
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Minimising conformational bias in fluoroprolines through vicinal difluorination†‡
Gert-Jan
Hofman
 ab,
Emile
Ottoy
b,
Mark E.
Light
a,
Bruno
Kieffer
c,
Ilya
Kuprov
a,
Jose C.
Martins
b,
Davy
Sinnaeve
*b and
Bruno
Linclau
*a
ab,
Emile
Ottoy
b,
Mark E.
Light
a,
Bruno
Kieffer
c,
Ilya
Kuprov
a,
Jose C.
Martins
b,
Davy
Sinnaeve
*b and
Bruno
Linclau
*a
aChemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: bruno.linclau@soton.ac.uk
bDepartment of Organic and Macromolecular Chemistry, Ghent University, Campus Sterre, S4, Krijgslaan 281, Ghent B-9000, Belgium. E-mail: Davy.Sinnaeve@UGent.be
cIGBMC – CNRS UMR 7104 – Inserm U 1258, 1 rue Laurent Fries, BP 10142, 67404 Illkirch, CEDEX, France
First published on 16th April 2018
Abstract
Monofluorination at the proline 4-position results in conformational effects, which is exploited for a range of applications. However, this conformational distortion is a hindrance when the natural proline conformation is important. Here we introduce (3S,4R)-3,4-difluoroproline, in which the individual fluorine atoms instil opposite conformational effects, as a suitable probe for fluorine NMR studies.
Proline is the only proteinogenic amino acid with a secondary amino group, resulting in the cis-peptide bond (Xaa–Pro) being significantly populated (Fig. 1a).1 The proline amino group is part of a pyrrolidine ring, and its five-membered ring pucker is thus closely connected with the backbone ϕ-dihedral angle. In addition, its cyclic nature inherently restricts this dihedral angle such that it enhances the importance of n → π* interactions between subsequent carbonyl groups in peptides. This in turn has further implications for peptide conformation and influences the Xaa–Pro cis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans ratio.1,2 These peculiar chemical features result in specific conformational and dynamical properties that are central to a number of biological mechanisms behind protein folding, protein aggregation or protein–protein interactions.3 The existence of peptidyl-prolyl cis–trans isomerases, a class of enzymes able to accelerate proline cis–trans isomerization, highlights the functional importance of this dynamical property in biology.4 Furthermore, post-translational modifications of the pyrrolidine ring by hydroxylation confer mechanical properties to proline-rich proteins such as collagen by further enhancing these n → π* interactions.3c Incorporation of a fluorine atom at the proline 4- (or γ-) position strongly affects both its dynamical and conformational properties. Because of the highly polar C–F bond, a destabilisation of the planar charged amide resonance structures results, which manifests itself in an increased amide isomerisation rate. Ring pucker is affected through the gauche effect, which is a favourable σC–H → σ*C–F hyperconjugation interaction.5 This stereoelectronic effect requires the C–H and C–F bonds to be antiperiplanar, and the stereogenicity of the fluorine substituent thus leads to one of the two puckers being favoured (Fig. 1a).1 Furthermore, C–F introduction affects the overall dipole moment, which also influences conformational stabilities (with a strong solvent effect).6
:trans ratio.1,2 These peculiar chemical features result in specific conformational and dynamical properties that are central to a number of biological mechanisms behind protein folding, protein aggregation or protein–protein interactions.3 The existence of peptidyl-prolyl cis–trans isomerases, a class of enzymes able to accelerate proline cis–trans isomerization, highlights the functional importance of this dynamical property in biology.4 Furthermore, post-translational modifications of the pyrrolidine ring by hydroxylation confer mechanical properties to proline-rich proteins such as collagen by further enhancing these n → π* interactions.3c Incorporation of a fluorine atom at the proline 4- (or γ-) position strongly affects both its dynamical and conformational properties. Because of the highly polar C–F bond, a destabilisation of the planar charged amide resonance structures results, which manifests itself in an increased amide isomerisation rate. Ring pucker is affected through the gauche effect, which is a favourable σC–H → σ*C–F hyperconjugation interaction.5 This stereoelectronic effect requires the C–H and C–F bonds to be antiperiplanar, and the stereogenicity of the fluorine substituent thus leads to one of the two puckers being favoured (Fig. 1a).1 Furthermore, C–F introduction affects the overall dipole moment, which also influences conformational stabilities (with a strong solvent effect).6
