
A multi-throughput multi-organ-on-a-chip system on a plate formatted pneumatic pressure-driven medium circulation platform†
T.
Satoh‡
a,
S.
Sugiura‡
 *a,
K.
Shin
a,
R.
Onuki-Nagasaki
a,
S.
Ishida
b,
K.
Kikuchi
c,
M.
Kakiki
c and
T.
Kanamori
a
*a,
K.
Shin
a,
R.
Onuki-Nagasaki
a,
S.
Ishida
b,
K.
Kikuchi
c,
M.
Kakiki
c and
T.
Kanamori
a
aBiotechnology Research Institute for Drug Discovery, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki, Japan. E-mail: shinji.sugiura@aist.go.jp
bDivision of Pharmacology, National Institute of Health Sciences, Tokyo, Japan
cDrug Metabolism and Pharmacokinetics Research, Eisai Co. Ltd., Tsukuba, Ibaraki, Japan
First published on 14th November 2017
Abstract
This paper reports a multi-throughput multi-organ-on-a-chip system formed on a pneumatic pressure-driven medium circulation platform with a microplate-sized format as a novel type of microphysiological system. The pneumatic pressure-driven platform enabled parallelized multi-organ experiments (i.e. simultaneous operation of multiple multi-organ culture units) and pipette-friendly liquid handling for various conventional cell culture experiments, including cell seeding, medium change, live/dead staining, cell growth analysis, gene expression analysis of collected cells, and liquid chromatography–mass spectrometry analysis of chemical compounds in the culture medium. An eight-throughput two-organ system and a four-throughput four-organ system were constructed on a common platform, with different microfluidic plates. The two-organ system, composed of liver and cancer models, was used to demonstrate the effect of an anticancer prodrug, capecitabine (CAP), whose metabolite 5-fluorouracil (5-FU) after metabolism by HepaRG hepatic cells inhibited the proliferation of HCT-116 cancer cells. The four-organ system, composed of intestine, liver, cancer, and connective tissue models, was used to demonstrate evaluation of the effects of 5-FU and two prodrugs of 5-FU (CAP and tegafur) on multiple organ models, including cancer and connective tissue.
Introduction
Recently, the cost of drug discovery has markedly increased,1 whereas the success rate of clinical trials has decreased.2 One important reason for this is that the results of animal experiments can't be directly extrapolated to clinical trials because of species differences. Therefore, there is increasing interest in in vitro cell-based assays using human-derived cultured cells. In particular, microphysiological systems (MPS), which include single organ-on-a-chip and multi-organ-on-a-chip (MOC) systems, are attracting attention as new culturing methods that reconstruct the three-dimensional tissue structure, blood flow, and mechanical movement in a specific organ on a microfluidic device to reproduce the organ function in vitro.3–5An MOC is a microfluidic device in which multiple organ models are interconnected via microchannels on a chip.6,7 MOCs using human-derived cells have the potential to provide physiological human responses in vitro and are attracting much attention as novel research tools in drug discovery and as alternatives to animal testing.4,5,8,9 Culture of multiple organ models was pioneered by Shuler et al., who connected milliliter-sized cell culture compartments in a device called a cell culture analog (CCA).10,11 Microfluidic technology allowed CCA devices to be downsized; a micro-CCA was successfully used to detect the toxicity of naphthalene, the reactive metabolites of which were generated in a liver model and exhibited a toxic effect in a lung model on the same device.12,13
MOCs can theoretically detect the toxicity and efficacy of chemicals while taking into account interactions between multiple organ models;14 for example, metabolism-dependent drug toxicity and anti-cancer efficacy have been demonstrated by combining liver and tumor models,15–17 neurotoxicity has been tested by combining liver and neuron models,18 and multi-organ toxicity has been tested in a four-organ system comprising cardiac, muscle, neuronal, and liver models.19 MOCs containing the organs related to ADMET (drug ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) bsorption,
bsorption, ![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) istribution,
istribution, ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) etabolism,
etabolism, ![[e with combining low line]](https://www.rsc.org/images/entities/char_0065_0332.gif) xcretion, and
xcretion, and ![[t with combining low line]](https://www.rsc.org/images/entities/char_0074_0332.gif) oxicology) screens have also been investigated rigorously to recapitulate dynamic drug behavior related to multiple organs.20–26 An MOC designed by using a physiologically based pharmacokinetic model is called a body-on-a-chip (BOC) and is expected to recapitulate the pharmacokinetics and pharmacodynamics of the whole animal or human body.27,28 Furthermore, an MOC or BOC created with patient-derived cells is expected to recapitulate patient-specific phenomena as an in vitro disease model.29 In response to the above-mentioned advantages and potential uses of MOCs and BOCs, a number of start-up companies have been launched to provide services using such devices in drug discovery.30
oxicology) screens have also been investigated rigorously to recapitulate dynamic drug behavior related to multiple organs.20–26 An MOC designed by using a physiologically based pharmacokinetic model is called a body-on-a-chip (BOC) and is expected to recapitulate the pharmacokinetics and pharmacodynamics of the whole animal or human body.27,28 Furthermore, an MOC or BOC created with patient-derived cells is expected to recapitulate patient-specific phenomena as an in vitro disease model.29 In response to the above-mentioned advantages and potential uses of MOCs and BOCs, a number of start-up companies have been launched to provide services using such devices in drug discovery.30
In MOCs and BOCs, multiple cell types are cultured in discrete microchambers interconnected via microchannels to perfuse or circulate the medium. In early MOCs, the medium was pumped or circulated by using an off-chip syringe pump17,21,23 or a peristaltic pump.15,24,27,31 Later, to avoid troublesome off-chip tube connection and reduce the circulating medium volume, built-in pneumatic peristaltic micropumps,18,25,32,33 magnetic stirrer-based pumps,20,34 and gravity-driven flow were developed for MOCs and BOCs.19,26,27,35–37 In an example of medium circulation on a chip that is not an MOC, a built-in pneumatic peristaltic micropump has also been used to circulate the medium for perfused three-dimensional liver tissue culture in a plate-formatted microfluidic device.38,39
Generally, before industrial application, the reliability of MOCs and BOCs should be assessed by using a wide range of compounds. For this purpose, a high-throughput device is likely to be required; therefore, MOCs capable of parallelized medium circulation under identical conditions are desirable. However, as far as we know, only two examples of parallelized medium circulation in MOCs have been reported;32,33 two sets of medium circulation networks with built-in micropumps were constructed on a single MOC in both of these examples. Further increasing parallelization by increasing the number of built-in micropumps would require complicated experimental set-ups. Therefore, development of a simple parallelized medium circulation in an MOC is still a big challenge.
Here, we report a plate-formatted multi-organ microfluidic device with parallelized medium circulation with simple system set-ups, namely, a multi-throughput multi-organ-on-a-plate system, which was developed by using the pneumatic pressure-driven medium circulation system that we previously developed for endothelial cell culture.40 Because the pneumatic pressure directly drives the liquid, built-in micropumps are not required.40–43 The advantage of using pneumatic pressure is that the pressure from a single pressure source can be easily distributed to multiple devices and multiple circulation culture units in each device (Fig. S1 in the ESI†). Our microplate-sized pneumatic pressure-driven multi-organ culture platform has a 4 × 4 culture chamber array, which can be used for an eight-throughput two-organ system or a four-throughput four-organ system by adopting the corresponding microfluidic plate. We demonstrate the utility of this system for evaluating anticancer drugs in two-organ and four-organ systems through various cell culture experiments including staining, growth analysis, gene expression analysis, and liquid chromatography–mass spectrometry (LC-MS) analysis.
