
Doping-induced enhancement of crystallinity in polymeric carbon nitride nanosheets to improve their visible-light photocatalytic activity†
Yuan-Yuan
Li
 a,
Bing-Xin
Zhou
a,
Hua-Wei
Zhang
a,
Shao-Fang
Ma
a,
Wei-Qing
Huang
*a,
Wei
Peng
a,
Wangyu
Hu
b and
Gui-Fang
Huang
*a
a,
Bing-Xin
Zhou
a,
Hua-Wei
Zhang
a,
Shao-Fang
Ma
a,
Wei-Qing
Huang
*a,
Wei
Peng
a,
Wangyu
Hu
b and
Gui-Fang
Huang
*a
aDepartment of Applied Physics, School of Physics and Electronics, Hunan University, Changsha 410082, China. E-mail: wqhuang@hnu.edu.cn; gfhuang@hnu.edu.cn
bSchool of Materials Science and Engineering, Hunan University, Changsha 410082, China
First published on 14th March 2019
Abstract
Structural defects can greatly inhibit electron transfer in two-dimensional (2D) layered polymeric carbon nitride (CN) unit, seriously lowering its utilization ratio of photogenerated charges during photocatalysis. Herein, we propose a new strategy based on intra-melon hydrogen bonding interactions in 2D CN frameworks to improve the crystallinity of CN. This concept was validated by removing some amino groups and connecting melon using codoped B and F atoms via a simple one-step sodium fluoroborate-assisted thermal treatment. The enhancement in crystallinity effectively promoted exciton dissociation and charge transfer in the CN nanosheets. Furthermore, the B/F dopants also improved the separation of photogenerated carriers by promoting charge capture. The highly efficient visible-light photocatalytic activity of the crystalline B/F-codoped CN nanosheets was demonstrated by degrading methyl orange, Rhodamine B, colorless phenol and tetracycline hydrochloride as models, where their degradation rate constant was more than 10, 5, 32 and 3 times higher than that of pure CN, respectively. Moreover, the B/F-codoped CN exhibited an excellent photoelectrocatalytic performance for the oxygen evolution reaction (OER), outperforming the precious-metal IrO2 catalyst. The simple and effective strategy proposed herein provides a direct route to engineer high crystallinity in 2D materials for tunable charge carrier separation and migration for electronic and optoelectronic applications.
Introduction
Semiconductor photocatalysis is attractive to solve the energy and environmental issues.1–3 Advances in this field largely rely on the development of low-cost, highly efficient and stable semiconductor photocatalysts. Among the various photocatalysts, polymeric carbon nitride (CN), as one of the most potential metal-free semiconductors, has drawn increasing attention because it possesses high thermal and chemical stability and an appealing electronic structure.4–7 Due to these unique properties CN has diverse photocatalytic applications, such as in H2 production,8 water oxidation,9 organic pollutant degradation,10 oxygen evolution reaction (OER),11 CO2 reduction12 and NO oxidation.13 However, pure CN suffers from limited visible-light absorption, high recombination rate of photoexcited charge carriers, low surface area and few active sites, which are the main factors responsible for its low photocatalytic activity.14–17Generally, pure CN routinely fabricated from the solid-state thermal condensation of nitrogen-rich precursors (such as melamine, cyanamide and dicyanamide) is composed of melon, which is a linear polymer of tri-s-triazine units with a certain amount of hydrogen.18–20 The incomplete deamination of the precursors primarily leaves some amino groups, and thus hydrogen bonds in the covalent bonding-dominated intralayer framework.21–23 These hydrogen bonds (as vincula) interconnect the polymeric melon units to maintain intralayer long-range atomic order patterns. However, hydrogen bonds greatly lower the crystallinity of CN, which hampers its performance due to the following reasons: (i) the large potential barrier across the hydrogen bond regions can impede the transport of charge carriers between the strands and (ii) the non-destructive CN nanosheets via hydrogen bond connections significantly decrease the active sites located at the sheet edges. Furthermore, pristine CN is essentially amorphous or semi-crystalline, with a large amount of surface un-condensed amino groups, which can serve as recombination centers for electron–hole pairs.24,25 Recently, improving the crystallinity of CN-based materials has been demonstrated to be an effective strategy to enhance their photocatalytic activity owing to the reduction in structural defects and improved charge transfer.26–28 To date, the approaches used for the synthesis of highly crystalline CN include molten salt routes at high temperature, microwave-alkali treatment, and thermal polymerization with an anodic aluminum oxide (AAO) membrane template.29–31 However, the crystalline CN obtained using these approaches shows inherent limitations, such as insufficient visible-light absorption, sparse active sites and low yield. Thus, CN is only applicable for photocatalysis when its visible light absorption, electronic conductivity, active site density, and crystallinity are synergistically optimized.32,33 Therefore, exploring new strategies to improve the crystallinity of CN to overcome the abovementioned drawbacks and simultaneously achieve a synergistic effect is still highly necessary.
Herein, for the first time, we developed a new strategy to improve the crystallinity of CN based on intra-melon hydrogen bonding interactions in 2D CN frameworks, which was validated by removing some amino groups and connecting melon using B/F dopants via a simple one-step sodium fluoroborate-assisted thermal treatment. Generally, heteroatom doping destroys the intrinsic skeleton of CN and lowers its crystallinity; however, high crystallinity (or low structural defect) is crucial for highly efficient charge transfer to ensure a high photocatalytic performance.34–38 Thus, sodium tetrafluoroborate (NaBF4) is selected as a representative to create B/F co-doped CN with a highly crystalline structure by breaking some hydrogen bonds and connecting the produced dissociated melon by B atoms. The enhancement in crystallinity in CN promoted charge transfer and improved the separation of electron–hole pairs. Furthermore, the introduction of B and F atoms in CN not only favored an enhancement in light scattering and specific surface area, but also increased the density of active sites. The optimized B/F co-doped CN (CN-B/F) nanosheets exhibited enhanced visible-light photocatalytic activity for the degradation of methyl orange (MO), with a rate constant about 10 times higher than that of pure CN. In addition, this material demonstrated superior universality for various pollutants and excellent photoelectrocatalytic performance for the oxygen evolution reaction (OER). The synthetic strategy introduced herein represents a simple and effective way to improve the synergistic optical absorption, active sites, carrier transfer, and photocatalytic properties of CN-based materials.
