
Photoinduced free radical promoted cationic polymerization 40 years after its discovery†
Mehmet Atilla
Tasdelen
 a,
Jacques
Lalevée
b and
Yusuf
Yagci
*c
a,
Jacques
Lalevée
b and
Yusuf
Yagci
*c
aDepartment of Polymer Engineering, Faculty of Engineering, Yalova University, 77100 Yalova, Turkey
bMembre Honoraire de l'Institut Universitaire de France (IUF) (promotion 2011) Institut de Science des Matériaux de Mulhouse, 15 rue Jean Starcky, 68057 Mulhouse Cedex, France
cIstanbul Technical University, Department of Chemistry, 34469 Istanbul, Maslak, Turkey. E-mail: yusuf@itu.edu.tr
First published on 30th December 2019
Abstract
This perspective article is prepared in the memory of Anthony Ledwith who discovered Free Radical Promoted Cationic Polymerization (FRPCP) as a versatile photoinitiating system for cationic polymerization about four decades ago. Mechanistically, distinct modes for the photochemical formation of electron donor radicals in the UV, Vis and NIR range that can be oxidized to reactive cations to initiate the cationic polymerization of oxiranes and vinyl monomers are discussed. The dual polymerization strategies that have been applied to combine FRPCP with the other modes of polymerization processes are presented.
Introduction
Photopolymerization has received remarkable interest over the last 50 years as it can bring many advantages including low energy consumption, high rates of polymerization, solvent-free formulations and wide adaptability over thermal polymerization.1,2 In addition, photopolymerization only occurs in the illuminated area, which results in both full temporal and spatial resolutions. These advantages enable the fabrication of films and complex three-dimensional objects for protective coating, adhesive, automotive, microcircuit, and semiconductor industries. The use of photosensitive resins in additive manufacturing techniques through stereolithography is an important strategy offering excellent spatial resolution and applications in many fields. Photopolymerization is typically a solidification process that transforms a monomer into a polymer by a chain reaction initiated by reactive species (radicals or cations and, in some cases, anions), which are generated from photosensitive compounds, namely photoinitiators, upon ultraviolet or visible light irradiation. Cationic photopolymerization provides some advantages including no oxygen inhibition, dark polymerization, low volume shrinkage, better clarity, abrasion, adhesion and chemical resistance, and less residual stress of cured materials compared to the free radical photopolymerization.3,4 Furthermore, a wide range of monomers such as vinyl ethers, oxiranes, epoxides, sulfides and acetals can be cationically polymerized via double bond addition or ring-opening reactions. Since the discovery of photochemically active onium salts by Crivello and co-workers in the seventies of the last century, cationic photopolymerization has been widely applied in many areas such as inks, films, and coatings on a variety of substrates including metal, paper, and wood.5–8 Intense efforts are also associated with the development of 3D printing resins based on the cationic polymerization of epoxides to reduce/avoid shrinkage and polymerization stress, both of which have huge negative impacts on the quality of the printed objects (shape fidelity, mechanical properties, etc.).9 In the case of a photoinitiated cationic polymerization, the onium salts homolytically or heterolytically decompose to yield a variety of reactive radical, radical–cation and cation intermediates upon UV irradiation. The cationic species can abstract hydrogen from a monomer or a solvent to generate a highly reactive super acid that is responsible for further initiation of cationic polymerization.10 However, the well-known cationic photoinitiators such as iodonium and sulfonium salts mainly absorb UV light between 200 and 320 nm, which restrict their practical applications (e.g. safe and eco-friendly Light Emitting Diodes, LEDs, characterized by emission wavelength >360 nm can hardly be used). One strategy to broaden the absorption range of the cationic photoinitiator is the addition of chromophores onto the aromatic groups of the onium salt.11 But this method requires multi-step synthetic and purification procedures that are of high cost and failed to result in the desired absorption characteristics. In 1978, during the early stages of my (Y. Yagci) PhD studies at Liverpool University, my supervisor Prof. Anthony Ledwith (Fig. 1) came to the lab with a sample bottle containing a small amount of diphenyl iodonium salt provided by Crivello and asked me to conduct two experiments on photoinitiated cationic polymerization of tetrahydrofuran at above 350 nm using iodonium salt in the presence and absence of 2,2-dimethoxy-2-phenyl acetophenone (DMPA). One experiment without DMPA did not work. With limited knowledge in the area at that time, I was very much disappointed with the result. When I told him that the polymerization proceeded only when the radical source was present in the system, he was extremely happy and then, we investigated the mechanistic details.12 This was the era of the sensitized photoinitiated cationic polymerization, which led to the development of many new initiating systems acting in a broad spectral range, which can be used in a wide range of industrial applications. | ||
| Fig. 1 Prof. Anthony Ledwith and Yusuf Yagci (1st year PhD student) at the MSc graduation ceremony of Liverpool University in March 1978. | ||
Following this discovery, several methodologies have been developed to activate the cationic photoinitiators by either energy transfer13,14 or electron transfer13–22 mechanisms between the onium salts with excited photosensitizers or free radicals and/or electron donor compounds in excited charge transfer complexes (CTCs). In all cases, the onium salts act as electron acceptors in charge transfer complexes and for long living excited states of photosensitizers and strong electron donating free radicals (also known as Free Radical Promoted Cationic Polymerization (FRPCP)).15 The electronically excited CTC16,17 of electron acceptor onium salts (pyridinium and sulfonium salts) and electron donor aromatic compounds (hexamethylbenzene and 1,2,4-trimethoxybenzene) undergoes an intermolecular electron transfer reaction resulting in an aromatic radical cation (Scheme 1a). A bathochromic shift of the absorption is usually observed for the CTC compared to the two separated compounds allowing cationic polymerization in the near UV and visible range. The photosensitization mechanism involves an electron transfer from the excited singlet state of a photosensitizer (anthracene, perylene and pyrene derivatives are the most efficient) to the onium salt (ammonium, phosphonium, sulfonium, iodonium and pyridinium), resulting in an unstable free radical and an aromatic cation radical, which is eligible to trigger the subsequent cationic polymerization (Scheme 1b). Notably, the rapid decomposition of the unstable free radical prevents the back electron transfer and makes the overall process essentially irreversible. Furthermore, the rate of initiation strongly relies on the quantum yield of the electron transfer process. Many photochemically formed radicals can be oxidized by onium salts to promote the generation of cations, which are considered as initiating species for cationic polymerization (Scheme 1c). The efficiency of the initiating system is directly related to the redox potentials of both free radicals and onium salts in this mechanism. The higher reduction potential of iodonium salts makes them very suitable for the oxidation of free radicals, whereas the use of sulfonium salts in this system is limited due to their low reduction potentials.