Experimental
Materials
Human colon carcinoma HCT-116 cells were obtained from RIKEN Cell Bank (Tsukuba, Japan) and were maintained in low-glucose Dulbecco's modified Eagle medium (DMEM) with sodium pyruvate and 10% fetal bovine serum (FBS). Human hepatoma HepaRG cells, Medium 620, and Medium 670 were obtained from KAC (Kyoto, Japan). Human colon carcinoma Caco-2 cells were obtained from RIKEN Cell Bank and maintained in MEM (Sigma, St. Louis, USA) with 20% FBS and 1% non-essential amino acids. Normal human diploid fibroblasts TIG-121 were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan) and maintained in MEM with 10% FBS. Capecitabine (CAP), tegafur (FT), propranolol, and niflumic acid were obtained from Sigma. 5-Fluorouracil (5-FU) was obtained from Wako Pure Chemical (Osaka, Japan). 5′-Deoxy-5-fluorocytidine (5′-DFCR) and 5′-deoxy-5-fluorouridine (5′-DFUR) were both obtained from Tokyo Chemical Industry (Tokyo, Japan). The cell proliferation assay reagent alamarBlue was obtained from Invitrogen (Carlsbad, USA). The LIVE/DEAD assay kit was obtained from Molecular Probes (Eugene, USA).A multi-throughput multi-organ-on-a-plate system
The concept of a multi-throughput multi-organ-on-a-plate system is illustrated in Fig. 1a with an example for a four-throughput four-organ system. The organs of interest in the human body were modeled by appropriate cell lines or tissue models, and the blood circulation system in the human body was modeled by a microfluidic network design on a microfluidic plate. The parallelized medium circulation in the microfluidic networks was generated in the culture device in which the microfluidic plate was assembled (Fig. 1b). The culture device was composed of an aluminum holder, stainless steel clips, a poly(dimethylsiloxane) (PDMS) microfluidic plate, a polycarbonate chamber plate, and glass and polycarbonate lid parts. The length and width of the holder were designed according to the standard plate format of the Society for Biomolecular Sciences. A 4 × 4 culture chamber array was formed by stacking the microfluidic plate and chamber plate on the holder and fastening them with small clips. Each culture chamber had a culture well (6 mm diameter and 2 mm depth) where cells could be cultured. Culture chambers were interconnected with microchannels on the microfluidic plate through the inlet and outlet in each chamber. For delivery of liquids, the elastomeric sheet and the lid, in which four parallel pneumatic pressure lines were fabricated, were placed on the chamber plate and fastened with large clips. One end of each pneumatic pressure line in the lid was equipped with an air-vent filter (Ekicrodisc 3CR, Nippon Genetics, Tokyo, Japan), which was further connected with outer tubing via a Luer-lock fitting to a pneumatic pressure control system, ASTF0401 (Engineering System, Matsumoto, Japan), by which pressure control was programmed (Fig. S1†).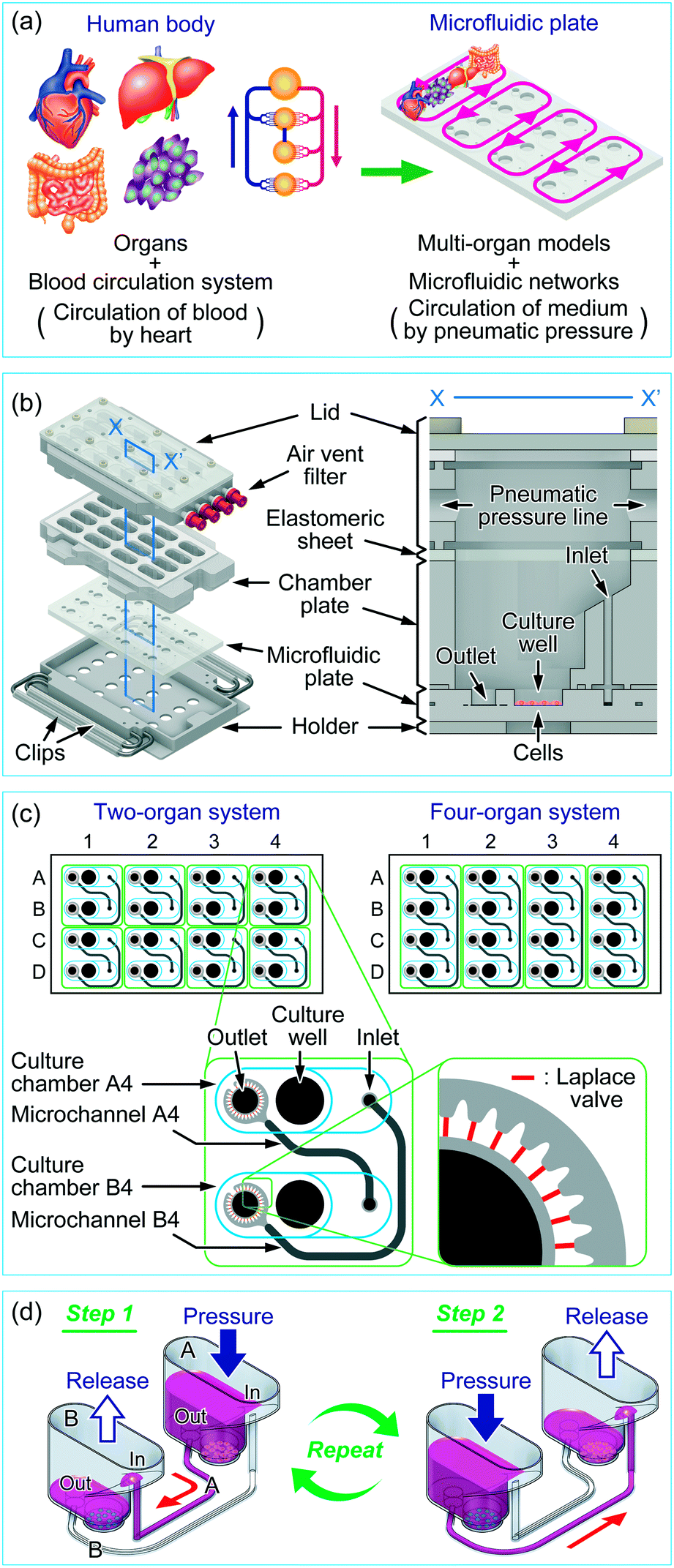 | ||
| Fig. 1 A multi-throughput multi-organ-on-a-plate system. (a) Concept of a multi-throughput multi-organ-on-a-plate system. (b) Projection view of a culture device containing 4 × 4 culture chambers and illustration of a culture chamber at X–X′ cross-section. (c) Designs of microfluidic networks in the microfluidic plates for an eight-throughput two-organ system and a four-throughput four-organ system. Closed circles indicate positions of holes opened to the top surface of the microfluidic plates. The dark- and light-shaded areas indicate microfluidic channels with deep and shallow depths, respectively. Areas enclosed by green lines indicate circulation culture units. Blue lines indicate walls of culture chambers. Thin red lines around the outlet indicate Laplace valves. (d) Process for medium circulation by pneumatic pressure in the two-organ system. Red arrows designate the direction of medium flow in the circulation unit. | ||
The designs of the microfluidic networks for a two-organ system and a four-organ system are shown in Fig. 1c. For two-organ and four-organ systems, a circulation culture unit was formed by connecting two or four culture chambers, respectively, through microchannels. As a result, the two- and four-organ systems had eight and four circulation units, respectively. Twenty-nine narrow microchannels called Laplace valves were radially placed around the outlet of each culture chamber.
Each culture chamber could be optionally equipped with a membrane insert for a barrier-type organ model such as the intestine. For this purpose, the top half of a commercial membrane insert (Transwell® permeable supports, 3470, 6.5 mm diameter, 0.4 μm pore-sized polyester membrane, Corning Costar, Cambridge, MA, USA) was removed (Fig. S2a in the ESI†), and the bottom half was fitted into the pocket (9.5 mm-diameter) of the culture chamber. With a membrane insert fitted into the pocket, there was a gap of 3 mm between the lower surface of the membrane and the bottom of the culture well to allow flow of medium (Fig. S2b in the ESI†); there was no flow on the top of the membrane.
The holder, clips, chamber plate, and lid were fabricated by machining. The microfluidic plates for the eight-throughput two-organ system and the four-throughput four-organ system were fabricated by injection molding of PDMS followed by oxygen plasma bonding; injection molding templates were fabricated by photolithography as reported previously.44
All parts of the culture device were autoclaved before cell culture. The membrane inserts were sterilized by soaking in 70% ethanol before use. The microfluidic plates and membrane inserts were single-use products; other parts were washed and reused.
