
Human brain organoid-on-a-chip to model prenatal nicotine exposure†
Yaqing
Wang
acd,
Li
Wang
ad,
Yujuan
Zhu
acd and
Jianhua
Qin
 *abc
*abc
aDivision of Biotechnology, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: jhqin@dicp.ac.cn; Fax: +86 411 84379059
bCenter for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai 200031, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dKey Laboratory of Separation Sciences for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 7th February 2018
Abstract
Nicotine has been recognized to trigger various neuronal disabilities in the fetal brain and long-lasting behavioral deficits in offspring. However, further understanding of fetal brain development under nicotine exposure is challenging due to the limitations of existing animal models. Here, we create a new brain organoid-on-a-chip system derived from human induced pluripotent stem cells (hiPSCs) that allows us to model neurodevelopmental disorders under prenatal nicotine exposure (PNE) at early stages. The brain organoid-on-a-chip system facilitates 3D culture, in situ neural differentiation, and self-organization of brain organoids under continuous perfused cultures in a controlled manner. The generated brain organoids displayed well-defined neural differentiation, regionalization, and cortical organization, which recapitulates the key features of the early stages of human brain development. The brain organoids exposed to nicotine exhibited premature neuronal differentiation with enhanced expression of the neuron marker TUJ1. Brain regionalization and cortical development were disrupted in the nicotine-treated organoids identified by the expressions of forebrain (PAX6 and FOXG1), hindbrain (PAX2 and KROX20) and cortical neural layer (preplate TBR1 and deep-layer CTIP2) markers. Moreover, the neurite outgrowth showed abnormal neuronal differentiation and migration in nicotine-treated brain organoids. These results suggest that nicotine exposure elicits impaired neurogenesis in early fetal brain development during gestation. The established brain organoid-on-a-chip system provides a promising platform to model neurodevelopmental disorders under environmental exposure, which can be extended for applications in brain disease studies and drug testing.
Introduction
The human brain is a highly complex organ with unique structures and functions that is vulnerable to environmental injury and toxicants during early development.1 Nicotine is a major component of inhaled tobacco smoke and is thought to elicit fetal brain dysfunction as a potential neuroteratogen2 [Fig. 1A]. Several studies have reported that prenatal nicotine exposure (PNE) during pregnancy may be associated with neurodevelopmental disorders and neurobehavioral impairments in the offspring, including attention deficit/hyperactivity disorder (AD/HD), cognitive dysfunction, and learning disabilities.3 Thus, nicotine exposure may give rise to long-lasting changes in the structure and function of the developing fetal brain.4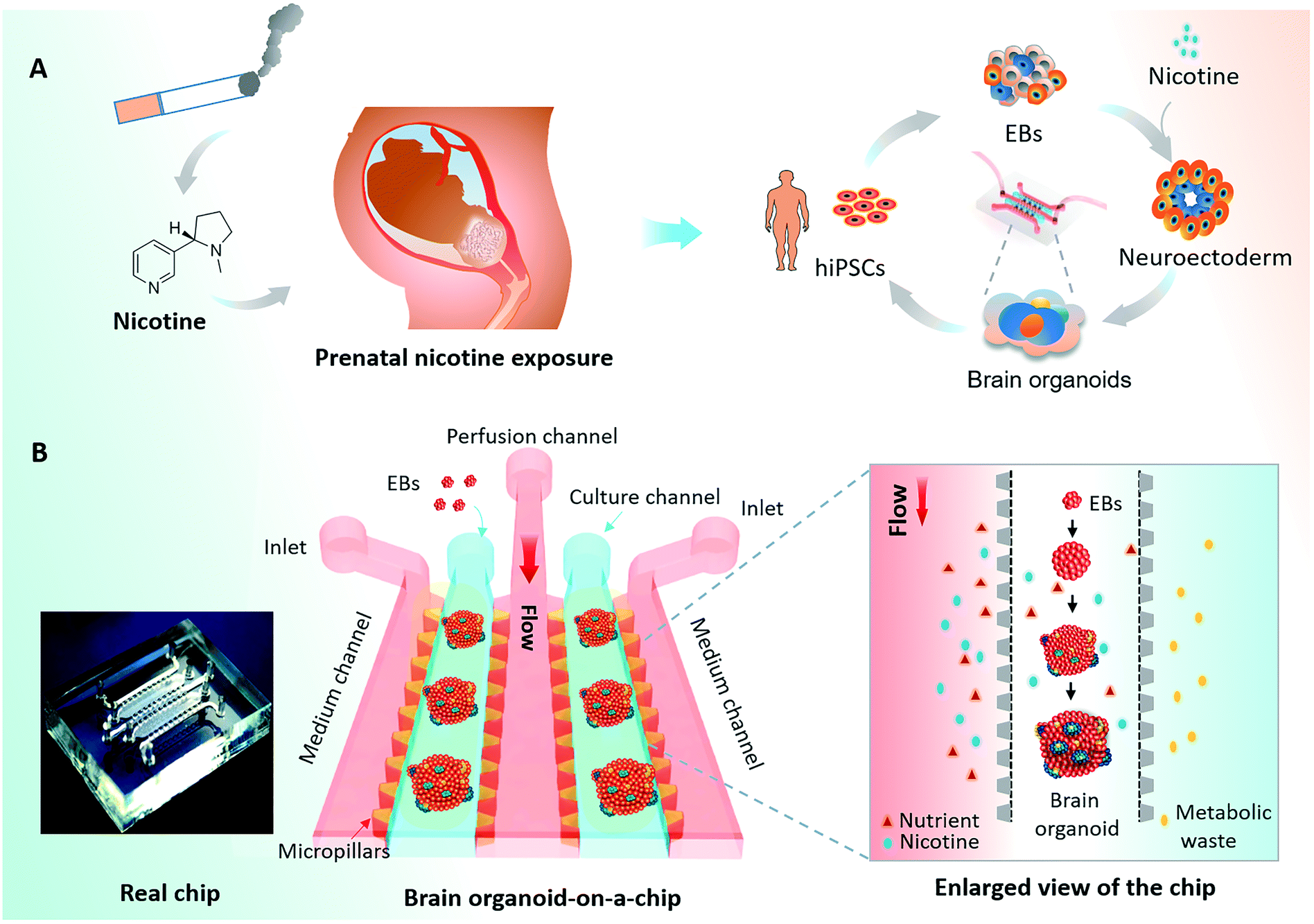 | ||
| Fig. 1 Schematic diagram of the brain organoid-on-a-chip system for modeling prenatal nicotine exposure (PNE). (A) Tobacco smoking during pregnancy is related to neurodevelopmental disorders in fetus with PNE. The process of hiPSCs-based brain organogenesis on a chip and its use to study the effects of nicotine exposure on brain organoid development. (B) The configuration of the brain organoid-on-a-chip device and enlarged view of the procedures for brain organoid generation on a chip. The brain organoid-on-a-chip system was established for EBs culture, neural differentiation and formation of brain organoids by integrating 3D Matrigel and fluid flow. By using this system, we explored the effects of nicotine exposure on brain development at early stages. | ||
Most studies on the effects of PNE on neurogenesis during brain development have been performed in animal models.5–8 Given the dramatic differences between humans and other mammalians in terms of histomorphology, intricate neurodevelopment, and spatiotemporal self-organization,9,10 it is difficult to acquire accurate information about the effects of PNE on human fetal brain development. Furthermore, the existing studies focused mostly on PNE-induced neurodevelopment disorders during the second and third trimesters.6,8,11,12 Few studies have investigated the effects of PNE on the developing fetal brain in the first trimester when the fetal brain is highly susceptible to external factors.
Brain organoids are the 3D multicellular clusters that derived from human induced pluripotent stem cells (hiPSCs) via self-renewal and self-organization. They can resemble the specific architecture and functions of the native brain, recapitulating specific characteristics of the early or even mid-prenatal brain.13,14 The in vitro brain organoid models provide the unique opportunity to obtain information about early brain development and elucidate the mechanisms of neurologic disorders.15–17 To date, brain organoid models have been used to study neurologic diseases, such as microcephaly13 and Zika virus infection,15 revealing their great potential to investigate the origins and pathology of psychiatric diseases.
