
Optimized synthesis of fluorinated dithienyl-diketopyrrolopyrroles and new copolymers obtained via direct heteroarylation polymerization†
Marius
Mamone
a,
Thomas
Bura
a,
Samuel
Brassard
a,
Eliane
Soligo
a,
Keqiang
He
b,
Yuning
Li
 *b and
Mario
Leclerc
*a
*b and
Mario
Leclerc
*a
aCanada Research Chair on Electroactive and Photoactive Polymers, Department of Chemistry, Université Laval, Quebec City, Quebec G1V 0A6, Canada. E-mail: mario.Leclerc@chm.ulaval.ca
bDepartment of Chemical Engineering, University of Waterloo, Waterloo, Ontario N2L 3G1, Canada
First published on 21st May 2020
Abstract
Diketopyrrolopyrrole-based (DPP-based) polymers are a very important class of materials for both organic solar cells (OSCs) and organic field-effect transistors (OFETs). Here we report an optimised synthesis of fluorinated dithienyl-diketopyrrolopyrrole (fDT-DPP) and three new highly fluorinated fDT-DPP-based materials obtained from direct heteroarylation polymerization (DHAP). This study confirms the properties of the fluorine atom in a polymer, activation of the α-position hydrogen on thiophene moieties, decreasing HOMO and LUMO energy, lower bandgaps and modification of the solubility and morphology of the materials.
Introduction
Since the first report on the synthesis of 3,6-diphenyl, 2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione by Farnum et al. in 1974,1 DPP-based π-conjugated polymers have been extensively studied due to their good performance in both organic solar cells (OSCs)2 and organic field-effect transistors (OFETs).3,4 Modifications of the aromatic moieties can highly modulate the electro-optical properties of the resulting copolymers and, in particular, dithienyl-DPP (DT-DPP) has attracted increasing interest.5,6 Fluorination of the conjugated backbone has also proven to be effective to fine-tune the properties of these organic materials.7–11 The electronegativity of the fluorine atom lowers both the HOMO and LUMO energy levels without creating additional steric hindrance.12,13Recently, our group has developed a fluorinated DT-DPP and reported fDT-DPP copolymers obtained via direct heteroarylation polymerization (DHAP), which showed very promising performance in OFET.14 The fDT-DPP was obtained by classical condensation of diisopropyl succinate with 4-fluoro-2-cyanothiophene as a key intermediate in 6 steps with a satisfactory 21% overall yield (Scheme 1, eqn (1)). Nevertheless, this synthetical pathway was not easily applicable to the large-scale preparation of this important monomer. Very recently, Wang et al. proposed a nonconventional stepwise synthesis of symmetrical and non-symmetrical fDPP and confirmed the properties of fDT-DPP copolymers (Scheme 1, eqn (2)).15 To fully study the potential of this monomer, we report here optimisation of its synthesis and application to new highly fluorinated DPP based copolymers.
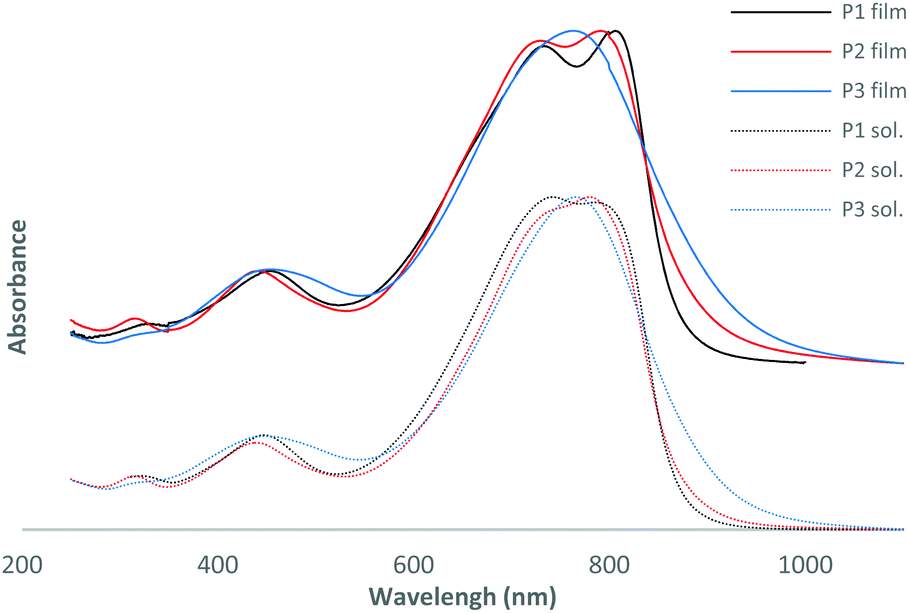 | ||