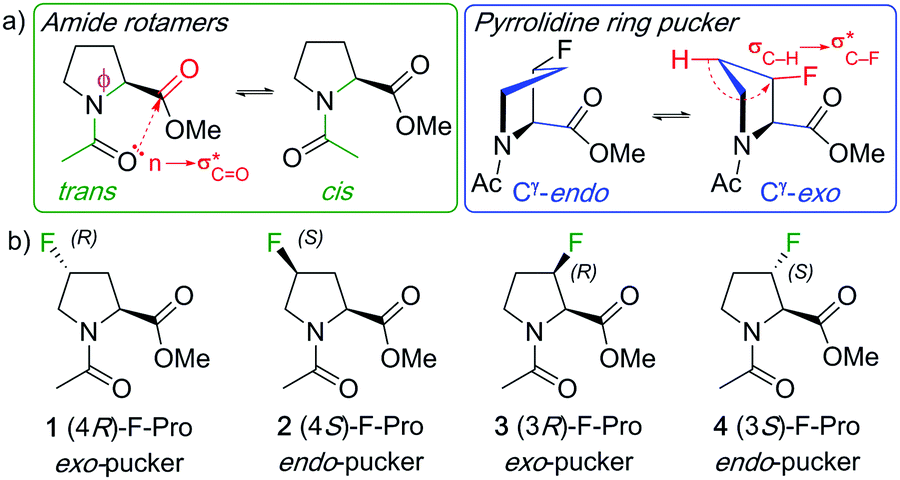 | ||
| Fig. 1 Illustration of amide rotamer and pyrrolidine pucker structures (a), and typical monofluorinated L-proline models with their conformational bias (b). | ||
N-Acylated proline esters such as 1–4 (Fig. 1b) are typical models to investigate the influence of fluorination on proline conformation.7 In a landmark study, Raines and Markley demonstrated, through NMR studies in 1,4-dioxane, that the exo-pucker is dominant in the (4R)-fluoroproline derivative 1 (75% population for the trans-isomer), while the endo-pucker is the most populated one for the 4S-isomer 2.7 Originally investigated for its effect on collagen stability,3c proline fluorination is now applied for a variety of purposes in the biosciences including stability and activity studies of peptides and proteins, protein engineering,8 as well as in medicinal chemistry applications.1,9
Fluorinated amino acids are of interest for biological 19F NMR applications, due to its high intrinsic sensitivity, its increased sensitivity to chemical exchange processes due to the broad chemical shift range, and the absence of background signals in biological fluids.10,11 Also, 19F labelling of single residues in a peptide or protein can afford easy access to site-specific information. Given the pivotal role of prolines in protein folding, stability and binding, and the biological importance of interactions with proline-rich motifs in signalling proteins,3a prolines would be a judicious choice to introduce a 19F reporter. Yet, while fluoroprolines have been a much-used tool to study peptide/protein structure and dynamics, or interactions between peptides and receptors,3b,9b,12 their potential use as 19F NMR probes has, to the best of our knowledge, never been exploited.
In many cases, the conformational bias caused by the common singly fluorinated prolines is a hindrance for biological 19F NMR purposes, as it distorts the natural proline pucker conformation and/or amide cis–trans equilibrium and thus the properties of the system under study. Instead, for this purpose, fluorinated analogues in which two fluorines are introduced in such positions as to offset each other's conformational bias would be of more interest. For example, by combining the 4S and 4R-fluoro motif to give 4,4-difluoroproline derivatives (e.g.5, Fig. 2), their respective stereoelectronic effects influencing pucker are expected to cancel out.1,13 While a detailed conformational analysis of 4,4-difluoroproline is yet to be published,1 initial analysis indicated that the exo- and endo-puckers of 4,4-difluoroproline are of similar energy, and that its preorganisational capacity is close to that of proline.14 However, a CF2-group in which the two fluorine atoms are diastereotopic is not an ideal 19F NMR reporter group because of the very large geminal F–F coupling (>200 Hz). This leads to severe J-modulation distortions in any NMR experiment involving, for instance, spin echoes, which is a key problem. Furthermore, since the fluorines typically have relatively close chemical shifts, strong second order effects are present, which complicate spectral interpretation. In addition, when introducing multiple fluoroproline probes, especially in similar environments (i.e. low-complexity peptide sequences such as polyprolines), the availability of a larger set of fluoroproline analogues with minimal conformational bias is desirable to prevent spectral overlap.