Typically, brain organoids are generated by the differentiation of multicellular aggregates from stem cells, termed embryoid bodies (EBs), in 3D suspended culture.13,15,18–22 However, the controlled microenvironment factors are critical for maintaining the development of brain organoids in vitro and the establishment of a realistic modeling of developmental disorders.23–26 Moreover, the existing approaches for organoid formation often require multiple steps and complicated instruments with high cost, which may lead to potential cells contamination and large consumption of culture media. Advances in organs-on-a-chip technology provide a promising platform to engineer the stem cell organoids with essential structural and physiological features in a controlled manner.27,28 The organs-on-a-chip systems can mimic the in vivo-like cellular microenvironment by providing precise control over niche factors such as 3D matrix, cell–cell interactions and mechanical fluidic cues. The organs-on-a-chip techniques have been recently developed to create a variety of biomimetic organ models, such as lung,29 liver,30 heart31 and neural networks.32
In this work, we proposed a new strategy to generate hiPSCs-derived brain organoids in a controlled manner by combining stem cell biology with organ-on-a-chip technology, thereby creating brain organoid-on-a-chip system, then applied it to investigate the impact of nicotine exposure on early neurodevelopment. The established system enabled 3D culture of EBs, in situ neural differentiation and formation of brain organoids under perfused cultures. The generated organoids were identified with brain-specific features in the aspects of neural differentiation, regional and cortical brain organization. The brain organoids exposed to nicotine were examined by immunohistochemical analysis and real-time PCR, indicating the impaired neurogenesis in developmental brain organoids under nicotine exposure. The presented brain organoid-on-a-chip system may provide a versatile and attractive platform to study neurodevelopmental disorders at early stages during gestation.
Materials and methods
Fabrication of the microfluidic chip
The microfluidic chip made of polydimethylsiloxane (PDMS) was fabricated using a conventional soft lithography procedure. The chip contained two layers (i.e., top and bottom). The top layer consisted of two parallel culture chambers and three separate medium channels. The height of the top layer channels was approximately 600 μm. Two parallel culture chambers (width: 2.5 mm; length: 14 mm) were separated by a central channel (width: 1 mm; length: 20 mm), which contained ten trapezoid pillar array structures at both sides of the channel for interconnection. The two parallel chambers were used for the 3D culture and formation of brain organoids. The central channel served as the fluid flow passageway with inlet and outlet holes of 0.8 mm in diameter, which were created with a sharp needle. The channels at the two sides of the chip were filled with cell culture medium. The top and bottom layers were molded by the PDMS pre-polymer containing a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) mixture of the PDMS precursor and curing agent. The pre-polymer in the channels was then polymerized by thermal curing in an 80 °C dry oven for 1 h. The top layer of the chambers was adhered to the plane PDMS layer after oxygen plasma treatment. Finally, the chips were autoclaved to restore the hydrophobicity of PDMS and kept sterile until use.
:1 (w/w) mixture of the PDMS precursor and curing agent. The pre-polymer in the channels was then polymerized by thermal curing in an 80 °C dry oven for 1 h. The top layer of the chambers was adhered to the plane PDMS layer after oxygen plasma treatment. Finally, the chips were autoclaved to restore the hydrophobicity of PDMS and kept sterile until use.
Generation of EBs
EBs are the 3D multicellular aggregates formed from the hiPSCs via 3D culture. The hiPSCs derived from skin fibroblasts of a healthy individual (provided as a gift from Dr. Ning Sun)33–37 were cultured in mTeSR1 medium in six-well plates coated with 1.5% Matrigel (BD Biosciences) in DMEM/F12 medium (Invitrogen). The medium was changed daily. Cells at 75% to 90% confluence were passaged at a 1:5 ratio by digestion with Accutase (Sigma-Aldrich). The pluripotency of the hiPSCs was regularly verified by cell morphology and immunostaining for the pluripotent marker.
Knockout serum replacement (KSR) medium was prepared using DMEM/F12 medium supplemented with 20% KSR (Invitrogen), 1% GlutaMAX (Invitrogen), 1% minimum essential media-nonessential amino acids (MEM-NEAA, Invitrogen), 0.2 mM 2-mercaptoethanol (Sigma-Aldrich), and 1% penicillin–streptomycin (Sigma). Dissociated hiPSCs (∼5 × 106 cells) were resuspended in KSR medium and immediately added to 10 μM ROCK inhibitor Y27632 (Millipore) and 4 ng ml−1 basic fibroblast growth factor (bFGF, Peprotech). Initially, the suspension of hiPSCs was seeded in a concave well (diameter: 800 μm) to form the EBs. The generated EBs were 100 to 300 μm in diameter. The EBs were then transferred to a low-adhesion plate, and fresh KSR medium was changed every other day. On day 5, the KSR medium was replaced with neural induction medium (NIM) that contained DMEM/F12, 1% GlutaMAX, 1% MEM-NEAA, 1 μg ml−1 heparin (Sigma), 1% N2 supplement (Invitrogen), and 1% penicillin–streptomycin. The EBs were then cultured for another 6 days with NIM.
Formation of brain organoids on chip and nicotine exposure
On day 11, approximately 300 EBs per milliliter were suspended in ice-cold Matrigel. Next, 40 to 50 μl of the EB–Matrigel mixture was carefully pipetted into each hydrogel channel of the microfluidic chip. The chip was then incubated at 37 °C for 25 to 30 min. EBs were immobilized on a chip by gelation of Matrigel, which enabled extended 3D culture and allowed in situ tracking without removal of EBs from the channels. A flexible polytetrafluoroethylene (PTFE) tube was then connected to the inlet of the central medium channel. This system provided a continuous flow of NDM at the rate of 25 μl h−1 using a syringe pump. EBs were further differentiated and organized into brain organoids under continuous perfusion conditions for 5 to 30 days in neural differentiation medium (NDM). Neural differentiation medium (NDM) was prepared using a mixture of DMEM/F12 and Neurobasal medium (Invitrogen) at a volume ratio of 1:1, supplemented with 1% B27 supplement, 0.5% N2 supplement, 0.2 mM 2-mercaptoethanol, 1% GlutaMAX, 0.5% MEM-NEAA, and 1% penicillin–streptomycin. Brain organoid growth was monitored by bright field images.
Brain organoids encapsulated on a chip were cultured with a continuous flow of NDM containing 1 μM or 10 μM nicotine for 5 or 14 days. The control group remained in normal NDM.
TUNEL assay
The apoptosis of cells within organoids was assessed using the TUNEL assay after 33 days of culture in the system. Tissue cryosections were treated with TUNEL kit reagents (Life Technologies) according to the manufacturer's instructions. After incubation with staining reagents, the samples were washed three times with PBS, and images were acquired using a confocal microscope. The viability of cells was quantified using ImageJ (NIH) to calculate the percentage of TUNEL positive cells.Cryosection and immunohistochemistry
On set days during the development period, brain organoids were fixed in 4% paraformaldehyde (PFA) for 25 to 30 min at room temperature. The fixed organoids were then washed with PBS three times, transferred to a 30% sucrose solution, and incubated at 4 °C overnight. The dehydrated tissues were then embedded in OCT compound (SAKURA), snap frozen, and stored at −80 °C. For immunofluorescence staining, 10 μm thick sections were obtained using a cryostat (Leica). Cryosections on adhesive slides were washed with PBS to remove excess OCT and permeabilized with 0.25% Triton X-100 diluted in PBS for 5 min at room temperature. The sections were blocked with 10% goat serum (Solarbio, SL1) for 1 hour and then incubated overnight at 4 °C with a primary antibody diluent. The following primary antibodies were used for immunocytochemical analysis: TUJ1 (mouse, 1:500, BioLegend 801201), SOX2 (rabbit, 1:500, Cell Signaling 3579), NESTIN (mouse, 1:500, Santa Cruz sc-20978), PAX6 (rabbit, 1:300, BioLegend PRB-278P), PAX2 (mouse, 1:500, Abnova H00005076-M01), ISL1 (mouse, 1:100, Thermo MA5-15515), CTIP2 (rat, Abcam ab18465, 1:500), TBR1 (rabbit, Abcam ab31940, 1:200), active Caspase 3 (rabbit, Abcam ab32042, 1:250). Samples were washed with PBS three times to remove excess primary antibody and then incubated with secondary antibody conjugated to Alexa Fluor 488 or 594 dyes (1:100) for 1 hour at room temperature. Cell nuclei were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Life Technologies, 1:4000) for 15 min. All images were photographed using a confocal microscope (Olympus). ImageJ (NIH) was used to merge images and to uniformly adjust the brightness or contrast.