Results and discussion
The production of highly crystalline CN-B/F involved two steps (Scheme 1). Firstly, pure CN was prepared via the polycondensation of melamine. It was found that the most obvious feature of this pure CN from other layered compounds is the existence of abundant hydrogen bonds and the residual amino groups in its covalent bonding-dominated intralayer framework. Secondly, highly crystalline CN-B/F material with a highly surface positive charge (Fig. S1†) were successfully produced by the thermal polymerization of pure CN and NaBF4, and the subsequent removal of NaBF4 by washing with distilled water. During the thermal polymerization process, the added NaBF4 played a multifold role: the first is that BF4− in the NaBF4 melts reacted with the amine groups; thus, breaking some of the amino groups; the second is that the B atoms could connect the dissociated melon produced by breaking some hydrogen bonds to form extended π-conjugated systems, which resulted in improved crystallinity; and the third is that NaBF4 provided B and F atoms for B/F co-doping in CN.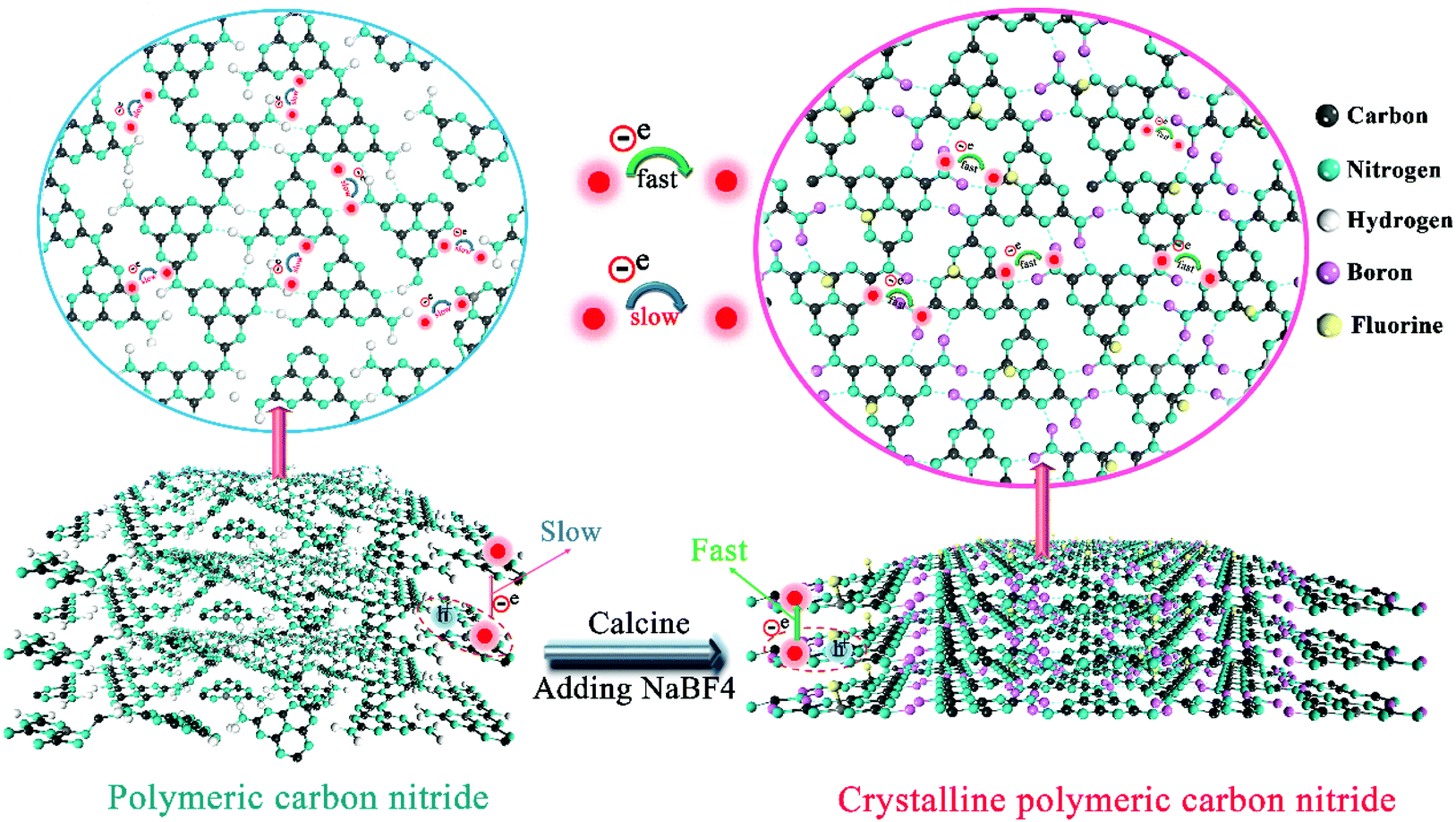 | ||
| Scheme 1 Preparation of highly crystalline B/F codoped polymeric carbon nitride (CN) via one-step fluoroborate-assisted strategy. The crystallinity of CN can be enhanced by removing some amino groups and connecting melon via codoped B and F atoms, which can promote photogenerated charge capture and transport. More importantly, the enhancement of crystallinity promotes efficient exciton dissociation and charge transfer; thus, promoting the charge separation and improving the photocatalytic activity of CN. Polymeric carbon nitride (left) and highly crystalline carbon nitride (right). | ||
To uncover the crystallinity–activity relationship, the crystal structures of the pure CN, CN-hot and a series of CN-B/F samples were characterized via XRD and the results are illustrated in Fig. 1a. All the samples showed two distinct peaks, which were present at different peak locations and had varying intensities. Two identical peaks located at 13.0° and 27.29° corresponding to the (100) interplanar packing of heptazine units and (002) π–π interlayer stacking motif were recorded for pure CN.39 Compared with pure CN, the weakening and widening of both diffraction peaks of CN-hot are related to the destruction of its structure after exfoliation.40 Meanwhile, the (002) diffraction peak of CN-hot slightly shifted toward a lower 2θ value, suggesting a larger stacking distance between nanosheets. In contrast, it was obvious that both peaks compared with pure CN strengthened and narrowed with the use of NaBF4, indicating the well-developed and more condensed crystal structure of the CN-B/F samples. Furthermore, the (002) peak, which is located at 27.29° with an interlayer distance of 0.325 nm in pure CN, slightly shifted to 27.53° for CN-B/F, corresponding to a interlayer spacing of 0.322 nm.41 This position change of the peak indicates a decreasing layer distance, which is most likely due to the enhanced interaction between layers.42 We noted that the (002) peak became wide and imperfect with an increase in the amount of NaBF4 from 0.2 g to 0.3 g in the CN-B/F samples, which may be owing to the excess NaBF4 breaking their crystal plane structure. The evident change in the stacking mode can be ascribed to the well-condensed polymers with a few structure defects. The enhancement in crystallinity in the B/F-CN nanosheets is expected to promote not only efficient exciton dissociation, but also charge-transfer efficiency in B/F-CN.