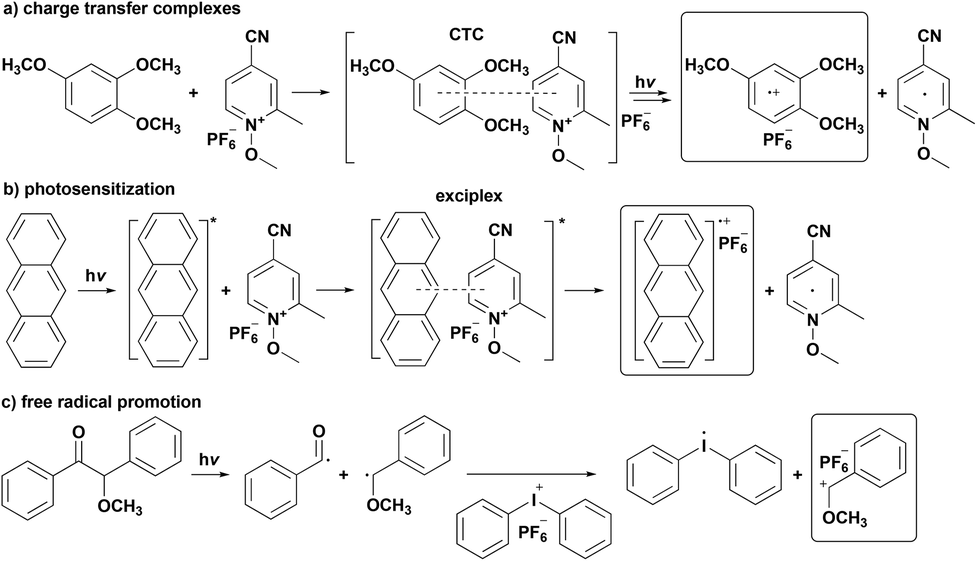 | ||
| Scheme 1 Indirect photoinitiation mechanisms for cationic polymerization; (a) charge transfer complex, (b) photosensitization and (c) free radical promotion. | ||
Various compounds including aromatic ketones, heterocyclic and fused-ring aromatic hydrocarbons, and organic dyes can be used as photosensitizers, whereas all conventional free radical photoinitiators are suitable for the activation of cationic polymerization. However, the use of some photosensitizers has been restricted due to their limited solubility in conventional monomers and toxicity of unmodified polynuclear aromatic hydrocarbons.18–23 On the other hand, the free radical photoinitiators offer many advantages including a wide range of commercial availability, good solubility in a number of commonly used monomers, wavelength flexibility covering the whole spectrum of UV and visible light, dissociation quantum yields and high reactivity of the produced radicals.
As mentioned above, the FRPCP system was first described in 1978 by Ledwith and Yagci.24–26 In this report, common free radical sources, whether obtained by thermal or photochemical routes can be used to promote the cationic polymerization of vinyl and cyclic ethers.24,27–29 The promotion of cationic polymerization was demonstrated by the photocrosslinking experiment of 1,2-epoxyethyl-3,4-epoxycyclohexane using DMPA as a free radical photoinitiator and bis(p-tolyl)iodonium hexafluorophosphate as an onium salt. The gel formation after only 10 min of UV irradiation was clear proof of the promoting effect of the free radical source. The dimethoxybenzyl radical generated from the cleavage of DMPA was an excellent choice for the FRPCP process i.e. this latter radical is electron rich, can be easily oxidized and the generated cation is highly efficient to react with the epoxy ring.
Since the discovery of the FRPCP system, various free radical generation mechanisms have been attempted to activate the onium salts. The basic idea is to generate radicals (e.g. from usual type I or type II radical photoinitiators), which should be oxidized by onium salts, the resulting cations being the polymerization initiation species. The nature of the radical formed by cleavage (type I) or by hydrogen abstraction (type II) is crucial to obtain excellent polymerization efficiency i.e. electron rich radicals are readily oxidized. In order to provide easily oxidizable radicals, additional accelerators such as benzyl alcohols, N-vinyl carbazole (NVC), aromatic tertiary amines and silanes have been developed. These compounds convert radicals having less oxidation capabilities into electron-rich radicals, which are readily oxidized by onium salts to form the corresponding cations.
Benzoin and DMPA are commercially available cleavage type photoinitiators generating two radical species unimolecularly upon UV light irradiation. In the case of benzoin, α-hydroxybenzyl (strong electron donor) and benzoyl (electron withdrawing) radicals are formed. The former radical is capable of reducing onium salts to yield corresponding carbocations for the cationic polymerizations of cyclic and vinyl ethers, whereas the benzoyl radical does not react with the onium salt efficiently30 but abstracts hydrogen from appropriate donors (e.g. accelerator or monomer).31 This indirectly generated radical is also responsible for reducing onium salts to form the corresponding carbocation.
Recently, allylic onium salts having an alternative initiating mechanism based on a two-step addition–fragmentation mechanism have attracted great attention.32 In this case, the addition of a photochemically formed radical with a double bond of allylonium salt generates an unstable radical cation intermediate that is subsequently fragmented into an onium radical cation. The resulting radical cation may directly initiate the polymerization of nucleophilic monomers or alternatively produce a protonic acid via hydrogen abstraction from the surrounding hydrogen donors (such as a solvent or monomer). The substitution of the allylic double bond (e.g., ethoxycarbonyl group) not only promotes the addition reaction of the double bond, but also facilitates the fragmentation reaction of the intermediate that exhibits enhanced initiation efficiency (Scheme 2).33–35
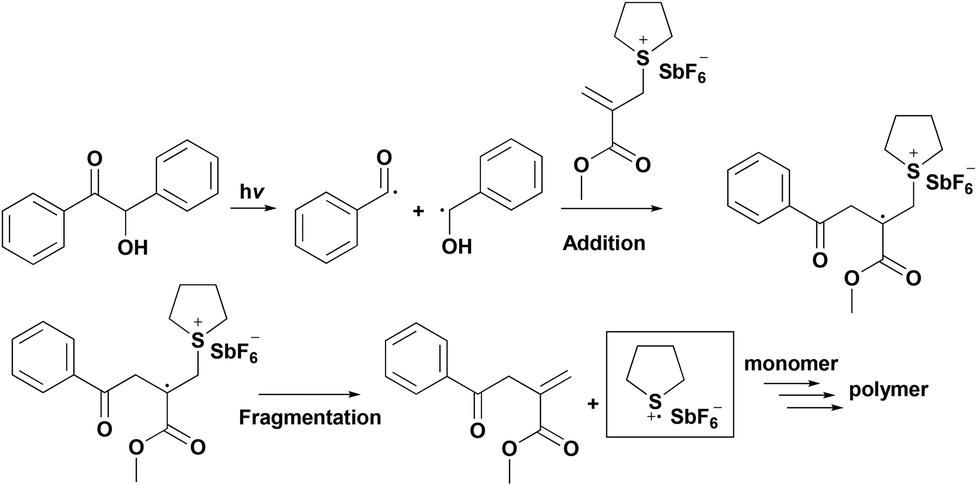 | ||
| Scheme 2 Addition–fragmentation mechanism for cationic photopolymerization. | ||
The addition–fragmentation type initiating system provides an important advantage by forming initiating species from all kinds of thermal and photosensitive free radical initiators in conjunction with allylonium salts. In contrast to the FRPCP system based on easily oxidizable radicals, the addition–fragmentation reactions have become a simple route to adjust the spectral response and initiation conditions of cationic polymerization. A variety of allylsulfonium, allylammonium, allyloxypyridinium and allylphosphonium salts have been utilized in the initiation of cationic polymerization via the addition–fragmentation mechanism.36–47
Visible-light induced FRPCP
The use of FRPCP to broaden and increase the spectral response of onium salts has been described by a number of investigators. The selection of suitable photoinitiators obviously allows the tuning of the absorption from the UV to visible wavelength range.48,49 In this context, the use of mild irradiation conditions using light emitting diodes (LEDs) is also a very attractive topic as these irradiation devices are eco-friendly (cheaper, safer, lower energy consumption, compact and used in 3D printing etc.) compared to traditional UV lamps (e.g. mercury bulbs).50 The main visible light type I photoinitiators are acylphosphine oxides, acyl germanes, titanocene and silyl glyoxylate that undergo different cleavages (C–P, C–Ge, Ti–C, C–C, respectively) (Scheme 3). Although monoacylphosphine oxides absorb visible light with a high extinction coefficient, their usages in the FRPCP system are limited. These photoinitiators are only active when the photochemically generated (through α-cleavage) benzoyl and phosphonyl radicals (they have high ionization potential and cannot be oxidized by onium salts51) undergo addition or abstraction reactions with a monomer or solvent. In order to overcome these limitations, a specially designed bisacylphosphine oxide by introducing a second benzoyl substituent adjacent to the phosphonyl group was tested in the FRPCP process. The absorption of bisacylphosphine oxides is shifted to longer wavelengths compared to monoacyl acylphosphine oxides due to conjugative carbonyl–phosphinoyl–carbonyl interactions. In contrast to monoacylphosphine oxides, the photochemically generated phosphinoyl radicals could be directly oxidized to the corresponding phosphonium ions that are capable of initiating the visible-light induced FRPCP of cyclic and vinyl ethers.51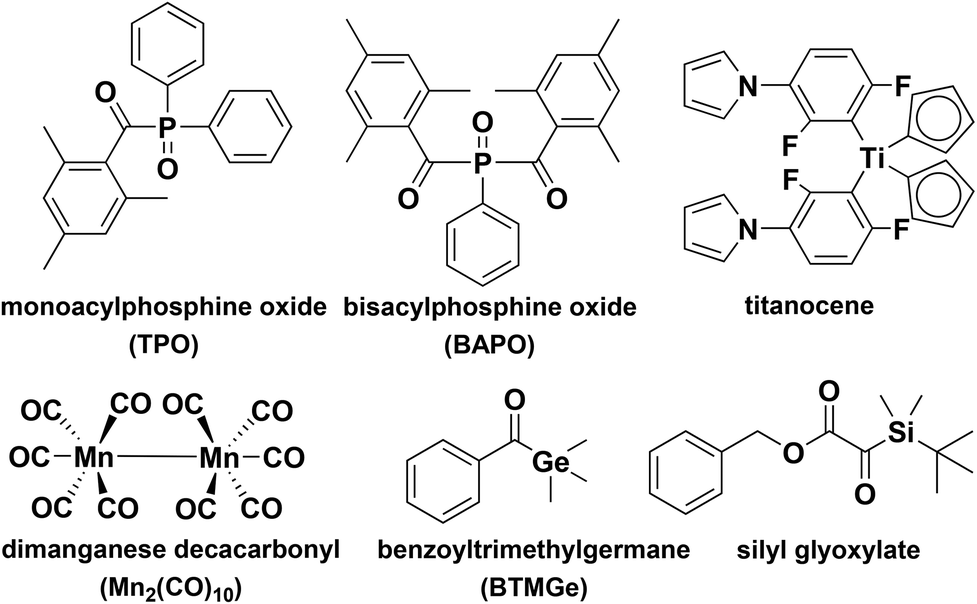 | ||
