140/10 | 540 | 9.43 | 47 | 2.36 | Inverted ITO/PEIE/AL/MoOx/Ag | 93 |
| PCDTBT/PC71BM | Miniemulsion | CF/water | SDS | 140/n.d. | 605 ± 87 | 3.79 ± 0.7 | 30.7 ± 3 | 0.7 ± 0.24 | Inverted ITO/ZnO/AL/MoO3/Ag | 14 |
| PTNT/PC71BM | Miniemulsion | o-Xylene/water | SDS | 140/4 | 870 ± 20 (890) | 4.58 ± 0.13 (4.73) | 39 ± 1 (39) | 1.56 ± 0.06 (1.65) | Inverted ITO/ZnO/AL/MoO3/Ag | 16 |
| TQ1/PC71BM | Miniemulsion | CF/water | SDS | 140/5 | 691 ± 39 (696) | 8.26 ± 0.92 (10.06) | 37.7 ± 2 (36) | 2.11 ± 0.21 (2.54) | Standard ITO/PEDOT–PSS/AL/Ca/Al | 74 |
| P(TBT-DPP)/ICBA | Miniemulsion | CF/20% EtOH in water | SDS | 150/10 | 430 ± 10 (440) | 10.49 ± 2.65 (12.73) | 47 ± 5 (47) | 2.16 ± 0.5 (2.63) | Standard PEN/PEDOT–PSS/AL/C60/Al | 94 |
| PDPP5T/PC71BM | Miniemulsion | CF/water | SDS | 150/10 | 540 | 11.59 | 49 | 3.07 (3.38) | Inverted ITO/ZnO/AL/MoOx/Ag | 95 |
| PBDTTPD/PC71BM | Miniemulsion | CB/water | SDS | 180/20 | 764 ± 136 (860) | 10.45 ± 0.64 (9.99) | 40 ± 6 (44) | 3.2 ± 0.8 (3.8) | Inverted ITO/ZnO/AL/MoOx/Ag | 15 |
| PBDTT-FTTE/PC61BM + PEG-C60 | Miniemulsion | CF/water | PEG-C60 | n.d. | 720 ± 10 | 13.6 ± 0.06 | 55.1 ± 0.09 | 5.29 (5.39) | Inverted ITO/MoO3/AL/LiF/Al | 37 |
| PCE10/o-IDTBR | Surfactant assisted nanoprecipitation | THF/water | F127 | 150/10 | 970 ± 30 | 12.01 ± 0.43 | 42.4 ± 2.0 | 4.94 ± 0.25 (5.19) | Inverted ITO/ZnO/AL/MoOxMoOx/Ag | 49 |
| PBQ-QF/o-IDTBR | Surfactant assisted nanoprecipitation | THF/water | F127 | 150/10 | 950 ± 30 | 13.09 ± 0.41 | 47.9 ± 4.3 | 5.96 ± 0.58 (6.52) | Inverted ITO/ZnO/AL/MoOx/Ag | 49 |
| PBQ-QF/ITIC | Surfactant assisted nanoprecipitation | THF/water | F127 | 150/10 | 850 ± 20 | 14.87 ± 0.30 | 52.7 ± 2.9 | 6.97 ± 0.53 (7.50) | Inverted ITO/ZnO/AL/MoOx/Ag | 49 |
In the following, we discriminate between active layers prepared from a dispersion of composite NPs or from a dispersion of a binary mixture of electron donor NPs and electron acceptor NPs.
:1, with a maximum PCE of 1.84% measured for a direct-structure OPV device. Such a ratio is similar to the ones usually observed in solution processed P3HT:PC61BM devices.58
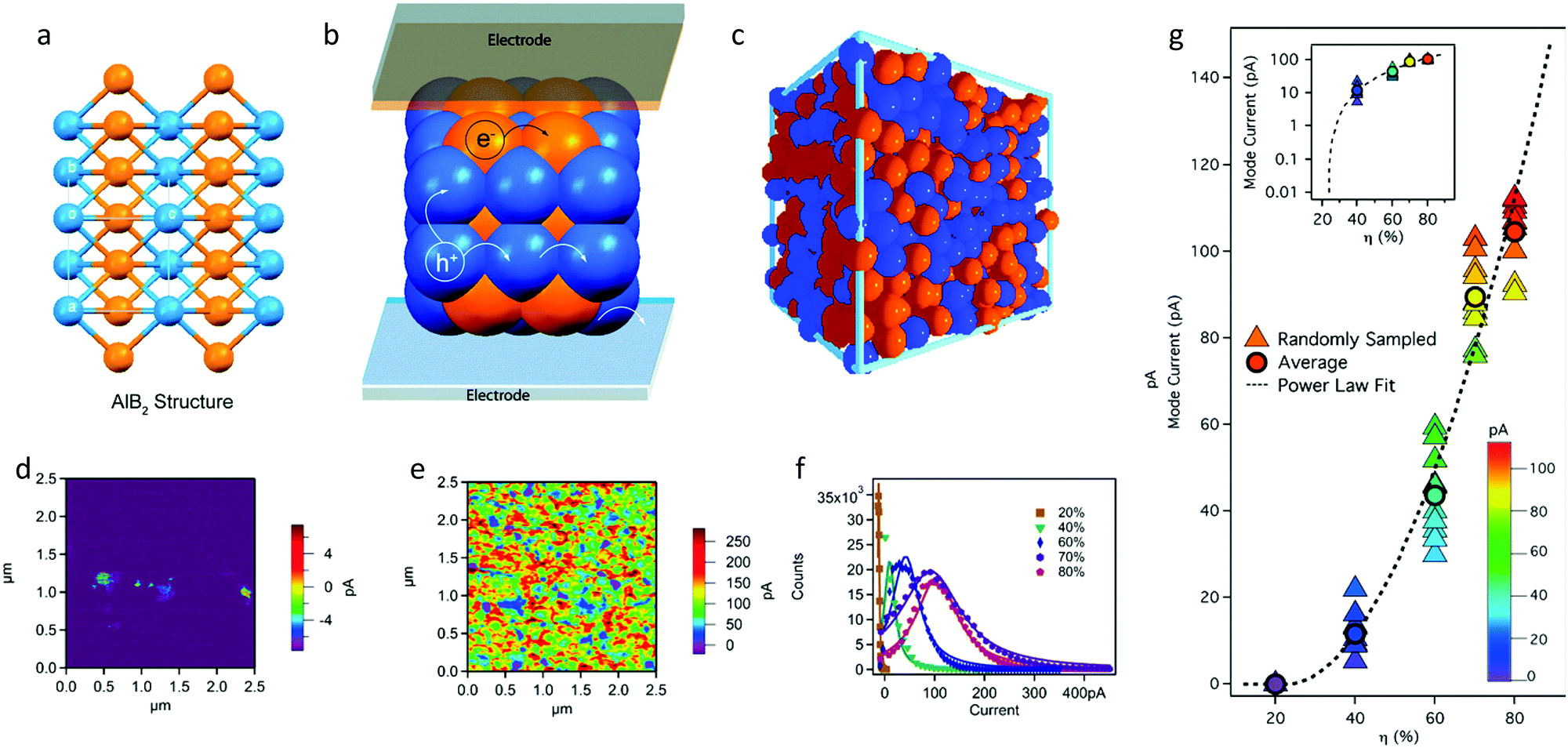 | ||
| Fig. 12 (a and b) Expanded view of the AlB2 structure showing continuous pathways for electron/hole transport to the cathode/anode, respectively, and (c) disordered assembly of NPs (RCP). Adapted with permission from ref. 66 (Copyright 2020 American Chemical Society). (d–g) Conductive AFM maps of binary P3HT:PS nanoparticle films at P3HT volume fractions (η) equal to (d) 20% and (e) 80%. (f) Pixel current histogram plots for five cAFM maps with varying η. (g) Mode current from current distributions as a function of η (dashed line) for ten randomly sampled subselections of each cAFM map, in closed triangle symbols, and the average in closed circle symbols. The semilog plot in the inset shows log mode current as a function of η. Adapted with permission from ref. 69 (Copyright 2016 American Chemical Society). | ||
After the deposition step, the random close-packed assembly of NPs still contains voids between the NPs and an annealing step is required to obtain a continuous compact film, by joining the NPs, thus increasing the contact area between the two different phases where exciton dissociation occurs and forming charge conduction pathways, leading to the final film morphology (Fig. 13b).
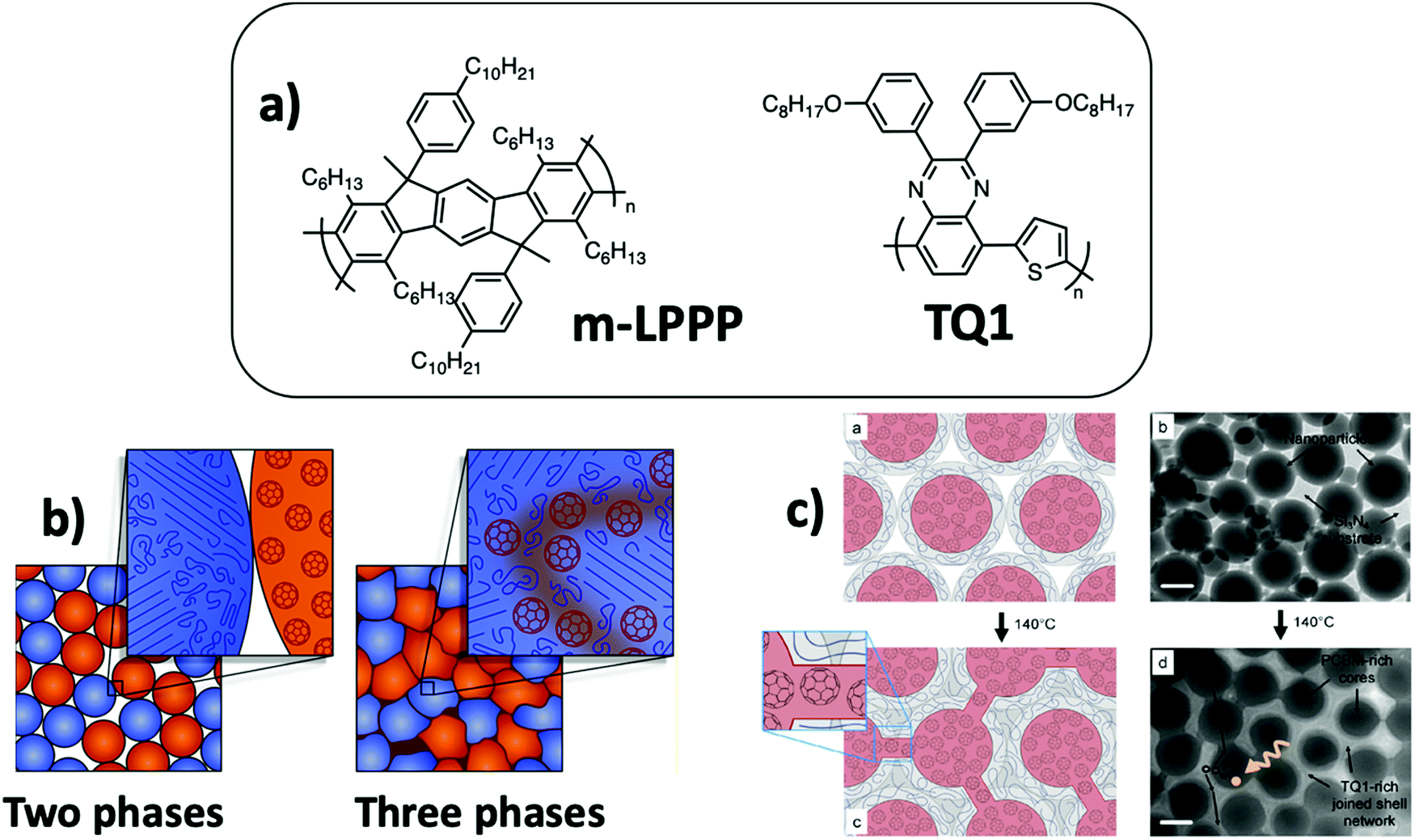 | ||
| Fig. 13 (a) m-LPPP and TQ1 chemical structures. (b) Schematic representation of the three phase film microstructure obtained by thermal annealing of the assembly of PCBM NPs and P3HT NPs comprising crystalline P3HT and amorphous P3HT subdomains. A third mixed phase is obtained by sintering and diffusion of PCBM in the P3HT amorphous domains. Adapted with permission from ref. 71 (Copyright 2018 American Chemical Society). (c) Schematic representation and TEM images of composite NPs made of a PCBM-rich core and an amorphous TQ1-rich shell. The mobility of the TQ1 polymer chains during thermal annealing slightly above its Tg allowed the movement of PCBM molecules and creation of bridging pathways between the PCBM-rich cores. Adapted with permission from ref. 74 (Copyright 2016 Elsevier B.V.). | ||
Kietzke et al.18 have prepared films from a mixture of separate NPs of two fluorescent polymers, one poly(p-phenylene)-type ladder polymer (m-LPPP, Fig. 13a) which does not show softening before its high decomposition temperature and one derivative of polyfluorene with Tg close to room temperature. The two polymers were selected to study the morphology of the films by energy-transfer experiments. After deposition by spin-coating, RCP NP assemblies were obtained with partial merging of the soft polymer particles. The film became smoother after annealing at 200 °C, and optical excitation at the absorption wavelength of polyfluorene resulted in quenching of the polyfluorene emission and charge transfer to the m-LPPP polymer, whose emission was increased by four, indicating an increase of the interfacial area between these two polymer phases during annealing. A similar phenomenon was observed for a binary mixture of PFB and F8BT NPs, showing increased energy transfer after annealing of the film slightly above the Tg of the polymers.18 Holmes et al.71 have prepared binary mixtures of PC61BM and P3HT NPs. They have shown using STXM and photoluminescence experiments that careful thermal annealing above the Tg of P3HT allowed the softening of the NPs, increasing the contact area between them and the diffusion of PC61BM into the amorphous parts of the P3HT phase, leading to a three-phase microstructure with P3HT crystalline domains, PC61BM domains and a mixed amorphous third phase containing more than 20% PC61BM (Fig. 13b). Diffusion of PC61BM into a P3HT phase had already been demonstrated in solvent-cast P3HT:PC61BM films.72,73 This third mixed phase, together with the high crystallinity of P3HT allowing a high diffusion length of excitons, resulted in an exciton dissociation efficiency as high as 60%, as compared to 37% before annealing.