Real-time PCR
Total mRNA was isolated from brain organoids or hiPSCs using Trizol reagent (TAKARA) The final mRNA concentration was adjusted to 500 ng μl−1. The RNA concentration and quality were determined using a Nanodrop (Thermo Fisher Scientific). The cDNA was produced from 1 μg RNA and then amplified using Ex Taq DNA polymerase (Takara) under the following reaction conditions: denaturation at 94 °C for 1 min, annealing at 58 °C for 45 s, and extension at 72 °C for 30 s, cycle number of 30. Primer pairs used were as follows: NESTIN forward: 5′-GAA ACA GCC ATA GAG GGC AAA-3′, reverse: 5′-TGG TTT TCC AGA GTC TTC AGT GA-3′; TUJ1 forward: 5′-CTC AGG GGC CTT TGG ACA TC-3′, reverse: 5′-CAG GCA GTC GCA GTT TTC AC-3′; PAX6 forward: 5′-AGT TCT TCG CAA CCT GGC TA-3′, reverse: 5′-ATT CTC TCC CCC TCC TTC CT-3′; FOXG1 forward: 5′-AGG AGG GCG AGA AGA AGA AC-3′, reverse: 5′-TGA ACT CGT AGA TGC CGT TG-3′; PAX2 forward: 5′-CTG GGC AGC AAC GTG TCA-3′, reverse: 5′-GAG TGG TGC TCG CCA TGT C-3′; ISL1 forward: 5′-GCT TTG TTA GGG ATG GGA AA-3′, reverse: 5′-ACT CGA TGT GAT ACA CCT TGG A-3′; KROX20 forward: 5′-TTG ACC AGA TGA ACG GAG TG-3′, reverse: 5′-CTT GCC CAT GTA AGT GAA GGT-3′; TBR1 forward: 5′-GAC TCA GTT CAT CGC CGT CA-3′, reverse: 5′-TCG TGT CAT AAT TAT CCC GAA ATC C-3′; CTIP2 forward: 5′-CAG AGC AGC AAG CTC ACG-3′, reverse: 5′-GGT GCT GTA GAC GCT GAA GG-3′; β-Actin forward: 5′-AAA TCT GGC ACC ACA CCT TC-3′, reverse: 5′-AGA GGC GTA CAG GGA TAG CA-3′.Neurite outgrowth assay
Brain organoids on a chip treated without or with nicotine (1 μM or 10 μM) on day 25 were transferred into PBS buffer and incubated with 1 mg ml−1 dispase for about 5 min at room temperature. The organoids were then gently mechanically disrupted into small tissue blocks by pipetting. The small tissue blocks were reseeded in Matrigel-coated plates and cultured in NDM with or without nicotine. The medium was changed every 2 days.Neurite outgrowth was traced in brain tissues attached to the plate on day 1, day 3 and day 5 by immunofluorescence staining for TUJ1 marker. Neurites projecting from at least eight brain tissues for each group (control and nicotine-treated group) were quantified. Various brain tissues were randomly selected. The length of the projected neurites was analyzed by Image J (NIH).
Statistical analysis
Statistical analysis of data is expressed as means ± SDs. P values were calculated using Student's t-test. The statistical significance thresholds were set at *p < 0.05, **p < 0.01, and ***p < 0.001. Cell counting for immunostaining images was analyzed with Image-Pro Plus 6.0.Results and discussion
Generation of hiPSCs-based brain organoids on chip
The process of hiPSCs-derived brain organoids formation often involves the production of embryoid bodies (EBs), neuroectoderm differentiation, and self-organization of brain organoids. To explore the effects of nicotine exposure on brain organoids development at early stages, we created a brain organoid-on-a-chip system to generate brain organoids in a controlled manner [Fig. 1A]. The brain organoid-on-a-chip system was designed with five parallel functional channels interconnected by micropillar structures, in which two culture channels permitted the culture of EBs by infusion with 3D Matrigel, and the central medium channel allowed the perfusion of fluid flow [Fig. 1B]. With this design, the system enabled 3D culture and organization of brain organoids under continuous perfusion cultures.To generate brain organoids on a chip, EBs were initially formed by the 3D culture of hiPSCs in an ultra-low attachment plate. EBs were then suspended in Matrigel, and the mixture was infused into the chip device on day 11. As shown in Fig. 2A, the neuroepithelial populations with large rosette-like structures were formed and rapidly expanded in 15 to 20 days. Finally, these neuroepithelial spheroids were rapidly developed into millimeter-sized complex brain organoids on a chip. To assess the growth of brain organoids, tissue morphology and viability were tested during the developmental period. The brain organoids formed large and polarized neuroepithelial surrounding a fluid-filled cavity with the characteristic expression of CD133 at the apical surface, reminiscent of the ventricular zone [Fig. 2B]. Minimal cell death was observed in the core of the brain organoids under perfused culture by TUNEL assay, indicating the high viability of the brain organoids on the chip with sufficient nutrient exchange [Fig. 2C]. These results suggest that the brain organoid-on-a-chip system offered a controlled cellular microenvironment for extended growth of EBs, in situ neural differentiation, and formation of brain organoids by incorporation of 3D Matrigel and mechanical fluid flow.
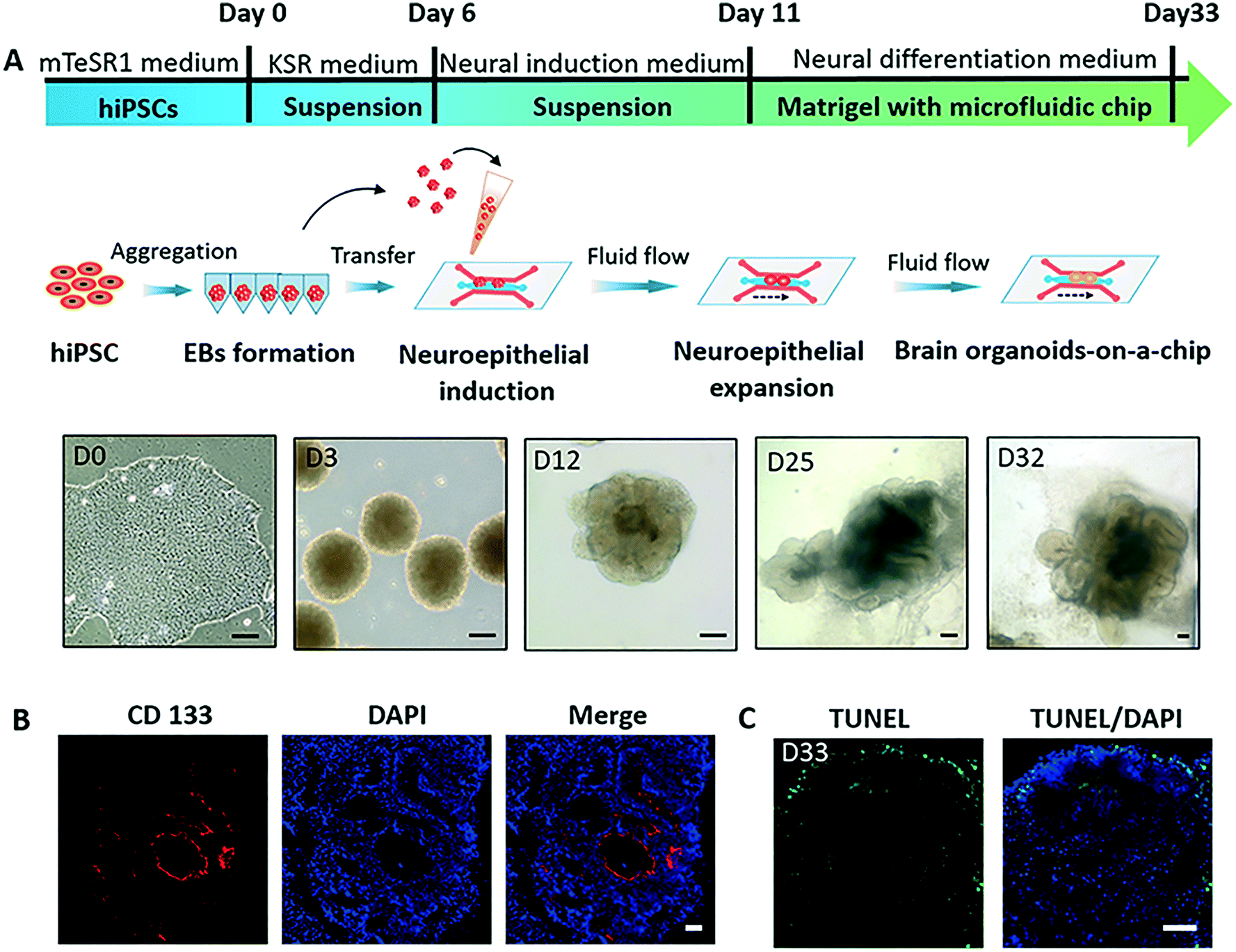 | ||
| Fig. 2 Generation of brain organoids derived from hiPSCs on chip. (A) Schematic procedure and representative microscopic images of hiPSCs-derived brain organoid development on a chip. Scale bar: 100 μm. (B) Immunohistochemical staining of frozen sections for the expression of apical marker CD133 in brain organoids on day 33. DAPI (blue) marks nuclei. Scale bars: 50 μm. (C) TUNEL staining of frozen sections showed a few dead cells within the brain organoids on day 33. Green fluorescence represents dead cells. DAPI (blue) marks nuclei. Scale bars: 50 μm. | ||