| Scheme 3 Example of visible light photoinitiators used in the FRPCP process. | ||
In the analogue process to phosphonyl radicals, Yagci and co-workers have developed a germane based photoinitiator, namely, benzoyltrimethylgermane, as an agent to reduce onium salts in the FRPCP process.52 Upon visible light photolysis of benzoyltrimethylgermane, trimethyl germyl radicals formed from the homolysis of carbon germanium bonds in the primary step undergo an electron transfer reaction yielding germanium cations capable of initiating cationic polymerization. The development of a visible light sensitive system based on acylgermanes in combination with diaryliodonium salts for the FRPCP was very useful for epoxides and highly reactive vinylic monomers such as ethers and N-vinyl carbazole at wavelengths up to 450 nm.52
The polysilanes were mentioned as capable of initiating FRPCP a long time ago.53,54 Indeed, silyl radicals can be generated by the cleavage of the Si–Si bonds under light exposure. As silyl radicals are being easily oxidized by iodonium salts, the related FRPCP process was shown to be quite efficient. Nevertheless, this strategy suffers from two drawbacks: (i) a low cleavage quantum yield and (ii) some side reactions, which are competitive to the silyl radical formation. Quite recently, the Yagci and Liska groups reported a new visible light cationic photoinitiating system by designing a silicon based photoinitiator tetrakis(2,4,6-trimethylbenzoyl)silane in conjunction with iodonium salts.55,56 This simple system was found to be effective for visible light initiated cationic polymerization of vinyl monomers such as isobutyl vinyl ether, N-vinylcarbazole and cyclohexene oxide. Because of its high extinction coefficient in the visible region and strong oxidation power, silyl-based FRPCP plays an important role in diverse applications requiring visible light irradiation. Furthermore, the resulting acyl silane terminated polymer could also be utilized as a polymeric photoinitiator for FRP of methyl methacrylate and FRPCP of cyclohexene oxide.
The organometallic photoinitiators (such as dimanganese decacarbonyl in conjunction with alkyl halides and bis(η-5-2,4-cyclopentadien-1-yl)-bis(2,6-difluoro-3-(1H-pyrrol-1-yl)-phenyl)titanium) are also utilized as free radical promoters providing the possibility of performing the cationic polymerization of cyclic and alkyl vinyl ethers. Upon absorption of visible light, the dimanganese decacarbonyl decomposes into manganese pentacarbonyl radicals that readily react with the terminal halide group yielding carbon-centered radicals oxidized by a suitable iodonium salt.57–59 In the case of titanocene type initiators, the generation of reactive cations and initiation mechanism are more complex. It is known that the fluorinated titanocene type photoinitiator yields no primary organic radicals (aryl and titanocene radicals) but rather titanium centered diradicals. In the presence of suitable iodonium and N-alkoxy pyridinium salts, electron transfer reactions essentially yield initiating cations. Although how the cations are generated is not fully known, the results suggest that the titanocene type photoinitiator played an important role in the initiation of cationic polymerization.60
Visible light induced photopolymerization reactions occur in the presence of organic molecules (dyes, highly conjugated thiophenes or coloured molecules) incorporating into an absorbing photoinitiating system and being able to act as photoinitiators or photosensitizers.61–64 FRPCP can be very efficient and can be performed even with low intensity sources. The decomposition of the iodonium salt in the presence of photosensitizers upon visible light irradiation has been largely studied in the literature.65–71 The key points of the researchers for highly efficient photoinitiating systems consists of looking for PS exhibiting: (i) suitable absorption (visible to near infrared region), (ii) high molar extinction coefficients, (iii) high reactivity for the photoreduction of the iodonium salt and low sensitivity to oxygen inhibition. To fulfill these issues, the hydrogen abstraction process was more recently proposed for the formation of silyl radicals. This approach consists of the incorporation of silanes (R3SiH) with a suitable hydrogen abstractor. The introduction of a silyl moiety on a given photoinitiator or photosensitizer also allows the design of efficient photoinitiating systems.65–75 It was shown that silyl radicals are highly efficient initiating structures towards acrylates in the free radical polymerization.76,77 Interestingly, they also revealed a good ability to sensitize the onium salt decomposition in FRPCP. Recent studies on the silyl radical chemistry have led to new developments for FRPCP in air and upon irradiation with various light sources. Some typical examples outlining the performance of organosilanes (e.g. tris(trimethylsilyl)silane) containing photoinitiators are given in Table 1 and the structures of different chemical agents are given in Scheme 4. A large tuning of the absorption range has been achieved using a three-component photoinitiating system allowing to cover the entire visible range: photosensitizers (commercial dyes, synthesized dyes, organophotoredox catalysts or commercial photoinitiators)/iodonium salts (such as Ph2I+PF6−)/silane (such as tris-(timethylsilyl)silane or R3SiH).