In several cases, it was shown that the use of composite NPs, together with mild thermal annealing, is a good way to control the size of the phase-separated domains, even when it was not possible by solution casting. As an example, PDPP-TNT (Fig. 14a), a diketopyrrolopyrrole-based electron donor polymer, and PC71BM processed in chloroform solutions led to phase segregated morphology with domain sizes reaching the micron scale (Fig. 14b). In contrast, films prepared from composite NPs allowed the morphology of the active layer to be controlled at the nanometer length scale (Fig. 14c) even if mild annealing (at 130 °C for 10 min) was necessary to join the NPs and obtain a working OPV device (1.99% at best).75 However, the control of the morphology of the film was lost at higher annealing temperature or longer annealing time, resulting in gross phase segregation of the fullerene outside the particles and a drop of the PCE.
 | ||
| Fig. 14 (a) PDPP-TNT chemical structure. AFM images of (b) an as-spun PDPP-TNT:PC71BM 1:2 blend film spincast from chloroform and (c) an as-spun PDPP-TNT:PC71BM 1:2 nanoparticulate film. Scale bars are 500 nm. Adapted with permission from ref. 75 (Copyright 2014 The Royal Society of Chemistry). | ||
This effect of coarse phase separation was also observed for P3HT:PC61BM composite NP films, at a lower annealing temperature of 140 °C for 4 min,76 probably due to the lower Tg of P3HT as compared to PDPP-TNT or TQ1. In addition, the authors demonstrated, by combining optical spectroscopic and microscopic (STXM and TEM) analyses of the thin films, that before annealing, the NP films already show evidence of greater phase segregation and interchain order within the NPs than in unannealed solution cast BHJ films. Consequently, coarse phase segregation occurs very quickly in NP films upon thermal annealing as compared to BHJ films. This clearly suggests that the initial state of the film constituents plays a preponderant role in the thermal annealing effect and thus influences the temperatures and times to be applied. Subsequently, using similar analysis methods and still based on the P3HT:PC61BM binary system, the same team77 showed the different impact of thermal annealing at 140 °C for 30 min, as a function of the NP preparation process, on the final film morphology. Indeed, while the composite P3HT:PC61BM NPs prepared by miniemulsion exhibit a core–shell structure, the NPs prepared by nanoprecipitation are fully blended. The OPV performances measured on the unannealed as-cast films are similar in both cases. However, upon thermal annealing the core–shell NPs undergo significant phase segregation, leading to micron-sized domains of near-pure PC61BM and P3HT, while the blended nanoprecipitated NPs melted together without any phase segregation, allowing the conservation of an optimum blended morphology. Consequently, the charge transport and device efficiency were improved upon annealing. A final PCE of 1.09% for the nanoprecipitated NPs was reported by the authors. As compared to standard P3HT:PC61BM spin-coated films, the lower PCE could be mainly attributed to the lower short-circuit current density (Jsc) and FF. It can thus be assumed that blended NPs, as obtained by nanoprecipitation, are less prone to coarse phase separation and consequently tolerate harder thermal annealing.
Gärtner et al.78 have illustrated this point clearly. Indeed, as seen previously, they produced P3HT:ICBA blended surfactant-free NPs by nanoprecipitation in MeOH and elaborated multilayer homogeneous thin-films of approximately 250 nm thickness. Subsequent thermal annealing (up to 200 °C) does not show any impact on P3HT crystallization as probed by UV-vis absorption spectroscopy. However, this thermal annealing is of crucial importance for the device efficiency, which increases significantly up to such temperatures well above the Tg of the organic materials (Fig. 15). In order to better understand the thermal annealing–OPV device efficiency relationship, the authors investigated the limiting recombination processes by performing intensity dependent photo-current density measurements. Upon thermal annealing, the limiting recombination process changes from bimolecular to monomolecular. Associated with a simultaneous decrease in film roughness and a significant increase in open circuit voltage (Voc) and FF, the authors suggested that thermal annealing increases the close packing of NPs and therefore facilitates the charge carrier transport and extraction. They thus reached a PCE as high as 4.1%, not so far from the standard of the system having the same composition but deposited from toxic solvent solutions.
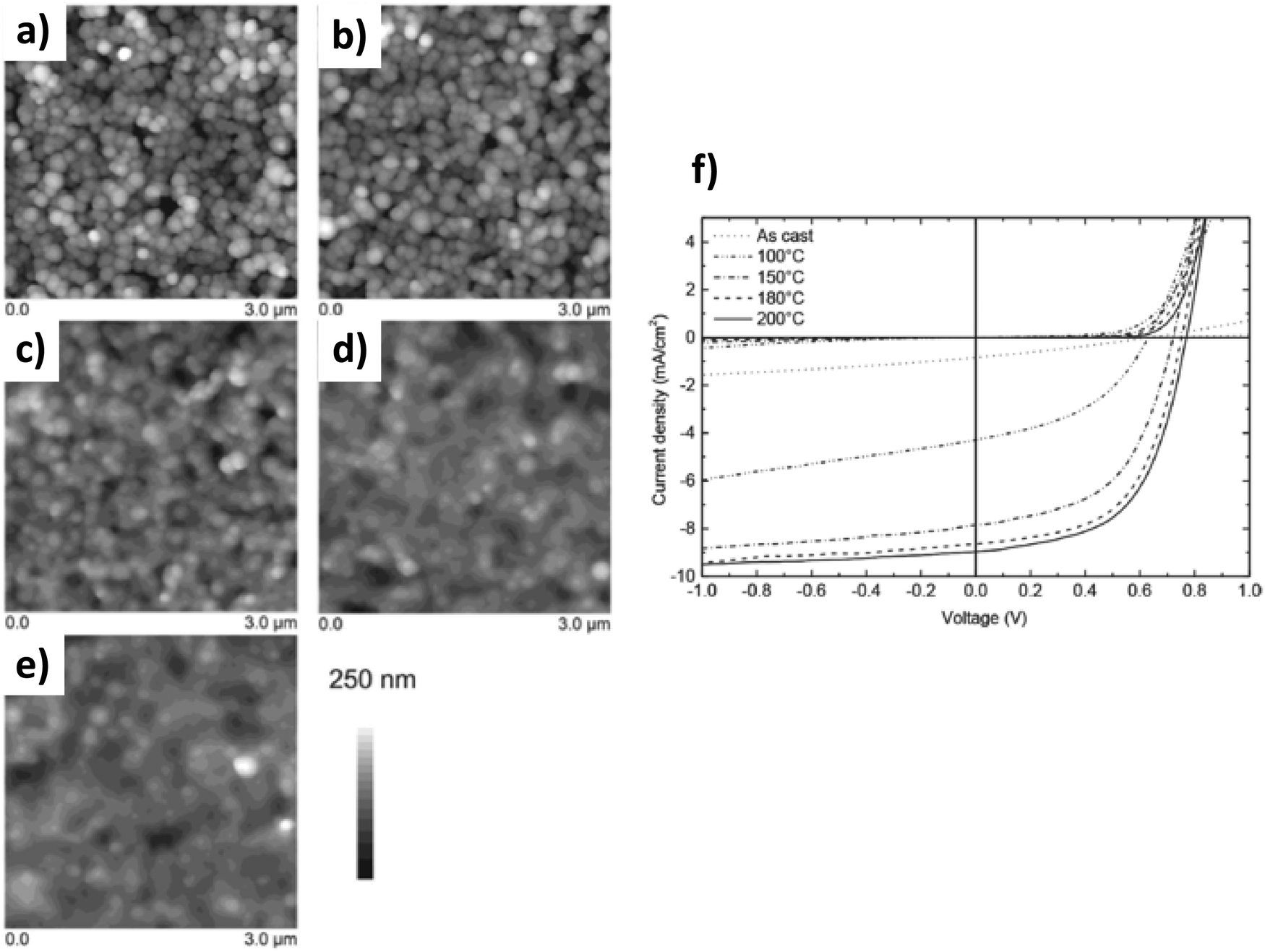 | ||
| Fig. 15 3 × 3 μm2 AFM images of the P3HT:ICBA nanoparticulate active layers spin coated from a 10 mg mL−1 dispersion in methanol: (a) as-cast photo-active layer; and (b) photo-active layer annealed for 10 min at 100 °C; (c) 150 °C; (d) 180 °C; and (e) 200 °C. The corresponding root mean square roughness Rq is (a) 43 nm, (b) 46 nm, (c) 30 nm, (d) 24 nm and (e) 23 nm, respectively. (f) Typical J–V curves of nanoparticulate solar cells with an inverted device architecture for different annealing temperatures. Adapted with permission from ref. 78 (Copyright 2018 Wiley-VCH Verlag). | ||
4. OPV cells
The development of an effective OPV device is not limited to the processing of a suitable photoactive thin film. Indeed, many chemical and physical parameters have to be taken into consideration, such as the donor:acceptor ratio, the interfacial layers, the type of electrodes or the nature of the optoelectronic components.
