
New exciplex systems composed of triazatruxene donors and N-heteroarene-cored acceptors†
Yuan-Cheng
Hu
a,
Zong-Liang
Lin
a,
Tzu-Chien
Huang
b,
Jhih-Wei
Lee
b,
Wei-Chih
Wei
a,
Tzu-Yu
Ko
a,
Chun-Yuan
Lo
a,
Deng-Gao
Chen
 a,
Pi-Tai
Chou
a,
Wen-Yi
Hung
*b and
Ken-Tsung
Wong
*ac
a,
Pi-Tai
Chou
a,
Wen-Yi
Hung
*b and
Ken-Tsung
Wong
*ac
aDepartment of Chemistry, National Taiwan University, Taipei, 10617, Taiwan. E-mail: kenwong@ntu.edu.tw
bDepartment of Optoelectronics and Materials Technology, National Taiwan Ocean University, Keelung 202, Taiwan. E-mail: wenhung@mail.ntou.edu.tw
cInstitute of Atomic and Molecular Science, Academia Sinica, Taipei, 10617, Taiwan
First published on 19th May 2020
Abstract
In this study, three triazatruxene-based molecules Tr-Me, Tr-Ph, and Tr-Tol with methyl, phenyl, and p-tolyl pendant groups, respectively, were intermixed with three acceptors 3P-T2T, 3P-T2P, and 3P-Pyr featuring phenylpyrazole peripherals and triazine, pyrimidine, and pyridine cores, respectively, to generate an array of nine blends to probe the feasibility of formation of an exciplex. The observed red-shifted emission compared to those of pristine donor and acceptor films is a good indication of exciplex formation. The exciplex emissions are mainly modulated by the features of N-heteroarene cores of acceptors, while the HOMOs of the donors are less influenced by the substituents. From the screening of these D:A combinations it was concluded that four exciplex-forming blends composed of Tr-Ph and Tr-Tol as donors and 3P-T2T and 3P-T2P as acceptors exhibit promising properties suitable for OLED applications. Among them, a green device employing a Tr-Tol:3P-T2P blend as the emitting layer performed with the best efficiency with a maximum external quantum efficiency (EQEmax) of up to 12.8%. These new exciplex-forming systems were further explored as cohosts for a newly developed fluorescent emitter (DT)2BTh2CN. The Tr-Ph:3P-T2P blend was found to serve as the best host to give a deep-red (EL λmax 671 nm, CIE (0.68, 0.31)) OLED device with an EQEmax of 5.52%. The data extracted from the transient photoluminescent studies shed light on the emission mechanism which is dominated by Förster energy transfer and simultaneously suffers from competing Dexter energy transfer quenching. Our results highlight the feasibility of using triazatruxene as the donor for exciplex formation as well as the tunability of the emission wavelength of exciplex-forming systems by rational structural modifications of both donors and acceptors.
Introduction
The strategy of efficiently harnessing excitons generated by the recombination of holes and electrons is crucial for high-efficiency organic light-emitting diodes (OLEDs). Among many approaches to molecular designs, thanks to the pioneering work reported by Adachi,1 organic materials that exhibit thermally activated delayed fluorescence (TADF) are emerging as cost-competitive alternatives to iridium-based phosphorescent emitters2 mainly attributable to their capability of achieving 100% internal quantum efficiency. The fundamental criterion for a molecule to perform TADF is a small energy gap (ΔEST) between the singlet (S1) and triplet (T1) excited states. A limited ΔEST allows the triplet excitons to be up-converted to singlet excitons through reverse intersystem crossing (rISC), giving “delayed” fluorescence in addition to intrinsic pump fluorescence. In principle, a small ΔEST can be feasibly realized by a molecule with a low overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). Many successful molecular designs have been reported: for example, a bipolar molecule incorporating a twisted π-bridge3 or a σ-bridge4 to separate electron-donor and electron-acceptor blocks. In considering the subtle balance between the limited HOMO/LUMO overlap and S1 to S0 transition probability, OLED devices employing TADF emitters that can achieve an external quantum efficiency (EQE) of over 36% have been reported.5–10 These exciting results verify the promising potential of organic TADF materials for giving high-efficiency OLEDs. Beyond the intramolecular charge transfer (ICT) approach, TADF can also be realized by the recombination of holes and electrons accumulated at the interface of a hole-transporting material (HTM) and an electron-transporting material (ETM) due to the large energy level off-sets.11 The HOMO of this interfacial excited state, also known as an exciplex, is located on the HTM, whereas the LUMO is positioned at the ETM, leading to a low exchange energy, and therefore a small ΔEST that is suitable for the rISC process. Since the first report of an exciplex showing TADF by Adachi in 2012,12 many exciplex-forming systems have been developed to act as the emitting layer (EML) for high-efficiency OLEDs with emissions spanning from blue,13 to green14 to red,15 and even to near infrared.16 In addition, exciplex-forming systems can also serve as hosts for fluorescent,17 phosphorescent,18 and TADF14 dopants for producing superior OLEDs. The versatile functions of exciplex-forming systems suggest promising potential for boosting research activities in material chemistry as well as OLED technology. Among various structures, carbazole-based HTMs are widely utilized as donors in exciplex studies.19–21 Similar to carbazole, triazatruxene with a more extended coplanar skeleton has been the core structure for designing good HTMs.22 More recently, triazatruxene was reported as serving as a donor moiety for a blue TADF material achieving a record-high performance.23 Triazatruxene derivatives also showed other promising optoelectronic applications, such as perovskite solar cells24 and organic field-effect transistors.25 However, to the best of our knowledge, triazatruxene has not been explored for a study of exciplex formation. This triggered our motivation to examine triazatruxene-based HTMs as donors for exciplex-forming systems. Many electron-deficient materials have been verified as good ETMs for exciplexes. In particular, triazine-centered materials are among the best ETMs for giving high-efficiency exciplex-based OLEDs.26 In this study, a previously reported ETM, 3P-T2T27 with a triazine core equipped with phenylpyrazole peripherals was selected as a model acceptor for blending with triazatruxene-cored HTMs. We envision that the replacement of the central N-heteroarene core from the triazine of 3P-T2T by the pyrimidine of 3P-T2P or the pyridine of 3P-Pyr is a feasible strategy for tuning the electronic properties of ETMs. In conjunction with the 3 different pendant groups (methyl, phenyl, and p-tolyl) of triazatruxene, nine possible exciplex-forming blends were characterized with various photophysical methods. Six new exciplex-forming systems were identified for further OLED applications. Among these D:A combinations, a device employing a blend of triazatruxene with a p-tolyl substituent (Tr-Tol) and 3P-T2P performed with the best efficiency with a maximum external quantum efficiency (EQEmax) of up to 12.8%. These D:A blends were further explored as the host for a tailor-made fluorescent emitter, (DT)2BTh2CN, to give deep-red OLEDs. An OLED device with EL λmax 671 nm, CIE (0.68, 0.31) and an EQEmax of 5.52% was achieved by using the Tr-Ph:3P-T2P blend as the cohost. Our results highlight the feasibility of tuning the features (emission wavelength and efficiency) of exciplex-forming systems by rational structural modifications that eventually lead to the best D:A blend for OLEDs.Results and discussions
The molecular structures and synthetic pathways of the triazatruxenes, 3P-T2T, 3P-T2P, 3P-Pyr, and (DT)2BTh2CN are depicted in Scheme 1. Tr-Me28 and Tr-Ph29 have been reported previously, while Tr-Tol was synthesized by a one-step reaction from the parent triazatruxene30 and 4-iodotoluene by a Pd-catalyzed Hartwig–Buchwald cross-coupling reaction. The model acceptor 3P-T2T was reported in our previous work,27 while 3P-T2P and 3P-Pyr were prepared by utilizing a Cu-catalyzed Ullmann cross-coupling reaction of 2,4,6-tris(3-bromophenyl)pyrimidine31 and 2,4,6-tris(3-bromophenyl)pyridine32 with pyrazole, respectively. In addition, the new fluorescent emitter (DT)2BTh2CN was synthesized by a one-step Suzuki–Miyaura reaction of reported 1,3-dibromobenzo[c]thiophene-5,6-dicarbonitrile33 and [4-[bis(4-methylphenyl)amino]phenyl]boronic acid. All newly synthesized molecules have been characterized with satisfactory spectroscopic data (see ESI†). | ||
| Scheme 1 Molecular structures and synthetic pathways of the triazatruxenes, 3P-T2T, 3P-T2P, 3P-Pyr, and (DT)2BTh2CN. | ||
The thermal stability of the material plays a crucial role in the fabrication of OLED devices with a vacuum-deposition process, which is analyzed by thermogravimetric analysis (TGA) under nitrogen. The new triazatruxene Tr-Tol exhibits a reliable thermal stability with a decomposition temperature relative to 5% weight loss (Td95) of 369 °C, which is higher than those of Tr-Me or Tr-Ph reported in the literature,28,29 mainly due to the higher molecular weight. The acceptors 3P-T2P and 3P-Pyr, on the other hand, show similar Td95 of 340 and 339 °C, respectively. The thermal stability of 3P-T2T (352 °C)27 is relatively higher than those of 3P-T2P or 3P-Pyr, which could be attributed to the higher molecular planarity due to the reduced steric interactions between peripheral aryl substitutions and the triazine core. The reduction of the nitrogen atom of the central heteroarene core leads 3P-T2P and 3P-Pyr to suffer from adjacent C–H/C–H bond repulsions, consequently giving them slightly twisted molecular conformations as well as decreased thermal stability. Nevertheless, from the aspect of optoelectronic applications, these materials possessed sufficiently high thermal stabilities, making them reliable for vacuum-deposition without the risk of decomposition during the high-temperature evaporation. The morphological stabilities of Tr-Tol, 3P-T2P and 3P-Pyr were examined with a differential scanning calorimeter (DSC). The observed glass transition temperature (Tg) was 64 °C for 3P-T2P and 67 °C for 3P-Pyr, whereas there was no phase transition detected for Tr-Tol up to 350 °C.
