
Hydrogels based on pH-responsive reversible carbon–nitrogen double-bond linkages for biomedical applications
Zhen
Zhang
ab,
Chaoliang
He
 *ab and
Xuesi
Chen
*ab
*ab and
Xuesi
Chen
*ab
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: clhe@ciac.ac.cn; xschen@ciac.ac.cn
bUniversity of Science and Technology of China, Hefei 230026, China
First published on 1st September 2018
Abstract
Covalently-crosslinked hydrogels are formed due to the generation of new covalent crosslinking bonds in situ, leading to the formation of covalently-linked networks. However, traditional polymerization might introduce potential toxicity issues. Recently, carbonyl-condensation reactions that form imine, hydrazone or oxime bonds have become important tools for the preparation of covalently-crosslinked hydrogels for biomedical applications. The formation of imine, hydrazone or oxime linkages occurs under mild conditions with water as the only byproduct. The reactions are reversible and pH-responsive, and are dependent on the chemical structure of the substrates. In this review, we focus on the utilization of the condensation reactions between nucleophiles and carbonyl groups as crosslinking strategies to prepare hydrogels with physiochemical properties sensitive to biologically relevant stimuli. A brief account of the formation mechanisms and stabilities of these linker moieties will help to understand how reactant structure controls the gelation and degradation of hydrogels. Natural and synthetic polymers that are commonly employed as building blocks and synthetic methods that are selected for decoration of different polymers with respective functional groups will be summarized. Then, the preparation and biomedical applications of the hydrogels based on imine/hydrazone/oxime formation will be presented respectively, with special focus on the developments in the past decade. This review will provide insight into the design of novel biocompatible, stimuli-responsive hydrogels based on dynamic linkages for various biomedical applications.
 Zhen Zhang | Zhen Zhang received his BS degree from the University of Science and Technology of China in 2014. He is now studying for his PhD degree under the supervision of Prof. Xuesi Chen in Key Laboratory of Polymer Ecomaterials (KLPE), Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences (CAS). His research interests are focused on the design and preparation of injectable hydrogels based on poly(amino acid)s and their applications in drug delivery and tissue engineering. |
 Chaoliang He | Chaoliang He received his PhD at Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences in 2007. After completing his post-doctoral research at Sungkyunkwan University in South Korea and University of Ottawa in Canada successively, he joined Changchun Institute of Applied Chemistry as an associate professor in 2010. He has been a full professor at CIAC since 2016. His research interests focus on the development of injectable polymeric hydrogels, and their applications in local drug delivery and regenerative medicine. |
 Xuesi Chen | Xuesi Chen received his PhD at Waseda University, Japan, in 1997. After completing his post-doctoral research at the University of Pennsylvania in USA, he was appointed as a full professor at Changchun Institute of Applied Chemistry, Chinese Academy of Sciences in 1999. His research interests focus on the design and preparation of biodegradable polymers, polymeric nanoparticles and hydrogels for biomedical applications, as well as the industrialization of biodegradable polymer materials and their composites. |
1. Introduction
Hydrogels are crosslinked networks of hydrophilic or amphiphilic polymers capable of retaining a large amount of water yet remaining insoluble and maintaining their three-dimensional structure.1 The high water content and porosity render the physical properties similar to native tissues, and provide the hydrogels excellent biocompatibility and capability of encapsulating cells and bioactive compounds. Hence, in recent years, hydrogels have been widely tested for applications in drug delivery and regenerative medicine.In hydrogels, the polymer chains are integrated through physical interactions2–4 or covalent bonds, affording physically- and covalently-crosslinked networks, respectively. The physically-crosslinked hydrogels, though formed under mild conditions, are typically weak and exhibit relatively low long-term stability. In contrast, covalently-crosslinked hydrogels, formed by chemical reactions, are generally identified by superior mechanical properties and durability. However, the compounds for chemical crosslinking reactions, such as initiators for radical polymerization, might introduce potential toxicity issues.
Click-type reactions have become crucial tools for the preparation of covalently-crosslinked hydrogels for biomedical applications.5 Biocompatible click reactions may allow the occurrence of crosslinking in situ in the presence of cells and bioactive molecules.6,7 Among the reactions that satisfy these requirements, carbonyl-condensation reactions, such as the reactions of hydrazone and oxime formation, possess some unique advantages.8 Bioconjugation kits SureLINK™ (KPL) that utilize hydrazone-bond formation are now commercially available, illustrating the important role of this type of bioconjugation method.
The Schiff base reaction was discovered by a German chemist, Hugo Schiff, in 1864. It refers to the reactions between carbonyl groups and primary amines, yielding imines containing a carbon-nitrogen double bond as the product. The formation of the imine proceeds under mild conditions with water as the only byproduct (Fig. 1). The reaction is reversible and pH-responsive, and is dependent on the chemical structure of the substrates. Hydrazones and oximes, structurally similar to imines, are condensation products of carbonyl groups with α-effect nucleophiles containing terminal primary amine groups next to nitrogen and oxygen atoms, respectively. In aqueous solutions, hydrazones and oximes are more stable than imines because of a mesomeric effect that decreases the electrophilicity of the carbon–nitrogen double-bond.
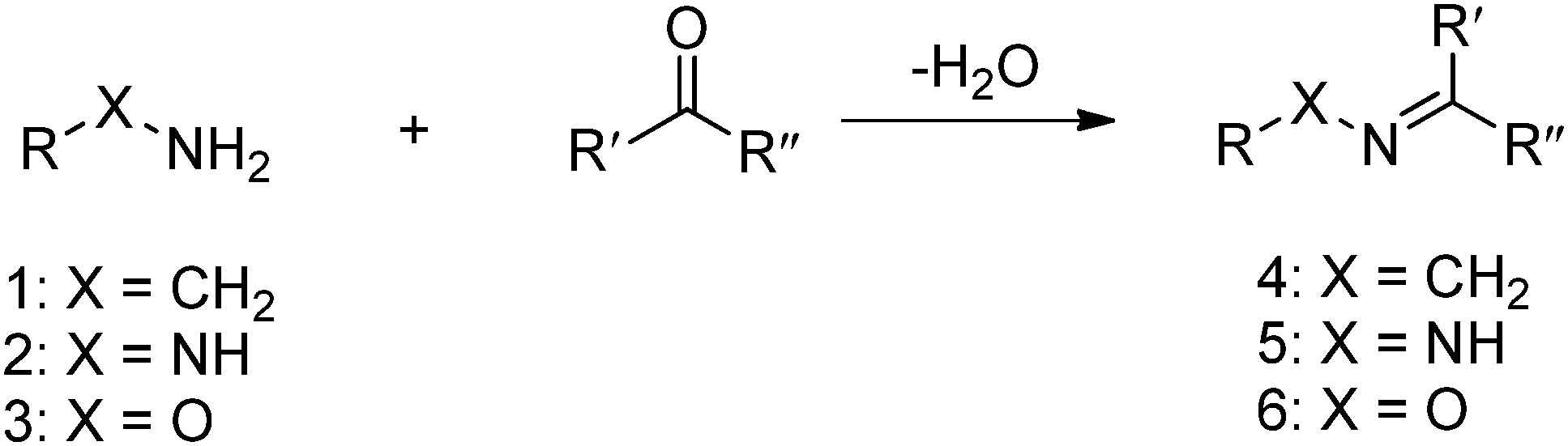 | ||
| Fig. 1 Formation of imine 4, hydrazones 5 or oxime 6 from the reaction between primary amine 1, hydrazine 2 or aminooxy 3 and aldehyde or ketone. | ||
Due to their simplicity and reversibility, carbonyl-condensation reactions have a pervasive influence on numerous biomedical fields, such as bioconjugation,9–11 nanomaterials12,13 and hydrogels.14,15 This review will highlight the utilization of the condensation reactions between nucleophiles and carbonyl groups as crosslinking strategies to prepare hydrogels with physiochemical properties responsive to biologically relevant environmental stimuli. After a brief account of the formation mechanisms and stabilities of the linker moieties, synthetic methods that incorporate nucleophiles or carbonyl groups into diverse polymers will be summarized. Then, the preparation and biomedical applications of the hydrogels based on imine/hydrazone/oxime formation will be discussed, respectively.
2. Imine/oxime/hydrazone: mechanisms of formation and stabilities
In order to utilize the reactions of carbonyls with nucleophiles in the preparation of hydrogels with desired gelation kinetics and stability, it is crucial to understand the underlying mechanisms. This section will provide insight into the equilibria and rates involved in the gelation and degradation of the hydrogels, which may be beneficial for the further development of efficient catalysts and substrates.The current understanding of the formation and hydrolysis of carbon–nitrogen double-bonds was founded by seminal studies on semicarbazone formation and hydrolysis,16 and by mechanism studies on pyridoxal phosphate enzymes.17 In particular, the elaborate kinetic analyses by Jencks elucidated the general mechanisms for the formation and hydrolysis of carbon–nitrogen double-bonds, where a carbinolamine intermediate is involved.18–21
The following section will outline the reactions between carbonyl groups and nucleophiles, and the hydrolysis reactions are obtained by reversing the order of the former reaction steps.
2.1 Mechanisms for the reactions
In general, the condensation reactions between nucleophiles and carbonyl groups start from proton-catalyzed attack of the nucleophiles to the carbonyl carbon atom. The obtained hemiaminal intermediate can undergo dehydration via proton transfer. The final deprotonation yields imine, oxime and hydrazone, respectively (Fig. 2).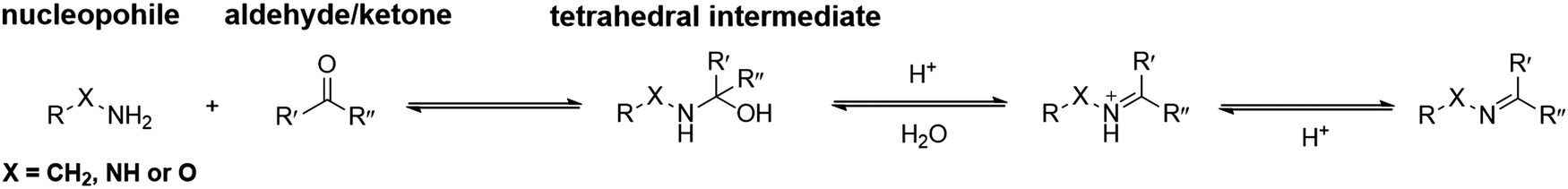 | ||
| Fig. 2 Standard mechanism for the formation of imine, hydrazone or oxime.31 | ||
At pH ranging from 3 to 7, the attack of strong nucleophiles on carbonyl groups is fast and the acid-catalyzed dehydration of the tetrahedral intermediate is usually the rate-determining step. The reaction rate can be accelerated by acid catalysis, though it slows down again if the pH is too low, due to the fact that protonation of terminal nitrogen atom attenuates the nucleophilicity. Under such conditions, the attack of the nucleophiles will become rate-determining. As a consequence, the reaction is the fastest if the acidity of the reaction medium is balanced between the acid-catalyzed dehydration of the hemiaminal intermediate and the protonation of unreacted α-effect nucleophiles. Typically, a pH of ca. 4.5 is preferred.
Among the condensation reactions between nucleophiles and carbonyl groups, the formation of hydrazones or oximes is listed as a click reaction in some cases due to their high reactivity, superior selectivity, and mild reaction conditions. These reactions have emerged as promising strategies to prepare hydrogels highly compatible with encapsulated living cells or bioactive compounds.22
However, many hydrazone and oxime linkages formed slowly at neutral pH with second-order rate constants below 0.01 M−1 s−1, which is lower than many of the cycloaddition-based reactions.23,24 Although the reaction rates for the formation of hydrazone and oxime can be greatly accelerated under general acid catalysis, physiological conditions are usually required for the crosslinking reactions in many biological applications. This makes it challenging to perform the reactions at neutral pH and low concentrations of crosslinking groups. Thus, the development of efficient catalysts and fast-reacting substrates has been a strong focus in recent work. Pioneering work by Jencks demonstrated the aniline-catalyzed semicarbazone formation, where aniline was used as a nucleophilic catalyst rather than a classical general acid catalyst.25 In the system, aniline first reacted with p-chlorobenzaldehyde to yield an imine, which was the rate-determining step. The formation of semicarbazone is fulfilled by subsequently rapid attack of semicarbazide on the imine (Fig. 3).26,27 The strong nucleophilicity makes aniline more reactive toward aldehyde than semicarbazide. The imine intermediate was presented at a relatively high concentration as a protonated form, rendering the attack of the semicarbazide on the imine several orders more rapid than on the pristine aldehyde. Although it involved more steps and intermediates, this pathway required less energy for the highest transition state barrier, thus speeding the reaction overall.