4.1. Donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :acceptor ratio
:acceptor ratio
A key parameter in OPVs is the optimum donor:acceptor (D:A) ratio to achieve the most ideal morphology, the right balance between charge carrier mobilities and the best PCE. This ratio usually depends mainly on the nature of the donor and acceptor components and, to a lesser extent, on the deposition parameters such as the solvent, the deposition temperature or the presence of additives. But very often this ratio evolves within a very narrow range of values. For example, when considering the P3HT:PC61BM binary blend, it is well established that a suitable ratio is around 1:0.8 by weight.58
Interestingly, a few articles show that this optimal ratio can be considerably modified when switching to active layers elaborated from NPs. This is particularly noticeable in the case of core–shell NPs, obtained by mini-emulsion.
Indeed, Holmes et al.25 investigated the OPV performances of core–shell NP-based thin films with varied D:A ratio. In particular, they highlight a PCE evolution, as a function of the PC61BM content, that is very different from the trend generally observed in P3HT:PC61BM BHJs. Indeed, with a high P3HT:PC61BM ratio ranging from 1:1 to 1:2, they observed a constant and relatively high PCE value, while it quickly decreases for a P3HT:PC61BM ratio below 1:1 and in particular for the observed 1:0.8 optimum ratio in standard BHJ solar cells. Using a combination of STXM and SEM microscopies, the authors observed that increasing the PC61BM proportion does not affect significantly the size of the PC61BM-rich core, which is still smaller than the exciton diffusion length, but it has a strong impact on the composition of the initially P3HT-rich shell, after thermal annealing. For the annealed 1:0.5 ratio, the joined shell phase consists of less than 20% PC61BM, which is below the minimum concentration required to achieve continuous PC61BM percolation pathways (percolation threshold) in the bulk film. When increasing the initial NP PC61BM content to a ratio over 1:1, the PC61BM shell fraction increases to values above 33%, approaching the optimum blend ratio required for balanced charge mobility. As such, they observed an uncommon increased PC61BM photocurrent contribution (up to 30%) for a P3HT:PC61BM blend ratio of 1:2.
By investigating two different binary systems, Dam et al.27 have demonstrated that the deviation from the optimal D:A ratio observed in BHJs can be directly correlated with the miscibility between the two components. Indeed, they investigated the morphology and device performance of NP-based OPV devices for two very different polymer:fullerene blends (i.e.P3HT:PC71BM and a more crystalline PSBTBT low band-gap (LBG) polymer blended with P71CBM, Fig. 16); the authors showed that the crystalline nature and the miscibility between (macro-)molecular components are key parameters to control the NP core and shell composition, called mesomorphology in their article. If both blends show similar core–shell NPs of comparable sizes (diameter around 24 nm) and a similar PC71BM-rich core volume of 20% of the NPs, STXM investigations allowed the authors to highlight the marked difference in core composition. Indeed, the core of the P3HT:PC71BM NPs is considerably richer in PCBM relative to that of the PSBTBT:PC71BM NPs (Fig. 16). Moreover, if the shell compositions are really close in both cases with a polymer fraction of around 0.7, this value for PSBTBT:PC71BM NPs is very far from the ideal composition of PSBTBT:PCBM blends (reported to be 0.4 in BHJs), whereas it is only 20% higher than the ideal case for P3HT:PCBM (0.56 in BHJs). Consequently, the authors suggest that the region in which charge generation occurs will be different in both cases. Thus, in the P3HT:PC71BM NPs, the charge generation is expected to be dominated by the shell region, while the opposite is true with the PSBTBT:PC71BM NPs. However, taking into account the relative volume proportion of the core and shell in these NPs (only 20% core in the NP volume), a much lower performance OPV device can be expected from the PSBTBT:PC71BM NPs. Indeed, if the two blends gave identical PCE values of 1.3%, the decrease in performance compared to the reference BHJ device is much greater in the PSBTBT:PC71BM NP case. Especially a Jsc divided by 2.6 has been measured in PSBTBT:PC71BM NPs compared to the same blend in BHJs, while it is divided by only 1.6 in the P3HT:PCBM case. These studies show that the D:A ratio can be tuned in order to optimize the shell composition of NPs.
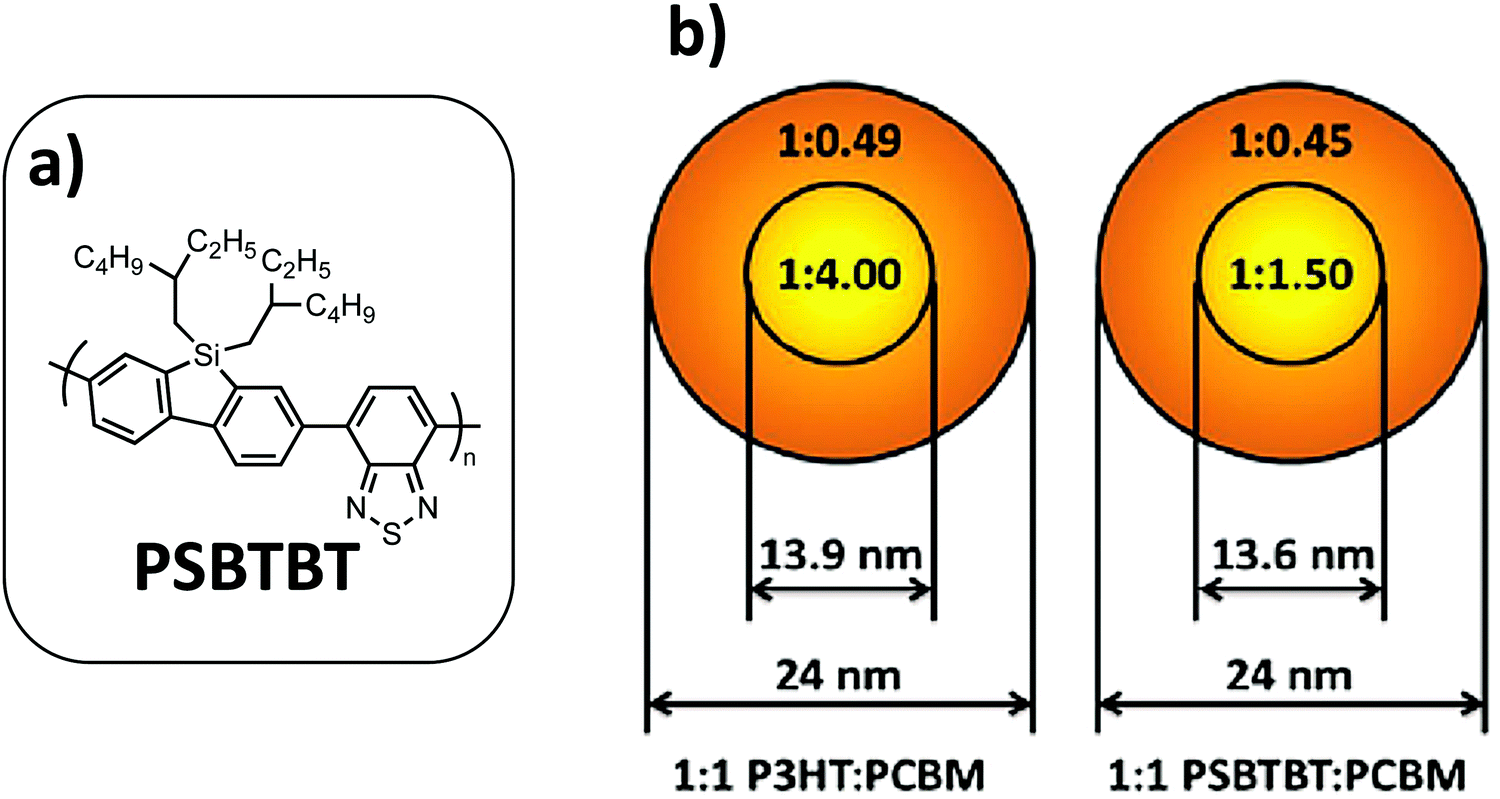 | ||
| Fig. 16 (a) Chemical structure of PSBTBT. (b) Schematic of small 1:1 P3HT:PC71BM and PSBTBT:PC71BM NPs showing the core and shell diameters and the polymer:PCBM ratio in the core and shell regions. Adapted with permission from ref. 27 (Copyright 2015 Elsevier B.V.). | ||
4.2. Vertical composition through the active layer
Interestingly, in addition to the manipulation of the D:A ratio, usually performed to optimize the power conversion efficiency of OPV devices, the colloid approach also enables one to finely tune the vertical composition of thin films, thanks to a multilayer deposition approach. It has been demonstrated for vapor deposited devices that a vertical gradient in donor concentration towards the anode and acceptor concentration towards the cathode was beneficial to limit charge recombination while driving the charge to the corresponding electrodes.79,80 However, the vertical morphology of a film processed from solutions is difficult to control and multilayer deposition is not possible without redissolving the previous layers. In the case of composite NPs of F8BT:PFB or P3HT:PC61BM, multilayered films (up to five layers) have been prepared by varying the composition of donor:acceptor NPs for each layer to 1:0, 2:1, 1:1, 1:2 and 0:1, thus improving the Voc in these devices.81 This sequential deposition strategy was also used by Gärtner et al.,53 who prepared new device architectures, introducing variable vertical composition (Fig. 17). Device A is a reference inverted device with ITO/ZnO used as a transparent electron-collecting electrode and PEDOT–PSS/Ag as a hole-collecting electrode. Device B includes a pristine P3HT NP layer on the top of the active layer (i.e. in contact with the hole-collecting electrode). Device C includes an additional ICBA layer below the active layer (i.e. on top of the hole-collecting electrode). Finally, device D is built to bring a favorable gradient composition in P3HT and in ICBA in the active-layer and a P3HT NP layer on the top of the active layer. The gradient in the active-layer has been introduced by synthesizing and depositing P3HT:ICBA NPs with different mixing ratios. Therefore, they sequentially applied P3HT:ICBA (1:3) NPs and P3HT:ICBA (1:1) NPs atop the ITO/ZnO cathode.