| Fig. 1 Normalized UV-vis absorption spectra of the polymers in thin films and in chloroform solution. | ||
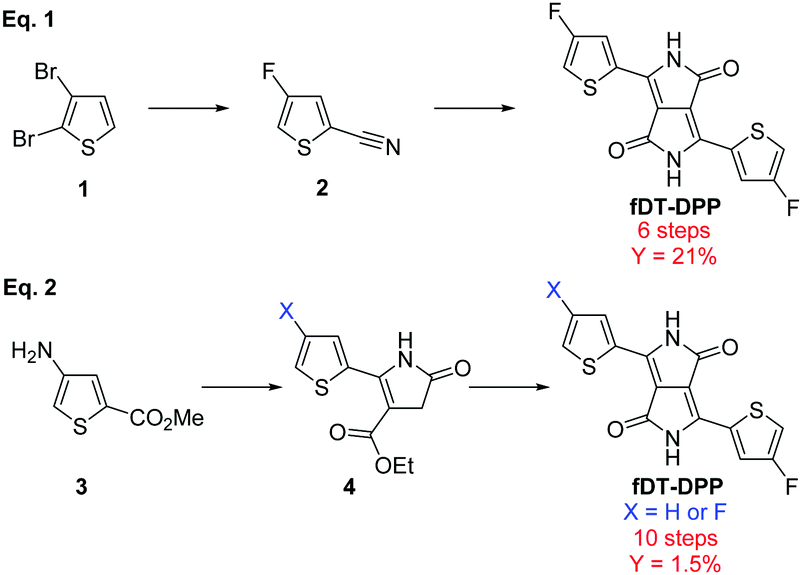 | ||
| Scheme 1 Previous synthesis of fDT-DPP by Leclerc's group (eqn (1)) and Wang's group (eqn (2)). | ||
Results and discussion
Optimization of fDT-DPP synthesis
As shown in Scheme 2 and based on our previous studies,14 3-bromo-2-(dimethyloctylsilyl)thiophene (5) was synthesized from a lithiation reaction of commercially available 2,3-dibromothiophene (1) in diethyl ether (at −78 °C) followed by addition of n-octyldimethylchlorosilane and isolated after reduced pressure distillation in 85% yield.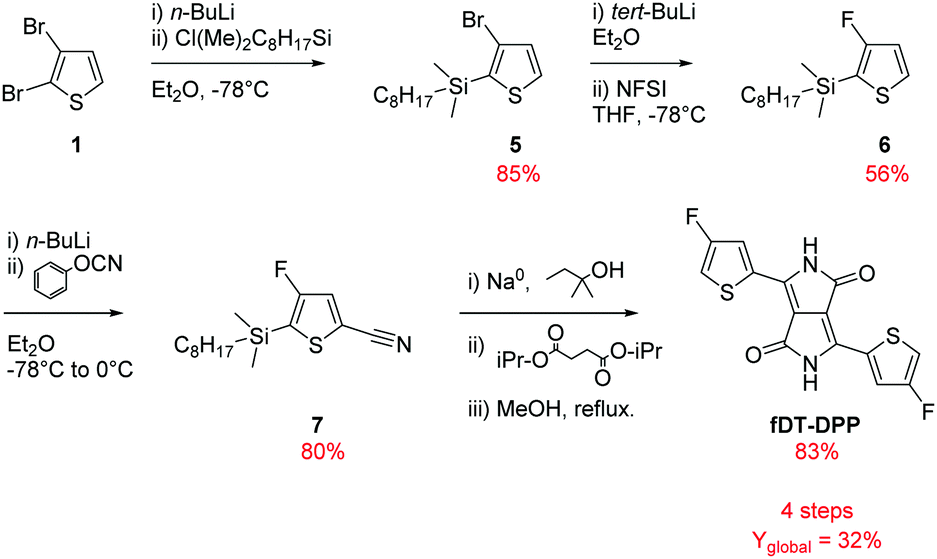 | ||
| Scheme 2 New fDT-DPP synthesis in 4 steps. | ||
The fluorination of compound (5) was the most delicate step in our previous synthesis due to the difficulty in separating hydrogenated and fluorinated thiophene by classical chromatography on silica gel. The reaction had to be realized at −100 °C to obtain a good selectivity for the fluorinated product which is very limiting for application in large scale synthesis. To solve these problems, tert-BuLi was used instead of n-BuLi for the monolithiation of compound (5) followed by a dropwise addition of a cold solution of NFSI in THF to increase the solubility of the fluorine donor. The reaction can now be realised at −78 °C, a more easily processable temperature, and a better conversion was observed due to the highest reactivity of tert-BuLi. The purification was also easier and the reaction can be applied at a larger scale with high purity and a very satisfying 56% yield.
As described later, compound (6) is also an important intermediate for the synthesis of various fluorinated bithiophene units.
Compound (6) could then be converted to compound (7) by a direct cyanation reaction with phenyl cyanate with 80% yield. This procedure reduces by one step the usual formylation/cyanation pathway. Finally, DPP was obtained by a classical cyclization with isopropyl succinate following an already described procedure.16 Due to the volatility of fluorinated thiophene, silylated compound (7) was directly used in this transformation and the product was deprotected in situ by quenching the reaction with dry MeOH. The desired fDT-DPP was isolated with 83% overall yield. These optimizations increased the global yield of the synthesis to 32% and reduced the number of steps to 4.
Design of fDT-DPP-based copolymers
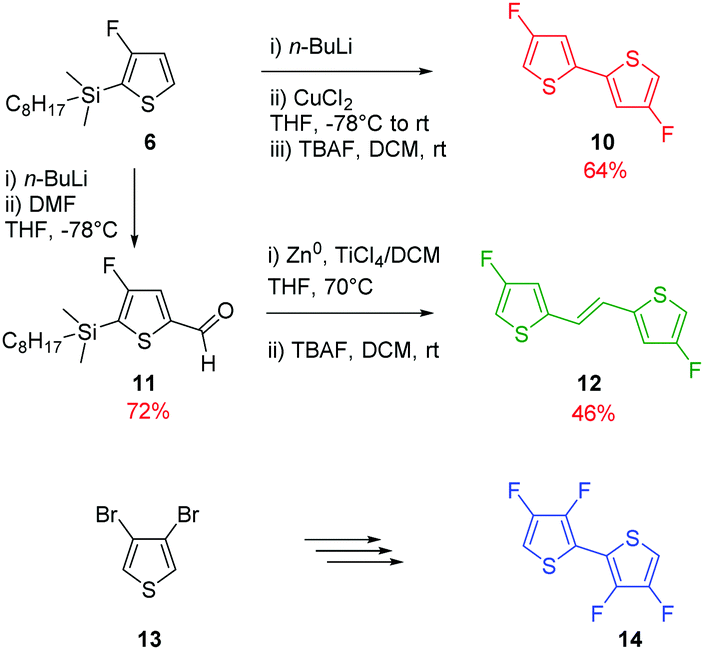
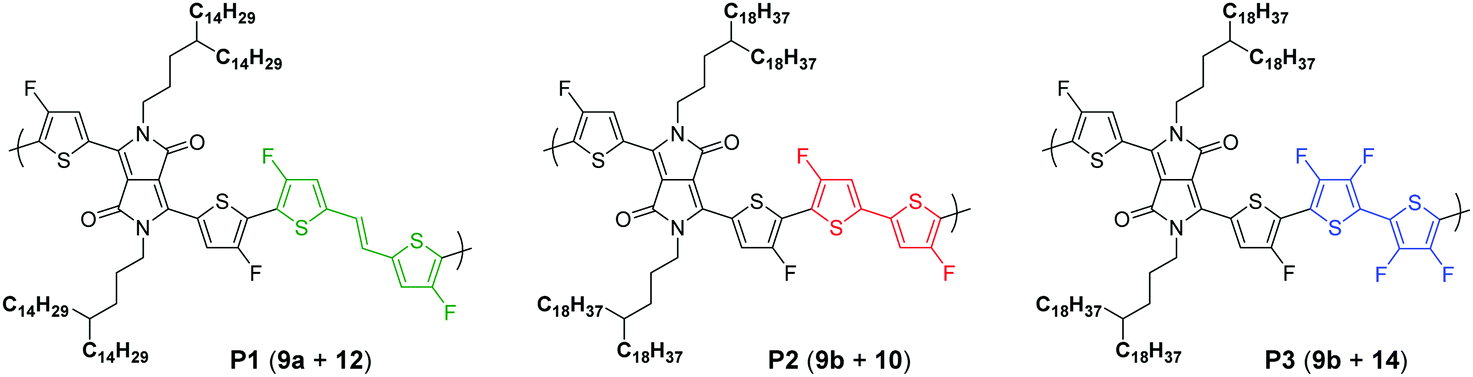
To pursue our study on fluorine atom effects on the properties of fDT-DPP-based copolymers, several new monomers (see Schemes 3 and 4) and conjugated copolymers (see Scheme 5) were designed and investigated for their properties in OFETs. Their structures were chosen according to a recent work published by Wang et al.17 on non-fluorinated DPP analogs.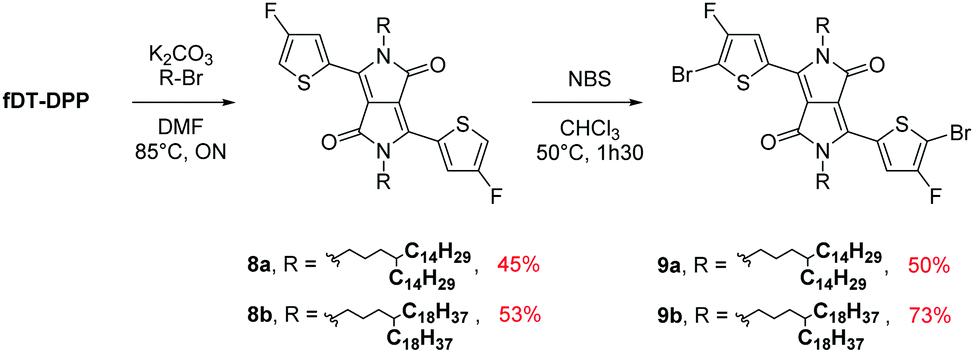 | ||