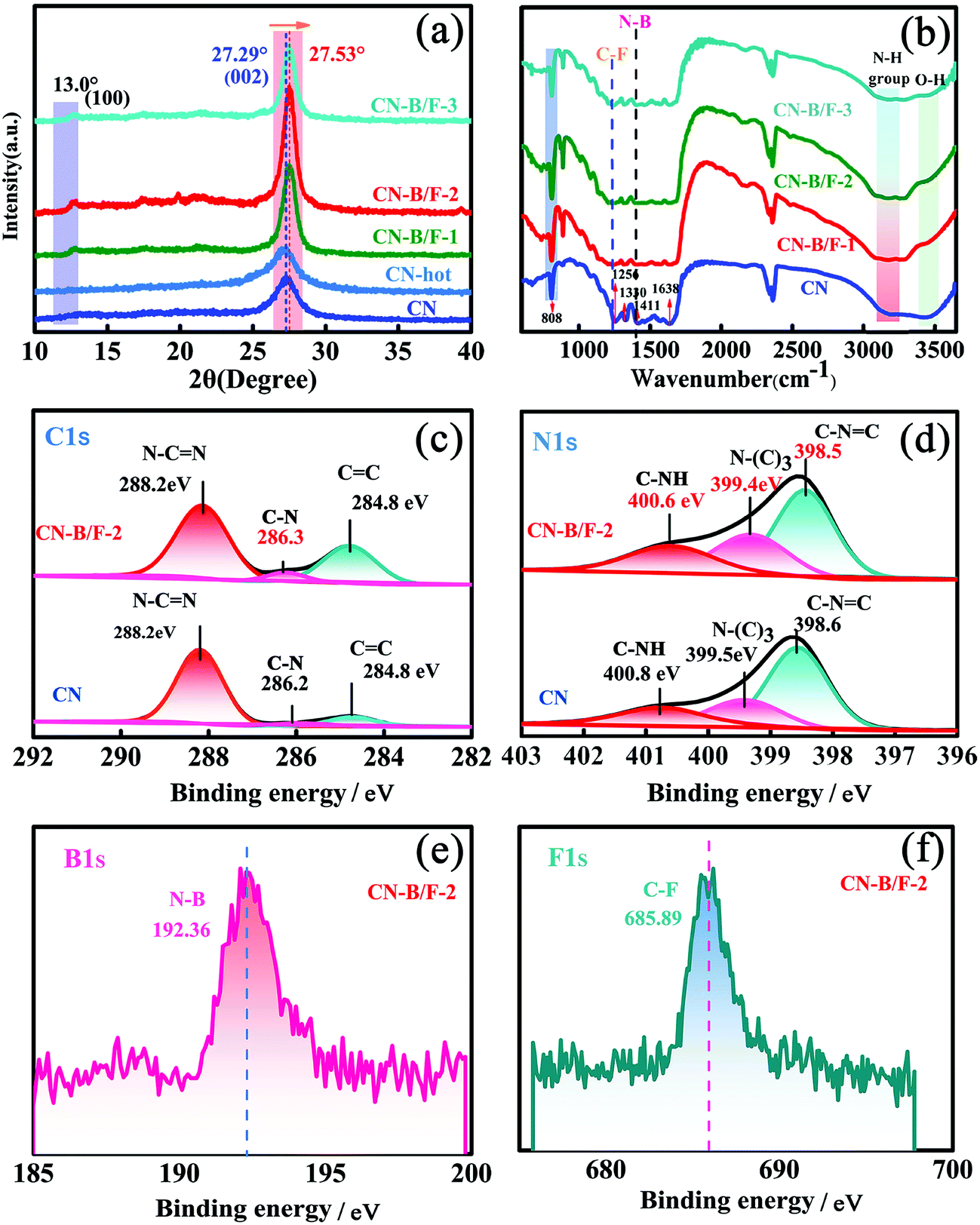 | ||
| Fig. 1 (a) XRD patterns of CN, CN-hot, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (b) FTIR spectra of CN, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (c) C 1s and (d) N 1s XPS spectra of CN and CN-B/F-2. (e) F 1s and (f) B 1s XPS of CN-B/F-2. | ||
The chemical structure of the samples was characterized via FTIR spectroscopy, as displayed in Fig. 1b. The FTIR spectrum of pure CN shows a peak at 802 cm−1, which is typical for the breathing vibration of the triazine units.43 The bands in the region ranging from 1242 to 1638 cm−1 (1242, 1320, 1409, 1560 and 1638 cm−1) are assigned to the C–NH–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration modes of the heterocycles,44,45 and the broad peaks between 3500 and 3000 cm−1 originate from the terminal amino groups (N–H) and hydroxy.46 For the CN-B/F samples, several distinct changes can be observed in their FTIR spectra. The first distinct change observed is that the peak intensity of the breathing vibration of the triazine units at 802 cm−1 increased. The next change is the development of two peaks at 1320 and 1225 cm−1, corresponding to the C–F and N–B bonds, respsectively.47,48 The emergence of the N–B bond reflects that B atoms may enter the C sites in the polymeric CN structures since BN is also an extremely stable covalent material.49 The other change is a decrease in the intensity of the N–H stretching peaks at 3000–3300 cm−1. These results suggest that the addition of NaBF4 during the synthesis of B/F-CN decreased the lattice defects, the concentration of N–H groups and introduced B and F atoms. It should be noted that NaBF4 decomposes at 380 °C, and thus B atoms may replace the H atoms of the amine groups during the thermal-polymerization process; thus, decreasing the lattice defects and generating high crystallinity. These results are consistent with the XRD measurements (Fig. 1a).
N stretching vibration modes of the heterocycles,44,45 and the broad peaks between 3500 and 3000 cm−1 originate from the terminal amino groups (N–H) and hydroxy.46 For the CN-B/F samples, several distinct changes can be observed in their FTIR spectra. The first distinct change observed is that the peak intensity of the breathing vibration of the triazine units at 802 cm−1 increased. The next change is the development of two peaks at 1320 and 1225 cm−1, corresponding to the C–F and N–B bonds, respsectively.47,48 The emergence of the N–B bond reflects that B atoms may enter the C sites in the polymeric CN structures since BN is also an extremely stable covalent material.49 The other change is a decrease in the intensity of the N–H stretching peaks at 3000–3300 cm−1. These results suggest that the addition of NaBF4 during the synthesis of B/F-CN decreased the lattice defects, the concentration of N–H groups and introduced B and F atoms. It should be noted that NaBF4 decomposes at 380 °C, and thus B atoms may replace the H atoms of the amine groups during the thermal-polymerization process; thus, decreasing the lattice defects and generating high crystallinity. These results are consistent with the XRD measurements (Fig. 1a).
To further investigate the effects of B, F codoping on the surface elemental composition of the CN-B/F sample, energy dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) measurements were performed. The C, N, B, F and N/C atomic ratios for pure CN and CN-B/F-2 determined from the two methods are listed in Tables S1–3.† The N/C atomic ratios for pure CN obtained from EDS and XPS are 1.29 and 1.48, respectively, which are close to the theoretical value for CN.50 It should be noted that the N/C atomic ratio in CN-B/F-2 decreased compared with pristine CN, as illustrated in Tables S1–3.† The decrease in the nitrogen content in CN-B/F-2 may be attributed to the preferential oxidation of nitrogen atoms during the calcination process, which is similar to that in a previous report.42 The atomic concentration of the doped B and F was determined to be 5.31% and 0.62% and 1.92% and 0.86% from EDS and XPS, respectively. The XPS survey in Fig. S2† display the elements of C, N, B and F. The C1s XPS spectra for pure CN contain three components located at 288.2, 286.2, and 284.8 eV, corresponding to sp2-hybridized carbon (N–CN), C–N and CC, respectively (Fig. 1c).51,52 The N 1s spectra of pure CN (Fig. 1d) were deconvoluted into three peaks at 400.8, 399.5, and 398.6 eV, which can be assigned to the tertiary nitrogen (N–(C)3) groups, positive charge localization in the heterocycles (C–NH) and sp2-hybridized nitrogen in the triazine rings (C–NC),53 respectively. Notably, there was an obvious shift in the N 1s and C 1s peaks for B/F-CN-2 compared to pure CN, suggesting that the electron structure of CN-B/F-2 changed. The B1S and F1S XPS spectra contained peaks located 192.36 and 685.89 eV (Fig. 1e and f), respectively, which are typical for N–B47 or C–F48 coordination. Based on these results together with the XRD and FTIR results (Fig. 1a and b, respectively), we can conclude that the enhancement in the crystallinity of CN is caused by the replacement of a portion of amine groups from the melon strands by B atoms during thermal treatment.
The morphologies of CN, CN-hot and CN-B/F were investigated via SEM analysis, as shown in Fig. 2a–f. Compared to the pure CN nanosheets obtained by the straightforward calcination of urea, the B/F-CN nanosheets progressively became loose and soft with an increase in NaBF4, which is attributed to the partial thermolysis of CN during the thermal treatment of pure CN and NaBF4. The loose nanosheets can provide a high specific surface area and a large number of edges as reaction active sites. The SEM-EDS elemental mapping over CN-B/F-2 and CN is presented in Fig. 2g and Fig. S3,† respectively. It was found that all the elements (C, N, B, and F) of CN-B/F-2 were uniformly dispersed. N2 physisorption measurements at 77 K were used to examine the effect of B, F codoping on the specific surface area and pore structure of the samples, as revealed by the nitrogen sorption isotherms (Fig. 2h) and derived pore size distributions (Fig. 2i). After the B and F modification, we noted that the Brunauer–Emmett–Teller (BET) surface area and the pore volume slightly increased, indicating that there was a high specific surface area exposed and abundant pores with a large number of the edges, which are beneficial for improving the photocatalytic activity.