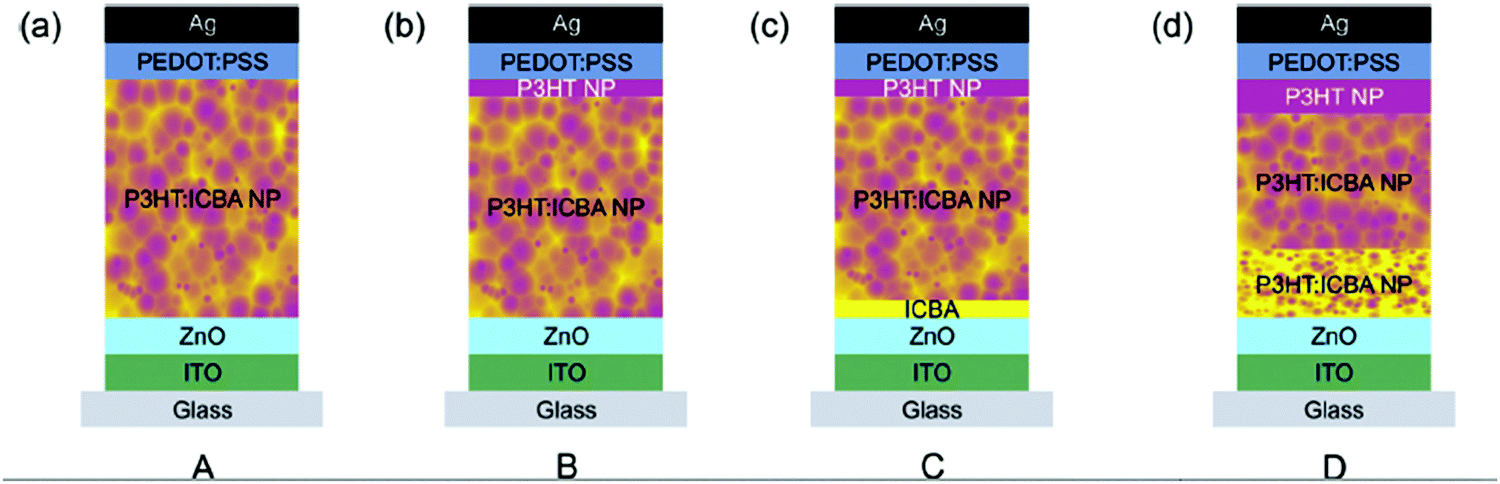 | ||
| Fig. 17 Device architectures: (a) reference device comprising an ITO/ZnO bottom cathode and a PEDOT:PSS/Ag top anode as well as a nanoparticulate P3HT:ICBA absorber layer. (b) Neat P3HT nanoparticles were introduced between the absorber layer and the PEDOT:PSS layer. The nanoparticulate P3HT top layer is not closed with its thickness of 15 nm representing the average layer thickness. The thickness of the entire absorber layer was reduced to match the overall amount of P3HT in configuration A. (c) A 15 nm nanoparticulate P3HT and a 20 nm ICBA interlayer were introduced. The overall amount of P3HT and ICBA approximately matches the reference device. (d) The vertical composition of the P3HT:ICBA layer was varied by subsequent deposition of nanoparticles with different mixing ratios ((1:3), (1:1), and neat P3HT). Adapted with permission from ref. 53 (Copyright 2016 The Royal Society of Chemistry). | ||
In order to keep similar photon harvesting properties, and to compare only the impact of the composition gradient on the OPV device performance, they used the same amount (mass) of photo-active P3HT, neglecting ICBA absorption and thin-film interference. By combining photovoltaic and light intensity dependent photocurrent measurements, the authors concluded that the vertical composition gradient introduced in devices B and C provides enhanced percolation paths for photo-generated charge carriers to the respective electrodes. The increased FF and Voc in these devices, with regards to the reference device A, combined with a FF that does not depend on the illumination intensity, support this hypothesis (see the discussion on mechanisms in Section 4.3). The PCE reaches 4.2% for both devices B and C, compared to a PCE of only 3.7% in device A. Device D exhibits unfortunately a lower PCE of only 3.9%, probably because of a thinner active layer, as confirmed by a significantly lower Jsc. However, no significant dependence of the FF on the illumination intensity between I = 250 W m−2 and I = 1000 W m−2 has been observed, highlighting again the probable enhanced percolation pathways for photo-generated charge carriers.
Such a multilayer approach paves the way towards tandem device development, which has already been demonstrated to be a promising approach for effectively managing the absorption properties of such OPV devices.82
4.3. Limiting mechanisms in the particular case of OPV devices made from NP dispersions
The most studied blend for OPV cells is based on an electron-donor polymer (P3HT) and an electron-acceptor small molecule (PC61BM).58 This reference blend has been therefore widely studied for NP solar cells, as we have seen throughout this manuscript. This blend is therefore a suitable reference for attempting to analyze the main factors limiting the efficiencies of OPV devices made from dispersions, in comparison with devices made from the same materials in solution. Two major mechanisms can hinder the OPV cell PCE. The first one is the free charge-carrier generation, which necessitates exciton dissociation, and the second one is free charge extraction. For both mechanisms, the specificity of NP active layers can be questioned. Further, separate P3HT and PC61BM NPs that are blended to form the active layer and intimate blends of P3HT and PC61BM in the same NP forming the active layer (composite NPs) can intuitively have different behaviors in terms of charge generation and charge extraction.In 2018, Al-Mudhaffer et al.83 used core–shell P3HT:PC61BM composite (1:1) NPs, prepared by miniemulsion, as the active layer of a direct structure solar cell (ITO/PEDOT–PSS/active layer/Ca/Al) and studied in detail the light-absorption and charge-extraction properties of their solar cells. They compare their NP solar cells with conventional bulk heterojunction (BHJ) solar cells ((P3HT:PC61BM) (1:0.8)) as a reference cell. On the one hand, a careful analysis of the optical properties of the cells led the authors to rule out plasmonic effects or light scattering by the nanoparticles as important mechanisms to explain different light absorption behavior. On the other hand, the spectral response of the OPV cells together with extraction of the different parameters to calculate the internal quantum efficiency led them to conclude that the main limiting parameter in their NP solar cells is the low exciton dissociation efficiency in that type of device, probably originating from the core–shell donor–acceptor morphologies in the active layer (Fig. 18).
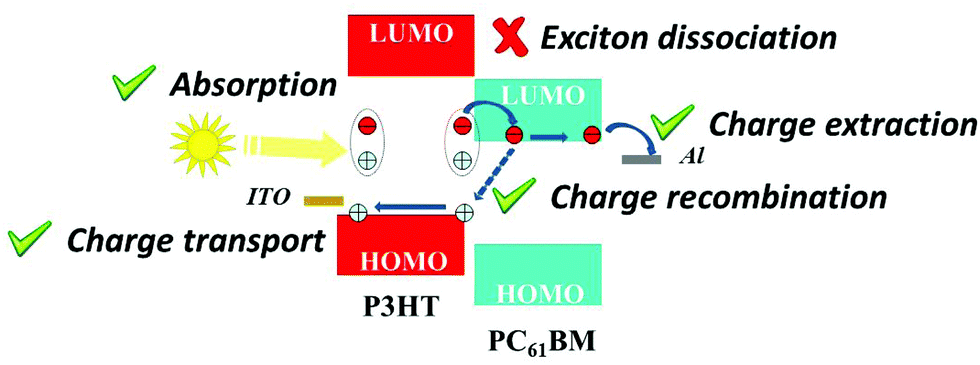 | ||
| Fig. 18 Schematic illustration of the different mechanisms leading to power conversion in BHJ and NP P3HT:PCBM solar cells. The exciton dissociation is a marked difference between BHJ and NP devices. Adapted with permission from ref. 83 (Copyright 2018 Elsevier B.V.). | ||
This conclusion is in contradiction with other reports on different systems. For instance, Gärtner et al.78 elaborated inverted solar cells (ITO/ZnO/active layer/MoO3/Al) based on composite NPs of P3HT and ICBA made by nanoprecipitation. They carefully investigated the photovoltaic parameters and the film morphology evolution as a function of thermal annealing treatments. They obtained, after an optimal annealing step, an efficiency comparable to BHJ solar cells elaborated from solution with similar architecture devices with, in particular, a high short-circuit current density (Jsc) and a high open-circuit voltage (Voc) but with negligible variations in the morphology of the film and in the crystallinity of the NPs during the thermal annealing. Their conclusion was that thermal annealing reduces the recombination of the free-charge carriers in NP devices but did not impact the free-charge generation, which was already efficient, even in non-annealed devices. Indeed, in the case of nanoprecipitation, composite NPs are uniform with segregated domains smaller than 10 nm. Consequently, the as deposited NP film morphology already resembles the ideal BHJ morphology with nanophase separation. With the same electron-donor polymer but using a non-fullerene acceptor (NFA), namely o-IDTBR (Fig. 10), Xie et al.49 elaborated inverted solar cells (ITO/ZnO/active layer/MoOx/Ag) with a composite NP active layer. Comparing NP devices with BHJ devices made of the same system, they extracted explicitly the free-charge carrier generation rate, which is equivalent in NP and BHJ solar cells. It seems therefore that using carefully elaborated NPs (and especially composite NPs) and optimized devices, the free-charge carrier generation via exciton dissociation is not the limiting factor in NP solar cells.
Once the free-charges are generated, the charge extraction may be problematic in NP solar cells. Different methods can be used to anticipate or to directly measure the charge-extraction efficiency. One of them is to measure the charge-carrier mobility.
Charge transport properties are a key concern in organic semiconducting materials for electronic applications and in particular for OPVs.84 The hole and electron mobilities should be high enough to use thick active layers, but they should also be well balanced to avoid internal electric field screening by space-charge zones. It is therefore crucial to check that charge transport occurs through a thin film obtained from NP dispersion deposition. Different devices can be used to probe the mobility, either along the substrate plane or in the out-of-plane direction. For example, Organic Field Effect Transistor (OFET) devices provide mobility values in the substrate plane,85 while Time Of Flight (TOF)86 devices or Space Charge Limited Current (SCLC)87 diodes allow one to estimate mobilities in a direction perpendicular to the electrodes (out-of-plane direction), which is the preferential direction of charge transport in multilayer OPV devices. However, one has to be careful when comparing mobility values extracted from different devices, using various architectures and elaborated in disparate conditions. For instance, OFET and TOF devices do not involve at all the same charge carrier density, which is much higher in OFET devices, nor the same active-layer thickness, which is much higher in TOF experiments. Such a difference usually results in a significantly higher OFET mobility value, except in the case of a very pronounced charge-transport anisotropy with a highly favored out-of-plane mobility for structural solid-state reasons.
Bag et al.61 estimated the hole transport mobilities in P3HT NP films using TOF measurements. They found hole mobilities on the same order of magnitude as for P3HT pristine thin films but the TOF measurements showed very dispersive characteristics. Decreasing the surfactant concentration led to less dispersive hole transport. A high surfactant concentration hindered the close packing of the NPs and increased the dispersive character of the hole transport in NP films. This behavior was further observed and explained by Han et al.,88 who analyzed their TOF measurements in the frame of a conventional set of diffusion and drift equations. The authors showed that the presence of surfactants in P3HT NP films increased the trap concentration as well as the trapping rate for charge carriers (in agreement with the observed dispersive hole transport). They get one step further in their analysis, showing that the detrapping rate in the presence of surfactants was really high, meaning that the surfactant trap state is associated with shallow trapping energy levels and low activation energy barriers so that the trapped holes can be easily detrapped to contribute to the hole current. The hole current is therefore more dispersive in the presence of surfactants and one order of magnitude lower than for surfactant-free NP films or drop-cast P3HT films, but the hole transport is not completely hindered in surfactant P3HT NP films. Another important conclusion of this study was that for drop-cast P3HT films or P3HT NPs where the NPs contained or not a high concentration of surfactants, the energetic or positional disorder was in the same range. All the conclusions drawn here are for TOF studies in P3HT NP films only and the situation can be very different in the active layer of solar cells.