| Scheme 3 Alkylation and bromination of fDT-DPP. | ||
 | ||
| Scheme 4 Preparation of fluorinated comonomers. | ||
 | ||
| Scheme 5 fDT-DPP-based copolymers synthesized by DHAP. | ||
Monomer preparation
Due to the known low solubility of fluorinated conjugated polymers and with the aim of making a relevant comparison with Wang's work,17 large branched 4-octadecyldocosyl and 4-pentadecylnonadecyl side chains were introduced on fDT-DPP in DMF in the presence of K2CO3 with 45 and 53% yield. Alkylated fDT-DPP was then brominated by N-bromosuccinimide at 50 °C in degassed chloroform to obtain the corresponding monomers (9) in 73 and 50% yields, respectively (Scheme 3).As reported in Scheme 4, monomer (10) and new monomer (12) were also easily prepared from compound (6) by direct oxidative coupling with CuCl2 with 64% yield or by formylation followed by a McMurry condensation with a 46% overall yield. 3,3′,4,4′-tetrafluoro-2,2′-bithiophene (14) can be prepared from 3,4-dibromothiophene following previously described procedures.18
Polymer synthesis
After optimizations (see the ESI†), copolymers P1, P2 and P3 (Scheme 4) could be prepared by direct (hetero)arylation polymerization (DHAP) using Pd(OAc)2 in a toluene/DMAc mixture at 120 °C for 16 h. After precipitation in methanol, the polymers were purified by successive Soxhlet extractions. The molecular weights of the resulting polymers were measured by high temperature size exclusion chromatography (SEC) with polystyrene as standard and 1,2,4-trichlorobenzene as the eluent at 110 °C and the results are listed in Table 1. Surprisingly, in all cases, Mn is significantly lower than the non-fluorinated analogs described by Wang et al.15 even with longer reaction times. Clearly, fDT-DPP monomers are highly reactive towards DHAP polymerization but the brominated ones are by far less reactive.14| Polymer | Mn (kDa) | Đ | Yield (%) | T d (°C) | λ max,abs (nm) film |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
HOMOb (eV) | LUMOcvb (eV) |
E
cvg
(eV) |
LUMOoptd (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| a The optical bandgaps (Eoptg) calculated from the film absorption onsets. b HOMO and LUMO energy levels were calculated according to EHOMO = −(4.80 + Eoxonset) eV and ELUMO = −(4.80 + Eredonset) eV, in which Eoxonset and Eredonset represent oxidation and reduction onset potentials of the polymers versus Fc/Fc+, respectively. c Calculated according to Ecvg = EHUMO − ELOMO. d Calculated according to EoptLUMO = EcvHOMO + Eoptg. | ||||||||||
| P1 | 16 | 2.7 | 81 | 390 | 806 | 1.43 | −5.35 | −3.43 | 1.92 | −3.92 |
| P2 | 23 | 1.5 | 74 | 395 | 791 | 1.41 | −5.51 | −3.54 | 1.97 | −4.10 |
| P3 | 22 | 1.8 | 82 | 400 | 763 | 1.31 | −5.60 | −3.62 | 1.98 | −4.29 |
The difference could also be explained by a higher rigidity induced by the fluorine atom on the DT-DPP moiety which reduces the solubility of the resulting copolymers.