Operating procedure for medium circulation
The medium circulation mechanism that we developed previously for the culture of endothelial cells under shear stress was applied to the multi-organ culture device, with modification. Previously, we used check valves to generate one-way medium circulation,40 whereas here we achieved semi-one-way circulation of medium between culture chambers by using different heights for the inlet and outlet of each culture chamber, Laplace valves, and sequential applied pressure. By using these modifications, which are described below, we realized a more convenient assembly without check valves.Fig. 1d shows the medium circulation process in the two-organ system, in which the circulation unit consists of two culture chambers and two microchannels. Two types of cells were cultured in the bottom of the culture wells in the culture chambers. The Laplace valve is a passive valve to stop the introduction of the gas phase into the microchannel owing to interfacial tension between the gas phase and medium at pressures below the Laplace pressure (a detailed explanation on the Laplace valve and calculation of Laplace pressure is available in Fig. S3 in the ESI†).40 When culture chamber A was pressurized at less than the Laplace pressure, the medium pooled in culture chamber A was transferred to culture chamber B through microchannels A and B (step 1). In this step, most of the medium (∼250 μL for the two-organ system) was transferred through microchannel A and a small volume of the medium (<20 μL) was transferred through microchannel B, because the inlet, which was the aperture to microchannel B, was higher than the outlet, which was the aperture to microchannel A. In both microchannels, the pressurized gas phase was finally stopped at the Laplace valves after the completion of medium transfer, with approximately 50 μL of medium remaining in the well to prevent exposure of the cells to air. Because, in this step, the contribution of microchannel B to medium transfer was less than 10% of that of microchannel A, a semi-one-way circulation system was possible. Medium transfer from culture chamber B into culture chamber A was performed in a similar manner by applying pressure to culture chamber B (step 2). In this step, the main path was switched to microchannel B. Repeating steps 1 and 2 achieved semi-one-way circulation. Medium circulation in the four-organ system was performed in a manner similar to that in the two-organ system by alternately pressurizing culture chambers A and C and B and D.
Flow characterization
Three lots of microfluidic plates were used (Table 1). The thickness of the microfluidic channels was measured from microscopic images of cross-sectional slices of the microfluidic plate. The Laplace pressure was measured by increasing the pressure in the culture chambers after the completion of medium transfer; the pressure just below that at which pressurized gas passed through the Laplace valves was determined as the Laplace pressure. The flow rate (mL min−1) of the medium in the two-organ system was measured with the microfluidic plate 2OP-1 at 26 °C. A volume of 300 μL of medium was loaded into each culture chamber using a micropipette. Medium circulation was performed by sequential pressurization at 4 kPa for 5 min for each step. We measured the time for medium transfer between each culture chamber by eye observation; the medium transfer could be easily noticed by color change in the outlet hole due to the red color of the media. The flow rate was calculated from the time for medium transfer. The above measurements were performed in triplicate in each culture chamber for three plates (9 measurements for each culture chamber).| Lot | System | Laplace valve | Depth [μm] | Laplace pressure [kPa] | |
|---|---|---|---|---|---|
| Channels around Laplace valvea | Main channelsb | ||||
| a Channels indicated by the light-shaded channels in Fig. 1c. b Channels indicated by the dark-shaded channels in Fig. 1c. | |||||
| 2OP-1 | Two organ | 27 ± 1 | 113 ± 2 | 298 ± 6 | 6.5 ± 0.1 |
| 2OP-2 | Two organ | 21 ± 1 | 96 ± 4 | 232 ± 5 | 5.2 ± 0.1 |
| 4OP-1 | Four organ | 23 ± 2 | 119 ± 3 | 294 ± 4 | 5.4 ± 0.2 |
The mixing and distribution of fluorescent dyes during medium circulation in the two-organ system was evaluated with microfluidic plate 2OP-2. Fluorescein isothiocyanate-labeled dextran (Mw: 70 kDa, Sigma) and tetramethylrhodamine-labeled dextran (Mw: 70 kDa, Sigma) were dissolved at concentrations of 0.02% and 0.1%, respectively, in MEM without phenol red (Sigma) containing 10% FBS. Three hundred microliters of medium was added to each culture chamber. The medium was transferred from culture chambers B and D to culture chambers A and C in odd steps of the medium circulation process at a pressure of 4 kPa and was returned in even steps. Fluorescence images of the residual medium in the wells were recorded for culture chambers B and D after odd steps and for culture chambers A and C after even steps. The medium circulation and the image acquisition were repeated for 20 steps. The fluorescence brightness over the whole area of the wells in each step was estimated by using ImageJ software (NIH, Bethesda, MD, USA).
Evaluation of the anticancer prodrug CAP in a two-organ system
We evaluated the metabolism-dependent anticancer prodrug CAP in a two-organ system composed of liver (HepaRG) and cancer (HCT-116) models. CAP is first metabolized to 5′-DFCR by carboxylesterase (CES); it is then converted into 5′-DFUR by cytidine deaminase (CDA) and then to 5-FU by thymidine phosphorylase (Fig. S4 in the ESI†).45 The culture devices were assembled with microfluidic plate 2OP-1. The wells of culture chambers A and C were coated with poly-L-lysine (Sigma) and CellMatrix type I-C (Nitta Gelatin, Osaka, Japan). The cultivation conditions of HepaRG (density, medium, pre-culture period before addition of drugs) were determined in accordance with the protocol provided by the provider (KAC). HepaRG cells were seeded at 6.4 × 104 cells per well in 50 μL of Medium 670 and were placed for 3.5 h in a 5% CO2 incubator at 37 °C for adhesion. Culture chambers A and C were then filled with 300 μL of Medium 670 and the cells were further incubated for 20.5 h. Medium 670 was replaced with Medium 620, and the medium was then changed every 2 days during an additional 10 day pre-culture period to induce maturation in hepatocyte-like cells. Wells of culture chambers B and D were coated with poly-L-lysine (Sigma) and fibronectin (Sigma), and then HCT-116 cells were seeded at 1.5 × 103 cells per well and placed overnight in the incubator for cell adhesion. Before the start of circulation culture, all microchannels were filled with Medium 670. All culture chambers were washed twice with 400 μL of Medium 670. Each culture chamber was filled with 300 μL of Medium 670 containing CAP; the final concentration of CAP was adjusted to 100 μM in the circulation unit. The lid was closed and the device was connected to the pressure tubing. Culture chambers A and C and B and D were pressurized alternately at 2.5 kPa at 5 min per step to achieve medium circulation. Circulation culture was continued for 71 h without any medium change.Viability staining of HepaRG cells and HCT-116 cells was carried out in quadruplicate on day 3 of circulation culture with the LIVE/DEAD assay kit. Proliferation of HCT-116 and HepaRG cells was evaluated using alamarBlue in quadruplicate on day 3 of circulation culture.
The concentrations of CAP and its metabolites in the medium were measured by using an LC-MS system consisting of TripleQuad 6500+ (AB Sciex, Framingham, MA, USA) and Nexera X2 (Shimadzu, Kyoto, Japan). Aliquots of the medium were collected on day 3 of culture and temporarily stored at −80 °C. Immediately before analysis, the samples were extracted with 70% acetonitrile/30% methanol including 0.01 μg mL−1 propranolol and 0.1 μg mL−1 niflumic acid as internal standards. LC-MS analysis was performed for quadruplicate samples from each experiment.
RNA extraction from HepaRG cells on day 0 and day 3 of circulation culture and the subsequent reverse transcription into cDNA were performed by using the RNeasy Midi Kit (QIAGEN, Hilden, Germany) and QuantiTect Reverse Transcription Kit (QIAGEN), respectively. Quantitative real-time polymerase chain reaction (RT-PCR) analysis was carried out for CES1, CES2, CDA, albumin (ALB), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by using a primer set from QuantiTect Primer Assays (QIAGEN) and a Thermal Cycler Dice PCR system (TP850, Takara Bio, Shiga, Japan). Primer information is available in Table S1 in the ESI.† Expression levels of mRNA were normalized to those of GAPDH and relative quantification was performed by using the ΔΔCt method. RT-PCR was performed for quadruplicate samples from each experiment.