 | ||
| Scheme 4 Photoinitiating systems using silyl radicals. | ||
| Photoinitiator | Onium salt | Wavelength (nm) |
|---|---|---|
| ITX: isopropylthioxanthone, OPCs: derivatives of pyrene, anthracene, naphthacene, and pentacene, dyes: based on diamine, oligophenylenevinylene, salphen ligands and polyazine skeletons, An–Si: 9,10-bis[(triisopropylsilyl)-ethynyl]anthracene, Ph2+PF6−: diphenyliodonium hexafluorophosphate, IPh2I+B(C6F5)4−: 4-isopropyl-4’-methyldiphenyliodonium tetrakis(pentafluorophenyl)borate. | ||
| Titanocene | Ph2I+PF6− | 390 < λ < 800 (ref. 65) |
| ITX or BAPO or TPO | Ph2I+PF6− | 390 < λ < 800 or 405 (ref. 66) |
| Acridinedione | Ph2I+PF6− | 390 < λ < 800 or 405 (ref. 67) |
| Perylene derivatives | IPh2I+B(C6F5)4− | 390 < λ < 800 or 532 (ref. 68) |
| Pyrene derivatives | Ph2I+PF6− or IPh2I+B(C6F5)4− | 390 < λ < 800 or 457 (ref. 69) |
| Pyridinium salt | Ph2I+PF6− | 390 < λ < 800 or 457 or 532 (ref. 70) |
| OPCs | Ph2I+PF6− | 300 < λ < 500 or 630 (ref. 71) |
| Dyes | Ph2I+PF6− | 390 < λ < 800 or 457 or 462 or 514 (ref. 72) |
| An–Si | Ph2I+PF6− | 390 < λ < 800 or 462 (ref. 73) |
Upon light exposure (Scheme 4), the excited PS (PS*) reacts with the iodonium salt to generate an aryl radical that interacts with silane R3SiH through a H-abstraction process (silane characterized by low Si–H bond dissociation energy should be preferred); the formed silyl radical R3Si˙ is oxidized into a silylium cation R3Si+. The feedback concerning a possible oxygen inhibition is ruled out as silyl radicals can consume oxygen and scavenge peroxyl radicals regenerating new silyls.
Photoinitiators are usually based on organic structures in industrial radiation curing application. Although except titanocenes and ferrocenium salt they were not industrially employed,78 the use of organometallic PIs was recently reported. The development of organic photoredox compounds (OPCs) as photoredox catalysts brings several advantages: (i) lower cost, (ii) in some cases, commercial availability, (iii) lower toxicity, (iv) better stability, (v) only low amounts required and (vi) good solubility. Metal based photoredox catalysts were recently proposed in different initiating systems for photopolymerization reactions under soft irradiation conditions.79–83 The OPCs play the role of a photoinitiator or a photosensitizer that would be recovered during the initiation step. In an oxidative cycle, the three-component systems (OPC/Iod/silane) generate silyliums that can initiate the cationic polymerization. These three-component systems (PS/Iod/silane) are really versatile and can easily work under very different experimental conditions. Interestingly, all the experiments are carried out under air conditions. Germanium hydride (R3GeH) is also known as a good hydrogen donor like silane. In fact, the use of R3GeH in combination with the silyl glyoxylate/iodonium salt was found to be very efficient to initiate the FRPCP of cationic monomers.84 Excellent final conversions (>60%) are reached upon blue LED light irradiation and in air.
The use of an additive in the visible light induced FRPCP process is certainly promising when the light absorption is driven by the photoinitiator or photosensitizer and a careful choice of the salt and the additive ensures an efficient formation of highly reactive initiating cations. The design of new systems, especially under soft irradiation conditions, remains a good challenge. In this context, the search for new additives that can be used in combination with iodonium salts is crucial. Aromatic amines, benzyl alcohol, NVC and 9H-carbazole-9-ethanol (CARET) were shown as very interesting additives to promote the cationic polymerization in three-component photoinitiating systems.85 It is well established that halogenated xanthene dyes such as eosin, erythrosine and rose bengal in combination with electron donating co-initiators such as aromatic amines act as efficient initiators for visible light induced free radical polymerization of acrylic monomers.25 Very elegantly, Neckers et al.86 have explored the use of these dyes and co-initiators in combination with onium salts as potential visible light initiating systems for cationic polymerization. The dye functions as an electron/photon acceptor with an amine to produce aminoalkyl radicals upon visible light irradiation. The onium salt is responsible for oxidizing these strong nucleophilic radicals to the carbocations, and the latter are capable of initiating cationic polymerization. The presence of a dye enables the sensitivity of the cationic initiator to be extended to a majority of visible wavelengths. The photosensitivity of the FRPCP process can be easily adjusted by selecting xanthene dyes with suitable absorption maxima.
Benzyl alcohol was also shown87 to be an efficient hydrogen donor for camphorquinone to generate free radicals upon visible light irradiation. The generated benzylic radicals are being easily oxidized by iodonium salts; therefore, the use of additional benzyl alcohol with a camphorquinone/iodonium salt system improves the efficiency of the FRPCP process. Interestingly, Crivello et al.88 reported the FRPCP of epoxides using a long wavelength type I photoinitiator (titanocene derivatives); the latter is characterized by good absorption bands at 405 and 480 nm with an appreciable tail absorption extending out to 560 nm. Titanocene complexes produce free radicals upon irradiation with visible light. The primary radical species interact with benzyl alcohols to abstract hydrogen atoms. The resulting benzyl radical species efficiently reduce diaryliodonium salts thereby generating oxycarbenium ions that can initiate the polymerization or fragment to form the corresponding aldehyde and a Brønsted superacid.88 This superacid can also subsequently initiate the ring-opening polymerization of a wide variety of epoxide monomers. In this connection, it is important to note that benzyl alcohol bearing electron donating substituents that stabilize the radical and the corresponding cations by resonance interaction would be predicted to accelerate the photopolymerization of the monomer.
The NVC was proposed as an additive in combination with a photoinitiator and an iodonium salt, and the photosensitivity to the visible region has been reported. However, a good performance was mainly obtained for a rather high light intensity. The NVC appeared as a cheap and efficient alternative to the silane: the same mechanism is observed except that Ar˙ adds to the NVC double bond (and formed Ar-NVC˙ that can be easily oxidized) instead of abstracting a hydrogen atom on the silane (Scheme 5).
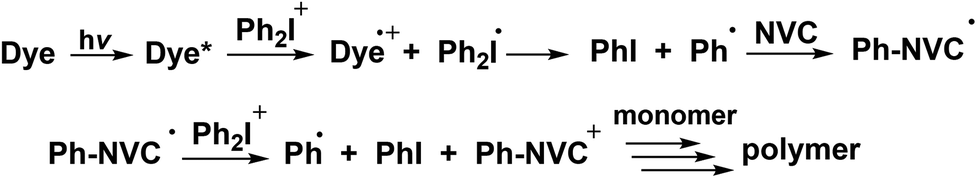 | ||
| Scheme 5 Proposed mechanism using NVC as an additive. | ||
The three-component system (dye/NVC/iodonium salt) is an elegant strategy that allows many possibilities. Following these studies, the possibility of using different dyes has been expanded, i.e. indanedione89 and perylene68 derivatives, and Michler's ketone.90 These dyes are based on a push–pull effect (related to an intramolecular donor/acceptor interaction and the favorable orientation of charge delocalization in the axis of the chromophore). They exhibit an unusual and remarkable broad absorption lying from the blue to the red, which allows polymerization reaction under various irradiation devices.
More recently, the CARET molecule was proposed as an interesting additive.91–93 Indeed, CARET is less toxic compared to the parent NVC. The CARET additive behaves as NVC through a reaction with aryl radicals formed in the photosensitizer/iodonium salt interaction. However, the process is basically different i.e. the addition of the aryl radical to the NVC double bond being changed for a hydrogen abstraction for CARET. Remarkably, CARET has a better additive effect than NVC for different systems suggesting that the hydrogen abstraction process is more favorable than the addition onto NVK for the aryl radicals.