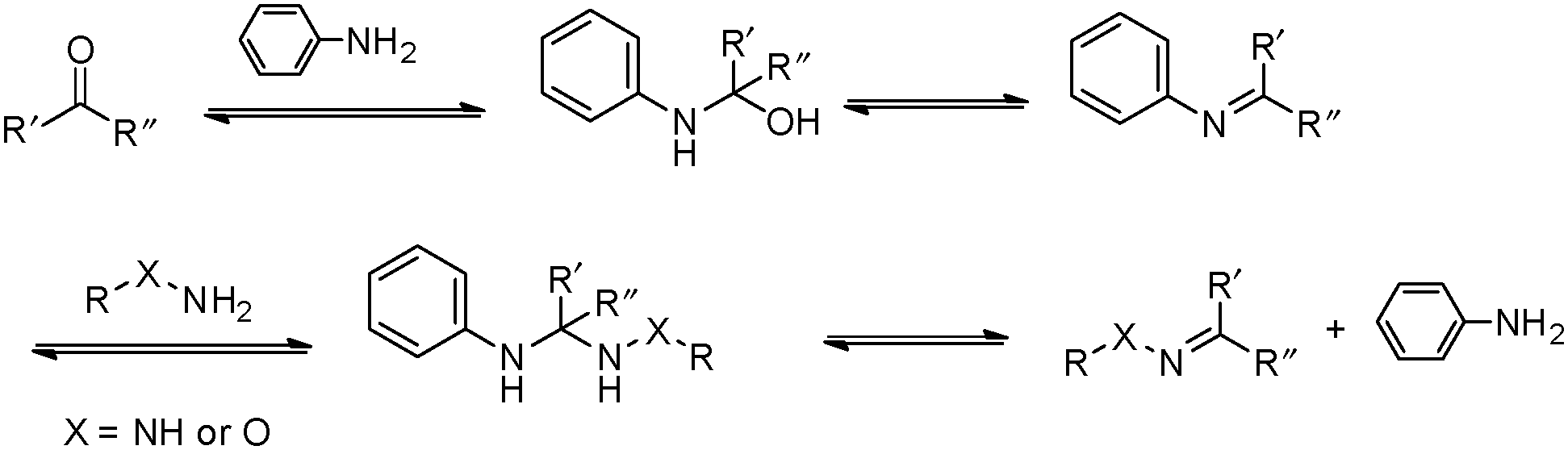 | ||
| Fig. 3 Mechanism for the aniline-catalyzed formation of hydrazone or oxime. Reproduced with permission from ref. 11. Copyright 2017 American Chemical Society. | ||
2.2 Stability
Compared with other crosslinking strategies, a unique feature of imine, hydrazone and oxime linkages is that they can be readily cleaved by water or another nucleophile. Moreover, the stability of the linkages can be tuned by the chemical structure of the substrate, as well as the catalyst.28–30The hydrolysis of carbon–nitrogen double-bonds proceeds by reversing the reaction sequences with the protonation of the sp2 nitrogen as the first step. The protonated products are highly susceptible to nucleophilic attack by water due to the enhanced electrophilicity of sp2 carbon. It is commonly observed that imines hydrolyze readily under aqueous conditions. Hydrazones show higher stability compared to imines, but are less stable than oximes. Mechanism studies by Raines showed that the inductive effects of the group X next to the sp2 nitrogen atom of the carbon–nitrogen double-bond directly influenced the stability of the respective linkages.31 In hydrazones and oximes, the electronegative heteroatom (N or O) diminishes the basicity of the sp2 nitrogen, therefore preventing its protonation. Particularly, it is found that the first-order hydrolysis rate constants of oximes are 600-fold lower than those of hydrazones, due to the higher electronegativity of oxygen [χp(O) = 3.5, χp(N) = 3.0]. Besides, the chemical structure of hydrazine also affects the stability of the resultant linkages. In comparison with isostructural hydrazones, the first-order hydrolysis rate constant of oxime was 160-fold lower than semicarbazone, 300-fold lower than acetylhydrazone, and 600-fold lower than methylhydrazone.
The nature of the carbonyl group also has a pronounced impact on the stability of the corresponding linkage. Typically, linkages obtained from ketones are more stable than the respective aldehyde derivatives. Richard et al. have shown the substituent effects on the equilibrium constants of imines formed by benzaldehyde and glycine.32 The study by Jencks showed that the equilibrium constants of oximes formed from different carbonyl compounds increase in the following order: acetone < cyclohexanone ∼ furfural ∼ benzaldehyde < pyruvic.18
3. Functionalization of polymers for crosslinking
In order to undergo an imine/hydrazone/oxime based crosslinking reaction, either a carbonyl group or a nucleophilic group needs to be introduced to the respective precursor polymer. Thereinto, primary amine groups can be found in a large number of native polymers; however, to incorporate other reactive groups, chemical modification is usually necessary. The following section will give a brief review of natural and synthetic polymers that are commonly employed as building blocks, and the strategies for decoration of different polymers with respective functional groups will be summarized.Chitosan
Chitosan, the partially deacetylated derivative of chitin, is a linear polycationic polysaccharide composed of randomly distributed β-(1,4)-linked D-glucosamine and N-acetyl-D-glucosamine (Fig. 4). The native properties of chitosan, such as excellent biocompatibility, moderate biodegradability, mucoadhesion, hemostatic and antimicrobial activities, make chitosan-based hydrogels attractive candidates for biomedical applications. The plenty of amine groups in chitosan provide the feasibility to create hydrogels via imine crosslinking, either with small-molecule dialdehyde-containing crosslinkers such as glutaraldehyde, or with a partially oxidized polysaccharide containing multiple aldehyde groups. In addition, several derivatives including glycol chitosan, N-succinyl chitosan, and carboxymethyl chitosan have also been used.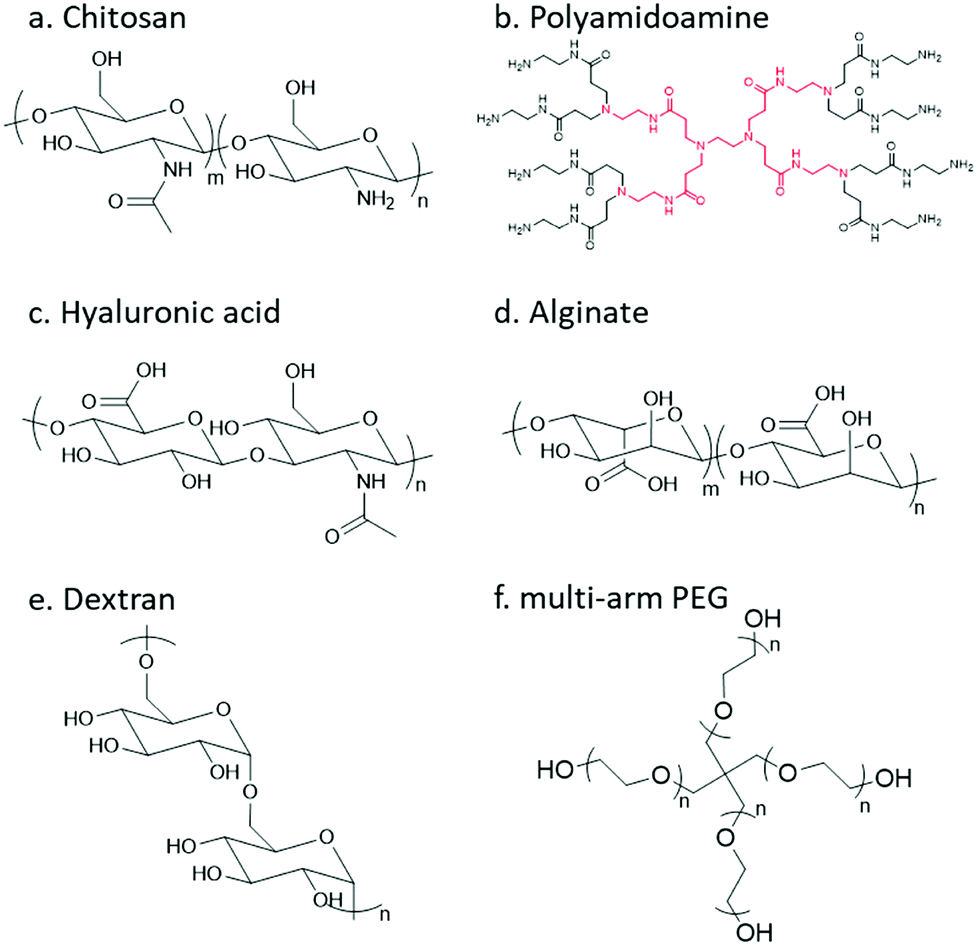 | ||
| Fig. 4 Natural and synthetic polymer building blocks. | ||
Dendrimer
Dendrimers are three-dimensional, highly orderly star-like oligomeric and polymeric macromolecules. In 1985, Tomalia et al. prepared polyamidoamine (PAMAM) dendrimers with precisely defined structures in a stepwise fashion. PAMAMs, consisting of repetitive branched subunits of amide and amine functionality, have a sphere-like shape overall with a large number of surface primary amine groups. Hydrogels can be easily obtained by mixing the PAMAMs with an oxidized polysaccharide to form imine linkages. The cationic surface of the dendritic system provides a convenient platform for attracting RNA via electrostatic interactions between the positively charged terminal amines of PAMAM dendrimers and negatively charged RNA phosphate.Hyaluronic acid
Hyaluronic acid (HA) is a non-sulfated glycosaminoglycan in the extracellular matrix (ECM) that is widely distributed throughout the connective, epithelial, and neural tissues. It is composed of repeating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine linked together with β-1,4 and β-1,3 glycosidic bonds. HA plays important roles in many biological processes, like matrix remodelling and angiogenesis, as well as participating in signalling pathways that direct cell adhesion, proliferation, differentiation, and migration.14Alginate
Alginate (Alg) is an anionic polysaccharide and consists of (1-4)-linked β-D-mannuronate with its C-5 epimer α-L-guluronate residues linked in different sequences or blocks. A challenge for the biomedical applications of alginate hydrogels in vivo lies in the fact that alginate is difficult to be degraded under physiological conditions, and the molecular weight is typically above the renal clearance threshold.33 The partial oxidation of alginate by sodium periodate not only introduces aldehyde groups, but also makes it biodegradable, making oxidized alginate an attractive material for hydrogel preparation.34Dextran
Dextran is a bacterial polysaccharide synthesized from sucrose by certain lactic-acid bacteria. The chemical structure of dextran includes main chains of D-glucopyranoses linked by α-1,6-linkages that can vary from 50 to 97% of total glucosidic bonds depending on the bacterial strains, with branching D-glucopyranoses linked by α-1,3-linkages. Dextran is subject to enzymatic degradation by dextranase in various parts of the body, including the liver, spleen, and colon.Poly(ethylene glycol)
Poly(ethylene glycol) (PEG) is the non-degradable polymer of ethylene oxide. Due to the hydrophilic and uncharged structure, it restricts protein adsorption to the highly hydrated layer. The excellent solubility and biocompatibility of PEG make it an ideal candidate for pharmaceutical and biomedical applications. Various PEG-containing formulations have been approved by the FDA. PEG can be modified with diverse functional groups via its terminal hydroxyl groups.3.1 Functionalization with carbonyl groups
The introduction of carbonyl groups to polymers can be easily carried out by coupling reactions with commercially available carbonyl-containing molecules (Fig. 5). Alternatively, an old but still widely applied strategy for functionalization of natural polysaccharides with aldehyde groups is the chemoselective oxidative cleavage of vicinal diols with sodium periodate. 1,2-Amino alcohols are susceptible to oxidative cleavage as well. Therefore, the periodate oxidation of a serine or threonine residue is a simple method for attaching the α-oxo-aldehyde group to the N-terminus of peptides or proteins.9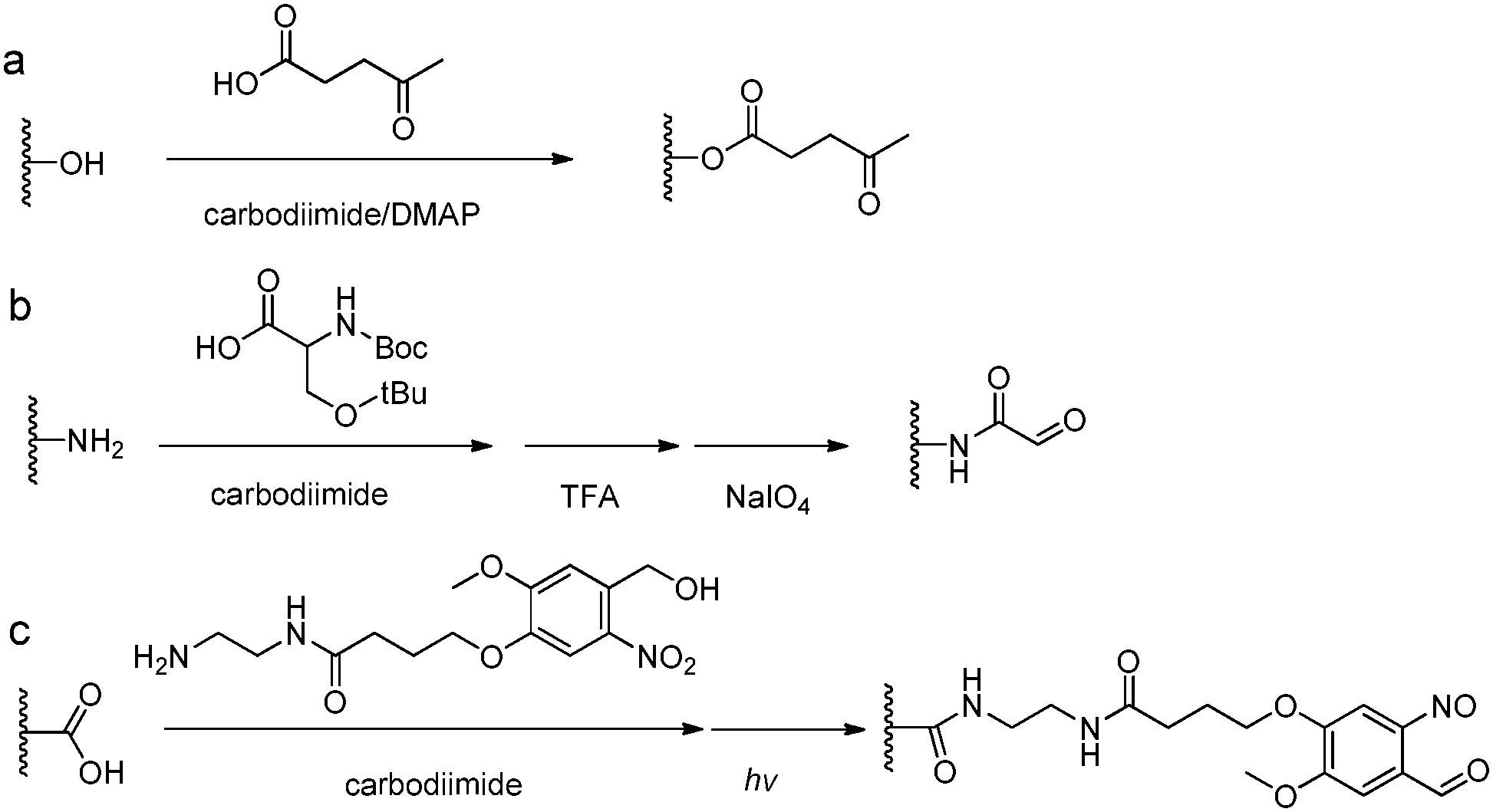 | ||
| Fig. 5 Synthetic methods for carbonyl functionalized polymers. (a) Carbodiimide coupling reactions with carbonyl-containing molecules; (b) periodate oxidation of β-amino alcohols or the Ser residue at the N-terminus;9 (c) phototriggered (λ = 365 nm) conversion of o-nitrobenzene to o-nitrobenzaldehyde.35–37 | ||
Oxidation-induced glycol cleavage is one of the most widely used methods for determining the constitution of polyoxygenated natural products.38 It was discovered by Malaprade in 1928,39 who observed that the periodate ion was able to rapidly oxidize polyols to give carbonyl products. Based on the original proposals of Criegee et al.,40 a typical cleavage reaction involves reversible formation of a cyclic complex or diester, followed by decomposing into the carbonyl products (Fig. 6). In addition to vicinal diols, other 1,2-dioxygenated groups, like 2-hydroxyaldehyde, 1,2-dicarbonyl compounds, α-hydroxy and α-keto acids and α-amino alcohols are oxidatively cleavable by periodate. It is worth mentioning that most of the common types of functional groups may remain stable under the conditions normally used in glycol-cleavage oxidations, except for thio derivatives. Aside from its role in the structure determination of polysaccharides, glycol-cleavage oxidation has now been applied widely to introduce aldehyde groups on various carbohydrate-containing macromolecules for various applications.
