
Intramolecular trapping of ammonium ylides with N-benzoylbenzotriazoles in aqueous medium: direct access to the pseudoindoxyl scaffold†
Lalita
Devi
ab,
Rashmi
Shukla
a and
Namrata
Rastogi
 *ab
*ab
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031, India. E-mail: namrata.rastogi@cdri.res.in
bAcademy of Scientific and Innovative Research, New Delhi, India
First published on 7th December 2018
Abstract
The present work documents an operationally simple, clean and practical method for accessing the 2,2-disubstituted indolin-3-one (pseudoindoxyl) scaffold. The rhodium carbenoid mediated reaction between N-o-alkylamino benzoylbenzotriazoles and aryl diazoacetates occurs smoothly in water and exploits the leaving group ability of the benzotriazole moiety to install the carbonyl function in the product. Other highlights of the methodology are a wide substrate scope and experimental practicality given the re-use of the benzotriazole byproduct for starting material preparation.
Introduction
The 2,2-disubstituted indolin-3-one core is found in several biologically active pseudoindoxyl spirocyclic alkaloids of plant and fungal origin such as (+)-aristotelone, ervaoffines, duocarmycins, (+)-austamide, brevianamides, etc. (Fig. 1).1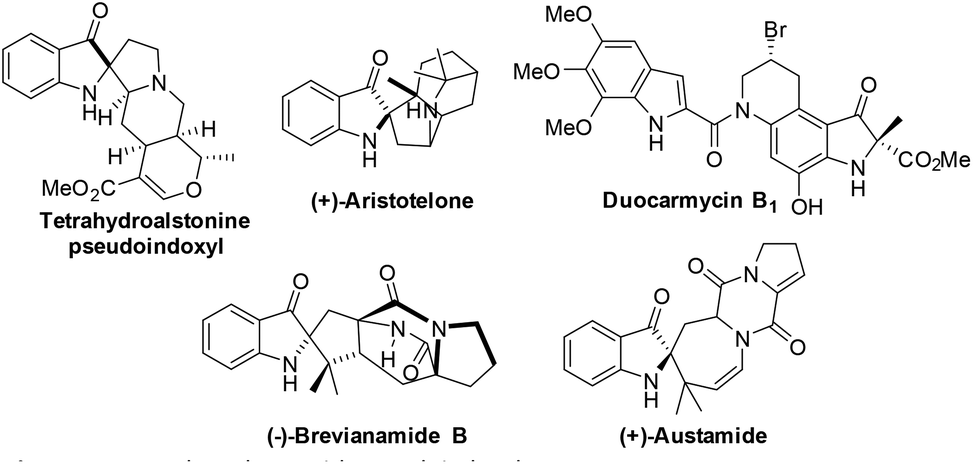 | ||
| Fig. 1 Natural products with the pseudoindoxyl core. | ||
This is also a key starting subunit for several natural molecules including hinkdentine A, isatisine A, grandilodine B, trigonoliimine C, etc.2 The 2,2-disubstituted-indolin-3-one oligomers have also been found to be efficient synthetic mimetics of the protein secondary structure.3
Several elegant protocols are known for the synthesis of 2,2-disubstituted indolin-3-ones. Gagosz and co-workers described a gold-carbenoid mediated Claisen rearrangement of 2-azido-alkynes for accessing indolin-3-ones bearing an allyl group at the 2-position.4 Xiao and co-workers subjected indoles to the photocatalytic aerobic oxidation/semipinacol rearrangement to afford 2,2-disubstituted indolin-3-ones.5 Okuma and co-workers synthesized a series of 2-phenylindolin-3-ones through the cycloaddition reaction of amino acid methyl esters with the in situ generated arynes.6 Fan and co-workers prepared several 2-acetoxy-indolin-3-ones from o-acyl anilines via the cyclization–acetoxylation