
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The core of the matter – arene substitution determines the coordination and catalytic behaviour of tris(1-phosphanyl-1′-ferrocenylene)arene gold(I) complexes†
Axel
Straube
 a,
Peter
Coburger
b,
Marvin
Michak
a,
Mark R.
Ringenberg
c and
Evamarie
Hey-Hawkins
*a
a,
Peter
Coburger
b,
Marvin
Michak
a,
Mark R.
Ringenberg
c and
Evamarie
Hey-Hawkins
*a
aInstitute of Inorganic Chemistry, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de; Web: https://anorganik.chemie.uni-leipzig.de/de/anorganik/ak-hey-hawkins/
bLaboratory of Inorganic Chemistry, ETH Zürich, CH-8093 Zürich, Switzerland
cInstitute of Inorganic Chemistry, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
First published on 15th October 2020
Abstract
Changing the aromatic core of C3-symmetric tris(ferrocenyl)arene-based tris-phosphanes has profound effects on their coordination behaviour towards gold(I). Depending on the arene (s-triazine, benzene, or trifluorobenzene), four different coordination modes can be distinguished and their preference has been rationalised using computational methods. The corresponding 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand-to-metal complexes, studied by variable-temperature NMR spectroscopy, revealed fluctional behaviour in solution. Given the presence of up to three or six ferrocenylene spacers per complex, their electrochemistry was investigated. The redox-responsive nature of the complexes can be advantageously exploited in the catalytic ring-closing isomerisation of N-(2-propyn-1-yl)benzamide, where the benzene-based 2:3 ligand-to-metal complex has been shown to display multiple activity states depending on the degree of (reversible) oxidation in a preliminary trial.
:1 ligand-to-metal complexes, studied by variable-temperature NMR spectroscopy, revealed fluctional behaviour in solution. Given the presence of up to three or six ferrocenylene spacers per complex, their electrochemistry was investigated. The redox-responsive nature of the complexes can be advantageously exploited in the catalytic ring-closing isomerisation of N-(2-propyn-1-yl)benzamide, where the benzene-based 2:3 ligand-to-metal complex has been shown to display multiple activity states depending on the degree of (reversible) oxidation in a preliminary trial.
Introduction
Modern-day ligand design is hardly imaginable without the inclusion of ferrocene.1–3 Pairing great stability with conformational flexibility, easy synthetic modifiability, the possibility for exploiting planar chirality, and reversible redox activity, ferrocene has attained the special place of perhaps the key organometallic building block.4,5 Next to numerous applications in asymmetric catalysis,6–8 ferrocene-containing ligands feature prominently in redox-switchable catalysis (RSC).9–11 Based on the concept of reversibly modifying the donor properties of a redox-active ligand and hence, in turn, the electronic nature of the coordinated metal,12,13 initial breakthroughs were achieved by Wrighton14 and Long.15 The latter report, the first to use ferrocene as the redox switch, incidentally also incorporated two pendant ferrocenyl termini into the ligand backbone (I, Chart 1). This is worth pointing out, as ligands built on multi-ferrocene-containing motifs are still comparably scarce, even without putting a focus on RSC. Notable examples include the BIFEP (II) and TRAP (III) ligand families,16,17 mesoionic carbene IV,18 terferrocene-based bis(terpyridine) V,19 and C2-symmetric tetradentate TriFer VI.20 Furthermore, a number of discrete multiferrocenyl compounds21–27 and ferrocenyl-containing dendrimers have been reported over the last years.28–31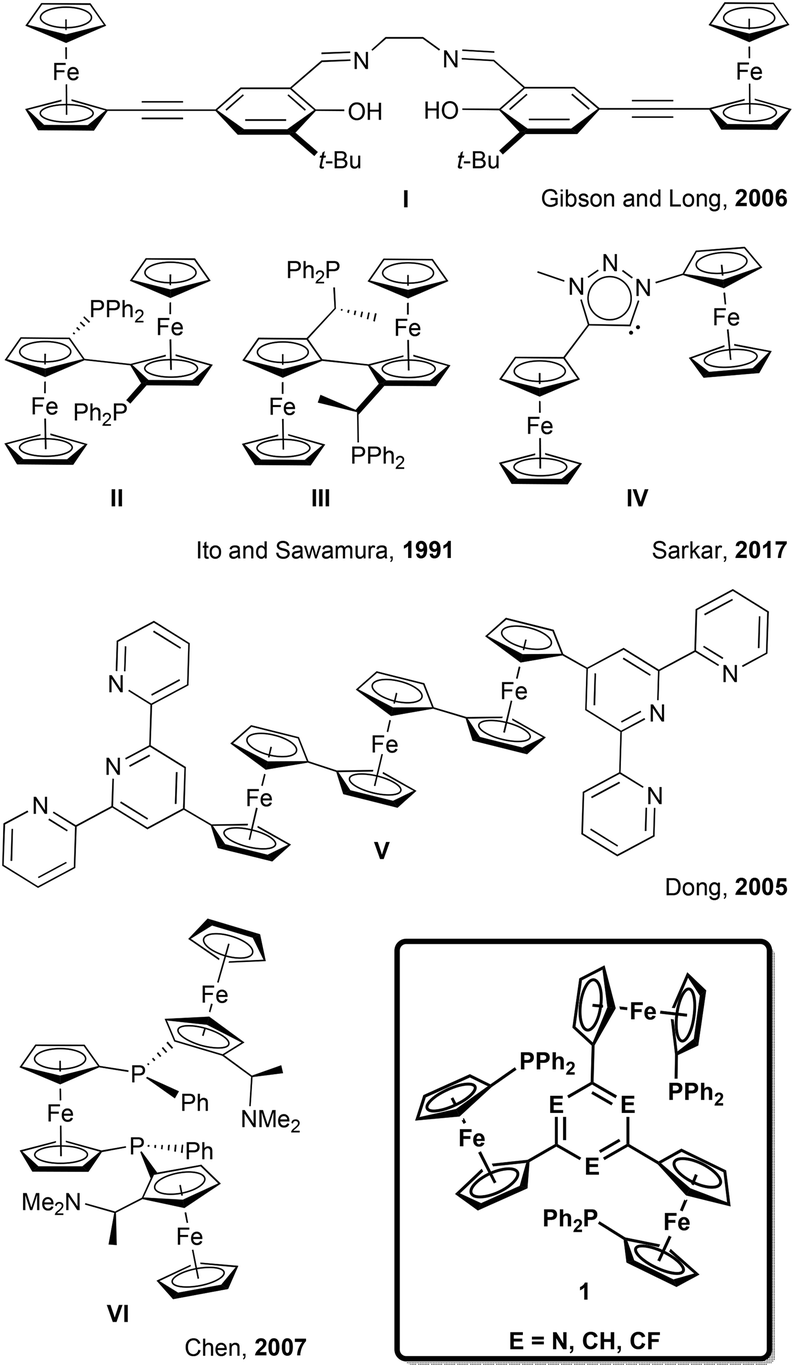 | ||
| Chart 1 Selected examples of mono- and multidentate ligands with two or more ferrocenyl and ferrocenylene groups in their backbone. | ||
Conceived as small-molecule analogues of redox-switchable dendrimers,32–34 we have recently reported the synthesis of a new family of tris-phosphanes 1 (Chart 1) incorporating a redox-active, C3-symmetric tris(ferrocenyl)arene backbone.35 Capable of forming both mono- and trinuclear gold(I) complexes (Chart 2), the latter were shown to act as four-state redox-switchable catalysts. The tris-phosphanes are prepared in a modular fashion, and s-triazine, benzene, and 1,3,5-trifluorobenzene have been used as central arenes.36
 | ||
| Chart 2 Previous work on the 1:1 ligand-to-metal coinage metal(I) complexes [1a(M)]OTf (left) and on the 1:3 ligand-to-metal chloridogold(I) complexes [1a–c(AuCl)3] (right), showcasing the impact of the arene core on various properties. | ||
As the groups of Lang and Santi have extensively shown, the nature of the arene core is decisive for the electrochemical response of the system both in terms of redox potentials and electronic communication, that is, the existence of mixed-valent oxidised states.21–23,37–49 In our case, 1:1 ligand-to-metal complexes of triazine derivative 1a (Chart 2, left) showed a distinct delocalised coinage metal–π(C3N3) interaction,50 while 1:3 ligand-to-gold complexes of 1a–c (Chart 2, right) demonstrated how the electron density of the respective core determined the electrochemical response both with respect to redox potential and reversibility. Furthermore, the arene cores were also found to impact the catalytic activity of these trinuclear gold(I) complexes in the ring-closing isomerisation of N-(2-propyn-1-yl)benzamide (2) to 5-methylene-2-phenyl-4,5-dihydrooxazole (3a).51 Further investigations on the role of the arenes on coordination abilities and catalytic efficacies were thus warranted, and the following contribution details some of our insights.
Results and discussion
Mononuclear complexes
Building on the observation of 1a forming trigonal-planar mononuclear complexes with the coinage metals,35 we first investigated the analogous reaction of benzene-based 1b with halide-free gold(I) precursor [Au(η2-nbe)3]OTf (nbe = nornbornene). Gold(I), in contrast to copper(I) and silver(I),52–54 tends to be coordinated by arenes in an η2 fashion with impact on asymmetric induction and catalyst stability.55–60 However, more delocalised gold–π(arene) interactions are also encountered and deemed important for catalysis and crystal engineering.50,61–65 Indeed, when a slight excess of tris-phosphane 1b was reacted with [Au(η2-nbe)3]OTf in CH2Cl2, 1H and 31P{1H} NMR spectra of the reaction mixture (Fig. S5†) suggested the formation of a C3v-symmetric mononuclear complex [1b(Au)]OTf with a similar, yet more broadened 31P{1H} NMR signal than [1a(Au)]OTf (δ([1a(Au)]OTf) = 26.8 ppm, ω1/2 = 1400 Hz; δ([1b(Au)]OTf) = 22.3 ppm, ω1/2 = 2130 Hz). Successful purification and isolation of [1b(Au)]OTf was achieved by concentrating a THF extract of the evaporated reaction mixture and storing it at −25 °C overnight. Single crystals suitable for X-ray diffraction analysis (XRD) were obtained from a saturated THF solution by slow cooling from 70 °C to 25 °C over two days. The molecular structure (Fig. 1, left) revealed only two of three diphenylphosphanyl groups to coordinate the gold(I) ion. This starkly contrasts the NMR spectroscopic features which, at 25 °C, suggested C3v symmetry.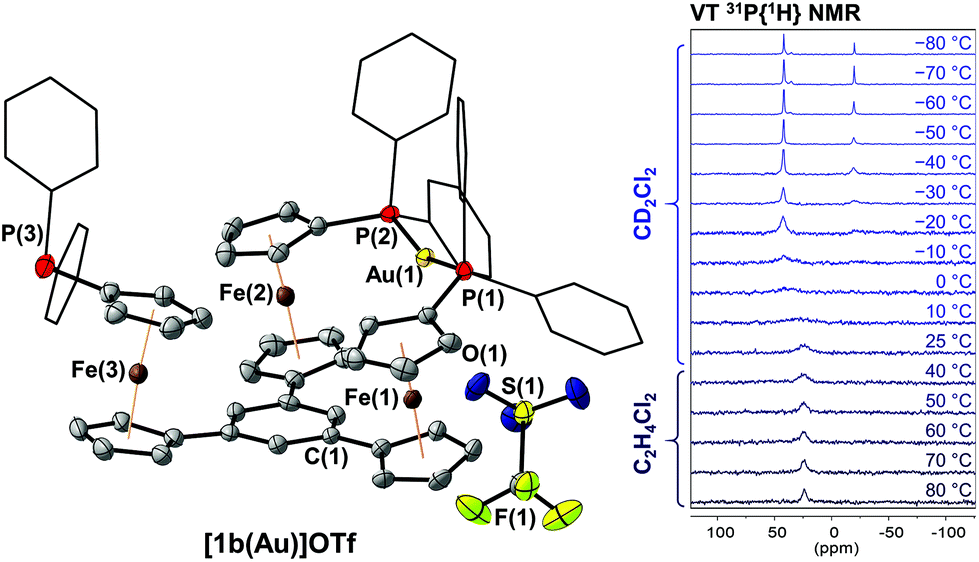 | ||
| Fig. 1 Left: Molecular structure of [1b(Au)]OTf, thermal ellipsoids set at 50% probability level. For clarity, P-bound phenyl rings are depicted in wireframe style, and co-crystallised solvent and hydrogen atoms are omitted. Right: Variable-temperature 31P{1H} NMR spectra of [1b(Au)]OTf in CD2Cl2 (−80 °C to 25 °C) and 1,2-C2H4Cl2 (40 °C to 80 °C), depicted with arbitrary scaling. | ||
Crystallographically characterised examples of poly-phosphane gold(I) complexes with pendant, uncoordinated phosphane groups are comparatively rare.66–68 The Au–P bond lengths of [1b(Au)]OTf (2.3025(8) Å and 2.3017(8) Å, Table S2†) and the close-to-linear P–Au–P bond angle of 161.31(4)° are similar to those observed for a closely related 2,6-bis(1-diphenylphosphanyl-1′-ferrocenylene)pyridine gold(I) complex by Siemeling and Štěpnička,69 a related 1,1′-bis(phosphanyl)ferrocene-derived dinuclear gold(I) complex reported by Cowie and Emslie,70 and other [Au(PR3)2]+ complexes.71–73 The triflate anion of [1b(Au)]OTf does not seem to strongly interact with the gold(I) centre, as the closest Au(1)⋯O(1) distance of 3.813(3) Å is just below the sum of the van der Waals radii.74
This P,P′ trans-spanning dicoordinate geometry, constructing a large, formally 12-membered macrocycle, is not just a solid-state phenomenon, as we were able to demonstrate by variable-temperature (VT) 1H and 31P{1H} NMR spectroscopy (Fig. 1, right, and Fig. S43 and S44†). Upon heating a 1,2-dichloroethane solution of [1b(Au)]OTf to 80 °C, the very broad 31P{1H} resonance partly sharpens, while cooling a CD2Cl2 solution from 25 °C down to −80 °C splits the resonance in two signals at 41.8 and −19.5 ppm with an integral ratio of 2:1. These chemical shifts are in line with those of free 1b (δ = −17.5 ppm)36 and 2,6-bis(1-diphenylphosphanyl-1′-ferrocenylene)pyridine gold(I) (δ = 42.6 ppm).69 The dynamic behaviour mirrors that of a similarly dicoordinating tris-phosphane by Wade and Gabbaï.66 In contrast to triazine-based [1a(Au)]OTf,35 the low-temperature 1H NMR spectra of [1b(Au)]OTf (Fig. S43†) do not give rise to well-defined sharp resonances. They are, however, in accordance with loss of the apparent C3v symmetry observed at and above room temperature which originates from fast coordination/dissociation equilibria.
