
Impact of molecular orientation on fluorescence emission enhancement in aggregates
P.
Srujana
and
T. P.
Radhakrishnan
 *
*
School of Chemistry, University of Hyderabad, Hyderabad – 500 046, India. E-mail: tpr@uohyd.ac.in; Fax: +91-40-2301-2460; Tel: +91-40-2313-4827
First published on 14th March 2018
Abstract
Enhanced fluorescence emission in molecular aggregates and solids has received wide attention in view of its extensive application potential. The uncommon effect of enhancement over quenching has largely been attributed to the rigidification of the molecule and its environment, and the restriction of intramolecular non-radiative excited state decay channels. The critical role of molecular orientations in hindering intermolecular energy transfer and the associated non-radiative excited state energy loss has been explored in recent studies. Further examples are demonstrated to argue for a closer look at the concomitant role of intra and intermolecular effects in realizing enhanced fluorescence in molecular aggregates.
Introduction
Fluorescent molecules find applications in sensing, displays and imaging, and as probes for chemical and biological processes. The selectivity and sensitivity to environmental factors make fluorescence-based detection significant from fundamental as well as application perspectives. In several advanced materials applications, fluorophores are used in the form of aggregates or thin films, conditions under which the fluorescence of most conventional dyes is prone to self-quenching. In bio-imaging for example, even at the low concentrations employed, the fluorophores tend to aggregate in hydrophobic pockets making them non-fluorescent. The need for fluorophores exhibiting bright emission in the aggregated/solid state is obvious. Early examples of strongly fluorescent solids include a polymorph of diketopyrrolopyrrole,1 thin films of ladder-type polymer poly(p-phenylene)2 and a platinum complex.3 Selected classes of molecules that have emerged later include, siloles,4 tetraphenylethenes,5 butadienes to diphenylbutadienes,6–8 distyrylbenzene to tetraphenyldistyrylbenzenes,9,10 anthracenes to 9,10-bis(arylvinyl)anthracenes11,12 and diaminodicyanoquinodimethanes (DADQs).13 Enhanced emission in the solid state has been described as aggregation-induced emission (AIE).14,15Mechanisms for enhanced emission in the aggregated state
The most widely discussed mechanism for enhanced emission in the solid state is based on the restriction of intramolecular motions (RIM) like rotations and vibrations resulting from the rigidification of the molecule and its environment.16 Computational studies have illustrated the role of low frequency modes in the non-radiative energy dissipation of the excited state of AIE molecules in the isolated state compared to that in the solid.17 A different perspective relates to the accessibility or not of a conical intersection between the ground and excited electronic states, leading to quenching or enhancement of emission in the isolated and aggregate molecules respectively.18 A recent study suggests that the enhanced emission in viscous solutions and the solid state of BF2-hydrazone based fluorescent molecular rotors arises from the suppression of internal conversion from emissive higher excited state to the dark lower excited state.19 Effect of the crystalline environment20 as well as steric/electronic factors on the excited state21 have also been discussed in the context of enhanced emission in the solid state.As hindrance to intramolecular relaxation and non-radiative excited state energy loss, as well as absence of effects like collisional quenching by solvent molecules should be manifested in all solids, observation of enhanced solid state emission only with select molecules, indicates that other factors related to molecular structure and assembly need to be addressed. Most of the earlier mechanistic studies have focused on specific molecules; it is important to analyse trends in solution-to-solid fluorescence efficiency enhancement (FEE) across a series of similar molecules to unravel the underlying factors. Excited state energy loss via intermolecular energy transfer is a critical factor that leads to fluorescence quenching. Even though the need to circumvent the detrimental effect of π-stacking to realize enhanced emission is often noted,22 there has been very few systematic and quantitative explorations of the correlation between molecular assembly and FEE.