Cyclic voltammetry was utilized to probe the electrochemical properties. The redox potentials of Tr-Me, Tr-Ph, Tr-Tol, 3P-T2T, 3P-T2P and 3P-Pyr were respectively measured and recorded with reference to a ferrocene/ferrocenium (Fc/Fc+) redox couple, as shown in Fig. S1 (ESI†). A reversible oxidation potential at 0.27, 0.37 and 0.35 V was observed for Tr-Me, Tr-Ph and Tr-Tol, respectively. Obviously, the electronic properties of triazatruxene can be modulated with the structural feature of the N-substitution, in which the methyl-substituted Tr-Me shows a lower oxidation potential compared to those of the aryl-substituted counterparts Tr-Ph and Tr-Tol. There is no surprise that Tr-Tol exhibits a slightly lower oxidation potential than Tr-Ph due to the electron-richer p-tolyl groups. With reference to the Fc/Fc+ redox couple, the HOMO energy levels were estimated as −5.07 eV (Tr-Me), −5.17 eV (Tr-Ph), and −5.15 eV (Tr-Tol). The respective ionization potentials of Tr-Me, Tr-Ph and Tr-Tol solid films were measured by AC-2 to be −4.96, −5.26 and −5.16 eV, respectively (Fig. S2 in ESI†). The trend observed by AC-2 measurement agrees with the results from CV measurement. In contrast to the triazatruxene-based donors, there was a reversible reduction potential at −1.82 and −2.15 V for the N-heteroarene-cored acceptors 3P-T2T and 3P-T2P, respectively, while an irreversible reduction potential at −2.43 V was observed for 3P-Pyr. Apparently, the number of nitrogen atoms embedded in the N-heteroarene core strongly governs the reduction behavior, in which the most electron-deficient triazine core of 3P-T2T shows a better propensity to be reduced compared to those of 3P-T2P or 3P-Pyr. The LUMO energy levels of 3P-T2T, 3P-T2P and 3P-Pyr were estimated to be −2.98, −2.65 and −2.37 eV, respectively, which are in line with the degree of electron deficiency in their central N-heteroarene core. Interestingly, the difference in the HOMO energy levels between triazatruxene-based donors is relatively small, whereas the shift in LUMO energy level is more apparent in N-heteroarene-cored acceptors. Therefore, we could reasonably speculate that the exciplex energy upon combining triazatruxene donors and N-heteroarene acceptors would be tuned mainly by the acceptor counterpart.
The photophysical behaviors of triazatruxene-based donors were studied by UV-vis absorption and photoluminescence (PL) spectroscopy, as shown in Fig. 1a. The data are summarized in Table 1. In diluted toluene solution at room temperature, the absorption spectra of Tr-Ph and Tr-Tol corresponding to the π–π* transition are similar, with a maximum absorption peak (λmax) centered around 317 nm, which is blue-shifted compared to that (λmax = 324 nm) of Tr-Me. In addition, two weak absorption peaks were observed at 332 nm and 350 nm for Tr-Ph and Tr-Tol, respectively, whereas Tr-Me shows slightly red-shifted absorptions at 337 and 354 nm, which are also ascribed to the π–π* transition in the conjugated π-electron system.28 The PL spectra of Tr-Ph and Tr-Tol show a maximum peak (PL λmax) centered at 391 nm, while the PL λmax of Tr-Me is red-shifted to 396 nm. The slightly red-shifted emission of Tr-Me from Tr-Ph is in line with the results reported in the literature.28,29 The absorption and emission spectra of the vacuum-deposited films of Tr-Me, Tr-Ph and Tr-Tol were acquired, as shown in Fig. S3a (ESI†). Obviously, compared to those of Tr-Ph and Tr-Tol, the absorption spectrum of Tr-Me film exhibits a significant red-shift and broad features, indicating strong intermolecular interactions in the solid state. In addition, all of these triazatruxenes show emissions with vibronic and red-shifted characteristics compared to those observed in dilute toluene solution. Parallel to the trend observed in the absorption spectra, the PL spectrum of Tr-Me shows an emission maximum centered at 414 nm, which is significantly red-shifted compared to those of Tr-Ph and Tr-Tol, implying a much stronger propensity for intermolecular aggregations in Tr-Me neat film than in Tr-Ph or Tr-Tol films due to the smaller methyl groups. The strong aggregations render the determination of the energy of the triplet excited state difficult. Therefore, the PL spectra of Tr-Me, Tr-Ph and Tr-Tol (10 wt%) doped in mCP were investigated, as shown in Fig. 1b. PL spectra without long wavelength tailing were detected, indicating the suppressed intermolecular interaction, as mCP was employed as a solid matrix. However, a slight red-shift in PL λmax (405 nm) relative to 393 nm for both Tr-Ph and Tr-Tol was still observed. Accordingly, the corresponding energies of the singlet state estimated from the emission onset are 3.35, 3.41, 3.41 eV for Tr-Me, Tr-Ph and Tr-Tol, respectively. The phosphorescence spectra of Tr-Me, Tr-Ph and Tr-Tol doped films were acquired at 77 K (Fig. 1b). Therefore, the triplet energy was estimated to be 2.90 eV for all these triazatruxene-based donors by the onset (429 nm) of phosphorescence. Apparently, the N-phenyl or N-p-tolyl rings are twisted with respect to the triazatruxene core, thereby preventing intermolecular π–π interactions in Tr-Ph and Tr-Tol molecules. Whereas, lacking intrinsic structural congestion, Tr-Me molecules form severe self-aggregations that implies a poor tendency for forming a uniform film mixed with acceptor materials.
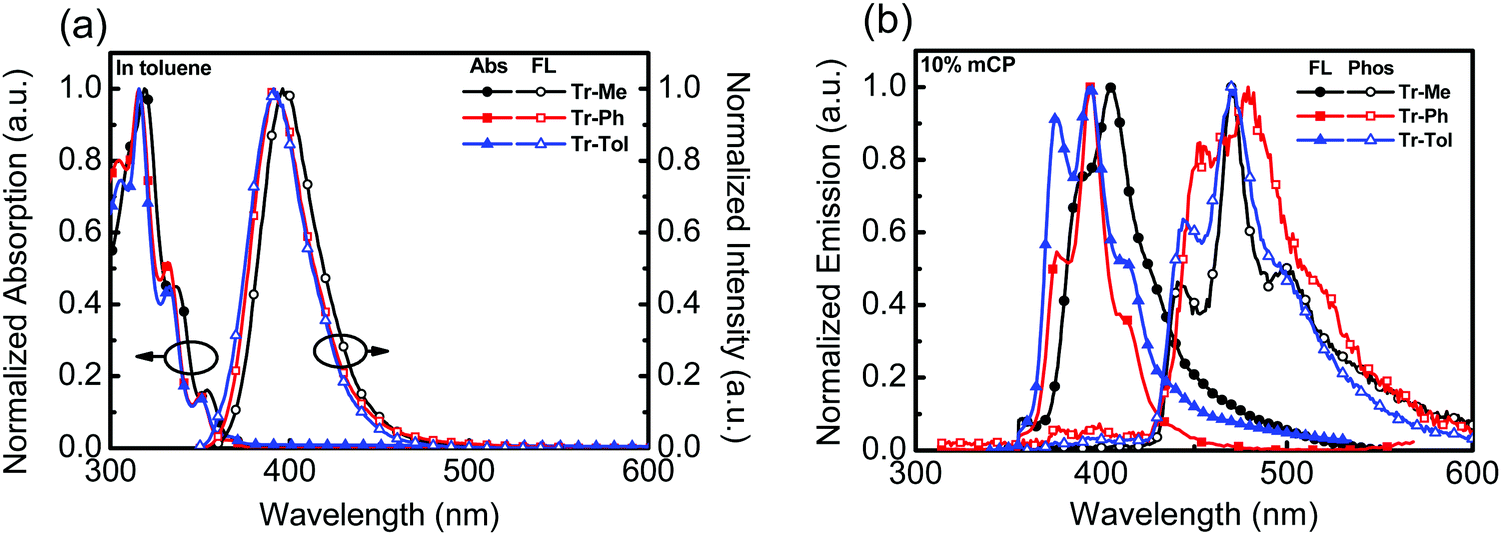 | ||
| Fig. 1 (a) UV-vis and PL of Tr-Me, Tr-Ph and Tr-Tol (10−5 M) in toluene, (b) the PL spectra of Tr-Me, Tr-Ph and Tr-Tol (10 wt%) doped in mCP measured at room temperature and 77 K. | ||
| Molecule | λ max sol./film (nm) | PL λmaxa sol./film/doped film (nm) | PLonset nm (eV)d | Phosonset nm (eV)e | E g (sol.) (eV) | HOMO (eV) | LUMO (eV) | IP (eV)f | T d (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in toluene at a concentration of about 10−5 M. b Calculated from potential vs. ferrocene/ferrocenium redox couple. c Calculated from the difference between HOMO/LUMO and corresponding optical bandgap. d Estimated from the onset of emission spectra from 10% materials doped in mCP at room temperature. e Estimated from the onset of emission spectra from the films of 10 wt% materials doped in mCP at 77 K. f Measured from AC-2. g Not available. h Reported value. | |||||||||
| Tr-Me | 319, 337, 354/325 | 396/414/406 | 371 (3.35) | 429 (2.90) | 3.37 | −5.07b | −1.70c | −4.96 | 327 |
| Tr-Ph | 316, 332, 350/319 | 391/397/394 | 364 (3.41) | 429 (2.90) | 3.31 | −5.17b | −1.86c | −5.26 | 363 |