Since 1,3,5-trifluorobenzene-derived tris-phosphane 1c falls between 1a and 1b regarding the electron density of the arene core, the reaction between 1c and [Au(η2-nbe)3]OTf was investigated next. A mononuclear 1:1 ligand-to-metal complex [1c(Au)]OTf with similar NMR spectral characteristics (Fig. S7–S10†) to [1a(Au)]OTf and [1b(Au)]OTf was obtained, again suggesting C3v symmetry in solution at 25 °C. Similar to [1b(Au)]OTf, [1c(Au)]OTf reveals a quickly equilibrating P,P′ trans-spanning coordination geometry in a multinuclear VT NMR experiment (Fig. S45–S47†); the C3-symmetric P,P′,P′′ coordination mode is apparently restricted to the s-triazine core of [1a(Au)]OTf (vide infra). Attempting purification of the slightly impure product, cooling a THF extract of [1c(Au)]OTf from 70 °C to room temperature over two days yielded a few single crystals suitable for XRD. Surprisingly, a one-dimensional coordination polymer (CP) {[(1c)2(Au)3](OTf)3}n with 2:3 ligand-to-metal stoichiometry had formed in the process (Fig. 2). The structure of {[(1c)2(Au)3](OTf)3}n (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) is characterised by the μ3-connecting, threefold κ1 coordination mode of ligand 1c, i.e., each phosphane moiety binding one of the three gold(I) ions. The three individual gold(I) centres are close-to-linearly coordinated by two diphenylphosphanyl groups each, and the P–Au–P bond angles (170.45(5)–172.72(5)°, cf. Table S3†) are closer to the ideal 180° than those found in [1b(Au)]OTf, most likely because the 1,3,5-tris(ferrocenyl)arene-derived ligand structure itself does not impose any steric restrictions anymore. The individual Au–P bond lengths range from 2.296(1) Å to 2.320(2) Å and do not show systematic variations according to their position in the polymeric structure. The individual, non-interacting CP strands propagate in parallel to the crystallographic (001) and along the (10) planes. Small channels (ca. 5.7 Å × 8.8 Å), filled with highly disordered THF molecules, are formed along the crystallographic a axis (Fig. S42†). Upon dissolution of {[(1c)2(Au)3](OTf)3}n in CD2Cl2, the polymeric structure seems to disintegrate; only broad signals are discernible in the respective 1H, 19F{1H}, and 31P{1H} NMR spectra (Fig. S11–S14†). This is in line with HR-ESI mass spectrometry showing fragments with m/z values for both monocationic 1:1 and tricationic 2:3 ligand-to-metal fragments.
) is characterised by the μ3-connecting, threefold κ1 coordination mode of ligand 1c, i.e., each phosphane moiety binding one of the three gold(I) ions. The three individual gold(I) centres are close-to-linearly coordinated by two diphenylphosphanyl groups each, and the P–Au–P bond angles (170.45(5)–172.72(5)°, cf. Table S3†) are closer to the ideal 180° than those found in [1b(Au)]OTf, most likely because the 1,3,5-tris(ferrocenyl)arene-derived ligand structure itself does not impose any steric restrictions anymore. The individual Au–P bond lengths range from 2.296(1) Å to 2.320(2) Å and do not show systematic variations according to their position in the polymeric structure. The individual, non-interacting CP strands propagate in parallel to the crystallographic (001) and along the (10) planes. Small channels (ca. 5.7 Å × 8.8 Å), filled with highly disordered THF molecules, are formed along the crystallographic a axis (Fig. S42†). Upon dissolution of {[(1c)2(Au)3](OTf)3}n in CD2Cl2, the polymeric structure seems to disintegrate; only broad signals are discernible in the respective 1H, 19F{1H}, and 31P{1H} NMR spectra (Fig. S11–S14†). This is in line with HR-ESI mass spectrometry showing fragments with m/z values for both monocationic 1:1 and tricationic 2:3 ligand-to-metal fragments.
 | ||
| Fig. 2 Top: Chemical structure of coordination polymer {[(1c)2(Au)3](OTf)3}n showing the repeating unit. Bottom: Molecular structure of {[(1c)2(Au)3](OTf)3}n in the solid state, thermal ellipsoids set at 50% probability level. For clarity, P-bound phenyl rings are depicted in wireframe style, and the three triflate anions as well as hydrogen atoms are omitted. The asymmetric unit of the unit cell is highlighted. | ||
Even though the formation of this CP was not intended and its preparation suffers from poor reproducibility, redox-active ferrocene-derived CPs have recently attained great interest for the insight into molecular architecture and self-assembly75–77 they provide as well as for their potential applications in catalysis,78,79 non-linear optics, and molecular magnetism.77 Following the classification of Mochida, {[(1c)2(Au)3](OTf)3}n is a one-dimensional main-chain CP derived from a neutral ligand.77 As such it is, to the best of our knowledge, the first CP to feature a ferrocene-derived tris-phosphane. Phosphane-based CPs are, in general, more elusive than analogues built from nitrogen- or sulfur-containing ligands,80 and often constructed around gold(I) with its tendency for simple coordination geometries and formative aurophilic interactions.81–83 Last but not least, the 2:3 ligand-to-metal stoichiometry of {[(1c)2(Au)3](OTf)3}n is also uncommon among CPs, a cadmium(II)-based CP by Tian and co-workers being the only other example.84
In light of 1a–c showing three different coordination modes for gold(I), this unexpected behaviour was studied by an energy decomposition analysis using density-functional theory (DFT) level calculations (for computational details, s. ESI, section 1.1†). Geometry optimisations and energy calculations for both the closed tricoordinate, C3-symmetric coordination mode (observed for [1a(Au)]OTf) and the open dicoordinate, Cs-symmetric coordination mode (observed for [1b(Au)]OTf in the solid state and in solution, and for [1c(Au)]OTf in solution) were conducted for gold(I) complexes of the three ligands 1a–c, excluding the triflate counterion in all cases (Fig. S54–S62†). The total coordination energy Ecoord, the energy gain upon combining the (native) tris-phosphane and a gold(I) ion, can be split into two contributions, namely the preparation energy Eprep (required to form the geometry of the coordinated ligand from the native tris-phosphane, representing steric contributions) and the interaction energy Einter (gained from the interaction between the isolated gold(I) ion and the pre-formed ligand, representing electronic contributions). Comparing the obtained values for all six complexes (Fig. 3, Tables 1 and S4†), the decisive role of the preparation energy becomes clear. Obviously, steric repulsion between the constituting arene fragments E (N|, C–H, C–F) and the ortho hydrogen atoms of the C-bound cyclopentadienyl rings governs the coordination geometry. The close-to co-planar arrangement of the three cyclopentadienyl rings and the arene core, a feature of the C3-symmetric closed geometry, is therefore less feasible, and open structures become more attractive. Correspondingly, the fluorine atoms, largest in this series, lead to the highest arene-cyclopentadienyl torsion φ both in the calculated and in the experimentally determined structures of 1c (Table 1). In terms of interaction energies Einter, the closed form is more favourable in all cases, likely owing to a higher coordination number (3 vs. 2) and attractive gold–π(arene) interactions increasing in strength from benzene over 1,3,5-trifluorobenzene to s-triazine. These findings might explain the formation of coordination polymer {[(1c)2(Au)3](OTf)3}n from [1c(Au)]OTf. As the experimentally determined φ values are the highest in the series of the three solid-state structures (Table 1), the μ3:κ1P,κ1P′,κ1P′′ coordination mode of 1c apparently accommodates the clashing ortho fluorine atoms and cyclopentadienyl ring protons most easily. For the borderline case of [1b(Au)]OTf, the conformation influence of the pendant ferrocenylphosphane was probed by comparing two extreme conformations (all-syn as found in the solid-state structures of both 1b and [1b(Au)]OTfvs. a syn,anti,anti arrangement; Fig. S65†). While this conformation was found to be significantly less stable than the all-syn conformer by about 50 kJ mol−1, the initial geometry of 1b was shown to be irrelevant for the outcome of the analysis in that the experimentally found all-syn structure of [1b(Au)]OTf was the most stable one.