Characterization of brain organoids on chip
Native brain development exhibits an orderly sequence of processes that involve the formation of distinct brain regions and hierarchical cortical structure by the radial organization of progenitor zones and neuronal migration.38–40 Therefore, we further examined the detailed features of neural differentiation, specific brain regionalization, and cortical organization in the brain organoids. Initially, the neural progenitor markers NESTIN and SOX2 were identified by immunohistochemical staining and revealed effective neural induction in brain organoids on day 33 [Fig. 3A]. Early brain regionalization during brain organoid development was identified by staining for specific forebrain (PAX6) and hindbrain (PAX2 and ISL1) markers, indicating the presence of both populations in brain organoids.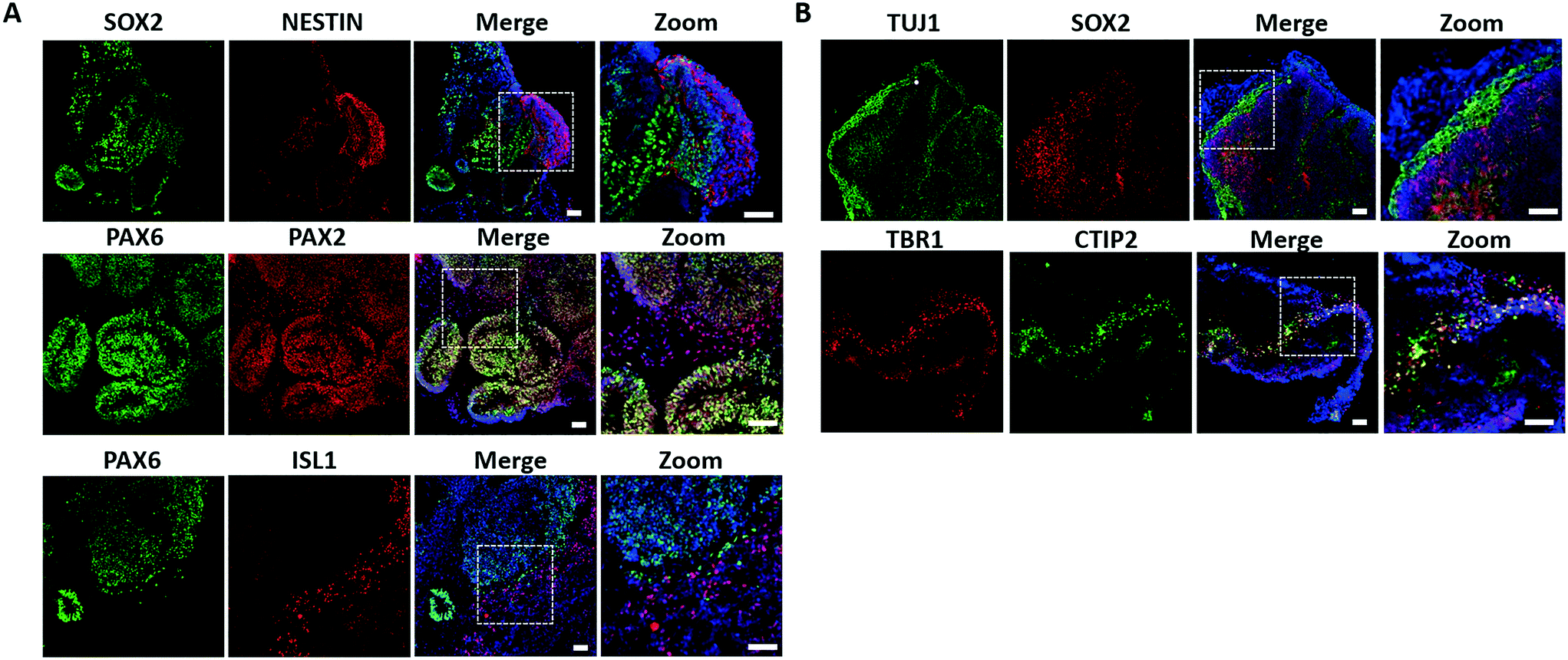 | ||
| Fig. 3 Characterization of hiPSCs-derived brain organoids on day 33. (A) Immunohistochemical staining was performed for the expression of neural progenitor markers (NESTIN, SOX2) and specific brain regions markers (forebrain, PAX6; hindbrain, PAX2 and ISL1) in brain organoids on day 33. (B) Expression of the neural progenitors (SOX2), neurons (TUJ1), and cortical layer markers (TBR1 and CTIP2) were identified in organoids by immunohistochemical analysis. Scale bars = 50 μm. | ||
As brain development proceeded, the hierarchical structure in the prefrontal cortex was formed by neuronal differentiation and migration. To examine the features of cortical neurogenesis, we stained for the early neurons (TUJ1), preplate (TBR1), and deep-layer neurons (CTIP2) in brain organoids on 33 days of differentiation [Fig. 3B]. Brain organoids displayed a large contiguous structure with a population of progenitors (SOX2+) and neuronal regions (TUJ1+) that resembled the heterogeneous regions. Furthermore, the early-born neuronal marker CTIP2 was located adjacent and peripheral to the TBR1 preplate, suggesting the initial formation of the cortical plate layer. Taken together, these results demonstrated the efficient organization and formation of brain organoids on the chip that recapitulated key features in the developing human fetal brain at early stages.
Effects of nicotine exposure on neuronal differentiation
Because the brain organoids replicated early fetal brain development, we next explored the effects of nicotine on neuronal differentiation using the brain organoid-on-a-chip system. The plasma nicotine concentration in heavy cigarette smokers reached values of 0.6 μM or higher.41,42 Thus, brain organoids were initially exposed to a physiological relevant concentration of nicotine (1 μM). Because the effects of nicotine on fetal brain development are dependent on concentration, exposure time, and target cells, we treated brain organoids with different doses of nicotine (0, 1 and 10 μM) for different time periods (0, 5 and 14 days). Moreover, the organoids were exposed to nicotine during the active period of proliferation of neuroepithelium and neurogenesis (from day 11).The neuronal differentiation within nicotine-exposed brain organoids was explored by immunofluorescence staining and quantifications for neural progenitors (SOX2) and neurons (TUJ1) on day 16 and day 25 [Fig. 4A and B]. The data showed a significant increase in the populations of TUJ1-positive neurons and no substantial changes in SOX2+ regions, suggesting premature neural differentiation in organoids treated with 1 or 10 μM nicotine for 5 days. With prolonged nicotine treatment for 14 days, the difference in neural differentiation became more visible. In addition, the organoids displayed a high level of TUJ1 expression with low-dose nicotine treatment for 5 days by real-time PCR assay, whereas no significant changes in NESTIN expression were observed [Fig. 4C]. Moreover, TUJ1 expression was greatly increased in tissue exposed to nicotine for 14 days, indicating the time-dependent effects of nicotine on neuronal differentiation during brain organoid development [Fig. 4D]. Furthermore, we examined the cell apoptosis in brain organoids with and without nicotine treatment by immunostaining for activated caspase 3 [Fig. S1†]. Caspase 3 is closely involved in cell apoptosis and neuronal death in neural diseases as a member of the caspase family. The data showed that nicotine induced significant cell apoptosis in brain organoids treated with different doses of nicotine (1 μM and 10 μM), and the effects were dose-time dependent. In addition, as shown in Fig. 4, the expressions of TUJ1 positive cells were higher in brain organoids with low-dose nicotine treatment than that with high-dose nicotine treatment. This may be explained by the high dose of nicotine induced increased cell apoptosis during organoid development. These results confirmed that nicotine exposure led to premature neuronal differentiation and cell apoptosis in brain organoids.