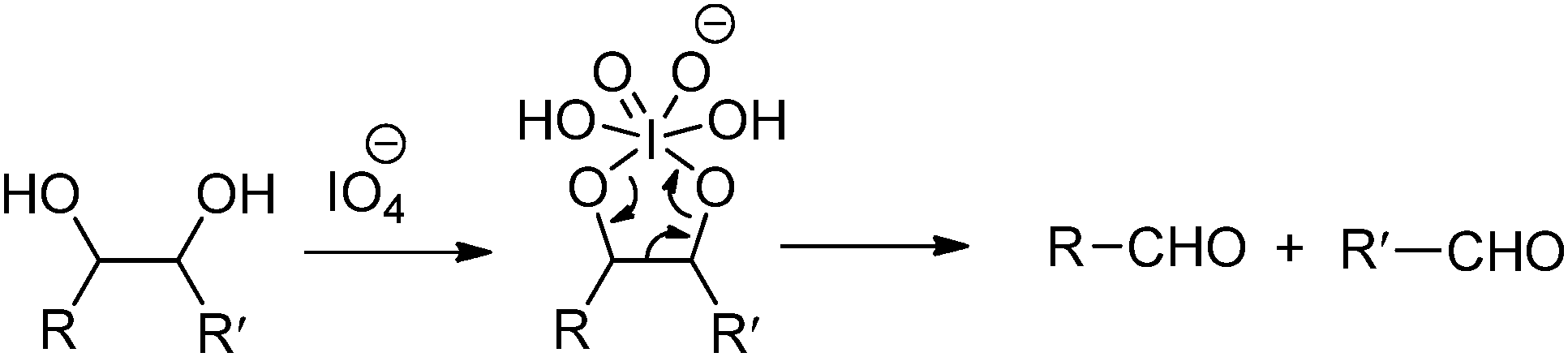 | ||
| Fig. 6 Periodate-mediated chemoselective oxidative cleavage of vicinal diols. | ||
The periodate-mediated oxidative cleavage of dextran proceeds in two steps, since there is 3,4,5-triol group in D-glucopyranose (Fig. 7).41 First, the periodate ion breaks the C–C bond between C2–C3 or C3–C4, yielding two aldehyde groups. Kinetic analysis by Ishak et al. suggested that the cleavage at the C3–C4 bond was 7.5 times faster than the cleavage at the C2–C3 bond.42 Further oxidation may occur at a similar rate, since the aldehyde derived from C3 has a vicinal hydroxyl group, and this carbon is expelled under formic acid. With regard to acid polysaccharides (HA and heparin), Scott et al. proposed that the intense negative electrostatic field of acid polysaccharides excludes negatively charged periodate ions from the domain of the polysaccharides, thus reducing the rates of oxidation.43 Oxidations of uncharged polysaccharides (dextran) are largely unaffected by change in pH.
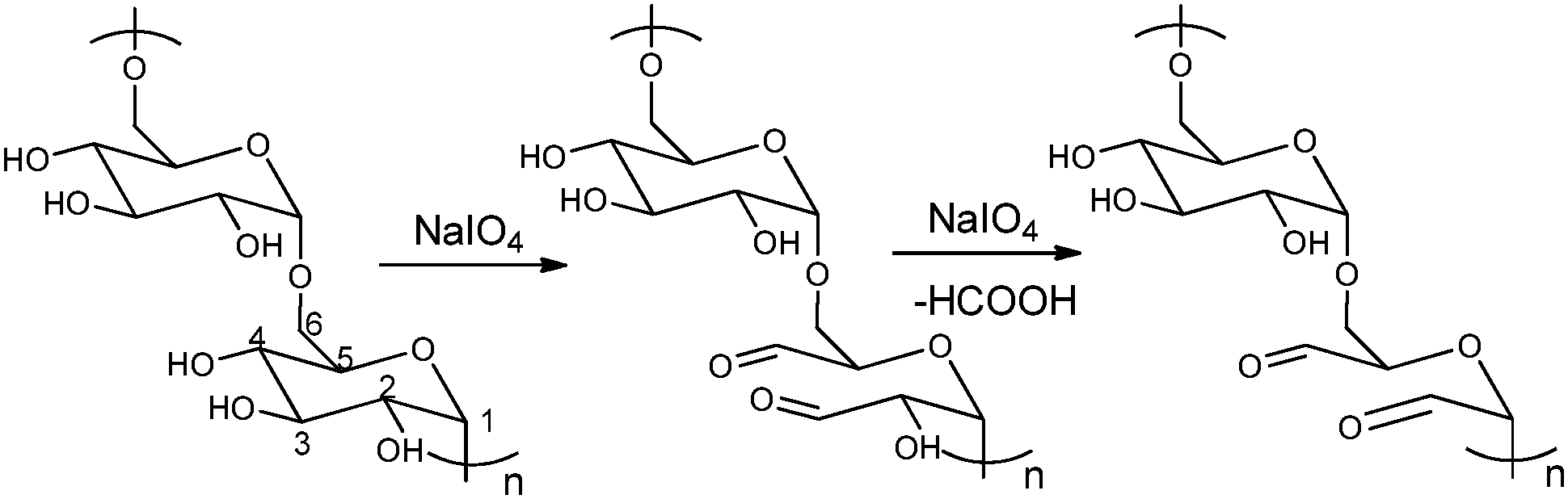 | ||
| Fig. 7 Dextran oxidation by sodium periodate. Reproduced with permission from ref. 41. | ||
3.2 Functionalization with hydrazine and aminooxy groups
Acylhydrazine (or hydrazide) functionalized polysaccharides with carboxylic side groups (Alg and HA) can be prepared by standard carbodiimide chemistry between the polysaccharides and hydrazine- or dihydrazide-containing compounds, such as carbodihydrazide (CDH), oxalyldihydrazide (ODH), and adipoyldihydrazide (ADH) (Fig. 8). Because both the terminal hydrazide moieties of dihydrazide can react with carboxylic groups, excess of dihydrazide, e.g., 5–10 fold of activated carboxylic group, was usually used to ensure the formation of a monosubstituted product. Besides, hydrazinolysis of various ester bonds is also widely used to introduce a hydrazide group to synthetic polymers. Alternatively, N,N,N′-tris(Boc)hydrazinoacetic acid can be used to prepare an aliphatic hydrazine functionalized polymer, followed by TFA deprotection. In contrast to canonically acid-catalyzed hydrazone formation, the modulus evolution of bis-aliphatic hydrazone hydrogel was most rapid at physiological pH 7.3, while both acidic and basic pH values slowed down the gelation rate.44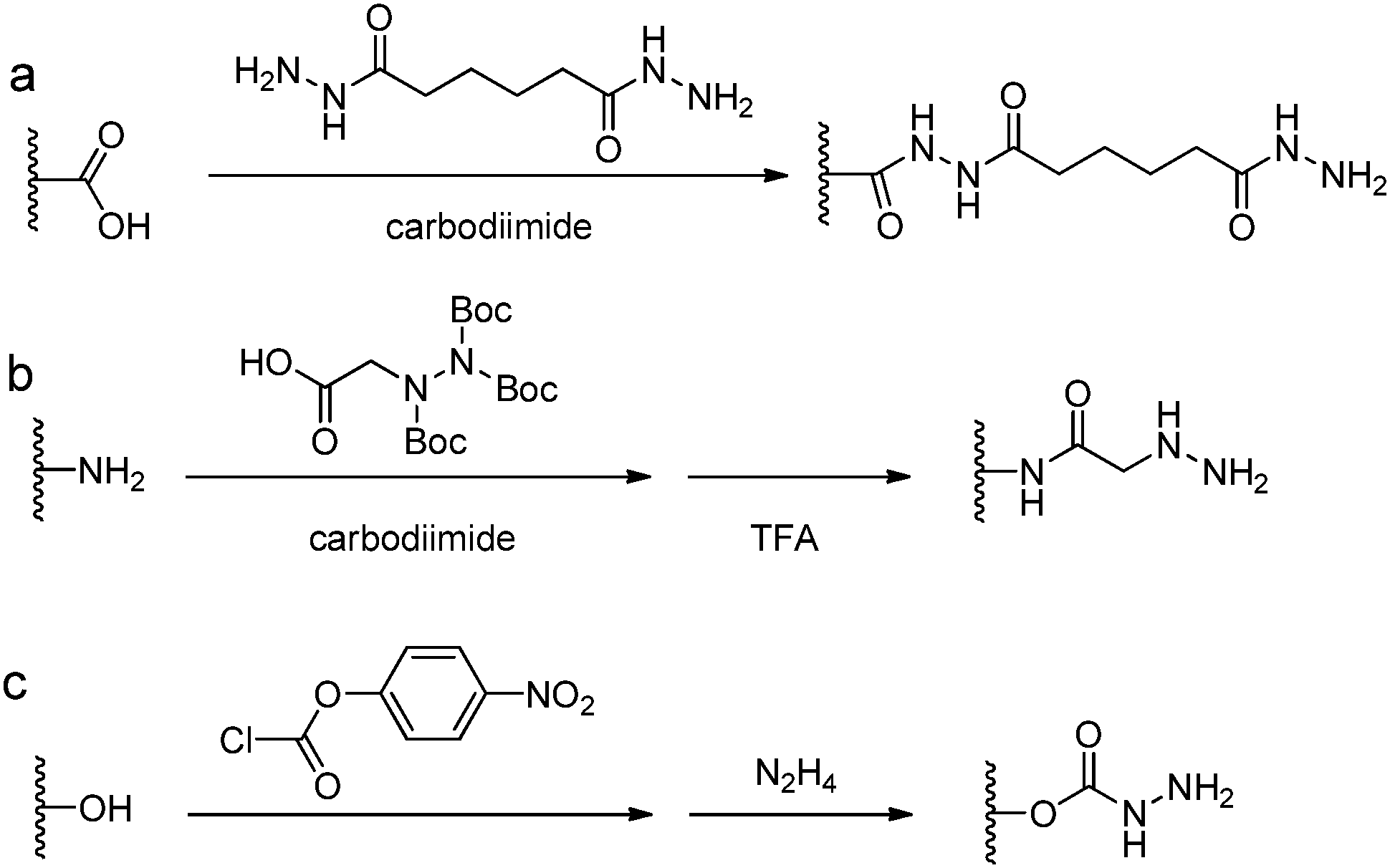 | ||
| Fig. 8 Synthetic methods for hydrazine functionalized polymers. (a) Carbodiimide coupling reactions with dihydrazide; (b) carbodiimide coupling reactions with N,N,N′-tris(Boc)hydrazinoacetic acid followed by TFA deprotection; (c) hydrazinolysis of the ester bond. | ||
The conversion of a hydroxyl group to aminooxy is usually carried out by Mitsunobu reaction with N-hydroxyphthalimide and subsequent hydrazinolysis (Fig. 9). In some cases, commercially available aminooxyacetic acid bearing a single tert-butyloxycarbonyl protecting group is introduced by a coupling method. However, the mono-protection of an aminooxy group does not ensure the complete protection of the nitrogen, yielding undesirable N-overacylated side products.45 To explore a highly clean chemistry to ensure the formation of well-defined molecules, 1-ethoxyethylidene has been developed as a new protecting group for preventing overacylation, which can be readily deprotected under mild acid conditions.46,47
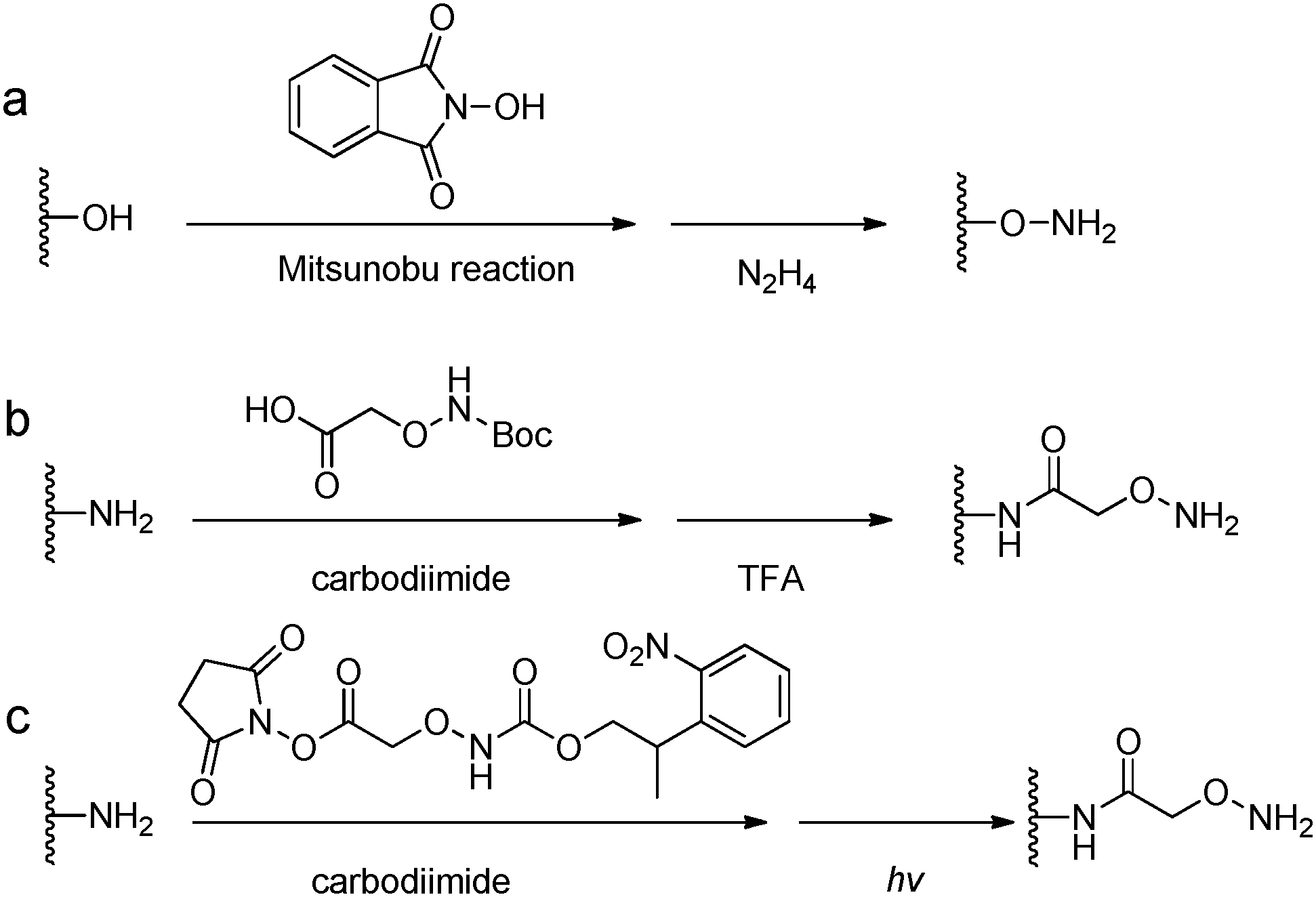 | ||
| Fig. 9 Synthetic methods for aminooxy functionalized polymers. (a) Mitsunobu reactions with N-hydroxyphthalimide followed by hydrazinolysis; (b) carbodiimide coupling reactions with (Boc-aminooxy)acetic acid followed by TFA deprotection. (c) Near UV irradiation (λ = 365 nm) cleavage of 2-(2-nitrophenyl)propyloxycarbonyl photocaged aminooxy.48,49 | ||
4. Hydrogels based on imine formation
Tan et al. reported a biocompatible and biodegradable composite hydrogel prepared via the Schiff-base reaction between N-succinyl-chitosan (S-CS) and oxidized HA (A-HA).50 The effects of the ratio of S-CS and A-HA on the gelation time, morphology, equilibrium swelling, compressive modulus, and in vitro degradation were examined. The adhesion of bovine chondrocytes on the surface of the hydrogels after 24 h of incubation showed a lower ratio than on a cell culture plate, and the cells maintained a polygonal morphology. The chondrocytes encapsulated within the hydrogel were found to be viable and exhibit an elliptical or round shape after 24 h.Zhang et al. developed multiresponsive and dynamic hydrogels by mixing benzaldehyde-terminated telechelic PEG with chitosan.51 The hydrogels formed rapidly within 60 s with solid contents of 4–8% through the formation of dynamic Schiff base linkages. The hydrogels exhibited self-healing ability and were sensitive to biochemical stimuli, such as pH, lysine, and pyridoxyl hydrochloride and papain (Fig. 10). Rhodamine B and lysozyme were encapsulated in the hydrogels as model drugs. Controlled release manners of the payloads were observed. Tseng et al. fabricated an injectable, self-healing hydrogel by mixing benzaldehyde-functionalized PEG with glycol chitosan as a scaffold for repairing the central nervous system.52 Murine neural stem cells (NSCs) encapsulated in the self-healing hydrogel proliferated twice as fast and expressed more neuronal marker genes compared with those in alginate gel. In a zebrafish embryo neural injury model, injection of the self-healing hydrogel with NSCs produced a marked healing effect. Huang et al. developed an injectable and self-healing hydrogel by mixing benzaldehyde-terminated 4-arm PEG with carboxymethyl chitosan.53 The hydrogel could rapidly heal and be easily extruded through a 20-gauze needle due to the dynamic and reversible Schiff base linkages. Human primary dermal fibroblasts encapsulated in the hydrogel maintained high viability for 1 week. In a rabbit liver incision, the hydrogels could effectively reduce the total blood loss and hemostasis time when applied on the bleeding site.