Terphenyl derivatives91 and copper photoredox catalyst bearing pyridine-pyrazole ligands94 were used as representative photosensitive systems exhibiting a good visible light absorption. The PS/CARET/iodonium salt-based three-component photoinitiating system initiates the FRPCP of the difunctional monomer ((3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate) upon exposure to visible light and allows high epoxy function conversions.
The chemical modification of a conventional photosensitizer is also a promising approach to obtain enhanced delocalization of the molecular orbitals and to improve light absorption properties (red-shifted wavelengths towards the visible/near IR region and with higher molar extinction coefficients) or the photochemical reactivity.95 Among commercially available photosensitizers, aromatic ketone (e.g. thioxanthone TX) has been largely exploited.96–98 TX derivatives conveniently absorbing in the 350–650 nm region are proposed for the cationic polymerization of vinyl ethers and epoxides. Interestingly, very soft irradiation conditions can be employed. Excellent polymerization profiles have been obtained from selected dye/iodonium salt combinations. TX derivatives possessing suitable hydrogen donors and oxidizing salts may be used to promote the cationic polymerization of appropriate monomers. A major advantage of these types of initiators is related to their one component nature. They can serve as both a triplet photosensitizer and a hydrogen donor. Moreover, due to the presence of hydrogen donating sites adjacent to the nitrogen atom of the carbazole group, such derivatives display a one-component nature and can act as an efficient photoinitiator in the visible light region for free radical and free radical promoted cationic photopolymerization without the need for an additional hydrogen donor. This novel photoinitiator initiates the free radical polymerization in the absence and presence of a hydrogen donor.96 In addition, thioxanthone-ethylcarbazole in combination with an iodonium salt initiates the cationic polymerization of various industrially important monomers in the visible range without the requirement of any additional compound.96 Light absorbing nanoparticles can also be used to promote cationic polymerization. Sangermano and co-workers recently reported that multi-walled carbon nanotubes can act as visible light photoinitiators to induce cationic polymerization.99 In this process, oxidizable radicals are produced by hydrogen abstraction from the monomer.
Near IR induced FRPCP
While an increasing number of systems have been reported for successful photopolymerization upon visible light (400–700 nm) irradiation over the past 10 years, only a few systems absorbing in the near-infrared (NIR) region (>750 nm) are reported for FRP processes. Compared with the conventional ultraviolet and visible region, the NIR has distinct advantages for industrial applications e.g. deeper penetration into the material, which provides access to thick and even filled cured systems. Classical NIR lamps are characterized by better efficiency in the conversion of energy to light i.e. they are eco-friendly with lower energy consumption and NIR LEDs are now accessible. Remarkably, a photoinitiating system for the cationic photopolymerization using NIR light was developed recently by Yagci et al.100 It was proposed that upconverting nanoparticles (UCNPs) as the emitter in combination with titanocene, and iodonium salt as the visible light photoinitiator and oxidant, respectively can act as the photosensitive system for cationic polymerization. The commercially available titanocene100 photoinitiator possesses strong absorption bands at 405 and 480 nm that can be activated by the blue or violet light emission of UCNP upon NIR irradiation (Scheme 6). | ||
| Scheme 6 NIR-photoinduced free radical promoted cationic polymerization of epoxy and vinyl monomers by using UCNP, titanocene, and Ph2I+PF6− in the absence and presence of benzyl alcohol. | ||
The initiation efficiency using the three component photoinitiating system was demonstrated for various monomers. Depending on the nature of monomers and the free radicals formed, two different mechanisms are feasible. Indeed, electron donor radicals can be generated by hydrogen abstraction from the monomer or from benzyl alcohol and can be oxidized to form cations able to initiate cationic polymerizations (Scheme 6).
In another study, Yagci et al. described a new approach for the NIR light activation using upconverting glass (UCG).101 This approach pertains to the laser irradiation of UCG at 975 nm in the presence of fluorescein and pentamethyldiethylene triamine. To perform the corresponding FRPCP of cyclohexene oxide, isobutyl vinyl ether and NVC; fluorescein, dimethyl aniline and diphenyliodonium hexafluorophosphate have been used as the sensitizer, co-initiator and oxidant, respectively. Iodonium salt promptly oxidizes amino alkyl radicals formed to the corresponding cations. Thus, cationic polymerization with an efficiency comparable to the conventional irradiation source is initiated.
Macromolecular engineering via FRPCP
The versatility of the FRPCP process has been revealed in the design and synthesis of various macromolecular architectures including block, graft and star copolymers which cannot be prepared by a single polymerization mechanism.102–104 The combination of two different polymerization methods by a mechanistic transformation approach is widely applied in polymer synthesis to merge structurally different blocks in one product. In this approach, the first block obtained by a certain polymerization is switched into another kind of species that is capable of initiating the second polymerization of the different monomer. Applying FRP of styrene followed by the FRPCP of cyclic or vinylic ethers enables one to obtain block copolymers containing two chemically distinct and frequently immiscible blocks that are covalently bound together.105,106 To exemplify, Yagci and co-workers also demonstrated the possibility of synthesizing graft copolymers by combining the ring-opening metathesis polymerization of ethylene and FRPCP of cyclohexene oxide (CHO).107 An analogous approach was