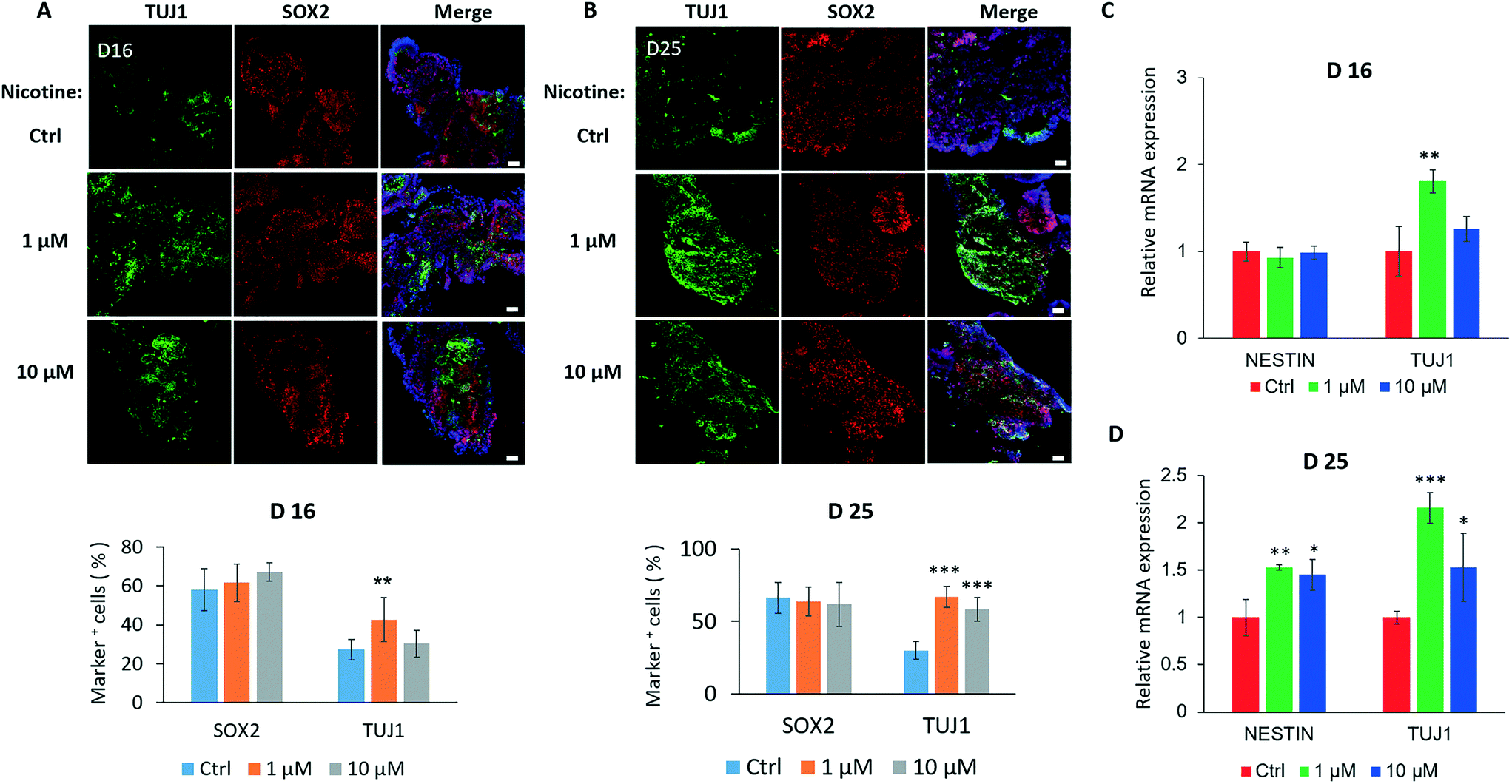 | ||
| Fig. 4 Nicotine-induced premature neuronal differentiation in brain organoids. (A and B) The expressions of TUJ1 and SOX2 were identified by immunohistochemical analysis and quantifications for the percentage of TUJ1+ and SOX2+ cells in brain organoids treated with and without nicotine (1 μM and 10 μM) on day 16 (A) and day 25 (B). Scale bars, 50 μm. The data represent means of 6 replicates ± SD. Student's t-test, **p < 0.01, ***p < 0.001. (C and D) NESTIN and TUJ1 expression by real-time PCR in brain organoids in the presence or absence of nicotine on day 16 (C) and day 25 (D). The fold changes were calculated by genes expression in organoids with nicotine treatment relative to that without nicotine treatment. The β-actin expression was used for normalization. Data are mean ± SD. Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Changes of brain regional organization and cortical development in brain organoids under nicotine exposure
As development proceeds in brain organoids, we further examined the effects of nicotine on the differentiation of specific brain regions. As shown in Fig. 5A, nicotine significantly inhibited proliferation of PAX6+ cells in organoids on day 16 and 25, but facilitated the development of the PAX2+ area compared to the control by immunohistochemistry analysis and quantifications. To analyze nicotine-induced changes in the regional development of the brain at the mRNA level, the expression levels of the forebrain (PAX6 and FOXG1) and hindbrain (KROX20) markers were measured by real-time PCR [Fig. 5C]. Consistently, brain organoids with nicotine treatment for 5 days exhibited decreased expression of PAX6 and FOXG1 in a dose-dependent manner and increased KROX20 expression. Moreover, the hindbrain markers PAX2 and ISL1 were also examined by real-time PCR, which displayed significant increased expression [Fig. S2A†]. With extended nicotine exposure, the trends of changes in the regional differentiation of the brain were almost constant [Fig. 5B and D and S2B†]. These data suggest a dysregulated development of forebrain and hindbrain populations in organoids under nicotine exposure. It is known that the inactivation or loss of transcription factor FOXG1 can lead to severe hypoplasia of the telencephalon by premature progenitor cell cycles,43 and KROX20 may play a key role in the regulation of pattern formation in the developing hindbrain.44,45 We thus speculated that nicotine-induced disruption of brain regionalization might be implicated in the dysregulation of the cell cycle and regional pattern formation. These alterations may further inhibit brain organogenesis during embryonic development.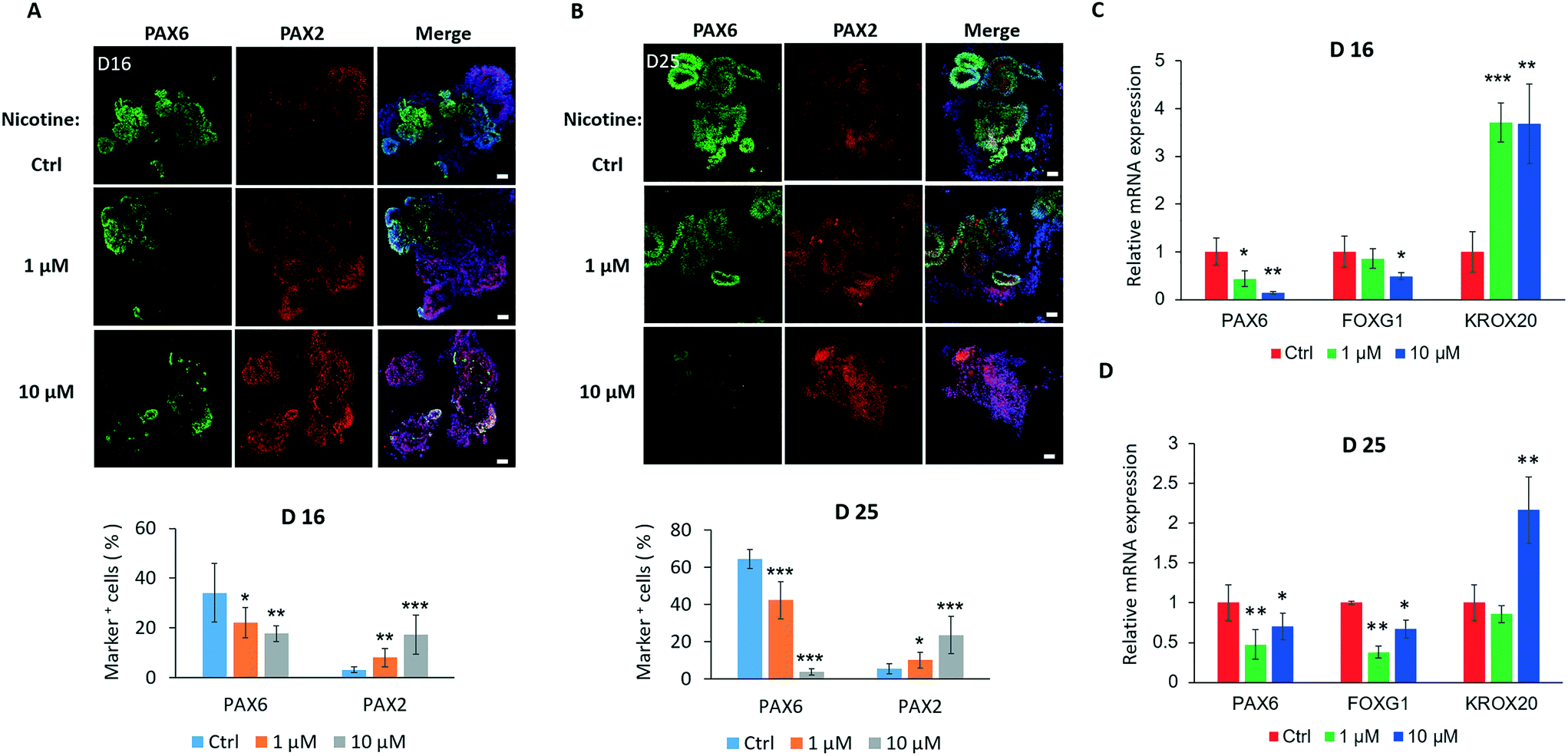 | ||
| Fig. 5 Impairment of brain regional organization in nicotine-exposed brain organoids. (A and B) Immunohistochemical staining for forebrain (PAX6) and hindbrain (PAX2) markers and quantifications for the percentage of PAX6+ and PAX2+ cells in brain organoids with and without nicotine treatment (1 μM and 10 μM) on day 16 (A) and day 25 (B). Scale bars, 50 μm. The data represent means of 6 replicates ± SD. Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001. (C and D) Expression of forebrain (PAX6, FOXG1) and hindbrain (KROX20) markers by real time-PCR in brain organoids with or without nicotine exposure on day 16 (C) and day 25 (D). The fold changes were normalized to β-actin. Data are mean ± SD. Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001. | ||