 | ||
| Fig. 2 Proposed difluorinated L-prolines featuring compensating conformational bias towards proline pucker. | ||
We wished to investigate the extent of compensating conformational bias in the proline ring by vicinal fluorine introduction at different ring carbons. For this, proline positions 3 and 4 are the most practical. Such analogues feature vicinal instead of geminal 19F–19F coupling, with 3JF–F ≪ 2JF–F. In addition, CHF groups have very different 19F chemical shift values than CF2 groups. This leads to 6 and 7 as analogues of interest (Fig. 2). There is some precedent for these structures: syntheses of the (3R,4S)-difluorinated proline motif,15 as in 6, and of the (3R,4R)-difluoromotif16 (not shown) have been reported. Unfortunately, many synthetic steps were required to convert 3,4-dehydroproline to Cbz-protected (3R,4S)-3,4-difluoro-L-proline16 and 3-deoxy-3-fluoro-1,2,5,6-di-O-isopropylidene-α-D-glucofuranose to N-benzyl protected (3R,4R)-3,4-difluoro-L-proline.16
Here we report a short synthesis of 7 from (4R)-hydroxypro-line, as well as NMR studies (amide cis:trans ratio and amide isomerisation rates) and preliminary theoretical calculations (ring pucker). The results are compared to the equivalent data of the 4,4-difluorinated derivative 5.14 Given 5 can be regarded as a combination of the 4R- and 4S-fluoroprolines 1 and 2, and the 3S,4R-difluorinated derivative as a combination of the 3R- and 4S-fluoroprolines 2 and 3, the corresponding conformational data of 1–3, all of which were synthesised using described methodology,17 were also obtained.
The synthesis of 7 is shown in Scheme 1, and employs a direct deoxyfluorination approach from the known18 3,4-diol, which was obtained from a suitably protected 3,4-dehydroproline. Marson has shown that bis-triflation of N-alkylated trans-3,4-dihydroxypyrrolidines, followed by TBAF treatment led to trans-3,4-difluoropyrrolidines with inversion of configuration in excellent yields.19 Although the methyl ester and N-acetyl groups present in 7 could have been introduced from the start, benzyl ester and Boc amine protection was chosen in order to make the process relevant for the synthesis of suitable peptide synthesis building blocks. Given 3,4-dehydroproline is expensive, cheap (4R)-hydroxyproline was employed, which was protected to give known208 (not shown). Elimination of 4-hydroxyproline's hydroxyl group is typically achieved via a two-step procedure involving alcohol activation, leading to a mixture of 3,4- and 4,5-alkene isomers.18a,b,21,22 Pleasingly, it was found that the one-pot Grieco elimination procedure23 starting from 8 gave the desired alkene 9 in excellent (10:1) selectivity. Dihydroxylation of 3,4-dehydroproline with OsO4 has been reported to form the all-cis-diol isomer in small quantities.18 However, with potassium osmate (0.3 mol%), the trans-diol obtained is as the only observable isomer.22b Direct conversion of the 3,4-diol to the required 3,4-difluoro motif was best achieved with nonafluorobutanesulfonyl fluoride (NFF) in combination with a triphenyldifluorosilicate salt,24 which led to 11 as the only 3,4-difluorinated stereomer in 24% yield. The enol sulfonate 12 was isolated in equal amounts, presumably through fluoride mediated E2 elimination of the corresponding bis-nonaflate intermediate. Only one regioisomer was isolated, which suggests that the bis-nonaflate intermediate preferentially adopted a Cγ-exo conformation, resulting in antiperiplanar disposition between the C3–H and C4–ONf bonds allowing for a smooth E2 reaction. Given this process essentially involves two separate deoxofluorination reactions, the obtained yield was deemed acceptable, as gram-scale quantities of 11 could be obtained. Finally, the protecting groups were replaced to give the 3,4-difluorinated model 7.