also applied for the synthesis of AB2 or A2B2 type star copolymers prepared by the combination of ring-opening polymerization (ROP) of epsilon-caprolactone (CL) and FRPCP of CHO. The ROP of CL initiated from two hydroxyl groups of 3-cyclohexene-1,1-dimethanol formed a well-defined two-arm PCL with a cyclohexene-functional group that converted to two benzoin groups by an azide/alkyne click reaction. Photolysis of the resulting macrophotoinitiator in the presence of 1-ethoxy-2-methylpyridinium hexafluorophosphate and CHO led to the corresponding A2B2 type star polymer.108 More recently, a modified FRPCP strategy was applied for the photoinitiated living cationic polymerization of vinyl or cyclic ethers which can be combined with atom transfer radical polymerization,109–111 reversible-addition–fragmentation-transfer polymerization112 and iniferter113,114 polymerization. All other examples of mechanistic transformation using the FRPCP process are summarized in Table 2.| Initiators | Wavelength (nm) | Copolymers |
|---|---|---|
| PSt: polystyrene, PCHO: poly(cyclohexene oxide), PECH: polyepichlorohydrin, PIBVE: poly(isobutyl vinyl ether), PBVE: poly(butyl vinyl ether), PCL: poly(ε-caprolactone), PLA: poly(D,L-lactide), PVK: poly(N-vinylcarbazole), PE: polyethylene, EMP+PF6−: 1-ethoxy-2-methylpyridinium hexafluorophosphate, Ph2I+PF6−: diphenyliodonium hexafluorophosphate,Mn2(CO)10: dimanganese decacarbonyl. | ||
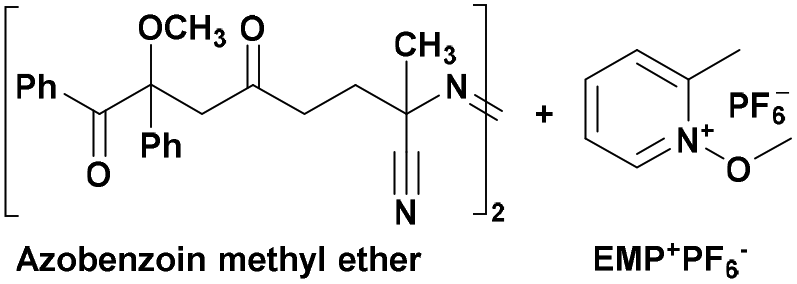
|
350 | PSt-b-PCHO105 |
| PCHO-b-PLC121,122 | ||

|
350 | PECH-b-PCHO123 |
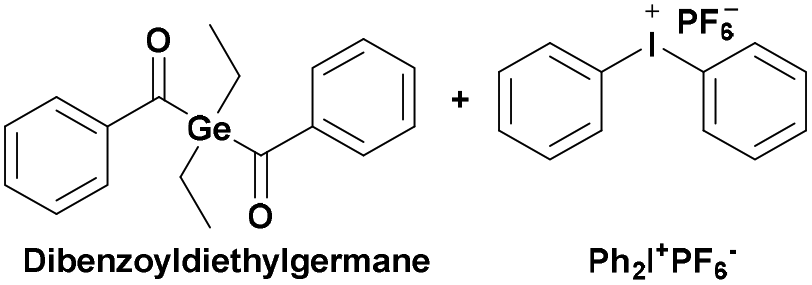
|
430–490 | PCHO-b-PSt124 |
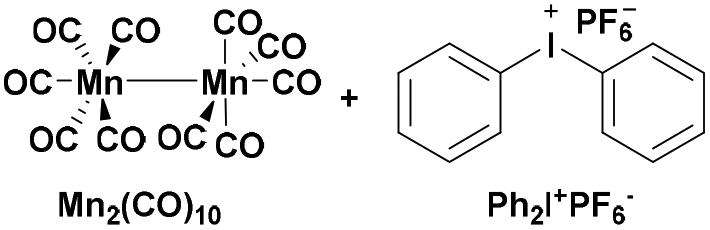
|
430 | PSt-b-PCHO106 |
| PSt-b-PIBVE106 | ||

|
430 | PSt-b-PCHO125 |

|
450 | PMMA-b-PIBVE126 |

|
450 | PMMA-b-PBVE127 |
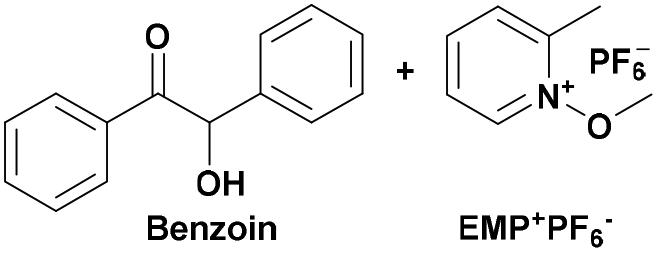
|
350 | PCHO-b-PSt128 |
| PCL-b-PLA129 | ||

|
400 | PVK-b-PCHO130 |

|
430 | PE-g-PCHO107 |

|
350 | PCL-(PCHO)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 131 131 |
| (PCL)2-(PCHO)2108 |
||
It should be pointed out that the electron transfer reactions used in FRPCP successfully adapted to the in situ formation of polymer/nanocomposites.115–118 In this strategy, metal salts were used to oxidize the radicals to the corresponding cations to initiate cationic polymerization and simultaneously reduced to the nanoparticles.
Finally, the FRPCP strategy can be well combined with the free radical polymerization for original access to interpenetrating polymer networks (IPNs) under mild conditions e.g. from the polymerization of epoxy/acrylate blends. Indeed, the photoinitiating systems presented above often generate initiating radicals and cations allowing simultaneous initiation of cationic and radical polymerization.119 IPNs are very interesting polymers combining the properties of both networks (e.g. lower shrinkage for usage in 3D printing).120
Conclusions
Following the discovery of FRPCP about four decades ago, tremendous efforts have been made to explore new initiating systems for cationic polymerization. Compared to the direct photoinitiating systems, FRPCP provides a significant advantage of wavelength selective chemistry and mild conditions to produce epoxy and vinyl based materials in the most suitable wavelength or temperature range desired for specific applications. The ability of the free radical photoinitiators to absorb and form oxidizable radicals provides the possibility of using the range of photoactive compounds from simple organic molecules to complex-structured nanomaterials. Although significant progress has been made, future research will focus on the development of environmentally friendly and refined systems acting at a higher wavelength with lower light intensity.Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- J. V. Crivello and E. Reichmanis, Chem. Mater., 2014, 26, 533–548 CrossRef CAS.
- Y. Yagci and I. Reetz, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Aust. J. Chem., 2011, 64, 982–991 CrossRef CAS.
- J. V. Crivello and J. L. Lee, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 479–503 CrossRef CAS.
- J. V. Crivello and J. L. Lee, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3951–3968 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- J. V. Crivello, Adv. Polym. Sci., 1984, 62, 1–48 CrossRef CAS.
- M. Abdallah, T.-T. Bui, F. Goubard, D. Theodosopoulou, F. Dumur, A. Hijazi, J.-P. Fouassier and J. Lalevée, Polym. Chem., 2019, 10, 6145–6156 RSC.
- S. Q. Shi, C. Croutxe-Barghorn and X. Allonas, Prog. Polym. Sci., 2017, 65, 1–41 CrossRef CAS.
- Y. Yagci, Y. Y. Durmaz and B. Aydogan, Chem. Rec., 2007, 7, 78–90 CrossRef CAS PubMed.
- Y. Yaǧci and A. Ledwith, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1911–1918 CrossRef.
- G. Hizal, Y. Yagci and W. Schnabel, Polymer, 1994, 35, 2428–2431 CrossRef CAS.
- D. J. Brunelle, Ring-Opening Polymerization. Mechanisms, Catalysis, Structure, Utility, Hanser Publishers, Munich, 1993 Search PubMed.
- Y. Yagci and W. Schnabel, Makromol. Chem., Macromol. Symp., 1992, 60, 133–143 CrossRef CAS.
- P. Garra, A. Caron, A. Al Mousawi, B. Graff, F. Morlet-Savary, C. Dietlin, Y. Yagci, J. P. Fouassier and J. Lalevee, Macromolecules, 2018, 51, 7872–7880 CrossRef CAS.
- K. Kaya, J. Kreutzer and Y. Yagci, ChemPhotoChem, 2019, 3, 1187–1192 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- X. D. Huang, B. J. McConkey, T. S. Babu and B. M. Greenberg, Environ. Toxicol. Chem., 1997, 16, 1707–1715 CAS.
- Y. Hua and J. V. Crivello, Macromolecules, 2001, 34, 2488–2494 CrossRef CAS.
- Y. Hua, F. Jiang and J. V. Crivello, Chem. Mater., 2002, 14, 2369–2377 CrossRef CAS.
- J. V. Crivello and F. Jiang, Chem. Mater., 2002, 14, 4858–4866 CrossRef CAS.
- J. V. Crivello and M. Jang, J. Photochem. Photobiol., A, 2003, 159, 173–188 CrossRef CAS.
- F. A. Abdul-Rasoul, A. Ledwith and Y. Yagci, Polymer, 1978, 19, 1219–1222 CrossRef CAS.
- A. Ledwith, Polymer, 1978, 19, 1217–1219 CrossRef CAS.
- Y. Yagci, Ph.D thesis, University of Liverpool, 1979.
- Y. Yagci, G. Hizal and A. Aydogan, Eur. Polym. J., 1985, 21, 25–27 CrossRef CAS.
- Y. Yagci, W. Schnabel and A. Ledwith, Eur. Polym. J., 1987, 23, 737–740 CrossRef CAS.
- F. A. Abdul-Rasoul, A. Ledwith and Y. Yagci, Polym. Bull., 1978, 1, 1–6 CAS.