The process of neocortical development during human gestation involves neurogenesis and neuronal subtype specification.46 Cortical neuron layers were examined to further explore the effects of nicotine exposure on brain organoid development. Nicotine was administered to brain organoids from days 35 to 40, during which the initial cortical organization was observed. We examined the expression of preplate marker TBR1 and deep-layer marker CTIP2 following a 5 day treatment of brain organoids with nicotine. Brain organoids displayed markedly decreased TBR1 expression after treatment with either 1 μM or 10 μM nicotine by immunohistochemical analysis [Fig. 6A] and quantifications [Fig. 6B]. Moreover, the effect of nicotine on the TBR1 preplate marker was dose-dependent. Conversely, a significant increase of CTIP2-positive cells was observed in organoids treated with either 1 μM or 10 μM nicotine. In addition, the expressions of TBR1 and CTIP2 in nicotine treated organoids were also examined by real-time PCR, which were consistent with the results in immunohistochemical analysis [Fig. 6C]. Notably, more increased expressions of CTIP2 positive cells were observed in brain organoids with low-dose nicotine treatment than that with high-dose nicotine treatment. This may be explained by the high dose of nicotine-induced decreased neuronal differentiation and increased cell apoptosis. These data show that nicotine exposure impaired cortical development in brain organoids, which might help to understand various postnatal cognitive dysfunctions, such as autism observed in individuals with PNE.
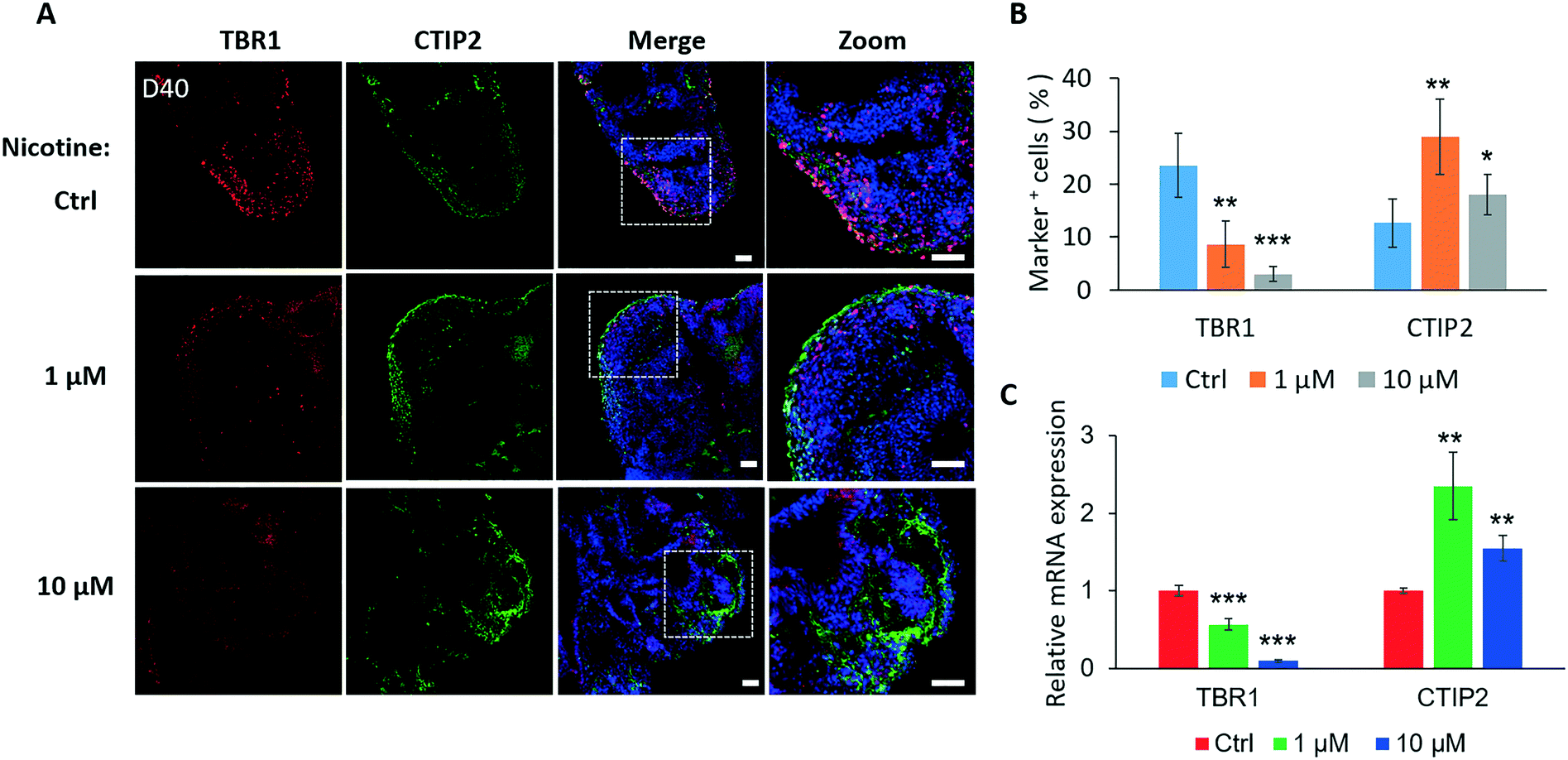 | ||
| Fig. 6 Nicotine disrupted the differentiation of cortical neuronal layers in brain organoids on day 40. (A) Immunohistochemical staining for preplate TBR1 and deep-layer neuron CTIP2 in brain organoids with or without nicotine treatment for 5 days. Scale bars, 50 μm. (B) Quantifications for the percentage of TBR1+ and CTIP2+ cells in sample images of brain organoids with and without nicotine treatment for 5 days. The data represent mean ± SD. Student's t-test, *p < 0.05, **p < 0.01, ***p < 0.001. (C) Relative expression of TBR1 and CTIP2 by real-time PCR revealed the increased expression of the deep cortical layer and reduced preplate differentiation. Data are mean ± SD. Student's t-test, **p < 0.01, ***p < 0.001. | ||
Abnormal neurite outgrowth in nicotine-exposed brain organoids
Neurite outgrowth is a fundamental process for neural behavior that involves neuronal differentiation and migration during brain development, which is essential for the communication of the nervous system.47 Because our results identified the influences of nicotine on cortical neuronal differentiation, further study was required to examine whether nicotine disturbed neurite outgrowth. To test neural projection, nicotine exposed organoids on day 25 were digested and gently mechanically disrupted into small tissue blocks by pipetting and seeded in Matrigel coated plates. As shown in Fig. 7A, the neurites rapidly projected from the periphery of small colonies and formed complex neural networks in radial orientation on the plate. The