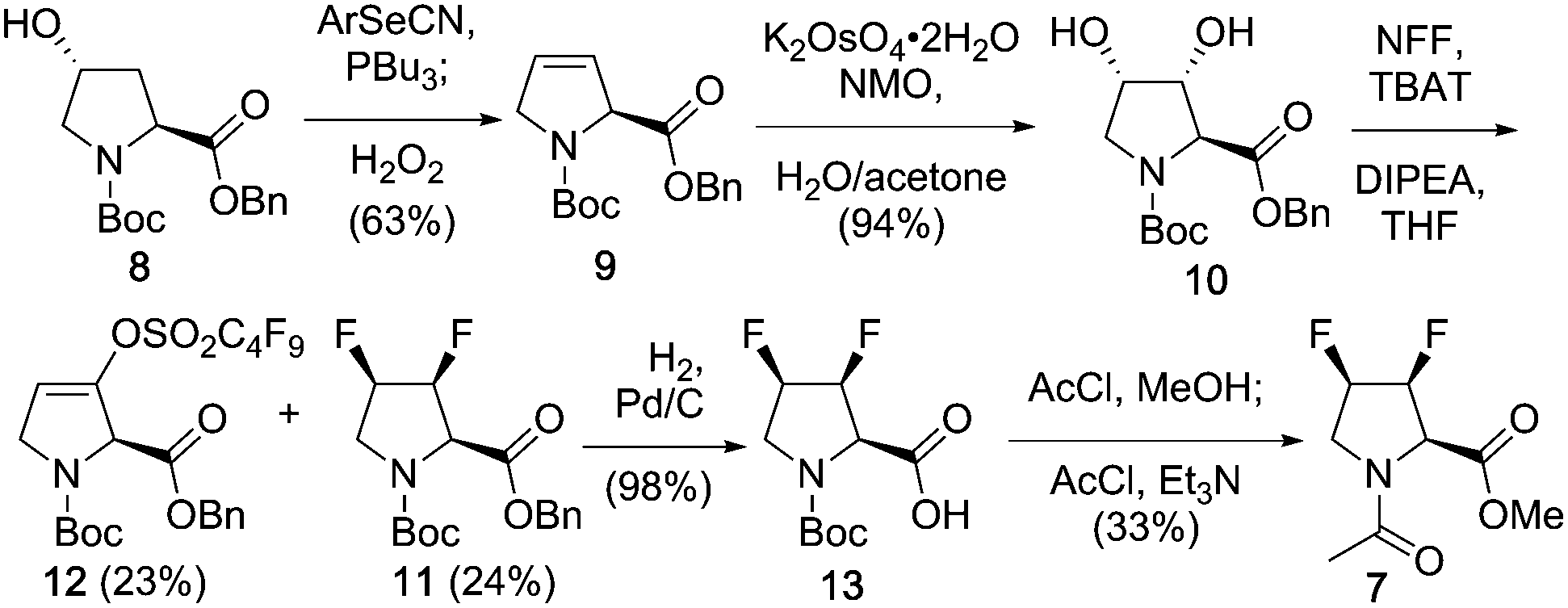 | ||
| Scheme 1 Synthesis of (3S,4R)-F2-Pro. | ||
Unambiguous assignment of the relative stereochemistry proving inversion of configuration in the fluorination step could be obtained by X-ray crystallographic analysis of 11 (Fig. 3), which crystallised in a perfect Cγ-exo pucker.§
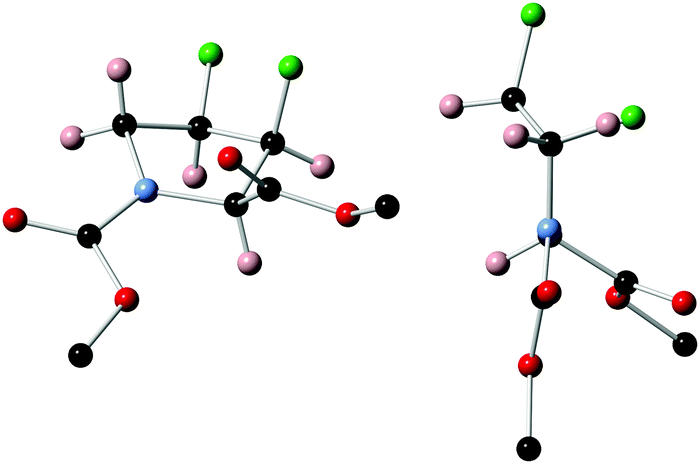 | ||
| Fig. 3 X-ray structure of 11 (Bn and tert-Bu group removed for clarity). | ||
Despite the expected acceleration of the cis–trans isomerisation rate, the exchange remains slow on the NMR time scale and both amide rotamers of 5 and 7 are always separately visible in the 1H and 19F NMR spectra. The difference in 19F chemical shift resonances of the cis rotamers is larger than that of the trans rotamers, both for 5 and 7 (Table 1). As expected the JF–F values for 5 are much larger than these of 7; the close chemical shifts of the fluorine atoms of trans-5 result in a roofed set of doublets (upon 1H decoupling).