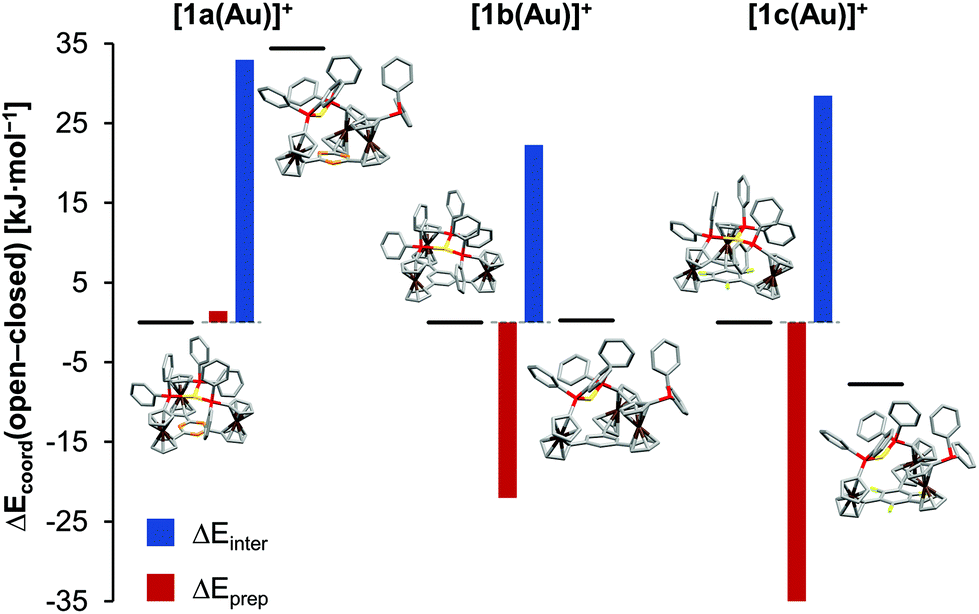 | ||
| Fig. 3 Total coordination energy difference ΔEcoord between open (dicoordinate) and closed (tricoordinate) forms of mononuclear complexes [1a–c(Au)]+ (black horizontal bars) and the respective contributions from interaction (ΔEinter, blue columns) and preparation (ΔEprep, red columns) energies obtained from calculations at the DFT level. | ||
| E coord | E prep | E inter | φ [°] | ||
|---|---|---|---|---|---|
| [kJ mol−1] | |||||
| a Torsion between arene-bound cyclopentadienyl rings of coordinating 1-diphenylphosphanyl-1′-ferrocenylene group and central arene. b Experimentally determined values for [1a(Au)]OTf.35 c Experimentally determined values for [1b(Au)]OTf. d Experimentally determined values for {[(1c)2(Au)3](OTf)3}n. | |||||
| [1a(Au)]+ | Open | −476.8 | +47.9 | −524.7 | 27.4, 28.3 |
| Closed | −511.2 | +46.5 | −557.7 | 8.9–9.2 (8.3–10.4)b | |
| Δ(o–c) | +34.4 | +1.4 | +33.0 | ||
| [1b(Au)]+ | Open | −508.8 | +18.0 | −526.8 | 32.3, 42.0 (11.6, 13.1)c |
| Closed | −509.1 | +40.0 | −549.1 | 13.8–14.0 | |
| Δ(o–c) | +0.3 | −22.0 | +22.3 | ||
| [1c(Au)]+ | Open | −499.1 | +25.3 | −524.4 | 32.9, 42.0 (27.4–34.1)d |
| Closed | −491.4 | +61.5 | −552.9 | 11.4–25.0 | |
| Δ(o–c) | −7.8 | −36.2 | +28.5 | ||
Intrigued by the possibility of modifying the coordination properties of tris(ferrocenylene)arene-based tris-phosphanes via the substitution pattern of the arene, we next introduced fluorine in the diphenylphosphanyl groups of 1a in form of the bis(pentafluorophenyl)phosphanyl moiety [P(PhF)2] (Chart 3). With regard to (redox-switchable) gold(I) catalysis, the P(PhF)2 moiety is of considerable interest due to its very low σ-donor capability.85,86 The resulting electron-poor gold(I) centres show great potential in gold(I)-catalysed multiple bond activation87–91 and could also be of interest in gold(I)-catalysed oxidative addition reactions.92 Besides catalyst design, the P(PhF) moiety has also found applications in the preparation of stimuli-responsive fluorescent metal complexes93 and organic materials with intriguing optoelectronic properties.94,95
 | ||
| Chart 3 Previously reported ferrocene-based bis(pentafluorophenyl)phosphanes VII and VIII, and bis(pentafluorophenyl)phosphane derivative 1aF. | ||
To date, ferrocene-based derivatives are rare, and only the 1,1′-diphenylphosphanylferrocene (dppf) analogue VII96–98 and a FcPHOX derivative VIII (Chart 3)99 have been prepared and used for complexation of palladium(II). Tris-phosphane 1aF can be prepared from 2,4,6-tris(1-bromo-1′-ferrocenylene)-1,3,5-triazine 4a, albeit in a low yield of only 15%, still highlighting the modular preparation of tris-phosphanes 1. The NMR chemical shifts of 1aF, an air- and moisture-stable, deep red microcrystalline solid, in 19F{1H} and 31P{1H} NMR spectroscopy match those of VII very well.96,100
Reacting 1aF with [Au(η2-nbe)3]OTf in CH2Cl2 resulted in a less clean conversion than anticipated, and only after several recrystallisation steps could a few crystals suitable for XRD be obtained. [1aF(Au)]OTf is the first crystallographically characterised example of a ferrocene-derived ligand featuring any P(PhF) group and, among the few reported gold(I) complexes of this donor, the only example of a bidentate phosphane.101–103 The solid-state structure of [1aF(Au)]OTf (Fig. 4, top) differs only slightly from that of [1b(Au)]OTf. Both complexes share the Cs-symmetric, P,P′-dicoordinate geometry with a pendant phosphane group. Apparently, the steric strain imposed by the H-to-F substitution on the P-bound phenyl rings outweighs the energy gain by the attractive gold(I)–π(C3N3) interaction.85 Even though a DFT-level geometry optimisation of the corresponding C3-symmetric tricoordinate gold(I) complex cation [1aF(Au)]closed+ initially suggested the synthetic accessibility of this coordination mode, the open form [1aF(Au)]open+ was found to be energetically far more favourable by 207 kJ mol−1 (section 4 of the ESI†). While [1aF(Au)]OTf shows marginally shorter Au–P bond lengths and a slightly increased P(1)–Au–P(2) bond angle (Table 2), the most notable difference to [1b(Au)]OTf lies in the much shorter Au⋯O(triflate) distance of only 3.08(1) Å, well below the sum of the van der Waals radii (3.82 Å),74 yet significantly above the sum of the covalent radii (2.02 Å).104 In a multinuclear VT NMR spectroscopy experiment of [1aF(Au)]OTf in CD2Cl2 (Fig. 5, left, and Fig. S51–S53†), no significant broadening of the triflate 19F{1H} resonance (*) is observed lowering the temperature from 25 °C to −60 °C, speaking against a strong interaction in solution. In contrast to [1b(Au)]OTf and [1c(Au)]OTf, the dicoordinate bonding mode is apparent from signals for the pendant phosphane group (#) already at 25 °C in both the 19F{1H} and the 31P{1H} NMR spectra (Fig. 5, left) and does not seem to be fluctional. The 19F{1H} NMR signals attributable to the coordinated P(PhF)2 groups of [1aF(Au)]OTf, very broad and hardly discriminable from the baseline at 25 °C, sharpen significantly and partly decoalesce (dashed lines) upon cooling. This points to free (A) vs. partly hindered rotation (B) about the P–C(PhF) bonds of the two P-bound pentafluorophenyl groups involved in coordination (Fig. 5, top left). The assignment of the resonances to either ring has been established through a 19F,19F COSY NMR experiment, but the relative orientation of the PhF rings is not clear. Warming the same solution to 45 °C does not result in apparent C3v but in a clearer Cs molecular symmetry for both nuclei.
 | ||
| Fig. 4 Molecular structures of [1aF(Au)]OTf (top) and [1aF(Ag)]OTf (bottom), thermal ellipsoids set at 30% probability level. For clarity, pentafluorophenyl rings of the coordinating phosphanes are depicted in wireframe style, and co-crystallised solvent and hydrogen atoms are omitted. | ||
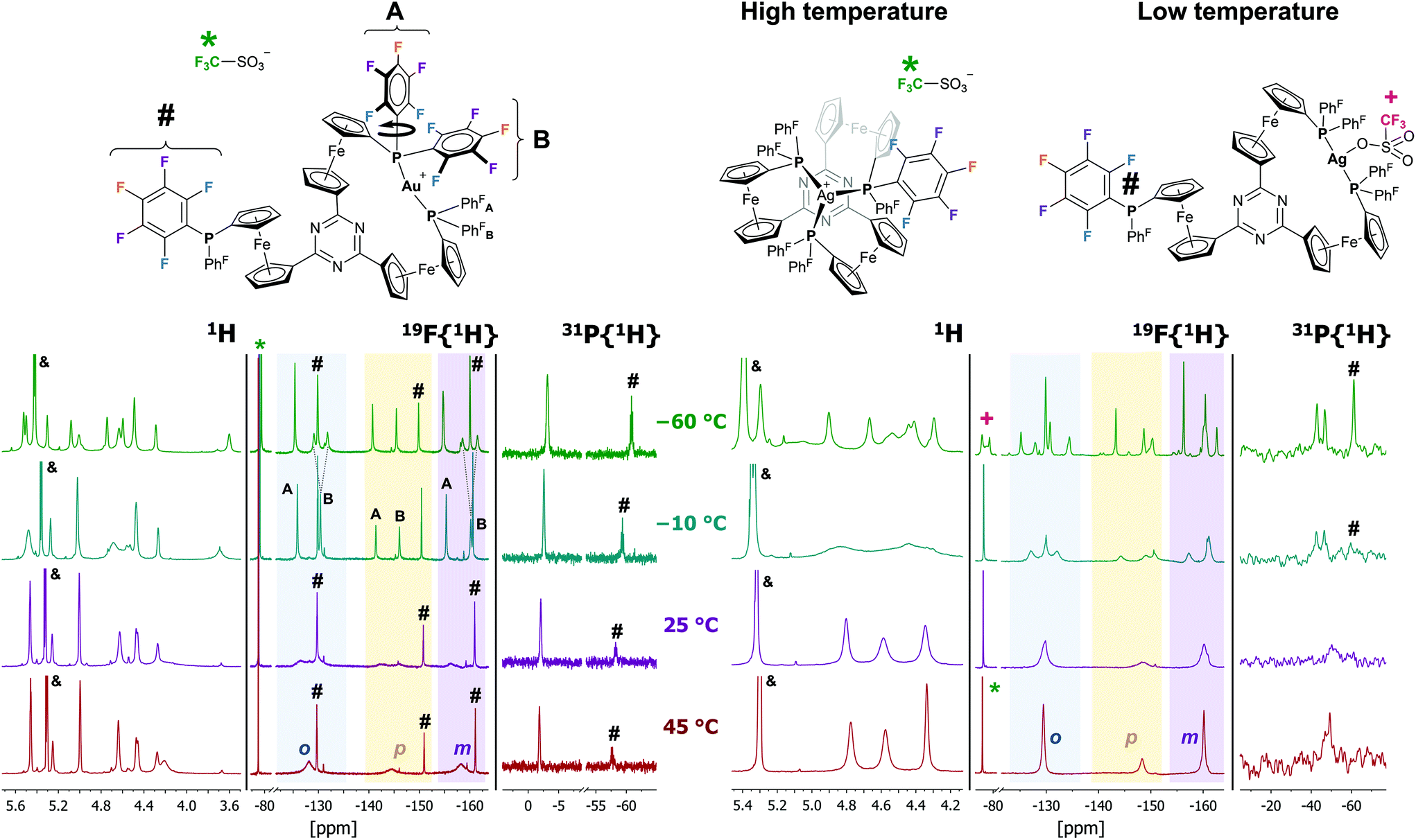 | ||
| Fig. 5 Sections of the variable-temperature 1H, 19F{1H}, and 31P{1H} NMR spectra of [1aF(Au)]OTf (left) and [1aF(Ag)]OTf (right) in CD2Cl2 (&: CHDCl2/CH2Cl2 resonance; *: triflate anion resonance; +: split triflate anion resonance of [1aF(Ag)]OTf; #: pendant P(PhF)2 group). The shaded areas in the 19F{1H} NMR spectra correspond to the spectral regions of the ortho (blue), para (orange), and meta fluorine substituents of the P(PhF)2 groups. | ||
The loss of symmetry is also apparent from the corresponding 1H NMR spectra. At 25 °C, the 1H NMR spectrum of [1aF(Au)]OTf is difficult to assign due to significant broadening of the signals. At −60 °C, 11 distinct signals with matching integrals for the 12 protons of a Cs-symmetric species can be identified (the signal at 4.45 ppm contributing with a relative integral of 2). The mononuclear composition in solution is also confirmed by HR-ESI mass spectrometry.