A few publications are devoted to the comparison of the charge carrier transport properties measured by TOF of P3HT:PC61BM thin films made either from separate NPs or composite NPs.68,89 Gehan et al. showed the existence of conductive pathways for holes through the bulk for separate as for composite NP films but with clearly more short pathways with low resistance for separate NP films. The TOF hole mobility was slightly lower in separate P3HT:PC61BM (1:1) NP films (8 × 10−5 cm2 (V s)−1) than in pure P3HT NP films (2 × 10−4 cm2 (V s)−1) with in both cases a weak dependence with the electric field. In composite NP films, even though the hole mobility at low electric field was comparable to the P3HT NP film one, a marked decrease of the hole mobility was observed at high electric field. Such a behavior is in agreement with an increase of the positional disorder in composite NP films.90 The very same conclusion was made by Han et al.89 They get even one step further, connecting the charge trap-site density to the charge transport pathway lengths. In composite NP films, each NP contains electron-transporting and hole-transporting materials, leading to continuous charge transport pathways with shorter charge hopping distances. In separate NP films, longer and more tortuous pathways for charge transport are anticipated. The charge-carrier trap-site density depends closely on the surfactant presence in the prepared NPs, highlighting the importance of surfactant removal strategies to obtain good charge-transport properties in NP films. In these two publications,68,89 a strong emphasis was put on device engineering for efficient photovoltaic devices based on NPs. In particular, the authors insisted on the influence of interfacial layers (electron transporting layers or ETLs and/or hole transporting layers or HTLs) to promote charge extraction. The authors highlighted the beneficial use of an ETL (a PC61BM layer) on top of the active layer and before the electron-collecting electrode deposition in a direct structure. This is also true for inverted structures and charge dynamics analysis on OPV cells is only valid if it is performed on optimized solar cells. A first rough estimate of the charge-carrier dynamics consists of studying the photovoltaic parameter variation as a function of light intensity. Gärtner et al.78 showed that for P3HT:ICBA NP solar cells in an inverted structure (ITO/ZnO/active layer/MoO3/Al), Jsc in thermally annealed optimized devices varied almost linearly with the light intensity. This behavior indicates the predominance of monomolecular charge-carrier recombination at high electric field (short-circuit conditions) as expected in efficient OPV systems where bimolecular recombination does not hinder charge-extraction.91 For a very similar system, Gärtner et al.53 observed a strong drop of the FF when the light intensity increases, showing that when the electric field decreases, the bimolecular recombination mechanism became more important. Further, Gärtner et al. changed the active-layer configuration to vary the percolation pathways toward the electrodes and favor charge extraction. They indeed showed that this strategy lowered the bimolecular recombination at low electric-field and ultimately improved the power conversion efficiency. The only way to efficiently study the charge-carrier dynamics is nevertheless a combination of transient photovoltage (TPV) and charge extraction (CE) as performed by Xie et al.49 on efficient OPV NP blends. Optimizing the NP elaboration as well as the device fabrication, they obtained a more than 5% efficiency with a composite NP active layer with P3HT and o-IDTBR. The measured recombination order (R) in the optimized NPs system is higher than two (R = 2.89) indicating the remaining presence of charge-carrier trap sites. However, it approaches the R measured for optimized (P3HT:o-IDTBR) BHJ solar-cells (R = 2.1) showing that efficient organic photovoltaic cells that are not limited by charge transport and extraction can be processed from NPs. This conclusion is confirmed by the high PCEs measured by the same authors on different systems and described below.
4.4. Device optimization through chemical selection
The recent progress observed in BHJ OPVs is mainly due to the development of device architectures (interlayers, electrodes, coating process, etc.) and photoactive materials. As seen in previous sections, due to their commercial availability and moderate costs, P3HT and PC61BM continue to be considered as reference systems for in-depth physico-chemical and physical studies on the structural and electrical properties of OPV active layers.58,96 However, their intrinsic optical and electronic properties limit the performance of OPV solar cells. One easy and known strategy to achieve higher efficiencies is to replace PC61BM with a fullerene bisadduct called ICBA. The ICBA derivative has indeed a LUMO level about 0.1–0.2 eV higher than PCBM, which improves Voc, and is also more soluble in organic solvents.97 Using this P3HT:ICBA binary system, Ulum et al.28 early described the elaboration of core–shell NPs with an ICBA-rich core and a P3HT-rich shell. Interestingly, as seen previously, ICBA being more miscible in P3HT than PC61BM, at all weight fractions they showed that a standard thermal annealing step led to a more blended morphology. Indeed, upon thermal annealing, the core–shell structure is lost, the particles merge together and the ICBA migrates from the core to the shell (Fig. 19). Further STXM observations support this scenario. This blended morphology provides enhanced charge carrier pathways and results in an improved FF after thermal annealing (around 55% against 35% for unannealed thin films). They thus recorded a best PCE of approximately 2.5% with an expected improved Voc of almost 800 mV. Later, using the same blend and optimizing both the device structure and active layer deposition process, a few teams reported PCEs more in line with the reference values obtained for BHJs, in the range of 4 to 4.5%.42,53,78 It should be noticed that in 2016 Sankaran et al.60 reported 1.1 cm2 active area solar cells deposited by doctor blading from the P3HT:ICBA binary system with a PCE of 3.4%, thereby showing the benefits of this approach in terms of process (see Table 2).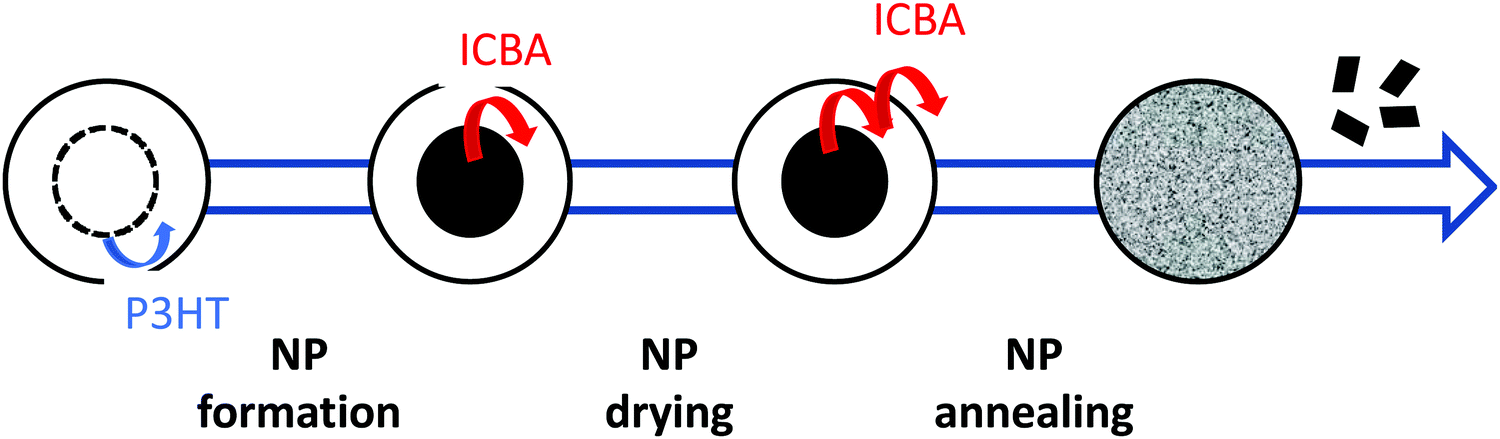 | ||
| Fig. 19 Schematic model of NP evolution during elaboration and thermal treatment. During NP formation, P3HT tends to migrate toward the shell due to its lower surface energy. A core–shell NP is obtained with a core containing 41% ICBA and a shell containing only 23% ICBA. After drying of the NPs (110 °C for 4 min), the ICBA shell composition increases to 34%, while the core composition remains the same. Finally, during thermal annealing at 150 °C for 15 min, ICBA continues to migrate toward the surface of the particles, resulting in homogeneous NPs with an average ICBA composition of 32%. Part of the ICBA is also lost from the NPs during annealing and small crystallites can be observed outside the NPs. Reprinted with permission from ref. 28 (Copyright 2018 Elsevier B.V.). | ||
Although progress has been made through the use of this ICBA fullerene derivative, the PCEs remain far from the standards nowadays measured in conventional BHJs. Indeed, the OPV field experienced a very recent and rapid development of photoactive materials with properties increasingly more appropriate for photon harvesting and charge carrier transport, such as low band-gap (LBG) polymers5,98 and non-fullerene acceptors (NFA).99 However, these materials, particularly the LBG polymers, generally have a much lower solubility than the reference P3HT.6,100 Obviously, this makes their dissolution tricky in low-boiling organic solvents. Consequently, NP synthesis and stable dispersion elaboration are challenging. This probably explains why very few studies have been done on LBG polymer NPs.
However, although exhibiting very low PCEs, it could be noticed that very early on, many groups studied other conjugated polymers, such as the PFB fluorene derivative previously discussed.18,59,70
Another extensively studied building-block for OPV application is diketopyrrolopyrrole (DPP). It exhibits indeed extraordinary charge absorption and transport properties associated with good solubility due to the presence of two alkyl side-chains.101 In this regard, it appears to be a scaffold of choice for preparing NP dispersions. Therefore, several studies have used DPP in combination with different comonomers to prepare NP based OPV devices (see the PDPP5T and PDPP-TNT structures in Fig. 20 and 14, respectively).75,93 However, the best PCE reported recently by Xie et al.95 is limited to 3.5%. Although promising, this work also highlights a limitation commonly encountered in NP-based devices, non-geminate recombination due to lower charge carrier mobilities and higher densities of traps (see Section 4.3 on limiting mechanisms).
 | ||
| Fig. 20 Chemical structures of diketyrrolopyrrole (DPP) and PDPP5T, and of benzodithiophene (BDT) and PBDTTPD. | ||
Recently, D’Olieslaeger et al.15 used another well-known building-block in OPVs, benzodithiophene (BDT).102 They described the fabrication of organic solar cells from mixed NPs elaborated by miniemulsion, using a mixture of PBDTTPD, a moderate band-gap BDT-based polymer (Fig. 20), and PC71BM, which reached best PCEs of 3.8% after a thermal annealing step. As already demonstrated on the reference P3HT:PC61BM system, this thermal annealing step, whose characteristics (time and temperature) must be adapted to each binary blend, has the effect of melting particles into a continuous interconnected film with enhanced continuous pathways for holes and electrons.
As seen in all the previously discussed examples, the NP-based BHJ approach is highly appealing but still suffers from a number of limitations: (i) in the miniemulsion case, the presence of surfactant and the predominantly core–shell nature of the synthesized NPs limit the charge carrier transport and extraction; and (ii) in the nanoprecipitation case, the control of the NP size is difficult and the resulting dispersions lack stability.
In this context, Xie et al.49 recently reported a very promising and innovative NP synthesis approach called the surfactant assisted nanoprecipitation technique as described in Section 2.2. This concept based on a non-ionic surfactant Pluronic F127 with temperature sensitive critical micelle concentration (cmc) was demonstrated to stabilize NPs. After preparation, surfactant stripping from the NPs was facilitated by its increased solubility at low temperature. This highly versatile approach enables the synthesis of high purity light-harvesting NPs by minimizing the amount of residual surfactant in the aqueous system. Consequently, in NP films with low surfactant amounts, electron donor polymers are able to reorganize and exhibit higher crystallinities and charge-carrier mobilities. In addition, the versatility of this approach is highlighted by the authors, who successfully obtained several composite polymer:NFA NPs. Thus, a champion PCE of 5.23% with a high FF of 65% was achieved for P3HT:o-IDTBR NP solar cells, being comparable to the devices processed from halogenated solvents. Moreover, NP-based devices using the low-bandgap polymer PBQ-QF (Fig. 10) further boosted the record PCE of water/alcohol-processed OPVs up to 7.5%. However, it is important to notice that this approach requires the use of organic semiconductor materials that are soluble in THF, which is not the case for most of them. Nevertheless, by overcoming the charge transport and recombination limits usually experienced in NP-based devices with traditional ionic surfactants, this approach paves the way towards a more systematic use of water-based dispersions in the OPV field.