| 5 | 7 | ||||||
|---|---|---|---|---|---|---|---|
| CDCl3 | D2O | CDCl3 | D2O | ||||
| δ (ppm) | cis | pro-S | −96.9 | −95.5 | F4 | −202.6 | −200.3 |
| pro-R | −101.6 | −104.7 | F3 | −207.2 | −208.5 | ||
| trans | pro-S | −98.4 | −98.4 | F4 | −206.7 | −203.3 | |
| pro-R | −99.3 | −102.2 | F3 | −208.9 | −210.4 | ||
| J F–F (Hz) | cis | 234.8 ± 0.1 | 235.8 ± 0.1 | 5.2 ± 0.1 | 5.7 ± 0.1 | ||
| trans | 234.1 ± 0.1 | 234.8 ± 0.1 | 4.2 ± 0.1 | 4.7 ± 0.1 | |||
Next, the conformational properties of 5 and 7 were determined, and compared with the non-fluorinated N-acetylated proline methyl ester, the 4,4-difluorinated derivative 5, and the monofluorinated prolines 1–3 (Table 2). The cis:trans ratio was measured with NMR for all compounds in CHCl3 and D2O. The ratios of both 5 and 7 in chloroform and water are quite similar to those of Ac-Pro-OMe, clearly cancelling out the marked biases seen in their respective monofluorinated progenitors. The trans-isomer appears in both chloroform and water slightly more favoured in 7 than in 5. Next, the kinetic rate constants of cis–trans isomerisation kcis–trans and ktrans–cis were determined in D2O by 2D 1H–1H or 19F–19F EXSY for 3, 5 and 7, while for Ac-Pro-OMe, 1 and 2, values were calculated based on results reported by Renner et al.13
|
trans:cis, 25 °C |
cis–trans kinetics (D2O), 35 °C (s−1) |
endo:exoj |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| CDCl3 | D2O | k cis–trans | k trans–cis | k ex | CHCl3 | H2O | |||
| trans | cis | trans | cis | ||||||
| a CIP prioritization changes with introduction of the second fluorine atom, so 5 must be compared with 1 and 2, and 7 with 2 and 3. b In good agreement with reported ratios by Siebler et al.6 c In good agreement with reported ratios by Shoulders et al.14 d In good agreement with reported ratios by Kim et al.25 e k ex is defined as kex = kcis–trans + ktrans–cis. f Calculated value based on Renner et al.13 g Experimental NMR value obtained using similar procedure as Renner et al.13 h Corresponding calculated values based on Renner et al. at 35 °C: 0.155 s−1 and 0.049 s−1.13 i Corresponding values reported by Thomas et al. at 37 °C using an alternative experimental procedure: 0.229 s−1 and 0.028 s−1.17c j DFT values, using the M06 functional with cc-pVDZ basis set and CHCl3 or water implicit solvent models. | |||||||||
| Ac-Pro-OMe | 79:21b |
82:18b |
0.031f | 0.007f | 0.038 | 81:19 |
90:10 |
66:34 |
83:17 |
| 5 (4,4) | 75:25 |
78:22c |
0.114 ± 0.006g,h | 0.034 ± 0.002g,h | 0.148 ± 0.008 | 64:36 |
93:7 |
77:23 |
89:11 |
| 7 (3S,4R)a | 79:21 |
83:17 |
0.119 ± 0.009g | 0.025 ± 0.002g | 0.144 ± 0.011 | 41:59 |
78:22 |
56:44 |
90:10 |
| 1 (4R) | 81:19b |
87:13b |
0.064f | 0.010f | 0.074 | 11:89 |
28:72 |
7:93 |
17:83 |
| 2 (4S) | 62:38b |