Studies on DADQs
We have developed several DADQ derivatives exhibiting strong fluorescence in the crystalline, nanocrystalline, colloidal and thin film forms;13,23,24 sensitivity of the FEE to the amorphous/crystalline nature of these solids has also been investigated.25,26 Focusing on the fluorophore DADQ, we have shown recently that subtle structural modifications with methyl and ethyl groups can systematically tune the molecular structure, which in turn exerts a systematic impact on the molecular assembly in the crystal lattice and the FEE across the series.27 Molecular electronic structure and lattice energy calculations provided insight into the likely impact of intra and intermolecular excited state energy loss pathways in the family of molecules. A detailed analysis of the relative energy transfer rates (Förster and Dexter modes) by considering the relative orientation of molecules and their transition dipoles in the crystals revealed the inverse correlation with the FEE across the series. A follow-up investigation explored a new DADQ derivative that could be crystallized in three different lattice structures exhibiting not only very large, but also significantly varying FEE values.28 As the molecular structure is identical and rigidity of the environment similar across the series, the intramolecular effect on fluorescence emission were found to be nearly the same. The molecular orientation once again plays the crucial role in tuning the FEE, allowing an unequivocal establishment of the inverse correlation with the relative energy transfer rates.Analysis of other classes of fluorophores
The general applicability of our model can be explored using families of crystals that satisfy some basic criteria: (i) a common fluorophore with a well-defined molecular plane and only peripheral differences should be present; (ii) the crystal structures and fluorescence quantum yields in solution and solid state are known; (iii) the crystals within a family show a systematic and significant variation of the fluorescence. We have identified two such families reported in the literature, based on 1,4,6,9-tetraalkylanthracenes (TAA)29 and bis(piperidyl)anthracenes (BPA)30 (Fig. 1); in the TAA family, we have considered only those crystals which show substantial variation in the fluorescence quantum yield. Table 1 lists the fluorescence quantum yield reported for the various crystals and the FEE value estimated based on the respective solution quantum yields. Trends in the energy transfer rates in the crystals within a series is estimated by considering the relative orientation of the transition dipoles (Förster mode) and the π-conjugated fluorophore moieties (Dexter mode) of the molecules within reasonable cut-off distances; the relevant expressions have been described earlier.27 Transition dipole moments were calculated using TD-DFT (B3LYP/6-31G*) computations31 and their relative orientations determined from the reported crystal structures. A mean molecular plane was defined using all the atoms in the