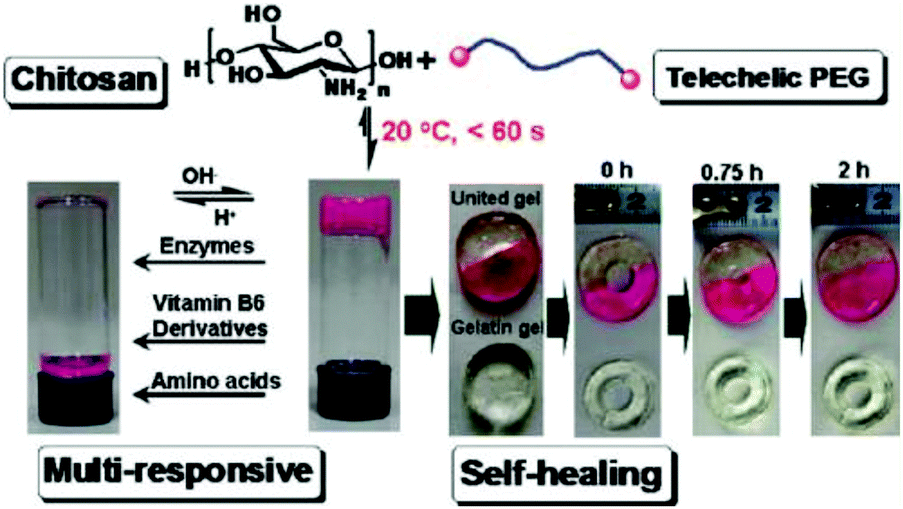 | ||
| Fig. 10 Multiresponsive and dynamic hydrogels by mixing benzaldehyde-terminated telechelic PEG with chitosan. Reproduced with permission from ref. 51. Copyright 2011 American Chemical Society. | ||
Zhao et al. designed an injectable amphiphilic hydrogel for delivering both hydrophilic doxorubicin hydrochloride (Dox) and hydrophobic paclitaxel (PTX).54 The hydrogel was synthesized via a Schiff-base reaction between glycol chitosan and benzaldehyde capped poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol). Incorporation of Dox and PTX had opposite effects on the gelation process in rheology tests. Sustained release profiles of the two drugs from the hydrogel were observed for up to two weeks, and the release rate could be accelerated by decreasing the pH from 7.4 to 6.8. Subcutaneous or intratumoral injection of the Dox incorporated hydrogels in healthy C57LB/6 mice resulted in much lower drug concentration in plasma, and consequently limited change in body weight and no obvious Dox-induced cardiotoxicity were observed. By intratumoral administration, the formulation of the drug-loaded hydrogel demonstrated efficient growth inhibition of subcutaneous B16F10 xenografts in mice.
Weng et al. prepared an in situ forming embolizing hydrogel from carboxymethyl chitosan and oxidized carboxymethyl cellulose for interventional therapies.55 The hydrogel was non-toxic to human umbilical vein endothelial cells, and had no obvious effect on the coagulation of human blood. Tetracycline and doxorubicin were loaded into the hydrogel, which could be released in a sustained manner. In a fusiform aneurysm model, the hydrogel precursor could be easily injected through a 5-F catheter and formed a gel filling the aneurysmal sac. In a rabbit renal model, effective occlusions of the renal artery and more distal arteries were achieved by embolization with hydrogel precursor without clogging.
Li et al. reported an injectable and biodegradable hydrogel for prevention of postoperative adhesion.56 The gelation was attributed to the Schiff-base reaction between N,O-carboxymethyl chitosan (NOCC) and oxidized HA. The hydrogel was non-cytotoxic and could be degraded within 2 weeks in vivo. The highly porous hydrogel could support the growth and proliferation of primary fibroblasts encapsulated in the hydrogel, but it was not favourable for the attachment of fibroblasts on the surface. Furthermore, in a rat model of sidewall defect-cecum abrasion, the hydrogel showed increased efficiency in preventing peritoneal adhesion formation, compared with normal saline and commercially available HA hydrogel.
Bu et al. constructed in situ hydrogels as hemostatic and antibacterial materials for emergency treatment of the wounded.57 Hydrogels with different amounts of Schiff-base linkages were prepared by tuning the ratios of 4-arm PEG amine, 4-arm PEG succinimidyl, and 4-arm PEG aldehyde. Vancomycin was incorporated in the hydrogels as an antimicrobial agent. The incorporation of Schiff bases improved the adhesiveness and made the release of vancomycin responsive to pH. In a rabbit infection model, no viable S. aureus (RN6390-GFP) was observed on the wounds treated by the hydrogels with vancomycin. In in vivo hemostatic tests on rabbits and pigs, the hydrogels showed an excellent hemostatic effect on severe lacerations and sealing capability on broken arteries.
Artzi et al. reported a series of studies on imine crosslinked hydrogels fabricated from oxidized dextran and amine-rich synthetic polymers, e.g. multi-arm PEG amine and PAMAM. First, imine crosslinked hydrogels composed of various ratios of star PEG amine and oxidized dextran were evaluated as potential soft tissue sealants.58 Systemic analyses of hydrogel formulations with various solid contents of oxidized dextran proved the aldehyde-mediated adhesion and the capacity to relax adhesive stress. Besides, a standard linear solid (SLS) model was proposed to describe the adhesive mechanics. Though the PEG/dextran hydrogels demonstrated the potential to adhere to intestinal tissue to a controllable extent, substantial differences in aldehyde affinity to different soft tissues demand the quantification and consideration of tissue properties for specific requirements. Moreover, excessive aldehydes required for adhesion can also induce tissue toxicity. In order to apply the adhesive hydrogels to the full range of target tissues and clinical needs, a series of PEG/dextran formulations featuring different dextran aldehyde solid contents were designed and evaluated for adhesive-tests with a range of soft tissues including the heart, lungs, liver, and duodenum of rats (Fig. 11).59 The results indicated that both tissue type and aldehyde content influence adhesive interactions. In particular, duodenal tissue possesses greater apparent aldehyde affinity and interfacial modulus compared to other tissues. Therefore, the adhesion was more stable and the hydrogel degraded at a lower rate. Additionally, subcutaneous implantation of the materials for nine days suggested that inflammation increased with aldehyde content, as exhibited by a thicker fibrous capsule as well as increased gelatinase activity.