5. Conclusion
In this article, we reviewed the NP dispersion strategy to elaborate the photoactive layer of bulk heterojunction (BHJ) organic solar cells. The main aim of using NPs in that case is to avoid the use of toxic solvents while still controlling the morphology at a nanometer scale.Miniemulsion and nanoprecipitation are the main strategies to produce single or composite organic semiconducting NPs (containing both the electron-donor and the electron-acceptor material). It has been shown that the internal morphology of the NPs depends greatly on the used technique. While the miniemulsion technique seems more versatile and produces stable NP dispersions, it leads more frequently to composite core–shell NPs. Moreover, removal of the surfactant is necessary to improve the active layer homogeneity and the OPV efficiency. In the case of nanoprecipitation, the two semi-conducting materials are better mixed with smaller domains inside the NP, their morphology being closer to the one of the active layers obtained by solvent casting. However, the NPs are less stable due to the absence of surfactant. To solve this problem, surfactant assisted nanoprecipitation was recently developed and appeared as a very promising route leading to a record PCE of 7.3%.49 Further, the morphology of the active layer obtained by the assembly of the synthesized NPs has also been discussed thoroughly as well as its influence on the charge-carrier dynamics. Specifically, the exciton dissociation efficiency and the charge-carrier transport and collection properties have been described in light of the specific photoactive-layer morphologies obtained from NP inks. Finally, different photovoltaic parameters including the Power Conversion Efficiency (PCE) of NP devices have been analyzed.
Research on OPV device elaboration from NP dispersions has made significant progress in the last decade and the PCE values reached by this approach are nowadays compatible with industrial criteria. Moreover, using NPs for the elaboration of the active layer of BHJ solar cells appeared to be an efficient way to finely tune the active-layer morphology. Further, it opens the way to interesting concepts like controlled vertical composition gradients in BHJ solar cells, which may lead to real breakthroughs in terms of efficiency. However, a lot of effort is still needed to get closer to the performance standards of solution processed organic solar cells. In particular, we believe that customized materials can be developed in order to better adapt the physico-chemical properties of organic semiconductors to the constraints of NP elaboration. In particular, the use of polar side chains would help to make such semiconducting materials more soluble and simultaneously organic semiconductor-based nanoparticles more stable in alcohol or water. Interestingly, driven by other applications like thermoelectricity, water photocatalysis, electrochemical energy storage or bioelectronics,103 such conjugated materials are currently undergoing significant development.104–106
We are therefore convinced that the NP approach is a major step not only toward the industrialization of organic photovoltaics, but also towards the use of these semiconducting polymer materials in many other applications, as illustrated recently by Kosco et al.107 in photocatalytic hydrogen production.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the French “Agence Nationale de la Recherche” for financial support through project FORCE ANR-17-CE09-0012.References
- V. Muteri, M. Cellura, D. Curto, V. Franzitta, S. Longo, M. Mistretta and M. L. Parisi, Review on life cycle assessment of solar photovoltaic panels, Energies, 2020, 13, 252 CrossRef CAS.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, 18% Efficiency organic solar cells, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- C. L. Chochos, M. Spanos, A. Katsouras, E. Tatsi, S. Drakopoulou, V. G. Gregoriou and A. Avgeropoulos, Current status, challenges and future outlook of high performance polymer semiconductors for organic photovoltaics modules, Prog. Polym. Sci., 2019, 91, 51–79 CrossRef CAS.
- R. Po, G. Bianchi, C. Carbonera and A. Pellegrino, “All That Glisters Is Not Gold”: An Analysis of the Synthetic Complexity of Efficient Polymer Donors for Polymer Solar Cells, Macromolecules, 2015, 48, 453–461 CrossRef CAS.
- N. Leclerc, P. Chávez, O. A. Ibraikulov, T. Heiser and P. Lévêque, Impact of Backbone Fluorination on π-Conjugated Polymers in Organic Photovoltaic Devices: A Review, Polymers, 2016, 8, 11 CrossRef PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Aggregation and morphology control enables multiple cases of high-efficiency polymer solar cells, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- S. Zhang, L. Ye, H. Zhang and J. Hou, Green-solvent-processable organic solar cells, Mater. Today, 2016, 19, 533–543 CrossRef CAS.
- C. Duan, K. Zhang, C. Zhong, F. Huang and Y. Cao, Recent advances in water/alcohol-soluble π-conjugated materials: new materials and growing applications in solar cells, Chem. Soc. Rev., 2013, 42, 9071–9104 RSC.
- Z. Ma, B. Zhao, Y. Gong, J. Deng and Z. Tan, Green-solvent-processable strategies for achieving large-scale manufacture of organic photovoltaics, J. Mater. Chem. A, 2019, 7, 22826–22847 RSC.
- S. Subianto, N. Dutta, M. Andersson and N. R. Choudhury, Bulk heterojunction organic photovoltaics from water-processable nanomaterials and their facile fabrication approaches, Adv. Colloid Interface Sci., 2016, 235, 56–69 CrossRef CAS PubMed.
- J. Pecher and S. Mecking, Nanoparticles of Conjugated Polymers, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- Z. Tian, J. Yu, C. Wu, C. Szymanski and J. McNeill, Amplified energy transfer in conjugated polymer nanoparticle tags and sensors, Nanoscale, 2010, 2, 1999 RSC.
- K. Landfester, R. Montenegro, U. Scherf, R. Güntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Semiconducting Polymer Nanospheres in Aqueous Dispersion Prepared by a Miniemulsion Process, Adv. Mater., 2002, 14, 651–655 CrossRef CAS.
- L. Parrenin, G. Laurans, E. Pavlopoulou, G. Fleury, G. Pecastaings, C. Brochon, L. Vignau, G. Hadziioannou and E. Cloutet, Photoactive Donor–Acceptor Composite Nanoparticles Dispersed in Water, Langmuir, 2017, 33, 1507–1515 CrossRef CAS PubMed.
- L. D’Olieslaeger, M. Pfannmöller, E. Fron, I. Cardinaletti, M. Van Der Auweraer, G. Van Tendeloo, S. Bals, W. Maes, D. Vanderzande, J. Manca and A. Ethirajan, Tuning of PCDTBT:PC71BM blend nanoparticles for eco-friendly processing of polymer solar cells, Sol. Energy Mater. Sol. Cells, 2017, 159, 179–188 CrossRef.
- X. Pan, A. Sharma, D. Gedefaw, R. Kroon, A. Diaz de Zerio, N. P. Holmes, A. L. D. Kilcoyne, M. G. Barr, A. Fahy, M. Marks, X. Zhou, W. Belcher, P. C. Dastoor and M. R. Andersson, Environmentally friendly preparation of nanoparticles for organic photovoltaics, Org. Electron., 2018, 59, 432–440 CrossRef CAS.
- O. Ghazy, Preparation and Characterization of P3H:PCBM Blend Nanoparticles and Their Plasmonic Modification, Macromol. Symp., 2015, 352, 25–32 CrossRef CAS.
- T. Kietzke, D. Neher, K. Landfester, R. Montenegro, R. Güntner and U. Scherf, Novel approaches to polymer blends based on polymer nanoparticles, Nat. Mater., 2003, 2, 408–412 CrossRef CAS PubMed.
- N. P. Holmes, K. B. Burke, P. Sista, M. Barr, H. D. Magurudeniya, M. C. Stefan, A. L. D. Kilcoyne, X. Zhou, P. C. Dastoor and W. J. Belcher, Nano-domain behaviour in P3HT:PCBM nanoparticles, relating material properties to morphological changes, Sol. Energy Mater. Sol. Cells, 2013, 117, 437–445 CAS.
- T. Kietzke, D. Neher, M. Kumke, O. Ghazy, U. Ziener and K. Landfester, Phase Separation of Binary Blends in Polymer Nanoparticles, Small, 2007, 3, 1041–1048 CrossRef CAS PubMed.
- J. J. Richards, C. L. Whittle, G. Shao and L. D. Pozzo, Correlating Structure and Photocurrent for Composite Semiconducting Nanoparticles with Contrast Variation Small-Angle Neutron Scattering and Photoconductive Atomic Force Microscopy, ACS Nano, 2014, 8, 4313–4324 CAS.
- K. B. Burke, A. J. Stapleton, B. Vaughan, X. Zhou, A. L. D. Kilcoyne, W. J. Belcher and P. C. Dastoor, Scanning transmission x-ray microscopy of polymer nanoparticles: probing morphology on sub-10 nm length scales, Nanotechnology, 2011, 22, 265710 CrossRef PubMed.
- C. M. Björström, A. Bernasik, J. Rysz, A. Budkowski, S. Nilsson, M. Svensson, M. R. Andersson, K. O. Magnusson and E. Moons, Multilayer formation in spin-coated thin films of low-bandgap polyfluorene:PCBM blends, J. Phys.: Condens. Matter, 2005, 17, L529–L534 CrossRef.
- X. Wang, T. Ederth and O. Inganäs, In Situ Wilhelmy Balance Surface Energy Determination of Poly(3-hexylthiophene) and Poly(3,4-ethylenedioxythiophene) during Electrochemical Doping–Dedoping, Langmuir, 2006, 22, 9287–9294 CrossRef CAS PubMed.
- N. P. Holmes, N. Nicolaidis, K. Feron, M. Barr, K. B. Burke, M. Al-Mudhaffer, P. Sista, A. L. D. Kilcoyne, M. C. Stefan, X. Zhou, P. C. Dastoor and W. J. Belcher, Probing the origin of photocurrent in nanoparticulate organic photovoltaics, Sol. Energy Mater. Sol. Cells, 2015, 140, 412–421 CrossRef CAS.
- K. Vakhshouri, D. R. Kozub, C. Wang, A. Salleo and E. D. Gomez, Effect of Miscibility and Percolation on Electron Transport in Amorphous Poly(3-Hexylthiophene)/Phenyl-C61-Butyric Acid Methyl Ester Blends, Phys. Rev. Lett., 2012, 108, 026601 CrossRef PubMed.
- H. F. Dam, N. P. Holmes, T. R. Andersen, T. T. Larsen-Olsen, M. Barr, A. L. D. Kilcoyne, X. Zhou, P. C. Dastoor, F. C. Krebs and W. J. Belcher, The effect of mesomorphology upon the performance of nanoparticulate organic photovoltaic devices, Sol. Energy Mater. Sol. Cells, 2015, 138, 102–108 CAS.
- S. Ulum, N. Holmes, M. Barr, A. L. D. Kilcoyne, B. B. Gong, X. Zhou, W. Belcher and P. Dastoor, The role of miscibility in polymer:fullerene nanoparticulate organic photovoltaic devices, Nano Energy, 2013, 2, 897–905 CAS.
- G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, Tuning Aggregation of Poly(3-hexylthiophene) within Nanoparticles, ACS Nano, 2012, 6, 10750–10758 CrossRef CAS PubMed.