| Tr-Tol | 316, 332, 350/319 | 391/394/394 | 364 (3.41) | 429 (2.90) | 3.31 | −5.15b | −1.84c | −5.16 | 369 |
| 3P-T2T | 267/268 | 397/419/440 | 395 (3.15) | 425 (2.92) | 3.58 | −6.56c | −2.98b | N/Ag | 352h |
| 3P-T2P | 261/259 | 361/380, 489/434 | 379 (3.28) | 430 (2.89) | 3.50 | −6.15c | −2.65b | N/A | 340 |
| 3P-Pyr | 258/259 | 367/367/396 | 349 (3.56) | 430 (2.89) | 3.63 | −6.00c | −2.37b | N/A | 339 |
The photophysical behaviors of N-heteroarene-cored acceptors were also studied by UV-vis absorption and PL spectroscopy, as shown in Fig. 2a. The data are summarized in Table 1. The absorption spectra of 3P-T2P and 3P-Pyr show similar results with the absorption peaks at around 259 and 325 nm, which could be attributed to the π–π* transition and n–π* transition,34,35 respectively. The absorption edge can be unambiguously assigned to determine the optical energy gap to be 3.58, 3.50 and 3.63 eV for 3P-T2T, 3P-T2P and 3P-Pyr, respectively. The emission with the PL λmax of 3P-T2P and 3P-Pyr in dilute CH2Cl2 solution is detected at 360 nm, which is much blue-shifted compared to that of 3P-T2T (395 nm) reported previously.27 The absorptions (Fig. S3b, ESI†) of 3P-T2P and 3P-Pyr films are similar with λmax at around 259 nm, which is slightly blue-shifted from the 268 nm of 3P-T2P film.27 Apart from 3P-T2T and 3P-Pyr, which show single emission peaks centered at 420 and 367 nm, respectively, in their emission spectra of neat films, the emission spectrum of 3P-T2P shows a weak emission at 380 nm corresponding to its typical molecular chromophore and a strong and broad emission centered at 490 nm ascribed to the excimer emission. The emission spectra of 3P-T2P, 3P-T2P and 3P-Pyr (10 wt%) blended in mCP were examined, as shown in Fig. 2b. The lower energy emission of 3P-T2P disappeared when it served as a guest dopant dispersed in an mCP host, verifying the possibility of forming excimer emission in pristine film. The respective PL λmax of 3P-T2P, 3P-T2P and 3P-Pyr blended films are distinguishable, indicating the effects of the electron-accepting power of the central N-heteroarenes and molecular rigidity on the singlet energy. The higher electron-withdrawing propensity of the triazine core together with the lack of C–H/C–H steric interactions leads 3P-T2T to have a red-shifted λmax of 395 nm as compared to the 379 nm of 3P-T2P and 349 nm of 3P-Pyr. The triplet energies of 3P-T2T, 3P-T2P and 3P-Pyr were determined to be 2.92, 2.89 and 2.89 eV, respectively, based on the onset energy of the phosphorescence spectra (Fig. 2b) obtained at 77 K.
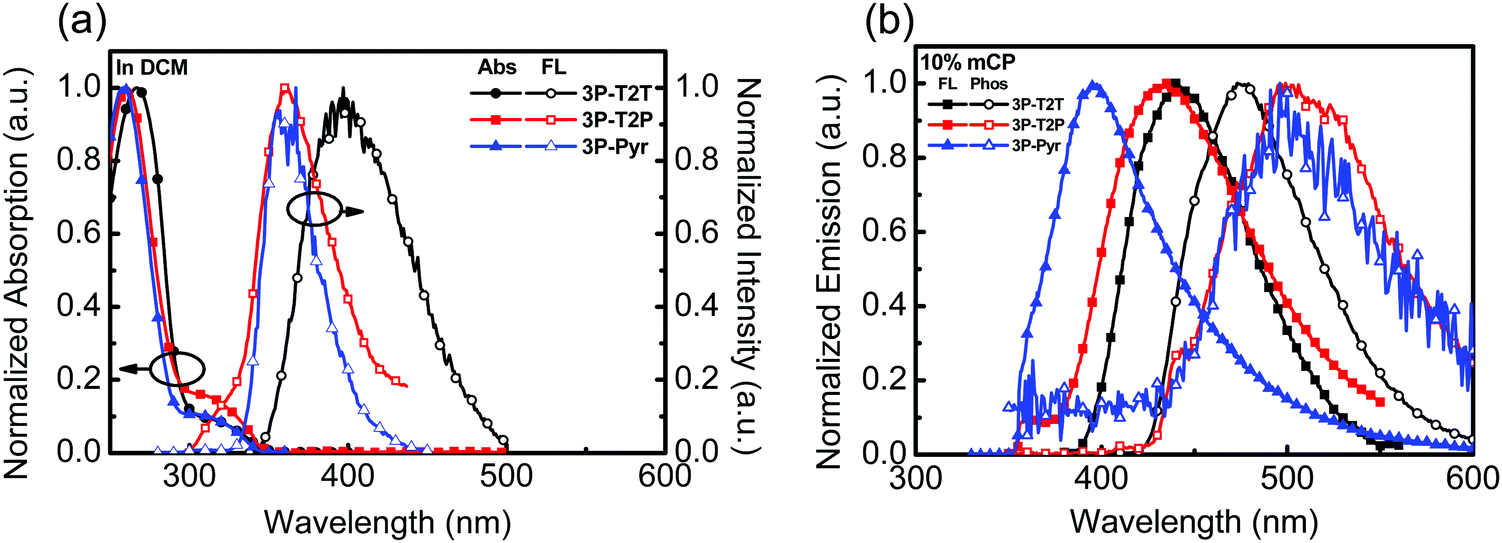 | ||
| Fig. 2 (a) UV-vis and PL of 3P-T2T, 3P-T2P and 3P-Pyr (10−5 M) in CH2Cl2, (b) the PL spectra of 3P-T2T, 3P-T2P and 3P-Pyr (10 wt%) doped in mCP measured at room temperature and 77 K. | ||
To explore the formation of exciplexes using Tr-Me, Tr-Ph and Tr-Tol as donors and 3P-T2T, 3P-T2P and 3P-Pyr as acceptors, an array of 9 vacuum-processed D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A (1:1) blends was prepared. The absorption spectra (Fig. S4, ESI†) of all blended films show strong absorption peaks around 260 and 320 nm, which can be considered to be the linear combination of composing donor and acceptor materials. The corresponding emission spectra are depicted in Fig. 3 and the data are summarized in Table 2. No significant new absorption peaks are found in these blends, indicating the lack of ground state interactions between donors and acceptors. In contrast, the blended films did exhibit distinct emission spectra different from their components. As compared to the individual emission of donor or acceptor, the red-shifted emission of blended film is the signature of forming an exciplex upon photoexcitation. The blends consisting of Tr-Me as donor mixed with acceptors 3P-T2T, 3P-T2P and 3P-Pyr gave emission peaks (Fig. 3a) centered at 551, 557 and 530 nm, respectively. In addition, the PL spectra of Tr-Me:3P-T2T and Tr-Me:3P-T2P blends show the residual emission of Tr-Me, indicating incomplete exciplex formation due to the self-segregation of less sterically hindered Tr-Me together with more coplanar acceptors 3P-T2T and 3P-T2P. However, the emission of individual D or A components disappeared in the Tr-Me:3P-Pyr blend, indicating that the twisted conformation of 3P-Pyr is beneficial for suppressing its self-segregation and also blocking the severe intermolecular interactions of Tr-Me molecules. The Tr-Ph-blended films composed of acceptors 3P-T2T, 3P-T2P and 3P-Pyr show emission (Fig. 3b) centered at 552, 527 and 482 nm, respectively. Finally, the blends formed with Tr-Tol as donor and 3P-T2T, 3P-T2P and 3P-Pyr as respective acceptors give emission peaks (Fig. 3c) centered at 554, 526 and 492 nm, respectively. In general, the Tr-Me-based blends exhibit red-shifted emissions compared to those counterpart blends with Tr-Ph and Tr-Tol as donors due to its higher lying HOMO energy level. Obviously, the energy of exciplex emission, typically correlated with the energy difference between donor (HOMO) and acceptor (LUMO),19 is governed mainly by the structural features of N-heteroarene centers. Time-resolved photoluminescence (TRPL) experiments, except for the incomplete exciplex formation of the blends with Tr-Me as donor, were conducted to probe the relaxation processes of these exciplex emissions (Fig. S5, ESI†). The results are summarized in Table 2. We speculate that the low electron deficiency of 3P-Pyr together with relatively weak donors Tr-Ph and Tr-Tol may not induce sufficient polarization for efficient rISC upon photoexcitation.36 The pump and delayed fluorescence spectra are provided in Fig. S6 (ESI†). The prompt emission acquired by an intensified charge couple detector at zero delay time and a gate width of 100 ns is attributed to fluorescence, while the delayed emission acquired at a delay time of 1 μs and gate width of 10 μs is ascribed to delayed fluorescence. The prompt and delayed emissions were recorded from the emission onsets to calculate the singlet–triplet splitting energy (ΔEST). Obviously, the large ΔEST of Tr-Ph: 3P-Pyr (0.34 eV) and Tr-Tol: 3P-Pyr (0.33 eV) make it hard to undergo an efficient rISC process that leads to low PLQY. To our delight, the TRPL of the 4 blends consisting of Tr-Ph and Tr-Tol as donors and 3P-T2T and 3P-T2P as acceptors showed rather evident delayed components in a microsecond timescale and moderate PLQYs ranging from 33 to 41%, indicating the possibility of producing better device characteristics due to superior TADF characters.