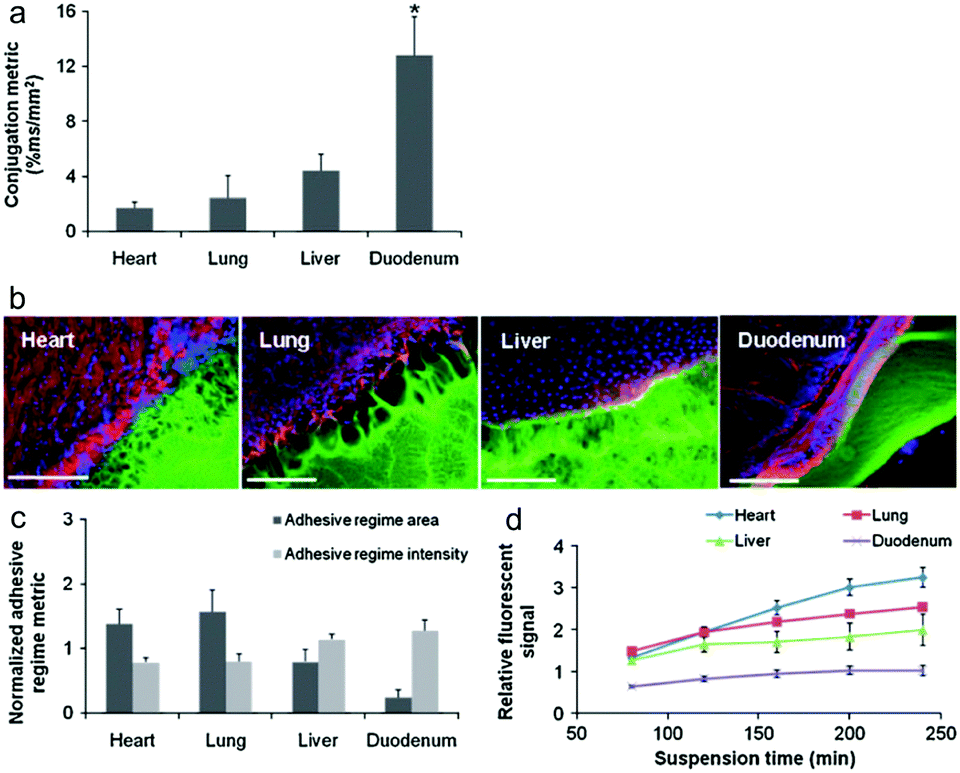 | ||
| Fig. 11 (a) The relative aldehyde reactivity of heart, lung, liver, and duodenal rat tissue were assessed through tissue-sample conjugation of fluorescent aldehyde-coated microspheres. Aldehyde reactivity was lowest in the lungs and heart, greater in the liver and highest in the duodenum (*p < 0.05 compared with liver and duodenum). (b) The interface between PEG:dextran (green) and various soft tissues, highlighted with rhodamine phalloidin (actin, red) and DAPI (cell nuclei, blue), varies with tissue. (c) Both the adhesive regime area (size normalized to interfacial length) and density (fluorescence intensity) demonstrate that tissues with high aldehyde reactivity form smaller and more continuous interfaces with PEG:dextran. *p < 0.05 compared with liver and duodenum. (d) Differential adhesive retention was seen when applied to different tissues. Adhesive is most stable when applied to the duodenum while those applied to the liver, lungs, and heart degrade faster and at an increasing rate. Degradation rate correlates with material affinity to the tissues (R = 0.9, p < 0.05). Reproduced with permission from ref. 59. | ||
Due to the reversibility and pH sensitivity of imine bonds, the performance of erodible PEG/dextran hydrogels relies on the in vivo retention time. In the follow-up work, a non-invasive imaging technique that tracks the erosion of PEG amine and oxidized dextran in vivo was developed.60 A fluorescent tag was covalently attached to PEG amine and the material erosion profile was calculated from the loss of fluorescence intensity using a non-invasive In Vivo Imaging System (IVIS). The results suggested that the erosion profile was biphasic, consisting of rapid sample loss at the first stage followed by a gradual degradation stage. More importantly, in vitro erosion of hydrogels correlated well with in vivo erosion, enabling the use of the former as a surrogate for the latter.
Subsequently, PAMAM dendrimers G5 were utilized to construct a family of adhesive materials with oxidized dextran, and their interactions within the small intestine were evaluated.61 Besides, to reveal the effect of pathological states on material performance, tissue–material interactions were assessed in inflammatory colitis and colon cancer models.62 Most recently, the application of this type of adhesive hydrogels as a depot for local cancer therapy was explored. First, a single application of nanoswitch loaded hydrogel to overcome cancer multidrug resistance before chemotherapeutic drug delivery was presented.63 Dark-gold nanoparticles, decorated with a 5-fluorouracil intercalated thiol–DNA hairpin containing a near-infrared dye and a thiol–DNA oligo containing a dark quencher, were embedded in the hydrogel as an ON/OFF nanoswitch. Upon specific hybridization of the DNA hairpin with MRP1 mRNA, the fluorescence is ON accompanying a concomitant 5-FU release. In a triple-negative breast cancer model, more than 90% tumor reduction was achieved with 80% MRP1 silencing. In a further work, a prophylactic hydrogel patch combined with siRNA, monoclonal antibody and phototherapy agent was reported for local colorectal cancer treatment (Fig. 12).64 Gold nanoparticle conjugates were embedded in the adhesive hydrogel patch to enhance their stability and provide a local delivery pattern. Among the nanoparticles with different shapes, the spherical ones were used to deliver siRNAs that silence Kras, and the rod shaped ones transduced near-infrared radiation into heat, causing the increase of local temperature as well as the release of Avastin. In the treatment of the mice with colorectal tumor xenografts, the triple-therapy patch led to the absence of tumor recurrence when implanted at the tumor site following resection, and caused complete tumor remission when implanted on the top of the tumor.
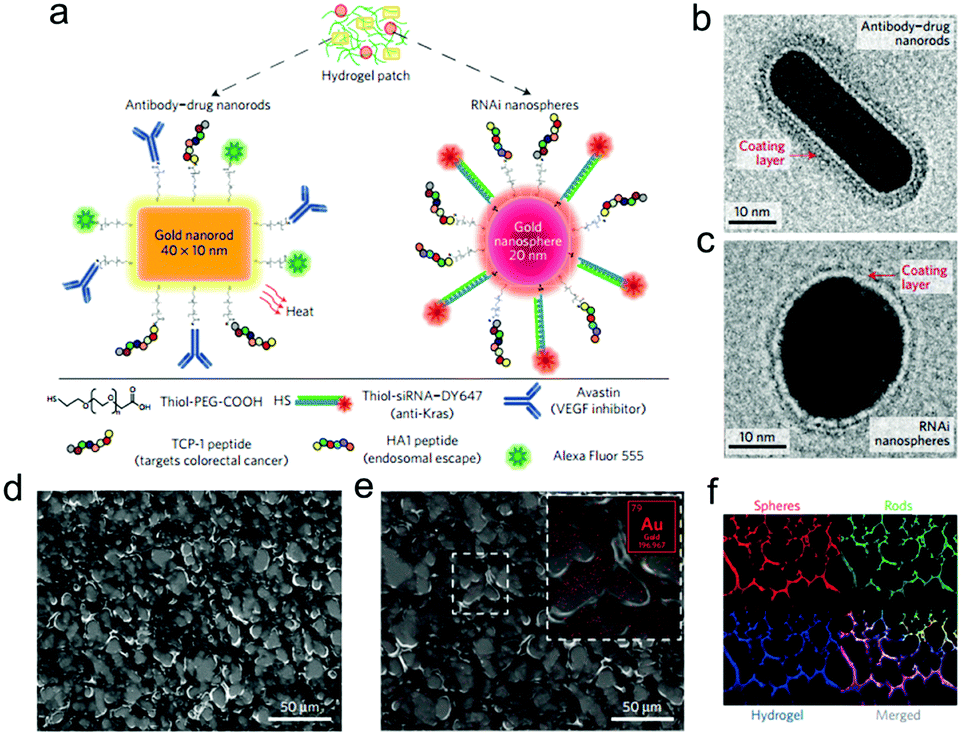 | ||
| Fig. 12 (a) Drug-gold nanorods and siRNA-gold nanospheres doped in implantable hydrogels for local drug/gene delivery and local hyperthermia. Transmission electron micrographs of (b) nanorods with an aspect ratio of 4.1 (40 × 10 nm) and (c) gold nanospheres with 20 nm diameter. The red arrows point to the negative staining of the coating layer. (d) High-resolution SEM and (e) SEM/energy-dispersive X-ray images of dextran-dendrimer hydrogel scaffolds. The gold content embedded in the hydrogel can be better appreciated in the magnified image (e, inset). (f) Cryosection of dextran-dendrimer adhesive hydrogel (12 μm thickness) where dextran aldehyde from the hydrogel is labelled with Alexa Fluor 405 (in blue), RNAi nanospheres are tagged with DY647 (in red) and drug gold nanorods are labelled with Alexa Fluor 555 (in green). Reproduced with permission from ref. 64. | ||
In a recent work by Chen and co-workers, injectable hydrogels based on imine crosslinking were prepared from polysaccharides and polypeptides as drug depots for local tumor treatment or anti-infective materials for wound healing. Nanogel-integrated, physical and chemical composite hydrogels (nPCGs) for co-delivery of doxorubicin (DOX), recombinant human interleukin-2 (IL-2), and recombinant human interferon-gamma (IFN-γ) were formed by ionic crosslinking of 4-arm poly(ethylene glycol)-b-poly(L-glutamic acid) (PPLG) with hydroxypropyl chitosan/4-arm poly(ethylene glycol)-b-poly(L-lysine) (HPCS/PPLL), as well as by the formation of imine bonds between the oxidized, cholesterol-bearing dextran (OCDEX) nanogels and HPCS/PPLL (Fig. 13).65 Combination of the imine bonds and ionic interactions resulted in multiply crosslinked networks, and led to enhanced strength and stability of the hydrogels. In a nude mice model bearing subcutaneous A549 xenografts, the treatment with peritumoral injection of nPCGs provided a marked tumor inhibition efficacy, compared to the treatment with the combination of free drugs or the hydrogel loaded with DOX or IL-2/IFN-γ. Gene expression analysis suggested that DOX, IL-2 and IFN-γ incorporated in the composite hydrogels provided a synergistic anticancer efficacy through the regulation of apoptosis-related genes in JAK/STAT pathways and mitochondrial pathways.
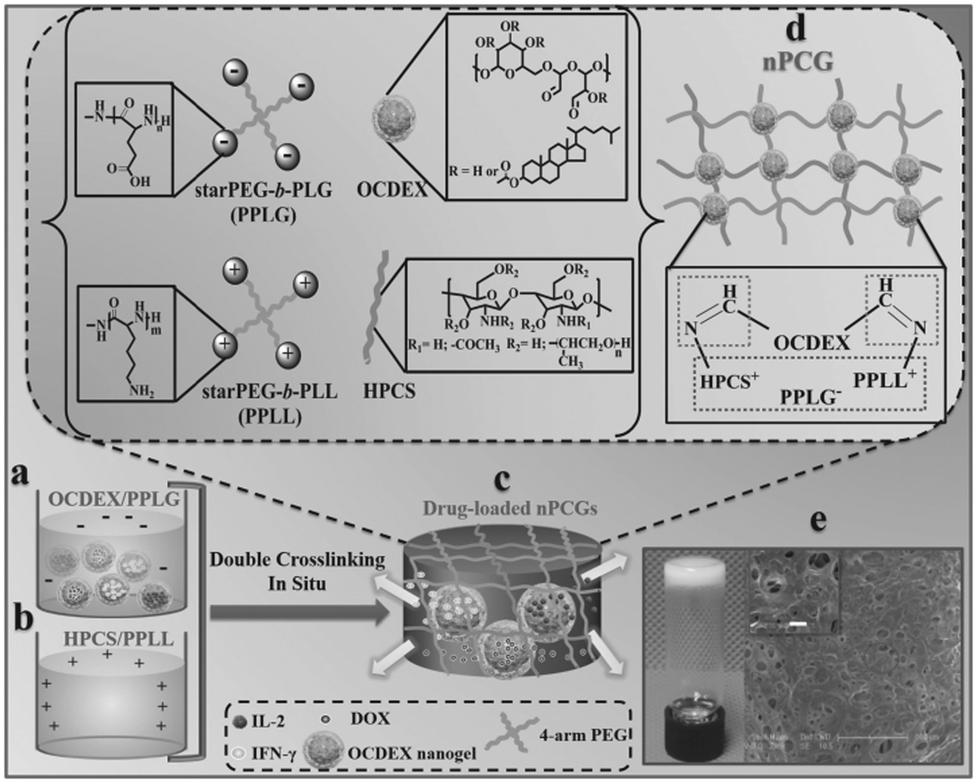 | ||
| Fig. 13 Schematic concept of nanogel-incorporated physical and chemical gels (nPCGs) as a delayed-release biosystem. (a) Negatively charged mixtures of drug-loaded oxidized cholesteryl-bearing dextran (OCDEX) nanogel solution containing 4-arm poly(ethylene glycol)-b-poly(l-glutamic acid) (PPLG) and (b) positively charged complexes hydroxypropyl chitosan/4-arm poly(ethylene glycol)-b-poly(l-lysine) (HPCS/PPLL) prepared in PBS at pH 7.4. (c) Drug-loaded hydrogels were formulated by the ionic crosslinking of negatively charged PPLG with positively charged polysaccharide/polypeptide composites (HPCS/PPLL) and the simultaneous Schiff-base reaction between amino-containing HPCS/PPLL complexes and aldehyde group-bearing nanogels containing IL-2, IFN-γ and DOX. (d) Schematic illustration for the formation of double-crosslinking networks of nPCG. (e) Optical image shows that the fabricated dual-crosslinking nPCG is white and opaque. SEM images exhibit that the micropores inside the hydrogel are all interconnected. Scale bar of the inset is 10 μm. Reproduced with permission from ref. 65. | ||
In a separate study by He and Chen et al., injectable hydrogels for the co-delivery of metformin (ME) and 5-fluorouracil (5-FU) were formulated by imine crosslinking of 4-formylbenzoic acid end-capped 4-arm PEG with PPLL.66 Both ME and 5-FU were released from the hydrogels in a controlled and pH-dependent manner. A single peritumoral injection of the hydrogel containing ME and 5-FU into BALB/c mice bearing a C26 xenograft displayed therapeutic activity superior to the free drugs or the hydrogel loaded with 5-FU, resulting from the combination of p53-mediated G1 arrest and apoptosis of C26 cells.