π-conjugated aromatic fluorophore and their relative orientations inferred from the crystal structure. The fluorophore in both the families is anthracene; its approximate length (9 Å) and width (5 Å) were taken as the cut-off distances for the molecular overlap, and all the near neighbours of a reference molecule considered for the transition dipole interactions. The spectral overlaps required in the energy transfer rate estimation32 were calculated using the normalized absorption and emission spectra reported for the solids. The relative energy transfer rates estimated for the two modes are collected in Table 1. Fig. 1 shows graphically, the relative energy transfer rates of the members of each family of crystals juxtaposed with the trends in the FEE values. The negative correlation between the two is clearly manifested; the FEE appears to be particularly sensitive to the Förster mode of energy transfer in these systems.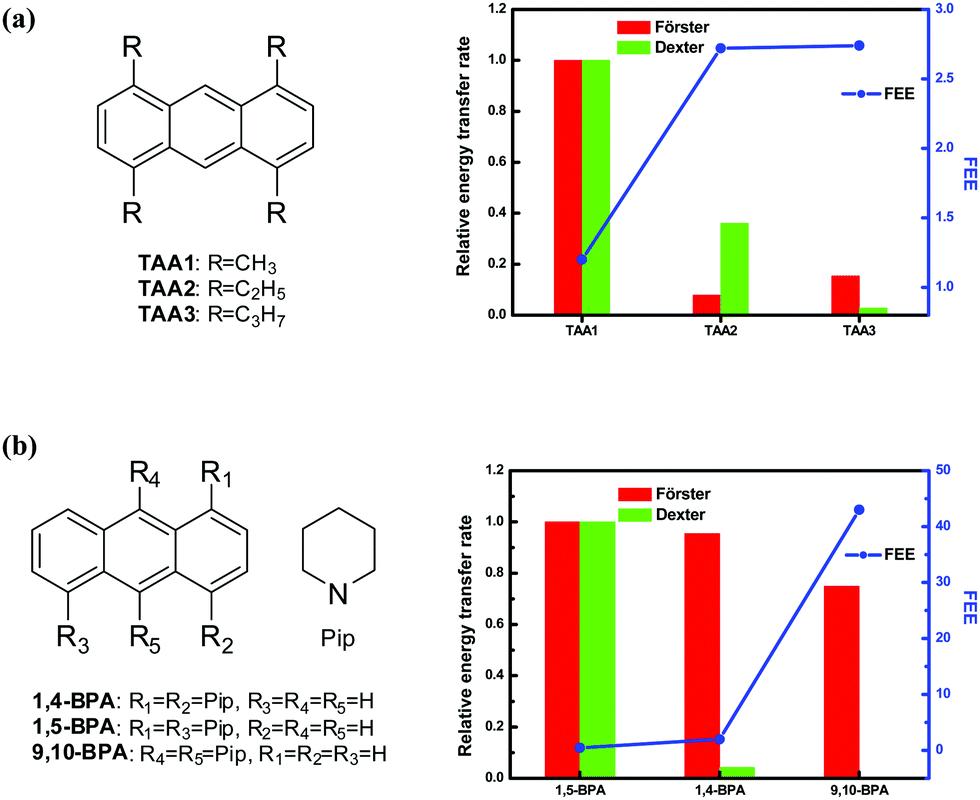 | ||
| Fig. 1 Molecular structure of the fluorophores (a) tetra(alkyl)anthracenes (TAA) and (b) bis(piperidyl)anthracenes (BPA),29,30 and the plot of relative energy transfer rates (Förster and Dexter modes); the FEE values are also plotted. | ||
| Crystal | ϕ (%) | FEE | Relative energy transfer rate | ||
|---|---|---|---|---|---|
| Solution | Solid | Förster | Dexter | ||
| TAA1 | 0.32 | 0.39 | 1.20 | 1.000 | 1.000 |
| TAA2 | 0.25 | 0.68 | 2.72 | 0.079 | 0.361 |
| TAA3 | 0.31 | 0.85 | 2.74 | 0.154 | 0.028 |
| 1,5-BPA | 0.77 | 0.34 | 0.44 | 1.000 | 1.000 |
| 1,4-BPA | 0.25 | 0.49 | 1.96 | 0.955 | 0.043 |
| 9,10-BPA | 0.02 | 0.86 | 43.00 | 0.750 | 0.000 |
Conclusion
Restriction of intramolecular structural relaxation and consequently non-radiative excited state decay is important for inducing enhanced fluorescence in molecular aggregates and crystals. The various studies highlighted establish unequivocally that the relative molecular orientations and the curtailment of intermolecular energy transfer are also critical in this context. A holistic evaluation of both intra and intermolecular factors is crucial for the effective design and development of molecular assemblies that can exhibit strong light emission useful in wide-ranging applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PS thanks the CSIR, New Delhi for a senior research fellowship. Financial support from the DST, New Delhi (J C Bose Fellowship) and infrastructure support from the DST, New Delhi (PURSE and FIST programs) and the UGC, New Delhi (CAS and UPE programs) are acknowledged with gratitude. We thank Prof. Chitoshi Kitamura (University of Shiga Prefecture, Japan) for providing the spectra of the TAA solids.References
- H. Langhals, T. Potrawa, H. Noth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 28, 478 CrossRef.
- U. Lemmer, S. Heun, R. F. Mahrt, U. Scherf, M. Hopmeier, U. Siegner, E. O. Göbel, K. Müllen and H. Bässler, Chem. Phys. Lett., 1995, 240, 373 CrossRef CAS.
- G. Y. Zheng and D. P. Rillema, Inorg. Chem., 1998, 37, 1392 CrossRef CAS PubMed.