71:29b |
0.037f | 0.015f | 0.052 | 97:3 |
99:1 |
99:1 |
99.5:0.5 |
| 3 (3R) | 84:16 |
89:11d |
0.141 ± 0.021g,i | 0.019 ± 0.003g,i | 0.159 ± 0.024 | 24:76 |
16:84 |
15:85 |
44:56 |
Since kcis–trans and ktrans–cis depend on the cis:trans ratios, we define their sum kex in order to compare isomerization kinetics between the different compounds. Clearly, every fluorinated compound shows accelerated isomerization kinetics compared to proline. When comparing the monofluorinated compounds, there are marked differences depending on the substitution patterns, even between 1 and 2, showing that inductive effects alone do not explain the change in rate constant. Both doubly fluorinated compounds 5 and 7 show a further increase in isomerization kinetics, and turn out to have very similar rate constants. Interestingly, while 5 shows faster kinetics than its monofluorinated progenitors 1 and 2, 7 unexpectedly has a slightly lower kex value than 3 (though markedly higher than 2).
Finally, the preference of the five-membered ring pucker was assessed by DFT, using the M06 functional with cc-pVDZ basis set and chloroform or water as implicit solvents (Table 2). It is clear that the monofluorinated compounds 1, 2 and 3 alter the pucker preference profoundly, while doubly fluorinated 5 and 7 display pucker ratios much more similar to the Pro model compound. In chloroform, 7 deviates more from Pro compared to 5, with higher preference for the exo-pucker. In water, the limited deviations to Pro for both 5 and 7 are similar and in opposite sense compared to the trans form.
In conclusion, we find that 3S,4R-difluoroproline, with a vicinal cis-difluoromotif, is a proline analogue featuring minimal conformational bias with respect to the cis–trans rotamers and ring pucker thanks to the offsetting stereoelectronic effects instilled by the individual fluorine atoms. While the 4,4-difluoroproline variant also displays such characteristics, the 3,4-difluoroproline is superior as a fluorinated proline marker for 19F spectroscopic studies of natural proline containing peptides and proteins due to the much more suitable NMR properties. Furthermore, we suggest that the very different chemical shifts of the 3,4- and 4,4-difluoroprolines offers opportunities for multi-proline labelling in peptides. The short, gram-scale synthesis assures convenient access to the 3S,4R-difluoro isomer, and the improvements for the hydroxyproline elimination and 3,4-dehydroproline dihydroxylation reactions, will be useful for synthetic proline chemistry. The 3S,4R-difluoroproline analogue is a valuable addition for applications of fluorinated prolines in medicinal chemistry and structural biology. Applications of these prolines in 19F NMR structural studies are in progress.