sequence mediated by hypervalent iodine reagents.7 Shimizu and co-workers reported an interesting aza-Brook rearrangement of α-imino esters followed by Dieckmann condensation of the resulting aluminium enolates for the construction of 2,2-disubstituted indolin-3-ones.8 An elegant cascade Fisher indolization-Claisen rearrangement sequence employing aryl hydrazines and allyloxyketones was developed by Yao and Xie.9 Recently, Zhu and co-workers reported a gold catalyzed heteroannulation reaction between α-amino imides and in situ generated arynes and further employed the strategy for the total synthesis of (+)-hinckdentine A successfully.10
Furthermore, the asymmetric synthesis of the pseudoindoxyl core has also garnered significant attention. For instance, several methods for the enantioselective synthesis of 2,2-disubstituted indolin-3-ones based on asymmetric alkylation of 2-monosubstituted indolin-3-ones/indol-3-ones via the Michael reaction, Mannich reaction, Henry reaction, etc., have been documented in the literature.11 Several groups reported enantioselective Friedal–Crafts alkylation of indoles with spiro indolin-3-ones,12a or with cyclic imines to access the pseudoindoxyl core.12b
The transition metal catalyzed X–H insertion reactions employing diazo compounds as the carbene precursor are among the most popular and practical methods for the construction of C–X bonds.13 In particular, the N–H and O–H insertion reactions have been developed extensively over other X–H insertion reactions, attributable to the significance of these functional groups in organic synthesis.14 Liang and co-workers reported a palladium catalyzed cross-coupling reaction of aryldiazoacetates with N-substituted-2-iodoanilines in the presence of carbon monoxide leading to the 2,2-disubstituted indolin-3-one scaffold.15 The reaction involves the formation of palladium carbenoid with aryldiazoacetates after palladium catalyzed carbonylation of aryl iodide with CO initially. The usual acyl migratory insertion and isomerization followed by the intramolecular nucleophilic substitution reaction affords the 2,2-disubstituted indolin-3-one. The limited substrate scope and moderate yields along with the use of CO as the source of the carbonyl group are major limitations of this otherwise elegant synthetic methodology. We