Polymer characterization
These polymers show good thermal stability with 5% weight loss temperature above 400 °C (see the ESI†). As described for the non-fluorinated analogues, broad thermal transitions between ∼−10 and 40 °C have been observed in the DSC curves, which are probably related to the melting of the long alkyl side chains (see the ESI†).19Optical absorption and electrochemical properties
As observed by Wang et al.,17 the more electron-rich nature of bis-thienylethene relative to the bithienyl group leads to a slightly red shifted maximum of absorption for P1 at 806 nm compared to 791 nm for P2 (see Table 1 and Fig. 1). It could also explain the decrease of energy levels moving from −5.35 to −5.51 eV for the HOMO and from −3.43 to −3.54 eV for the LUMO for P1 and P2, respectively. As expected, addition of a fluorine atom on the conjugated backbone decreased the energy level. If one compares P2 and the non-fluorinated DT-DPP analogue P4F2T-C40,17 we can see that the position of the fluorine atom on the skeleton has very little impact on the optical and electrochemical properties of the polymer. In fact, the absorbance maxima are similar, 791 and 799 nm, the HOMO and LUMO energy levels have slightly changed and the electrochemical bandgaps are very close.Interestingly, the addition of extra fluorine atoms has significant effects. The absorption maximum of P3 is blue-shifted by 28 nm in comparison to P2 and the HOMO and LUMO energy levels are both lowered. One can notice a decreased optical band gap for P3 and no secondary absorption shoulder in the UV-vis absorption spectrum in film or in solution.
Film morphology
The film microstructures play important roles in determining the device performances. Therefore, two-dimensional grazing-incidence X-ray diffraction (2D-GIXD) (see the ESI†) was performed and the results are depicted in Fig. 2.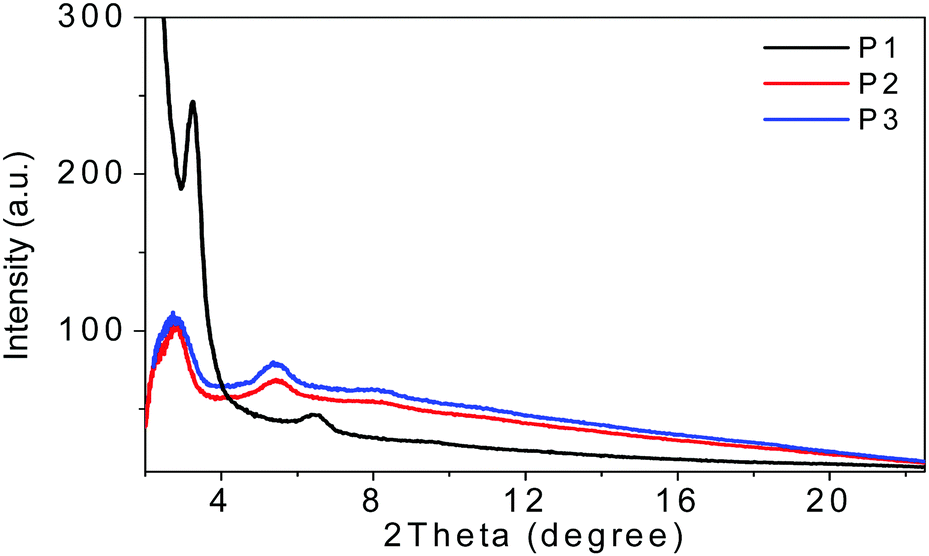 | ||
| Fig. 2 The GIXD out of plane line-cut profiles of P1, P2, and P3 films on SiO2/Si substrates annealed at 200 °C for 20 min in an argon-filled glove box. | ||
From the out of plane line-cuts, (100) and (200) diffraction peaks were observed for spin-coated thin films of all these three polymers, indicating that all of them adopt edge-on packing with respect to the substrate plane in thin films, which was preferred for charge transportation in OFET devices. The lamella packing d-spacings of P1, P2 and P3 were 27.2, 32.2 and 31.9 Å, respectively. P1 showed a higher (100) diffraction peak than that of P2 or P3, suggesting that P1 has much higher crystallinity compared to the other two polymers, which was beneficial for charge transport in the thin films. Unfortunately, no in-plane diffraction peaks were found in the films of all these three polymers, indicative of their lower packing order in the in plane direction, which may be an explanation for the moderate mobility of ca. 10−3–10−2 cm2 V−1 s−1 for these polymers, which will be discussed in the next section.
Semiconducting properties
Bottom gate and bottom contact OFET devices were fabricated to evaluate the semiconducting properties of the polymers.The mobility (μ) was calculated in the saturation regime and the results are summarized in Table 2. All polymers exhibited ambipolar properties with slightly better performances in the p-channel operation. Among them, P1, which carries a fluorinated bis-thienylethene group, showed the highest hole and electron mobility of 4.10 × 10−2 and 7.63 × 10−3 cm2 V−1 s−1, respectively, in agreement with the 2D-GIXD results. The highly fluorinated polymer, P3, showed the lowest hole and electron mobility of 2.03 × 10−3 and 2.92 × 10−4 cm2 V−1 s−1, respectively, which were an order of magnitude lower than those of P1 and P2. This may have resulted from the lowest solubility (∼2–3 mg ml−1) of P3, which would have a negative influence on the quality of the film. It should be noted that they all showed ideal transfer curves with a linear relationship between the square root of drain current (IDA1/2) and gate voltage (VG) (see ESI†), implying that the mobility values obtained are reliable. On the other hand, the ratios of μh/μe of P1, P2 and P3 were 5.37, 8.89 and 6.95, respectively. P1 showed the most balanced hole and electron transport properties. The above results indicated that P1 has the best OFET performance among the three polymers, which may be due to the introduction of a vinyl group improving the planarity of the polymer backbone, which is consistent with the most red-shifted absorption spectra.