average neurite lengths under different conditions were quantified to assess the effects of nicotine on neurite outgrowth [Fig. 7B]. Notably, the group treated with low-dose nicotine showed increased neurite length in the first 5 days, indicating that nicotine at a low concentration triggered neuronal outgrowth. This result might be explained by premature neuronal differentiation under low-dose nicotine exposure in brain organoids. In contrast, neurite length was significantly reduced after treatment with high-dose nicotine (10 μM) as time was extended, which suggested a disturbed neurite outgrowth. These data suggest the impairment of neural differentiation and outgrowth in the developing brain under PNE.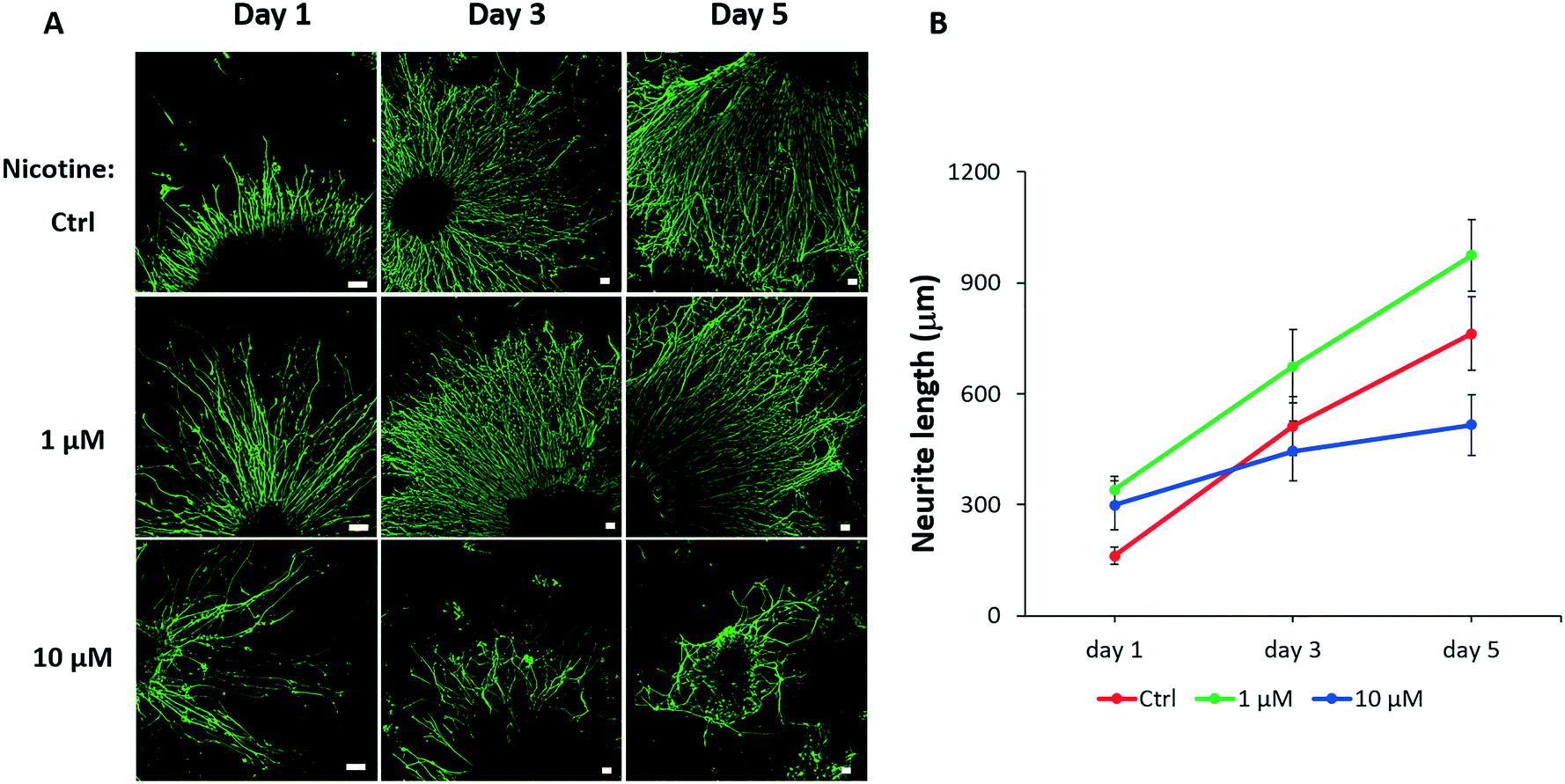 | ||
| Fig. 7 Abnormal neurite outgrowth induced by nicotine exposure. (A) Immunocytochemical staining of neurons (TUJ1) projecting from the periphery of tissue blocks in the control and nicotine-exposed brain organoids at 1, 3 and 5 days after seeding on Matrigel-coated plates. Scale bars, 50 μm. (B) Quantified analysis of neurite length by Image J software. The data represent mean ± SD. Student's t-test. More than eight tissue colonies for each sample were randomly analyzed. | ||
Conclusion
We presented a simple and robust organ-on-a-chip system that allowed to generate hiPSCs-derived brain organoids and to model neurological disorders under prenatal nicotine exposure for the first time. The generated brain organoids displayed essential well-defined brain features, reminiscent of fetal brain development at early stages. In this model, nicotine exposure led to premature neuronal differentiation, disrupted brain regional organization, abnormal cortical development and neuronal outgrowth, mimicking impaired neurogenesis in the human fetal brain with PNE during early gestation. The obvious advantages of the established system lie in its capabilities for perfused 3D culture and self-organization of brain organoids in a controlled cellular microenvironment, and additional functions for in situ tracking and real-time imaging, overcoming the potential limitations of conventional approaches.This work provides a proof-of-concept to create a human brain organoid-on-a-chip to probe impaired neurogenesis under environmental toxins exposure. The created system combines stem cell biology with organs-on-a-chip technology, which may open up new avenues for engineering stem cell organoids with enhanced functions, flexibility and controllability. In near future, this system can be incorporated with more organs-on-a-chip elements to greatly improve the maturity and functions of brain organoids and expand its applications in studying brain development, toxicity predictions and neurological diseases.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16020900, XDPB0305), National Nature Science Foundation of China (No. 91543121, 31671038, 81573394, 31600784), International Science and Technology Cooperation Program of China (2015DFA00740), and Key Laboratory of Separation Science for Analytical Chemistry (Dalian Institute of Chemical Physics, Chinese Academy of Sciences). We thank Professor Ning Sun (Fudan University, China) for kindly providing the human induced pluripotent stem cells.References
- D. A. Feinberg and A. S. Mark, Radiology, 1987, 163, 793–799 CrossRef CAS PubMed
.