Evaluation of three drugs in a four-organ system
We evaluated three drugs, namely, CAP, FT, and 5-FU, in a four-organ system composed of intestine (Caco-2 cells), liver (HepaRG cells), cancer (HCT-116 cells), and connective tissue (TIG-121 cells) models. FT is an anticancer prodrug that is metabolized to 5-FU by mainly CYP2A6 in the liver and exhibits an anticancer effect (Fig. S4 in the ESI†).46,47 The culture devices were assembled with microfluidic plate 4OP-1. The bottom halves of the membrane inserts were coated with poly-L-lysine (Sigma) and CellMatrix type I-C and attached to the culture chambers. Caco-2 cells were seeded at 2.0 × 105 cells per insert on the apical side (top side of membrane) of the insert (Fig. S2b in the ESI†). The volume of the medium was adjusted to 200 μL and 700 μL at the apical and basolateral sides of the membrane inserts. The cells were cultured for 21 days for maturation.48 The trans-epithelial electrical resistance (TEER) of the Caco-2 cell monolayers formed in the membrane insert was measured in quadruplicate by using a Millicell ERS-2 system (Millipore) according to the manufacturer's instructions. TEER measurement was carried out for the membrane inserts in the device with the lid removed in a tissue culture hood (Fig. S2c†). Membrane inserts containing Caco-2 cell monolayers with TEER values greater than 500 Ω cm2 were transferred into culture chamber A. HepaRG cells and HCT-116 cells were seeded at 6.4 × 104 and 1.5 × 103 cells per well into culture chambers B and C, respectively, and were cultured in the same manner as in the two-organ system. TIG-121 cells were seeded at 1.5 × 103 cells per well into culture chamber D, which was pre-coated with poly-L-lysine (Sigma) and fibronectin, and then pre-cultured in the same way as HCT-116 cells. Before the start of circulation culture, all microchannels were filled with Medium 670. All culture chambers were washed twice with 400 μL of Medium 670. All culture chambers were filled with 300 μL of Medium 670, and 200 μL of Medium 670 containing 700 μM CAP, FT, or 5-FU was loaded to the apical side of the membrane inserts in chamber A. Since the total volume of medium in each circulation unit was 1400 μL (the apical side of the membrane insert in chamber A, 200 μL; the basolateral side of chamber A and chambers B, C, and D, each 300 μL), the average concentration of each drug in the circulation unit was 100 μM if the drug was homogeneously distributed in the circulation unit after circulation culture. Circulation culture was performed for 72 h without any medium change in the same way as in the two-organ system.Microscope images of all cell lines, except Caco-2, were recorded on day 3 of culture. TEER values of Caco-2 cell monolayers were measured in quadruplicate on day 0 and day 3 of circulation culture. Proliferation of all the four cell lines was evaluated using alamarBlue in quadruplicate on day 3 of circulation culture. The concentrations of the drugs and their metabolites in the medium on day 3 of circulation culture were determined by using the LC-MS system.
Microscopy
Bright field and fluorescence microscope images were recorded by using an IX71 fluorescence microscope (Olympus, Tokyo, Japan) with an Orca Flash 4.0 complementary metal-oxide-semiconductor (CMOS) camera (Hamamatsu Photonics, Hamamatsu, Japan) and MetaMorph software version. 7.8.8.0 (Molecular Devices, Sunnyvale, CA, USA). Bright field microscope images were also recorded by using a CKX41 phase-contrast microscope (Olympus) with a DP25 camera (Olympus) and CellSens Standard software version 1.12 (Olympus).Statistical analysis
Statistical comparisons were performed using two-way ANOVA with Bonferroni post-tests using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA). P values less than 0.05 were classed as statistically significant.Results
Pneumatic pressure-driven multi-organ-on-a-plate system
A perspective view of the fabricated culture device is shown in Fig. 2a. Pneumatic pressure was applied to the device through the sterilized air vent filters, which enabled us to maintain sterile conditions inside the culture device over the entire process of cell culture. Because the connections to the pneumatic pressure lines were easily detachable and the lid was easily removed, cell seeding and medium change were performed by using a micropipette in a tissue culture hood. Different types of microfluidic plates were fabricated for the two-organ and four-organ systems (Fig. 2b and c). Three lots of microfluidic plates (two for the two-organ system, and one for the four-organ system) were used, and slight differences in depths of microchannels were found among them (Table 1). Fig. 2d shows the bottom half of the membrane insert attached to the pocket of the culture chamber. | ||