| p-Channel | n-Channel | μ h/μe | |||
|---|---|---|---|---|---|
| μ max (μavg) (cm2 V−1 s−1) | I on/off | μ max (μavg) (cm2 V−1 s−1) | I on/off | ||
| All polymer films were annealed at 200 °C for 15 min under argon. | |||||
| P1 | 4.10 × 10−2 (3.74 × 10−2) | 104 | 7.63 × 10−3 (6.38 × 10−3) | 104 | 5.37 |
| P2 | 1.92 × 10−2 (1.33 × 10−2) | 105 | 2.16 × 10−3 (1.40 × 10−3) | 103 | 8.89 |
| P3 | 2.03 × 10−3 (1.46 × 10−3) | 103 | 2.92 × 10−4 (2.06 × 10−4) | 103 | 6.95 |
Conclusions
In summary, we successfully optimized the fDT-DPP synthesis by reducing the number of steps and increasing the total yield to 32%. The monomer could be used in the preparation of three new highly fluorinated copolymers by DHAP. Characterization of these polymers confirms the fluorine effect on the electro-optical properties of materials. P3 showed the most significant decreases in the HOMO and LUMO energy levels and a bandgap diminution. The low solubility of this highly fluorinated polymer also induced morphology changes that explained low hole and electron mobilities. These ambipolar copolymers with strong absorption between 600 and 800 nm could also be utilized as p-type or even n-type semiconductors in various organic solar cells.Experimental
3-Bromo-2-(dimethyloctylsyilyl)thiophene (5)
2,3-Dibromothiophene (20 g, 82.66 mmol) is put in a dry 1000 ml round-bottom flask with 200 ml of anhydrous diethyl ether. The solution is then cooled down to −80 °C, before dropwise addition of 2.5 M n-butyllithium solution (36.4 ml, 90.9 mmol, 1.1 eq.). The mixture is then left to react for 20 minutes before adding n-octyldimethylchlorosilane (18.81 g, 90.9 mmol, 1.1 eq.). The mixture can then be left to react overnight while heating up to RT. The product can be purified by a silica plug followed by distillation (110–115 °C, 0.35 mm Hg). The substance obtained is a transparent liquid (23.43 g, 85%). 1H NMR 500 MHz (CDCl3) δ (ppm): 7.46 (d, J =4.8 Hz, 1H) 7.11 (d, J = 4.8 Hz, 1H), 1.34–1.24 (m, 12H), 0.92–0.87 (m, 5H), 0.39 (s, 6H). NMR data are identical as previously described.133-Fluoro-2-(dimethyloctylsyilyl)thiophene (6)
3-Bromo-2-(dimethyloctylsyilyl)thiophene (11.81 g, 35.42 mmol, 1 eq.) is put in a 250 ml dry round-bottom flask with 125 ml of anhydrous diethyl ether, which is then cooled to −78 °C. A 1.7 M tert-butyllithium solution in pentane (25 ml, 42.5 mmol, 1.2 eq.) is added dropwise, and then the mixture is stirred for 30 minutes at −80 °C. A precooled solution of N-fluorobenzenesulfonimide (13.4 g, 42.5 mmol, 1.2 eq.) in 275 ml of anhydrous tetrahydrofuran was added dropwise via a canula maintaining a temperature of −78 °C. The mixture is then left to react overnight while returning to RT. The solution is neutralized by addition of a saturated NH4Cl solution and extracted three times with Et2O. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The crude product can be purified by a silica column followed by a C18 column, yielding the desired product as a transparent liquid (5.40 g, 56%). NMR data are identical as previously described in the literature.131H NMR 500 MHz (CDCl3) δ (ppm): 7.39 (dd, J = 5.0, 2.8 Hz, 1H), 6.85 (dd, J = 5.0, 1.6 Hz, 1H), 1.34–1.25 (m, 12H), 0.90–0.87 (m, 3H), 0.81–0.78 (m, 2H), 0.31 (s, 6H). 19F NMR 500 MHz (CDCl3) δ (ppm): −121 (s, 1F). NMR data are identical as previously described.134-Fluoro-5-(dimethyloctylsyilyl)-2-thiophenecarbonitrile (7)
3-Fluoro-2-(dimethyloctylsyilyl)thiophene (6) (4.74 g, 17.4 mmol, 1 eq.) is put in a round-bottom flask with 500 ml anhydrous diethyl ether, then cooled to −80 °C. A n-butyllithium solution (2.5 M in hexanes, 7.7 ml, 19.1 mmol, 1.1 eq.) is added dropwise. The mixture is then stirred for 15 minutes at −80 °C, then 30 minutes at 0 °C. The flask is cooled back down to −80 °C before adding neat phenyl cyanate (2.71 g, 22.6 mmol, 1.3 eq.) and stirring at this temperature for 30 minutes, then for 2 hours at 0 °C. The mixture can then be left to react overnight while returning to RT. The reaction is quenched by adding a saturated NH4Cl solution and then extracted three times with Et2O. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The product can be isolated by silica column, yielding a transparent liquid (4.14 g, 80%). 1H NMR 500 MHz (CDCl3) δ (ppm): 7.34 (d, J = 1.6 Hz, 1H), 1.34–1.25 (m, 12H), 0.90–0.87 (m, 3H), 0.84–0.80 (m, 2H), 0.34 (s, 6H). 19F NMR 500 MHz (CDCl3) δ (ppm): 118 (s, 1F). HRMS (ESI) m/z calcd for C15H24F1N1S1Si1 [M + H]+ 298.1456, found 298.1445.3,6-Bis(4-fluorothiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (fDT-DPP)
20 ml of 2-methyl-2-butanol is put in a dry twin-neck round-bottom flask equipped with a condenser, then heated up to 105 °C. Sodium (0.432 g, 18.8 mmol, 4 eq.) is added piecemeal to the liquid, which is then stirred until complete digestion. 4-Fluoro-5-(dimethyloctylsyilyl)-2-thiophenecarbonitrile (2.934 g, 9.86 mmol, 2.1 eq.) is added rapidly, then diisopropyl succinate (0.950 g, 4.70 mmol, 1 eq.) is added dropwise. The mixture is then left to react overnight at 110 °C. The solution is then cooled down to 60 °C before adding methanol and stirring for 1 hour. The mixture is then neutralized by adding acetic acid (1.34 ml, 23.5 mmol, 5 eq.). The mixture can be filtered by Büchner and washed with methanol. The product is obtained as a purple solid (1.31 g, 83%). 19F NMR (470 MHz, DMSO, 60 °C) δ (ppm) −126.98 (s, 2F). 1H NMR (500 MHz, DMSO, 60 °C) δ (ppm): 11.19 (s, 2H), 8.02 (d, J = 1.7 Hz, 2H), 7.60 (app. t, J = 1.4 Hz, 2H).3,6-Bis(4-fluorothiophen-2-yl)-2,5-bis(4-tetradecyloctadecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8a)