:A (1:1) blends was prepared. The absorption spectra (Fig. S4, ESI†) of all blended films show strong absorption peaks around 260 and 320 nm, which can be considered to be the linear combination of composing donor and acceptor materials. The corresponding emission spectra are depicted in Fig. 3 and the data are summarized in Table 2. No significant new absorption peaks are found in these blends, indicating the lack of ground state interactions between donors and acceptors. In contrast, the blended films did exhibit distinct emission spectra different from their components. As compared to the individual emission of donor or acceptor, the red-shifted emission of blended film is the signature of forming an exciplex upon photoexcitation. The blends consisting of Tr-Me as donor mixed with acceptors 3P-T2T, 3P-T2P and 3P-Pyr gave emission peaks (Fig. 3a) centered at 551, 557 and 530 nm, respectively. In addition, the PL spectra of Tr-Me:3P-T2T and Tr-Me:3P-T2P blends show the residual emission of Tr-Me, indicating incomplete exciplex formation due to the self-segregation of less sterically hindered Tr-Me together with more coplanar acceptors 3P-T2T and 3P-T2P. However, the emission of individual D or A components disappeared in the Tr-Me:3P-Pyr blend, indicating that the twisted conformation of 3P-Pyr is beneficial for suppressing its self-segregation and also blocking the severe intermolecular interactions of Tr-Me molecules. The Tr-Ph-blended films composed of acceptors 3P-T2T, 3P-T2P and 3P-Pyr show emission (Fig. 3b) centered at 552, 527 and 482 nm, respectively. Finally, the blends formed with Tr-Tol as donor and 3P-T2T, 3P-T2P and 3P-Pyr as respective acceptors give emission peaks (Fig. 3c) centered at 554, 526 and 492 nm, respectively. In general, the Tr-Me-based blends exhibit red-shifted emissions compared to those counterpart blends with Tr-Ph and Tr-Tol as donors due to its higher lying HOMO energy level. Obviously, the energy of exciplex emission, typically correlated with the energy difference between donor (HOMO) and acceptor (LUMO),19 is governed mainly by the structural features of N-heteroarene centers. Time-resolved photoluminescence (TRPL) experiments, except for the incomplete exciplex formation of the blends with Tr-Me as donor, were conducted to probe the relaxation processes of these exciplex emissions (Fig. S5, ESI†). The results are summarized in Table 2. We speculate that the low electron deficiency of 3P-Pyr together with relatively weak donors Tr-Ph and Tr-Tol may not induce sufficient polarization for efficient rISC upon photoexcitation.36 The pump and delayed fluorescence spectra are provided in Fig. S6 (ESI†). The prompt emission acquired by an intensified charge couple detector at zero delay time and a gate width of 100 ns is attributed to fluorescence, while the delayed emission acquired at a delay time of 1 μs and gate width of 10 μs is ascribed to delayed fluorescence. The prompt and delayed emissions were recorded from the emission onsets to calculate the singlet–triplet splitting energy (ΔEST). Obviously, the large ΔEST of Tr-Ph: 3P-Pyr (0.34 eV) and Tr-Tol: 3P-Pyr (0.33 eV) make it hard to undergo an efficient rISC process that leads to low PLQY. To our delight, the TRPL of the 4 blends consisting of Tr-Ph and Tr-Tol as donors and 3P-T2T and 3P-T2P as acceptors showed rather evident delayed components in a microsecond timescale and moderate PLQYs ranging from 33 to 41%, indicating the possibility of producing better device characteristics due to superior TADF characters.
 | ||
| Fig. 3 Emission spectra of (a) Tr-Me:acceptor, (b) Tr-Ph:acceptor and (c) Tr-Tol:acceptor intermixed films. | ||
| Donor | Acceptor | PLQYa (%) | PLmax/onset (nm) | ΔESTb (eV) | TRPLc | |||
|---|---|---|---|---|---|---|---|---|
| A 1 | τ 1 (μs) | A 2 | τ 2 (ns) | |||||
|
a The PLQY for the blend were measured with an integrating sphere (Hamamatsu C9920-02).
b ΔEST are calculated from the energy difference between the emission onsets of prompt and delayed emissions, as shown in Fig. S6 (ESI).
c The TRPL profiles were measured under an ambient atmosphere, and the decay components were fitted with two exponential decay models as I(τ) = A1exp(−t/τ1) + A2exp(−t/τ2), as shown in Fig. S5 (ESI).
d Derived optical energy gap (eV) from 1242.4/PLonset (nm).
|
||||||||
| Tr-Ph | 3P-T2T | 39 | 553/479 (2.59)d | 0.10 | 0.11 | 1.95 | 0.89 | 33.1 |
| 3P-T2P | 41 | 526/456 (2.73) | 0.18 | 0.09 | 1.77 | 0.91 | 52.0 | |
| 3P-Pyr | 8 | 480/405 (3.07) | 0.34 | 0.06 | 0.07 | 0.94 | 25.6 | |
| Tr-Tol | 3P-T2T | 33 | 553/480 (2.59) | 0.10 | 0.10 | 1.73 | 0.90 | 25.8 |
| 3P-T2P | 40 | 525/457 (2.73) | 0.10 | 0.09 | 2.39 | 0.91 | 56.4 | |
| 3P-Pyr | 10 | 491/405 (3.07) | 0.33 | 0.04 | 1.0 | 0.96 | 29.0 | |
Device
To investigate the electroluminescence (EL) from the exciplexes, OLEDs were fabricated with the device architecture ITO/4 wt% ReO3:donor (60 nm)/donor (15 nm)/D:A (1:1, 20 nm)/acceptor (10 nm)/CN-T2T (40 nm)/Liq (0.5 nm)/Al (100 nm). ITO and Liq/Al are the anode and the cathode, respectively (Fig. 4). Due to the severe self-aggregation of Tr-Me, only Tr-Ph and Tr-Tol were selected as donors to blend with acceptors 3P-T2T, 3P-T2P and 3P-Pyr. The mixtures of D and A (1:1 weight ratio) were introduced as the EML. The 4 wt% rhenium oxide (ReO3) doped in the donor was used as the hole injection layer (HIL).37 The hole mobility of Tr-Ph and Tr-Tol determined by the time-of-flight technique is ca. 10−4 cm2 V−1 s−1, as shown in Fig. S7 (ESI†), making donors Tr-Ph and Tr-Tol good hole-transporting layers. A previously reported 3′,3′′′,3′′′′′-(1,3,5-triazine-2,4,6-triyl)tris-([1,1′-biphenyl]-3-carbonitrile) (CN-T2T)38 with a high triplet level (ET = 2.82 eV), suitable HOMO/LUMO (−6.7/−2.78 eV) and high electron mobility (μe = 10−4 cm2 V−1 s−1 order) was selected as the electron-transporting layer. The additional 10 nm acceptor was introduced as a hole-blocking layer (HBL) to prevent undesirable exciplex formation between donor and CN-T2T.