| Fig. 2 The fabricated culture device and microfluidic plate. (a) The fabricated culture device in the assembled state and with the lid removed. The culture chambers were filled with medium. (b) The fabricated microfluidic plates. The microfluidic networks are indicated by the colored solution. The pale color of the solution in the Laplace valves reflects the shallow depth of those areas. (c) Enlargement of the culture unit and Laplace valves. (d) View of the modified membrane insert, and its attachment to the pocket of the culture chamber. The insert was filled with medium. | ||
Flow characterization
The average flow rate of the medium in microchannels in the two-organ system under 4 kPa pressure was 0.68 ± 1.4 mL min−1 (Fig. 3a). Mixing of two fluorescent dyes in the medium was evaluated during medium circulation in the two-organ system by analyzing fluorescence images of the wells after completion of medium transfer at each step, in which a constant volume of the medium remained, as described in the Experimental section. The images indicated that a uniform distribution of the dyes in the wells was gradually generated during medium circulation (Fig. S5 in the ESI†); the relative brightness of the images gradually converged to a constant value (Fig. 3b), and the values of relative brightness after 10 steps were within ±10% of those after 20 steps.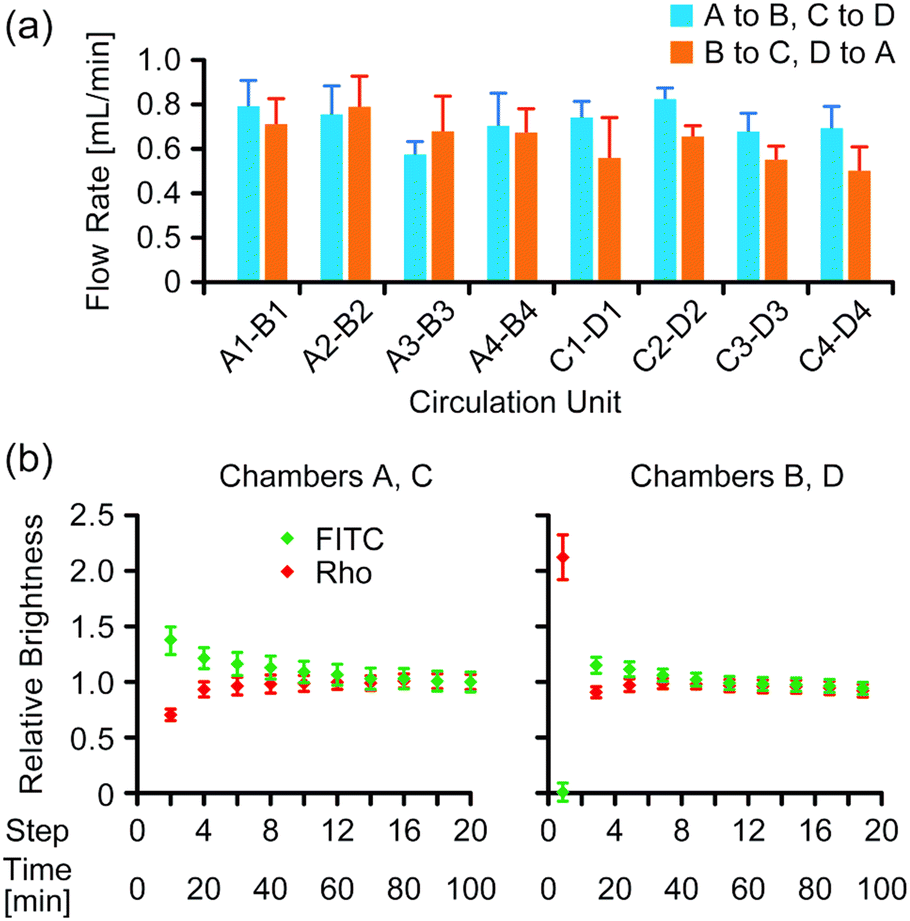 | ||
| Fig. 3 Flow characterization in the two-organ system. (a) Flow rate of the medium in the two-organ system at 4 kPa of pressure (n = 9, mean ± S.D.). (b) Relative brightness of fluorescence analyzed from image data of wells (n = 6, mean ± S.D.). | ||
Evaluation of the anticancer prodrug CAP in the two-organ system
The effect of the anticancer prodrug CAP was evaluated in the two-organ system with HepaRG cells as a liver model and HCT-116 cells as a cancer model. Both cell types were seeded and cultured separately in culture chambers A and C, and B and D, respectively, in advance, and then connected circulation culture was performed with a medium containing 100 μM CAP for 3 days (Fig. 4a). As a comparison, experiments were performed under the same conditions without HepaRG cells or without CAP, or both.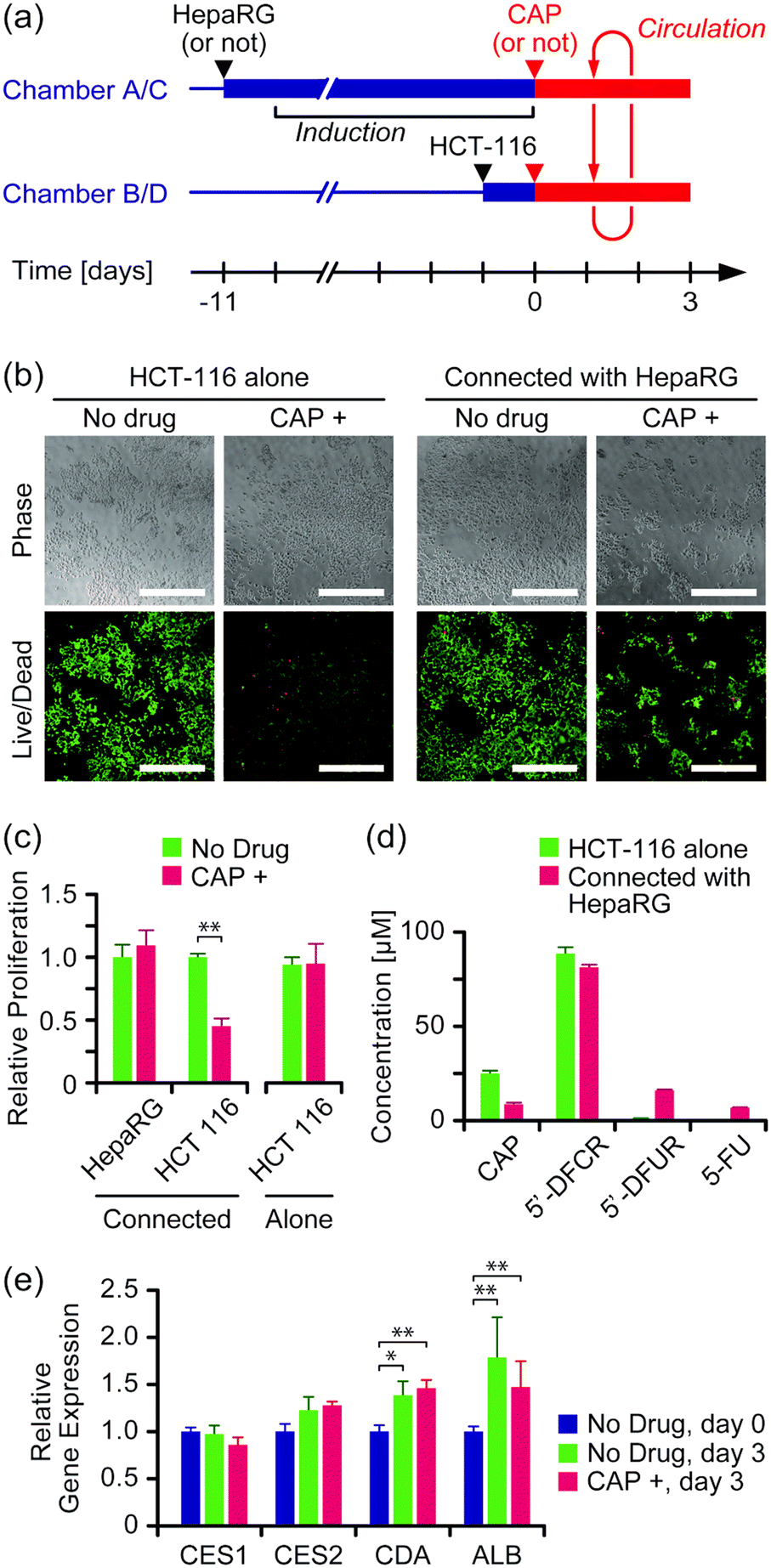 | ||
| Fig. 4 Evaluation of the anticancer prodrug CAP in the two-organ system. (a) Experimental schedule. (b) Viability assay of HCT-116 cells on day 3 of culture. Green and red colors indicate live and dead cells, respectively. Scale bars: 500 μm. Enlarged images are available in Fig. S6 in the ESI.† (c) Proliferation of HepaRG and HCT-116 cells on day 3 of culture (n = 4, mean ± S.D., *** P < 0.001). (d) Concentrations of CAP and its metabolites in the medium on day 3 of culture (n = 4, mean ± S.D.). (e) Gene expression in HepaRG cells on day 0 and day 3 of culture (n = 4, mean ± S.D., * P < 0.05; ** P < 0.01). | ||
Fig. 4b shows microscope images of HCT-116 cells stained with the LIVE/DEAD assay kit on day 3 of culture. Cell proliferation was determined by alamarBlue assay (Fig. 4c). Significant growth inhibition induced by CAP was observed in HCT-116 cells that were connected with HepaRG cells (P < 0.05); growth of HCT-116 cells was inhibited to 45% ± 6% by the presence of CAP compared to without CAP. In contrast, no significant growth inhibition by CAP was observed with HCT-116 cells alone. LC-MS measurement revealed that more than 80% of CAP was converted into its first metabolite, 5′-DFCR, regardless of the presence of HepaRG cells (Fig. 4d). The second metabolite, 5′-DFUR, and 5-FU were detected at 16 μM and 7 μM, respectively, when HCT-116 cells were connected with HepaRG cells, whereas negligible concentrations of 5′-DFUR and 5-FU were detected with HCT-116 cells alone. The expression of genes encoding enzymes involved in CAP metabolism, i.e., CES1, CES2, and CDA, in HepaRG cells was analyzed (Fig. 4e). Expression of CDA and ALB slightly but significantly increased after circulation culture for 3 days, regardless of the presence of CAP. No significant differences were observed in the expression levels of the above genes between the group with CAP and that without CAP (P > 0.05).