Since we were unable to obtain satisfactorily pure samples and sufficient amounts of [1aF(Au)]OTf for further analyses or applications, we turned our attention to the reaction of 1aF with silver(I) triflate. In the corresponding complex of 1a, the silver(I)–π(C3N3) interaction had been found stronger than for the gold(I) analogue.35 While the formation and isolation of [1aF(Ag)]OTf proved much more facile, the solid-state molecular structure (Fig. 4, bottom) is analogous to that of the gold(I) congener. As previously noted for isostructural pairs of silver(I) and gold(I) complexes, the M–P bonds (Table 2) are longer for the silver(I) complex.35,71,105 The P(1)–Ag(1)–P(2) bond angle deviates more strongly from the ideal 180°, paralleled by a shorter Ag(1)⋯N(1) distance of 3.89(1) Å ([1aF(Au)]OTf: 4.14(1) Å). Moreover, a significantly closer contact between the triflate anion and the silver(I) cation (d(Ag(1)⋯O(1)) = 2.57(1) Å) can be discerned. While this distance is still well above the sum of the covalent radii (2.11 Å),104 its deviation from the sum of the van der Waals radii (4.03 Å) is far greater than in [1aF(Au)]OTf.
[1aF(Ag)]OTf is, to the best of our knowledge, the first silver complex of a pentafluorophenylphosphane (one example of a corresponding phosphanido complex, formed by C6F5 group transfer from AgC6F5 to a PCl2 moiety, has been reported by Schulz and co-workers).106 Despite their similar structures, the silver(I) complex deviates from the gold(I) analogue in its (VT) NMR-spectroscopic features (Fig. 5, right, and Fig. S48–S50†).
Above 5 °C, [1aF(Ag)]OTf appears C3v-symmetric in CD2Cl2 solution in the corresponding 1H and 31P{1H} NMR spectra (Fig. 5, top right, and Fig. S48, S49†); coalescence in the 19F{1H} NMR spectra occurs only at higher temperatures. Upon cooling, the 31P{1H} resonance decoalesces into a 2:1 pattern indicative of the P,P′-dicoordinate bonding mode, and the signal for the silver-bound phosphane further splits into a doublet due to unresolved 1J coupling with both 107/109Ag isotopes. The close silver-triflate contact of the molecular structure is also observed in the 19F{1H} NMR spectra below −30 °C due to a strong splitting of the triflate-CF3 resonance (+), clearly setting [1aF(Ag)]OTf apart from [1aF(Au)]OTf. Furthermore, the low-temperature 1H and 19F{1H} NMR spectra of [1aF(Ag)]OTf are more complex, and no unambiguous assignments could be made.
Trinuclear complexes
Incited by the adventitious formation of {[(1c)2(Au)3](OTf)3}n and the presence of pendant phosphane groups in the mononuclear complexes of 1b and 1c in contrast to those of 1a, 1a–c were reacted with [Au(η2-nbe)3]OTf in a 2:3 ligand-to-metal ratio similar to that found for CP {[(1c)2(Au)3](OTf)3}n. Indeed, both 1a and 1b reacted to form well-defined trinuclear complexes [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3 which could be crystallised from CH2Cl2 and toluene and fully characterised. In contrast, despite testing variations of stoichiometry as well as different purification and crystallisation protocols, discrete trinuclear gold(I) complexes of 1c could not be obtained and only ill-defined, potentially oligomeric products with varying 31P{1H} NMR signals around 40 ppm were formed.
The molecular structures of [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3 (Fig. 6 top and centre) can be understood as two mononuclear, P,P′-dicoordinate complexes linked by a third gold(I) ion coordinated by the remaining phosphane. Thus, even though the ligand-to-metal stoichiometry of 2:3 is formally identical to that of {[(1c)2(Au)3](OTf)3}n, the discrete trinuclear complexes contain 1a and 1b in a different, μ2:κ1P,κ2P′,P′′ coordination mode. In general, trinuclear gold(I) complexes formed from two tris-phosphanes (or other P-donors) are relatively scarce in the literature and mostly contain linear Au3 chains.107–111 The particular coordination mode observed here has not yet been reported for solid-state structures deposited in the Cambridge Structural Database.112 As a result of the packing, which does not involve aurophilic interactions or close contacts between the individual tricationic molecules, small and large channels are formed along the crystallographic a axis (Fig. 6, bottom). The smaller channels (approx. diameter of 5.3 Å) are occupied by the severely disordered triflate anions. Only a single one in the structure of [(1a)2(Au)3](OTf)3 could be modelled, showing a weak Au(1)⋯O(1) contact (3.46(2) Å). The presence of three triflate anions per trication is, however, confirmed by 19F{1H} NMR spectroscopy and CHN analysis. Additionally, the larger channels (approx. 6.8 Å × 9.9 Å) are filled with heavily disordered solvent molecules. In line with the solvent stabilising the porous structure, drying the crystalline materials obtained from CH2Cl2/toluene under reduced pressure leads to a visible and sudden loss of crystallinity, leaving fine powders after drying.
 | ||
| Fig. 6 Molecular structures of [(1a)2(Au)3](OTf)3 (top) and [(1b)2(Au)3](OTf)3 (centre). Thermal ellipsoids set at 50% probability level. For clarity, P-bound phenyl groups are depicted in wireframe style, and co-crystallised solvent, anions, and hydrogen atoms are omitted. Bottom: Space-filling model of [(1b)2(Au)3](OTf)3 viewed along the crystallographic a axis; crystallographic axes of the unit cell are shown, solvent molecules are omitted. | ||
The discrete trinuclear complexes can be understood as a blend of mononuclear [1b(Au)]OTf and CP {[(1c)2(Au)3](OTf)3}n, particularly regarding the P–Au–P bond angles which, for the central gold(I) ion, are closer to the ideal 180° than for the outer gold(I) ions (Tables 3 and S3†). [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3 maintain their structure in CD2Cl2 solution as evident from HR-ESI mass spectrometry through signals for the tricationic species, and from 1H and 31P{1H} NMR spectroscopy (Fig. S15ff†). The two distinct 31P{1H} resonances also demonstrate the influence of the P–Au–P bond angle on the exact chemical shift – more acute angles result in a slight deshielding.
| [(1a)2(Au)3] (OTf)3 | [(1b)2(Au)3] (OTf)3 | {[(1c)2(Au)3] (OTf)3}n | ||
|---|---|---|---|---|
| Au(m)–P [Å] | Au(1) | 2.301(2) | 2.295(2) | 2.303(1) |
| 2.302(2) | 2.298(2) | 2.304(1) | ||
| Au(2) | 2.303(2) | 2.299(2) | 2.305(3) | |
| 2.303(2) | 2.298(2) | 2.320(3) | ||
| Au(3) | 2.302(3) | 2.302(2) | 2.296(1) | |
| 2.304(3) | 2.300(1) | 2.299(1) | ||
| P–Au(m)–P [°] | Au(1) | 171.96(8) | 173.33(6) | 170.45(5) |
| Au(2) | 164.05(8) | 162.44(7) | 171.4(1) | |
| Au(3) | 162.92(8) | 164.85(6) | 172.72(5) | |
These results corroborate the decisive role of the arene core constituent E and, as demonstrated for 1aF, the role of the P-bound substituents in determining the coordination mode of the tris-phosphanes towards gold(I) as schematically summarised in Fig. 7.
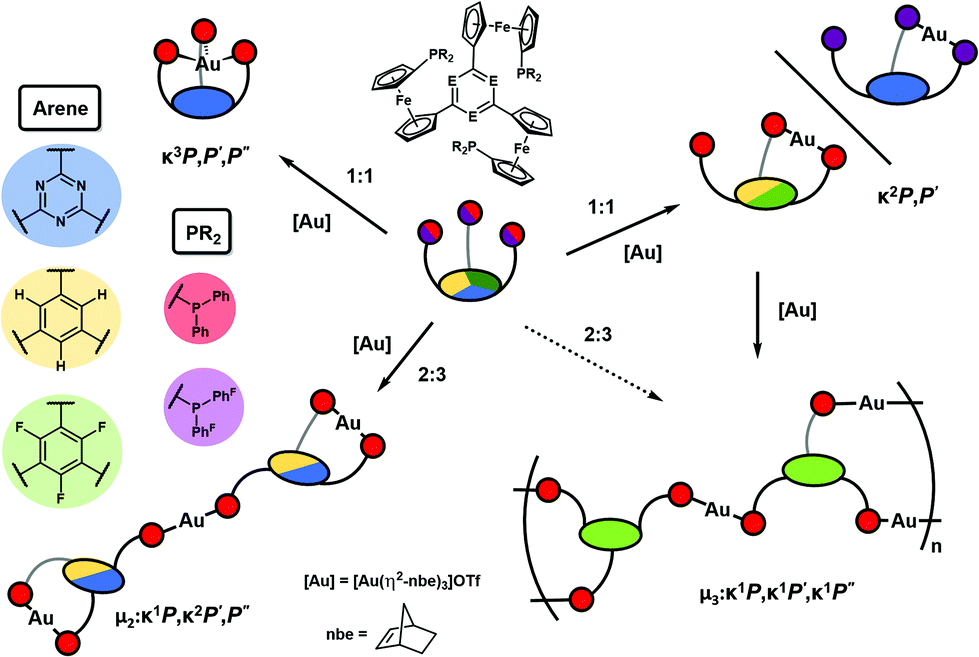 | ||
| Fig. 7 Schematic depiction of the different coordination modes observed for tris-phosphanes 1a–c and 1aF depending on the arene core constituent E and on the phosphanyl moiety PR2. For clarity, charges and triflate anions have been omitted from the depicted structures. The dashed arrow represents the indirect access to the coordination polymer from 1c. | ||
Electrochemistry and redox-switchable catalysis
As trinuclear chloridogold(I) complexes of 1a–c have shown high potential for multi-state redox-switchable catalysis due to their stepwise, well-separated, and reversible trifold oxidation,36 the electrochemical and, following, catalytic characterisation of the previously described gold(I) complexes were also of interest. The electrochemistry of tris-phosphanes 1a–c and [1a(Au)]OTf has already been studied,35,36 and [1a(Au)]OTf in particular has shown unexpected behaviour involving chemical follow-up reactions induced by electrochemical oxidation (EC mechanism). Because the isolation of sufficient amounts of pure [1c(Au)]OTf had not been successful and since the CP {[(1c)2(Au)3](OTf)3}n is likely ill-defined in solution, we first focused on well-defined [1b(Au)]OTf, [(1b)2(Au)3](OTf)3, and [(1a)2(Au)3](OTf)3 in BF4−- and tetrakis[3,5-bis(trifluoromethylphenyl)]borate-based (BArF4−) supporting electrolytes (SE). The corresponding cyclic voltammograms are shown in Fig. 8.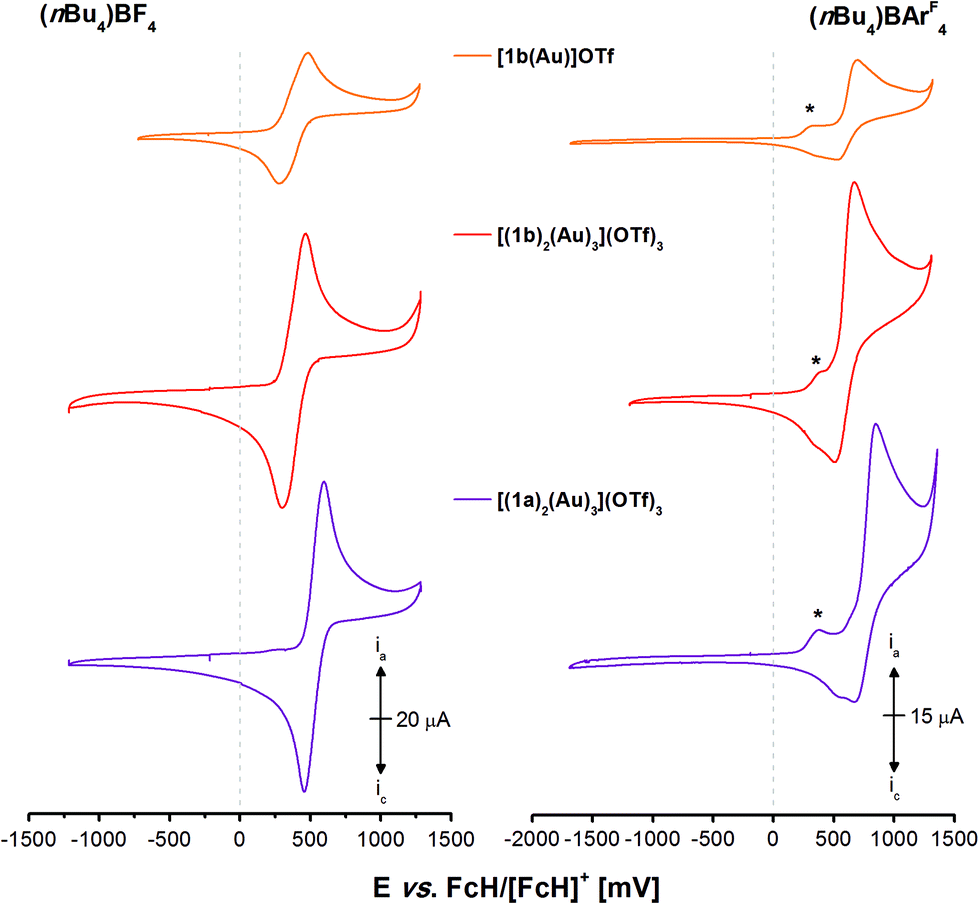 | ||
| Fig. 8 Cyclic voltammograms of 1 mmol L−1 gold(I) complexes [1b(Au)]OTf (orange), [(1b)2(Au)3](OTf)3 (red), and [(1a)2(Au)3](OTf)3 (purple) in 0.1 mol L−1 supporting electrolyte solutions ((nBu4N)BF4 (left) and (nBu4N)BArF4 (right) in CH2Cl2). Traces have been recorded at 100 mV s−1 (working electrode: glassy carbon, counter electrode: Pt wire) at room temperature. The first oxidations (*) in the BArF4-based SE are irreversible when addressed separately. The second of three measured cycles are shown. | ||
Quite unexpectedly, given the pendant diphenylphosphane group of [1b(Au)]OTf and the well-documented involvement of free phosphorus-centred lone pairs of electrons in ferrocene oxidation,113–116 the oxidation of [1b(Au)]OTf (orange trace) in the BF4-based SE (Fig. 8, left) was found quasireversible (cf. section 5.1 of the ESI†).117,118 Assuming that the fluctional coordination behaviour observed in the VT NMR experiments renders all three ferrocenylene moieties equivalent on the cyclic voltammetry timescale and hence not bearing lone pairs of electrons available for follow-up chemistry, all three ferrocenylene groups are most likely oxidised at the same potential. Similar behaviour has been reported for 1,3,5-tris(ferrocenyl)benzene37,119 and its 1,1′-substituted derivatives, yet not for free 1b itself.36 Further support comes from hexa-ferrocene derivatives [(1b)2(Au)3](OTf)3 (red trace) and [(1a)2(Au)3](OTf)3 (purple trace) which also yield one quasireversible redox event under the same conditions, as all phosphanes are permanently involved in gold(I) coordination. Twice-as-high currents ia/c suggest the simultaneous oxidation of all six ferrocenylene groups. The decisive role of the high mobility and of the potential to form tight ion pairs of the BF4− anions becomes apparent when the SE is changed to the much more weakly coordinating BArF4− anions (Fig. 8, right).120 The oxidations of all three complexes follow the same pattern of a low-current, irreversible first oxidation (*) followed by a much stronger second, also not fully reversible, oxidation event (for corresponding potentials, cf. Table 4). On reversing the scan direction, two close-spaced reductions take place, less reversible for the triazine than for the benzene core. In general and in line with our expectations, the less electron-rich triazine leads to appreciably higher oxidation/reduction potentials for its gold(I) complex (with the notable exception for the weak first oxidation (*)), while the similar coordination modes of all complexes result in overall very similar electrochemical characteristics.
| (nBu4N)BF4 | (nBu4N)BArF4 | ||||
|---|---|---|---|---|---|
| E 0 (ΔEp)b [mV] | E ox1 [mV] | E ox2 [mV] | E red1 [mV] | E red2 [mV] | |
| a All potentials reported vs. the FcH/[FcH]+ couple at a glassy carbon working electrode (scan rate 100 mV s−1) in anhydrous supporting electrolytes, measured under a blanket of nitrogen. b Splitting between the anodic (oxidation) and cathodic (reduction) peaks. c Shoulder in cyclic voltammograms of [1b(Au)]OTf and [(1b)2(Au)3](OTf)3. | |||||
| [1b(Au)]OTf | 371 (204) | 335 | 702 | 523 | 368 |
| [(1b)2(Au)3](OTf)3 | 382 (162) | 400 | 671 | 509 | 339 |
| [(1a)2(Au)3](OTf)3 | 527 (141) | 380 | 847 | 659 | 512 |
The electrochemistry of 1aF and the two mononuclear complexes [1aF(Ag)]OTf (Fig. 9, Fig. S66 and S67†) and [1aF(Au)]OTf (Fig. S68,† measured at a lower concentration due to a limited amount of available material; all potentials in Table S6†) is, in contrast, very different from that of their non-fluorinated counterparts. Free 1aF (light green) can be reversibly oxidised in three well-separated steps in the BArF4-based SE (1a–c only feature a first reversible oxidation and need to be protected by BH3 groups to see the same pattern),36 in line with reports for dppf derivative VII (Chart 3) by the Gusev group.98 Gusev and co-workers speculated that this reversibility originated from steric protection of potential P-centred radicals from dimerisation. While the steric demand of fluorine is indeed greater than that of hydrogen,85 we believe the lowered energy of the lone pair of electrons at the P atoms to be a more likely explanation.113,114,116,121,122 Accordingly, the Mulliken spin densities of monocations [1a]+ and [1aF]+ calculated at the DFT level are distributed differently, with [1aF]+ showing a four times greater spin density (0.012) summed up over all three P atoms than [1a]+ (0.003). In the BF4-based SE, the oxidations are not reversible anymore, and much like for 1a, the first oxidation induced a delayed reduction.
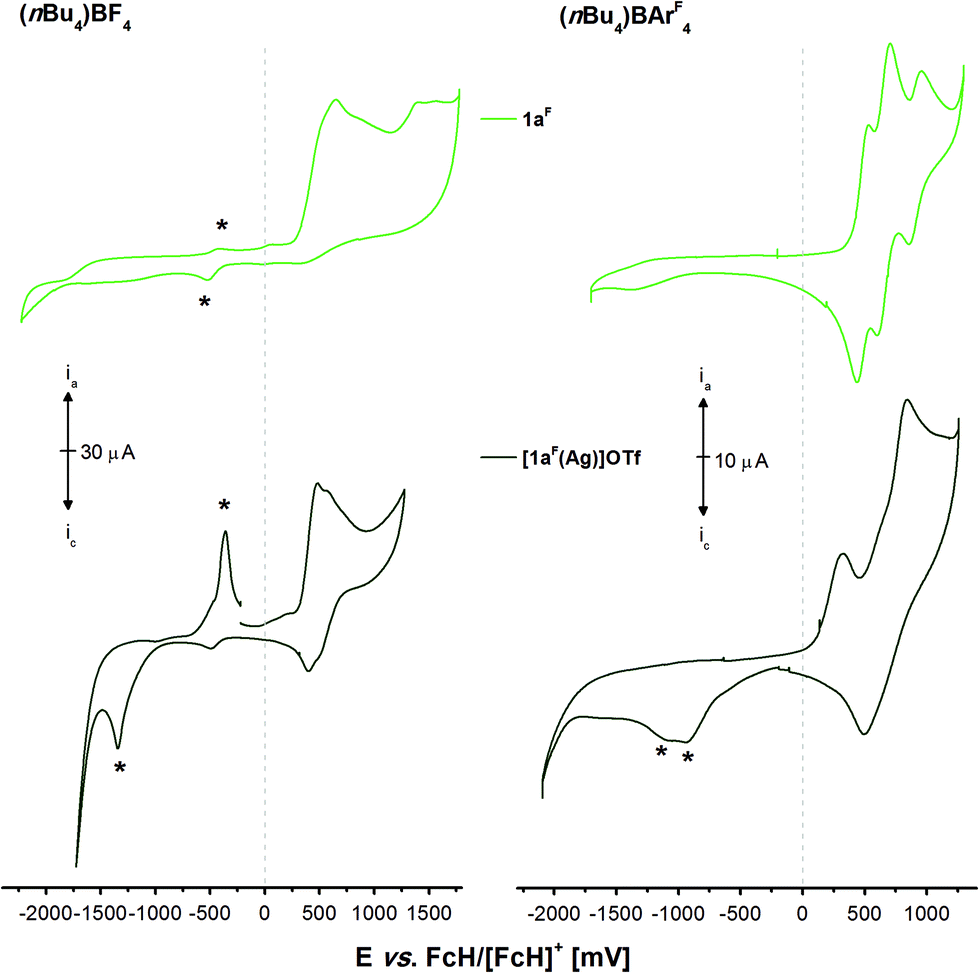 | ||
| Fig. 9 Cyclic voltammograms of 1 mmol L−11aF (light green) and [1aF(Ag)]OTf (dark green) in 0.1 mol L−1 supporting electrolyte solutions ((nBu4N)BF4 (left) and (nBu4N)BArF4 (right) in CH2Cl2). Traces have been recorded at 100 mV s−1 (working electrode: glassy carbon, counter electrode: Pt wire) at room temperature. The second of three cycles are shown and asterisks (*) mark oxidation/reduction events which depend on prior oxidation in the first cycle (cf. Fig. S66 and S67†). | ||
In the BF4-based SE, neither the two complexes nor free 1aF are reversibly oxidisable, and all compounds show oxidation-induced delayed reductions (and re-oxidations; Fig. 8, *). This behaviour has previously been observed for 1a and its coinage metal complexes as well. A higher degree of reversibility is observed in the BArF4-based SE; yet, similar to [1b(Au)]OTf, the oxidations are not completely reversible and, in the case of [1aF(Ag)]OTf, delayed reductions are observed again. Given their complex redox chemistry and unsatisfactory yields and purity, [1aF(Ag)]OTf and [1aF(Au)]OTf were not considered for the following redox-switchable catalysis (RSC) investigations.
As a suitable model reaction for testing the previously presented mono- and trinuclear complexes in RSC, the ring-closing isomerisation of propargylic amide 2 to oxazoline 3a (Scheme 1)18,123–126 was studied by time-resolved 1H NMR spectroscopy vs. an internal standard (1,3,5-trimethoxybenzene) in CD2Cl2. While trinuclear chloridogold(I) complexes of 1a–c exclusively yielded 3a with an exocyclic methylene group,36 other (redox-switchable) gold(I/III) catalysts are known to also catalyse the aromatisation of 3a to oxazole 3b in a subsequent step.51,127
 | ||
| Scheme 1 Gold(I)-catalysed ring-closing isomerisation of N-(2-propyn-1-yl)benzamide (2) to oxazoline 3a and potential subsequent aromatisation to oxazole 3b, including oxidants 5a and 5b and reductant 6 used in redox switching experiments. | ||
[1a(Au)]OTf – which had not yet been tested in catalysis – and [1b(Au)]OTf, at a concentration of 3 mol% gold(I) with respect to 2, were found to be catalytically inactive (Fig. 10, phase i). Most likely, steric hindrance arising from the κ3P,P′,P′′ coordination mode in [1a(Au)]OTf prevents the substrate from approaching the cationic gold(I) centre which otherwise would be expected to show some, albeit low, activity (vide infra). Given the fluctional coordination behaviour of [1a(Au)]OTf and the resulting apparent C3v-symmetric tricoordinate geometry on the NMR time scale, the substrate might be unable to approach the gold(I) ion on the catalytic time scale, too. Attempting to rationalise this assumption, the buried volume %Vbur, a measure for the steric bulk of a ligand at and around the coordinated metal originally developed for N-heterocyclic carbenes,128,129 was calculated for both coordination modes (bi- and tridentate). Based on the available solid-state structures and using the SambVca web application by Cavallo and co-workers,130 the C3-symmetric coordination of the three phosphanes results in a %Vbur of almost 90%, while the bidentate bonding mode in [1b(Au)]OTf corresponds to a significantly smaller %Vbur of about 67% (cf. ESI, section 7.2†). Due to the fast coordination-dissociation equilibrium of [1b(Au)]OTf, the effective buried volume in solution is likely closer to the 90% of [1a(Au)]OTf.
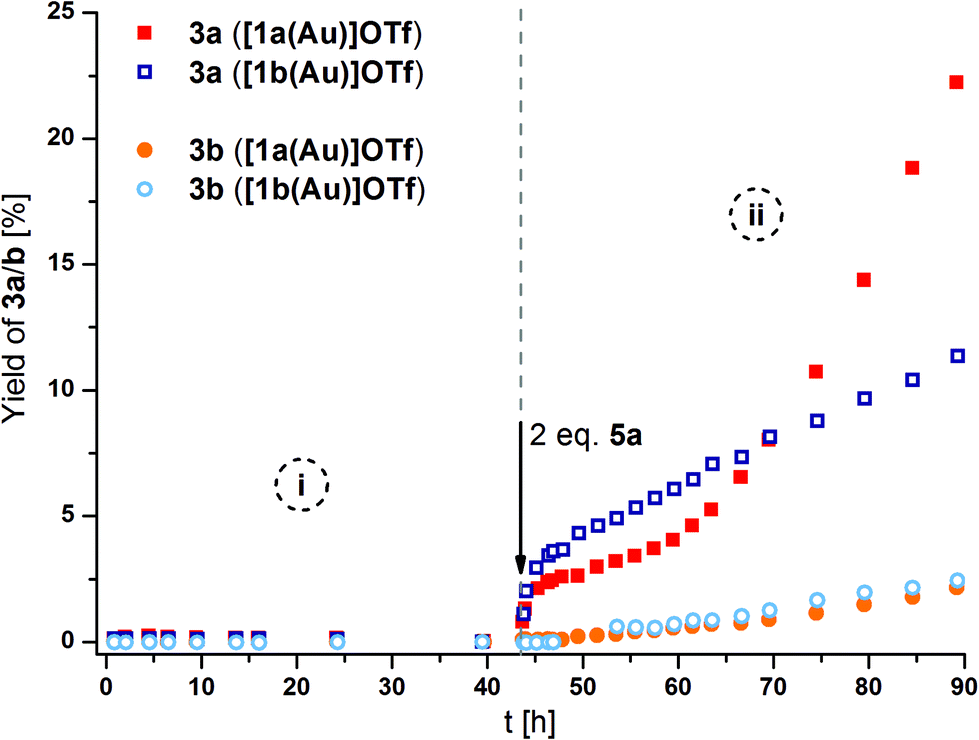 | ||
| Fig. 10 Reaction profiles for ring-closing isomerisation of 2 to 3a (squares) by [1a(Au)]OTf (solid symbols) and [1b(Au)]OTf (hollow symbols), also showing the formation of the aromatic follow-up product 3b (circles) before (i) and after (ii) oxidation with 2 eq. 5a (with respect to catalyst; 3 mol% Au, [2]0 = 60 mmol L−1, CD2Cl2, 25 °C). For corresponding 1H NMR spectra and TOF regression plots, s. Fig. S73, S75, and S76.† | ||
In line with the room-temperature oxidation of [1a(Au)]OTf following a two-electron-process,35 2 eq. of oxidant 5a (Scheme 1), containing the very weakly coordinating teflonate anion (tetrakis(tert-butoxy)aluminate, TEF),131,132 were added to the respective reaction mixtures. While the conversion of 2 to 3a (Fig. 10, squares) was indeed initiated upon oxidation (phase ii), the resulting catalytic activities as expressed by the turn-over-frequencies (TOF) were very low ([1a(Au)]OTf: 0.79 ± 0.02 h−1; [1b(Au)]OTf: 0.18 ± 0.01 h−1, cf. Table S7 and Fig. S75, S76†). Next to unusual reaction profiles – a steep initial increase followed by a flattening for [1b(Au)]OTf and an inverse-sigmoidal progression for [1a(Au)]OTf – both catalytic runs also produced detectable amounts of aromatic follow-up product 3b (Fig. 10, spheres).
The fate of [1a(Au)]OTf upon oxidation had already been studied in some detail and was revealed to involve electron transfer from P to FeIII after an initial iron(II)-centred oxidation, leading to the formation of a phosphane oxide species due to adventitious traces of water.35 Oxidation-induced reactivity, potentially involving substrate 2 and generating new, catalytically active species, is thus quite likely in this case, too.
The stepwise oxidation of [1b(Au)]OTf with 5a in CD2Cl2 was followed by a multinuclear NMR experiment, including the attempted reduction with decamethylferrocene (6) (Fig. S92–S94†). The appearance of several 31P{1H} signals centred at about 43 ppm with small but noticeable changes between the addition of one and two equivalents of 5a indicates the generation of P,P′-dicoordinate species. Regeneration of [1b(Au)]OTf by addition of 6 was not observed in neither the 1H nor the 31P{1H} NMR spectra, even though crystalline [6](TEF) was isolated from the reaction mixture after work-up. Furthermore, the 19F{1H} NMR signal of the triflate anion is strongly broadened suggesting its involvement in the follow-up chemistry. Spectroelectrochemical (SEC) measurements in the BArF4-based SE at 25 °C, −50 °C, and −80 °C (Fig. S69 and S70†), contrasting the cyclic voltammetry experiments in the same SE, showed good reversibility of the likely FeII-centred first oxidation and a less reversible second oxidation at 25 °C which became more reversible at −50 °C. All taken together, these partly conflicting and complex observations disqualify [1a(Au)]OTf and [1b(Au)]OTf for further applications in RSC.
We thus focused our attention on the tri-gold, hexa-ferrocene complexes [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3, expecting them to show a more predictable and rewarding catalytic profile. The buried volumes %Volbur for the central (73%) and peripheral (66%, cf. section 7.2 of the ESI†) gold(I) centres are similar to that of [1b(Au)]OTf and likely profit from the fixed coordination geometry (i.e., no pendant phosphane group). Indeed, both complexes displayed low but discernible catalytic activity in their native states (Fig. 11, top, hollow symbols). At this low activity level (phase iii), the TOF of both catalysts are similar and, over long time periods (native species), virtually identical (cf. Table S7†).
 | ||
| Fig. 11 Top: Reaction profiles for ring-closing isomerisation of 2 to 3a by [(1a)2(Au)3](OTf)3 (squares) and [(1b)2(Au)3](OTf)3 (circles) in native (hollow) and oxidised/reduced (solid) state (3 mol% Au, [2]0 = 60 mmol L−1, CD2Cl2, 25 °C). Arrows indicate the addition of additives for the redox-switched traces. Bottom: Comparison of turn-over frequencies (TOF) determined from linear fits (For corresponding 1H NMR spectra and TOF regression plots, s. Fig. S74 and S77–80†) referring to reaction phases iii–iv. | ||
Upon addition of two equivalents of 5a (phase iv), an immediate increase in activity took place ([(1a)2(Au)3](OTf)3 + 2 5a: 10.7×, [(1b)2(Au)3](OTf)3 + 2 5a: 12.3×), triazine-based [(1a)2(Au)3](OTf)3 (+2 5a) clearly outperforming (TOF = 4.2 ± 0.1 h−1) benzene-based [(1b)2(Au)3](OTf)3 (+ 2 5a) (TOF = 1.5 ± 0.1 h−1). As the increased activity is usually explained based on the increase in the Lewis acidity of gold(I) through oxidation of iron(II) to iron(III),18,124,127,133 reduction of the presumed ferrocenium species should regenerate the initial low activity. Thus, 2.2 equivalents of 6 were added to the reaction mixtures (phase v). In both cases, the TOF moderately decreased ([(1a)2(Au)3](OTf)3 + 2 5a + 2.2 6: 0.67×, [(1b)2(Au)3](OTf)3 + 2 5a + 2.2 6: 0.55×), yet without fully returning to the initial low activities.134 When 2.2 equivalents of 5a were added (phase vi), the activity of re-oxidised [(1a)2(Au)3](OTf)3 slightly surpassed the pre-reduction value by a factor of 1.1, while re-oxidised [(1b)2(Au)3](OTf)3 showed a 3-fold increase of TOF with respect to phase iv.
To gain more insight into the redox-switching behaviour, SEC measurements of [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3 in the BArF4-based SE were conducted (Fig. 12 and S71, S72†), the BArF4− anion being a suitable substitute for the teflonate anion concerning its low ion-pairing properties and inertness.132 In some contrast to the results from the CV (vide supra), the oxidation of [(1b)2(Au)3](OTf)3 (Fig. 12, left) was found to be fully reversible under these conditions, even though the reduction potential had to be applied for an extended time. The UV/Vis signature relates to an iron-centred oxidation,135–137 and the broad band centred at about 820 nm likely relates to a ligand-to-metal charge transfer between the cyclopentadienyl rings and iron(III).37 When [(1a)2(Au)3](OTf)3 is oxidised at potentials greater than 1 V (vs. FcH/[FcH]+, Fig. 12 top right), the intensely purple species formed cannot be reduced anymore, while oxidation below 1 V leads to a slightly different UV/Vis trace and allows for a reduction, however requiring a cathodic potential of −1 V. In both cases, the UV/Vis spectrum obtained after oxidation strongly resembles that obtained from oxidising [1a(Au)]OTf in the same SE, a process which was found to generate very reactive species from intramolecular electron transfer after an initial iron-centred oxidation.35 Following the oxidation of [(1a)2(Au)3](OTf)3 with two equivalents of 5a by multinuclear NMR spectroscopy (Fig. S95†), good yet delayed reducibility by 2.2 equivalents of 6 was observed. Using BF4-based oxidant 5b in the same stoichiometric ratio (Fig. S96†), a different spectral fingerprint was observed, and addition of 6 did not regenerate the initial spectral features. Treating [(1b)2(Au)3](OTf)3 with two equivalents of 5b (Fig. S97†), full reversibility upon addition of 6 was found. Studying 5b as a substitute for 5a – anions are known to play important roles in gold(I) catalysis89,138,139 – was, however, complicated by its poor solubility in CH2Cl2/CD2Cl2, as exact dosing of the required small amounts was found to be impossible.
 | ||
| Fig. 12 Plots obtained from UV/Vis spectroelectrochemical measurements of [(1a)2(Au)3](OTf)3 (right) and [(1b)2(Au)3](OTf)3 (left) in 0.1 mol L−1 (nBu4N)BArF4 in CH2Cl2 at 25 °C. Arrows highlight the position and direction of evolving/disappearing peaks. | ||
Owing to its more attractive redox features, especially as observed in SEC, a series of catalytic runs in which [(1b)2(Au)3](OTf)3 was oxidised by one to six equivalents of 5a was conducted (Fig. 13). When left oxidised, the doubly oxidised species (light green hollow circles) performs with a TOF of 5.4 ± 0.1 h−1 (32.6–41.0 h), similar to the TOF of 4.5 ± 0.1 h−1 observed after re-oxidation in the previous experiment (Fig. 11). Using only one equivalent of 5a, an approximately halved TOF for the same conversion interval (30–75%) of 2.4 ± 0.1 h−1 results. When three or more equivalents of 5a are added to [(1b)2(Au)3](OTf)3, the increment in TOF is by far greater than expected from the addition of one and two equivalents. When the reaction profiles obtained from adding four and six equivalents of 5a to [(1b)2(Au)3](OTf)3 are followed more closely by using smaller time intervals for the measurements (Fig. 13, top right), close-to-identical TOF for both cases result (shaded bars in Fig. 13, bottom). Relating these findings to the steric characterisation of the gold(I) centres by their respective buried volume (vide supra), it thus seems possible that the first two oxidations might be localised at the central ferrocenylene moieties (Fe(1) and Fe(4)), in turn activating the more buried central gold(I) ion Au(1). In the third and fourth oxidation, the chelated outer gold(I) centres Au(2) and Au(3), characterised by a higher substrate approachability, might followingly be activated, resulting in a sharp activity increase. In the absence of substrate, stepwise addition of up to six equivalents of 5a to [(1b)2(Au)3](OTf)3 resulted in a broadening and subsequent disappearance of the ferrocenylene and benzene-core signals in the corresponding 1H NMR spectra, while the 31P{1H} NMR signals, next to being strongly broadened, became successively shifted to higher field (Fig. S98 and S99†). Upon addition of 6.6 eq. of 6, the original spectral features of [(1b)2(Au)3](OTf)3 were almost fully recovered with some delay. Since no significant changes between the addition of four and six equivalents of 5a are observed in the corresponding 31P{1H} NMR spectra, it is possible that the fourfold oxidation is the limit of the oxidation potential of 5a under these conditions.
 | ||
| Fig. 13 Top left: Reaction profiles for ring-closing isomerisation of 2 to 3a by [(1b)2(Au)3](OTf)3 (3 mol% Au, [2]0 = 60 mmol L−1, CD2Cl2, 25 °C) oxidised by 1–6 eq. of 5a at the indicated time (arrow). The insert shows a magnified time period, and lines connecting the symbols are for visual guidance only. Top right: Reaction profiles for ring-closing isomerisation of 2 to 3a by [(1b)2(Au)3](OTf)3 (conditions as above), oxidised by four (orange/grape) and six (mustard) eq. of 5a directly before the start of the 1H NMR monitoring (viii). To one run (grape), 4.4 eq. of reductant 6 were added at the indicated time (arrow). Reaction phases ix–xi correspond to that run only. For TOF regression plots, s. Fig. S81–S89† referring to reaction phases vii and viii–xi. | ||
Thus, 4.4 eq. of 6 were added to a final run of [(1b)2(Au)3](OTf)3 oxidised by 4 eq. of 5a (purple hollow diamonds in Fig. 13, top right), and a decrease of activity to 0.27 ± 0.08 h−1 (phase xi) results after prolonged waiting (>6 h after addition of 6), mirroring the observations from SEC and NMR spectroscopy. Immediately after the addition of 6 (phase x), the TOF is reduced to about a third (14 ± 1 h−1) of the previous value (41 ± 1 h−1).
While all catalytic activities are far from record-breaking,123 the highly oxidised forms of both [(1a)2(Au)3](OTf)3 and [(1b)2(Au)3](OTf)3 convert 2 faster than tris(chloridogold(I)) complexes of 1a–c at the same gold(I) concentration.36
Their intriguing behaviour warrants a more detailed study on the corresponding oxidation/reduction mechanisms in RSC, and we are currently working on understanding the switching behaviour as well as investigating the use of 1a–c for different catalytic transformations.
Conclusions
C 3-Symmetric tris-phosphanes 1a–c and a perfluorophenyl derivative of 1a, 1aF, have been studied with respect to their coordination chemistry towards gold(I). Owing to steric strain arising from the clashing ortho substituents of the arene and the cyclopentadienyl rings, the tricoordinate bonding mode is reserved for the triazine derivative [1a(Au)]OTf, while 1:1 complexes of 1b and 1c prefer a fluctional dicoordinate bonding mode. Steric strain can further be mitigated by adopting a μ3:κ1P,κ1P′,κ1P′′ coordination mode, observed for 1c forming a one-dimensional coordination polymer with gold(I) in a 2:3 ligand-to-metal ratio. This very ratio can also be used to form discrete 2:3 complexes of 1a and 1b which, in contrast to the 1:1 complexes, show promising properties in the redox-switchable ring-closing isomerisation of propargylic amide 2 including multi-state activity behaviour.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Studienstiftung des deutschen Volkes (doctoral fellowships to P. C. and A. S.), the DFG (He 1376/51-1), and the Graduate School BuildMoNa is gratefully acknowledged. M. R. R. gratefully acknowledges funding provided by Universität Stuttgart, Stuttgart, Germany. The authors thank Dr Eckhard Bill (MPI CEC, Mülheim (Ruhr)) for supporting EPR and Mössbauer spectroscopy measurements, M. Sc. Nils Rotthowe and Prof. Rainer Winter (Universität Konstanz) for recording the room temperature-SEC spectra of [1b(Au)]OTf, and M. Sc. Luis Dütsch and Prof. Manfred Scheer for the generous donation of 5a.Notes and references
- Ferrocenes. Ligands, Materials and Biomolecules, ed. P. Štěpnička, J. Wiley, Chichester, England, Hoboken, NJ, 2008 Search PubMed.
- Ferrocenes. Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni and T. Hayashi, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 1995 Search PubMed.
- R. Horikoshi, Coord. Chem. Rev., 2013, 257, 621 CrossRef CAS.
- D. Astruc, Eur. J. Inorg. Chem., 2017, 2017, 6 CrossRef CAS.
- P. Štěpnička, Eur. J. Inorg. Chem., 2017, 2017, 215 CrossRef.
- Chiral Ferrocenes in Asymmetric Catalysis, ed. L.-X. Dai and X.-L. Hou, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef.
- L.-X. Dai, T. Tu, S.-L. You, W.-P. Deng and X.-L. Hou, Acc. Chem. Res., 2003, 36, 659 CrossRef CAS.
- J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415 CrossRef CAS.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969 CrossRef CAS.
- Y. Ryu, G. Ahumada and C. W. Bielawski, Chem. Commun., 2019, 55, 4451 RSC.
- A. M. Allgeier, C. S. Slone, C. A. Mirkin, L. M. Liable-Sands, G. P. A. Yap and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 550 CrossRef CAS.
- A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894 CrossRef.
- I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617 CrossRef CAS.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef CAS.
- M. Sawamura, H. Hamashima and Y. Ito, Tetrahedron: Asymmetry, 1991, 2, 593 CrossRef CAS.
- M. Sawamura, A. Yamauchi, T. Takegawa and Y. Ito, J. Chem. Soc., Chem. Commun., 1991, 45, 874 RSC.
- L. Hettmanczyk, L. Suntrup, S. Klenk, C. Hoyer and B. Sarkar, Chem. – Eur. J., 2017, 23, 576 CrossRef CAS.
- T.-Y. Dong, K. Chen, M.-C. Lin and L. Lee, Organometallics, 2005, 24, 4198 CrossRef CAS.
- W. Chen, P. J. McCormack, K. Mohammed, W. Mbafor, S. M. Roberts and J. Whittall, Angew. Chem., Int. Ed., 2007, 46, 4141 CrossRef CAS.
- S. Santi, A. Bisello, R. Cardena and A. Donoli, Dalton Trans., 2015, 44, 5234 RSC.
- A. Donoli, A. Bisello, R. Cardena, C. Prinzivalli and S. Santi, Organometallics, 2013, 32, 1029 CrossRef CAS.
- Y.-K. Lim, S. Wallace, J. C. Bollinger, X. Chen and D. Lee, Inorg. Chem., 2007, 46, 1694 CrossRef CAS.
- M. S. Inkpen, S. Scheerer, M. Linseis, A. J. P. White, R. F. Winter, T. Albrecht and N. J. Long, Nat. Chem., 2016, 8, 825 CrossRef CAS.
- L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148 RSC.
- Y. Yu, A. D. Bond, P. W. Leonard, U. J. Lorenz, T. V. Timofeeva, K. P. C. Vollhardt, G. D. Whitener and A. A. Yakovenko, Chem. Commun., 2006, 2572 RSC.
- A. Kasprzak, A. Kowalczyk, A. Jagielska, B. Wagner, A. M. Nowicka and H. Sakurai, Dalton Trans., 2020, 49, 9965–9971 RSC.
- D. Astruc, C. Ornelas and J. Ruiz, Acc. Chem. Res., 2008, 41, 841 CrossRef CAS.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2011, 133, 629 CrossRef CAS.
- D. Astruc, J. Leather Sci. Eng., 2020, 2, 3534 Search PubMed.
- C. Dengiz, B. Breiten, J.-P. Gisselbrecht, C. Boudon, N. Trapp, W. B. Schweizer and F. Diederich, J. Org. Chem., 2015, 80, 882 CrossRef CAS.
- P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311 CrossRef CAS.
- P. Neumann, H. Dib, A. Sournia-Saquet, T. Grell, M. Handke, A.-M. Caminade and E. Hey-Hawkins, Chem. – Eur. J., 2015, 21, 6590 CrossRef CAS.
- J. Popp, A.-M. Caminade and E. Hey-Hawkins, Eur. J. Inorg. Chem., 2020, 17, 1654 CrossRef.
- A. Straube, P. Coburger, M. R. Ringenberg and E. Hey-Hawkins, Chem. – Eur. J., 2020, 26, 5758 CrossRef CAS.
- A. Straube, P. Coburger, L. Dütsch and E. Hey-Hawkins, Chem. Sci., 2020, 11, 10657–10668 RSC.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Organometallics, 2012, 31, 6761 CrossRef CAS.
- A. Preuß, M. Korb, D. Miesel, T. Rüffer, A. Hildebrandt and H. Lang, Dalton Trans., 2019, 48, 14418 RSC.
- R. K. Al-Shewiki, M. Korb, A. Hildebrandt, S. Zahn, S. Naumov, R. Buschbeck, T. Rüffer and H. Lang, Dalton Trans., 2019, 48, 1578 RSC.
- S. Santi, L. Orian, A. Donoli, A. Bisello, M. Scapinello, F. Benetollo, P. Ganis and A. Ceccon, Angew. Chem., Int. Ed., 2008, 47, 5331 CrossRef CAS.
- S. Rossi, A. Bisello, R. Cardena, L. Orian and S. Santi, Eur. J. Org. Chem., 2017, 5966 CrossRef CAS.
- S. Rossi, A. Bisello, R. Cardena and S. Santi, Organometallics, 2018, 37, 4242 CrossRef CAS.
- S. Steffens, M. H. Prosenc, J. Heck, I. Asselberghs and K. Clays, Eur. J. Inorg. Chem., 2008, 1999 CrossRef CAS.
- J. M. Speck, M. Korb, A. Schade, S. Spange and H. Lang, Organometallics, 2015, 34, 3788 CrossRef CAS.
- A. Hildebrandt, D. Schaarschmidt and H. Lang, Organometallics, 2011, 30, 556 CrossRef CAS.
- M. Korb, U. Pfaff, A. Hildebrandt, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2014, 2014, 1051 CrossRef CAS.
- A. Hildebrandt and H. Lang, Dalton Trans., 2011, 40, 11831 RSC.
- D. Miesel, A. Hildebrandt and H. Lang, Curr. Opin. Electrochem., 2018, 8, 39 CrossRef CAS.
- G. Filipczyk, S. W. Lehrich, A. Hildebrandt, T. Rüffer, D. Schaarschmidt, M. Korb and H. Lang, Eur. J. Inorg. Chem., 2017, 2017, 263 CrossRef.
- I. Caracelli, J. Zukerman-Schpector and E. R. T. Tiekink, Gold Bull., 2013, 46, 81 CrossRef CAS.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef CAS.
- A. M. Wright, B. J. Irving, G. Wu, A. J. H. M. Meijer and T. W. Hayton, Angew. Chem., Int. Ed., 2015, 54, 3088 CrossRef CAS.
- R. T. Stibrany, C. Zhang, T. J. Emge, H. J. Schugar, J. A. Potenza and S. Knapp, Inorg. Chem., 2006, 45, 9713 CAS.
- C. Fan, C. Ma, C. Chen, F. Chen and Q. Liu, Inorg. Chem. Commun., 2003, 6, 491 CAS.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569 CrossRef CAS.
- Q.-S. Li, C.-Q. Wan, R.-Y. Zou, F.-B. Xu, H.-B. Song, X.-J. Wan and Z.-Z. Zhang, Inorg. Chem., 2006, 45, 1888 CrossRef CAS.
- E. Herrero-Gómez, C. Nieto-Oberhuber, S. López, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455 CrossRef.
- P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem. – Eur. J., 2010, 16, 5324 CrossRef.
- Q.-L. Ni, X.-F. Jiang, T.-H. Huang, X.-J. Wang, L.-C. Gui and K.-G. Yang, Organometallics, 2012, 31, 2343 CrossRef CAS.
- C. Griebel, D. D. Hodges, B. R. Yager, F. L. Liu, W. Zhou, K. J. Makaravage, Y. Zhu, S. G. Norman, R. Lan, C. S. Day and A. C. Jones, Organometallics, 2020, 39(14), 2665–2671 CrossRef CAS.
- E. R. T. Tiekink and J. Zukerman-Schpector, CrystEngComm, 2009, 11, 1176 RSC.
- M. A. Cinellu, M. Arca, F. Ortu, S. Stoccoro, A. Zucca, A. Pintus and L. Maiore, Eur. J. Inorg. Chem., 2019, 2019, 4784 CAS.
- S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912 CrossRef CAS.
- M. R. Holmes, J. F. Manganaro, C. L. Barnes and B. W. Gung, J. Organomet. Chem., 2015, 795, 18 CrossRef CAS.
- H. Teller, S. Flügge, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 1949 CrossRef CAS.
- C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2011, 50, 7369 CrossRef CAS.
- E. J. Derrah, C. Martin, S. Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Dalton Trans., 2012, 41, 14274 RSC.
- V. Rampazzi, J. Roger, R. Amardeil, M.-J. Penouilh, P. Richard, P. Fleurat-Lessard and J.-C. Hierso, Inorg. Chem., 2016, 55, 10907 CrossRef CAS.
- U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Štěpnička, Z. Anorg. Allg. Chem., 2011, 637, 1824 CrossRef CAS.
- B. E. Cowie and D. J. H. Emslie, Organometallics, 2018, 37, 1007 CrossRef CAS.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006 CrossRef CAS.
- E. M. Barranco, O. Crespo, M. C. Gimeno, A. Laguna, P. G. Jones and B. Ahrens, Inorg. Chem., 2000, 39, 680 CrossRef CAS.
- K. Škoch, I. Císařová and P. Štěpnička, Chem. – Eur. J., 2015, 21, 15998 CrossRef.
- S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC.
- R. Horikoshi, T. Tominaga and T. Mochida, Cryst. Growth Des., 2018, 18, 5089 CrossRef CAS.
- L. A. Wilkinson, T. T. C. Yue, E. Massey, A. J. P. White and N. J. Long, Dalton Trans., 2018, 48, 72 RSC.
- R. Horikoshi and T. Mochida, Eur. J. Inorg. Chem., 2010, 2010, 5355 CrossRef.
- R. Shekurov, M. Khrizanforov, T. Gerasimova, Z. Yamaleeva, K. Ivshin, A. Lakomkina, I. Bezkishko, A. Kononov, O. Sinyashin, Y. Budnikova, O. Kataeva and V. Miluykov, Molecules, 2020, 25, 939 CrossRef CAS.
- V. Khrizanforova, R. Shekurov, V. Miluykov, M. Khrizanforov, V. Bon, S. Kaskel, A. Gubaidullin, O. Sinyashin and Y. Budnikova, Dalton Trans., 2020, 49, 2794 RSC.
- S. L. James, Chem. Soc. Rev., 2009, 38, 1744 RSC.
- M. Streitberger, A. Schmied and E. Hey-Hawkins, Inorg. Chem., 2014, 53, 6794 CrossRef CAS.
- R. Hoy, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2018, 47, 14515 RSC.
- R. J. Puddephatt, Coord. Chem. Rev., 2001, 216–217, 313 CrossRef CAS.
- J. Su, L. Yao, J. Zhang, S. Yuan, F. Xie, Y. Ding, M. Zhao, S. Wang, H. Li, S. Zhang, J. Wu and Y. Tian, New J. Chem., 2016, 40, 97 RSC.
- C. L. Pollock, G. C. Saunders, E. C. M. S. Smyth and V. I. Sorokin, J. Fluor. Chem., 2008, 129, 142 CrossRef CAS.
- B. G. Anderson and J. L. Spencer, Chem. – Eur. J., 2014, 20, 6421 CrossRef CAS.
- P. H. Lee, S. Kim, A. Park, B. C. Chary and S. Kim, Angew. Chem., Int. Ed., 2010, 49, 6806 CrossRef CAS.
- C. Shu, L. Li, C.-B. Chen, H.-C. Shen and L.-W. Ye, Chem. - Asian J., 2014, 9, 1525 CrossRef CAS.
- J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2493 CrossRef.
- S.-H. Shin, Bull. Korean Chem. Soc., 2005, 26, 1925 CrossRef CAS.
- Y. H. Lee and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 6444 CrossRef CAS.
- Y. Yang, L. Eberle, F. F. Mulks, J. F. Wunsch, M. Zimmer, F. Rominger, M. Rudolph and A. S. K. Hashmi, J. Am. Chem. Soc., 2019, 141, 17414 CrossRef CAS.
- M. Nishikawa, Y. Wakita, T. Nishi, T. Miura and T. Tsubomura, Dalton Trans., 2015, 44, 9170 RSC.
- Y. Ren, Y. Dienes, S. Hettel, M. Parvez, B. Hoge and T. Baumgartner, Organometallics, 2009, 28, 734 CrossRef CAS.
- Y. Ren, A. Orthaber, R. Pietschnig and T. Baumgartner, Dalton Trans., 2013, 42, 5314 RSC.
- O. V. Gusev, T. Y. A. Peganova, A. M. Kalsin, N. V. Vologdin, P. V. Petrovskii, K. A. Lyssenko, A. V. Tsvetkov and I. P. Beletskaya, Organometallics, 2006, 25, 2750 CrossRef CAS.
- A. M. Kalsin, N. V. Vologdin, T. A. Peganova, P. V. Petrovskii, K. A. Lyssenko, F. M. Dolgushin and O. V. Gusev, J. Organomet. Chem., 2006, 691, 921 CrossRef CAS.
- O. V. Gusev, M. G. Peterleitner, A. M. Kal'sin and N. V. Vologdin, Russ. J. Electrochem., 2003, 39, 1293 CrossRef CAS.
- N. J. Adamson, H. Jeddi and S. J. Malcolmson, J. Am. Chem. Soc., 2019, 141, 8574 CrossRef CAS.
- Please note that 19F{1H} NMR chemical shifts for VII have been reported with reference to δ(CF3COOH) = 0 ppm, while chemical shifts reported in this publication follow the IUPAC convention of δ(CFCl3) = 0 ppm, resulting in Δδ = −76.6 ppm .
- H. W. Chen and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m50–m52 CrossRef CAS.
- Y. Dienes, U. Englert and T. Baumgartner, Z. Anorg. Allg. Chem., 2009, 635, 238 CrossRef CAS.
- G. Moreno-Alcántar, H. Hernández-Toledo, J. M. Guevara-Vela, T. Rocha-Rinza, Á. Martín Pendás, M. Flores-Álamo and H. Torrens, Eur. J. Inorg. Chem., 2018, 2018, 4413 CrossRef.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- M. Streitberger, A. Schmied, R. Hoy and E. Hey-Hawkins, Dalton Trans., 2016, 45, 11644 RSC.
- A. Schulz, A. Villinger and A. Westenkirchner, Inorg. Chem., 2014, 53, 3183 CrossRef CAS.
- W. Schuh, H. Kopacka, K. Wurst and P. Peringer, Chem. Commun., 2001, 2186 RSC.
- C. Hollatz, A. Schier and H. Schmidbaur, Inorg. Chem. Commun., 1998, 1, 115 CrossRef CAS.
- D. Li, C.-M. Che, S.-M. Peng, S.-T. Liu, Z.-Y. Zhou and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1993, 189 RSC.
- G. S. M. Tong, S. C. F. Kui, H.-Y. Chao, N. Zhu and C.-M. Che, Chem. – Eur. J., 2009, 15, 10777 CrossRef CAS.
- P. Sevillano, M. E. García, A. Habtemariam, S. Parsons and P. J. Sadler, Met.-Based Drugs, 1999, 6, 211 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- M. J. Verschoor-Kirss, O. Hendricks, C. M. Verschoor, R. Conry and R. U. Kirss, Inorg. Chim. Acta, 2016, 450, 30 CrossRef CAS.
- A. Louati, M. Gross, L. Douce and D. Matt, J. Organomet. Chem., 1992, 438, 167 CrossRef CAS.
- K. D. Reichl, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2013, 135, 9354 CrossRef CAS.
- F. Barrière, R. U. Kirss and W. E. Geiger, Organometallics, 2005, 24, 48 CrossRef.
- H. Baumgärtel, Electrochemistry. A Guide for Newcomers, De Gruyter, Berlin/Boston, 2019 Search PubMed.
- M. V. Mirkin, Determination of Electrode Kinetics in Handbook of Electrochemistry, ed. C. G. Zoski, Elsevier, Oxford, 2007, pp. 639–660 Search PubMed.
- M. Iyoda, T. Kondo, T. Okabe, H. Matsuyama, S. Sasaki and Y. Kuwatani, Chem. Lett., 1997, 26, 35 CrossRef.
- F. Barrière and W. E. Geiger, J. Am. Chem. Soc., 2006, 128, 3980 CrossRef.
- A. G. Furneaux, N. A. Piro, R. Hernández Sánchez, K. M. Gramigna, N. Fey, M. J. Robinson, W. S. Kassel and C. Nataro, Dalton Trans., 2016, 45, 4819 RSC.
- D. A. Durfey, R. U. Kirss, C. Frommen and W. Feighery, Inorg. Chem., 2000, 39, 3506 CrossRef CAS.
- S. Vanicek, M. Podewitz, J. Stubbe, D. Schulze, H. Kopacka, K. Wurst, T. Müller, P. Lippmann, S. Haslinger, H. Schottenberger, K. R. Liedl, I. Ott, B. Sarkar and B. Bildstein, Chem. – Eur. J., 2018, 24, 3742 CrossRef CAS.
- S. Klenk, S. Rupf, L. Suntrup, M. van der Meer and B. Sarkar, Organometallics, 2017, 36, 2026 CrossRef CAS.
- L. Hettmanczyk, D. Schulze, L. Suntrup and B. Sarkar, Organometallics, 2016, 35, 3828 CrossRef CAS.
- P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019, 55, 4615 RSC.
- M. Rigo, L. Hettmanczyk, F. J. L. Heutz, S. Hohloch, M. Lutz, B. Sarkar and C. Müller, Dalton Trans., 2016, 46, 86 RSC.
- S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef.
- A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872 CrossRef CAS.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490 CrossRef CAS.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982 CrossRef CAS.
- A. Feyrer, M. K. Armbruster, K. Fink and F. Breher, Chem. – Eur. J., 2017, 23, 7402 CrossRef CAS.
- It should be noted that, while the TOF is an oversimplified approximation of catalytic activity, trying to fit the reaction profiles by first- or second-order kinetics through the respective curve transformation did not yield meaningful profiles for the corresponding linear fits .
- H. B. Gray, Y. S. Sohn and N. Hendrickson, J. Am. Chem. Soc., 1971, 93, 3603 CrossRef.
- D. M. Duggan and D. N. Hendrickson, Inorg. Chem., 1975, 14, 955 CrossRef CAS.
- S. Hartmann, R. F. Winter, B. M. Brunner, B. Sarkar, A. Knödler and I. Hartenbach, Eur. J. Inorg. Chem., 2003, 2003, 876 CrossRef.
- D. Zuccaccia, A. Del Zotto and W. Baratta, Coord. Chem. Rev., 2019, 396, 103 CrossRef CAS.
- A. Zhdanko and M. E. Maier, ACS Catal., 2014, 4, 2770 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, detailed spectral and crystallographic characterisation, DFT calculations, electrochemical data, catalysis plots. CCDC 2021517–2021522. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0dt02743j |
| This journal is © The Royal Society of Chemistry 2020 |