In a two-neck flask equipped with a condenser, 3,6-bis(4-fluorothiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (fDT-DPP) (443 mg, 1.32 mmol, 1 eq.) was stirred at r.t. in dry DMF (9 ml, 0.15 M). 15-(3-Bromopropyl)triacontane (2.09 g, 3 eq.) was added to the mixture followed by dry K2CO3 (546 mg, 3 eq.) then the mixture was stirred at 100 °C for 24 h. After cooling at r.t., the reaction was quenched by the addition of 1 M HCl solution and extracted three times with Et2O. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The solid residue was purified by chromatography on silica gel with Hexane/CHCl3, 70 to 100%, to afford the desired product as a deep purple solid (730 mg, 45%). 19F NMR (470 MHz, CDCl3) δ −125.87 (s, 2F). 1H NMR (500 MHz, CDCl3) 8.71 (d, J = 1.7 Hz, 2H), 7.10–6.87 (m, 2H), 4.05–3.96 (m, 5H), 1.76–1.65 (m, 4H), 1.39–1.15 (m, 127H), 0.93–0.83 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 160.76, 158.61 (d, J = 260.4 Hz), 139.24 (d, J = 2.8 Hz), 128.22 (d, J = 9.4 Hz), 123.94 (d, J = 27.5 Hz), 109.04 (d, J = 21.5 Hz), 108.14, 42.66, 37.25, 33.63, 32.08, 30.58, 30.23, 29.87, 29.83, 29.53, 27.14, 26.82, 22.83, 14.24. HRMS (APPI) m/z calcd for C78H134F2N2O2S2 [M + H]+ 1233.9928, found 1233.9942.3,6-Bis(4-fluorothiophen-2-yl)-2,5-bis(4-octadecyldocosyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8b)
In a two-neck flask equipped with a condenser, 3,6-bis(4-fluorothiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (fDT-DPP) (797 mg, 2.37 mmol, 1 eq.) was stirred at r.t. in dry DMF (16 ml, 0.15 M). 19-(3-Bromopropyl)heptatriacontane (3.35 g, 2.2 eq.) was added in the mixture followed by dry K2CO3 (720 mg, 2.2 eq.) and then the mixture was stirred at 100 °C for 24 h. After cooling at r.t., the reaction was quenched by addition of 1 M HCl solution and extracted three times with Et2O. The organic layer was washed with water and brine, dried over anhydrous MgSO4, filtered and concentrated under vacuum. The solid residue was purified by chromatography on silica gel with Hexane/CHCl3, 70 to 100%, to afford the desired product as a deep purple solid (1.82 g, 53%).19F NMR (470 MHz, CDCl3) δ −125.87 (s, 2F). 1H NMR (500 MHz, CDCl3) 8.71 (d, J = 1.7 Hz, 2H), 7.03 (d, J = 1.7 Hz, 2H), 4.08–3.91 (m, 4H), 1.77–1.61 (m, 4H), 1.24 (d, J = 5.3 Hz, 148H), 0.88 (t, J = 6.9 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 161.00, 158.7 (d, J = 260.8 Hz), 139.45 (d, J = 2.9 Hz), 128.26 (d, J = 9.1 Hz), 123.95 (d, J = 27.7 Hz), 109.16 (d, J = 21.5 Hz), 108.30, 77.41, 77.16, 76.91, 42.72, 37.27, 33.65, 32.09, 30.61, 30.24, 29.88, 29.83, 29.53, 27.21, 26.83, 22.85, 14.27. HRMS (APPI) m/z calcd for C94H166F2N2O2S2 [M + H]+ 1458.2386, found 1458.2410.
3,6-Bis(5-bromo-4-fluorothiophen-2-yl)-2,5-bis(4-tetradecyloctadecyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (9a)
To a solution of (8b) (870 mg, 0.705 mmol, 1 eq.) in degassed CHCl3 (14 ml, 0.05 M) under nitrogen was added portionwise freshly crystallized NBS (376 mg, 3 eq.) and the solution was stirred at 50 °C in the dark for 1.5 h. Water was added and the mixture was extracted with chloroform. The organic layer was washed with water and brine, dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by chromatography in silica gel with hexane/CHCl3, 50 to 80%, followed by recrystallization from hexane to give the desired product as a purple solid (491 g, 50%). 19F NMR (470 MHz, CDCl3) δ −124.24 (s, 2F). 1H NMR (500 MHz, CDCl3) 8.64 (s, 2H), 3.98–3.91 (m, 4H), 1.73–1.62 (m, 4H), 1.24 (q, J = 8.3, 5.7 Hz, 123H), 0.91–0.79 (m, 12H). 13C NMR (126 MHz, CDCl3) δ 160.45, 156.53 (d, J = 262.1 Hz), 138.47 (d, J = 3.3 Hz), 127.45 (d, J = 9.1 Hz), 123.18 (d, J = 26.5 Hz), 108.12, 98.88 (d, J = 22.9 Hz), 42.54, 36.95, 33.37, 31.84, 30.26, 29.99, 29.58, 29.28, 26.91, 26.59, 22.60, 14.02. HRMS (APPI) m/z calcd for C78H132Br2F2N2O2S2 [M + H]+ 1391.8130, found 1391.8140.3,6-Bis(5-bromo-4-fluorothiophen-2-yl)-2,5-bis(4-octadecyldocosyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (9b)
To a solution of (8a) (1.89 g, 1.3 mmol, 1 eq.) in degassed CHCl3 (36 ml, 0.05 M) under nitrogen was added portionwise freshly crystallized NBS (484 mg, 2.1 eq.) and the solution was stirred at 50 °C in the dark for 1.5 h. Water was added and the mixture was extracted with chloroform. The organic layer was washed with water and brine, dried over MgSO4, filtered and concentrated under vacuum. The residue was purified by chromatography in silica gel with hexane/CHCl3, 50 to 80%, followed by recrystallization from hexane to give the desired product as a purple solid (1.52 g, 73%). 19F NMR (470 MHz, CDCl3) δ −124.22 (s, 2F). 1H NMR (500 MHz, CDCl3) δ 8.64 (s, 2H), 4.00–3.81 (m, 4H), 1.71–1.62 (m, 4H), 1.39–1.15 (m, 148H), 0.88 (t, J = 6.9 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ 160.77, 156.88 (d, J = 257.0 Hz), 138.77 (d, J = 3.1 Hz), 127.88 (d, J = 9.3 Hz), 123.38 (d, J = 26.5 Hz), 108.60, 99.00 (d, J = 22.9 Hz), 77.41, 77.16, 76.91, 42.87, 37.36, 33.80, 32.10, 30.74, 30.27, 29.88, 29.83, 29.52, 27.29, 26.94, 22.84, 14.21. HRMS (APPI) m/z calcd for C94H164Br2F2N2O2S2 [M + H]+ 1616.0582, found 1616.0680.4,4′-Difluoro-2,2′-bithiophene (10)