Evaluation of three anticancer drugs in the four-organ system
The anti-cancer effects of prodrugs of 5-FU (i.e., CAP and FT) and 5-FU itself were investigated in the four-organ system (Fig. 5a). Four cell lines – Caco-2, HepaRG, HCT-116, and TIG-121 – were used as models of intestine, liver, cancer, and connective tissue, respectively. Caco-2 cells were pre-cultured for 21 days on membrane inserts to form matured monolayers with TEER values higher than 500 Ω cm2. Other cell lines were separately cultured in advance in the three culture chambers in each circulation unit. Each drug was loaded to the apical side of the membrane inserts, and circulation culture was performed for 3 days.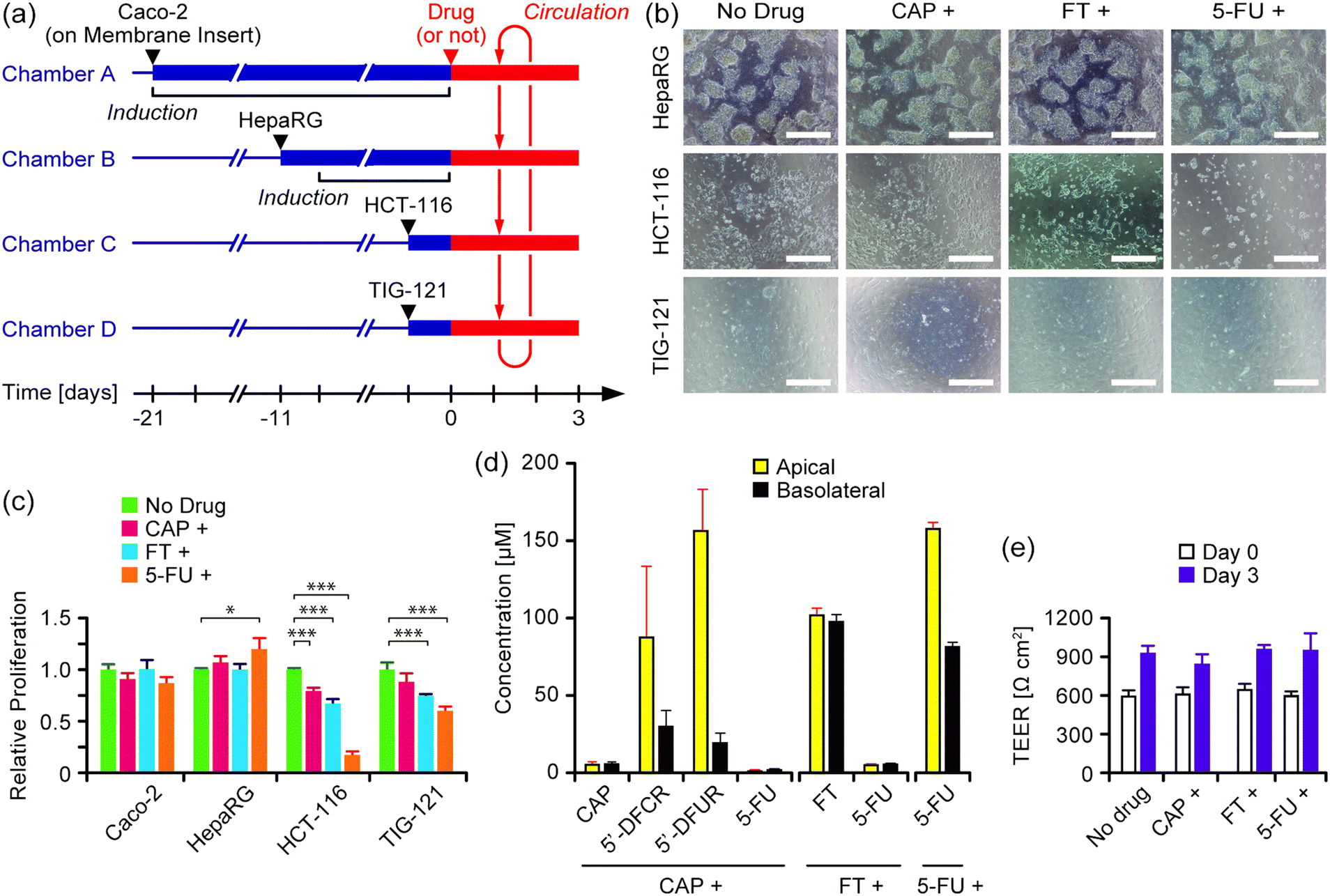 | ||
| Fig. 5 Evaluation of three anticancer drugs in the four-organ system. (a) Experimental schedule. (b) Microscope images of the cells on day 3. Scale bars: 500 μm. Enlarged images are available in Fig. S7 in the ESI.† (c) Proliferation of the cells on day 3 (n = 3 or 4, ±S.D.). Asterisks indicate significant differences (* P < 0.05; ** P < 0.001). (d) Concentrations of the drugs and their metabolites in the medium on day 3 of culture (n = 3 or 4, ±S.D.). (e) TEER values of Caco-2 cell monolayers on day 0 and day 3 of culture (n = 3 or 4, ±S.D.). | ||
The microscope images of HCT-116 cancer cells on day 3 of culture indicated that the cell density of those with exposure to 5-FU was considerably lower than that of those under the other conditions (Fig. 5b). The alamarBlue assay demonstrated that the growth of HCT-116 cells on day 3 of culture was decreased to 79% ± 3%, 67% ± 4%, and 17% ± 3% with exposure to CAP, FT, and 5-FU, respectively, compared with that under drug-free conditions (Fig. 5c). A similar tendency in growth inhibition was observed in the non-cancer TIG-121 cells, although the inhibitory effect was less than that in HCT-116 cells. In contrast, no significant growth inhibition or cell death was observed in Caco-2 or HepaRG cells. The distribution of drugs and their metabolites on day 3 of culture was assessed by LC-MS analysis (Fig. 5d). Three days after CAP was loaded to the apical side of the membrane inserts, the concentration of CAP remaining on both the apical and basolateral (i.e., circulating medium) sides of the monolayers was 6 μM, suggesting that 94% of CAP had been metabolized, decomposed, or absorbed. Two intermediate metabolites, 5′-DFCR and 5′-DFUR, were detected at markedly higher concentrations in the apical side compared to the basolateral side, whereas the concentration of 5-FU was very low (∼2 μM) on both sides of the monolayers. In contrast to CAP, 3 days after FT was loaded to the apical side of the membrane inserts, the concentration of FT was equally high (∼94 μM) on both sides of the membrane inserts, suggesting that (i) most of the FT remained intact, with only 6% being converted into 5-FU, and (ii) FT was freely transported through the Caco-2 cell monolayers in the membrane inserts. When 5-FU itself was applied, the 5-FU concentration was 158 ± 4 μM and 82 ± 3 μM on the apical and basolateral sides, respectively, after 3 days. The amount of 5-FU that permeated through the Caco-2 cell monolayer was calculated to be 76% when taking into account the ratio between the medium volumes of the apical and basolateral sides (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6). The TEER values of the Caco-2 monolayers increased in all culture conditions after the circulation culture, indicating the absence of cell death (Fig. 5e).
:6). The TEER values of the Caco-2 monolayers increased in all culture conditions after the circulation culture, indicating the absence of cell death (Fig. 5e).
Discussion
Here, we reported a multi-throughput multi-organ-on-a-plate system and demonstrated its efficacy for evaluating anticancer drugs in two-organ and four-organ systems. The technique of circulating the medium by pneumatic pressure has advantages for a high-throughput system, because pneumatic pressure can be easily branched and the number of devices and culture units can be increased using a single control system.40–43 Sterile culture conditions were maintained by pressurization through the air vent filter, whereas liquid handling – including cell seeding, medium change, staining, and collection of medium and cells – was conveniently performed by opening the lid in the tissue culture hood. The use of the assembly-type culture device with 4 × 4 culture chamber arrays and exchangeable microfluidic plates allowed us to construct an eight-throughput two-organ system and a four-throughput four-organ system on the same device. The 6 mm diameter of the culture wells is consistent with those in 96-well plates; thus, general experimental protocols for cell-based assays for microplates are applicable to our culture device. In addition, membrane inserts are optionally attachable in the culture chambers for barrier-type organs. Therefore, our multi-organ-on-a-plate system is applicable to multi-organ experiments such as those currently performed for cell-based assays.There have been a couple of reports on four-organ devices.19,25 Our multi-throughput multi-organ-on-a-plate system firstly realized multi throughput of multi-organ experiments with more than three organ models. In addition, unlike multi-organ circulation culture devices reported previously,18,20,25,32–34 our system does not require a built-in micropump; therefore, the structure of the culture device is simple and can achieve higher throughput. There has also been a pumpless multi-throughput multi-organ device using gravity-driven flow.49 Unlike multi-organ devices with gravity-driven flow,19,26,27,35–37,49 most of which adopted a reciprocating flow of medium, our system achieved multi-throughput semi-one-way circulation of medium in a single device.
We have demonstrated the evaluation of three drugs (5-FU, FT, and CAP). For this experiment, mixing of culture media in each chamber is essential, and semi-one-way circulation is theoretically not necessary. However, the platform technology capable of semi-one-way medium circulation should be important to recapitulate the blood flow in our body, especially for applying shear stress and modeling the physiological circulation network for precise pharmacokinetic study.
The three lots of microfluidic plates had slightly different microchannel dimensions and Laplace pressure (Table 1). Some variation in flow rates between individual microchannels in the 2OP-1 plates was probably due to distortion of the PDMS during assembly (Fig. 3a). The depth of the Laplace valve is an important parameter to control the flow rate. The depth of microfluidic plate 2OP-1 and that of 2OP-2 was 27 and 21 μm, respectively (Table 1). Considering the analytical solutions to Navier–Stokes equations for the rectangular microchannel,50 this difference theoretically corresponds to a 100% difference in flow rate if the device was operated under the same pressure condition. However, we could use the same pressure condition (4 kPa) for all plates because we have adopted a long enough time (5 min) during each pressurization step. Actually, the step cycle for medium circulation (5 min) was long enough compared to the flow rate (0.5 to 0.8 mL min−1 to deliver 0.5 mL of medium). It is possible to operate in as short a cycle time as 1 min for the continuous medium flow. However, we chose 5 min as the cycle time to assure the complete medium transfer in each cycle in the case that the structure of the narrow microchannel in the Laplace valve is slightly different or the flow rate is decreased by the debris in the culture media. The flow rate during medium circulation is adjustable by changing both the step cycle and the pressure, as reported in our previous study,40 within the pressure range below the Laplace pressure. Uniform mixing of fluorescent dyes in the medium after 10 cycles of medium circulation was indicated by the distribution of fluorescent dyes (Fig. S5 in the ESI†). The results suggest that the cells cultured in the connected culture chambers were similarly exposed to the compounds in the medium. We note that the flow rate and diffusional mixing at 37 °C should be faster than those measured at room temperature (shown in Table 1) because of the difference in viscosity and diffusion coefficient.