- M. Marks, N. P. Holmes, A. Sharma, X. Pan, R. Chowdhury, M. G. Barr, C. Fenn, M. J. Griffith, K. Feron, A. L. D. Kilcoyne, D. A. Lewis, M. R. Andersson, W. J. Belcher and P. C. Dastoor, Building intermixed donor–acceptor architectures for water-processable organic photovoltaics, Phys. Chem. Chem. Phys., 2019, 21, 5705–5715 RSC.
- F. Almyahi, T. R. Andersen, N. A. Cooling, N. P. Holmes, M. J. Griffith, K. Feron, X. Zhou, W. J. Belcher and P. C. Dastoor, Optimisation of purification techniques for the preparation of large-volume aqueous solar nanoparticle inks for organic photovoltaics, Beilstein J. Nanotechnol., 2018, 9, 649–659 CrossRef CAS PubMed.
- J. Cho, S. Yoon, K. M. Sim, Y. J. Jeong, C. E. Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, Universal selection rule for surfactants used in miniemulsion processes for eco-friendly and high performance polymer semiconductors, Energy Environ. Sci., 2017, 10, 2324–2333 RSC.
- J. Cho, K. H. Cheon, H. Ahn, K. H. Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, High Charge-Carrier Mobility of 2.5 cm2 V−1 s−1 from a Water-Borne Colloid of a Polymeric Semiconductor via Smart Surfactant Engineering, Adv. Mater., 2015, 27, 5587–5592 CrossRef CAS PubMed.
- B. Tan, Y. Li, M. F. Palacios, J. Therrien and M. J. Sobkowicz, Effect of surfactant conjugation on structure and properties of poly(3-hexylthiophene) colloids and field effect transistors, Colloids Surf., A, 2016, 488, 7–14 CrossRef CAS.
- F. D. Fleischli, N. Ghasdian, T. K. Georgiou and N. Stingelin, Tailoring the optical properties of poly(3-hexylthiophene) by emulsion processing using polymeric macrosurfactants, J. Mater. Chem. C, 2015, 3, 2065–2071 RSC.
- Y. J. Kim and B. Lee, Unique p–n Heterostructured Water-Borne Nanoparticles Exhibiting Impressive Charge-Separation Ability, ChemSusChem, 2018, 11, 1628–1638 CAS.
- Y. J. Kim, R. D. Schaller and H. C. Fry, Control of Shell Morphology in p–n Heterostructured Water-Processable Semiconductor Colloids: Toward Extremely Efficient Charge Separation, Small, 2019, 15, 1803563 CrossRef PubMed.
- S. Subianto, R. Balu, L. de Campo, A. Sokolova, N. K. Dutta and N. R. Choudhury, Sulfonated Thiophene Derivative Stabilized Aqueous Poly(3-hexylthiophene):Phenyl-C61-butyric Acid Methyl Ester Nanoparticle Dispersion for Organic Solar Cell Applications, ACS Appl. Mater. Interfaces, 2018, 10, 44116–44125 CrossRef CAS PubMed.
- D. Horn and J. Rieger, Organic nanoparticles in the aqueous phase—theory, experiment, and use, Angew. Chem., Int. Ed., 2001, 40, 4330–4361 CrossRef CAS PubMed.
- F. Wang, M.-Y. Han, K. Y. Mya, Y. Wang and Y.-H. Lai, Aggregation-Driven Growth of Size-Tunable Organic Nanoparticles Using Electronically Altered Conjugated Polymers, J. Am. Chem. Soc., 2005, 127, 10350–10355 CAS.
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Synthesis, Properties, and Electronic Applications of Size-Controlled Poly(3-hexylthiophene) Nanoparticles, Langmuir, 2010, 26, 13056–13061 CrossRef CAS PubMed.
- C. Xie, X. Tang, M. Berlinghof, S. Langner, S. Chen, A. Späth, N. Li, R. H. Fink, T. Unruh and C. J. Brabec, Robot-Based High-Throughput Engineering of Alcoholic Polymer: Fullerene Nanoparticle Inks for an Eco-Friendly Processing of Organic Solar Cells, ACS Appl. Mater. Interfaces, 2018, 10, 23225–23234 CrossRef CAS PubMed.
- G. Prunet, L. Parrenin, E. Pavlopoulou, G. Pecastaings, C. Brochon, G. Hadziioannou and E. Cloutet, Aqueous PCDTBT:PC(71)BM Photovoltaic Inks Made by Nanoprecipitation, Macromol. Rapid Commun., 2017, 1700504 Search PubMed.
- S. N. Clafton, D. M. Huang, W. R. Massey and T. W. Kee, Femtosecond Dynamics of Excitons and Hole-Polarons in Composite P3HT/PCBM Nanoparticles, J. Phys. Chem. B, 2013, 117, 4626–4633 CrossRef CAS PubMed.
- K. N. Schwarz, S. B. Farley, T. A. Smith and K. P. Ghiggino, Charge generation and morphology in P3HT:PCBM nanoparticles prepared by mini-emulsion and reprecipitation methods, Nanoscale, 2015, 7, 19899–19904 RSC.
- S. Gärtner, A. J. Clulow, I. A. Howard, E. P. Gilbert, P. L. Burn, I. R. Gentle and A. Colsmann, Relating Structure to Efficiency in Surfactant-Free Polymer/Fullerene Nanoparticle-Based Organic Solar Cells, ACS Appl. Mater. Interfaces, 2017, 9, 42986–42995 CrossRef PubMed.
- S. Chambon, C. Schatz, V. Sébire, B. Pavageau, G. Wantz and L. Hirsch, Organic semiconductor core–shell nanoparticles designed through successive solvent displacements, Mater. Horiz., 2014, 1, 431–438 RSC.
- A. Palacio Valera, C. Schatz, E. Ibarboure, T. Kubo, H. Segawa and S. Chambon, Elaboration of PCBM Coated P3HT Nanoparticles: Understanding the Shell Formation, Front. Energy Res., 2019, 6, 146 CrossRef.
- C. Xie, T. Heumüller, W. Gruber, X. Tang, A. Classen, I. Schuldes, M. Bidwell, A. Späth, R. H. Fink, T. Unruh, I. McCulloch, N. Li and C. J. Brabec, Overcoming efficiency and stability limits in water-processing nanoparticular organic photovoltaics by minimizing microstructure defects, Nat. Commun., 2018, 9, 5335 CrossRef CAS PubMed.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, High-efficiency and air-stable P3HT-based polymer solar cells with a new non-fullerene acceptor, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- M. Bag, T. S. Gehan, L. A. Renna, D. D. Algaier, P. M. Lahti and D. Venkataraman, Fabrication conditions for efficient organic photovoltaic cells from aqueous dispersions of nanoparticles, RSC Adv., 2014, 4, 45325–45331 RSC.
- Y. He, H.-Y. Chen, J. Hou and Y. Li, Indene–C60 Bisadduct: A New Acceptor for High-Performance Polymer Solar Cells, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- S. Gärtner, S. Reich, M. Bruns, J. Czolk and A. Colsmann, Organic solar cells with graded absorber layers processed from nanoparticle dispersions, Nanoscale, 2016, 8, 6721–6727 RSC.
- F. Almyahi, T. R. Andersen, A. Fahy, M. Dickinson, K. Feron, W. J. Belcher and P. C. Dastoor, The role of surface energy control in organic photovoltaics based on solar paints, J. Mater. Chem. A, 2019, 7, 9202–9214 RSC.
- J. A. Bartelt, J. D. Douglas, W. R. Mateker, A. E. Labban, C. J. Tassone, M. F. Toney, J. M. J. Fréchet, P. M. Beaujuge and M. D. McGehee, Controlling Solution-Phase Polymer Aggregation with Molecular Weight and Solvent Additives to Optimize Polymer-Fullerene Bulk Heterojunction Solar Cells, Adv. Energy Mater., 2014, 4, 1301733 CrossRef.
- S. F. Hoefler, T. Rath, N. Pastukhova, E. Pavlica, D. Scheunemann, S. Wilken, B. Kunert, R. Resel, M. Hobisch, S. Xiao, G. Bratina and G. Trimmel, The effect of polymer molecular weight on the performance of PTB7-Th:O-IDTBR non-fullerene organic solar cells, J. Mater. Chem. A, 2018, 6, 9506–9516 RSC.
- Z. Zheng, O. M. Awartani, B. Gautam, D. Liu, Y. Qin, W. Li, A. Bataller, K. Gundogdu, H. Ade and J. Hou, Efficient Charge Transfer and Fine-Tuned Energy Level Alignment in a THF-Processed Fullerene-Free Organic Solar Cell with 11.3% Efficiency, Adv. Mater., 2017, 29, 1604241 CrossRef PubMed.
- M. T. Dang, L. Hirsch and G. Wantz, P3HT:PCBM, Best Seller in Polymer Photovoltaic Research, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS PubMed.
- A. Stapleton, B. Vaughan, B. Xue, E. Sesa, K. Burke, X. Zhou, G. Bryant, O. Werzer, A. Nelson, A. L. David Kilcoyne, L. Thomsen, E. Wanless, W. Belcher and P. Dastoor, A multilayered approach to polyfluorene water-based organic photovoltaics, Sol. Energy Mater. Sol. Cells, 2012, 102, 114–124 CrossRef CAS.
- S. Sankaran, K. Glaser, S. Gärtner, T. Rödlmeier, K. Sudau, G. Hernandez-Sosa and A. Colsmann, Fabrication of polymer solar cells from organic nanoparticle dispersions by doctor blading or ink-jet printing, Org. Electron., 2016, 28, 118–122 CrossRef CAS.
- M. Bag, T. S. Gehan, D. D. Algaier, F. Liu, G. Nagarjuna, P. M. Lahti, T. P. Russell and D. Venkataraman, Efficient Charge Transport in Assemblies of Surfactant-Stabilized Semiconducting Nanoparticles, Adv. Mater., 2013, 25, 6411–6415 CrossRef CAS PubMed.
- S. K. Karunakaran, G. M. Arumugam, W. Yang, S. Ge, S. N. Khan, X. Lin and G. Yang, Recent progress in inkjet-printed solar cells, J. Mater. Chem. A, 2019, 7, 13873–13902 RSC.
- T. R. Andersen, T. T. Larsen-Olsen, B. Andreasen, A. P. L. Böttiger, J. E. Carlé, M. Helgesen, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jørgensen and F. C. Krebs, Aqueous Processing of Low-Band-Gap Polymer Solar Cells Using Roll-to-Roll Methods, ACS Nano, 2011, 5, 4188–4196 CrossRef CAS PubMed.
- T. T. Larsen-Olsen, T. R. Andersen, B. Andreasen, A. P. L. Böttiger, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jørgensen and F. C. Krebs, Roll-to-roll processed polymer tandem solar cells partially processed from water, Sol. Energy Mater. Sol. Cells, 2012, 97, 43–49 CrossRef CAS.
- F. Zhao, C. Wang and X. Zhan, Morphology Control in Organic Solar Cells, Adv. Energy Mater., 2018, 8, 1703147 CrossRef.
- J. A. Labastide, M. Baghgar, I. Dujovne, Y. Yang, A. D. Dinsmore, B. G. Sumpter, D. Venkataraman and M. D. Barnes, Polymer Nanoparticle Superlattices for Organic Photovoltaic Applications, J. Phys. Chem. Lett., 2011, 2, 3085–3091 CrossRef CAS.
- L. A. Renna, C. J. Boyle, T. S. Gehan and D. Venkataraman, Polymer Nanoparticle Assemblies: A Versatile Route to Functional Mesostructures, Macromolecules, 2015, 48, 6353–6368 CrossRef CAS.