To a solution of (4-fluorothiophen-2-yl)dimethyl(octyl)silane (6) (2.476 g, 9.09 mmol, 1 eq.) in dry THF under argon at −78 °C was added dropwise nBuLi (2.5 M in hexane, 4 ml, 1.1 eq.). The mixture was stirred for 30 min at r.t. then cooled at −78 °C. Dry CuCl2 (1.47 g, 1.2 eq.) was added in one shot and the solution was warmed to r.t. and stirred overnight. A 1 M HCl solution was added to quench the reaction and the resulting mixture was extracted with Et2O. The organic layer was washed with water and brine then dried over anhydrous MgSO4. The mixture was filtrated, and the solvent was evaporated by vacuum. The residue was filtered on a short pad of silica with hexane and concentrated under vacuum. The residue was dissolved in DCM (0.8 M) and TBAF-3H2O (2.1 g, 2 eq.) was added and the mixture was stirred at r.t. for 2 h. Water was added, and the solution was extracted with DCM. The organic layer was washed with water and brine and dried over anhydrous MgSO4, filtered and concentrated under vacuum. The residue was purified by chromatography on silica gel with hexane, followed by recrystallization from MeOH to give (10) as a white solid (589 g, 64%). m. p. = 61 °C, 19F NMR (470 MHz, CDCl3) δ −126.16 (s, 2F). 1H NMR (500 MHz, CDCl3) 6.93 (d, J = 1.7 Hz, 2H), 6.63 (t, J = 1.5 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 157.65 (d, J = 259.3 Hz), 135.74 (dd, J = 9.1, 2.0 Hz), 113.92 (d, J = 27.5 Hz), 103.03 (d, J = 21.3 Hz). HRMS (APPI) m/z calcd for C8H4F2S2 [M]+ 201.9717, found 201.9737.5-(Dimethyl(octyl)silyl)-4-fluorothiophene-2-carbaldehyde (11)
3-Fluoro-2-(dimethyloctylsilyl)thiophene (6) (2 g, 7.35 mmol, 1 eq.) was put in a dry 250 ml round-bottom flask with a magnetic stirrer under an argon atmosphere, then 40 ml of dry THF was added. The mixture was cooled to −80 °C, then 2.5 M n-butyllithium (3.1 ml, 7.72 mmol, 1.05 eq.) was added dropwise. The mixture was stirred for 30 minutes at −80 °C, and then dimethyl formamide (0.740 ml, 9.56 mmol, 1.3 eq.) was added dropwise, then the mixture was stirred overnight and allowed to reach room temperature. The reaction mixture was then neutralized with a saturated NH4Cl solution, then extracted with hexanes, washed with water and brine, and dried over MgSO4. The solvent was then removed under vacuum. The product was purified by column chromatography on silica gel, the eluent being a hexanes/diethyl ether mix (90/10), affording the compound as a colorless oil (Y = 80%). The spectral data was identical with the reported values.1319F NMR (470 MHz, CDCl3) δ −116.6 (s, 1F). 1H NMR (500 MHz, CDCl3) 9.80 (s, 1H), 7.47 (d, J = 1.7 Hz, 1H), 1.39–1.09 (m, 12H), 0.94–0.68 (m, 5H), 0.35 (s, 6H).(E)-1,2-Bis(4-fluorothiophen-2-yl)ethene (12)
Activated Zn0 (0.78 g, 12 mmol, 2.4 eq.) was put in a dry three-necked 250 ml flask along with 30 ml of dry THF and put under an argon atmosphere. The reaction medium was cooled to 0 °C, followed by a slow addition of neat TiCl4 (1.14, 6 mmol, 1.2 eq.) and then stirred for 1 h at 65 °C. The mixture was then cooled to room temperature, and a solution of 4-fluoro-5-(dimethyloctylsilyl)-2-thiophenecarboxaldehyde (1.5 g, 5.00 mmol, 1 eq.) dissolved in dry THF was inserted in the reaction medium through a canula. The resulting mixture was then stirred overnight at a temperature of 70 °C. The product was then extracted with hexanes, washed with water and brine, and dried over MgSO4. The solvent was evaporated under vacuum. The crude product was dissolved in 50 ml of dichloromethane, TBAF·3H2O (3.16 g, 2 eq.) was added and the mixture was stirred at room temperature for 2 h. The mixture was extracted with hexanes and washed with water and brine. The solvent was then removed under vacuum. The product was purified with column chromatography on silica gel with a hexanes/ethyl acetate mix (98/2) as the eluent, yielding a white powder (Y = 46%). m. p. = 124 °C, 19F NMR (470 MHz, CDCl3) δ −126.7 (s, 2F). 1H NMR (500 MHz, CDCl3) δ 6.90 (s, 2H), 6.82 (dd, J = 1.6, 0.7 Hz, 2H), 6.61–6.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 157.87 (d, J = 258.7 Hz), 140.16 (d, J = 8.4 Hz), 121.42 (d, J = 2.4 Hz), 115.75 (d, J = 26.8 Hz), 102.65 (d, J = 21.4 Hz). HRMS (APPI) m/z calcd for C10H6F2S2 [M*] 227.9879, found 227.9864.3,3′,4,4′-Tetrafluoro-2,2′-bithiophene (14)
The product was obtained from 3,4-dibromothiophene following the described literature procedure8 as a white solid (230 mg, 22% overall yield in 5 steps). m. p. = 96 °C, 19F NMR (470 MHz, CDCl3) δ −134.98 (d, J = 12.5 Hz, 2F), −136.39 (d, J = 12.4 Hz, 2F). 1H NMR (500 MHz, CDCl3) δ 6.78–6.75 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 145.44 (dd, J = 261.8, 19.2 Hz), 142.16 (dd, J = 266.6, 20.9 Hz), 110.93 (ddd, J = 6.9, 5.0, 3.3 Hz), 102.66 (dt-like, J = 15.8, 4.2 Hz). HRMS (APPI) m/z calcd for C8H2F4S2 [M]+ 237.9529, found 237.9562.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Natural Sciences and Engineering Research Council of Canada (NSERC), Université Laval (APOGEE, Sentinelle Nord) and the Canadian Institute for Advanced Research (CIFAR). We also want to thank Mathieu Mainville for DFT calculations.Notes and references
- D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Attempted reformatskii reaction of benzonitrile, 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]pyrrole. A lactam analogue of pentalene, Tetrahedron Lett., 1974, 15, 2549–2552 CrossRef.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Design of Diketopyrrolopyrrole (DPP)-Based Small Molecules for Organic-Solar-Cell Applications, Adv. Mater., 2017, 29, 1600013 CrossRef PubMed.