In the two-organ system, CAP was successively metabolized to 5′-DFCR, 5′-DFUR, and 5-FU in the presence of HepaRG liver cells. We have confirmed that the conversion of 5′-DFCR into 5′-DFUR occurred in the HepaRG cells alone (data not shown). The conversion of 5′-DFUR into 5-FU probably occurred in the cancer cells, as reported previously (Fig. S4†).45 The metabolite 5-FU probably inhibited the growth of HCT-116 cancer cells (Fig. 4c and d); production of 5-FU and growth inhibition were not observed with HCT-116 alone. The cell lines used in the two-organ experiment – HepaRG and HCT-116 cells – required different media and schedules for pre-culture. Our system allowed us to perform separate static cultures in the culture chambers of the device before starting the connected circulation culture (Fig. 4a). In addition, RT-PCR revealed that the expression level of genes encoding metabolic enzymes related to the activation of CAP, including CES1, CES2, and CDA, was stable in HepaRG cells independent of the presence of CAP during the circulation culture (Fig. 4e). With these results taken together, we consider that our two-organ system successfully reproduced the metabolism-dependent anticancer effect of CAP. Regarding the increased gene expression of CDA and ALB of HepaRG after 3 days of co-culture, there should be a possibility of shear stress and crosstalk with HCT-116 having an effect. The detailed effect of co-culture can be investigated using our system in the future.
In the four-organ system, we could simultaneously evaluate intestinal absorption, hepatic metabolism, and growth inhibition in cancer and connective tissue models. Caco-2 cells were incubated on the membrane inserts, and maturation of cell monolayers was confirmed from the TEER values before drug testing, like commercially available membrane inserts. We evaluated the effects of three anticancer drugs, CAP, FT, and 5-FU, in the four-organ system. The growth inhibitory effect on HCT-116 cancer cells was in the order 5-FU > FT > CAP (Fig. 5b and c); this is consistent with the concentration of 5-FU detected by LC-MS analysis, in which 82, 6, and 2 μM of 5-FU were detected on the basolateral side (i.e., in circulating medium) following the addition of 5-FU, FT, and CAP, respectively (Fig. 5d). Growth inhibition of TIG-121 normal fibroblasts showed the same order as that of HCT-116 cells (Fig. 5c); however, the inhibition was lower than that observed for HCT-116 cells, possibly because the proliferation of TIG-121 cells is lower than that of HCT-116 cells before treatment.
In the preliminary experiment, we have also examined the degradation and absorption of 5-FU, FT, and CAP in our system (Fig. S8 in the ESI†). Although it is very difficult to distinguish degradation and absorption, we have confirmed that the absorption of the parent and degraded compounds was not major on the PDMS microfluidic plate without cells during 3 days of medium circulation.
Growth inhibition of HCT-116 cells by CAP in the four-organ system was lower than that found in the two-organ system. LC-MS revealed that 94% of CAP was converted into its metabolites (Fig. 5d). Although large concentrations of 5′-DFCR and 5′-DFUR were detected on the apical side, they were found at only 30 and 20 μM, respectively, on the basolateral side. The conversions from CAP to 5′-DFCR and 5′-DFUR in the apical side medium were probably due to hydrolysis and metabolic activity in the Caco-2 cells. Considering the volume of the medium in the apical side (200 μL in the apical side of chamber A) and basolateral side (1200 μL in the basolateral side of chamber A and chambers B, C, and D), the parent and metabolic compounds were detected at 120 nmol in total in our system. This amount corresponds to 86% of the amount of CAP that was added in the apical side on day 0 (700 μM × 200 μL = 140 nmol). Additionally, although the volume of the circulated medium in the four-organ system was double that in the two-organ system, the metabolic activity of HepaRG cells was the same as that in the two-organ system; therefore, the concentrations of 5′-DFUR and 5-FU produced by HepaRG cells and HCT-116 cells were diluted to half of those in the two-organ system.
After FT was loaded to the apical side of the membrane inserts, FT was found at the same concentration in both sides of the membrane inserts, indicating that FT could permeate freely through the Caco-2 cell monolayers on the membrane inserts. However, the rate of FT metabolism was low in HepaRG cells, with only 6 μM 5-FU being generated. When 5-FU itself was loaded to the apical side, 76% of the 5-FU permeated through the Caco-2 monolayers, and the permeated 5-FU inhibited the growth of HCT-116 and TIG-121 cells.
In our four-organ system, we were able to evaluate the effects of the three anticancer drugs at a concentration of 100 μM. The maximum plasma concentrations of the three drugs reported in clinical studies are 81, 46, and 11 μM for 5-FU, FT, and CAP, respectively;51–53 therefore, the concentrations of parent and metabolite compounds detected on the basolateral side were similar to, or higher than, those in the clinical studies. To detect the effects of anticancer drugs at physiological concentrations, scaled design of organ models will be required. As discussed in previous studies,54–57 by adjusting scaling parameters (sizes of organ modules, volumes, and flow rates) on the basis of pharmacokinetic modeling, it should be possible to recapitulate the physiological pharmacokinetic response in MOCs.52–55 Design of microfluidic networks with physiological organ scaling by using our multi-organ-on-a-plate system will be important for the future application of this technique.
Conclusions
We developed a multi-throughput multi-organ-on-a-plate system using a pneumatic pressure-driven medium circulation platform. The developed culture device, composed of a 4 × 4 array of culture chambers, was applied to eight-throughput two-organ and four-throughput four-organ systems by using different microfluidic plates. The sequential applied pressure enabled us to circulate the medium in the multiple circulation culture units simultaneously; medium components, as measured with fluorescent dyes, were mixed within a reasonable time to achieve uniform distribution of components. In the two-organ system, we successfully detected the metabolism-dependent anticancer effect of CAP in a culture system composed of HCT-116 and HepaRG cells. In the four-organ system, we successfully evaluated the effects of three anticancer drugs on multiple organ functions, including intestinal absorption, hepatic metabolism, and growth inhibition in cancer and connective tissue. Our pneumatic pressure-driven multi-throughput multi-organ-on-a-plate system possesses the following advantages for application to drug discovery: simultaneous operation of multiple multi-organ culture units, design flexibility of the microfluidic network, a pipette-friendly liquid handling interface, and applicability to experimental protocols and analytical methods widely used in microplates. Therefore, we believe that our multi-organ culture platform will be an advantageous research tool for drug discovery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. and T. S. proposed the concept. T. S. and S. S. designed the microfluidic devices. K. S., S. S., and S. I. designed the cell culture experiment. T. S. fabricated the microfluidic devices. R. N. and K. S performed the flow characterization. K. K. and M. K. performed the LC-MS analysis. T. S., K. S., R. N., and S. S. analyzed the experimental data. S. S. and T. K. supervised the study. T. S. and S. S. wrote the manuscript. All the authors reviewed and provided comments on the manuscript.Notes and references
- J. W. Scannell, A. Blanckley, H. Boldon and B. Warrington, Nat. Rev. Drug Discovery, 2012, 11, 191–200 CrossRef CAS PubMed
.
- F. Pammolli, L. Magazzini and M. Riccaboni, Nat. Rev. Drug Discovery, 2011, 10, 428–438 CrossRef CAS PubMed
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed
- E. W. Esch, A. Bahinski and D. Huh, Nat. Rev. Drug Discovery, 2015, 14, 248–260 CrossRef CAS PubMed
- U. Marx,
et al.
, Altex, 2016, 33, 272–321 Search PubMed
- J. H. Sung, M. B. Esch, J.-M. Prot, C. J. Long, A. Smith, J. J. Hickman and M. L. Shuler, Lab Chip, 2013, 13, 1201–1212 RSC
- M. L. Shuler, Lab Chip, 2017, 17, 2345–2346 RSC
- C. Y. Chan,
et al.