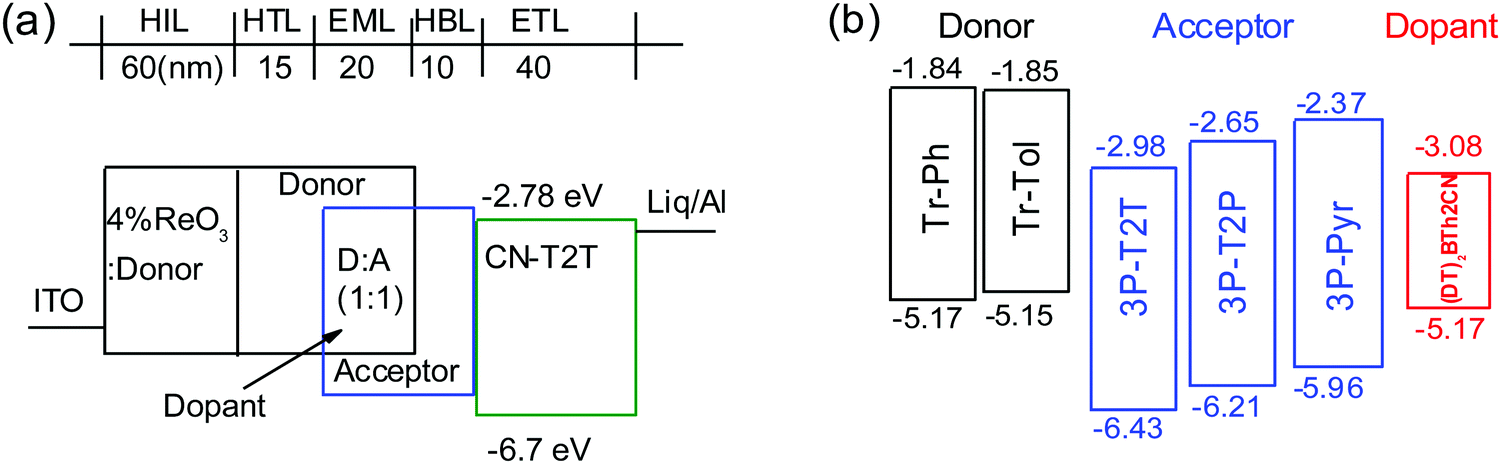 | ||
| Fig. 4 (a) Schematic exciplex-based OLED structure, and (b) energy levels of donors, acceptors, and fluorescent emitter used in this study. | ||
Fig. 5 depicts the current density–voltage–luminance (J–V–L) characteristics, device efficiencies, and EL spectra. The key characteristics are summarized in Table 3. Fig. 4b shows a schematic energy level of the donors and acceptors as well as the fluorescent emitter used in this work. The HOMO levels of Tr-Ph and Tr-Tol are similar (−5.17 and −5.15 eV). However, the LUMO levels of 3P-Pyr (−2.37 eV) > 3P-T2P (−2.65 eV) > 3P-T2T (−2.98 eV) will strongly influence the emissive property of the resultant exciplex. Devices A1 and B1 use the EML where 3P-T2T was selected as the acceptor to blend with Tr-Ph and Tr-Tol, respectively. The electrons are injected from CN-T2T to 3P-T2T without any energy barrier, leading to a low turn-on voltage (Von) of 2.0 eV and a high current density. Device A1 gives a luminance (L) of 84318 cd m−2 at 6.0 V (1309 mA cm−2) with an EL peak at 558 nm and CIE coordinates of (0.43, 0.53) and the maximum EQE, current efficiency (CE), and power efficiencies (PE) are 10.3%, 29.8 cd A−1 and 37.5 lm W−1. The extremely low driving voltage of 2.7 V at 1000 cd m−2 means that the carriers are injected smoothly into the HOMO/LUMO levels of the exciplex EML without any energy barrier. Device A1 retains an EQE of 9.9% at a high luminance of 1000 cd m−2. In addition, device A1 exhibits better efficiency than device B1 that exhibits EQE, CE, and PE of 9.7%, 27.3 cd A−1, and 35.7 lm W−1 and CIE coordinates of (0.44, 0.53). This result is consistent with the PLQYs measured for the thin films of Tr-Ph:3P-T2T (39%) and Tr-Tol:3P-T2T (33%) blends. Obviously, similar to the previously reported results regarding 3P-T2T39–41 as a good acceptor with other donors for efficient exciplex systems, the triazatruxene-based molecules Tr-Ph and Tr-Tol are also good donors, which can be further utilized to examine the new exciplex-forming systems with other acceptors.
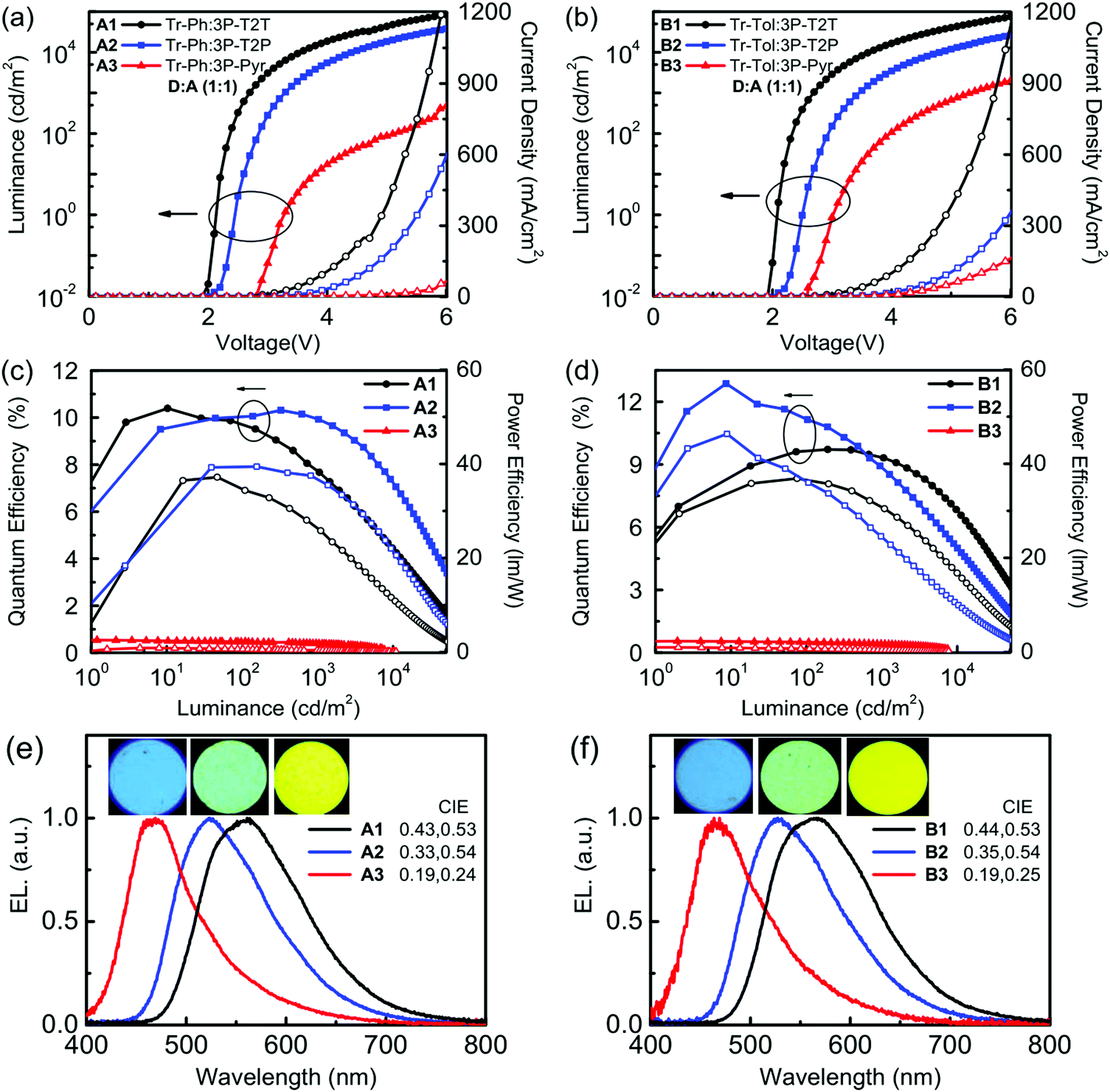 | ||
| Fig. 5 (a)/(b) Current density–voltage–luminance (J–V–L) characteristics, (c)/(d) external quantum efficiencies (EQE) and power efficiencies (PE) as a function of luminance, and (e)/(f) EL spectra of Tr-Ph and Tr-Tol based exciplex devices. | ||
| EML | V on [V] | L/J at 6 V [cd m−2/mA cm−2] | EQE/CE/PEmax [%/cd A−1/lm W−1] | At 1000c cd m−2 [%/cd A−1/lm W−1 V−1] | λ max [nm] | CIE [x, y] | |
|---|---|---|---|---|---|---|---|
| a The notation A and B indicate the donors Tr-Ph and Tr-Tol, respectively. The notation 1–3 indicate the acceptors 3P-T2T, 3P-T2P and 3P-Pyr, respectively. The notation r indicates 10 wt% (DT)2BTh2CN as dopant. b Turn-on voltage at which emission became detectable. c The values of EQE, CE, PE and driving voltages of device at 1000 cd m−2. | |||||||
| A1 | Tr-Ph:3P-T2T | 2.0 | 84318/1309 |
10.3/29.8/37.5 | 9.92/28.7/33.4/2.7 | 558 | 0.43, 0.53 |
| A2 | Tr-Ph:3P-T2P | 2.2 | 37975/601 |
10.4/30.8/37.2 | 7.67/22.7/21.6/3.3 | 522 | 0.33, 0.54 |
| A3 | Tr-Ph:3P-Pyr | 2.8 | 463/64 | 0.54/0.91/0.86 | 0.40/0.67/0.31/6.7 | 469 | 0.19, 0.24 |
| B1 | Tr-Tol:3P-T2T | 2.0 | 75082/1157 |
9.7/27.3/35.7 | 9.5/26.6/32.2/2.6 | 566 | 0.44, 0.53 |
| B2 | Tr-Tol:3P-T2P | 2.2 | 26280/360 |
12.8/38.3/46.3 | 8.9/26.6/24.6/3.4 | 526 | 0.35, 0.54 |
| B3 | Tr-Tol:3P-Pyr | 2.6 | 2008/164 | 0.55/0.97/0.98 | 0.42/0.74/0.44/5.3 | 467 | 0.19, 0.25 |
| 10 wt% (DT)2BTh2CN doped in cohost | |||||||
| A1r | Tr-Ph:3P-T2T | 1.8 | 3165/1961 | 4.64/1.24/1.96 | 1.07/0.28/0.20/4.5 | 674 | 0.69, 0.31 |
| A2r | Tr-Ph:3P-T2P | 2.3 | 966/410 | 5.52/1.66/2.09 | 0.75/0.22/0.11/6.1 | 671 | 0.68, 0.31 |
| B1r | Tr-Tol:3P-T2T | 1.8 | 3666/3078 | 3.48/0.98/1.47 | 0.68/0.19/0.13/4.5 | 669 | 0.69, 0.31 |
| B2r | Tr-Tol:3P-T2P | 2.3 | 782/276 | 4.42/1.72/2.16 | 0.64/0.25/0.12/6.4 | 667 | 0.67, 0.32 |
Devices A2 and B2 using the exciplex-based EML composed of 3P-T2P as acceptor and Tr-Ph and Tr-Tol as donors, respectively, exhibit green emissions, which are blue-shifted compared to those of the devices employing 3P-T2T as an acceptor due to the higher LUMO energy of 3P-T2P. The lower current density and higher Von for 3P-T2P-based devices are due to the energy barrier (0.13 eV) for the electron injection from CN-T2T to 3P-T2P. Device B2 based on the Tr-Tol:3P-T2P blend as the EML shows the best performance among the devices reported in this work, giving a Von of 2.2 V and maximum efficiencies as high as 12.8% (EQE), 38.3 cd A−1 (CE), and 46.3 lm W−1 (PE) with CIE coordinates of (0.35, 0.54). Correspondingly, the device with the Tr-Ph:3P-T2P blend shows lower maximum efficiencies (10.4%, 30.8 cd A−1, 37.2 lm W−1) with CIE coordinates of (0.33, 0.54). The superior efficiency obtained in devices A2 and B2 agrees with the observed better PLQYs of exciplex-forming systems with 3P-T2P as the acceptor (Table 2). Devices A3 and B3 use 3P-Pyr as an acceptor and Tr-Ph and Tr-Tol as a donor, respectively, as the EML for making exciplexes. The higher LUMO level (−2.37 eV) of 3P-Pyr leads the exciplex to have blue emission with CIE coordinates of (0.19, 0.24–0.25). The low J–V performance and high Von are because of the larger energy barrier (0.41 eV, see Fig. 4a and b) between CN-T2T and 3P-Pyr that impedes the electron injection to the EML. Together with the low fluorescence quantum yield of 8% for Tr-Ph:3P-Pyr and 10% for Tr-Tol:3P-Pyr, these give the low EQE (<1%) of 3P-Pyr-based blue devices.