In separate studies by the same group, injectable polysaccharide-based hydrogels were prepared via the Schiff-base crosslinking between carboxymethyl chitosan (CMC) and oxidized dextran (ODex).67 The hydrogels displayed fast gelation, in vitro degradability, and cytocompatibility with L929 cells. When used to treat SD rats with deep second-degree burn wound, the CMC/ODex hydrogel led to nearly complete wound closure and the regeneration of skin appendages at 21 days. In another work by the same group, ceftriaxone sodium (CTX) was loaded in the hydrogels for preventing local infections.68 The CTX-loaded hydrogels showed a sustained drug release manner over 2 weeks in vitro and displayed markedly enhanced anti-infective effects both in a subcutaneous infection model and a cecal ligation and puncture model.
5. Hydrogels based on hydrazone formation
Hudson et al. developed an injectable hydrogel for local antifungal treatment.69 Amphotericin B (AmB) was conjugated to oxidized dextran (ODex), yielding a water-soluble polymer with antifungal efficacy. The hydrogel was formed by mixing hydrazide-modified carboxymethylcellulose with AmB-conjugated ODex through formation of hydrazone bonds. The in vitro release media of the hydrogel retained antifungal activity for 11 days, and the gel displayed Candida inhibition efficacy for 3 weeks. No apparent tissue toxicity and adhesion was observed in the murine peritoneum for 33 days.Varghese et al. developed a strategy to synthesize hydrazone crosslinked hydrogel via mixing aldehyde-modified HA and hydrazide-conjugated HA for in vivo bone augmentation.70 The aldehyde modified HA was prepared by selective side chain oxidation of 3-amino-1,2-propanediol conjugated HA with NaIO4 for 5 min, which was mixed with adipohydrazide modified HA to form a hydrogel within 30 s. The controlled release of recombinant human bone morphogeneic protein-2 (rhBMP-2) from the hydrogel lasted for 28 days in vitro. Subperiosteal injection of hydrogel as an rhBMP-2 carrier showed that newly formed bone was firmly fixed to the native bond in 8 weeks and the total volume was correlative with the amount of rhBMP-2 incorporated within the hydrogel. Later on, three types of hydrazide functionalized HA were used to develop an ECM mimetic hydrogel.71 The CDH, ODH, and ADH derivatives of HA were prepared by carbodiimide coupling with 10-fold excess of dihydrazides to ensure the formation of a monosubstituted product. The CDH-hydrogel demonstrated superior stability as CDH based hydrazone linkages were 15-fold more stable at pH 5 than those based on ODH and ADH. In vivo evaluation of the CDH-hydrogel with a low dose of rhBMP-2 in a rat subcutaneous model after 7 weeks showed bone regeneration with an oriented collagen matrix and blood vessel formation.
Purcell et al. prepared a polysaccharide-based hydrogel for on-demand matrix metalloproteinase (MMP) inhibition (Fig. 14).72 HA was modified to contain either aldehyde (ALD) or hydrazide (HYD) functional groups to form the hydrogels. Acting as a heparin mimetic, dextran sulphate (DS) was oxidized and incorporated into the hydrogels to immobilize encapsulated heparin-binding recombinant tissue inhibitor of MMPs (rTIMP-3). MMP sensitivity was introduced into the hydrogels by using an MMP-cleavable peptide GGRMSMPV as a linkage between the hydrazide group and HA. The hydrogels could be locally injected into myocardium, and degraded in myocardial infarction (MI) regions with elevated MMP activity after 14 days. Encapsulated rTIMP-3 was bound to DS through electrostatic interactions and released as the hydrogels were degraded by active MMPs. In a porcine model of myocardial infarction, local and on-demand delivery of rTIMP-3 from the hydrogels to regions of MMP overexpression effectively inhibited MMP activity and adverse left ventricular remodelling.
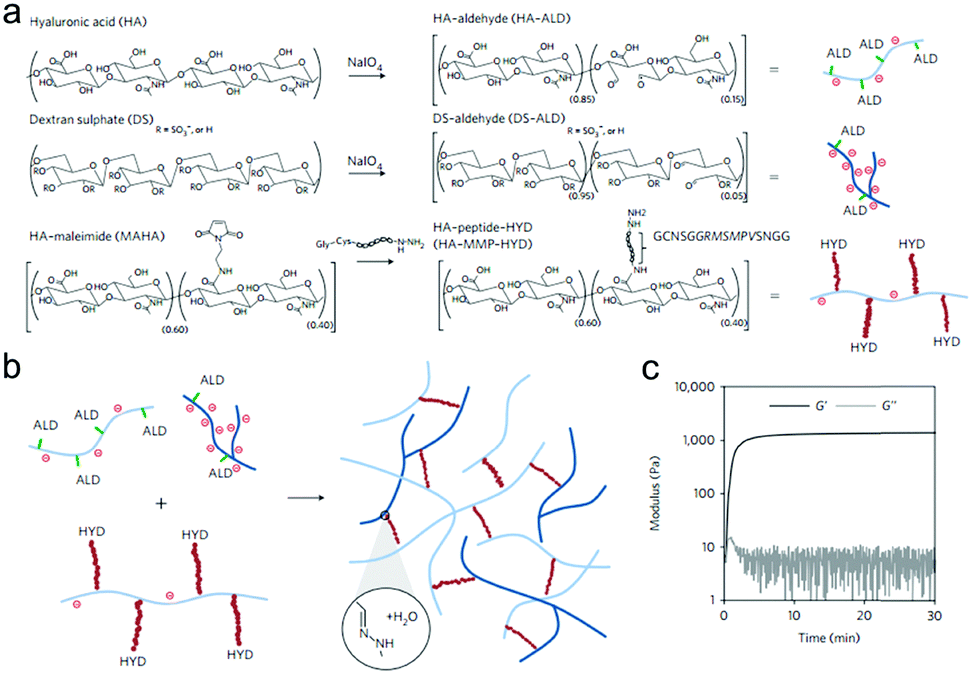 | ||
| Fig. 14 The fabrication of injectable and MMP-sensitive hydrogels for therapeutic delivery. (a) HA and DS polymers were modified with ALD groups through periodate oxidation of diols along the polysaccharide backbone. ALD modifications for HA and DS were ∼15% and 5% of the repeat disaccharides, respectively. A separate HA batch was modified with complementary HYD groups through conjugation of the peptide GCNSGGRMSMPVSNGG–HYD where C is a thiol-containing cysteine to couple to a MAHA via a thiol–maleimide click reaction, HYD is the terminal HYD for hydrogel crosslinking, NS is a hydrophilic spacer, and GGRMSMPV is the MMP-cleavable sequence. (b) Hydrogel crosslinking was designed through hydrazone bond formation of complementary ALD and HYD groups to form hydrogels under physiological conditions. (c) On mixing ALD (2.4 wt% HA-ALD, 1.4 wt% DS-ALD) and HYD (3.2 wt% HA-MMP-HYD) polymers, robust hydrogels formed rapidly as evidenced by development of storage (G′) and loss (G′′) moduli within minutes. Reproduced with permission from ref. 72. | ||
Liu et al. designed a hydrazone crosslinked sericin/dextran injectable hydrogel with in vivo real-time noninvasive trackability.73 Degraded sericin, a natural photoluminescent protein from silk, was functionalized with hydrazide groups by adipodihydrazide conjugation. The hydrogel exhibited weight-correlated, transdermal, and in vivo stable fluorescence with the emission wavelength at 530 nm that enabled long-time in vivo tracking over 70 days. The in vivo evaluation of the hydrogel as a sustained drug carrier was performed by a single peritumoral injection of hydrogel containing doxorubicin (Dox) in a B16-F10 melanoma-bearing mouse model. The DOX-loaded hydrogel efficiently suppressed the tumor growth and improved the survival rate in comparison to the free DOX.
Dahlmann et al. developed an in situ hydrogelation system based on the aldehyde- and hydrazide-derivatives of Alg and HA for myocardial tissue engineering.74 Hydrazide functional groups on Alg and HA were transferred from the corresponding carboxylate groups using carbodiimide chemistry. The rheological properties and swelling ratio could be easily adjusted by varying the degree of derivatization, concentration and composition of hydrogel precursors. Application of the hydrogels for the generation of bioartificial cardiac tissue was performed by mixing the cardiomyocyte-enriched neonatal rat heart cells in hydrogel precursors with human type I collagen.
The nature of the reactive group plays an important role in the stability of the corresponding linkage, which further determines the degradation rate and stress relaxation profiles of hydrogels. McKinnon et al. performed small molecule kinetic studies to understand the formation and stress relaxation characteristics of the bis-aliphatic hydrazone hydrogel.44 8-arm PEG-OH was oxidized to the corresponding aliphatic aldehyde with a Swern oxidation procedure, and 4-arm PEG-NH2 was coupled with triboc-hydrazinoacetic acid followed by TFA deprotection. In contrast to canonically acid-catalyzed hydrazone formation, the modulus evolution of a bis-aliphatic hydrazone hydrogel was most rapid at physiological pH 7.3, while both acidic and basic pH values slowed down the gelation rate. A stress relaxation test confirmed that the rate of stress relaxation increased as the rate of hydrazone hydrolysis increased at more acidic pH values. Small molecule kinetic studies suggested that the rates of gelation and gel relaxation were correlated closely with the rates of bond formation and hydrolysis, respectively. Subsequently, a covalently adaptable PEG hydrogel was developed via the crosslinking of aliphatic hydrazine-functionalized multi-arm PEG with either benzaldehyde- or aliphatic aldehyde-functionalized multi-arm PEG.75 The physiological modulus and stress relaxation profile can be precisely controlled by changing the ratio of aliphatic aldehyde to benzaldehyde. The studies on the hydrogels decorated with benzaldehyde-terminated KGRGDS peptide for Myoblast C2C12 encapsulation demonstrated that these covalently adaptable hydrogels allowed cytoskeletal rearrangement and extension.
Boehnke et al. reported hydrazone crosslinked hydrogels and the degradation was controllable by the introduction of mixed hydrazone and oxime crosslinks.76 PEG-hydrazide was synthesized by carbodiimide coupling of 8-arm PEG-COOH with ADH or CDH. PEG-aldehyde was prepared by Williamson ether synthesis using 8-arm PEG-OH with 2-bromo-1,1-dithioxyethane, followed by hydrolysis in acid. Hydrazone-based hydrogels were formed by mixing PEG-hydrazide with PEG-aldehyde in phosphate buffer. PEG-ADH gels swelled rapidly over 2 days before dissolving completely in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. Encapsulation of mouse mesenchymal stem cells (MSCs) in gels containing 0.5 mM RGD further accelerated the degradation rate. The stability of the hydrazone hydrogels could be enhanced by employing PEG-CDH or introducing PEG-AO to form oxime linkages. Mouse MSCs encapsulated in the hydrogels exhibited high viability and rounded morphologies at all hydrazone/oxime ratios in spite of the presence of 1 mM RGD.
Hozumi et al. developed an injectable hydrogel with slow degradation rate, consisting of CDH-modified gelatin and HA monoaldehyde.77 The monoaldehyde was prepared from conjugation of 3-amino-1,2-propanediol followed by oxidation. The hydrogel degraded much more slowly than the hydrogels comprising adipic dihydrazide-modified gelatin or HA dialdehyde obtained by directly oxidizing HA with NaIO4. The thoracic aorta was removed from a rat and embedded in the hydrogels. It was found that cells migrated from the aorta and a network structure was formed after 7 days.
Double network and interpenetrating network hydrogels based on hydrazone linkages were developed. Wang et al. reported an injectable double network hydrogel formed by hydrazine modified elastin-like protein (ELP) and oxidized HA.78 Upon heating up to the transition temperature of 25.6 °C, secondary physical crosslinking formed via thermal phase segregation of ELP, resulting in an increased storage modulus and a tenfold slower erosion rate compared to the control. After syringe needle delivery, MSCs encapsulated within the hydrogel showed a cell viability of 90% even at the highest flow rate and maintained their differentiation potential toward multiple lineages. Lou et al. reported a stress relaxing interpenetrating network (IPN) hydrogel system based on collagen I and hydrazone crosslinked HA that mimics the viscoelasticity and fibrillarity of extracellular matrix in tissues (Fig. 15).79 The IPN hydrogels were prepared by mixing collagen and HA-hydrazine with HA-aliphatic aldehyde (ALD) or HA-benzaldehyde (BLD). The two-stage stress relaxation profile of the IPN hydrogels stemmed from the combination of the realignment of collagen fibers and the dissociation of hydrazone crosslinks. Human MSCs cultured in the 3D IPNs for three days demonstrated that the IPN hydrogels promoted cell spreading, fiber remodelling, and focal adhesion.