, Lab Chip, 2013, 13, 4697–4710 RSC
- A. Polini, L. Prodanov, N. S. Bhise, V. Manoharan, M. R. Dokmeci and A. Khademhosseini, Expert Opin. Drug Discovery, 2014, 9, 335–352 CrossRef CAS PubMed
- L. M. Sweeney, M. L. Shuler, J. G. Babish and A. Ghanem, Toxicol. In Vitro, 1995, 9, 307–316 CrossRef CAS PubMed
- M. L. Shuler, A. Ghanem, D. Quick, M. C. Wong and P. Miller, Biotechnol. Bioeng., 1996, 52, 45–60 CrossRef CAS PubMed
-
A. Sin, G. T. Baxter and M. L. Shuler, in Proc. SPIE 4560, 2001, pp. 98–101 Search PubMed
- A. Sin, K. C. Chin, M. F. Jamil, Y. Kostov, G. Rao and M. L. Shuler, Biotechnol. Prog., 2004, 20, 338–345 CrossRef CAS PubMed
- K. M. Fabre, C. Livingston and D. A. Tagle, Exp. Biol. Med., 2014, 239, 1073–1077 CrossRef CAS PubMed
- J. H. Sung and M. L. Shuler, Lab Chip, 2009, 9, 1385–1394 RSC
- Z. Y. Li, Y. Q. Guo, Y. Yu, C. Xu, H. Xu and J. H. Qin, Integr. Biol., 2016, 8, 1022–1029 RSC
- M. S. Jie, H. F. Li, L. Y. Lin, J. Zhang and J. M. Lin, RSC Adv., 2016, 6, 54564–54572 RSC
- E.-M. Materne,
et al.
, J. Biotechnol., 2015, 205, 36–46 CrossRef CAS PubMed
- C. Oleaga,
et al.
, Sci. Rep., 2016, 6, 20030 CrossRef CAS PubMed
- H. Nakayama, H. Kimura, K. Komori, T. Fujii and Y. Sakai, J. Rob. Mechatronics, 2007, 19, 544–549 CrossRef
- Y. Imura, K. Sato and E. Yoshimura, Anal. Chem., 2010, 82, 9983–9988 CrossRef CAS PubMed
- P. M. van Midwoud, M. T. Merema, E. Verpoorte and G. M. M. Groothuis, Lab Chip, 2010, 10, 2778–2786 RSC
- Y. Imura, E. Yoshimura and K. Sato, Anal. Sci., 2012, 28, 197–199 CrossRef CAS PubMed
- J. M. Prot,
et al.
, Biotechnol. Bioeng., 2014, 111, 2027–2040 CrossRef CAS PubMed
- I. Maschmeyer,
et al.
, Lab Chip, 2015, 15, 2688–2699 RSC
- A. Choe, S. K. Ha, I. Choi, N. Choi and J. H. Sung, Biomed. Microdevices, 2017, 19, 4 CrossRef PubMed
- J. H. Sung, C. Kam and M. L. Shuler, Lab Chip, 2010, 10, 446–455 RSC
- J. H. Sung, B. Srinivasan, M. B. Esch, W. T. McLamb, C. Bernabini, M. L. Shuler and J. J. Hickman, Exp. Biol. Med., 2014, 239, 1225–1239 CrossRef PubMed
- A. K. Capulli, K. Tian, N. Mehandru, A. Bukhta, S. F. Choudhury, M. Suchyta and K. K. Parker, Lab Chip, 2014, 14, 3181–3186 RSC
- B. Zhang and M. Radisic, Lab Chip, 2017, 17, 2395–2420 RSC
- C. Zhang, Z. Q. Zhao, N. A. A. Rahim, D. van Noort and H. Yu, Lab Chip, 2009, 9, 3185–3192 RSC
- I. Wagner,
et al.
, Lab Chip, 2013, 13, 3538–3547 RSC
- J. R. Coppeta,
et al.
, Lab Chip, 2017, 17, 134–144 RSC
- H. Nakayama, H. Kimura, T. Fujii and Y. Sakai, J. Biosci. Bioeng., 2014, 117, 756–762 CrossRef CAS PubMed
- M. B. Esch, H. Ueno, D. R. Applegate and M. L. Shuler, Lab Chip, 2016, 16, 2719–2729 RSC
- P. G. Miller and M. L. Shuler, Biotechnol. Bioeng., 2016, 113, 2213–2227 CrossRef CAS PubMed
- H. Lee, D. S. Kim, S. K. Ha, I. Choi, J. M. Lee and J. H. Sung, Biotechnol. Bioeng., 2017, 114, 432–443 CrossRef CAS PubMed
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. M. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC
- U. Sarkar,
et al.
, Drug Metab. Dispos., 2015, 43, 1091 CrossRef CAS PubMed
- T. Satoh, G. Narazaki, R. Sugita, H. Kobayashi, S. Sugiura and T. Kanamori, Lab Chip, 2016, 16, 2339–2348 RSC
- S. Sugiura, J. Edahiro, K. Kikuchi, K. Sumaru and T. Kanamori, Biotechnol. Bioeng., 2008, 100, 1156–1165 CrossRef CAS PubMed
- S. Sugiura, K. Hattori and T. Kanamori, Anal. Chem., 2010, 82, 8278–8282 CrossRef CAS PubMed
- K. Hattori, S. Sugiura, T. Kanamori and J. Lab, Automation, 2013, 18, 437–445 CrossRef CAS PubMed
- Y. N. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 551–575 CrossRef
- C. M. Walko and C. Lindley, Clin. Ther., 2005, 27, 23–44 CrossRef CAS PubMed
- T. Komatsu, H. Yamazaki, N. Shimada, M. Nakajima and T. Yokoi, Drug Metab. Dispos., 2000, 28, 1457–1463 CAS
- M. Kobayakawa and Y. Kojima, OncoTargets Ther., 2011, 4, 193–201 CrossRef CAS PubMed
- S. Yamashita, Y. Tanaka, Y. Endoh, Y. Taki, T. Sakane, T. Nadai and H. Sezaki, Pharm. Res., 1997, 14, 486–491 CrossRef CAS
- J. Y. Kim, D. A. Fluri, J. M. Kelm, A. Hierlemann, O. Frey and J. Lab, Automation, 2014, 20, 274–282 CrossRef PubMed
-
F. M. White, Viscous Fluid Flow, McGraw-Hill Companies, Inc, Boston, 2006 Search PubMed
- N. Chirstophidis, F. J. Vajda, I. Lucas, O. Drummer, W. J. Moon and W. J. Louis, Clin. Pharmacokinet., 1978, 3, 330–336 CrossRef CAS PubMed
- K. Shirao,
et al.
, J. Clin. Oncol., 2004, 22, 3466–3474 CrossRef CAS PubMed
- B. Reigner,
et al.
, Cancer Chemother. Pharmacol., 1999, 43, 309–315 CrossRef CAS PubMed
- C. Moraes, J. M. Labuz, B. M. Leung, M. Inoue, T.-H. Chun and S. Takayama, Integr. Biol., 2013, 5, 1149–1161 RSC
- J. P. Wikswo, E. L. Curtis, Z. E. Eagleton, B. C. Evans, A. Kole, L. H. Hofmeister and W. J. Matloff, Lab Chip, 2013, 13, 3496–3511 RSC
- H. E. Abaci and M. L. Shuler, Integr. Biol., 2015, 7, 383–391 RSC
- C. Maass, C. L. Stokes, L. G. Griffith and M. Cirit, Integr. Biol., 2017, 9, 290–302 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: The layout of the pneumatic pressure lines for the pneumatic pressure-driven multi-throughput multi-organ-on-a-plate system (Fig. S1), membrane inserts for barrier-type organs (Fig. S2), the schematic of the structure and function of a “Laplace valve” (Fig. S3), metabolic pathways of capecitabine and tegafur (Fig. S4), composite images from overhead views of culture wells for the characterization of the mixing process (Fig. S5), enlarged images of Fig. 4b and 5b (Fig. S6 and S7), degradation and absorption of drugs on PDMS microfluidic plates (Fig. S8), and a list of primers used in the RT-PCR (Table S1) are available. See DOI: 10.1039/c7lc00952f |
| ‡ These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2018 |