The highly efficient exciplex system can be employed as the cohost for improving the efficiencies of OLEDs42 because the triplet excitons of the exciplex can be up-converted back to singlets by way of the efficient rISC process. Then, the singlet excitons of the exciplex cohost can transfer to the singlet state of the fluorescent dopant via a Förster energy transfer (FRET) pathway, making high-efficiency fluorescent OLEDs accessible.11 To realize an effective FRET, a good spectral overlap between the emission of the host and the absorption of the dopant is required. Fig. 6(a) displays the good overlaps between the absorption spectrum of the newly developed fluorescent dopant (DT)2BTh2CN (Scheme 1) and the PL spectra of exciplex cohosts (Tr-Ph/Tr-Tol:3P-T2T and Tr-Ph/Tr-Tol:3P-T2P), ensuring that efficient energy transfer from the exciplex to the dopant is feasible.
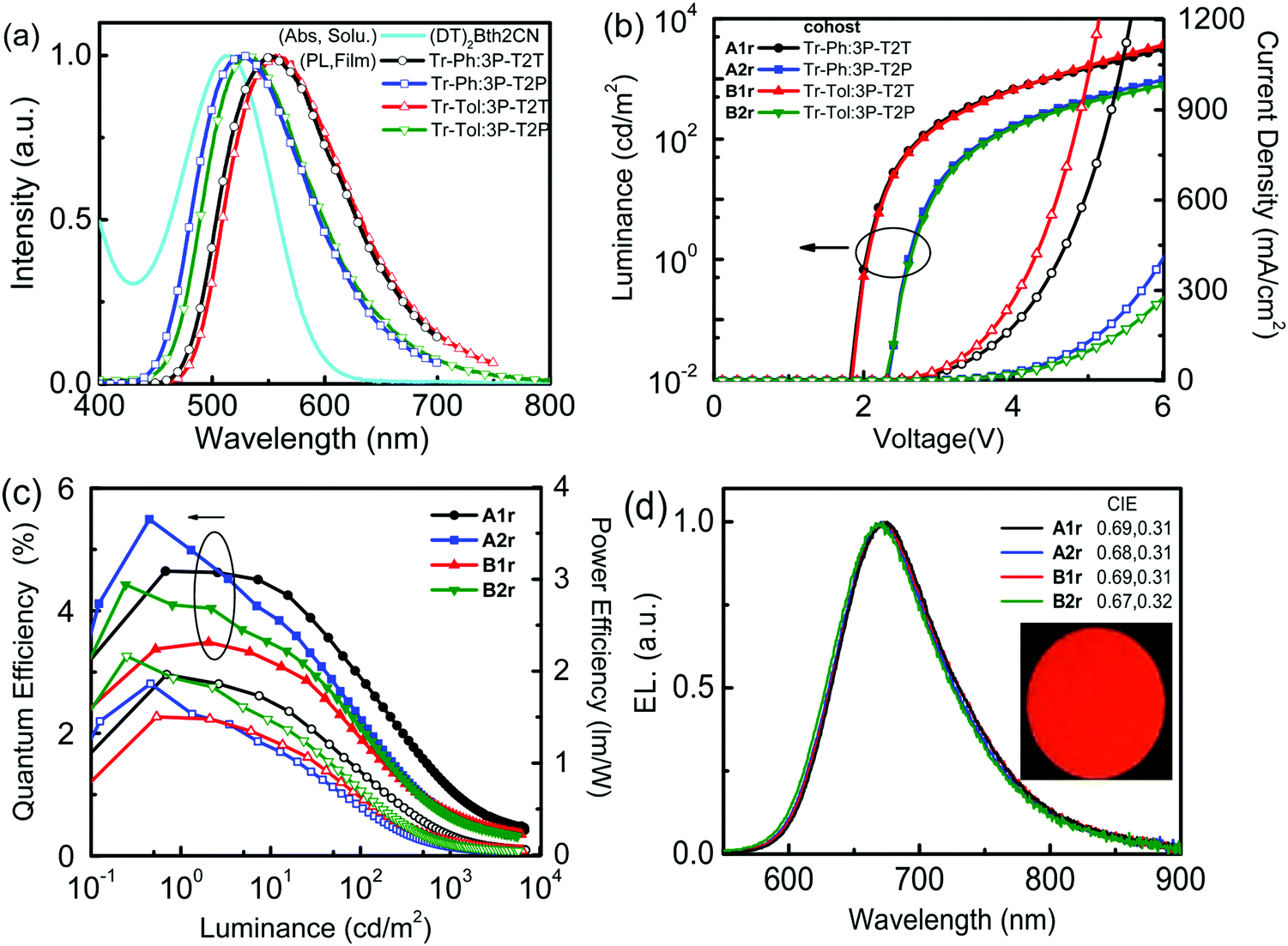 | ||
| Fig. 6 (a) The PL spectra of the exciplex cohost and the absorption spectrum of the dopant (DT)2BTh2CN. (b) J–V–L characteristics, (c) EQE and PE as a function of luminance and (d) EL spectra of 10 wt% (DT)2BTh2CN doped devices. | ||
The exciplex-forming systems were further exploited as cohosts for a red fluorescent dopant (DT)2BTh2CN. The photophysical and electrochemical properties of (DT)2BTh2CN are depicted in Fig. S8 (ESI†), and the data together with the thermal stability are summarized Table S1 (ESI†). First, the devices employing the best exciplex Tr-Tol:3P-T2P blend as the cohost incorporated with various doping concentrations of (DT)2BTh2CN were fabricated to optimize the EL performance (Fig. S9, ESI†). The device doped with 1 wt% (DT)2BTh2CN exhibits maximum EQE and PE of 5.5% and 8.4 lm W−1, respectively. However, at such a low doping concentration, the EL spectrum shows residual emission from the exciplex cohost with an EL peak at 520 nm.43 The device doped with 5 wt% (DT)2BTh2CN displays better energy transfer; however, a weak emission from the exciplex cohost still remains. This device shows the EL emission peak centered at 660 nm and CIE coordinates of (0.64, 0.35), but a lower maximum EQE (4.39%) compared to the 1 wt% doped device. When the doping concentration increases to 10 wt%, the device retains a comparable EQE (4.42%) and exhibits EL solely from the dopant but with the peak further red-shifted to 672 nm and CIE coordinates of (0.67, 0.32), indicating complete energy transfer. The color-shifting is because of the intermolecular interactions and strong local polarization field.44 The reduced device efficiency found in the devices with higher doping concentrations of (DT)2BTh2CN can be plausibly ascribed to the triplet–triplet Dexter energy transfer that leads to direct exciton quenching by the non-emissive triplets of the fluorescent dopant. To balance the efficient Förster energy transfer for giving pure emission of (DT)2BTh2CN and the competitive quenching process by the inevitable Dexter energy transfer, 10 wt% (DT)2BTh2CN was selected as the doping concentration for the exciplex cohosts (Tr-Ph/Tr-Tol:3P-T2T and Tr-Ph/Tr-Tol:3P-T2P) to investigate the performance of the deep-red fluorescent devices. Fig. 6(b–d) depict the EL characteristics of these deep-red fluorescent devices, and the key data are summarized in Table 3. All 10 wt% (DT)2BTh2CN-doped devices exhibit pure emission from the dopant with the EL peak centered at ca. 670 nm. Among them, the Tr-Ph:3P-T2P-hosted device A2r shows the best performance, achieving maximum EQE of 5.52%, CE of 1.66 cd A−1 and PE of 2.09 lm W−1 with CIE coordinates of (0.68, 0.31). The 5.52% EQE based on a Tr-Ph:3P-T2T cohost is higher than the theoretical limit of 5% typically anticipated for a conventional fluorescence emitter. The device performances are in good agreement with the PLQYs of the (DT)2BTh2CN emission extracted from the exciplex-forming cohosts as in the data summarized in Table S2 (ESI†). TRPL measurements were further performed on the (DT)2BTh2CN-doped exciplex cohosts (Tr-Ph/Tr-Tol:3P-T2T and Tr-Ph/Tr-Tol:3P-T2P) to probe the working mechanism. The data are summarized in Table S2 (ESI†). As indicated, the (DT)2BTh2CN-dispersed films show a dominant prompt decay component with a lifetime of around 7 ns together with a delay component with a lifetime of around 0.13 μs. The delay component only counted for about 1% of the whole relaxation process when Tr-Ph was applied as the donor in the blended film. In the case of Tr-Tol utilized as the donor for exciplex-forming systems, the delay components are as low as only 0.2% of the overall relaxation process. Accordingly, the EL from the exciplex-forming cohosts doped with 10 wt% (DT)2BTh2CN is mainly contributed by the Förster energy transfer, but simultaneously suffers from the inferior triplet-harvesting of the exciplex due to the competing Dexter energy transfer quenching in the presence of high dopant concentration.