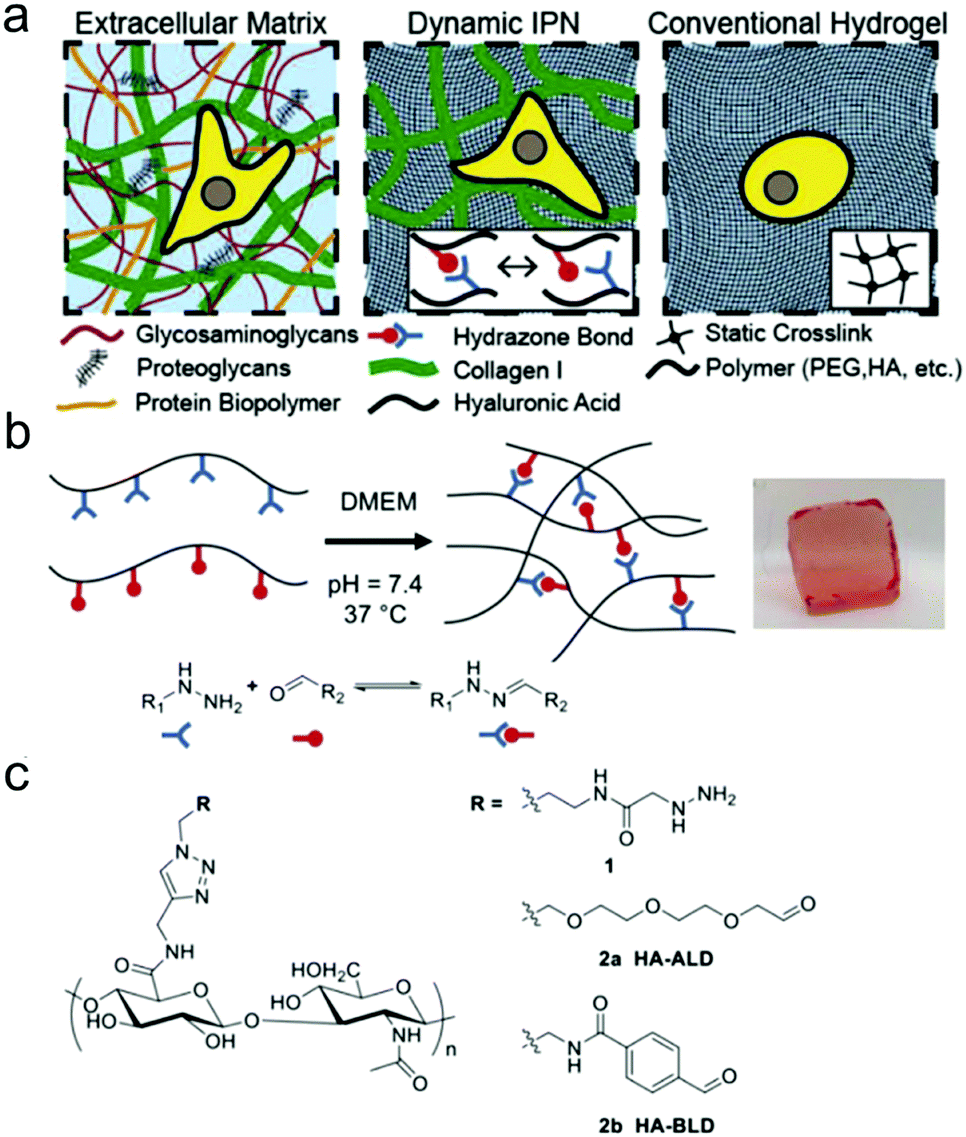 | ||
| Fig. 15 HA-collagen interpenetrating networks (IPNs) mimic the fibrillarity and viscoelasticity of ECM. (a) Illustrations of composition and network structure of natural ECM, dynamic HA-collagen IPN, and conventional statically crosslinked hydrogel. (b) Formation of a viscoelastic HA single network (SN) hydrogel via dynamic covalent hydrazone crosslinking (phenol red used to monitor pH). (c) Chemical structures of modified HAs. Reproduced with permission from ref. 79. | ||
Recently, light was exploited to achieve precise control over temporal and spatial signals for cell encapsulation and patterning of biological signals within hydrogels. Azagarsamy et al. reported PEG hydrogels based on photodriven hydrazone formation, where aldehyde was formed by a photocleavage reaction (Fig. 16).35 A photoreactive 2-nitrobenzylalcohol moiety, when exposed to UV-visible light (365–405 nm), formed 2-nitrobenzaldehyde and reacted with hydrazine to produce hydrazone crosslinked PEG hydrogels. The gelation kinetics and storage modulus could be tuned by the light intensity and duration time. The light-driven hydrazone chemistry was further exploited to precisely pattern fluorescein-labeled cell adhesive RGD peptide in spatially defined regions within the hydrogel. Meanwhile, Yang et al. demonstrated a tissue-integratable phototriggered gelation strategy based on the formation of o-nitrobenzaldehyde moieties from o-nitrobenzene (NB) upon 365 nm light irradiation, leading to quick crosslinking with nucleophilic groups in the hydrogel precursors and surrounding tissue surfaces.36 NB and CDH were conjugated to the backbone of HA via carbodiimide chemistry, forming the photogenerated aldehyde (HA-NB) and hydrazide (HA-CDH) components, respectively. A rheological experiment using a dynamic time sweep on a photorheometer showed the gel point rapidly crossed over at 27 s with final G′ of 160 Pa at 220 s for a 2.5 wt% HA-NB/HA-CDH hydrogel. In a tissue defect model in SD rat dorsal skin, the in situ formed hydrogel on the wound surface exhibited good tissue integration and faster initial wound healing at 7 d than preformed hydrogel and saline.
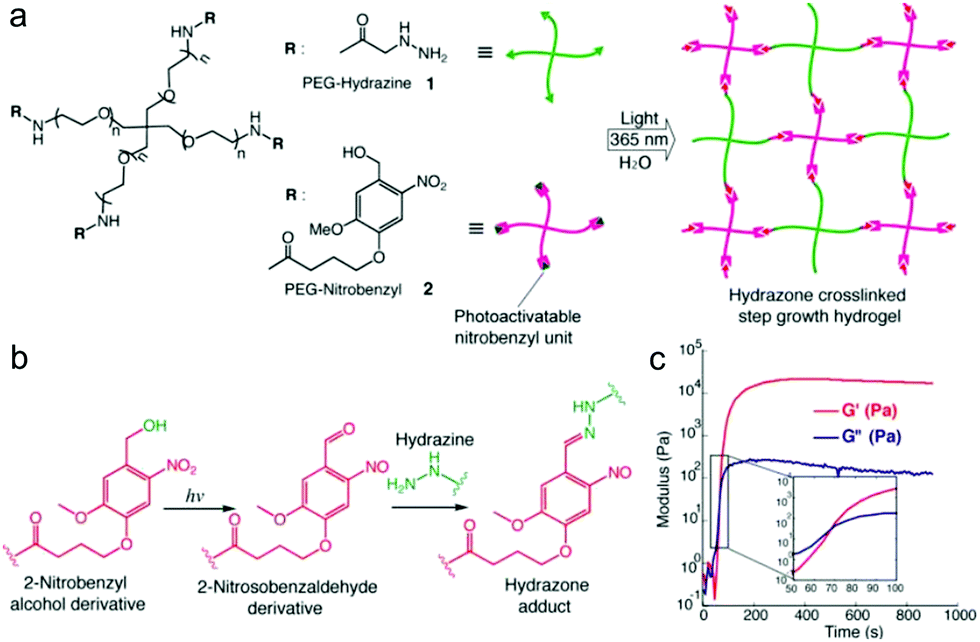 | ||
| Fig. 16 Photodriven hydrazone-based hydrogel formation. (a) Molecular structures of 4-arm PEG macromers: PEG hydrazine 1 and PEG nitrobenzyl 2. 10 wt% hydrogels were formed in PBS at 25 °C and pH 7.5 upon exposing equimolar concentrations of 1 and 2 (5 mM, 5 wt%) to 365 nm light wavelength at 20 mW cm−2. (b) Photoinduced formation of the 2-nitrosobenzaldehyde from 2-nitrobenzyl alcohol and immediate capturing of the aldehyde by hydrazine to form the hydrazone bond. (c) Evolution of the storage modulus (G′) with time when a solution of 1 and 2 was exposed to 365 nm light wavelength at 20 mW cm−2. Inset indicates the crossover point between G′ and G′′. Reproduced with permission from ref. 35. | ||
6. Hydrogels based on oxime formation
Grover et al. prepared oxime-linked hydrogels by mixing 8-arm aminooxy-capped PEG (AO-PEG) with glutaraldehyde.80 The rheological properties, water content and swelling ratio could be tuned by changing the polymer concentration and the stoichiometric ratio of aldehyde to aminooxy. The gelation time could be easily tuned from 5 min to 30 min by adjusting the pH from 6.0 to 7.2. Mouse MSCs within the gels functionalized with ketone-modified integrin binding peptide (RGD) remained viable and exhibited a rounded morphology for 7 days. Subsequently, the same group investigated the injectable PEG hydrogels formed by oxime chemistry for delivery to the heart.81 4-arm ketone-PEG (ket-PEG) was synthesized by coupling PEG with levulinic acid. PEG hydrogels crosslinked by oxime bonds were prepared by mixing the two types of 4-arm PEG derivatives. In vitro gelation rates could be tuned from 30 min to 50.3 h by adjusting the pH value from 4.0 to 7.4, allowing injection through a catheter. Subcutaneous dorsal injections in Sprague Dawley rats at pH values of 4.0, 7.4, and 10.5 resulted in gelation after 20 min, suggesting that the in vitro gelation rates were irrelevant to in vivo gelation. The difference is likely due to the effects of proteins or enzymes within the tissues in vivo. Gelation of oxidized polysaccharides, such as HA and Alg, with AO-PEG demonstrated the versatility of oxime chemistry for the formation of injectable hydrogel. Recently, the same group used 8-arm PEG containing electron deficient benzaldehyde and 8-arm AO-PEG to prepare oxime crosslinked hydrogels with the ability to adhere to different cardiac tissues ex vivo.82 The hydrogels could prevent the adhesion of 3T3 fibroblasts and RAW macrophages. The elution products did not change the cell morphology and metabolic activity. The type of cardiac tissue and the ratio of aldehyde![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :aminooxy (1:1, 1:3, 3:1) directly impacted the adhesion ability and retention time.
:aminooxy (1:1, 1:3, 3:1) directly impacted the adhesion ability and retention time.
Becker et al. fabricated a type of PEG hydrogel via oxime ligation and incorporation of functional peptides within the hydrogel postgelation.83 A 4-arm aminooxy crosslinker was produced by esterification of pentaerythritol with (Boc-aminooxy)acetic acid and subsequent deprotection of the Boc group. Benzaldehyde-functionalized linear PEG was synthesized by a carbodiimide coupling reaction of PEG with 4-formylbenzoic acid. The gelation kinetics and storage modulus could be tuned by adjusting the pH from 1.5 to 7.6 and by using an aniline catalyst. The largest storage modulus appeared at pH 4.7, and the time to reach the gel point and complete gelation reduced as the pH value decreased. Besides, the 2-arm aminooxy extender with azide or alkene handles was used for further conjugation or patterning of RGD within the hydrogel postgelation. The formation of mechanically distinguishable hydrogels was achieved by a kinetically controlled oxime crosslinking reaction.84 A series of hydrogels in phosphate-citrate buffer with different pH (pH 5.7–7.1) and buffer concentration ([buffer] = 10–100 mM) were prepared by mixing 4-arm ket-PEG with 4-arm aminooxy cross-linker. Studies of small amplitude oscillatory shear rheology and small-angle neutron scattering (SANS) showed the formation of hydrogels with a range of storage moduli by tuning the kinetics of network formation with pH and buffer strength. This led to the change in the mesh size and phase correlation length in the scaffold microstructure.
Farahani et al. exploited photomediated oxime ligation for hydrogel fabrication and post-gelation modification (Fig. 17).49 2-(2-Nitrophenyl)propyloxycarbonyl (NPPOC) was utilized as a photocage for aminooxy groups. Aqueous solutions containing multi-arm PEG end-functionalized with either ONH-NPPOC or benzaldehyde groups gelled rapidly upon UV exposure (λ = 365 nm). The gelation rate and storage moduli were highly dependent on the PEG concentration, UV intensity, aniline concentration and pH value. NIH/3T3 fibroblasts encapsulated within hydrogels exhibited high viability over 3 days.
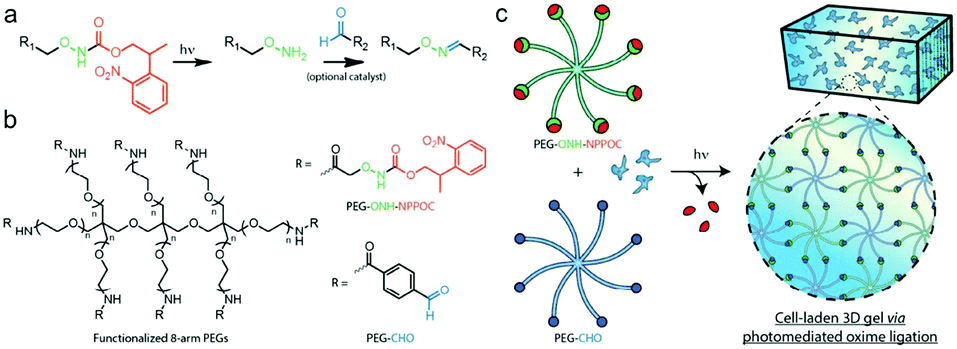 | ||
| Fig. 17 (a) Alkoxyamines (–ONH2, green) protected by an NPPOC photocage (red) can be utilized for photomediated oxime ligation. Upon UV irradiation (λ = 365 nm), the alkoxyamine is freed for reaction with a benzaldehyde (–CHO, blue) to form a stable oxime linkage. (b) 8-arm PEG crosslinkers end-functionalized with either ONH-NPPOC or benzaldehyde moieties provide complementary monomeric units that can be used for hydrogel photopolymerization. (c) Aqueous solutions containing PEG-ONH-NPPOC and PEG-CHO hydrogel precursors undergo step-growth polymerization via photomediated oxime ligation. Cells included in the polymer solution are encapsulated within bulk 3D hydrogels. Reproduced with permission from ref. 49. | ||
7. Conclusions
In the past several years, carbonyl-condensation reactions have emerged as versatile strategies to construct functional hydrogels for biomedical applications (Table 1). The simplicity, reversibility, and pH-responsiveness of carbonyl-condensation reactions provide the hydrogels unique properties as functional extracellular matrices for tissue engineering or as injectable depots for sustained release of bioactive compounds. The hydrolytic stability of carbon–nitrogen double-bonds can be effectively tailored by structural changes in the adjacent moieties. Consequently, the degradation rates and stress relaxation profiles of the hydrogels can span several orders of magnitude.| Crosslinking types | Nucleophilic group-containing blocks | Carbonyl group-containing blocks | Properties | Applications | Ref. |
|---|---|---|---|---|---|
| Imine | Glycol chitosan | PEG-BLD | Self-healing property | Repair of central nerve system | 52 |
| Carboxymethyl chitosan | HA-ALD | — | Prevention of peritoneal adhesion | 56 | |
| PEG-b-PLL | PEG-BLD | pH-responsive release | Localized drug delivery | 66 | |
| PEG-NH2 | ODex | Adhesiveness to soft tissues | Soft tissue sealants | 58–60 | |
| Hydrazone | HA-MMP-HYD | HA-ALD | Responsiveness to MMP activity | MMPs inhibition | 72 |
| PEG-HYD | PEG-ALD/BLD | Stress relaxation properties | C2C12 encapsulation | 75 | |
| ELP-HYD | HA-ALD | Injectability | MSC encapsulation | 78 | |
| HA-HYD | HA-ALD/BLD | Stress relaxation properties | MSC encapsulation | 79 | |
| HA-CDH | HA-NB | Tissue integration | Wound healing | 36 | |
| Oxime | AO-PEG | Ket-PEG | Injectability | Cardiac catheter delivery | 81 |
| AO-PEG | PEG-BLD | Adhesiveness to cardiac tissue | Prevention of cardiac adhesion | 82 | |
| 4-arm aminooxy linker | Ket-PEG | Tuneable G′ by pH and buffer strength | — | 84 | |
| PEG-ONH-NPPOC | PEG-BLD | Gelation upon UV exposure | Protein patterning | 49 | |
Compared with many cycloaddition-based reactions, the formation of hydrazone and oxime linkages was generally regarded to be relatively slow under neutral conditions. Recent studies have provided new insights to develop fast-reacting substrates and efficient catalysts, which can be exploited to construct hydrogels for applications with special requirements. Notably, the catalysts speed up both forward and reverse reactions, thus making them also useful in accelerating degradation and relaxation profiles.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (projects 51622307, 21574127, and 51520105004), and the Youth Innovation Promotion Association CAS is gratefully acknowledged.References
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef PubMed
.