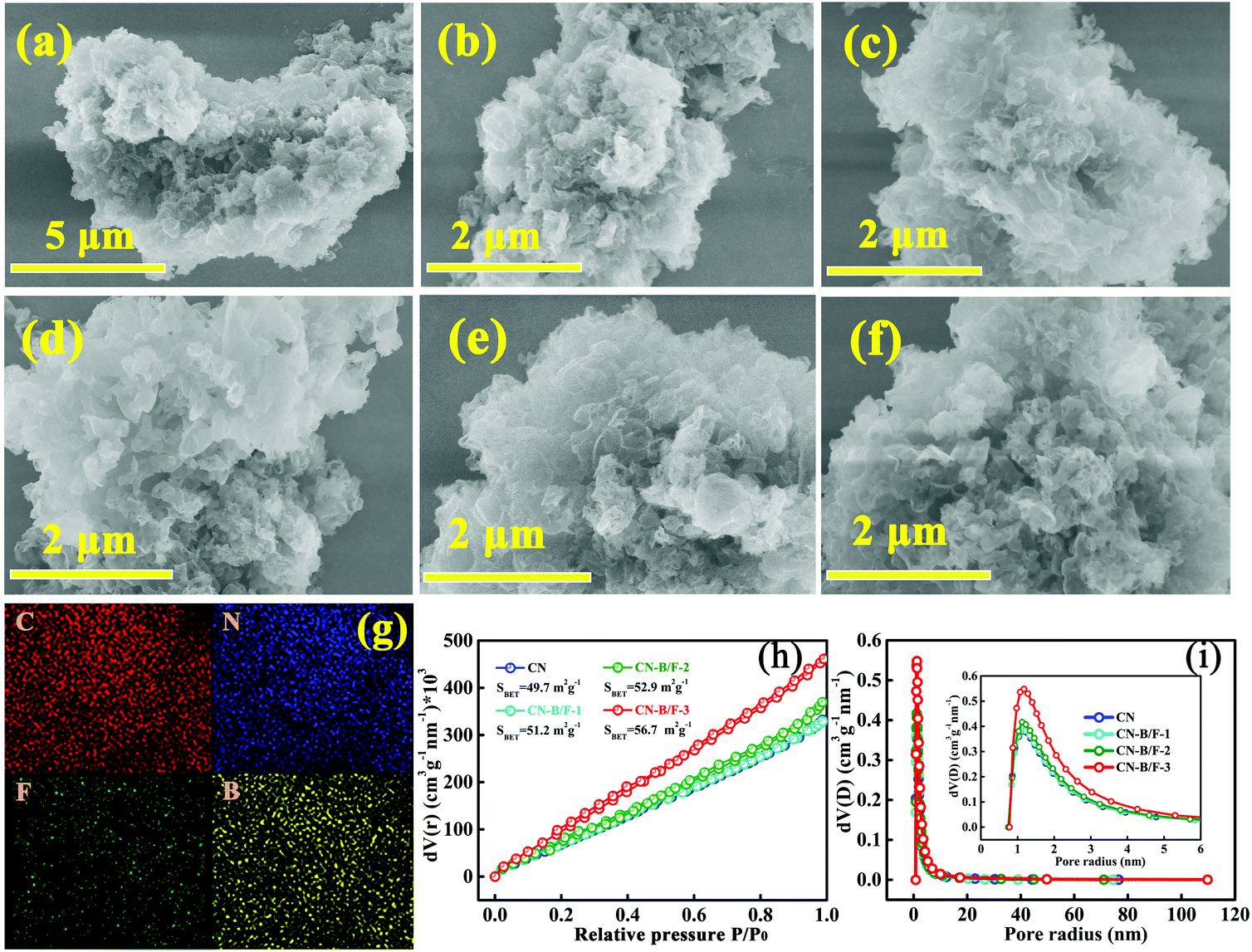 | ||
| Fig. 2 SEM images of (a and b) CN and (c–f) CN-hot, CN-B/F-1, CN-B/F-2, and CN-B/F-3, respectively. (g) SEM-EDS elemental mapping of CN-B/F-2. (h) N2 adsorption–desorption isotherms and (i) pore-size distribution curves of CN, CN-B/F-1, CN-B/F-2 and CN-B/F-3. | ||
The B and F introduced into the CN nanosheets altered the optical properties and light harvesting ability of the samples. It is clear that as the content of NaBF4 increased, the CN-B/F samples progressively became pale yellow compared with the milky CN. Fig. 3a shows the UV-vis absorption spectra and calculated bandgaps for all the samples. The absorption spectrum of pure CN consists of a strong intrinsic absorption band showing a steep edge with the extrapolated onset of 435 nm and a weak absorption band starting from the onset to the end at around 700 nm. It was found that a progressive redshift in the absorption edge for CN-B/F was achieved with an increase in NaBF4, which is ascribed to the incorporation of B and F element into the CN structure. The corresponding band gap energy decreased from 2.85 eV (pure CN) to 2.79 eV (B/F-CN-0.3). The conduction band (CB) and valence band (VB) positions are normally closely related to the reduction and oxidation capacity of semiconductors.54 Therefore, the flat potential of the CN-B/F samples was estimated using the traditional Mott-Schottky methods (Fig. 3b and c), where an estimated flat band potential was directly used as the CB potential.55 It was observed that the CB of CN-B/F-0.2 is −1.28 V vs. Ag/AgCl (Ph = 7), which is slightly positive compared with that of the pure CN (−1.54 V vs. Ag/AgCl). The relative CB and VB of the polymers (vs. RHE) are presented in Fig. 3d and Table S4.† It was noted that the CB slightly decreased to −0.63 V compared with pure CN (−0.89 V), which is still negative enough for proton reduction. However, the VB of CN-B/F-0.2 is 0.22 V more positive than that of pure CN. This means that CN-B/F-0.2 possess stronger oxidizing ability than pure CN.56
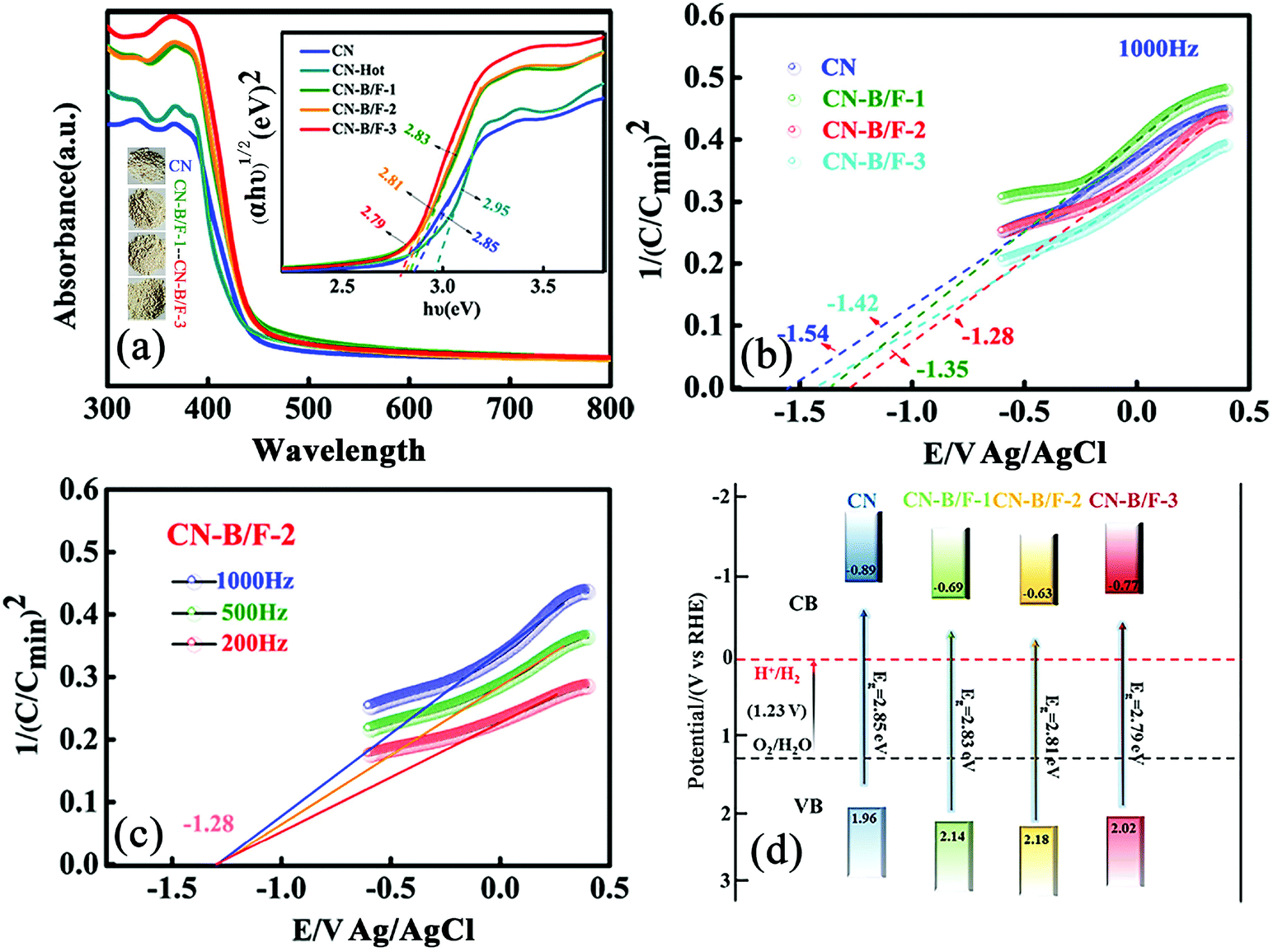 | ||
| Fig. 3 (a) UV-vis absorption spectra and band gaps of CN, CN-hot, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (b) Mott–Schottky plots of CN, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (c) Mott–Schottky plots of CN-B/F-2 with different measurement frequencies. (d) Schematic band structures of CN, CN-B/F-1, CN-B/F-2 and CN-B/F-3. | ||
The effective transfer of photo-generated charge carriers sensitively depends on the structural defects in semiconductor photocatalysts. The enhanced crystallization of the CN-B/F samples can effectively suppress electron–hole recombination, as revealed by their weakened PL peak (Fig. 4a). The PL emission intensity at 350 nm excitation for pure CN showed an intense fluorescence signal. Compared with pure CN, the PL signal of CN-hot displayed a blue shift, which can be attributed the quantum confinement effect associated with the decreased size of CN.57 Whereas, the PL of CN-B/F exhibited a slight red shift compared to pure CN, corresponding to the UV-vis absorption results, owing to the incorporation of B and F elements into the CN structure.58 The PL emission intensity of CN-B/F dramatically decreased compared to that of pure CN. Most likely, the high crystallization of CN facilitated charge carrier migration from the bulk to the interface without recombination. Furthermore, the shortened layer distance in the highly crystalline CN accelerated the charge transfer over the plane and layers and the singlet excitons to dissociate.28 The improved charge transport was classically certified by the increased photocurrent and decreased hemicycle radius from electrochemical impedance spectroscopy (EIS). A low photocurrent response was observed for the pure CN electrode under visible light irradiation (Fig. 4b). In contrast, the enhanced photocurrent response of the CN-B/F samples reflects their improved charge separation.59 As shown in Fig. 4c, it can be found that the CN-B/F samples showed a smaller semicircle in the intermediate-frequency compared to that of pure CN, suggesting that the highly crystalline structure can suppress radiative electron–hole recombination by promoting the transfer of electrons.
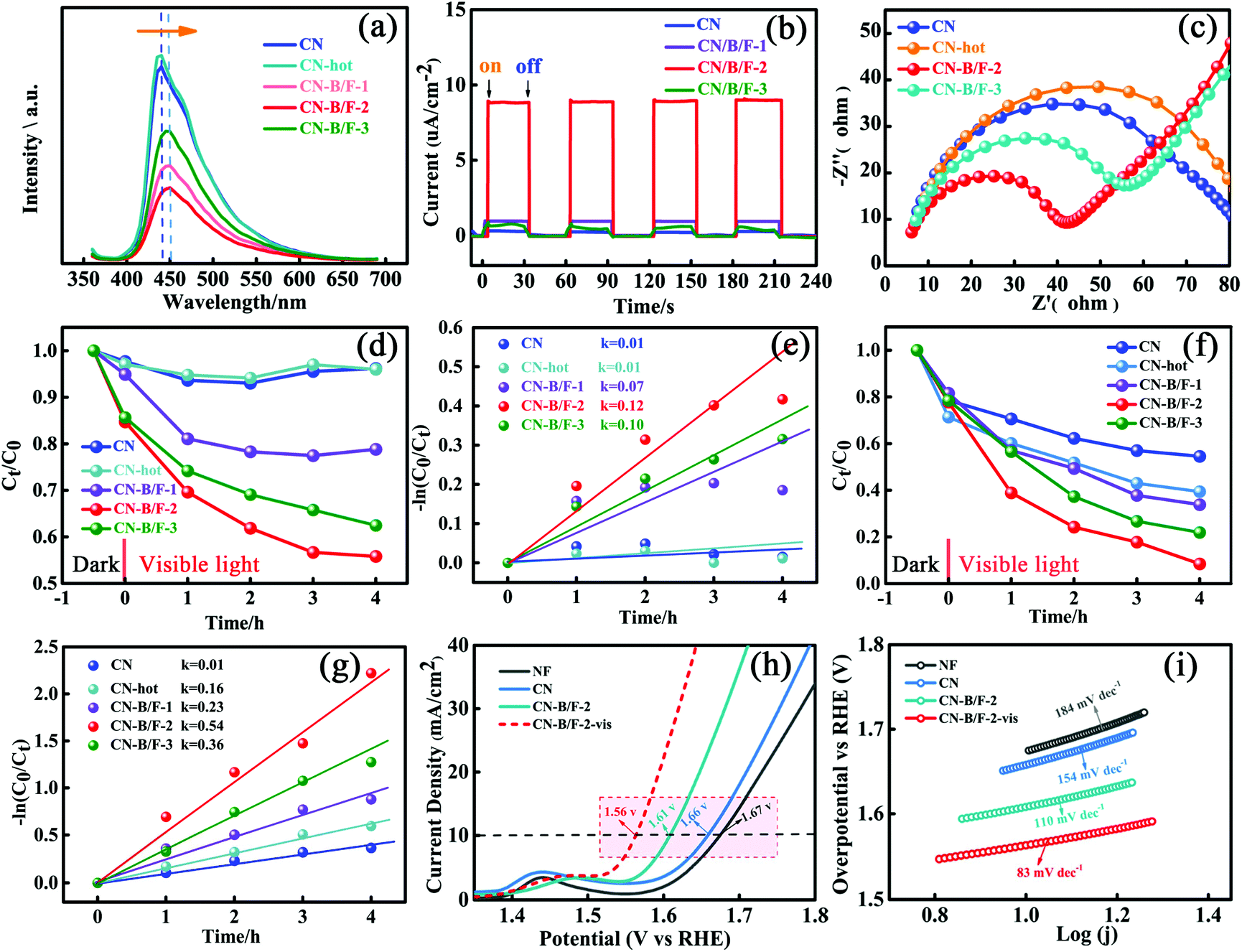 | ||
| Fig. 4 (a) PL spectra of CN, CN-hot, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (b) Transient photocurrent response and (c) EIS Nyquist plots of CN, CN-B/F-1, CN-B/F-2 and CN-B/F-3. (d) MO (10 mg L−1) degradation and (e) slopes for the degradation with (10 mg) CN, CN-hot, CN-B/F-1, CN-B/F-2 and CN-B/F-3 under visible light. (f) RhB (10 mg L−1) degradation and (g) slope for degradation with (10 mg) CN, CN-hot, CN-B/F-1, CN-B/F-2 and CN-B/F-3 under visible light. (h) Linear sweep voltammetry curves and (i) Tafel plots of NF, CN, CN-B/F-2 and CN-B/F-2-vis. | ||
As is known, MO and RhB are typical dyes, which are widely used in various fields and representative organic pollutants in wastewater; thus, they were chosen as model pollutants. The photocatalytic degradation experiments were conducted under visible light irradiation, in which MO with a negative surface potential was selected as the first model pollutant. An instantaneous variation in MO concentration versus time was observed in the presence of the different samples in the dark and under visible light irradiation (Fig. 4d). All the photocatalysts showed adsorption and photocatalytic activities towards MO. It was found that the adsorption of MO (10 mg L−1, 80 mL) for 10 mg CN-hot sample was almost same for pure CN after reaching adsorption–desorption equilibrium in the dark. Notably, the CN-B/F samples showed a higher adsorption capacity compared to pure CN (2.3%) and CN-hot (2.9%) owing to their microcosmic structure consisting of shaggy nanosheets and more positive surface potential (Fig. S1†).60 Moreover, the CN-B/F samples exhibited superior photocatalytic activity. Under visible light irradiation, it was observed that the degradation efficiency of MO initially increased, then decreased with an increase in NaBF4. The best degradation efficiency was obtained with CN-B/F-2, as shown in Fig. 4d. After 4 h of visible light irradiation, 21.2%, 44.2%, 37.5%, 4.0% and 3.8% of MO was degraded using CN-B/F-1, CN-B/F-2, CN-B/F-3, CN-hot and pure CN, respectively. It is worth noting that the surface area and number of pores in the samples increased with the doping content and CN-B/F-3 displayed the largest surface area and most pores. Meanwhile, CN-B/F-2 exhibited the highest photocatalytic activity since the efficiency of photocatalysts depends on the synergistic effects of several factors such as light absorption, active sites exposed and the separation of photoinduced electron–hole pairs. The superior photocatalytic activity of CN-B/F-2 can be attributed to the high crystallinity of CN-B/F, which promotes the transfer and separation of photoinduced electron–hole charges.
Fig. 4e shows the linear relationship of ln(Ct/C0) versus time. The rate constant, k, value was calculated using the first-order model, as expressed by the following formula
| ln(C0/Ct) = kt | (1) |
To further verify the photocatalytic degradation activity of the samples, RhB, which is an organic pollutant with no surface charge, was also selected as a model contaminant. Fig. 4f shows that the adsorption capacity of RhB over the CN-B/F samples was slightly lower than that over CN-hot. This may be ascribed to the more positive surface potential of CN-B/F, which is disadvantageous for the adsorption of RhB owing to the coulombic repulsion interaction between CN-B/F and the N(C2H5)2+ cation in RhB solution. RhB could be further decomposed under light irradiation. In the case of pure CN, about 45.5% RhB was removed after visible light irradiation for 4 h. However, the photoactivity of CN-B/F-2 was remarkably enhanced, and RhB was completely disintegrated after 4 h irradiation. Furthermore, it was found that the degradation rate for CN-B/F-2 (0.54 h−1) was more than 5 times that of CN (0.10 h−1) in Fig. 4g, which is better than that of most of the reported CN-based catalysts (Table S5†). Besides the chosen colored organic pollutants as model contaminants, colorless phenol and tetracycline hydrochloride (TC) were also selected as model pollutants for excluding the occurrence of sensitization (Fig. S4†). Obviously, CN-B/F-2 exhibited much higher photocatalytic activity compared to pure CN. Furthermore, the reaction rate for the degradation of TC by CN-B/F-2 was about 4-fold higher than that of pure CN.
Stability and reusability are also significant issues for the practical application of photocatalysts besides enhanced photocatalytic activity. Therefore, five consecutive cycles of photo-degradation for MO over CN-B/F-2 under visible light were carried out by collecting and re-using the samples. After each adsorption and photo-degradation experiment, the CN-B/F-2 sample was centrifuged and rinsed with DI water several times. Fig. 5a shows the adsorption and degradation curves of MO in five consecutive applications with CN-B/F-2 nanosheets. No apparent decrease in the photocatalytic activity of CN-B/F-2 was observed after five repetitive runs, ensuring that the synthesized CN-B/F-2 sample exhibited robust stability for the photocatalytic degradation of organic contaminants.
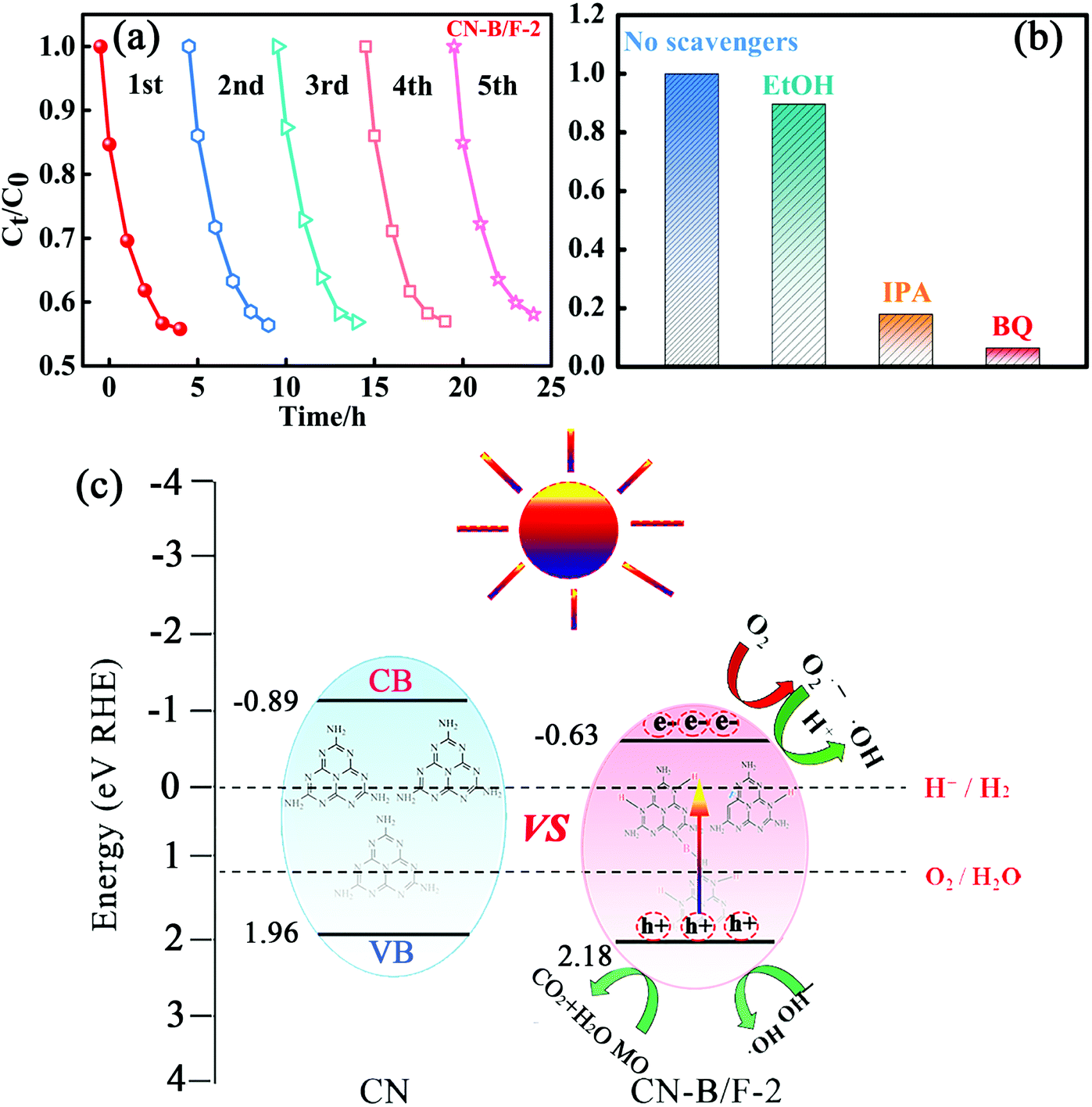 | ||
| Fig. 5 (a) Cycling runs for the photocatalytic degradation of MO in the presence of CN-B/F-2 under visible light irradiation. (b) Photocatalytic performance of CN-B/F-2 in the presence of various quenchers. (c) Schematic illustration of the band structures and the photogenerated charge transfer process of CN-B/F-2. | ||
To further elucidate the effect of the optimization on catalysis in general, the OER activity of CN-B/F-2 and pure CN were studied via (photo)-electrochemical measurements conducted in a typical three-electrode cell in 1 M KOH at a scan rate of 20 mV s−1 (Fig. 4h). The overpotential at the current density of 10 mA cm−2 is generally used to evaluate the performance of electrocatalysts.61 As expected, CN-B/F-2 afforded this current density at a small overpotential (η) of 380 mV, which was lower than that for bulk CN of 430 mV. Interestingly, under visible-light irradiation, the η for CN-B/F-2 was remarkably reduced to 330 mV, reflecting its effective photo-responsive OER behavior. The high OER activity of CN-B/F-2 is superior to that of most transition metal alloys, including IrO2 (470 mV).62 As shown in Fig. 4i, the Tafel slope of CN-B/F-2 is smaller than that of pure CN. Moreover, the Tafel slope further decreased under light irradiation. CN-B/F-2-vis exhibited a low Tafel slope of 83.0 mV dec−1. These results suggest that the CN-B/F samples have greatly enhanced photocatalytic performance for the degradation of organic pollutants and OER activity under visible light irradiation.
Generally, the photogenerated holes and electrons originating from the CB and VB and the subsequently produced ˙O2− and ˙OH radicals are potential reactive species for the photocatalytic oxidation of organic molecules.61 The trapping experiments were applied to identify the main active species during the photocatalytic process in the presence of the CN-B/F-2 sample, as shown in Fig. 5b. Only a slight decrease in photodegradation activity of MO for CN-B/F-2 was observed with the addition of 0.05 mM C2H6O as a quencher of holes, suggesting that hole radicals play a minor role in the degradation of MO under visible-light irradiation.63 In contrast, the photocatalytic activity of CN and PCNx-0.5 was largely suppressed by the addition of BQ and IPA, indicating that the ˙O2− and ˙OH radicals play a vital role in the photocatalytic process and serve as the principle oxidative species.
A tentative mechanism was proposed for the high photocatalytic activity of the highly crystalline CN-B/F-2 under light irradiation, as illustrated in Fig. 5c. The efficiency of photocatalysts largely depends on the synergistic effects of three factors as follows: (i) light absorption, (ii) number of active sites, and (iii) separation of photoinduced electron–hole pairs. Based on the experimental results given above, the photocatalytic activity for organic pollutant degradation and OER activity over CN-B/F was greatly enhanced, which can be ascribed to several factors. The introduction of B and F atoms can improve not only light harvesting owing to a decrease in the band gap energy, but also the electric conductivity. The fluffy nanosheets of CN-B/F provide abundant active sites. In addition, the high positive surface potential of CN-B/F has superior adsorption ability for negatively charged pollutants. Especially, the high crystallinity of CN-B/F promotes the transfer and separation of photoinduced electron–hole charges, as evidenced by the optical measurements demonstrated above. Under visible light irradiation, the photoinduced electrons on the VB are excited and transferred to the CB; thus, leaving positive holes on the VB. In contrast, the high recombination of electron–hole pairs in the pure CN results in lower photoexcited charge transport to the surface of the catalysts. In the presence of the highly crystalline CN-B/F-2, the electrons quickly transfer to the nanosheet surface and promote the separation of electrons and holes, thereby improving the photocatalytic activity. The photoinduced electrons on the CB of B/F-CN-2 will reduce the adsorbed O2 on its surface to superoxide radicals (˙O2−), which can further oxidize water to produce hydroxide radicals (˙OH).64 Therefore, it is understandable that ˙OH and ˙O2− radicals are crucial oxidizing species responsible for the degradation of MO, as confirmed by the above active species trapping experiments (Fig. 5a). Therefore, the significantly improved photocatalytic activity of the highly crystalline CN-B/F-2 can be attributed to the synergistic effects of its electronic structure and morphology.
Conclusions
In conclusion, we prepared highly crystalline CN-B/F based on the new strategy of engineering intra-melon hydrogen bonding interactions in 2D CN frameworks. The well-developed, highly crystalline CN-B/F was synthesized via a thermal polymerization process in the presence of thermally polymerized CN and NaBF4. The highly crystalline structure of the obtained CN-B/F is mainly caused by the partial breaking of the amino groups and connecting of the produced dissociated melon by B and F atoms. The highly crystalline CN-B/F displayed improved exciton dissociation and charge transport ability. Moreover, the introduction of B and F atoms into CN-B/F increased visible light harvesting and suppressed charge recombination by facilitating photoelectron capture; thus, improving the photocatalytic activity of CN. Compared with previous routes for the preparation of highly crystalline CN structures, our one-step method is more facile, green and effective, and it also can be extended to prepare other 2D materials. Thus, this work provides a new design for highly crystalline 2D materials and may advance the utilization of functionalized 2D materials in the vast fields associated with catalysis, energy and environmental science.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (no. 51772085 and U1830138).References
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Guo, J. Li, Y. Yuan, L. Li, M. Zhang, C. Zhou and Z. Lin, Angew. Chem., 2016, 128, 14913–14917 CrossRef.
- F.-X. Xiao and B. Liu, Adv. Mater. Interfaces, 2018, 5, 1701089 Search PubMed.
- G. G. Liu, T. Wang, H. B. Zhang, X. G. Meng, D. Hao, K. Zhang, P. Li, T. Kako and J. H. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561–13565 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 47, 1917–1933 CrossRef PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. Wu, H. Wang, W. Tu, Y. Liu, S. Wu, Y. Z. Tan and J. W. Chew, Appl. Catal., B, 2018, 233, 58–69 CrossRef CAS.
- Y. Wu, H. Wang, W. Tu, S. Wu, Y. Liu, Y. Z. Tan, H. Luo, X. Yuan and J. W. Chew, Appl. Catal. B, 2018, 229, 181–191 CrossRef CAS.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443–446 RSC.
- L. Ge, Z. Peng, W. Wang, F. Tan, X. Wang, B. Su, X. Qiao and P. K. Wong, J. Mater. Chem. A, 2018, 6, 16421–16429 RSC.
- X.-H. Li, X. Wang and M. Antonietti, ACS Catal., 2012, 2, 2082–2086 CrossRef CAS.
- H. Zhou, P. Li, J. Liu, Z. Chen, L. Liu, D. Dontsova, R. Yan, T. Fan, D. Zhang and J. Ye, Nano Energy, 2016, 25, 128–135 CrossRef CAS.
- S. Wan, M. Ou, Q. Zhong, S. Zhang and W. Cai, Adv. Opt. Mater., 2017, 5, 1700536 CrossRef.
- S. Yu, J. Li, Y. Zhang, M. Li, F. Dong, T. Zhang and H. Huang, Nano Energy, 2018, 50, 383–392 CrossRef CAS.
- S. Pal, S. Bayan and S. K. Ray, Nanoscale, 2018, 10, 19203–19211 RSC.
- J. Li, Z. Zhang, W. Cui, H. Wang, W. Cen, G. Johnson, G. Jiang, S. Zhang and F. Dong, ACS Catal., 2018, 8, 8376–8385 CrossRef CAS.
- Y. Wu, H. Wang, Y. Sun, T. Xiao, W. Tu, X. Yuan, G. Zeng, S. Li and J. W. Chew, Appl. Catal., B, 2018, 227, 530–540 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Yuan, L. Zhang, J. Xing, M. I. B. Utama, X. Lu, K. Du, Y. Li, X. Hu, S. Wang, A. Genc, R. Dunin-Borkowski, J. Arbiol and Q. Xiong, Nanoscale, 2015, 7, 12343–12350 RSC.
- H. Wang, Y. Wu, M. Feng, W. Tu, T. Xiao, T. Xiong, H. Ang, X. Yuan and J. W. Chew, Water Res., 2018, 144, 215–225 CrossRef CAS PubMed.
- Y. Zhao, Y. Wang, X. Liu, J. Liu, B. Han, X. Hu, F. Yang, Z. Xu, Y. Li and S. Jia, Angew. Chem., Int. Ed., 2018, 57, 5765–5771 CrossRef PubMed.
- L. Jing, R. Zhu, D. L. Phillips and J. C. Yu, Adv. Funct. Mater., 2017, 27, 1703484 CrossRef.
- Y. Wu, H. Wang, W. Tu, Y. Liu, Y. Z. Tan, X. Yuan and J. W. Chew, J. Hazard. Mater., 2018, 347, 412–422 CrossRef CAS PubMed.
- U. Diebold, Nat. Chem., 2011, 3, 271–272 CrossRef CAS PubMed.
- K. Maeda, Adv. Mater., 2014, 26, 4920–4935 CrossRef PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. R. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53, 11001–11005 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- Y. Guo, J. Li, Y. Yuan, L. Li, M. Zhang, C. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 14693–14697 CrossRef CAS PubMed.
- X.-H. Li, J. Zhang, X. Chen, A. Fischer, A. Thomas, M. Antonietti and X. Wang, Chem. Mater., 2011, 23, 4344–4348 CrossRef CAS.
- M. J. Bojdys, J.-O. Mueller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- C. G. Gomez, A. M. Silva, M. C. Strumia, L. B. Avalle and M. I. Rojas, Nanoscale, 2017, 9, 11170–11179 RSC.
- F. Zeng, W.-Q. Huang, J.-H. Xiao, Y.-Y. Li, W. Peng, W. Y. Hu, K. Li and G.-F. Huang, J. Phys. D: Appl. Phys., 2019, 52, 025501 CrossRef.
- Q. Fan, J. Liu, Y. Yu, S. Zuo and B. Li, Appl. Surf. Sci., 2017, 391, 360–368 CrossRef CAS.
- D. Gao, Y. Liu, M. Song, S. Shi, M. Si and D. Xue, J. Mater. Chem. C, 2015, 3, 12230–12235 RSC.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian, Q. Pan and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1830–1834 CrossRef CAS PubMed.
- D. Zheng, X.-N. Cao and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS PubMed.
- S. Patnaik, G. Swain and K. M. Parida, Nanoscale, 2018, 10, 5950–5964 RSC.
- Y. Li, R. Jin, Y. Xing, J. Li, S. Song, X. Liu, M. Li and R. Jin, Adv. Energy Mater., 2016, 6, 1601273 CrossRef.
- L.-R. Zou, G.-F. Huang, D.-F. Li, J.-H. Liu, A.-L. Pan and W.-Q. Huang, RSC Adv., 2016, 6, 86688–86694 RSC.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- M. Wu, J. Zhang, B.-B. He, H.-W. Wang, R. Wang and Y.-S. Gong, Appl. Catal., B, 2019, 241, 159–166 CrossRef CAS.
- D. Jing, Q. Liu, Z. Zhang, L. Xin, J. Zhao, S. Cheng, B. Zong and W. L. Dai, Appl. Catal., B, 2015, 165, 511–518 CrossRef.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6, 3921–3931 CrossRef CAS.
- F. He, G. Chen, J. Miao, Z. Wang, D. Su, S. Liu, W. Cai, L. Zhang, S. Hao and B. Liu, ACS Energy Lett., 2016, 1, 969–975 CrossRef CAS.
- Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- Z. Lin and X. Wang, ChemSusChem, 2014, 7, 1547–1550 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- Q. Han, Z. Cheng, B. Wang, H. Zhang and L. Qu, ACS Nano, 2018, 12, 5221–5227 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Z. Zeng, F.-X. Xiao, H. Phan, S. Chen, Z. Yu, R. Wang, N. Thuc-Quyen and T. T. Y. Tan, J. Mater. Chem. A, 2018, 6, 1700–1713 RSC.
- J. Zhang and F.-X. Xiao, J. Mater. Chem. A, 2017, 5, 23681–23693 RSC.
- Z. Zeng, Y.-B. Li, S. Chen, P. Chen and F.-X. Xiao, J. Mater. Chem. A, 2018, 6, 11154–11162 RSC.
- W. Xing, C. Li, G. Chen, Z. Han, Y. Zhou, Y. Hu and Q. Meng, Appl. Catal., B, 2017, 203, 65–71 CrossRef CAS.
- C. Ye, J.-X. Li, Z.-J. Li, X.-B. Li, X.-B. Fan, L.-P. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, ACS Catal., 2015, 5, 6973–6979 CrossRef CAS.
- J. Ji, J. Wen, Y. Shen, Y. Lv, Y. Chen, S. Liu, H. Ma and Y. Zhang, J. Am. Chem. Soc., 2017, 139, 11698–11701 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- F.-X. Xiao and B. Liu, Nanoscale, 2017, 9, 17118–17132 RSC.
- B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188–195 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- M. Tariq, W. Q. Zaman, W. Sun, Z. Zhou, Y. Wu, L.-m. Cao and J. Yang, ACS Sustainable Chem. Eng., 2018, 6, 4854–4862 CrossRef CAS.
- Q. Qiao, K. Yang, L.-L. Ma, W.-Q. Huang, B.-X. Zhou, A. L. Pan, W. Y. Hu, X. Fan and G.-F. Huang, J. Phys. D: Appl. Phys., 2018, 51, 275302 CrossRef.
- Y.-Y. Li, S.-F. Ma, B.-X. Zhou, W.-Q. Huang, X. Fan, X. Li, K. Li and G.-F. Huang, J. Phys. D: Appl. Phys., 2019, 52, 105502 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr00229d |
| This journal is © The Royal Society of Chemistry 2019 |