- T. S. Gehan, M. Bag, L. A. Renna, X. Shen, D. D. Algaier, P. M. Lahti, T. P. Russell and D. Venkataraman, Multiscale Active Layer Morphologies for Organic Photovoltaics Through Self-Assembly of Nanospheres, Nano Lett., 2014, 14, 5238–5243 CrossRef CAS PubMed.
- L. A. Renna, M. Bag, T. S. Gehan, X. Han, P. M. Lahti, D. Maroudas and D. Venkataraman, Tunable Percolation in Semiconducting Binary Polymer Nanoparticle Glasses, J. Phys. Chem. B, 2016, 120, 2544–2556 CrossRef CAS PubMed.
- T. Kietzke, D. Neher, M. Kumke, R. Montenegro, K. Landfester and U. Scherf, A Nanoparticle Approach To Control the Phase Separation in Polyfluorene Photovoltaic Devices, Macromolecules, 2004, 37, 4882–4890 CrossRef CAS.
- N. P. Holmes, M. Marks, J. M. Cave, K. Feron, M. G. Barr, A. Fahy, A. Sharma, X. Pan, D. A. L. Kilcoyne, X. Zhou, D. A. Lewis, M. R. Andersson, J. van Stam, A. B. Walker, E. Moons, W. J. Belcher and P. C. Dastoor, Engineering Two-Phase and Three-Phase Microstructures from Water-Based Dispersions of Nanoparticles for Eco-Friendly Polymer Solar Cell Applications, Chem. Mater., 2018, 30, 6521–6531 CrossRef CAS.
- D. Chen, F. Liu, C. Wang, A. Nakahara and T. P. Russell, Bulk Heterojunction Photovoltaic Active Layers via Bilayer Interdiffusion, Nano Lett., 2011, 11, 2071–2078 CrossRef CAS PubMed.
- N. D. Treat, M. A. Brady, G. Smith, M. F. Toney and E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Interdiffusion of PCBM and P3HT Reveals Miscibility in a Photovoltaically Active Blend, Adv. Energy Mater., 2011, 1, 82–89 CrossRef CAS.
- N. P. Holmes, M. Marks, P. Kumar, R. Kroon, M. G. Barr, N. Nicolaidis, K. Feron, A. Pivrikas, A. Fahy, A. D. de, Z. Mendaza, A. L. D. Kilcoyne, C. Müller, X. Zhou, M. R. Andersson, P. C. Dastoor and W. J. Belcher, Nano-pathways: Bridging the divide between water-processable nanoparticulate and bulk heterojunction organic photovoltaics, Nano Energy, 2016, 19, 495–510 CrossRef CAS.
- B. Vaughan, E. L. Williams, N. P. Holmes, P. Sonar, A. Dodabalapur, P. C. Dastoor and W. J. Belcher, Water-based nanoparticulate solar cells using a diketopyrrolopyrrole donor polymer, Phys. Chem. Chem. Phys., 2014, 16, 2647–2653 RSC.
- S. Ulum, N. Holmes, D. Darwis, K. Burke, A. L. David Kilcoyne, X. Zhou, W. Belcher and P. Dastoor, Determining the structural motif of P3HT:PCBM nanoparticulate organic photovoltaic devices, Sol. Energy Mater. Sol. Cells, 2013, 110, 43–48 CrossRef CAS.
- D. Darwis, N. Holmes, D. Elkington, A. L. David Kilcoyne, G. Bryant, X. Zhou, P. Dastoor and W. Belcher, Surfactant-free nanoparticulate organic photovoltaics, Sol. Energy Mater. Sol. Cells, 2014, 121, 99–107 CrossRef CAS.
- S. Gärtner, M. Christmann, S. Sankaran, H. Röhm, E.-M. Prinz, F. Penth, A. Pütz, A. E. Türeli, B. Penth, B. Baumstümmler and A. Colsmann, Eco-Friendly Fabrication of 4% Efficient Organic Solar Cells from Surfactant-Free P3HT:ICBA Nanoparticle Dispersions, Adv. Mater., 2014, 26, 6653–6657 CrossRef PubMed.
- S. Heutz, P. Sullivan, B. M. Sanderson, S. M. Schultes and T. S. Jones, Influence of molecular architecture and intermixing on the photovoltaic, morphological and spectroscopic properties of CuPc–C60 heterojunctions, Sol. Energy Mater. Sol. Cells, 2004, 83, 229–245 CrossRef CAS.
- W. Tress, K. Leo and M. Riede, Effect of concentration gradients in ZnPc:C60 bulk heterojunction organic solar cells, Sol. Energy Mater. Sol. Cells, 2011, 95, 2981–2986 CAS.
- B. Vaughan, A. Stapleton, E. Sesa, N. P. Holmes, X. Zhou, P. C. Dastoor and W. J. Belcher, Engineering vertical morphology with nanoparticulate organic photovoltaic devices, Org. Electron., 2016, 32, 250–257 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Organic and solution-processed tandem solar cells with 17.3% efficiency, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- M. F. Al-Mudhaffer, M. J. Griffith, K. Feron, N. C. Nicolaidis, N. A. Cooling, X. Zhou, J. Holdsworth, W. J. Belcher and P. C. Dastoor, The origin of performance limitations in miniemulsion nanoparticulate organic photovoltaic devices, Sol. Energy Mater. Sol. Cells, 2018, 175, 77–88 CrossRef CAS.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- G. Bratina and E. Pavlica, Characterisation of charge carrier transport in thin organic semiconductor layers by time-of-flight photocurrent measurements, Org. Electron., 2019, 64, 117–130 CrossRef CAS.
- J. C. Blakesley, F. A. Castro, W. Kylberg, G. F. A. Dibb, C. Arantes, R. Valaski, M. Cremona, J. S. Kim and J.-S. Kim, Towards reliable charge-mobility benchmark measurements for organic semiconductors, Org. Electron., 2014, 15, 1263–1272 CrossRef CAS.
- X. Han, M. Bag, T. S. Gehan, D. Venkataraman and D. Maroudas, Analysis of hole transport in thin films and nanoparticle assemblies of poly(3-hexylthiophene), Chem. Phys. Lett., 2014, 610–611, 273–277 CrossRef CAS.
- X. Han, M. Bag, T. S. Gehan, D. Venkataraman and D. Maroudas, Analysis of Charge Transport and Device Performance in Organic Photovoltaic Devices with Active Layers of Self-Assembled Nanospheres, J. Phys. Chem. C, 2015, 119, 25826–25839 CrossRef CAS.
- H. Bässler, Charge Transport in Disordered Organic Photoconductors a Monte Carlo Simulation Study, Phys. Status Solidi B, 1993, 175, 15–56 CrossRef.
- L. Liu and G. Li, Investigation of recombination loss in organic solar cells by simulating intensity-dependent current–voltage measurements, Sol. Energy Mater. Sol. Cells, 2011, 95, 2557–2563 CrossRef CAS.
- E. B. L. Pedersen, M. C. Pedersen, S. B. Simonsen, R. G. Brandt, A. P. L. Böttiger, T. R. Andersen, W. Jiang, Z. Y. Xie, F. C. Krebs, L. Arleth and J. W. Andreasen, Structure and crystallinity of water dispersible photoactive nanoparticles for organic solar cells, J. Mater. Chem. A, 2015, 3, 17022–17031 RSC.
- F. J. M. Colberts, M. M. Wienk and R. A. J. Janssen, Aqueous Nanoparticle Polymer Solar Cells: Effects of Surfactant Concentration and Processing on Device Performance, ACS Appl. Mater. Interfaces, 2017, 9, 13380–13389 CrossRef CAS PubMed.
- N. A. D. Yamamoto, M. E. Payne, M. Koehler, A. Facchetti, L. S. Roman and A. C. Arias, Charge transport model for photovoltaic devices based on printed polymer: Fullerene nanoparticles, Sol. Energy Mater. Sol. Cells, 2015, 141, 171–177 CrossRef CAS.
- C. Xie, A. Classen, A. Späth, X. Tang, J. Min, M. Meyer, C. Zhang, N. Li, A. Osvet, R. H. Fink and C. J. Brabec, Overcoming Microstructural Limitations in Water Processed Organic Solar Cells by Engineering Customized Nanoparticulate Inks, Adv. Energy Mater., 2018, 8, 1702857 CrossRef.
- O. Cardozo, S. Farooq, A. Stingl and N. Fraidenraich, Investigation of performance of P3HT:PCBM organic photovoltaic module under real operating conditions, Sol. Energy, 2019, 190, 543–548 CrossRef.
- M. Lenes, G.-J. A. H. Wetzelaer, F. B. Kooistra, S. C. Veenstra, J. C. Hummelen and P. W. M. Blom, Fullerene Bisadducts for Enhanced Open-Circuit Voltages and Efficiencies in Polymer Solar Cells, Adv. Mater., 2008, 20, 2116–2119 CrossRef CAS.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Low bandgap semiconducting polymers for polymeric photovoltaics, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- S. Dey, Recent Progress in Molecular Design of Fused Ring Electron Acceptors for Organic Solar Cells, Small, 2019, 15, 1900134 CrossRef PubMed.
- O. A. Ibraikulov, C. Ngov, P. Chávez, I. Bulut, B. Heinrich, O. Boyron, K. L. Gerasimov, D. A. Ivanov, S. Swaraj, S. Méry, N. Leclerc, P. Lévêque and T. Heiser, Face-on orientation of fluorinated polymers conveyed by long alkyl chains: a prerequisite for high photovoltaic performances, J. Mater. Chem. A, 2018, 6, 12038–12045 RSC.
- Q. Liu, S. E. Bottle and P. Sonar, Developments of Diketopyrrolopyrrole-Dye-Based Organic Semiconductors for a Wide Range of Applications in Electronics, Adv. Mater., 2020, 32, 1903882 CrossRef CAS PubMed.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Molecular Design of Benzodithiophene-Based Organic Photovoltaic Materials, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Conjugated Polymers in Bioelectronics, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- Z. Hu, Z. Wang, X. Zhang, H. Tang, X. Liu, F. Huang and Y. Cao, Conjugated Polymers with Oligoethylene Glycol Side Chains for Improved Photocatalytic Hydrogen Evolution, iScience, 2019, 13, 33–42 CrossRef CAS PubMed.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Design and evaluation of conjugated polymers with polar side chains as electrode materials for electrochemical energy storage in aqueous electrolytes, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- B. Xu, X. Yi, T.-Y. Huang, Z. Zheng, J. Zhang, A. Salehi, V. Coropceanu, C. H. Y. Ho, S. R. Marder, M. F. Toney, J.-L. Brédas, F. So and J. R. Reynolds, Donor Conjugated Polymers with Polar Side Chain Groups: The Role of Dielectric Constant and Energetic Disorder on Photovoltaic Performance, Adv. Funct. Mater., 2018, 28, 1803418 CrossRef.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Enhanced photocatalytic hydrogen evolution from organic semiconductor heterojunction nanoparticles, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- T. T. Larsen-Olsen, B. Andreasen, T. R. Andersen, A. P. L. Böttiger, E. Bundgaard, K. Norrman, J. W. Andreasen, M. Jørgensen and F. C. Krebs, Simultaneous multilayer formation of the polymer solar cell stack using roll-to-roll double slot-die coating from water, Sol. Energy Mater. Sol. Cells, 2012, 97, 22–27 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |