
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjugated molecules for colourimetric and fluorimetric sensing of sodium and potassium†
Zachary S.
Parr
 and
Christian B.
Nielsen
*
and
Christian B.
Nielsen
*
Materials Research Institute and School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London E1 4NS, UK. E-mail: c.b.nielsen@qmul.ac.uk
First published on 25th June 2020
Abstract
We present a series of conjugated molecules for colourimetric and fluorimetric sensing of alkali metal cations Na+ and K+. The dye molecules are based on an electron-rich bithiophene core bridged by an oligoethylene glycol bridge capable of selectively binding either sodium or potassium. The molecules are further functionalised with electron-rich or electron-deficient moieties to modulate the band gap and thus shift the absorbance across the visible range. We observe distinct colour changes and high selectivity for alkali metal cations at millimolar concentrations. We moreover see highly selective fluorimetric responses with the molecules exhibiting either turn-on, turn-off or ratiometric sensing behaviour. We finally demonstrate a dual optical sensor capable of distinguishing between sodium and potassium in solution.
Introduction
Colourimetric measurement of physical properties is commonplace, with every practicing chemist having at some point employed universal indicator paper as a simple method to measure pH. Commonly, pH paper contains a series of dyes, which exhibit colour change dependant on pH allowing for visual determination of acidity of a solution. In order to examine biological samples, dyes have been employed in microscopy for many years and recent advancements in fluorescent materials have been employed in confocal, fluorescence as well as two-photon microscopy with a number of selective fluorescent probes having been developed.1,2Two of the most common intra- and extracellular biologically active cations are the sodium and potassium ions, associated with a range of biological processes including propagation of nerve impulses, muscle contraction, and regulation of blood pressure.3 A deviation from a narrow typical concentration range of 100–200 mM for Na+ in serum and 4–5 mM for K+ in the blood and 140 mM in the intracellular fluid for a healthy individual leads to serious health conditions,4 thus defining a concentration range on the order of 1–100 mM where accurate and selective measurement of sodium and potassium ions is desirable. Alkali metal ion sensing has previously been carried out electrochemically, with either ion-selective membranes on organic electrochemical transistors5 or with crown ether functionalised polymers6 and commercially with ion-selective electrodes. Optical sensors are generally characterised by a slightly simpler operation over ion-selective electrochemical sensors which require careful calibration, temperature and pH control.7
Typically, solution phase optical sensors for Na+ and K+ and other metal ions have been based on size-selective heteroatom-containing macrocycles such as the crown ether,8,9 azacrown,10,11 cryptand12 or calixarene13,14 as the binding unit. The binding unit is covalently bonded to a chromophore or fluorophore which exhibits a change in absorbance or fluorescence properties as a result of the binding event. The attractiveness of optical sensors is their relative ease of application, high selectivity and high concentration range for quantification. Further, ratiometric sensing, whereby a change in analyte concentration results in a decrease of one optical transition and an increase of a second transition, provide a more robust measurement of concentration. The alternative being an on/off sensor where only depletion or enhancement of an optical event is observed upon change in analyte concentration. Ratiometric sensors eliminate some device-to-device variation and background noise due to the self-referencing measurement of the probe.1 Vanishingly few Na+ and K+ sensors show ratiometric colourimetric detection in the visible range of the spectrum and even fewer fluorescent materials show ratiometric detection.15–17 Furthermore, if absorbance and emission can be pushed into the 650–950 nm range or the near infrared over 1100 nm then biological sensing becomes more relevant due to the transparency window of tissue18 as well as being beyond autofluorescence emission wavelength for almost all common endogenous fluorophores.19 It is moreover advantageous for excitation wavelengths to be red-shifted to mitigate the effect of high energy photon damage to cells.
Conjugated small molecules have been explored extensively as active materials in organic electronic applications from field-effect transistors to photovoltaics, light-emitting diodes and electrochromic devices and more recently biological sensing.20–24 Common to these materials is the synthetic flexibility that allows for inclusion of various structural moieties to control solubility, solid state morphology, colour, selectivity to biological analytes, and electronic properties, and is one of the standout advantages of organic electronic materials. Recently, it was demonstrated that a conjugated polymer which included either a 17-crown-5 or a 20-crown-6 ether moiety was capable of ratiometric detection of Na+ and K+ in the visible range in both solution and solid state.25,26 Upon binding of an alkali metal ion, a backbone twist occurred breaking conjugation and thereby resulting in ratiometric colour change in the visible spectrum. This concept has more recently been extended to small conjugated molecules showing ratiometric colourimetric response to Na+ ions.27 Here, we present a series of related small conjugated molecules and show how rational molecular design allows for both colourimetric and fluorimetric sensing of sodium and potassium. While all colourimetric responses are ratiometric in nature, we show how different structural motifs can be used to alter fluorimetric responses from ratiometric to turn-off and turn-on. As a proof-of-concept, we furthermore show how access to optical sensor molecules with spectral complementarity allows for mixtures with dual Na+/K+ sensing capabilities.
Synthesis
In an effort to create soluble donor–acceptor small molecule ion sensors, we initially synthesised 2,2′-bithiophene tethered at the 3 and 3′ positions by a polyether bridge,28 followed by a series of cross coupling reactions to afford the optical ion sensing materials; full experimental details are available in the ESI† and described in brief here and in Scheme 1. The sodium-binding unit 1-Na was brominated to afford 2-Na, after which Suzuki coupling with 4-(n-dodecyl)phenylboronic acid pinacol ester afforded M1 in 57% yield as a yellow solid. To afford a second all-donor potassium sensor, 2-K was reacted under Stille conditions with 5-dodecyl-5′-trimethylstannyl-2,2′-bithiophene to afford M2 as an orange solid in 58% yield. We then developed a series of donor–acceptor materials by introducing either 2,1,3-benzothiadiazole (BT) or 2,3,5,6-tetrafluorophenylene moieties flanking the bithiophene core through stannylation of 1-Nain situ followed by reaction with excess acceptor unit under Stille conditions. Reaction with 4,7-dibromo-2,1,3-benzothiadiazole afforded M3 in a yield of 12% as a purple solid, while reaction with 1,4-dibromo-2,3,5,6-tetrafluorphenylene gave M4 in a yield of 25% as a yellow solid. M3 and M4 were further reacted under Stille conditions with 5-dodecyl-5′-trimethylstannyl-2,2′-bithiophene to afford molecules M5 and M6 as blue and orange powders respectively in good yields of 99% and 89% respectively. Thus, a total of six uniquely coloured materials were synthesised as sodium and potassium ion-selective chromo- and fluoroionophores.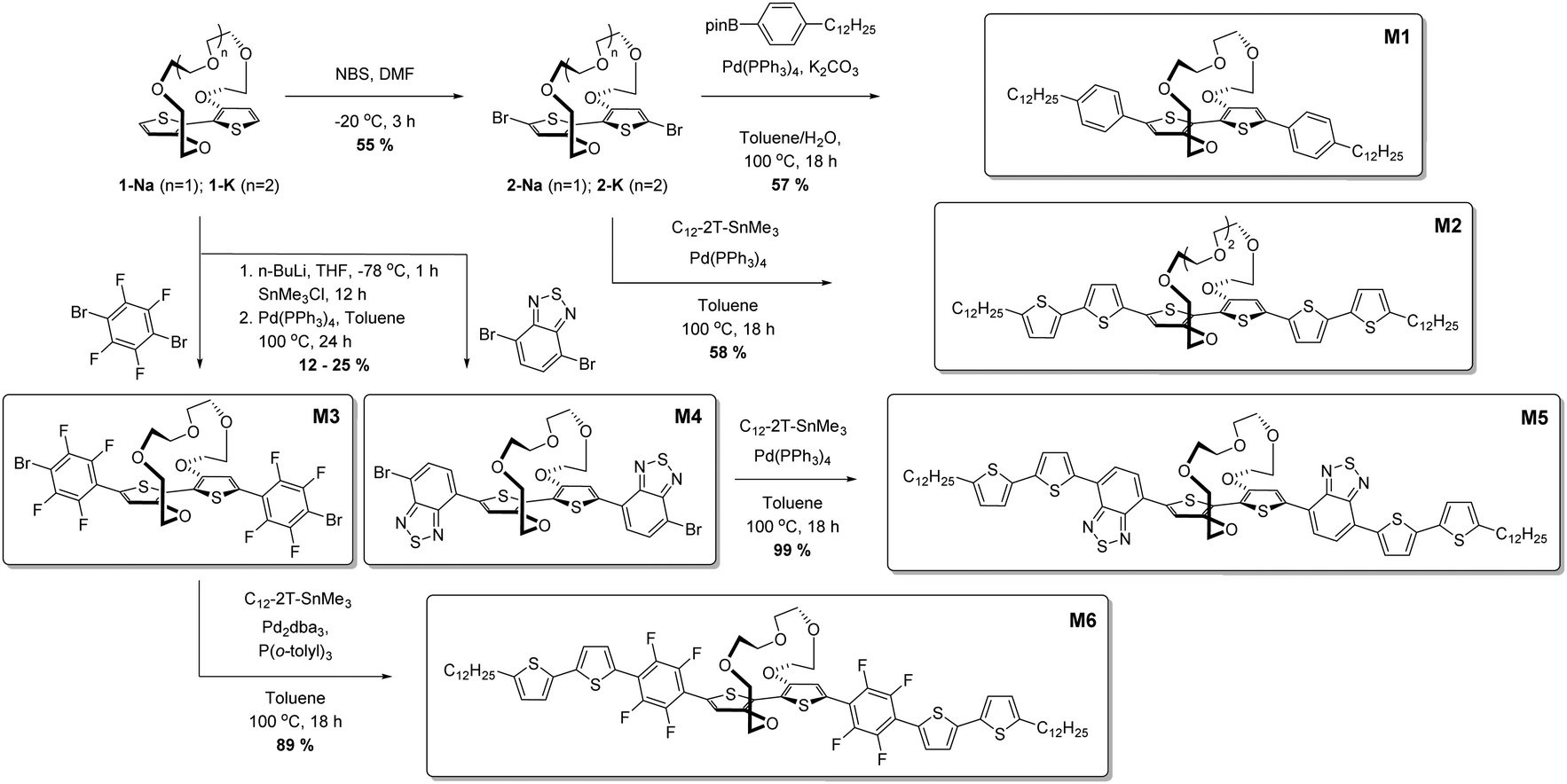 | ||
| Scheme 1 Synthetic scheme outlining the modular synthesis of molecular ion sensors M1–M6via stepwise palladium-mediated cross-coupling reactions; C12-2T-SnMe3 = 5-dodecyl-5′-trimethylstannyl-2,2′-bithiophene. | ||
Results and discussion
Initial characterisation was carried out using UV-vis spectroscopy. The molecules were dissolved in chloroform or tetrahydrofuran (THF) and titrated with a solution of NaBPh4 or KBPh4. Chloroform and THF were chosen to emphasise the behaviour of these novel sensor molecules in solvents miscible and immiscible with water. In the ion-free (unbound) form, the molecules absorb across most of the visible spectrum appearing from yellow to orange through to blue and purple in solution (Fig. 1a). Upon binding with Na+ (M1, M3, M4, M5, and M6) or K+ (M2) in solution, there is a quenching of the dominant optical absorption band and a simultaneous growth of a new blue-shifted (29–88 nm) absorption feature from the alkali metal ion complex, consistent with previous reports for this binding unit (Fig. 1c).25–27 This blue-shift in absorbance is rationalised from a twist in the central S–C–C–S dihedral angle of the ion selective bithiophene core upon Na+ or K+ binding. The conjugation along the backbone is disrupted as the dihedral twists as much 70° as has been demonstrated by previous crystal structures and theoretical calculations (Fig. 1b).26 Representative titration plots can be seen in Fig. 2 for M2 and M5 with the remaining plots depicted in Fig. S1–S12 in the ESI† and all data summarised in Table 1.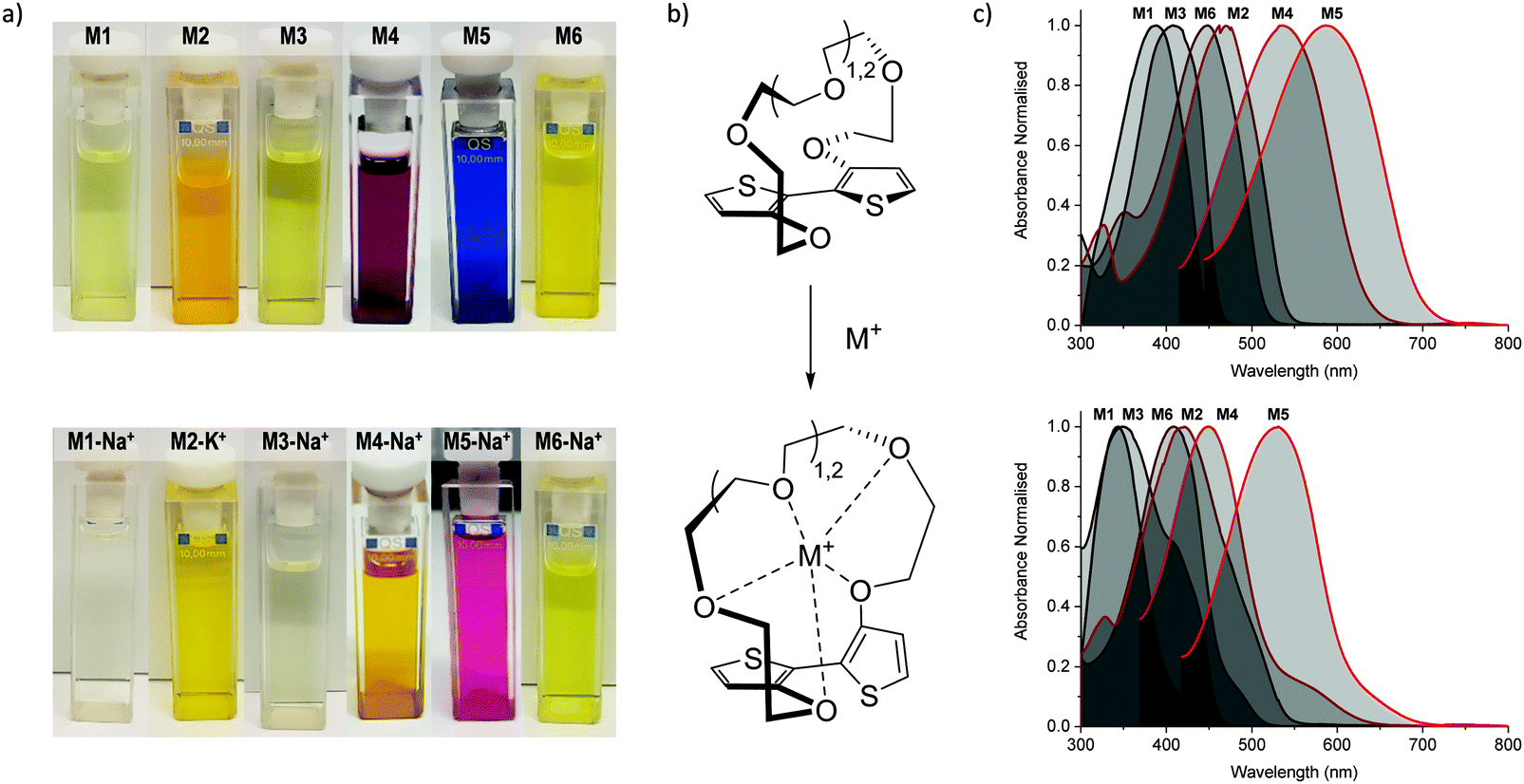 | ||
| Fig. 1 (a) Photographs of the molecules (top) and their alkali metal ion complexes (bottom) in chloroform solution; (b) illustration of backbone twist of crown ether tethered bithiophene sensing unit adapted from ref. 26; (c) collated UV-vis absorption spectra for the molecules (top) and their alkali metal ion complexes (bottom) in chloroform solution at approximately 10−5 M concentration at room temperature. | ||
 | ||
| Fig. 2 (a) Representative UV-vis absorption spectra in tetrahydrofuran solution for M2 upon titration with aliquots of 0.002 M KBPh4 in tetrahydrofuran (left) and for M5 upon titration with aliquots of 0.005 M NaBPh4 in tetrahydrofuran (right); (b) photographs of M5 titration series with NaBPh4 in tetrahydrofuran. All data recorded at room temperature. | ||
| λ max M (nm) |
λ
max M-Na+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (nm) a (nm) |
λ
max M-K+a (nm) |
Δλmaxa (nm) | ε Mb (103 cm−1 M−1) | K d (mM) | LODa (nM) | LOQa (nM) | |
|---|---|---|---|---|---|---|---|---|
| a Data obtained from tetrahydrofuran solution at room temperature. b Extinction coefficients (ε) determined in chloroform solution at room temperature. | ||||||||
| M1 | 397 | 338 | — | 59 | 32.6 | 1.45 | 70 | 220 |
| M2 | 468 | — | 400 | 68 | 72.8 | 0.13 | 90 | 300 |
| M3 | 412 | 347 | — | 65 | 31.6 | 34.4 | 1100 | 3700 |
| M4 | 540 | 452 | — | 88 | 43.0 | 6.81 | 240 | 790 |
| M5 | 595 | 531 | — | 64 | 83.5 | 0.33 | 180 | 590 |
| M6 | 453 | 424 | — | 29 | 123.0 | 18.2 | 590 | 1960 |
The molecules can be divided into three categories. M1 and M2 are all-donor molecules comprising only phenylene and/or thiophene building blocks. In solution, M1 is pale yellow, turning colourless upon titration with Na+ caused by a 44 nm blue-shift (Fig. 1a). The increased conjugation length of the sexithiophene backbone in M2 causes a red-shift of the absorption maximum (λmax) from 397 nm for M1 to 468 nm for M2. When titrated with KBPh4, the λmax blue-shifts 49 nm as depicted in Fig. 2a. The K+ sensing unit (1-K) was employed for M2 in order to have complementary Na+ and K+ molecular sensors available. The second set of molecules (M3 and M6) are weak donor–acceptor systems containing electron-deficient tetrafluorophenylene units with additional extension of conjugation in M6 with flanking bithiophene moieties. M3 displays a λmax at 412 nm with a significant 65 nm blue-shift upon ion-sensing to form the colourless M3-Na+ complex. The π-extended M6 also appears yellow with a λmax of 453 nm in solution (Fig. 1a); however, it displays the smallest blue-shift of 29 nm when undergoing complexation to form M6-Na+. The third set of molecules are stronger donor–acceptor systems where the central 1-Na core is flanked by highly electron-deficient 2,1,3-benzothiadiazole groups, thus affording M4 and its bithiophene-extended derivative M5. These red-shifted materials achieved the highest visual contrast of the series upon sensing of sodium ions. In solution, M4 is purple with a λmax of 540 nm; the colour changes from purple to yellow when the M4-Na+ complex forms (Fig. 1a). This molecule displayed the largest blue-shift of 88 nm upon complexation and displayed two isosbestic points at 481 nm and 392 nm indicating a direct conversion from M4 to M4-Na+ without interfering side-reactions. M5 is blue when ion-free and when complexed with Na+ has a magenta hue (Fig. 1a). The shift in λmax was 64 nm from 595 nm to 531 nm upon Na+ sensing as depicted in Fig. 2a. It should be noted that the presence of sodium ions can be detected visually at concentrations below 1 mM using a 10−5 M solution of M5 as illustrated in Fig. 2b.
The affinities of the sensor molecules for Na+ and K+ were investigated at room temperature and quantified using the Benesi–Hildebrand method to afford the dissociation constants, Kd, listed in Table 1 with further details available in the ESI† (Fig. S12–S23).29,30 The limit of detection (LOD) and limit of quantification (LOQ) were quantified from the absorbance of the complex (M-Na+ or M-K+) relative to the absorbance of the ion-free sensor molecule (M) at their respective absorption maxima during titration experiments (Table 1 and Fig. S24–S34, ESI†). We observe slightly lower Kd values in chloroform than in THF, indicating stronger affinities, likely due to the ability of THF to coordinate ions in solution thus competing with the binding of the molecule.31 Deviations from linearity in the Benesi–Hildebrand and LOD/LOQ plots obtained in chloroform lead us to focus mainly on the alkali metal ion affinities in THF in the following. We observe that the dissociation constant drops markedly as the conjugation length of the sensor molecule increases with Kd of M6 (18.2 mM) lower than that of M3 (34.4 mM) and similarly Kd of M5 (0.33 mM) lower than that of M4 (6.81 mM). We hypothesise that this is caused by the fact that breaking of the π-conjugation upon complexation is more unfavourable in a shorter π-conjugated molecule than in a longer molecule as the highest occupied molecular orbital would be significantly more localised in the first instance. Interestingly, the fluorinated molecules, M6 and M3, exhibit the highest Kd values followed by the brominated sensor molecule M4, which are all significantly higher than that of the non-halogenated molecules. It is likely that the halogen lone pairs and the partial negative charge on the polarised C–X bonds compete as binding sites for the alkali metal ions and therefore reduce the binding efficiency of the crown ether moiety.32 For all the sensor molecules, the alkali metal ion binding is at least an order of magnitude weaker than for the parent crown ethers 15-crown-5 (Kd < 0.1 mM) and 18-crown-6 (Kd < 0.01 mM).33 This allows for a reversible process as the unbound state can be regenerated upon addition of either 15-crown-5 or 18-crown-6. Characteristic of π-conjugated materials, the sensor molecules show very high extinction coefficients of 32600–123000 M−1 cm−1 with the longer molecules M2, M5 and M6 showing the highest values as expected. Sensor molecules M2, M4, M5 and M6 are highly bench stable having been stored as solids for six months under ambient conditions without any change in UV-vis spectra, while small changes are seen for M1 and M3 possible due to oxidation and/or aggregation (Fig. S35–S40, ESI†).
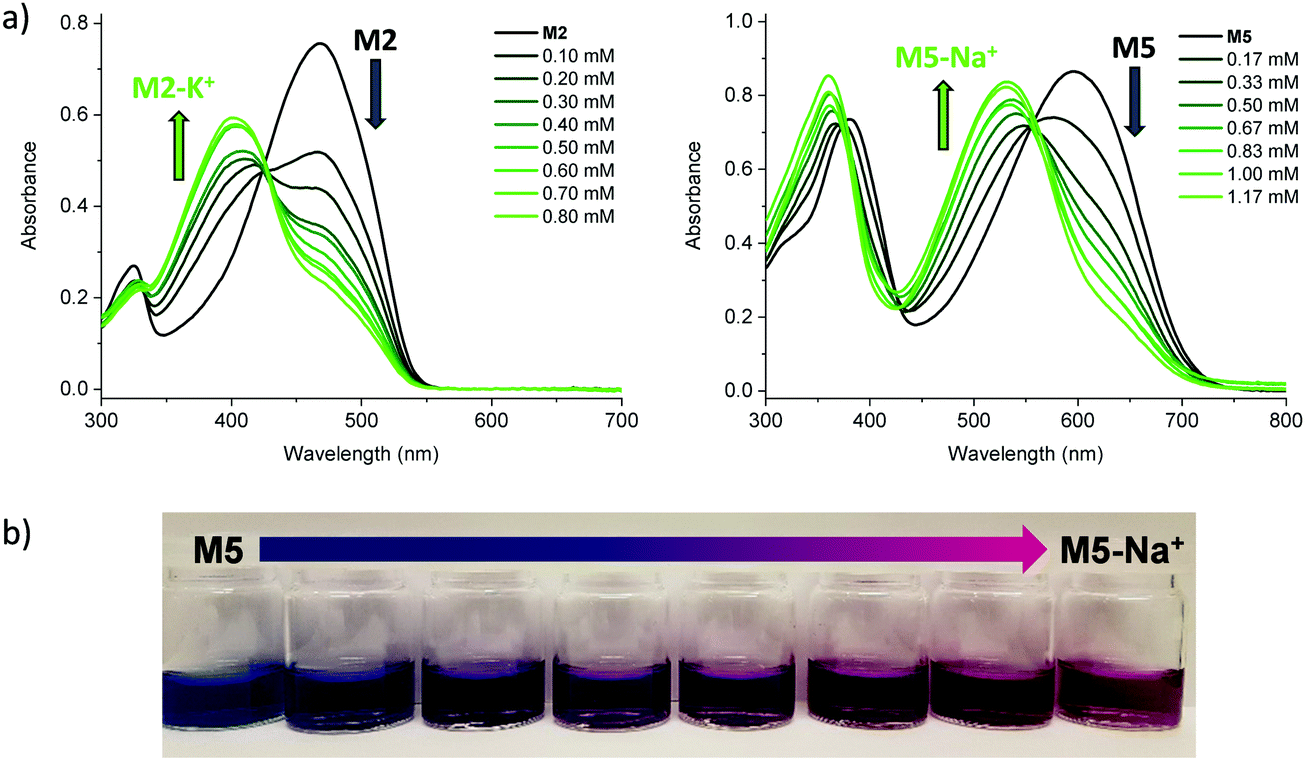
We subsequently explored the emissive properties of the sensor molecules by exciting at the isosbestic point identified from the UV-vis titration experiments (Fig. 3 and Fig. S41–S50, ESI†). In line with the colourimetric studies, the sensor molecules were examined in both tetrahydrofuran and chloroform with our main focus devoted to the behaviour in tetrahydrofuran due to more reliable extraction of sensing parameters and tetrahydrofuran generally solvating alkali metal salts better than chloroform. Upon titration with NaBPh4 in tetrahydrofuran, M1 exhibited a decrease in fluorescence intensity (turn-off), with a slight shift in emission wavelength (λem) from 476 nm to 459 nm. With KBPh4, M2 showed a similar decrease in fluorescence intensity with a slight broadening and blue-shift of the emission band. The second set of sensor molecules, M4 and M5, showed a significant fluorescence enhancement (turn-on) upon complexation with Na+. This turn-on fluorimetric response also had the advantage of being significantly shifted towards the red compared to the other materials, with the λem being 712 nm and 733 nm for the ion-free sensor molecules in solution and 650 nm and 661 nm upon Na+ complexation for M4 and M5, respectively. The molecules behave similarly in chloroform exhibiting strong fluorescence turn-on (Fig. S46 and S48, ESI†). It is likely that there is significant charge transfer character from donor to acceptor moiety in these molecules, which in the ion-free form quenches the emission. Complexation with sodium, on the other hand, is expected to disrupt effective charge transfer and thus enhance emission. While M3 did not fluoresce, M6 showed a ratiometric fluorimetric response to titration with sodium ions. Upon titration with NaBPh4 in THF, a decrease in the emission peak at λem 530 nm occurred with a concurrent increase in intensity of a second fluorescence feature at 480 nm; the ratiometric response is even clearer in chloroform as illustrated in Fig. 3. The selectivity of all the molecules to Na+ and K+ examined by colourimetry and fluorimetry is summarized in Fig. 4 (details in Fig. S64–S73, ESI†). As expected from the crown ether ring size, M1, M3, M4, M5 and M6 were all highly selective to Na+ with M2 being moderately selective to K+. The Stokes shifts (Table 2) for the unbound molecules were on the order of 3000–4000 cm−1 with the shorter molecules M1 and M4 showing the larger Stokes shifts and the longer and more conformationally locked molecules showing lower values.34 The metal ion complexes have significantly larger Stokes shifts with values approaching 8000 cm−1, again with the shorter sensor molecules with more conformational freedom having the larger values.35,36 Quantum yields (Table 2) for the unbound molecules were lowest for the bromine-containing molecules M3 (no fluorescence detected) and M4, while strong donor–acceptor character (M4 and M5) was also seen to promote non-radiative emission. The longer molecules M2 and M6 showed relatively high quantum yields suggesting that the bulky tethered crown ether effectively prevents intermolecular interactions and the associated non-radiative processes.
 | ||
| Fig. 3 (a) Representative fluorescence spectra of M1 (1.7 × 10−5 M) and M5 (1.1 × 10−5 M) in tetrahydrofuran and M6 in tetrahydrofuran (1.0 × 10−5 M) and chloroform (2.3 × 10−5 M) upon titration with aliquots of NaBPh4; (b) photographs of representative unbound sensor molecules and their alkali metal ion complexes in chloroform solution upon excitation at 365 nm. All data recorded at room temperature. | ||
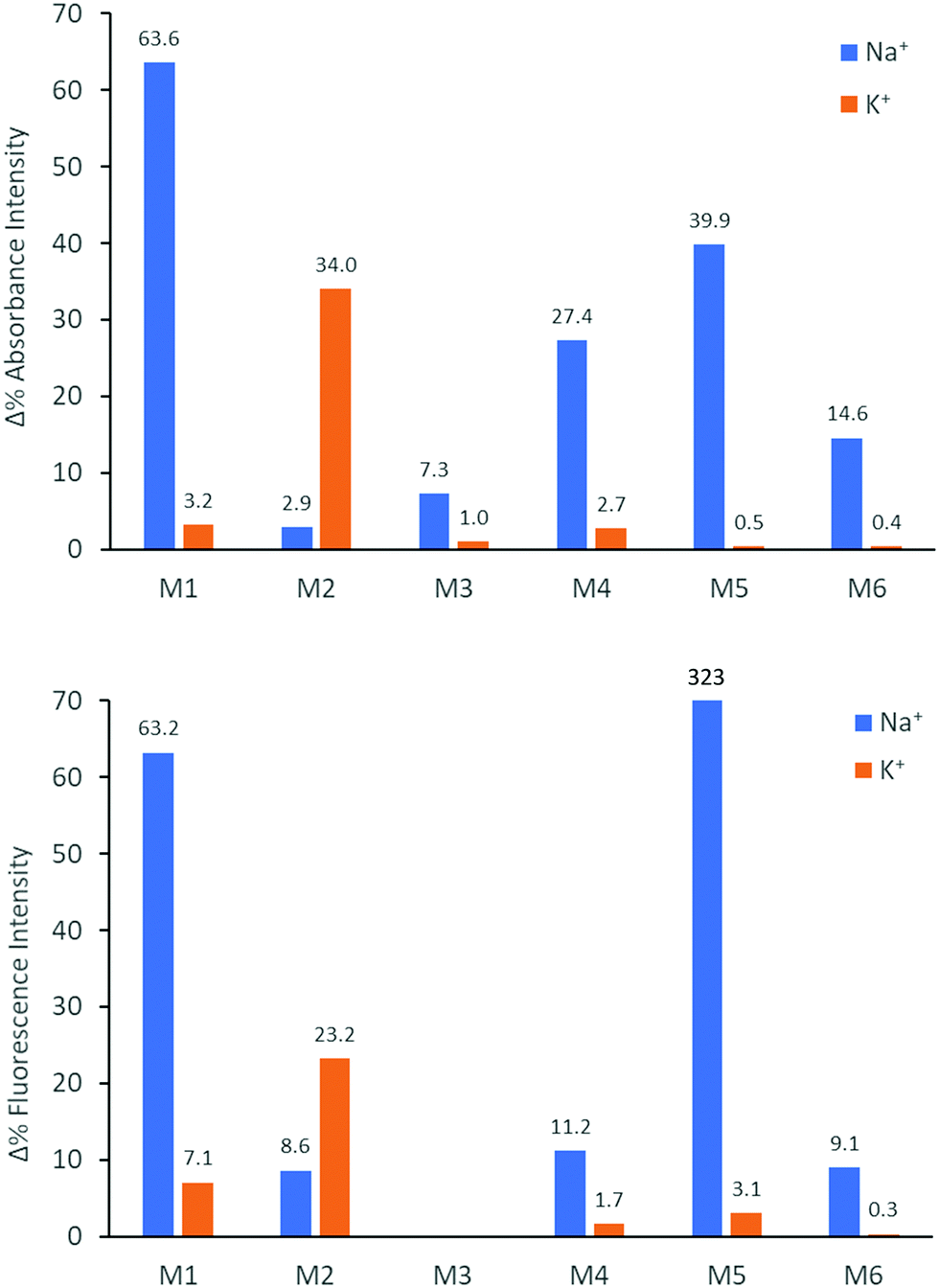 | ||
| Fig. 4 Colorimetric (top) and fluorimetric (bottom) selectivity of the sensor molecules for Na+ and K+ (added as BPh4 salts at 0.33 μM concentration) measured at 10−5 M sensor molecule concentration in tetrahydrofuran at room temperature. The change in emission is 323% for M5 upon addition of Na+, the axis was cropped for clarity. | ||
| λ em Ma (nm) |
λ
em M-Na+a (nm) |
λ
em M-K+a (nm) |
Stokes shift Ma (cm−1) | Stokes shift M-Na+/M-K+a (cm−1) |
Φ M | Φ M-Na+/M-K+ | |
|---|---|---|---|---|---|---|---|
| a Data obtained from tetrahydrofuran solution at room temperature at approximately 10−5 M concentration (see Fig. S41–S50, ESI) with excitation wavelengths 362 nm (M1), 426 nm (M2), 481 nm (M4), 556 nm (M5) and 427 nm (M6). b Quantum yields (Φ) determined in either dichloromethane (M2) or chloroform (M1, M4–M6) solution at room temperature (see Fig. S51–S63, ESI). | |||||||
| M1 | 476 | 459 | — | 4180 | 7800 | 0.06 | 0.01 |
| M2 | 546 | — | 542 | 3050 | 6550 | 0.12 | 0.12 |
| M4 | 712 | 650 | — | 4470 | 6740 | 0.005 | 0.02 |
| M5 | 733 | 661 | — | 3160 | 3700 | 0.006 | 0.02 |
| M6 | 524 | 528 | — | 2990 | 4650 | 0.36 | 0.07 |
Having confirmed alkali metal ion selectivity and the ability of the sensor molecules to give visual responses to Na+ and K+, the two most complementary sodium and potassium sensor molecules in terms of spectral response were investigated as a blend. Employing M2 as the K+ selective chromophore and M5 as the Na+ selective chromophore, a 1:1 blend of the two materials was used to create a colorimetric sensor for sodium and potassium ions (Fig. 5). The two molecules have similar molar extinction coefficients and exhibit a large separation in λmax for both unbound and bound species in solution and thus make for an optimal dual sensor system. The blended M2:M5 solution was greenish when ion-free and turned blue upon addition of KBPh4 and orange/red upon addition of NaBPh4.
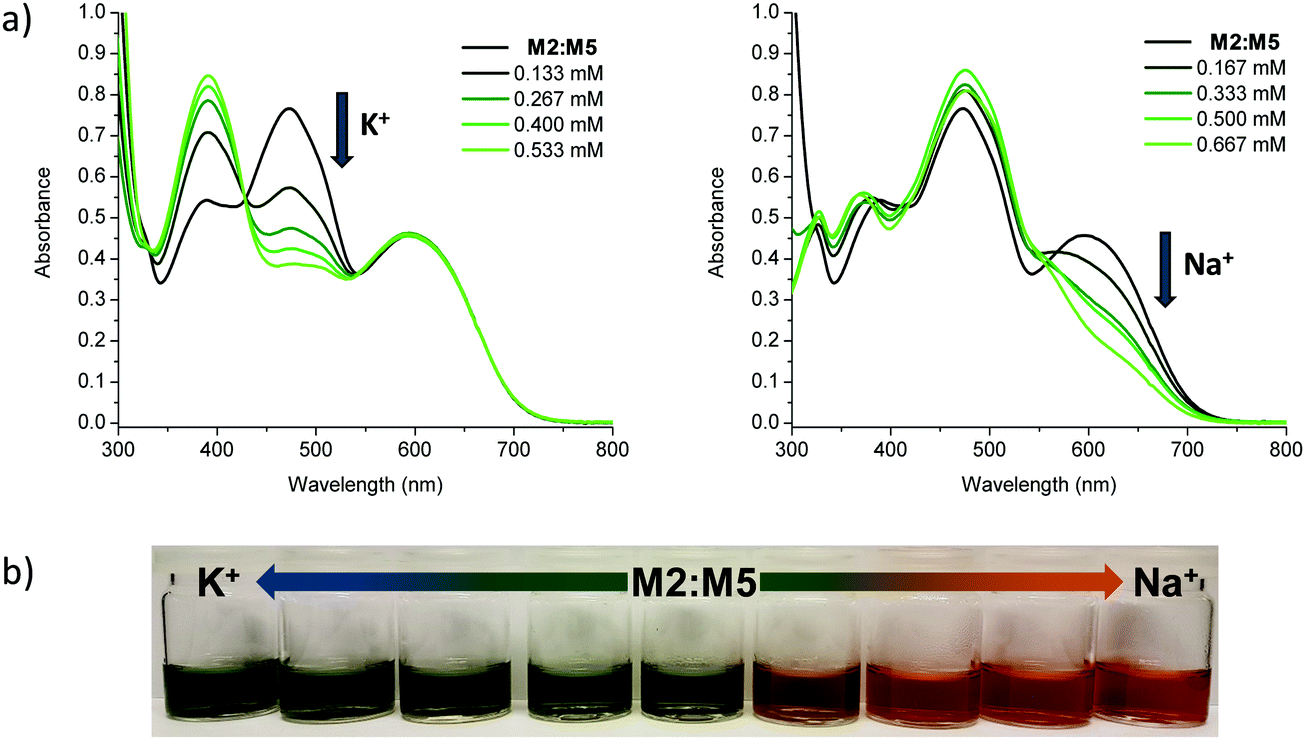 | ||
| Fig. 5 (a) UV-vis absorption spectra of the M2:M5 blend at approximately 3 × 10−5 M in tetrahydrofuran titrated with aliquots of KBPh4 (left) and NaBPh4 (right) in tetrahydrofuran; (b) photographs of M2:M5 titration series with KBPh4 and NaBPh4 in tetrahydrofuran. All data recorded at room temperature. | ||
Conclusions
In conclusion, we have shown the design and modular synthesis of six novel ratiometric sensor molecules, capable of selective colourimetric and fluorimetric sensing of sodium and potassium cations. By rational design and choice of peripheral π-conjugated donor or acceptor moieties, we have created ion-selective sensor molecules with colours covering the entire visible spectrum in solution. This ability to navigate the colour palette was demonstrated with relatively short π-conjugated sensor molecules (M1 and M3) that go from coloured to colourless upon sensing and longer π-conjugated sensors such as M5 that exhibit a very clear colour change from blue to magenta. Spectral complementarity was furthermore utilised to create a dual sodium and potassium sensor that turned blue in the presence of potassium and orange/red in the presence of sodium cations. Effective alkali metal ion binding – realised by a tethered crown ether that can be varied in size to host either sodium or potassium ions – resulted in low dissociation constants, in particular for the non-halogenated sensor molecules with extended π-conjugation, M2 and M5. The ratiometric colourimetric response, due to the breaking of π-conjugation upon ion complexation, has furthermore enabled low detection and quantification limits in the 100 nM range well within a typical physiological concentration range. We further examined the potential of these molecules as fluorimetric sensors. Depending on the peripheral π-conjugated segments, the molecular sensors exhibited either turn-on, turn-off or ratiometric fluorimetric sensing of Na+ and K+ in both tetrahydrofuran and chloroform. The all-donor materials M1 and M2 displayed turn-off fluorimetric responses, while the strong donor–acceptor systems with 2,1,3-benzothiadiazole (M4 and M5) exhibited turn-on fluorimetric responses. M6, on the other hand, with weak donor–acceptor character owing to the weakly electron-withdrawing tetrafluorophenylene moiety, showed clear ratiometric fluorimetric response. We believe that these efforts to understand structure–property relations in π-conjugated sensor molecules will help pave the way for the development of efficient molecular sensors. In this context, we have laid out several molecular design strategies including all-donor and weak and strong donor–acceptor type π-conjugated systems for selective colorimetric and fluorimetric sensing of alkali metal cations at biological relevant concentrations. While this proof-of-principle study was carried out with peripheral alkyl chains impairing good solubility and thus sensing behaviour in tetrahydrofuran and chloroform, recent work has demonstrated aqueous activity with π-conjugated molecules bearing ethylene glycol-based chains.37 Further biological relevance can thus be sought through further synthetic design in particular through substituting the peripheral alkyl chains with more polar counterparts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Academy of Medical Sciences & Wellcome Trust (SBF002/1158) and the Materials Research Institute for financial support.References
- S.-H. Park, N. Kwon, J.-H. Lee, J. Yoon and I. Shin, Synthetic ratiometric fluorescent probes for detection of ions, Chem. Soc. Rev., 2020, 49, 143–179 RSC.
- S. Singha, Y. W. Jun, S. Sarkar and K. H. Ahn, An Endeavor in the Reaction-Based Approach to Fluorescent Probes for Biorelevant Analytes: Challenges and Achievements, Acc. Chem. Res., 2019, 52, 2571–2581 CrossRef CAS PubMed.
- H. R. Pohl, J. S. Wheeler and H. E. Murray, in Interrelations between Essential Metal Ions and Human Diseases, ed. A. Sigel, H. Sigel and R. Sigel, Springer, Dordrecht, 2013, 29–47 Search PubMed.
- R. Milo, R. Phillips and N. Orme, Cell Biology by the Numbers, CRC Press/Taylor & Francis, New York, 2015 Search PubMed.
- K. Schmoltner, J. Kofler, A. Klug and E. J. W. List-Kratochvil, Electrolyte-Gated Organic Field-Effect Transistor for Selective Reversible Ion Detection, Adv. Mater., 2013, 25, 6895–6899 CrossRef CAS PubMed.
- S. Wustoni, C. Combe, D. Ohayon, M. H. Akhtar, I. McCulloch and S. Inal, Membrane-Free Detection of Metal Cations with an Organic Electrochemical Transistor, Adv. Funct. Mater., 2019, 29, 1904403 CrossRef CAS.
- K. Thajee, L. Wang, K. Grudpan and E. Bakker, Colorimetric ionophore-based coextraction titrimetry of potassium ions, Anal. Chim. Acta, 2018, 1029, 37–43 CrossRef CAS PubMed.
- P. Nandhikonda, M. P. Begaye and M. D. Heagy, Highly water-soluble, OFF–ON, dual fluorescent probes for sodium and potassium ions, Tetrahedron Lett., 2009, 50, 2459–2461 CrossRef CAS.
- J. Qiu, Y. Zhang, C. Dong, Y. Huang, L. Sun, H. Ruan, H. Wang, X. Li and A. Wu, Rapid colorimetric detection of potassium ions based on crown ether modified Au NPs sensor, Sens. Actuators, B, 2019, 281, 783–788 CrossRef CAS.
- A. Thibon and V. C. Pierre, A Highly Selective Luminescent Sensor for the Time-Gated Detection of Potassium, J. Am. Chem. Soc., 2009, 131, 434–435 CrossRef CAS PubMed.
- J. Li, D. Yim, W.-D. Jang and J. Yoon, Recent progress in the design and applications of fluorescence probes containing crown ethers, Chem. Soc. Rev., 2017, 46, 2437–2458 RSC.
- R. D. Carpenter and A. S. Verkman, Function-Oriented Synthesis of a Didesmethyl Triazacryptand Analogue for Fluorescent Potassium Ion Sensing, Eur. J. Org. Chem., 2011, 1242–1248 CrossRef CAS PubMed.
- J. P. Malval, I. Leray and B. Valeur, A highly selective fluorescent molecular sensor for potassium based on a calix[4]bisazacrown bearing boron-dipyrromethene fluorophores, New J. Chem., 2005, 29, 1089–1094 RSC.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Revisiting Fluorescent Calixarenes: From Molecular Sensors to Smart Materials, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- V. V. Martin, A. Rothe, Z. Diwu and K. R. Gee, Fluorescent sodium ion indicators based on the 1,7-diaza-15-crown-5 system, Bioorg. Med. Chem. Lett., 2004, 14, 5313–5316 CrossRef CAS PubMed.
- M. Taki, H. Ogasawara, H. Osaki, A. Fukazawa, Y. Sato, K. Ogasawara, T. Higashiyama and S. Yamaguchi, A red-emitting ratiometric fluorescent probe based on a benzophosphole P-oxide scaffold for the detection of intracellular sodium ions, Chem. Commun., 2015, 51, 11880–11883 RSC.
- G. Song, R. Sun, J. Du, M. Chen and Y. Tian, A highly selective, colorimetric, and environment-sensitive optical potassium ion sensor, Chem. Commun., 2017, 53, 5602–5605 RSC.
- A. M. Smith, M. C. Mancini and S. Nie, Second window for in vivo imaging, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed.
- M. Monici, Cell and tissue autofluorescence research and diagnostic applications, Biotechnol. Annu. Rev., 2005, 11, 227–256 CAS.
- H. Sirringhaus, 25th Anniversary Article: Organic Field-Effect Transistors: The Path Beyond Amorphous Silicon, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- G. M. Farinola and R. Ragni, Electroluminescent materials for white organic light emitting diodes, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, All-Polymer Solar Cells: Recent Progress, Challenges, and Prospects, Angew. Chem., Int. Ed., 2019, 58, 4129–4142 CrossRef CAS PubMed.
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Conjugated Polymers in Bioelectronics, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- M. J. Marsella and T. M. Swager, Designing conducting polymer-based sensors: selective ionochromic response in crown ether-containing polythiophenes, J. Am. Chem. Soc., 1993, 115, 12214–12215 CrossRef CAS.
- A. Giovannitti, C. B. Nielsen, J. Rivnay, M. Kirkus, D. J. Harkin, A. J. P. White, H. Sirringhaus, G. G. Malliaras and I. McCulloch, Sodium and Potassium Ion Selective Conjugated Polymers for Optical Ion Detection in Solution and Solid State, Adv. Funct. Mater., 2016, 26, 514–523 CrossRef CAS.
- M. Moser, K. J. Thorley, F. Moruzzi, J. F. Ponder, I. P. Maria, A. Giovannitti, S. Inal and I. McCulloch, Highly selective chromoionophores for ratiometric Na+ sensing based on an oligoethyleneglycol bridged bithiophene detection unit, J. Mater. Chem., 2019, 7, 5359–5365 CAS.
- M. A. M. Leenen, T. Meyer, F. Cucinotta, H. Thiem, R. Anselmann and L. De Cola, Synthesis, characterization, and field-effect transistor performance of poly[2,6-bis(3-tridecanoxythiophen-2-yl)benzo[1,2-b;4,5-b′]dithiophene], J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1973–1978 CrossRef CAS.
- Y. Shiraishi, S. Sumiya, Y. Kohno and T. Hirai, A Rhodamine–Cyclen Conjugate as a Highly Sensitive and Selective Fluorescent Chemosensor for Hg(II), J. Org. Chem., 2008, 73, 8571–8574 CrossRef CAS PubMed.
- S. Goswami, K. Aich, S. Das, A. K. Das, A. Manna and S. Halder, A highly selective and sensitive probe for colorimetric and fluorogenic detection of Cd2+ in aqueous media, Analyst, 2013, 138, 1903–1907 RSC.
- A. Shrivastava and V. Gupta, Methods for the determination of limit of detection and limit of quantitation of the analytical methods, Chronicles Young Sci., 2011, 2, 21 CrossRef.
- E. Magnusson and N. W. Moriarty, Binding Patterns in Single-Ligand Complexes of NH3, H2O, OH−, and F− with First Series Transition Metals, Inorg. Chem., 1996, 35, 5711–5719 CrossRef CAS PubMed.
- R. M. Izatt, J. S. Bradshaw, S. A. Nielsen, J. D. Lamb, J. J. Christensen and D. Sen, Thermodynamic and kinetic data for cation-macrocycle interaction, Chem. Rev., 1985, 85, 271–339 CrossRef CAS.
- X. Liu, Z. Xu and J. M. Cole, Molecular Design of UV-vis Absorption and Emission Properties in Organic Fluorophores: Toward Larger Bathochromic Shifts, Enhanced Molar Extinction Coefficients, and Greater Stokes Shifts, J. Phys. Chem. C, 2013, 117, 16584–16595 CrossRef CAS.
- C. Zhu, S. Li, M. Luo, X. Zhou, Y. Niu, M. Lin, J. Zhu, Z. Cao, X. Lu, T. Wen, Z. Xie, P. V. R. Schleyer and H. Xia, Stabilization of anti-aromatic and strained five-membered rings with a transition metal, Nat. Chem., 2013, 5, 698–703 CrossRef CAS PubMed.
- Y. Zems, A. G. Moiseev and D. F. Perepichka, Convenient Synthesis of a Highly Soluble and Stable Phosphorescent Platinum Porphyrin Dye, Org. Lett., 2013, 15, 5330–5333 CrossRef CAS PubMed.
- Z. S. Parr, R. B. Rashid, B. D. Paulsen, B. Poggi, E. Tan, M. Freeley, M. Palma, I. Abrahams, J. Rivnay and C. B. Nielsen, Semiconducting Small Molecules as Active Materials for p-Type Accumulation mode Organic Electrochemical Transistors, Adv. Electron. Mater., 2020 DOI:10.1002/aelm.202000215.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00157k |
| This journal is © the Partner Organisations 2020 |