hereby report a simple strategy to achieve the synthesis of the 2,2-disubstituted indolin-3-one core via the Rh2(OAc)4 catalyzed reaction of diazoesters with o-amino benzoylbenzotriazoles in aqueous medium. The reaction takes advantage of the benzotriazole group's leaving abilities to install the carbonyl function after the nucleophilic addition of the transient ammonium ylide species onto the carbonyl carbon (Scheme 1).16
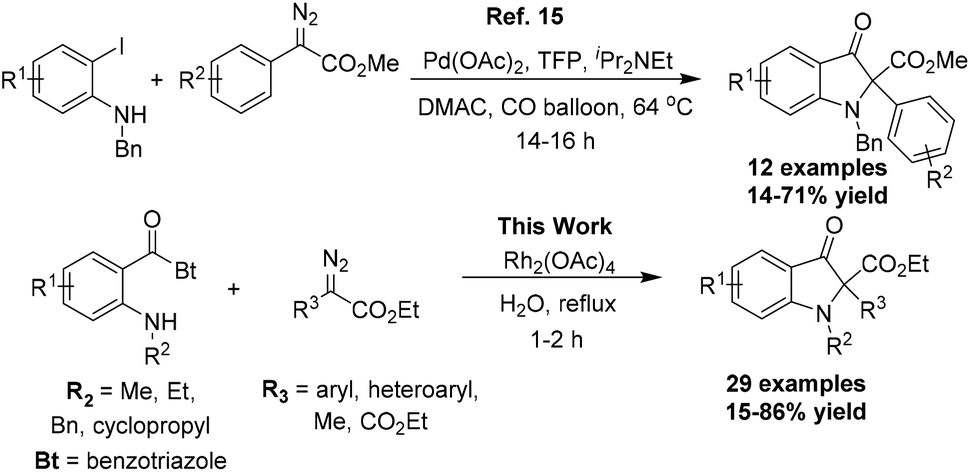 | ||
| Scheme 1 Pseudoindoxyl core from diazo compounds. | ||
Results and discussion
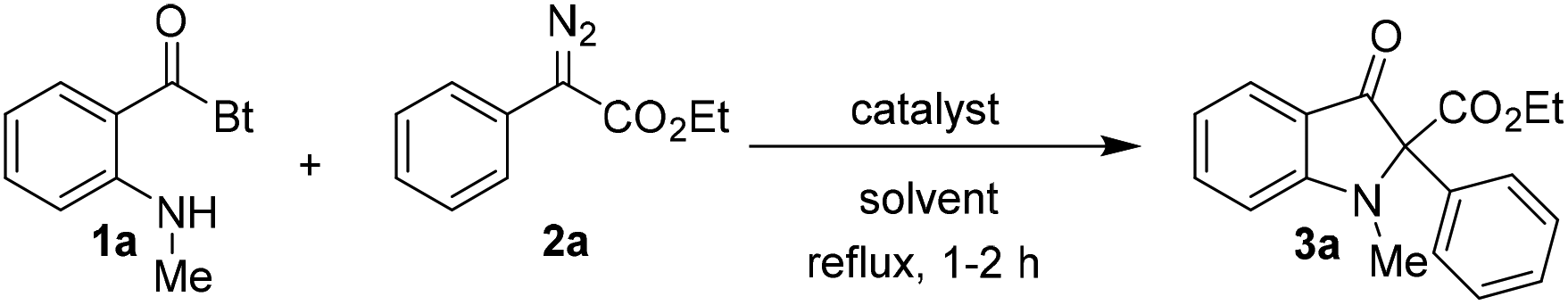
For preliminary investigations, we selected ethyl 2-diazo-2-phenylacetate 2a as the carbene precursor and N-o-methylamino benzoylbenzotriazole 1a as the nucleophile in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, after an initial unsuccessful attempt to use o-amino benzoylbenzotriazole in the reaction.17
:1 ratio, after an initial unsuccessful attempt to use o-amino benzoylbenzotriazole in the reaction.17
The reaction carried out in toluene in the presence of Rh2(OAc)4 at room temperature was ineffective but provided the expected product in moderate yield at reflux temperature (entries 1 and 2; Table 1). There was only a slight enhancement of yield upon increasing the amount of 2a to 1.5 equivalents; however yield increased enormously upon doubling the amount of 2a in the reaction (entries 3 and 4). Furthermore, screening of catalysts revealed that Cu(acac)2 works equally well in the reaction, whereas Pd(OAc)2 resulted in a much lower product yield (entries 5 and 6). Since Rh2(OAc)4 proved to be the better catalyst in terms of yield so far, it was used for the optimization of the solvent used in the reaction. The reaction when carried out with Rh2(OAc)4 in DCE provided 3a in 85% NMR yield (entry 7). The catalyst Rh2(OAc)4 worked efficiently in aqueous medium as well affording 3a in 88% NMR yield but surprisingly the product was obtained in poor yield when Cu(acac)2 was used as the catalyst in H2O (entries 8 and 9).18 Although the product yield was approximately 7% better in the reaction carried out with Rh2(OAc)4 in toluene as compared to the reaction in the aqueous medium (compare entries 4 and 8), water was selected as the reaction solvent due to its innocuous character. Moreover, the product was isolated in excellent yield upon reducing the catalyst loading to half in the aqueous reaction (entry 11). Therefore, the 1:2 ratio of substrates 1 and 2 in the presence of 0.5 mol% of Rh2(OAc)4 in water at reflux temperature was established as the optimum condition for the reaction.
|
|
||||
|---|---|---|---|---|
| Entry |
1a:2a |
Catalyst (mol % w.r.t. 2a) | Solvent | 3a (%) |
| a Unless otherwise noted, all reactions were performed with 0.5 mmol of 1a, specified amount of 2a and catalysts in 5 mL of solvent at reflux temperature. b NMR yields. c No reaction at room temperature. d Isolated yield in parentheses. | ||||
| 1 | 1:1 |
Rh2(OAc)4 (1) | Toluene | NRc |
| 2 | 1:1 |
Rh2(OAc)4 (1) | Toluene | 58 (51)d |
| 3 | 1:1.5 |
Rh2(OAc)4 (1) | Toluene | 64 |
| 4 | 1:2 |
Rh2(OAc)4 (1) | Toluene | 95 |
| 5 | 1:2 |
Cu(acac)2 (1) | Toluene | 93 (80)d |
| 6 | 1:2 |
Pd(OAc)2 (1) | Toluene | 65 |
| 7 | 1:2 |
Rh2(OAc)4 (1) | DCE | 85 |
| 8 | 1:2 |
Rh2(OAc)4 (1) | H2O | 88 |
| 9 | 1:2 |
Cu(acac)2 (1) | H2O | 26 |
| 10 | 1:2 |
Rh2(OAc)4 (1.5) | H2O | 75 |
| 11 | 1:2 |
Rh2(OAc)4 (0.5) | H2O | 96 (86)d |
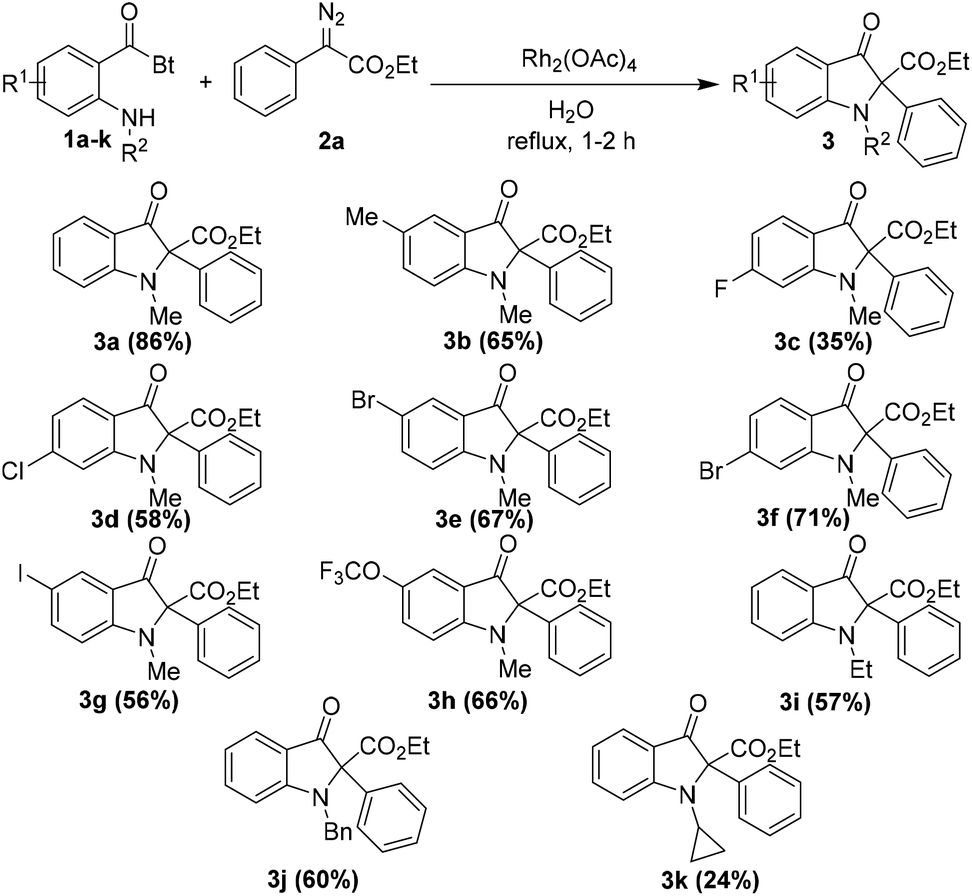
After optimizing the reaction conditions, we decided to examine the scope of o-amino benzoylbenzotriazoles 1 in the reaction. A variety of differently substituted o-amino benzoylbenzotriazoles 1, bearing electron withdrawing as well as electron donating substituents, were reacted with ethyl 2-diazo-2-phenylacetate 2a under the optimized reaction conditions (Table 2).
| a Reaction conditions: 1 (0.5 mmol), 2a (1.0 mmol), Rh2(OAc)4 (0.005 mmol), H2O (5 mL), reflux |
|---|
|
It was satisfying to note that the desired products were isolated in high yields in all the cases except with the fluorine substituted o-amino benzoylbenzotriazole 1c. Additionally, different substitutions on the amine nitrogen including the ethyl group and benzyl group were tolerated well and the corresponding products 3i and 3j were isolated in 57% and 60% yields, respectively. However, the cyclopropyl substitution on nitrogen resulted in lower yield of the product 3k, viz. 24%. Notably, this is quite a clean and practical reaction methodology since the byproducts are nitrogen gas and benzotriazole, which could be isolated conveniently and reused for the synthesis of the starting material.
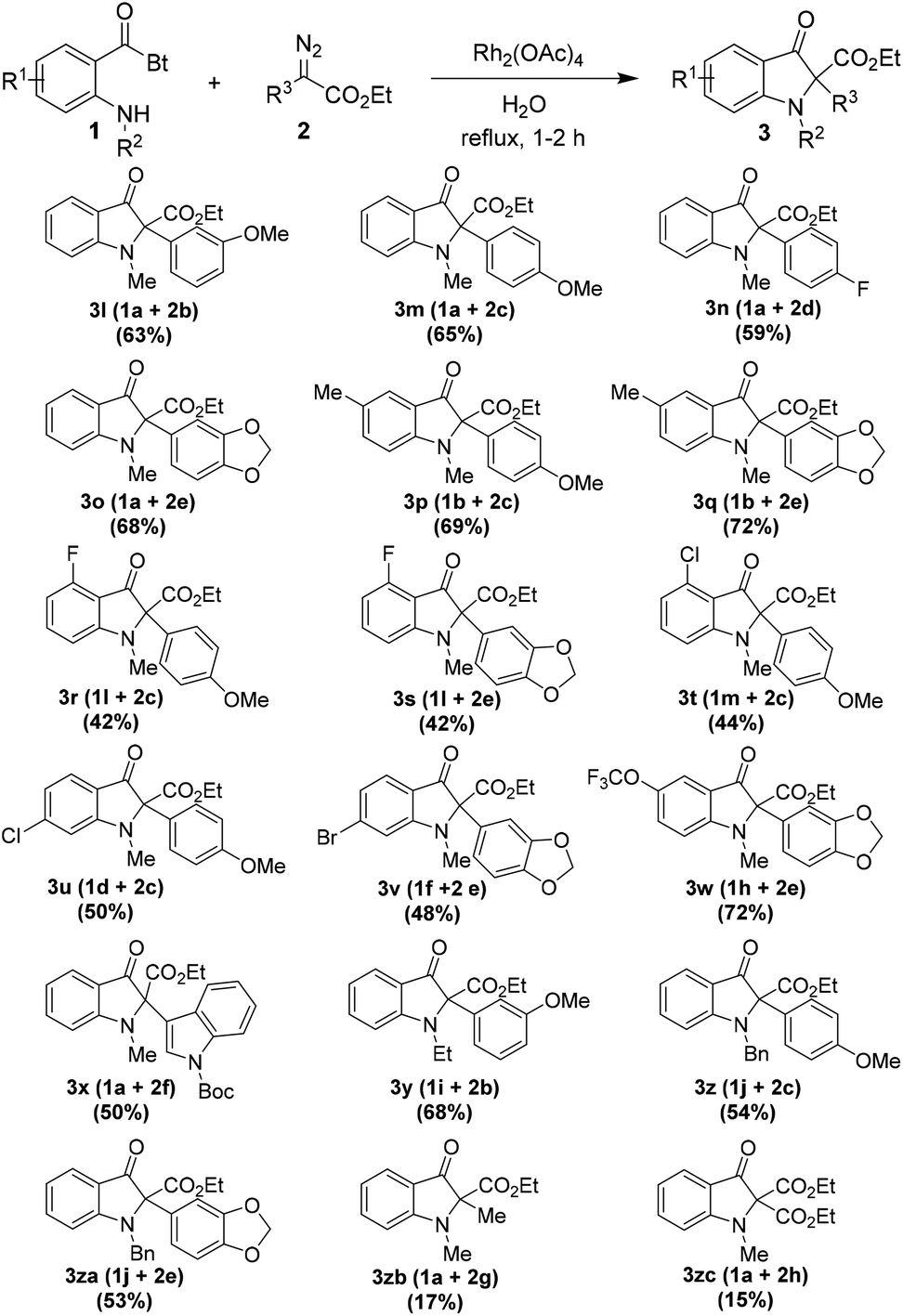
After utilizing the wide range of o-amino benzoylbenzotriazoles 1 in the reaction, we aimed to investigate the scope of diazoacetates 2 in the reaction (Table 3). Therefore, variously substituted diazophenylacetates were prepared from the corresponding phenylacetates via the diazo transfer reaction.19
| a Reaction conditions: 1 (0.5 mmol), 2 (1.0 mmol), Rh2(OAc)4 (0.005 mmol), H2O (5 mL), reflux |
|---|
|
The reaction of several o-amino benzoylbenzotriazoles 1 with a range of diazoacetates 2, bearing substituents with different electronic characters, proceeded smoothly under the reaction conditions. The products were isolated in moderate to good yields ranging from 42% to 72%. Notably, the heteroaryldiazoacetate 2f too participated well in the reaction with 1a providing product 3x in 50% yield. Furthermore, the reaction of N-o-methylamino benzoylbenzotriazole 1a with ethyl 2-diazopropanoate 2g as well as diethyl 2-diazomalonoate 2h (with acceptor–acceptor group substitution on diazo carbon) furnished the desired products 3zb and 3zc, respectively, albeit in low yields.
Mechanistically, the reaction plausibly follows the carbenoid generation → ylide formation → nucleophilic addition → proton transfer and elimination sequence (Scheme 2). The rhodium carbenoid A derived from diazoacetate 2 upon interaction with the lone pair of electrons on nitrogen of sec-amine 1 results in the generation of rhodium ylide species B.20 The transient rhodium ylide B dissociates to provide the resonance stabilized ammonium ylide C. The intramolecular nucleophilic addition of the ylide C onto the amide carbonyl group followed by “delayed proton transfer” and benzotriazole elimination ultimately leads to the 2,2-disubstituted indolin-3-one product 3.21
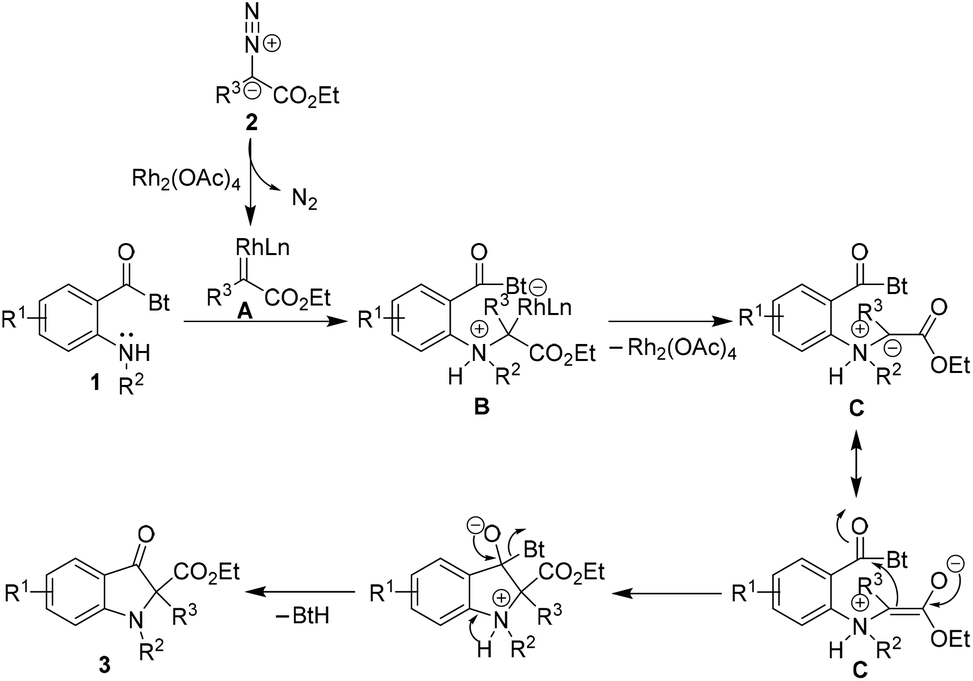 | ||
| Scheme 2 Plausible mechanism. | ||
Conclusions
In summary, we devised an efficient strategy for the synthesis of the 2,2-disubstituted indolin-3-one or pseudoindoxyl scaffold through the Rh-carbenoid mediated reaction between N-o-alkylamino benzoylbenzotriazoles and aryl diazoacetates. The method elegantly exploits the leaving group ability of the benzotriazole moiety to lodge the carbonyl function in the products. The reaction takes place in aqueous medium and offers a wide substrate scope in terms of both the reaction partners. Furthermore, this methodology offers a clean reaction since other than nitrogen gas, the only byproduct is benzotriazole which was isolated and reused for preparing the starting material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LD and RS thank the CSIR, New Delhi and DST, New Delhi, respectively for the Ph. D. and project fellowships. We thank the SAIF division of CSIR-CDRI for the analytical support. CDRI Communication No: 9775.Notes and references
- (a) C. Niemann and J. W. Kessel, J. Org. Chem., 1966, 31, 2265–2269 CrossRef CAS; (b) D. L. Boger, J. A. McKie, T. Nishi and T. Ogiku, J. Am. Chem. Soc., 1997, 119, 311–325 CrossRef CAS; (c) H. Takayama, H. Ishikawa, M. Kurihara, M. Kitajima, N. Aimi, D. Ponglux, F. Koyama, K. Matsumoto, T. Moriyama, L. T. Yamamoto, K. Watanabe, T. Murayama and S. Horie, J. Med. Chem., 2002, 45, 1949–1956 CrossRef CAS; (d) R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127–139 CrossRef CAS PubMed; (e) B.-Q. Tang, W.-J. Wang, X.-J. Huang, G.-Q. Li, L. Wang, R.-W. Jiang, T.-T. Yang, L. Shi, X.-Q. Zhang and W.-C. Ye, J. Nat. Prod., 2014, 77, 1839–1846 CrossRef CAS PubMed.
- (a) K. Higuchi, Y. Sato, M. Tsuchimochi, K. Sugiura, M. Hatori and T. Kawasaki, Org. Lett., 2009, 11, 197–199 CrossRef CAS; (b) A. Karadeolian and M. A. Kerr, J. Org. Chem., 2010, 75, 6830–6841 CrossRef CAS; (c) B. N. Reddy and C. V. Ramana, Chem. Commun., 2013, 49, 9767–9769 RSC; (d) S. Han and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 10768–10771 CrossRef CAS PubMed; (e) X. B. Qi, H. L. Bao and U. K. Tambar, J. Am. Chem. Soc., 2011, 133, 10050–10053 CrossRef CAS PubMed; (f) Y. H. Liu and W. W. McWhorter, J. Am. Chem. Soc., 2003, 125, 4240–4252 CrossRef CAS PubMed; (g) C. Y. Wang, Z. L. Wang, X. N. Xie, X. T. Yao, G. Li and L. S. Zu, Org. Lett., 2017, 19, 1828–1830 CrossRef CAS.
- P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566–4567 CrossRef CAS PubMed.
- A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354–7358 CrossRef CAS PubMed.
- W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648–4652 CrossRef CAS.
- K. Okuma, N. Matsunaga, N. Nagahora, K. Shiojia and Y. Yokomori, Chem. Commun., 2011, 47, 5822–5824 RSC.
- Y. Sun and R. Fan, Chem. Commun., 2010, 46, 6834–6836 RSC.
- M. Shimizu, Y. Takao, H. Katsurayama and I. Mizota, Asian J. Org. Chem., 2013, 2, 130–134 CrossRef CAS.
- Z. Xia, J. Hu, Y.-Q. Gao, Q. Yao and W. Xie, Chem. Commun., 2017, 53, 7485–7488 RSC.
- R. O. Torres-Ochoa, T. Buyck, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 5679–5683 CrossRef CAS PubMed.
- (a) L. Li, M. Han, M. Xiao and Z. Xie, Synlett, 2011, 1727–1730 CAS; (b) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC; (c) M. Rueping, R. Rasappan and S. Raja, Helv. Chim. Acta, 2012, 95, 2296–2303 CrossRef CAS; (d) C.-Y. Jin, Y. Wang, Y.-Z. Liu, C. Shen and P.-F. Xu, J. Org. Chem., 2012, 77, 11307–11312 CrossRef CAS PubMed; (e) A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. G. Ruano and J. Alemán, Chem. Commun., 2012, 48, 9759–9761 RSC; (f) Y.-L. Zhao, Y. Wang, J. Cao, Y.-M. Liang and P.-F. Xu, Org. Lett., 2014, 16, 2438–2441 CrossRef CAS PubMed; (g) T.-G. Chen, P. Fang, X.-L. Hou and L.-X. Dai, Synthesis, 2015, 134–140 CAS; (h) R. Dalpozzo, Adv. Synth. Catal., 2017, 359, 1772–1810 CrossRef CAS.
- (a) Q. Yin and S.-L. You, Chem. Sci., 2011, 2, 1344–1348 RSC; (b) M. Rueping, S. Raja and A. Núñez, Adv. Synth. Catal., 2011, 353, 563–568 CrossRef CAS.
- For selected reviews on X–H insertion reactions, see: (a) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577–6605 CrossRef CAS; (b) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed; (c) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- For recent examples of N–H and O–H Insertion reactions: (a) Z. Wang, X. Bi, Y. Liang, P. Liao and D. Dong, Chem. Commun., 2014, 50, 3976–3978 RSC; (b) X. Xu, C. Li, Z. Tao and Y. Pan, Adv. Synth. Catal., 2015, 357, 3341–3345 CrossRef CAS; (c) W. W. Tan and N. Yoshikai, Chem. Sci., 2015, 6, 6448–6455 RSC; (d) W. Yuan, L. Eriksson and K. J. Szabý, Angew. Chem., Int. Ed., 2016, 55, 8410–8415 CrossRef CAS PubMed; (e) R. P. Pandit, S. H. Kim and Y. R. Lee, Adv. Synth. Catal., 2016, 358, 3586–3599 CrossRef CAS; (f) O. A. Davis, R. A. Croft and J. A. Bull, J. Org. Chem., 2016, 81, 11477–11488 CrossRef CAS PubMed; (g) Z.-S. Chen, X.-Y. Huang, L.-H. Chen, J.-M. Gao and K. Ji, ACS Catal., 2017, 7, 7902–7907 CrossRef CAS; (h) A. C. Hunter, S. C. Schlitzer, J. C. Stevens, B. Almutwalli and I. Sharma, J. Org. Chem., 2018, 83, 2744–2752 CrossRef CAS PubMed.
- P.-X. Zhou, Z.-Z. Zhou, Z.-S. Chen, Y.-Y. Ye, L.-B. Zhao, Y.-F. Yang, X.-F. Xia, J.-Y. Luoa and Y.-M. Liang, Chem. Commun., 2013, 49, 561–563 RSC.
- (a) A. R. Katritzky, A. Pastor, M. Voronkov and D. Tymoshenko, J. Comb. Chem., 2001, 3, 167–170 CrossRef CAS PubMed; (b) A. R. Katritzky and B. V. Rogovoy, Chem. – Eur. J., 2003, 9, 4586–4593 CrossRef CAS; (c) M. Li, B. Zhang and Y. Gu, Green Chem., 2012, 14, 2421–2428 RSC; (d) A. K. Chaturvedi and N. Rastogi, Synthesis, 2015, 47, 249–255 CAS; (e) M. M. D. Pramanik and N. Rastogi, Org. Biomol. Chem., 2016, 14, 1239–1243 RSC; (f) A. K. Chaturvedi and N. Rastogi, J. Org. Chem., 2016, 81, 3303–3312 CrossRef CAS PubMed.
- The use of o-amino benzoylbenzotriazole in the reaction resulted into a complex mixture of products, whereas the desired product formed in traces (6% NMR yield) with N-methyl benzoyl chloride as starting substrate.
- (a) N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, J. Org. Chem., 2006, 71, 5489–5497 CrossRef CAS PubMed; (b) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS PubMed; (c) H. F. Srour, P. L. Maux, S. Chevance, D. Carrié, N. L. Yondre and G. Simonneaux, J. Mol. Catal. A: Chem., 2015, 407, 194–203 CrossRef CAS; (d) K. Ramakrishna and C. Sivasankar, Org. Biomol. Chem., 2017, 15, 2392–2396 RSC.
- (a) D. B. Ramachary, V. V. Narayana and K. Ramakumar, Tetrahedron, 2008, 49, 2704–2709 CrossRef CAS; (b) M. Presset, D. Mailhol, Y. Coquerel and J. Rodriguez, Synthesis, 2011, 2549–2552 CAS; (c) B. J. Deadman, R. M. O'Mahony, D. Lynch, D. C. Crowley, S. G. Collins and A. R. Maguire, Org. Biomol. Chem., 2016, 14, 3423–3431 RSC; (d) G. T. Potter, G. C. Jayson, G. J. Miller and J. M. Gardiner, J. Org. Chem., 2016, 81, 3443–3446 CrossRef CAS PubMed.
- (a) A. Padwa, Helv. Chim. Acta, 2005, 88, 1357–1374 CrossRef CAS; (b) A. Padwa, B. Cheng and Y. Zou, Aust. J. Chem., 2014, 67, 343–353 CrossRef CAS; (c) A. DeAngelis, R. Panish and J. M. Fox, Acc. Chem. Res., 2016, 49, 115–127 CrossRef CAS PubMed.
- X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c8ob02683a |
| This journal is © The Royal Society of Chemistry 2019 |