- Z. Zhao, B. Hea and B. Z. Tang, Chem. Sci., 2015, 6, 5347 RSC.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- N. S. S. Kumar, S. Varghese, C. H. Suresh, N. P. Rath and S. Das, J. Phys. Chem. C, 2009, 113, 11927 CAS.
- Y. Ezhumalai, T. H. Wang and H. F. Hsu, Org. Lett., 2015, 17, 536 CrossRef CAS PubMed.
- R. Davis, N. S. S. Kumar, S. Abraham, C. H. Suresh, N. P. Rath, N. Tamaoki and S. Das, J. Phys. Chem. C, 2008, 112, 2137 CAS.
- J. Gierschner, L. Lüer, B. M. Medina, D. Oelkrug and H. J. Egelhaaf, J. Phys. Chem. Lett., 2013, 4, 2686 CrossRef CAS.
- J. Freudenberg, F. Rominger and U. H. F. Bunz, Chem. – Eur. J., 2015, 21, 16749 CrossRef CAS PubMed.
- S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G. I. Konishi, J. Am. Chem. Soc., 2016, 138, 8194 CrossRef CAS PubMed.
- F. Li, N. Gao, H. Xu, W. Liu, H. Shang, W. Yang and M. Zhang, Chem. – Eur. J., 2014, 20, 9991 CrossRef CAS PubMed.
- S. Jayanty and T. P. Radhakrishnan, Chem. – Eur. J., 2004, 10, 791 CrossRef CAS PubMed.
- A. Qin and B. Z. Tang, Aggregation-Induced Emission, Wiley, Chichester, 2014 Search PubMed.
- B. Liu, A. Pucci and T. Baumgartner, Mater. Chem. Front., 2017, 1, 1689 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- T. Zhang, Y. Jiang, Y. Niu, D. Wang, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2014, 118, 9094 CrossRef CAS PubMed.
- Q. Li and L. Blancafort, Chem. Commun., 2013, 49, 5966 RSC.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83 CrossRef CAS PubMed.
- D. Presti, L. Wilbraham, C. Targa, F. Labat, A. Pedone, M. C. Menziani, I. Ciofini and C. Adamo, J. Phys. Chem. C, 2017, 121, 5747 CAS.
- J. Shi, L. E. A. Suarez, S. Yoon, S. Varghese, C. Serpa, S. Y. Park, L. Lüer, D. Roca-Sanjuán, B. Milián-Medina and J. Gierschner, J. Phys. Chem. C, 2017, 121, 23166 CAS.
- Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- A. Patra, Ch. G. Chandaluri and T. P. Radhakrishnan, Nanoscale, 2012, 4, 343 RSC.
- B. Balaswamy, L. Maganti, S. Sharma and T. P. Radhakrishnan, Langmuir, 2012, 28, 17313 CrossRef CAS PubMed.
- C. G. Chandaluri and T. P. Radhakrishnan, Angew. Chem., Int. Ed., 2012, 51, 11849 CrossRef CAS PubMed.
- P. Srujana and T. P. Radhakrishnan, Angew. Chem., Int. Ed., 2015, 54, 7270 CrossRef CAS PubMed.
- P. Srujana, T. Gera and T. P. Radhakrishnan, J. Mater. Chem. C, 2016, 4, 6510 RSC.
- P. Srujana and T. P. Radhakrishnan, Chem. – Eur. J., 2018, 24, 1784 CrossRef CAS PubMed.
- C. Kitamura, Y. Abe, N. Kawatsuki, A. Yoneda, K. Asada, T. Kobayashi and H. Naito, Mol. Cryst. Liq. Cryst., 2007, 474, 119 CrossRef CAS.
- S. Sasaki, K. Igawa and G. Konishi, J. Mater. Chem. C, 2015, 3, 5940 RSC.
- M. J. Frisch et al. , Gaussian 09 (Revision C.01), Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- P. Ceroni, The Exploration of Supramolecular Systems and Nanostructures by Photochemical Techniques, Lecture Notes in Chemistry, Springer Science & Business Media, 2011, vol. 78, ch. 2 Search PubMed.
| This journal is © the Partner Organisations 2018 |