We thank the University of Southampton for funding. The Research Foundation – Flanders (FWO) is indebted for a research project to J. C. M. and D. S. (3G011015), PhD and postdoctoral fellowships to E. O. and D. S., and staff exchange funding (FWO-WOG Multimar). The EPSRC is thanked for a partial PhD grant to G.-J. H. (EPSRC-DTG EP/M50662X/1) and instrument funding (core capability EP/K039466/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. W. Newberry and R. T. Raines, Top. Heterocycl. Chem., 2017, 48, 1 Search PubMed
.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838 CrossRef CAS PubMed
-
(a) B. K. Kay, M. P. Williamson and P. Sudol, FASEB J., 2000, 14, 231 CrossRef CAS PubMed
- S. F. Gothel and M. A. Marahiel, Cell. Mol. Life Sci., 1999, 55, 423 CrossRef CAS PubMed
- C. Thiehoff, Y. P. Rey and R. Gilmour, Isr. J. Chem., 2016, 57, 92 CrossRef
- C. Siebler, B. Maryasin, M. Kuemin, R. S. Erdmann, C. Rigling, C. Grünenfelder, C. Ochsenfeld and H. Wennemers, Chem. Sci., 2015, 6, 6725 RSC
- M. L. DeRider, S. J. Wilkens, M. J. Waddell, L. E. Bretscher, F. Weinhold, R. T. Raines and J. L. Markley, J. Am. Chem. Soc., 2002, 124, 2497 CrossRef CAS PubMed
- C. Odar, M. Winkler and B. Wiltschi, Biotechnol. J., 2015, 10, 427 CrossRef CAS PubMed
-
(a) C. Held, H. Hubner, R. Kling, Y. A. Nagel, H. Wennemers and P. Gmeiner, ChemMedChem, 2013, 8, 772 CrossRef CAS PubMed
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132 CrossRef CAS
-
(a) H. Chen, S. Viel, F. Ziarelli and L. Peng, Chem. Soc. Rev., 2013, 42, 7971 RSC
-
(a) T. Steiner, P. Hess, J. H. Bae, B. Wiltschi, L. Moroder and N. Budisa, PLoS One, 2008, 3, e1680 Search PubMed
- C. Renner, S. Alefelder, J. H. Bae, N. Budisa, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2001, 40, 923 CrossRef CAS PubMed
- M. D. Shoulders, K. J. Kamer and R. T. Raines, Bioorg. Med. Chem. Lett., 2009, 19, 3859 CrossRef CAS PubMed
-
U. Hommel, E. L. J. Lorthiois, J. K. Maibaum, N. Ostermann, S. A. Randl and A. Vulpetti, Novartis Pat., WO2014/002057, 2014 Search PubMed
- Z. Liu, S. F. Jenkinson, T. Vermaas, I. Adachi, M. R. Wormald, Y. Hata, Y. Kurashima, A. Kaji, C.-Y. Yu, A. Kato and G. W. J. Fleet, J. Org. Chem., 2015, 80, 4244 CrossRef CAS PubMed
-
(a) N. Panasik, E. S. Eberhardt, A. S. Edison, D. R. Powell and R. T. Raines, Int. J. Pept. Protein Res., 1994, 44, 262 CrossRef CAS PubMed
-
(a) J. K. Robinson, V. Lee, T. D. W. Claridge, J. E. Baldwin and C. J. Schofield, Tetrahedron, 1998, 54, 981 CrossRef CAS
- C. M. Marson and R. C. Melling, Chem. Commun., 1998, 1223 RSC
- T. R. Webb and C. Eigenbrot, J. Org. Chem., 1991, 56, 3009 CrossRef CAS
- H. Bernard, G. Bulow, U. E. W. Lange, H. Mack, T. Pfeiffer, B. Schafer, W. Seitz and T. Zierke, Synthesis, 2004, 2367 CAS
-
(a) H. Rueger and M. H. Benn, Can. J. Chem., 1982, 60, 2918 CrossRef
- P. A. Grieco, S. Gilman and M. Nishizawa, J. Org. Chem., 1976, 41, 1485 CrossRef CAS
- X. Q. Zhao, W. P. Zhuang, D. S. Fang, X. W. Xue and J. M. Zhou, Synlett, 2009, 779 CrossRef CAS
- W. Kim, K. I. Hardcastle and V. P. Conticello, Angew. Chem., Int. Ed., 2006, 45, 8141 CrossRef CAS PubMed
Footnotes |
| † Raw NMR data files are available. Please see DOI: 10.5258/SOTON/D0475 |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and characterisation data, copies of spectra of all compounds, determination of kinetic and thermodynamic parameters. CCDC 1531555. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc01493k |
| § Crystal data C17H21F2NO4, Mr = 341.35, orthorhombic, Pna21 (No. 33), a = 9.3962(3) Å, b = 10.8390(3) Å, c = 33.4374(9) Å, α = β = γ = 90°, V = 3405.45(17) Å3, T = 100(2) K, Z = 8, Z′ = 2, μ(MoKα) = 0.108, 24677 reflections measured, 8447 unique (Rint = 0.0563) which were used in all calculations. The final wR2 was 0.1201 (all data) and R1 was 0.0703 (I > 2(I)). |
| This journal is © The Royal Society of Chemistry 2018 |