- J. Ren, Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, F. Ricci and I. Willner, Chem. Sci., 2015, 6, 4190–4195 RSC
- W. Guo, C.-H. Lu, R. Orbach, F. Wang, X.-J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73–78 CrossRef PubMed
- J. S. Kahn, Y. Hu and I. Willner, Acc. Chem. Res., 2017, 50, 680–690 CrossRef PubMed
- Z. Zhang, C. He, Q. Xu, X. Zhuang and X. Chen, Acta Polym. Sin., 2018, 1, 99–108 Search PubMed
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef PubMed
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef PubMed
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chemistry, 2014, 20, 34–41 CrossRef PubMed
- O. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef PubMed
- S. M. Agten, P. E. Dawson and T. M. Hackeng, J. Pept. Sci., 2016, 22, 271–279 CrossRef PubMed
- D. K. Kolmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef PubMed
- F. della Sala and E. R. Kay, Angew. Chem., Int. Ed., 2015, 54, 4187–4191 CrossRef PubMed
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC
- B. H. Ye, L. Meng, L. H. Li, N. Li, Z. W. Li, R. W. Li, Z. W. Cai and C. R. Zhou, Acta Polym. Sin., 2016, 134–148 Search PubMed
- J. B. Conant and P. D. Bartlett, J. Am. Chem. Soc., 1932, 54, 2881–2899 CrossRef
- D. E. Metzler, M. Ikawa and E. E. Snell, J. Am. Chem. Soc., 1954, 76, 648–652 CrossRef
- W. P. Jencks, J. Am. Chem. Soc., 1959, 81, 475–481 CrossRef
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 832–837 CrossRef
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 4319–4328 CrossRef
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843–2848 CrossRef
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef PubMed
- A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., 2006, 45, 7581–7584 CrossRef PubMed
- M. Cai, Y. Han, Q. Zhang and S. Luo, Chin. J. Org. Chem., 2018, 38, 1–10 CrossRef
- S. Karmakar, E. M. Harcourt, D. S. Hewings, A. F. Lovejoy, D. M. Kurtz, T. Ehrenschwender, L. J. Barandun, C. Roost, A. A. Alizadeh and E. T. Kool, Nat. Chem., 2015, 7, 752–758 CrossRef PubMed
- X. Yang, G. Liu, L. Peng, J. Guo, L. Tao, J. Yuan, C. Chang, Y. Wei and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703174 CrossRef
- J. Lou, F. Liu, C. D. Lindsay, O. Chaudhuri, S. C. Heilshorn and Y. Xia, Adv. Mater., 2018, 30, 1705215 CrossRef PubMed
- J. Kalia and R. T. Raines, Angew. Chem., 2008, 47, 7523–7526 CrossRef PubMed
- J. Crugeiras, A. Rios, E. Riveiros and J. P. Richard, J. Am. Chem. Soc., 2009, 131, 15815–15824 CrossRef PubMed
- A. Alshamkhani and R. Duncan, J. Bioact. Compat. Polym., 1995, 10, 4–13 CrossRef
- K. H. Bouhadir, K. Y. Lee, E. Alsberg, K. L. Damm, K. W. Anderson and D. J. Mooney, Biotechnol. Prog., 2001, 17, 945–950 CrossRef PubMed
- M. A. Azagarsamy, I. A. Marozas, S. Spans and K. S. Anseth, ACS Macro Lett., 2016, 5, 19–23 CrossRef
- Y. Yang, J. Zhang, Z. Liu, Q. Lin, X. Liu, C. Bao, Y. Wang and L. Zhu, Adv. Mater., 2016, 28, 2724–2730 CrossRef PubMed
- Z. Ming, J. Fan, C. Bao, Y. Xue, Q. Lin and L. Zhu, Adv. Funct. Mater., 2018, 28, 1706918 CrossRef
-
A. S. Perlin, in Advances in Carbohydrate Chemistry and Biochemistry, ed. D. Horton, 2006, vol. 60, pp. 183–250 Search PubMed
- L. Malaprade, C. R. Hebd. Seances Acad. Sci., 1928, 186, 382–385 Search PubMed
- R. Criegee, L. Kraft and B. Rank, Justus Liebigs Ann. Chem., 1933, 507, 159–197 CrossRef
- J. Maia, L. Ferreira, R. Carvalho, M. A. Ramos and M. H. Gil, Polymer, 2005, 46, 9604–9614 CrossRef
- M. F. Ishak and T. J. Painter, Carbohydr. Res., 1978, 64, 189–197 CrossRef
- J. E. Scott and R. J. Harbinso, Histochemie, 1968, 14, 215–220 CrossRef
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Chem. Mater., 2014, 26, 2382–2387 CrossRef
- J. Brask and K. J. Jensen, J. Pept. Sci., 2000, 6, 290–299 CrossRef PubMed
- V. Duléry, O. Renaudet and P. Dumy, Tetrahedron, 2007, 63, 11952–11958 CrossRef
- S. Foillard, M. O. Rasmussen, J. Razkin, D. Boturyn and P. Dumy, J. Org. Chem., 2008, 73, 983–991 CrossRef PubMed
- C. A. DeForest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef PubMed
- P. E. Farahani, S. M. Adelmund, J. A. Shadish and C. A. DeForest, J. Mater. Chem. B, 2017, 5, 4435–4442 RSC
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Biomaterials, 2009, 30, 2499–2506 CrossRef PubMed
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef PubMed
- T.-C. Tseng, L. Tao, F.-Y. Hsieh, Y. Wei, I.-M. Chiu and S.-H. Hsu, Adv. Mater., 2015, 27, 3518–3524 CrossRef PubMed
- W. Huang, Y. Wang, Y. Chen, Y. Zhao, Q. Zhang, X. Zheng, L. Chen and L. Zhang, Adv. Healthcare Mater., 2016, 5, 2813–2822 CrossRef PubMed
- L. Zhao, L. Zhu, F. Liu, C. Liu, D. Shan, Q. Wang, C. Zhang, J. Li, J. Liu, X. Qu and Z. Yang, Int. J. Pharm., 2011, 410, 83–91 CrossRef PubMed
- L. Weng, N. Rostambeigi, N. D. Zantek, P. Rostamzadeh, M. Bravo, J. Carey and J. Golzarian, Acta Biomater., 2013, 9, 8182–8191 CrossRef PubMed
- L. Li, N. Wang, X. Jin, R. Deng, S. Nie, L. Sun, Q. Wu, Y. Wei and C. Gong, Biomaterials, 2014, 35, 3903–3917 CrossRef PubMed
- Y. Bu, L. Zhang, J. Liu, L. Zhang, T. Li, H. Shen, X. Wang, F. Yang, P. Tang and D. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 12674–12683 CrossRef PubMed
- T. M. Shazly, N. Artzi, F. Boehning and E. R. Edelman, Biomaterials, 2008, 29, 4584–4591 CrossRef PubMed
- N. Artzi, T. Shazly, A. B. Baker, A. Bon and E. R. Edelman, Adv. Mater., 2009, 21, 3399–3403 CrossRef PubMed
- N. Artzi, N. Oliva, C. Puron, S. Shitreet, S. Artzi, A. Bon Ramos, A. Groothuis, G. Sahagian and E. R. Edelman, Nat. Mater., 2011, 10, 704–709 CrossRef PubMed
- N. Oliva, S. Shitreet, E. Abraham, B. Stanley, E. R. Edelman and N. Artzi, Langmuir, 2012, 28, 15402–15409 CrossRef PubMed
- N. Oliva, M. Carcole, M. Beckerman, S. Seliktar, A. Hayward, J. Stanley, N. M. A. Parry, E. R. Edelman and N. Artzi, Sci. Transl. Med., 2015, 7, 272ra11 CrossRef PubMed
- J. Conde, N. Oliva and N. Artzi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1278–E1287 CrossRef PubMed
- J. Conde, N. Oliva, Y. Zhang and N. Artzi, Nat. Mater., 2016, 15, 1128–1138 CrossRef PubMed
- X. Wu, C. He, Y. Wu, X. Chen and J. Cheng, Adv. Funct. Mater., 2015, 25, 6744–6755 CrossRef
- X. Wu, C. He, Y. Wu and X. Chen, Biomaterials, 2016, 75, 148–162 CrossRef PubMed
- Z. Li, B. Yuan, X. Dong, L. Duan, H. Tian, C. He and X. Chen, RSC Adv., 2015, 5, 94248–94256 RSC
- Z. Li, C. He, B. Yuan, X. Dong and X. Chen, Macromol. Biosci., 2017, 17, 1600347 CrossRef PubMed
- S. P. Hudson, R. Langer, G. R. Fink and D. S. Kohane, Biomaterials, 2010, 31, 1444–1452 CrossRef PubMed
- E. Martinez-Sanz, D. A. Ossipov, J. Hilborn, S. Larsson, K. B. Jonsson and O. P. Varghese, J. Controlled Release, 2011, 152, 232–240 CrossRef PubMed
- O. P. Oommen, S. Wang, M. Kisiel, M. Sloff, J. Hilborn and O. P. Varghese, Adv. Funct. Mater., 2013, 23, 1273–1280 CrossRef
- B. P. Purcell, D. Lobb, M. B. Charati, S. M. Dorsey, R. J. Wade, K. N. Zellars, H. Doviak, S. Pettaway, C. B. Logdon, J. A. Shuman, P. D. Freels, J. H. Gorman, 3rd, R. C. Gorman, F. G. Spinale and J. A. Burdick, Nat. Mater., 2014, 13, 653–661 CrossRef PubMed
- J. Liu, C. Qi, K. Tao, J. Zhang, J. Zhang, L. Xu, X. Jiang, Y. Zhang, L. Huang, Q. Li, H. Xie, J. Gao, X. Shuai, G. Wang, Z. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 6411–6422 CrossRef PubMed
- J. Dahlmann, A. Krause, L. Moeller, G. Kensah, M. Moewes, A. Diekmann, U. Martin, A. Kirschning, I. Gruh and G. Draeger, Biomaterials, 2013, 34, 940–951 CrossRef PubMed
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef PubMed
- N. Boehnke, C. Cam, E. Bat, T. Segura and H. D. Maynard, Biomacromolecules, 2015, 16, 2101–2108 CrossRef PubMed
- T. Hozumi, T. Kageyama, S. Ohta, J. Fukuda and T. Ito, Biomacromolecules, 2018, 19, 288–297 CrossRef PubMed
- H. Wang, D. Zhu, A. Paul, L. Cai, A. Enejder, F. Yang and S. C. Heilshorn, Adv. Funct. Mater., 2017, 27, 1605609 CrossRef
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef PubMed
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef PubMed
- G. N. Grover, R. L. Braden and K. L. Christman, Adv. Mater., 2013, 25, 2937–2942 CrossRef PubMed
- G. N. Grover, J. Garcia, M. M. Nguyen, M. Zanotelli, M. M. Madani and K. L. Christman, Adv. Healthcare Mater., 2015, 4, 1327–1331 CrossRef PubMed
- F. Lin, J. Yu, W. Tang, J. Zheng, A. Defante, K. Guo, C. Wesdemiotis and M. L. Becker, Biomacromolecules, 2013, 14, 3749–3758 CrossRef PubMed
- Z. K. Zander, G. Hua, C. G. Wiener, B. D. Vogt and M. L. Becker, Adv. Mater., 2015, 27, 6283–6288 CrossRef PubMed
| This journal is © the Partner Organisations 2018 |