Conclusion
For the first time, three triazatruxenes (Tr-Me, Tr-Ph, Tr-Tol) were examined as donors for exciplex formation with N-heteroarene-cored acceptors (3P-T2T, 3P-T2P, 3P-Pyr). As compared to those of pristine donor and acceptor films, these nine D:A combinations were found to show red-shifted emissions that are the signatures of forming exciplexes. The emission energies of the exciplexes corresponding to the interfacial energy difference between the HOMO (donor) and the LUMO (acceptor) are governed mainly by the features of N-heteroarene cores of acceptors, while the HOMOs of triazatruxene donors are less sensitive to the substituents. The red-shifted absorption and emission spectra of Tr-Me pristine film indicate the evident intermolecular interactions due to the less hindered methyl substitutions. The severe self-aggregation together with the coplanar feature of acceptors prevent the complete exciplex formation of Tr-Me:3P-T2T and Tr-Me:3P-T2P blends, as indicated by the presence of the residual emission of Tr-Me. The inferior exciplex formation of Tr-Me can be suppressed by combining the acceptor 3P-Pyr with twisted conformation. To our surprise, the blends using 3P-Pyr as acceptor and Tr-Ph, Tr-Tol as the respective donors gave low PLQYs due to the insufficient polarization effect for promoting efficient rISC. Systematic examination of the device characteristics concluded that the four blends composed of Tr-Ph and Tr-Tol as donors and 3P-T2T and 3P-T2P as acceptors are good EMLs for OLED applications. Among them, the Tr-Tol:3P-T2P blend gave a high-efficiency green device with an EQEmax of up to 12.8%, agreeing with the observed superior PLQY upon photoexcitation. These new exciplex-forming systems were further exploited as cohosts to fabricate deep-red devices with a tailor-made fluorescent dopant (DT)2BTh2CN. The deep-red device based on the 10 wt% (DT)2BTh2CN doped Tr-Ph:3P-T2P blend as the EML gave EL λmax 671 nm and CIE (0.68, 0.31) solely from the dopant and an EQEmax of 5.52%, which is consistent with the highest PLQY observed in the exciplex-hosted (DT)2BTh2CN films. The TRPL studies on the exciplex-hosted (DT)2BTh2CN films provide information for proposing a reasonable working mechanism in an EL device. The limited proportion of the delayed component in the whole relaxation process of (DT)2BTh2CN doped in exciplex-forming blends implies that the EL is mainly contributed by the singlet-to-singlet Förster energy transfer without the aid of triplet-harvesting from the exciplex cohost due to the competitive triplet-to-triplet quenching of Dexter energy transfer occurring in the presence of a high dopant concentration. Our results reveal that the characteristics of exciplex-forming systems can be feasibly tuned by systematic structural variations implemented on donor and/or acceptor components and the versatile functions of an exciplex can be achieved in OLED applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Ministry of Science and Technology (Grant No. MOST 107-2113-M-002-019-MY3 and 106-2112-M-019-002-MY3).References
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Thermally activated delayed fluorescence from Sn4+-porphyrin complexes and their application to organic light emitting diodes-a novel mechanism for electroluminescence, Adv. Mater., 2009, 21, 4802–4806 CrossRef CAS PubMed.
- M. Ikai and S. Tokito, Highly efficient phosphorescence from organic light-emitting devices with an exciton-block layer, Appl. Phys. Lett., 2001, 79, 156–158 CrossRef CAS.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Efficient up-conversion of triplet excitons into a singlet state and its application for organic light emitting diodes, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. V. Voorhis, M. A. Baldo and T. M. Swager, Thermally activated delayed fluorescence materials based on homoconjugation effect of donor–acceptor triptycenes, J. Am. Chem. Soc., 2015, 137, 11908–11911 Search PubMed.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Purely organic electroluminescent material realizing 100% conversion from electricity to light, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Diboron compound-based organic light-emitting diodes with high efficiency and reduced efficiency roll-off, Nat. Photonics, 2018, 12, 235–240 CrossRef CAS.
- D. Zhang, M. Cai, Z. Bin, Y. Zhang, D. Zhang and L. Duan, Highly efficient blue thermally activated delayed fluorescent OLEDs with record-low driving voltages utilizing high triplet energy hosts with small singlet–triplet splittings, Chem. Sci., 2016, 7, 3355–3363 RSC.
- W. Zeng, H.-Y. Lai, W.-K. Lee, M. Jiao, Y.-J. Shiu, C. Zhong, S. Gong, T. Zhou, G. Xie, M. Sarma, K.-T. Wong, C.-C. Wu and C. Yang, External quantum efficiency for orange–red organic light emitting diodes by employing thermally activated delayed fluorescence emitters composed of 1,8-naphthalimide-acridine hybrids, Adv. Mater., 2018, 30, 1704961 Search PubMed.
- S.-W. Li, C.-H. Yu, C.-L. Ko, T. Chatterjee, W.-Y. Hung and K.-T. Wong, Cyanopyrimidine–carbazole hybrid host materials for high-efficiency and low-efficiency roll-off TADF OLEDs, ACS Appl. Mater. Interfaces, 2018, 10, 12930–12936 CrossRef CAS PubMed.
- X.-K. Liu, Z. Chen, C.-J. Zheng, M. Chen, W. Liu, X.-H. Zhang and C.-S. Lee, Nearly 100% triplet harvesting in conventional fluorescent dopant-based organic light-emitting devices through energy transfer from exciplex, Adv. Mater., 2015, 27, 2025–2030 CrossRef CAS PubMed.
- K. Goushi and C. Adachi, Efficient organic light-emitting diodes through up-conversion from triplet to singlet excited states of exciplexes, Appl. Phys. Lett., 2012, 101, 023306 CrossRef.
- V. Jankus, C.-J. Chiang, F. Dias and A. P. Monkman, Deep blue exciplex organic light-emitting diodes with enhanced efficiency; p-type or e-type triplet conversion to singlet excitons?, Adv. Mater., 2013, 25, 1455–1459 CrossRef CAS PubMed.
- B. S. Kim and J. Y. Lee, Engineering of mixed host for high external quantum efficiency above 25% in green thermally activated delayed fluorescence device, Adv. Funct. Mater., 2014, 24, 3970–3977 CrossRef CAS.
- M. Zhang, K. Wang, C.-J. Zheng, D.-Q. Wang, Y.-Z. Shi, H. Lin, S.-L. Tao, X. Li and X.-H. Zhang, Development of red exciplex for efficient OLEDs by employing a phosphor as a component, Front. Chem., 2019, 7, 16 Search PubMed.