- H. Baumann, U. Müller, D. Pfeifer and H. J. Timpe, J. Prakt. Chem., 1982, 324, 217–226 CrossRef CAS.
- Y. Yaḡci, J. Borbely and W. Schnabel, Eur. Polym. J., 1989, 25, 129–131 CrossRef.
- Y. Yagci and I. Reetz, React. Funct. Polym., 1999, 42, 255–264 CrossRef CAS.
- P. Monecke, W. Schnabel and Y. Yagci, Polymer, 1997, 38, 5389–5395 CrossRef CAS.
- I. Reetz, V. Bacak and Y. Yagci, Polym. Int., 1997, 43, 27–32 CrossRef CAS.
- Y. Yagci and A. Onen, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 3621–3624 CrossRef CAS.
- B. Yoneyman, A. Onen and Y. Yagci, Des. Monomers Polym., 2001, 4, 381–390 CrossRef CAS.
- U. Kucuktonbekici, M. Degirmenci and Y. Yagci, Turk. J. Chem., 2002, 26, 793–800 CAS.
- Y. Hepuzer, U. Kucuktonbekici and Y. Yagci, J. Photochem. Photobiol., A, 2000, 130, 71–74 CrossRef CAS.
- M. Onciu, A. Onen and Y. Yagci, Polym. Int., 2001, 50, 144–147 CrossRef CAS.
- A. Onen and Y. Yagci, Macromolecules, 2001, 34, 7608–7612 CrossRef.
- A. Onen and Y. Yagci, Polymer, 2001, 42, 6681–6685 CrossRef CAS.
- L. Atmaca, A. Onen and Y. Yagci, Eur. Polym. J., 2001, 37, 677–682 CrossRef CAS.
- A. Onen and Y. Yagci, Macromol. Chem. Phys., 2001, 202, 1950–1954 CrossRef CAS.
- Y. Yagci, S. Yildirim and A. Onen, Macromol. Chem. Phys., 2001, 202, 527–531 CrossRef CAS.
- V. Bacak, I. Reetz, Y. Yagci and W. Schnabel, Polym. Int., 1998, 47, 345–350 CrossRef CAS.
- S. Yurteri, A. Onen and Y. Yagci, Eur. Polym. J., 2002, 38, 1845–1850 CrossRef CAS.
- L. Atmaca, I. Kayihan and Y. Yagci, Polymer, 2000, 41, 6035–6041 CrossRef CAS.
- C. Dietlin, S. Schweizer, P. Xiao, J. Zhang, F. Morlet-Savary, B. Graff, J. P. Fouassier and J. Lalevee, Polym. Chem., 2015, 6, 3895–3912 RSC.
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevee, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- J. V. Crivello and M. Sangermano, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 343–356 CrossRef CAS.
- C. Dursun, M. Degirmenci, Y. Yagci, S. Jockusch and N. J. Turro, Polymer, 2003, 44, 7389–7396 CrossRef CAS.
- Y. Y. Durmaz, N. Moszner and Y. Yagci, Macromolecules, 2008, 41, 6714–6718 CrossRef CAS.
- D. Yücesan, H. Hostoygar, S. Denizligil and Y. Yagci, Angew. Makromol. Chem., 1994, 221, 207–216 CrossRef.
- Y. Yagci, I. Kminek and W. Schnabel, Eur. Polym. J., 1992, 28, 387–390 CrossRef CAS.
- M. Mitterbauer, M. Haas, H. Stüger, N. Moszner and R. Liska, Macromol. Mater. Eng., 2017, 302, 1600536 CrossRef.
- E. Sari, M. Mitterbauer, R. Liska and Y. Yagci, Prog. Org. Coat., 2019, 132, 139–143 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen and Y. Yagci, Polym. Int., 2016, 65, 1001–1014 CrossRef CAS.
- M. Ciftci, M. A. Tasdelen and Y. Yagci, Polym. Chem., 2014, 5, 600–606 RSC.
- Y. Yagci and Y. Hepuzer, Macromolecules, 1999, 32, 6367–6370 CrossRef CAS.
- M. Degirmenci, A. Onen, Y. Yagci and S. P. Pappas, Polym. Bull., 2001, 46, 443–449 CrossRef CAS.
- J. P. Fouassier, F. Morlet-Savary, J. Lalevee, X. Allonas and C. Ley, Materials, 2010, 3, 5130–5142 CrossRef CAS PubMed.
- B. Aydogan, Y. Yagci, L. Toppare, S. Jockusch and N. J. Turro, Macromolecules, 2012, 45, 7829–7834 CrossRef CAS.
- B. Aydogan, G. E. Gunbas, A. Durmus, L. Toppare and Y. Yagci, Macromolecules, 2010, 43, 101–106 CrossRef CAS.
- B. Aydogan, A. S. Gundogan, T. Ozturk and Y. Yagci, Macromolecules, 2008, 41, 3468–3471 CrossRef CAS.
- M. Tehfe, J. Lalevée, X. Allonas and J. Fouassier, Macromolecules, 2009, 42, 8669–8674 CrossRef CAS.
- M.-A. Tehfe, J. Lalevee, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, N. Blanchard, C. Fries, B. Graff, X. Allonas, F. Louërat and J. P. Fouassier, Eur. Polym. J., 2010, 46, 2138–2144 CrossRef CAS.
- M.-A. Tehfe, F. Dumur, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2013, 214, 1052–1060 CrossRef CAS.
- M.-A. Tehfe, F. Dumur, E. Contal, B. Graff, F. Morlet-Savary, D. Gigmes, J.-P. Fouassier and J. Lalevée, Polym. Chem., 2013, 4, 1625–1634 RSC.
- M.-A. Tehfe, A. Zein-Fakih, J. Lalevée, F. Dumur, D. Gigmes, B. Graff, F. Morlet-Savary, T. Hamieh and J.-P. Fouassier, Eur. Polym. J., 2013, 49, 567–574 CrossRef CAS.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, Macromolecules, 2012, 45, 1746–1752 CrossRef CAS.
- J. Lalevée, F. Dumur, M.-A. Tehfe, A. Zein-Fakih, D. Gigmes, F. Morlet-Savary, B. Graff and J.-P. Fouassier, Polymer, 2012, 53, 4947–4954 CrossRef.
- M.-A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J.-P. Fouassier, ACS Macro Lett., 2011, 1, 198–203 CrossRef.
- D. Wang, P. Garra, F. Szillat, J. P. Fouassier and J. Lalevée, Macromolecules, 2019, 52, 3351–3358 CrossRef CAS.
- D. Wang, F. Szillat, J. P. Fouassier and J. Lalevée, Macromolecules, 2019, 52, 5638–5645 CrossRef CAS.
- J. Lalevée, N. Blanchard, M. El-Roz, B. Graff, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 4180–4186 CrossRef.
- J. Lalevée, A. Dirani, M. El-Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2003–2010 CrossRef.
- A. F. Cunningham and V. Desobry, in Radiation Curing in Polymer Science and Technology, ed. J. P. Fouassier, Springer Netherlands, Dordrecht, 1993, vol. II, pp. 323–373 Search PubMed.
- J. Lalevée, N. Blanchard, M.-A. Tehfe, F. Morlet-Savary and J. P. Fouassier, Macromolecules, 2010, 43, 10191–10195 CrossRef.
- J. Lalevée, N. Blanchard, M.-A. Tehfe, M. Peter, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1986–1991 RSC.
- J. Lalevée, N. Blanchard, M. A. Tehfe, M. Peter, F. Morlet-Savary and J. P. Fouassier, Macromol. Rapid Commun., 2011, 32, 917–920 CrossRef.