- I. Kang, H. J. Yun, D. S. Chung, S. K. Kwon and Y. H. Kim, Record High Hole Mobility in Polymer Semiconductors via SideChain Engineering, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- H. J. Yun, S. J. Kang, Y. Xu, S. O. Kim, Y. H. Kim, Y. Y. Noh and S. K. Kwon, Dramatic Inversion of Charge Polarity in Diketopyrrolopyrrole-Based Organic Field-effect Transistors via Simple Nitrile Group Substitution, Adv. Mater., 2014, 26, 7300–7307 CrossRef CAS PubMed.
- H. Choi, S.-J. Ko, T. Kim, P.-O. Morin, B. Walker, B. H. Lee, M. Leclerc, J. Y. Kim and A. J. Heeger, Small-Bandgap Polymer Solar Cells with Unprecedented Short-Circuit Current Density and High Fill Factor, Adv. Mater., 2015, 27, 3318–3324 CrossRef CAS PubMed.
- M. Lee, T. Kim, H. Van, T. Nguyen, H. W. Cho, K.-K. Lee, J.-H. Choi, B. S. Kim and J. Y. Kim, Regio-regular alternating diketopyrrolopyrrolebased D1–A–D2–A terpolymers for the enhanced performance of polymer solar cells, RSC Adv., 2019, 9, 42096–42109 RSC.
- N. Leclerc, P. Chávez, O. A. Ibraikulov, T. Heiser and P. Lévêque, Impact of Backbone Fluorination on π-Conjugated Polymers in Organic Photovoltaic Devices: A Review, Polymers, 2016, 8, 11 CrossRef PubMed.
- T. Olla, O. A. Ibraikulov, S. Ferry, O. Boyron, S. Méry, B. Heinrich, T. Heiser, P. Levéque and N. Leclerc, Benzothiadiazole Halogenation Impact in Conjugated Polymers, a Comprehensive Study, Macromolecules, 2019, 52, 8006–8016 CrossRef CAS.
- C. Sun, F. Pan, S. Chen, R. Wang, R. Sun, Z. Shang, B. Qiu, J. Min, M. Lv, L. Meng, C. Zhang, M. Xiao, C. Yang and Y. Li, Achieving Fast Charge Separation and Low Nonradiative Recombination Loss by Rational Fluorination for HighEfficiency Polymer Solar Cells, Adv. Mater., 2019, 31, 1905480 CrossRef CAS PubMed.
- Z. Fei, P. Boufflet, S. Wood, J. Wade, J. Moriarty, E. Gann, E. L. Ratcliff, C. R. McNeill, H. Sirringhaus, J.-S. Kim and M. Heeney, Influence of Backbone Fluorination in Regioregular Poly(3-alkyl-4-fluoro)thiophenes, J. Am. Chem. Soc., 2015, 137(21), 6866–6879 CrossRef CAS PubMed.
- B. Jiang, C. C. Du, M. J. Li, K. Gao, L. Kou, M. Chen, F. Liu, T. P. Russell and H. Wang, Synthesis of fluorinated diphenyldiketopyrrolopyrrole derivatives as new building blocks for conjugated copolymers, Polym. Chem., 2016, 7, 3311 RSC.
- J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, A high mobility DPP-based polymer obtained via direct (hetero)arylation polymerization, Polym. Chem., 2015, 6, 278–282 RSC.
- T. Bura, S. Beaupré, M.-A. Légaré, O. A. Ibraikulov, N. Leclerc and M. Leclerc, Theoretical Calculations for Highly Selective Direct Heteroarylation Polymerization: New Nitrile-Substituted Dithienyl-Diketopyrrolopyrrole-Based Polymers, Molecules, 2018, 23, 2324 CrossRef PubMed.
- T. Bura, S. Beaupré, O. A. Ibraikulov, M.-A. Legaré, J. Quinn, P. Levéque, T. Heiser, Y. Li, N. Leclerc and M. Leclerc, New Fluorinated Dithienyldiketopyrrolopyrrole Monomers and Polymers for Organic Electronics, Macromolecules, 2017, 50, 7080–7090 CrossRef CAS.
- X. Wang, B. Jiang, C. Du, X. Ren, Z. Duan and H. Wang, Fluorinated dithienyl-diketopyrrolopyrrole: a new building block for organic optoelectronic materials, New J. Chem., 2019, 43, 16411 RSC.
- Y. Zou, D. Gendron, R. Badrou-Aïch, A. Najari, Y. Tao and M. Leclerc, A High-Mobility Low-Bandgap Poly(2,7-carbazole) Derivative for Photovoltaic Applications, Macromolecules, 2009, 42(8), 2891–2894 CrossRef CAS.
- Y. Gao, J. Bai, Y. Sui, Y. Han, Y. Deng, H. Tian, Y. Geng and F. Wang, High Mobility Ambipolar Diketopyrrolopyrrole-Based Conjugated Polymers Synthesized via Direct Arylation Polycondensation: Influence of Thiophene Moieties and Side Chains, Macromolecules, 2018, 51, 8752–8760 CrossRef CAS.
- K. Kawashima, T. Fukuhara, Y. Suda, Y. Suzuki, T. Koganezawa, H. Yoshida, H. Ohkita, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 10265–10275 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, A BDOPV-Based Donor–Acceptor Polymer for High-Performance n-Type and Oxygen-Doped Ambipolar Field-Effect Transistors, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00218f |
| This journal is © the Partner Organisations 2020 |