- Y. Hu, Y.-J. Yu, Y. Yuan, Z.-Q. Jiang and L.-S. Liao, Exciplex-based organic light-emitting diodes with near-infrared emission, Adv. Opt. Mater., 2020, 1901917 Search PubMed.
- T. Zhang, B. Zhao, B. Chu, W. Li, Z. Su, X. Yan, C. Liu, H. Wu, Y. Gao, F. Jin and F. Hou, Simple structured hybrid WOLEDs based on incomplete energy transfer mechanism: from blue exciplex to orange dopant, Sci. Rep., 2015, 5, 10234 Search PubMed.
- S. Lee, K.-H. Kim, D. Limbach, Y.-S. Park and J.-J. Kim, Low roll-off and high efficiency orange organic light emitting diodes with controlled co-doping of green and red phosphorescent dopants in an exciplex forming co-host, Adv. Funct. Mater., 2013, 23, 4105–4110 CrossRef CAS.
- M. Sarma and K.-T. Wong, Exciplex: an intermolecular charge-transfer approach for TADF, ACS Appl. Mater. Interfaces, 2018, 10, 19279–19304 CrossRef CAS PubMed.
- N. R. A. Amin, K. K. Kesavan, S. Biring, C.-C. Lee, T.-H. Yeh, T.-Y. Ko, S.-W. Liu and K.-T. Wong, A comparative study via photophysical and electrical characterizations on interfacial and bulk exciplex-forming systems for efficient organic light-emitting diodes, ACS Appl. Electron. Mater., 2020, 2, 1011–1019 CrossRef.
- M. Guzauskas, D. Volyniuk, A. Tomkeviciene, A. Pidluzhna, A. Lazauskasc and J. V. Grazulevicius, Dual nature of exciplexes: exciplex-forming properties of carbazole and fluorene hybrid trimers, J. Mater. Chem. C, 2019, 7, 25–32 RSC.
- S. W. Shelton, T. L. Chen, D. E. Barclay and B. Ma, Solution-processable triindoles as hole selective materials in organic solar cells, ACS Appl. Mater. Interfaces, 2012, 4, 2534–2540 CrossRef CAS PubMed.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Highly efficient blue thermally activated delayed fluorescence emitters based on symmetrical and rigid oxygen-bridged boron acceptors, Nat. Photonics, 2018, 13, 540–546 Search PubMed.
- K. Rakstys, A. Abate, M. I. Dar, P. Gao, V. Jankauskas, G. Jacopin, E. Kamarauskas, S. Kazim, S. Ahmad, M. Grätzel and M. K. Nazeeruddin, Triazatruxene-based hole transporting materials for highly efficient perovskite solar cells, J. Am. Chem. Soc., 2015, 137, 16172–16178 Search PubMed.
- S. Mula, T. Han, T. Heiser, P. Lévêque, N. Leclerc, A. P. Srivastava, A. R. Carretero and G. Ulrich, Hydrogen bonding as a supramolecular tool for robust OFET devices, Chem. – Eur. J., 2019, 25, 8304–8312 CrossRef CAS PubMed.
- J.-H. Lee, S.-H. Cheng, S.-J. Yoo, H. Shin, J.-H. Chang, C.-I. Wu, K.-T. Wong and J.-J. Kim, An exciplex forming host for highly efficient blue organic light emitting diodes with low driving voltage, Adv. Funct. Mater., 2015, 25, 361 CrossRef CAS.
- H.-F. Chen, T.-C. Wang, S.-W. Lin, W.-Y. Hung, H.-C. Dai, H.-C. Chiu, K.-T. Wong, M.-H. Ho, T.-Y. Cho, C.-W. Chen and C.-C. Lee, Peripheral modification of 1,3,5-triazine based electron-transporting host materials for sky blue, green, yellow, red, and white electrophosphorescent devices, J. Mater. Chem., 2012, 22, 15620–15627 RSC.
- G. W. Kim, R. Lampande, D. C. Choe, H. W. Bae and J. H. Kwon, Efficient hole injection material for low operating voltage blue fluorescent organic light emitting diodes, Thin Solid Films, 2015, 589, 105–110 CrossRef CAS.
- D. H. Huh, G. W. Kim, G. H. Kim, C. Kulshreshtha and J. H. Kwon, High hole mobility hole transport material for organic light-emitting devices, Synth. Met., 2013, 180, 79–84 Search PubMed.
- M. Franceschin, L. G. Satriani, A. Alvino, G. Ortaggi and A. Bianco, Study of a convenient method for the preparation of hydrosoluble fluorescent triazatruxene derivatives, Eur. J. Org. Chem., 2010, 134–141 CrossRef CAS.
- S.-J. Su, C. Cai and J. Kido, Three-carbazole-armed host materials with various cores for RGB phosphorescent organic light-emitting diodes, J. Mater. Chem., 2012, 22, 3447–3456 RSC.
- A. G. Martinez, A. H. Fernandez, F. M. Jimenez, A. G. Fraile, L. R. Subramanian and M. Hanack, On the mechanism of the reaction between ketones and trifluoromethanesulfonic anhydride. An improved and convenient method for the preparation of pyrimidines and condensed pyrimidines, J. Org. Chem., 1992, 57, 1627–1630 CrossRef.
- J. D. Douglas, G. Griffini, T. W. Holcombe, E. P. Young, O. P. Lee, M. S. Chen and J. M. J. Fréchet, Functionalized isothianaphthene monomers that promote quinoidal character in donor–acceptor copolymers for organic photovoltaics, Macromolecules, 2012, 45, 4069–4074 CrossRef CAS.
- S.-J. Su, H. Sasabe, T. Takeda and J. Kido, Pyridine-containing bipolar host materials for highly efficient blue phosphorescent OLEDs, Chem. Mater., 2008, 20, 1691–1693 CrossRef CAS.
- Y. Watanabe, D. Yokoyama, T. Koganezawa, H. Katagiri, T. Ito, S. Ohisa, T. Chiba, H. Sasabe and J. Kido, Control of molecular orientation in organic semiconductor films using weak hydrogen bonds, Adv. Mater., 2019, 31, 1808300 CrossRef PubMed.
- M. Wang, Y.-H. Huang, K.-S. Lin, T.-H. Yeh, J. Duan, T.-Y. Ko, S.-W. Liu, K.-T. Wong and B. Hu, Revealing the cooperative relationship between spin, energy, and polarization parameters toward developing high-efficiency exciplex light-emitting diodes, Adv. Mater., 2019, 31, 1904114 CrossRef CAS PubMed.
- D.-S. Leem, H.-D. Park, J.-W. Kang, J.-H. Lee, J. W. Kim and J.-J. Kim, Low driving voltage and high stability organic light-emitting diodes with rhenium oxide-doped hole transporting layer, Appl. Phys. Lett., 2007, 91, 011113 CrossRef.
- W.-Y. Hung, P.-Y. Chiang, S.-W. Lin, W.-C. Tang, Y.-T. Chen, S.-H. Liu, P.-T. Chou, Y.-T. Hung and K.-T. Wong, Balance the carrier mobility to achieve high performance exciplex OLED using a triazine-based acceptor, ACS Appl. Mater. Interfaces, 2016, 8, 4811–4818 CrossRef CAS PubMed.
- C.-J. Shih, C.-C. Lee, Y.-H. Chen, S. Biring, G. Kumar, T.-H. Yeh, S. Sen, S.-W. Liu and K.-T. Wong, Exciplex-forming cohost for high efficiency and high stability phosphorescent organic light emitting diodes, ACS Appl. Mater. Interfaces, 2018, 10, 2151–2157 CrossRef CAS PubMed.
- W.-Y. Hung, G.-C. Fang, Y.-C. Chang, T.-Y. Kuo, P.-T. Chou, S.-W. Lin and K.-T. Wong, Highly efficient bilayer interface exciplex for yellow organic light-emitting diode, ACS Appl. Mater. Interfaces, 2013, 5, 6826–6831 CrossRef CAS PubMed.
- Y.-C. Lo, T.-H. Yeh, C.-K. Wang, B.-J. Peng, J.-L. Hsieh, C.-C. Lee, S.-W. Liu and K.-T. Wong, High-efficiency red and near-infrared organic light-emitting diodes enabled by pure organic fluorescent emitters and an exciplex-forming cohost, ACS Appl. Mater. Interfaces, 2019, 11, 23417–23427 CrossRef CAS PubMed.
- Q. Wang, Q.-S. Tian, Y.-L. Zhang, X. Tang and L.-S. Liao, High-efficiency organic light-emitting diodes with exciplex hosts, J. Mater. Chem. C, 2019, 7, 11329–11360 RSC.
- B. Zhao, T. Zhang, B. Chu, W. Li, Z. Su, H. Wu, X. Yan, F. Jin, Y. Gao and C. Liu, Highly efficient red OLEDs using DCJTB as the dopant and delayed fluorescent exciplex as the host, Sci. Rep., 2015, 5, 10697 Search PubMed.
- K.-H. Kim, C.-K. Moon, J. W. Sun, B. Sim and J.-J. Kim, Triplet harvesting by a conventional fluorescent emitter using reverse intersystem crossing of host triplet exciplex, Adv. Opt. Mater., 2015, 3, 895–899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00188k |
| This journal is © the Partner Organisations 2020 |