- J. Lalevée, N. Blanchard, M.-A. Tehfe, M. Peter, F. Morlet-Savary and J. P. Fouassier, Polym. Bull., 2012, 68, 341–347 CrossRef.
- J. Lalevée, M. Peter, F. Dumur, D. Gigmes, N. Blanchard, M. A. Tehfe, F. Morlet-Savary and J. P. Fouassier, Chem. – Eur. J., 2011, 17, 15027–15031 CrossRef.
- J. Kirschner, M. Bouzrati-Zerelli, J. P. Fouassier, J. M. Becht, J. E. Klee and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1420–1429 CrossRef CAS.
- J. P. Fouassier and J. Lalevée, RSC Adv., 2012, 2, 2621–2629 RSC.
- Y. Bi and D. C. Neckers, Macromolecules, 1994, 27, 3683–3693 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866–875 CrossRef CAS.
- J. V. Crivello, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 474–483 CrossRef CAS.
- M.-A. Tehfe, F. d. r. Dumur, B. Graff, D. Gigmes, J.-P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 3332–3341 CrossRef CAS.
- H. Mokbel, F. Dumur, B. Graff, C. R. Mayer, D. Gigmes, J. Toufaily, T. Hamieh, J. P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2014, 215, 783–790 CrossRef CAS.
- A. A. Mousawi, C. Dietlin, B. Graff, F. Morlet-Savary, J. Toufaily, T. Hamieh, J. P. Fouassier, A. Chachaj-Brekiesz, J. Ortyl and J. Lalevée, Macromol. Chem. Phys., 2016, 217, 1955–1965 CrossRef CAS.
- A. Al Mousawi, F. Dumur, P. Garra, J. Toufaily, T. Hamieh, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2017, 50, 2747–2758 CrossRef CAS.
- A. Al Mousawi, D. M. Lara, G. Noirbent, F. Dumur, J. Toufaily, T. Hamieh, T.-T. Bui, F. Goubard, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2017, 50, 4913–4926 CrossRef CAS.
- A. Al Mousawi, A. Kermagoret, D.-L. Versace, J. Toufaily, T. Hamieh, B. Graff, F. Dumur, D. Gigmes, J. P. Fouassier and J. Lalevee, Polym. Chem., 2017, 8, 568–580 RSC.
- S. Dadashi-Silab, C. Aydogan and Y. Yagci, Polym. Chem., 2015, 6, 6595–6615 RSC.
- D. Tunc and Y. Yagci, Polym. Chem., 2011, 2, 2557–2563 RSC.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS.
- H. Mokbel, F. Dumur, C. R. Mayer, F. Morlet-Savary, B. Graff, D. Gigmes, J. Toufaily, T. Hamieh, J.-P. Fouassier and J. Lalevée, Dyes Pigm., 2014, 105, 121–129 CrossRef CAS.
- M. Sangermano, D. Rodriguez, M. C. Gonzalez, E. Laurenti and Y. Yagci, Macromol. Rapid Commun., 2018, 39, 1800250 CrossRef PubMed.
- Z. Li, J. Zhu, X. Guan, R. Liu and Y. Yagci, Macromol. Rapid Commun., 2019, 40, 1900047 CrossRef PubMed.
- A. Kocaarslan, S. Tabanli, G. Eryurek and Y. Yagci, Angew. Chem., 2017, 129, 14699–14702 CrossRef.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- G. Yilmaz and Y. Yagci, J. Photopolym. Sci. Technol., 2018, 31, 719–725 CrossRef CAS.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- Y. Yagci, A. Onen and W. Schnabel, Macromolecules, 1991, 24, 4620–4623 CrossRef CAS.
- M. U. Kahveci, G. Acik and Y. Yagci, Macromol. Rapid Commun., 2012, 33, 309–313 CrossRef CAS PubMed.
- M. Ciftci, S. Kork, G. Xu, M. R. Buchmeiser and Y. Yagci, Macromolecules, 2015, 48, 1658–1663 CrossRef CAS.
- Z. Uyar, M. Durgun, M. S. Yavuz, M. B. Abaci, U. Arslan and M. Degirmenci, Polymer, 2017, 123, 153–168 CrossRef CAS.
- M. Ciftci, Y. Yoshikawa and Y. Yagci, Angew. Chem., Int. Ed., 2017, 56, 519–523 CrossRef CAS.
- A. E. Muftuoglu, I. Cianga, S. Yurteri and Y. Yagci, J. Appl. Polym. Sci., 2004, 93, 387–394 CrossRef CAS.
- A. B. Duz and Y. Yagci, Eur. Polym. J., 1999, 35, 2031–2038 CrossRef CAS.
- A. Puglisi, E. Murtezi, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 7307–7310 RSC.
- M. Ciftci and Y. Yagci, Macromol. Rapid Commun., 2018, 39, 1800464 CrossRef PubMed.
- M. A. Tasdelen, M. Degirmenci, Y. Yagci and O. Nuyken, Polym. Bull., 2003, 50, 131–138 CrossRef CAS.
- Y. Yagci, O. Sahin, T. Ozturk, S. Marchi, S. Grassini and M. Sangermano, React. Funct. Polym., 2011, 71, 857–862 CrossRef CAS.
- M. Sangermano, Y. Yagci and G. Rizza, Macromolecules, 2007, 40, 8827–8829 CrossRef CAS.
- Y. Yagci, M. Sangermano and G. Rizza, Macromolecules, 2008, 41, 7268–7270 CrossRef CAS.
- Y. Yagci, M. Sangermano and G. Rizza, Polymer, 2008, 49, 5195–5198 CrossRef CAS.
- P. Garra, M. l. Carré, F. d. r. Dumur, F. Morlet-Savary, C. l. Dietlin, D. Gigmes, J.-P. Fouassier and J. Lalevée, Macromolecules, 2018, 51, 679–688 CrossRef CAS.
- J. P. Fouassier and J. Lalevée, Polymers, 2014, 6, 2588–2610 CrossRef.
- I. Serhatli, G. Galli, Y. Yagci and E. Chiellini, Polym. Bull., 1995, 34, 539–546 CrossRef CAS.
- G. Galli, E. Chiellini, Y. Yagci, E. I. Serhatli, M. Laus and A. S. Angeloni, Macromol. Symp., 1996, 107, 85–97 CrossRef CAS.
- Y. Hepuzer, Y. Yagci, T. Biedron and P. Kubisa, Angew. Makromol. Chem., 1996, 237, 163–171 CrossRef CAS.
- Y. Y. Durmaz, M. Kukut, N. Moszner and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4793–4799 CrossRef CAS.
- M. Degirmenci, O. Izgin, A. Acikses and N. Genli, React. Funct. Polym., 2010, 70, 28–34 CrossRef CAS.
- K. Kaya, M. Seba, T. Fujita, S. Yamago and Y. Yagci, Polym. Chem., 2018, 9, 5639–5643 RSC.
- H. Braun, Y. Yagci and O. Nuyken, Eur. Polym. J., 2002, 38, 151–156 CrossRef CAS.
- M. Degirmenci and N. Genli, Polym. Int., 2010, 59, 859–866 CAS.
- M. Degirmenci, N. Benek and M. Durgun, Polym. Bull., 2017, 74, 167–181 CrossRef CAS.
- M. Kamachi, H. Q. Guo and A. Kajiwara, Macromol. Chem. Phys., 2002, 203, 991–997 CrossRef CAS.
- Z. Uyar, M. Degirmenci, N. Genli and A. Yilmaz, Des. Monomers Polym., 2017, 20, 42–53 CrossRef CAS PubMed.
Footnote |
| † Dedicated to the memory of Prof. Anthony Ledwith. |
| This journal is © The Royal Society of Chemistry 2020 |