- J. R. Pauly and T. A. Slotkin, Acta Paediatr., 2008, 97, 1331–1337 CrossRef PubMed
- L. S. Pagani, Neurosci. Biobehav. Rev., 2014, 44, 195–205 CrossRef CAS PubMed
- M. Ernst, E. T. Moolchan and M. L. Robinson, J. Am. Acad. Child. Adolesc. Psychiatry., 2001, 40, 630–641 CAS
- J. Zhu, X. Zhang, Y. Xu, T. J. Spencer, J. Biederman and P. G. Bhide, J. Neurosci., 2012, 32, 9410–9418 CrossRef CAS PubMed
- T. Alkam, H. C. Kim, M. Hiramatsu, T. Mamiya, Y. Aoyama, A. Nitta, K. Yamada and T. Nabeshima, Behav. Brain Res., 2013, 239, 80–89 CrossRef CAS PubMed
- T. Schneider, N. Ilott, G. Brolese, L. Bizarro, P. J. Asherson and I. P. Stolerman, Neuropsychopharmacology, 2011, 36, 1114–1125 CrossRef CAS PubMed
- Y. Aoyama, K. Toriumi, A. Mouri, T. Hattori, E. Ueda, A. Shimato, N. Sakakibara, Y. Soh, T. Mamiya, T. Nagai, H. C. Kim, M. Hiramatsu, T. Nabeshima and K. Yamada, Neuropsychopharmacology, 2016, 41, 578–589 CrossRef CAS PubMed
- R. S. Hill and C. A. Walsh, Nature, 2005, 437, 64–67 CrossRef CAS PubMed
- P. Rakic, Nat. Rev. Neurosci., 2009, 10, 724–735 CrossRef CAS PubMed
- J. D. Thomas, M. E. Garrison, C. J. Slawecki, C. L. Ehlers and E. P. Riley, Neurotoxicol. Teratol., 2000, 22, 695–701 CrossRef CAS PubMed
- L. Jiang and L. W. Role, J. Neurophysiol., 2008, 99, 1988–1999 CrossRef CAS PubMed
- M. A. Lancaster, M. Renner, C. A. Martin, D. Wenzel, L. S. Bicknell, M. E. Hurles, T. Homfray, J. M. Penninger, A. P. Jackson and J. A. Knoblich, Nature, 2013, 501, 373–379 CrossRef CAS PubMed
- C. Luo, M. A. Lancaster, R. Castanon, J. R. Nery, J. A. Knoblich and J. R. Ecker, Cell Rep., 2016, 17, 3369–3384 CrossRef CAS PubMed
- X. Qian, H. N. Nguyen, M. M. Song, C. Hadiono, S. C. Ogden, C. Hammack, B. Yao, G. R. Hamersky, F. Jacob, C. Zhong, K. J. Yoon, W. Jeang, L. Lin, Y. Li, J. Thakor, D. A. Berg, C. Zhang, E. Kang, M. Chickering, D. Nauen, C. Y. Ho, Z. Wen, K. M. Christian, P. Y. Shi, B. J. Maher, H. Wu, P. Jin, H. Tang, H. Song and G. L. Ming, Cell, 2016, 165, 1238–1254 CrossRef CAS PubMed
- G. Quadrato, J. Brown and P. Arlotta, Nat. Med., 2016, 22, 1220–1228 CrossRef CAS PubMed
- C. T. Lee, J. Chen, A. A. Kindberg, R. M. Bendriem, C. E. Spivak, M. P. Williams, C. T. Richie, A. Handreck, B. S. Mallon, C. R. Lupica, D. T. Lin, B. K. Harvey, D. C. Mash and W. J. Freed, Neuropsychopharmacology, 2017, 42, 774–784 CrossRef CAS PubMed
- A. M. Pasca, S. A. Sloan, L. E. Clarke, Y. Tian, C. D. Makinson, N. Huber, C. H. Kim, J. Y. Park, N. A. O'Rourke, K. D. Nguyen, S. J. Smith, J. R. Huguenard, D. H. Geschwind, B. A. Barres and S. P. Pasca, Nat. Methods, 2015, 12, 671–678 CrossRef CAS PubMed
- M. Eiraku, K. Watanabe, M. Matsuo-Takasaki, M. Kawada, S. Yonemura, M. Matsumura, T. Wataya, A. Nishiyama, K. Muguruma and Y. Sasai, Cell Stem Cell, 2008, 3, 519–532 CrossRef CAS PubMed
- T. Kadoshima, H. Sakaguchi, T. Nakano, M. Soen, S. Ando, M. Eiraku and Y. Sasai, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20284–20289 CrossRef CAS PubMed
- J. Mariani, M. V. Simonini, D. Palejev, L. Tomasini, G. Coppola, A. M. Szekely, T. L. Horvath and F. M. Vaccarino, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12770–12775 CrossRef CAS PubMed
- M. A. Lancaster and J. A. Knoblich, Nat. Protoc., 2014, 9, 2329–2340 CrossRef CAS PubMed
- D. A. Feinberg and A. S. Mark, Radiology, 1987, 163, 793–799 CrossRef CAS PubMed
- J. M. Rutkowski and M. A. Swartz, Trends Cell Biol., 2007, 17, 44–50 CrossRef CAS PubMed
- M. A. Swartz and M. E. Fleury, Annu. Rev. Biomed. Eng., 2007, 9, 229–256 CrossRef CAS PubMed
- N. J. Abbott, Neurochem. Int., 2004, 45, 545–552 CrossRef CAS PubMed
- D. Huh, Y. S. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed
- A. Schepers, C. Li, A. Chhabra, B. T. Seney and S. Bhatia, Lab Chip, 2016, 16, 2644–2653 RSC
- C. Xu, L. Wang, Y. Yu, F. Yin, X. Zhang, L. Jiang and J. Qin, Biomater. Sci., 2017, 5, 1810–1819 RSC
- J. M. Peyrin, B. Deleglise, L. Saias, M. Vignes, P. Gougis, S. Magnifico, S. Betuing, M. Pietri, J. Caboche, P. Vanhoutte, J. L. Viovy and B. Brugg, Lab Chip, 2011, 11, 3663–3673 RSC
- L. Wang, C. Xu, Y. Zhu, Y. Yu, N. Sun, X. Zhang, K. Feng and J. Qin, Lab Chip, 2015, 15, 4283–4290 RSC
- Q. Wang, H. Yang, A. Bai, W. Jiang, X. Li, X. Wang, Y. Mao, C. Lu, R. Qian, F. Guo, T. Ding, H. Chen, S. Chen, J. Zhang, C. Liu and N. Sun, Biomaterials, 2016, 105, 52–65 CrossRef CAS PubMed
- N. Sun, N. J. Panetta, D. M. Gupta, K. D. Wilson, A. Lee, F. Jia, S. Hu, A. M. Cherry, R. C. Robbins, M. T. Longaker and J. C. Wu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15720–15725 CrossRef CAS PubMed
- Y. Zhu, L. Wang, F. Yin, Y. Yu, Y. Wang, M. J. Shepard, Z. Zhuang and J. Qin, Integr. Biol., 2017, 9, 968–978 RSC
- Y. Zhu, L. Wang, H. Yu, F. Yin, Y. Wang, H. Liu, L. Jiang and J. Qin, Lab Chip, 2017, 17, 2941–2950 RSC
- Z. Molnar and A. Pollen, Development, 2014, 141, 11–16 CrossRef CAS PubMed
- H. Tabata, S. Yoshinaga and K. Nakajima, Exp. Brain Res., 2012, 216, 161–168 CrossRef PubMed
- L. H. Tsai and J. G. Gleeson, Neuron, 2005, 46, 383–388 CrossRef CAS PubMed
- R. Satta, E. Maloku, A. Zhubi, F. Pibiri, M. Hajos, E. Costa and A. Guidotti, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16356–16361 CrossRef CAS PubMed
- V. I. Pidoplichko, M. DeBiasi, J. T. Williams and J. A. Dani, Nature, 1997, 390, 401–404 CrossRef CAS PubMed
- M. N. Manuel, B. Martynoga, M. D. Molinek, J. C. Quinn, C. Kroemmer, J. O. Mason and D. J. Price, Neural Dev., 2011, 6, 9 CrossRef PubMed
- T. Seitanidou, S. Schneider-Maunoury, C. Desmarquet, D. G. Wilkinson and P. Charnay, Mech. Dev., 1997, 65, 31–42 CrossRef CAS PubMed
- P. Gilardi, S. Schneider-Maunoury and P. Charnay, Biochimie, 1991, 73, 85–91 CrossRef CAS PubMed
- C. Englund, A. Fink, C. Lau, D. Pham, R. A. Daza, A. Bulfone, T. Kowalczyk and R. F. Hevner, J. Neurosci., 2005, 25, 247–251 CrossRef CAS PubMed
- S. C. Vernes, P. L. Oliver, E. Spiteri, H. E. Lockstone, R. Puliyadi, J. M. Taylor, J. Ho, C. Mombereau, A. Brewer, E. Lowy, J. Nicod, M. Groszer, D. Baban, N. Sahgal, J. B. Cazier, J. Ragoussis, K. E. Davies, D. H. Geschwind and S. E. Fisher, PLoS Genet., 2011, 7, e1002145 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc01084b |
| This journal is © The Royal Society of Chemistry 2018 |